
Modern Genome Editing Technologies in Huntington’s Disease Research
Abstract
The development of new revolutionary technologies for directed gene editing has made it possible to thoroughly model and study NgAgo human diseases at the cellular and molecular levels. Gene editing tools like ZFN, TALEN, CRISPR-based systems, NgAgo and SGN can introduce different modifications. In gene sequences and regulate gene expression in different types of cells including induced pluripotent stem cells (iPSCs). These tools can be successfully used for Huntington’s disease (HD) modeling, for example, to generate isogenic cell lines bearing different numbers of CAG repeats or to correct the mutation causing the disease. This review presents common genome editing technologies and summarizes the progress made in using them in HD and other hereditary diseases. Furthermore, we will discuss prospects and limitations of genome editing in understanding HD pathology.
INTRODUCTION
Huntington’s disease (HD) is a severe neurodegenerative disorder caused by the autosomal dominant mutation in the first exon of the HTT gene encoding huntingtin protein. HD patients usually carry more than 36 CAG repeats in HTT as compared to 16–20 repeats in healthy individuals. Mutant HTT contains an expanded polyglutamine stretch that leads to protein misfolding and formation of aggregates. These aggregates potentially affect a wide range of molecular and cellular processes resulting in selective death of striatal neurons and cortical neurons [1]. The main HD symptoms are movement disorders, cognitive impairment, and psychiatric disturbances. These problems develop in middle age (35–45 years) in the majority of cases. Additionally, there is a correlation between age at onset and repeat number. A higher number of CAG repeats lead to an earlier manifestation and more severe forms of the disease [2].
Although the mutation was identified more than 20 years ago, disease pathways are not fully elucidated and only symptomatic treatment is available. One reason for the slow progress was the lack of an efficient tool to study the effects of mutant huntingtin on cellular and molecular processes and the role of other genes in the progression of the disease.
Transcriptome analysis, bioinformatic assays and RNAi are usually used to explore the mechanisms of disease development. Nevertheless such methods can’t fully reveal all connections and interactions between genes, proteins and cellular organelles during disease progression. Modern gene editing approaches such as ZFN (zinc finger nuclease) [3], TALEN (transcription activator-like effector nuclease) [4], CRISPR-based systems (clustered regularly interspaced short palindromic repeats [5–7], NgAgo-gDNA (DNA-guided endonuclease Argonaute Natronobacterium gregoryi) [8] and SGN (structure-guided nuclease) [9] uncover new opportunities for scientific research due to the ability of directly changing DNA sequences and then altering the expression of target genes. Firstly, these tools enable the introduction of expanded CAG tracts in normal cells or correction of the mutation in patient-derived cells and thus to create isogenic cell lines with identical genetic background. Isogenic cell lines provide the ideal platform for study of disease mechanisms and cell-based high-throughput compound screening [10, 11]. Secondly, molecular tools allow researchers to perform multiplex analysis and functional screening of genes involved in neurodegenerative processes. Finally, cells with corrected HTT are a promising source for autologous cell therapy, and mutant allele-specific gene editing is a promising tool for gene therapy of HD in vivo.
Here, we review modern molecular tools for gene editing, recent results and future directions of using them in HD studies.
GENOME EDITING TOOLS
Gene editing tools have been widely used in recent years. The use of these tools has facilitated knockout or knock-in of target loci, and thus, to study the function of genes and gene networks and regulatory sequences, to create models of hereditary diseases, and to develop methods for their treatment. These tools have various structures which mediate functional parameters such as specificity, efficiency of delivery and gene editing, sequence limitations, assembly cost, etc (Fig. 1). This section briefly describes the main components of such tools, as well as the advantages and disadvantages to edit genes.
ZFN
ZFN was constructed by Kim and colleagues in 1996 as a chimeric nuclease consisting of two parts [3]. The first part is a DNA-binding box consisting of 3-4 zinc finger domains (ZFPs). Each ZFP binds to 3 nucleotides so a set of 3-4 ZFPs in each subunit provides sufficient specificity for site-specific DNA binding. The second part is the nuclease domain of endonuclease FokI. Nuclease domains are identical and can introduce double-strand breaks (DSBs) after the dimerization of two closely located ZFN subunits [12] (Fig. 2). This tool became the basis for gene editing in cultured human cells including pluripotent stem cells [13], animals and plant cells [14, 15]. However, ZFNs have some disadvantages. There is the complexity and high cost of DNA-binding domain construction to target the DNA site and a high off-target probability due to single-nucleotide substitutions or incorrect interaction of domains (Fig. 1).
TALEN
Transcription activator-like effector nuclease (TALEN) has an artificial DNA-binding domain and DNA-cleavage domain endonuclease FokI like ZFN. DNA-binding domains consist of monomers containing tandem repeats of 34 amino acids and two of them (on 12th and 13th positions) are high variable (repeat variable diresidue, RVD) and responsible for nucleotide recognition. There are RVDs such as Asn and Ile (NI), Asn and Gly (NG), two Asn (NN), His and Asp (HD) which bind to A, T, G, C nucleotides respectively in the target DNA site. The first amino acid is needed to stabilize spatial conformation and the second amino acid interacts with a specific nucleotide. DNA binding domains of two subunits are associated to opposite DNA chains at a distance of 12-24bp (spacer sequence) andC-terminal FokI domains dimerize and cleave the target DNA site (Fig. 2) [16]. Although this tool demonstrates higher efficiency and specificity than ZFN, TALEN requires T at the 5’ end of the target that limits options of target sequences (Fig. 1).
CRISPR-based systems
Clustered regularly interspaced short palindromic repeats/ CRISPR associated protein 9 (CRISPR/Cas9) was firstly reported in 2012 [17]. Originally it is a bacterial analog of the immune system against phage infection but researchers adapted it for directed modification of eukaryotic genomes. All CRISPR/Cas systems are divided into three main types (I-III), and at least 10 subtypes. In genome engineering II-A type system is currently the most widely used from Streptococcus pyogenes (SpCas9) [18, 19]. CRISPR/Cas9 consists of two elements as well as ZFN and TALEN. However unlike TALEN and ZFN, CRISPR/Cas9 functions as a monomer. There is a DNA-binding single-guide RNA (sgRNA), which is complementary to the target site (18–20bp) in the genome and Cas9 nuclease that introduces DSB into the target. To cleave DNA, SpCas9 requires not only RNA-DNA complex but also the presence of a special PAM sequence (5’-NGG-3’) in the 3’-end of the target (Fig. 2) that can vary for Cas9 from other bacteria, for example, Cas9 from Streptococcus thermophilus and Neisseria meningitides require 5’-NGGNG-3’ and 5’-NNNNGATT-3’ PAMs respectively [20]. Due to design simplicity and low cost, CRISPR/Cas9 has become an indispensable tool for genetic manipulations. The system can efficiently and specifically target any site in the genome, and has been widely used in plants and animals in vitro and in vivo and in human cells [21–23] and has enabled generation of human iPSC-based genetic disease models [24–26].
CRISPR/Cas9 allows genome editing with high efficiency but can make unwanted modifications at off-target sites [27]. The off-target effects may cause genomic instability and disrupt the functionality of otherwise normal genes, and this reason restricts application of CRISPR/Cas9 in biomedicine and the clinic [28]. In January 2016, it was reported that a new modified Cas9 nuclease had been created [6]. Specificity of Cas9 was improved by substitutions in Cas9 sequence which blocked some hydrogen bonds between Cas9 and target DNA strand. Such modifications altered energetics of the Cas9-sgRNA complex and thus reduced off-target binding and subsequently off-target effects that were confirmed by next-generation sequencing.
Another direction to increase the specificity of CRISPR system is the application of Cas9 orthologs, which require other PAM-sequences and have higher cleavage specificity and smaller size of nuclease encoding genes. Such orthologs were found in Neisseria meningitides (NM-Cas9), Streptococcus thermophilus (ST1-Cas9) [29], and Streptococcus aureus (SaCas9) [30]. A CRISPR-based tool with high specificity is CRISPR-Cpf1 of Francisella novicida U112 [7]. Cpf1 is an endonuclease, which participates in a bacterial immune system like Cas9. Similar to CRISPR/Cas9, it forms RNA-DNA complexes that cleave a target site. Cpf1 significantly differs from Cas9 in structure and function. Firstly, gRNA of Cpf1 has a simpler structure than gRNA of Cas9 that could simplify the design and delivery of genome-editing tool. Secondly, the nuclease requires 5’-TTN PAM to form RNA-DNA complex and subsequently to cleave target DNA (Fig. 2). Moreover, Cpf1 introduces a staggered DNA DSBs with 4 or 5-nt 5′ overhangs compared to blunt ends generated by Cas9 [31]. Overhangs allow programming the desired sequence and integration of new DNA in proper orientation using NHEJ-based gene insertion avoiding homology-directed recombination (HDR). It is very important for non-dividing cells in which HDR occurs very rarely. It was shown that Cpf1 could allow up to two mismatches in the 3′ PAM-distal region, but not in the 5′ PAM-proximal region when Cas9 admits up to five mismatches in the target sequence. Therefore Cpf1 is more precise than Cas9, and using recombinant Cpf1 ribonucleoproteins entirely abolished off-target effects [7].
NgAgo
The next gene editing tool was developed by Gao and colleagues in 2016 [8]. It is endonuclease Argonaute derived from Natronobacterium gregoryi (NgAgo) that uses 5’-phosphorylated short single-stranded 24nt DNA (ssDNA) as a guide and doesn’t require an additional specific PAM sequence in order to cleave a target site. So this feature provides a wide range of targets while having a low tolerance to off-target sites. Furthermore 5’-phosphorylated short ssDNAs due to a very small number of endogenous 5’-phosphorylated ssDNAs contribute to the high specificity of NgAgo (Fig. 2). Another important point contributing towards specificity is ‘one-guide-faithful’ rule, that is, once complexed with gDNA NgAgo can’t swap the guide. In the first case, such ssDNAs are very rare in mammalian cells, and in the second case NgAgo can be guided by once loaded gDNA. Finally, ssDNAs are easily synthesized and a researcher can choose the required amount for the experiment.
SGN
Also in 2016 a promising structure-guided nuclease (SGN) was engineered consisting of flap endonuclease-1 (FEN-1) that recognizes DNA-DNA 3’-flap structures and nuclease domain FokI [9]. FEN-1 is involved in replication and DNA repair catalyzing deletion of 3’ unpaired flap by phosphodiester. This 3’-flap forms due to competing between newly synthesized DNA and displaced region. Actually, it was hypothesized that FEN-1 guided by >20nt ssDNA forming an unpaired 3’-flap with target site in common with cleavage domain FokI can be an efficient tool for gene modifications. SGN functions as a dimer like ZFN and TALEN but without binding second subunit to opposite DNA strand (one FokI bound to the target site and recruits another FokI molecule) and introduces DSB at a distance of 9nt from the flap (Fig. 2). However, SGN has substantial defects such as low efficiency, off-target effects, and cell toxicity due to the fact that SGN can recognize and cleave “endogenous” 3’-flap structures (Fig. 1). Therefore, additional improvements are required to minimize or eliminate these drawbacks.
As NgAgo and SGN are the most recent technologies and only two reports were published further studies are needed to confirm applicability for gene editing in cells.
POSSIBLE APPLICATIONS OF GENOME EDITING TOOLS IN HD STUDIES
Gene editing tools are indispensable components for the generation of targeted DSBs that are repaired through one of two major DNA-repair pathways –nonhomologous end-joining (NHEJ) in the absence of homologous donor template or homology-directed repair (HDR) in the presence of the donor template (plasmids, single stranded oligonucleotides). NHEJ pathway is error-prone and leads to the generation of insertion and/or deletions (indels) in the target DNA sequence. So targeted DSBs repaired by NHEJ can be used to disrupt desired gene sequences and to shift reading frame resulting in the synthesis of nonfunctional protein. Alternatively, in HDR artificial exogenous homologous sequence serves as a template for the recovery of native DNA structure and DSBs can stimulate this process [32]. HDR allows inserting any sequence in the target site, for example, restriction site, reporter gene. Moreover it allows correcting or introducing certain mutations that enable modeling hereditary diseases.
Correction/insertion of mutation causing disease
Initially, cells from healthy individuals were used as a negative control in studying the disease and experimental results obtained in such studies were not fully reliable. Targeted gene editing enables the generation of isogenic cell lines which provide an ideal healthy control for disease modeling, drug-screening and searching for new mutations involved in disease pathogenesis. Additionally, corrected patient-derived cells can be used in cell replacement therapy. A promising cell source for both approaches is iPSCs. On one hand, these cells can be generated by the reprogramming of somatic cells and can be further differentiated into various cell types that enable comprehensive disease study and drug-screening. On the other hand, iPSCs can be obtained during a patient’s life, “corrected” and transplanted in the damaged tissue. The first patient-specific iPSC line of HD containing 72 CAG repeats was obtained in 2008 by Park and colleagues [33] and since this publication, the number of HD patient-derived iPSC lines has been expanded [34].
In an unprecedented study performed by An et al., mutant HTT in induced pluripotent stem cells (iPSCs) derived from an HD patient was corrected by the replacement of the expanded CAG tract with a normal repeat using homologous recombination. Subsequently corrected iPSCs were differentiated into DARPP-32-positive striatal neurons in vitro and in vivo in mice. Correction of HTT normalized signaling pathways dysregulated in HD (TGF-β, cadherin, caspase activation, and BDNF) and reversed disease phenotypes such as susceptibility to cell death and altered mitochondrial bioenergetics in neural stem cells. Thus researchers first generated an isogenic HD model using only homologous recombination [35].
The first successful study using genome editing tool for gene correction was published in 2007 [36]. Researchers used ZFNs and donor sequence to correct a point mutation in hematopoietic stem cells in the IL2RG gene that causes leukemia in X-linked SCID (severe combined immunodeficiency) in patients treated by retroviral therapy. Such an approach could be useful for HTT editing wherein donor template would contain a normal number of CAG repeats (16–20 CAG).
In order to obtain isogenic cell lines from healthy cells, a disease causing mutation may be inserted into the genome of these cells. CRISPR/Cas9 was applied to create a set of isogenic HD cell lines bearing various length CAG tracts (21, 72, 97 CAG) (Fig. 2) [37]. It should be noted that donor plasmids with expanded CAG repeats were generated using PCR-products of mutant HTT from HD patients. Expanded CAG repeats were efficiently introduced in HEK293 cells and iPSCs, and the insertion was confirmed by PCR analysis, Western and Southern blotting.
Another study was reported by Malakhova and colleagues in 2016 [38]. The feature of this study is the donor plasmid bearing 215 CAG repeats which was built by the Golden Gate cloning method [39]. PCR-product from normal HTT with 16 CAG repeats was cloned into plasmid and after a series of restriction/ligation rounds using type IIS restriction enzyme and ligase was prolonged up to 215 CAG repeats. This expanded CAG tract was introduced into the genome of HEK293 cell line using CRISPR/Cas9. Mutant cell clones demonstrated decreased proliferation rate and increased apoptosis. Moreover, in this research group, embryonic fibroblasts with 215 CAG repeats in HTT were generated applying CRISPR/Cas9 and donor template. Subsequently, these cells were reprogrammed to a pluripotent state, and iPSCs showed decreased viability and proliferation rate and HTT aggregation after exposure to proteasome inhibitor (Zakian lab, unpublished data).
One of the most important aspects in genome editing is the structure of target site nucleotide sequence. It is known that the promoter region and the first exon of HTT are GC-rich sequences [40]. In this case, NgAgo has advantages for targeting GC-rich loci over other tools including the CRISPR-system. In fact, RNAs but not DNAs are prone to form secondary structures, which may disturb binding and normal conformation of Cas9-sgRNA. Indeed, Cas9-sgRNA was significantly less efficient than NgAgo-gDNA in cleaving GC-rich loci [8].
Disruption or deletion of mutant allele
Mutant allele inactivation can benefit dominantly inherited disorders, particularly HD. TALEN was used for allele-specific HTT correction in human HD adult fibroblasts by Fink and colleagues in 2016 [41]. They applied special TALEN containing obligate-heterodimeric variant of endonuclease FokI that by necessitating complementary binding of the two TALEN monomers cleaves DNA [42, 43]. Furthermore, such design considers the CAG number required for each monomer binding and spacer sequence for FokI dimerization and DNA cleavage. Thus TALEN can cleave only sequences containing more than 15 CAG repeats. This avoids DSBs in short (normal) CAG tracts. There was a significant decrease in protein aggregation and mutant HTT expression in HD fibroblasts after the transfection of TALEN-encoding vector.
Although such contraction of the mutant allele are appropriate, it was found that DSBs caused both contractions and expansions of CAG tracts when researchers used ZFN and CRISPR/Cas9 targeted to the CAG repeat tract [44]. In contrast, Cas9 D10A mutant (Cas9 nickase) induced single strand breaks within the CAG tract which resulted in mainly repeat contractions due to the activation of an alternative mechanism of DNA repair. Authors proposed that the type of DNA damage plays a key role in determining direction of DNA repair. Thus these results pave the way for shortening expansions in polyglutamine diseases and a possible path for therapy.
Another strategy to inactivate the mutant allele used haplotype-specific CRISPR/Cas9 [45]. The strategy was based on using mutant allele-specific single-nucleotide polymorphisms (SNPs) for PAM of CRISPR/Cas9. Researchers simultaneously used two sgRNAs that depend on PAM sites generated by SNPs in the mutant allele. This resulted in excision of ∼44 kb sequence including promoter region, transcription start site, and expanded CAG tract in the mutant HTT allele without altering the normal allele. The excision prevented the transcription of mutant HTT. The approach applied in this study may have a broad applicability in disorders with diverse disease haplotypes. These cells can be reprogrammed and differentiated into desired cell type and used in one of the previously mentioned ways. Moreover, this study clearly showed how allele-specific SNPs can be used for targeted knockout of the mutant allele and such an approach is applicable for many inherited disorders.
Inactivation of the mutant allele is more attractive due to the need of only gene editing tool delivery. Moreover, it is much easier to introduce DSBs that are repaired by more efficient NHEJ compared to homologous recombination. Genome editing tools based on the CRISPR system and NgAgo are most suitable for this purpose because they have high efficiency. However, it is necessary to take into account the specificity of the tools so careful selection of target site and more accurate molecular tools (high-fidelity CRISPR/Cas9, CRISPR-Cpf1, NgAgo) are required.
Modification of genes involved in disease pathogenesis
HD is a monogenic Mendelian neurodegenerative disorder and genetic risk of HD entirely depends on the length of the expanded repeat. However, it is known that HD patients carrying CAG repeat tracts with equal length have a variable age of disease onset indicating the considerable epistatic effects of genetic background [46]. So identification of modifier genes can reveal or confirm molecular pathways implicated in pathophysiological processes in HD. Moreover, confirmed modifier genes provide new targets for disease therapy.
More than 100 genes are known to be involved in disease progression [47, 48] which were identified using transcriptome, proteome and bioinformatic analyses. There are several potential HD modifiers such as huntingtin-associated protein 1 (HAP1), ATG7 (autophagy-related gene 7), ubiquitin carboxy-terminal hydrolase L1 (UCHL1), PCG-1α (PPARGC1A), adrenosinergic A2A receptor (ADORA2A), neuropeptide Y and its receptor NPY2R (NPY, NPY2R), and many others [49–55]. It was shown that single-nucleotide polymorphisms (SNPs) in these genes may substantially alter HD phenotype. For example, functional assays demonstrated that SNP substituting methionine (M441) for threonine (T441) at amino acid 441 in HAP1 affects the interaction between mutant HTT and HAP1. M441-HAP1 interacts with mutant HTT more tightly than does T441-HAP1, hence decreases soluble HTT degraded products and defends against HTT-mediated toxicity [50]. Minor nucleotide substitution in the transcribed region of PPARGC1A is associated with the delay of age at onset, the minor allele of SNP in the promoter region of the gene results in an earlier onset of HD [53]. Polymorphisms in these genes influence onset age in HD but functional and molecular mechanisms remain unknown.
In order to study how these genes influence HD pathogenesis, gene editing tools can be used in two directions. The first way is the generation of isogenic cell lines that differ from each other by a specific SNP in the gene modifier. These SNPs can be introduced or corrected like expanded CAG repeats. Therefore such isogenic cell lines could provide the platform for studies of the effects of SNPs in gene modifiers on disease pathogenesis and candidates for new drug targets in disease therapy.
The second way is to knockout the modifier gene to show how deletion of the gene protein influences the course of the disease. Any of the molecular tools can be used for this purpose. Special attention should be paid to the CRISPR-system. Due to the simplicity of programming the CRISPR in order to modify almost any genomic locus, this tool can be used to study gene function on a genome-wide scale. Researchers from Cambridge University created a genome-scale library of sgRNAs (64,751 unique sgRNAs) targeting 18,080 genes for genome-scale CRISPR/Cas9 knockout (GeCKO) to identify genes essential for cell viability and drug-resistance in cancer [23]. Indeed, such an approach could easily and efficiently reveal functions of multiple genes and determine the role of each of them in HD in a short time.
In both cases, the most useful is the CRISPR-system. The advantage of this system is that sgRNA can be integrated into the genome using lentiviruses and transcribed when it is needed. However given the problem of off-target effects, a new system for multiplex gene editing based of NgAgo-gDNA may soon be developed. But here the problem of delivering gDNA is worth considering. A possible solution to this problem is the delivery of complexes NgAgo-gDNA into cells.
REGULATION OF GENE EXPRESSION AND EPIGENOME EDITING
Regulation of gene expression by up- or down-regulation is an alternative way of interrogation of gene function. Moreover it can be useful for treatment by repression of mutant protein production and activation of genes combating pathological processes. There are several regulators of gene expression based on genome editing tools. Their DNA-binding domains serve for targeted regulation. Nuclease domains are inactive and replaced by different effectors such as transcription activators and repressors and chromatin modifiers.
Gene expression effectors
ZFPs can be repressors of transcription in the absence of additional effector domains. In 2012, Garriga-Canut and colleagues demonstrated excellent results using ZFPs as mutant HTT repressors by steric hindrance of RNA polymerase complex progression. They constructed multifinger ZF×Hunt containing different numbers of fingers (from 4 to 18) that were complementary to CAG triplet. It was shown that longer ZF×Hunt has a higher affinity for double-strand CAG probes. ZF×Hunt demonstrated 5-fold reduction in EGFP expression after the repression of a reporter gene containing polyCAG. Interestingly, preferential repression of longer CAG tracts was observed. Consequently, ZF×Hunt-Kox-1 constructs were created for stronger repression where Kox-1 is a repressor domain. ZF×Hunt-Kox-1 inhibited only the mutant allele (up to 80% mRNA and 95% protein) in mouse and human HD cell lines without affecting the healthy allele and other CAG-rich genes such as atrophin-1, ataxin-1,2,3,7, calcium channel α1A subunit and TATA binding protein. Finally analysis in vivo was performed using designed AAV vectors encoding ZF×Hunt-Kox-1. R6/2 mice were stereotaxically infected and after two weeks mutant HTT mRNA was reduced in the striatum by 40% on average, and there was no toxic effect. Furthermore, motor and behavioral assessment showed improved motor coordination and absence of clasping behavior and there was no difference between treated and wild type mice. Thus, these results showed that zinc fingers can successfully, safely and specifically repress mutant HTT in vitro and in vivo. This study uncovered new prospects for the use of zinc finger technology as an approach for HD therapy in biomedicine [56].
There are several effectors based on the TALEN system. For targeted activation of gene expression, DNA-binding TALE-domain fused with VP64 (transcription activator domain) is used (TALE-TF) that attracts endogenous activators, and KRAB (Krüppel associated box) and SRDX (ethylene-responsive element binding factor-associated amphiphilic repression motif repressor) domains are used for repression of the targeted gene [57].
Transcription repression of the mutant HTT allele was also performed using TALEs combined with the repressor domain [58, 59]. TALEs were constructed targeting three SNPs that preferentially occurred independently of each other in the mutant allele in or near the promoter region of HTT (45% of the HD population). Three SNP-targeted TALEs were combined with a transcriptional repressor KRAB. SNP-specific TALE-KRAB showed efficient binding and didn’t demonstrate cell toxicity. It indicates that TALE-KRAB is able to safely bind and regulate gene expression. Decreased HTT aggregation (Western blot) and significant mutant allele knockdown (SNP genotyping assay) were detected. Most importantly, normal expression of the healthy allele was found, which indicates specificity of the TALE repressor. In the future, the researchers plan to perform experiments in the mouse model in vivo, as well as on neurons in vitro. Thus, this study exhibits great capabilities and opportunities of TALE as a transcription effector for targeted allele manipulation [41]. Allele-specific knockdown like allele-specific knockout is a promising path for the therapy of hereditary diseases.
Another therapeutic application is the up-regulation of genes involved in ablation misfolding proteins. It is known that heat shock proteins (HSPs) provide a first line of defense against aberrant proteins in such diseases as Alzheimer’s, Parkinson’s and Huntington’s disease and gain-of-function of these genes could reverse disease phenotype at least at the early stages of the disease [60]. More research is required in order to understand exactly which HSPs improve neuroprotection and through what mechanisms they act.
In 2014, Konermann and colleagues developed a genome-scale transcriptional activation system based on CRISPR/Cas9 complex (synergistic activation mediator, SAM) [61]. They created a system, which consisted of catalytically inactive Cas9 (dCas9)-VP64 fusion protein and MS2-p65-HSF1 transcription activator domain and upgraded sgRNA containing aptamers to facilitate the recruitment of effector domains to the Cas9 complex. Like the GeCKO system it is also possible to create a library of sgRNAs targeting multiple genes. So, the CRISPR-system can be successfully used for multiplex activation (SAM) or repression (dCas9-KRAB) [62], particularly repression of the mutant HTT allele and activation of HSPs.
Targeted epigenetic editing
The emergence of gene editing tools allows not only manipulation of the genome, but also modifications of the epigenome. Epigenome editing tools are built upon nucleotide sequence recognition (DNA-binding domains and sgRNAs) and effector domains responsible for DNA or histone modifications (DNA (de)methylation, DNA hydroxymethylation, histone (de)acetylation, (de)methylation and (de)phosphorylation). There are several ZFN, TALE and CRISPR-based tools fused with effectors of DNA methylation (DNMT3A), demethylation (TDG) and hydroxymethylation (Tet1) (Table 1) [63].
The first epigenome editing tool was ZFN-based and was a popular instrument due to efficient target binding and small size [69–71]. However, a new system relying on TALEs was developed, which received greater interest due to simplicity of design and targeting [72, 74]. But this system has also a limitation, namely, sensitivity to CpG methylation and thus it became unsuitable for promoter region modifications in mammalian cells [81]. Further, a new platform for DNA methylation based on CRISPR was constructed, which combines the positive features such as ease of targeting sgRNAs and insensitivity to CpG methylation [76, 82].
DNA methylation is known to regulate a myriad of cellular processes in mammalian cells and alterations in DNA methylation are frequently involved in psychiatric and neurological diseases such as schizophrenia, Alzheimer’s and Huntington’s disease. For example, DNA hypermethylation underlies fragile X-chromosome syndrome caused by the expansion of trinucleotide repeat CGG in the promoter region of FMR1 leading to hypermethylation of the gene and significant suppression of gene expression [88]. Therefore, targeted DNA methylation editing tools have the potential to facilitate our understanding of functional roles of DNA methylation and demethylation in specific contexts and enable control of gene expression and functionality fordisease therapy.
Histone modifications are also important regulators of gene expression thus affecting such processes as development, cell-fate decision, epigenetic inheritance and pathogenesis of neurological and psychiatric diseases [89]. There are many types of histone modifications –histone acetylation, methylation, phosphorylation, etc. –which directly (gene) or indirectly (enhancer) influence gene expression. Previously, it was not possible to precisely modify histones and combine such modifications which consequently limited our understanding of functional relationships of histone modifications. To address this problem programmable histone TALE-modifiers have been developed that specifically and efficiently reverse the expression of target genes (Table 1) [78, 79]. Furthermore, a CRISPR-based tool for targeted histone acetylation was designed leading to directed acetylation and subsequent target-gene activation [80].
Direct histone modification can be very useful for studying causal relationships between epigenetic changes and development of neurodegenerative diseases, particularly histone hypoacetylation which is the most frequent alteration in these disorders. Histone hypoacetylation is found in amyotrophic lateral sclerosis, Parkinson’s and Huntington’s disease and Friedreich’s ataxia. Moreover, many other changes of histone profiles in neurological diseases have been reported such as histone acetylation and phosphorylation alterations in Alzheimer’s disease and epilepsy, and H3K9 hypertrimethylation in Huntington’s disease and Friedreich’s ataxia [90]. It suggests the idea that targeted epigenetic editing can also be a promising therapeutic agent for the treatment of neurodegenerative disorders.
Comparing the prospects of the two platforms based on TALEs and CRISPR it is easy to see that CRISPR-based epigenome editing tools are much more promising. The first advantage is guide RNA programming almost any genome site whereas the design of DNA-binding domains for TALE-based editing is more difficult. Another advantage is the possibility to simultaneously modify DNA or histones in different loci and thus to carry out multiplex epigenome editing.
CONCLUSION
In recent years, significant progress in the study of molecular mechanisms and modeling of genetic diseases has been achieved. An important role was played by the rapid development of tools for directed genome editing, and ease of construction and low cost have made them widely used. It is worth noting that the choice of the tool depends on goals of researchers, as each tool has its own specific characteristics and functional features. First artificial nucleases ZFN and TALEN are inferior to the CRISPR system in many aspects such as design simplicity, high efficiency and specificity and ease of delivery. However, the latest tools NgAgo and SGN have not yet been proven, and perhaps in the future will be able to compete with the CRISPR system.
The contributions that genome editing technologies can make particularly to the HD field are substantial and may impact on our understanding of HD and the therapeutic directions being pursued. Although, there are still few researchers using these tools in the study of HD, the potential of these tools is just beginning to unfold for mutant HTT targeted therapy and creation of cell lines bearing desired mutations. Researchers using these tools will be able to confirm and verify any hypothesis in HD and answer the question why and how do striatal neurons primarily die?
FUNDING
The funding for this research was provided by the Russian Science Foundation project No 16-15-10128.
CONFLICT OF INTEREST
The authors have no conflict to declare.
REFERENCES
[1] | Reiner A , Dragatsis I , Dietrich P . Genetics and neuropathology of Huntington’s disease. Int Rev Neurobiol. (2011) ;98: :325–72. doi: 10.1016/B978-0-12-381328-2.00014-6 |
[2] | Zhang N , Bailus BJ , Ring KL , Ellerby LM . iPSC-based drug screening for Huntington’s disease. Brain Res. (2016) ;1638: :42–56. doi: 10.1016/j.brainres.2015.09.020 |
[3] | Kim YG , Cha J , Chandrasegaran S . Hybrid restriction enzymes: Zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci U S A. (1996) ;93: :1156–60. |
[4] | Miller JC , Tan S , Qiao G , Barlow KA , Wang J , Xia DF , et al. A TALE nuclease architecture for efficient genome editing. Nat Biotechnol. (2011) ;29: :143–8. doi: 10.1038/nbt.1755 |
[5] | Cong L , Ran FA , Cox D , Lin S , Barretto R , Habib N , et al. Multiplex genome engineering using CRISPR/Cas systems. Science. (2013) ;339: :819–23. doi: 10.1126/science.1231143 |
[6] | Kleinstiver BP , Pattanayak V , Prew MS , Tsai SQ , Nguyen NT , Zheng Z , et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. (2016) ;529: :490–5. doi: 10.1038/nature16526 |
[7] | Kim D , Kim J , Hur JK , Been KW , Yoon S-H , Kim J-S . Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat Biotechnol. (2016) ;34: :863–8. doi: 10.1038/nbt.3609 |
[8] | Gao F , Shen XZ , Jiang F , Wu Y , Han C . DNA-guided genome editing using the Natronobacterium gregoryi Argonaute. Nat Biotechnol. (2016) ;34: :768–73. doi: 10.1038/nbt.3547 |
[9] | Xu S , Cao S , Zou B , Yue Y , Gu C , Chen X , et al. An alternative novel tool for DNA editing without target sequence limitation: The structure-guided nuclease. Genome Biol. (2016) ;17: :186. doi: 10.1186/s13059-016-1038-5 |
[10] | Liu P-Q , Tan S , Mendel MC , Murrills RJ , Bhat BM , Schlag B , et al. Isogenic human cell lines for drug discovery: Regulation of target gene expression by engineered zinc-finger protein transcription factors. J Biomol Screen. (2005) ;10: :304–13. doi: 10.1177/1087057104272663 |
[11] | Freude K , Pires C , Hyttel P , Hall V . Induced pluripotent stem cells derived from Alzheimer’s disease patients: The promise, the hope and the path ahead. J Clin Med. (2014) ;3: :1402–36. doi: 10.3390/jcm3041402 |
[12] | Osborn MJ , DeFeo AP , Blazar BR , Tolar J . Synthetic zinc finger nuclease design and rapid assembly. Hum Gene Ther. (2011) ;22: :1155–65. doi: 10.1089/hum.2011.072 |
[13] | Yao Y , Nashun B , Zhou T , Qin L , Qin L , Zhao S , et al. Generation of CD34+ cells from CCR5-disrupted human embryonic and induced pluripotent stem cells. Hum Gene Ther. (2012) ;23: :238–42. doi: 10.1089/hum.2011.126 |
[14] | Cantos C , Francisco P , Trijatmiko KR , Slamet-Loedin I , Chadha-Mohanty PK . Identification of “safe harbor” loci in indica rice genome by harnessing the property of zinc-finger nucleases to induce DNA damage and repair. Front Plant Sci. (2014) ;5: :302. doi: 10.3389/fpls.2014.00302 |
[15] | Zhang W , Wang D , Liu S , Zheng X , Ji H , Xia H , et al. Multiple copies of a linear donor fragment released in situ from a vector improve the efficiency of zinc-finger nuclease-mediated genome editing. Gene Ther. (2014) ;21: :282–8. doi: 10.1038/gt.2013.83 |
[16] | Christian M , Cermak T , Doyle EL , Schmidt C , Zhang F , Hummel A , et al. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics. (2010) ;186: :757–61. doi: 10.1534/genetics.110.120717 |
[17] | Jinek M , Chylinski K , Fonfara I , Hauer M , Doudna JA , Charpentier E . A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. (2012) ;337: :816–21. doi: 10.1126/science.1225829 |
[18] | Makarova KS , Haft DH , Barrangou R , Brouns SJJ , Charpentier E , Horvath P , et al. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol. (2011) ;9: :467–77. doi: 10.1038/nrmicro2577 |
[19] | Makarova KS , Aravind L , Wolf YI , Koonin EV . Unification of Cas protein families and a simple scenario for the origin and evolution of CRISPR-Cas systems. Biol Direct. (2011) ;6: :38. doi: 10.1186/1745-6150-6-38 |
[20] | Anders C , Niewoehner O , Duerst A , Jinek M . Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature. (2014) ;513: :569–73. doi: 10.1038/nature13579 |
[21] | Xue W , Chen S , Yin H , Tammela T , Papagiannakopoulos T , Joshi NS , et al. CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature. (2014) ;514: :380–4. doi: 10.1038/nature13589 |
[22] | Wu Y , Zhou H , Fan X , Zhang Y , Zhang M , Wang Y , et al. Correction of a genetic disease by CRISPR-Cas9-mediated gene editing in mouse spermatogonial stem cells. Cell Res. (2015) ;25: :67–79. doi: 10.1038/cr.2014.160 |
[23] | Shalem O , Sanjana NE , Hartenian E , Shi X , Scott DA , Mikkelsen TS , et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. (2014) ;343: :84–7. doi: 10.1126/science.1247005 |
[24] | Xie F , Ye L , Chang JC , Beyer AI , Wang J , Muench MO , et al. Seamless gene correction of β-thalassemia mutations in patient-specific iPSCs using CRISPR/Cas9 and piggyBac. Genome Res. (2014) ;24: :1526–33. doi: 10.1101/gr.173427.114 |
[25] | Wang G , McCain ML , Yang L , He A , Pasqualini FS , Agarwal A , et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat Med. (2014) ;20: :616–23. doi: 10.1038/nm.3545 |
[26] | Li HL , Fujimoto N , Sasakawa N , Shirai S , Ohkame T , Sakuma T , et al. Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Reports. (2015) ;4: :143–54. doi: 10.1016/j.stemcr.2014.10.013 |
[27] | Fu Y , Foden JA , Khayter C , Maeder ML , Reyon D , Joung JK , et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. (2013) ;31: :822–6. doi: 10.1038/nbt.2623 |
[28] | Zhang X-H , Tee LY , Wang X-G , Huang Q-S , Yang S-H . Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol Ther Acids. (2015) ;4: :e264. doi: 10.1038/mtna.2015.37 |
[29] | Esvelt KM , Mali P , Braff JL , Moosburner M , Yaung SJ , Church GM . Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat Methods. (2013) ;10: :1116–21. doi: 10.1038/nmeth.2681 |
[30] | Ran FA , Cong L , Yan WX , Scott DA , Gootenberg JS , Kriz AJ , et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. (2015) ;520: :186–91. doi: 10.1038/nature14299 |
[31] | Zetsche B , Gootenberg JS , Abudayyeh OO , Slaymaker IM , Makarova KS , Essletzbichler P , et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. (2015) ;163: :759–71. doi: 10.1016/j.cell.2015.09.038 |
[32] | Perez C , Guyot V , Cabaniols J-P , Gouble A , Micheaux B , Smith J , et al. Factors affecting double-strand break-induced homologous recombination in mammalian cells. Biotechniques. (2005) ;39: :109–15. |
[33] | Park I-H , Arora N , Huo H , Maherali N , Ahfeldt T , Shimamura A , et al. Disease-specific induced pluripotent stem cells. Cell. (2008) ;134: :877–86. doi: 10.1016/j.cell.2008.07.041 |
[34] | Tousley A , Kegel-Gleason KB . Induced pluripotent stem cells in Huntington’s disease research: Progress and opportunity. J Huntingtons Dis. (2016) ;5: :99–131. doi: 10.3233/JHD-160199 |
[35] | An MC , Zhang N , Scott G , Montoro D , Wittkop T , Mooney S , et al. Genetic correction of Huntington’s disease phenotypes in induced pluripotent stem cells (2012) ;11: :253–63. doi: 10.1016/j.stem.2012.04.026 |
[36] | Lombardo A , Genovese P , Beausejour CM , Colleoni S , Lee Y-L , Kim KA , et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol. (2007) ;25: :1298–306. doi: 10.1038/nbt1353 |
[37] | An MC , O’Brien RN , Zhang N , Patra BN , De La Cruz M , Ray A , et al. Polyglutamine disease modeling: Epitope based screen for homologous recombination using CRISPR/Cas9 system. PLoS Curr. (2014) ;6: :1–19. doi: 10.1371/currents.hd.0242d2e7ad72225efa72f6964589369a |
[38] | Malakhova AA , Sorokin MA , Sorokina AE , Malankhanova TB , Mazurok NA , Medvedev SP , Zakian SM . Genome editing approach for generation of isogenic cell lines modelling Huntington’s disease in vitro. Genes Cells. (2016) ;11: :106–13. |
[39] | Engler C , Marillonnet S . Golden Gate cloning. Methods Mol. Biol. (2014) ;1116: :119–31. doi: 10.1007/978-1-62703-764-8 9 |
[40] | Coles R , Caswell R , Rubinsztein DC . Functional analysis of the Huntington’s disease (HD) gene promoter. Hum Mol Genet. (1998) ;7: :791–800. doi: 10.1093/HMG/7.5.791 |
[41] | Fink KD , Deng P , Gutierrez J , Anderson JS , Torrest A , Komarla A , et al. Allele-specific reduction of the mutant huntingtin allele using transcription activator-like effectors in human Huntington’s disease fibroblasts. Cell Transplant. (2016) ;25: :677–86. doi: 10.3727/0963z6916X690863 |
[42] | Cade L , Reyon D , Hwang WY , Tsai SQ , Patel S , Khayter C , et al. Highly efficient generation of heritable zebrafish gene mutations using homo- and heterodimeric TALENs. Nucleic Acids Res. (2012) ;40: :8001–10. doi: 10.1093/nar/gks518 |
[43] | Doyon Y , Vo TD , Mendel MC , Greenberg SG , Wang J , Xia DF , et al. Enhancing zinc-finger-nuclease activity with improved obligate heterodimeric architectures. NatMethods. (2011) ;8: :74–9. doi: 10.1038/nmeth.1539 |
[44] | Cinesi C , Aeschbach L , Yang B , Dion V . Contracting CAG/CTG repeats using the CRISPR-Cas9 nickase. Nat Commun. (2016) ;7: :13272. doi: 10.1038/ncomms13272 |
[45] | Shin JW , Kim K-H , Chao MJ , Atwal RS , Gillis T , MacDonald ME , et al. Permanent inactivation of Huntington’s disease mutation by personalized allele-specific CRISPR/Cas9. Hum Mol Genet. (2016) ;ddw286. doi: 10.1093/hmg/ddw286 |
[46] | Summers KM . Relationship between genotype and pheno-type in monogenic diseases: Relevance to polygenic diseases. Hum Mutat. (1996) ;7: :283–93. doi: 10.1002/(SICI)1098-1004(1996)7:4<283::AID-HUMU1>3.0.CO;2-A. |
[47] | Mastrokolias A , Ariyurek Y , Goeman JJ , van Duijn E , Roos RAC , van der Mast RC , et al. Huntington’s disease biomarker progression profile identified by transcriptome sequencing in peripheral blood. Eur J Hum Genet. (2015) ;23: :1349–56. doi: 10.1038/ejhg.2014.281 |
[48] | Lee J-M , Wheeler VC , Chao MJ , Vonsattel JPG , Pinto RM , Lucente D , et al. Identification of genetic factors that modify clinical onset of Huntington’s disease. Cell. (2015) ;162: :516–26. doi: 10.1016/j.cell.2015.07.003 |
[49] | Metzger S , Bauer P , Tomiuk J , Laccone F , Didonato S , Gellera C , et al. The S18Y polymorphism in the UCHL1 gene is a genetic modifier in Huntington’s disease. Neurogenetics. (2006) ;7: :27–30. doi: 10.1007/s10048-005-0023-z |
[50] | Metzger S , Rong J , Nguyen H-P , Cape A , Tomiuk J , Soehn AS , et al. Huntingtin-associated protein-1 is a modifier of the age-at-onset of Huntington’s disease. Hum Mol Genet. (2008) ;17: :1137–46. doi: 10.1093/hmg/ddn003 |
[51] | Metzger S , Saukko M , Van Che H , Tong L , Puder Y , Riess O , et al. Age at onset in Huntington’s disease is modified by the autophagy pathway: Implication of the V471A polymorphism in Atg7. Hum Genet. (2010) ;128: :453–9. doi: 10.1007/s00439-010-0873-9 |
[52] | Dhaenens C-M , Burnouf S , Simonin C , Van Brussel E , Duhamel A , Defebvre L , et al. A genetic variation in the ADORA2A gene modifies age at onset in Huntington’s disease. Neurobiol Dis. (2009) ;35: :474–6. doi: 10.1016/j.nbd.2009.06.009 |
[53] | Che HVB , Metzger S , Portal E , Deyle C , Riess O , Nguyen HP . Localization of sequence variations in PGC-1α influence their modifying effect in Huntington disease. Mol Neurodegener. (2011) ;6: :1. doi: 10.1186/1750-1326-6-1 |
[54] | Kloster E , Saft C , Akkad DA , Epplen JT , Arning L . Association of age at onset in Huntington disease with functional promoter variations in NPY and NPY2R. J Mol Med (Berl). (2014) ;92: :177–84. doi: 10.1007/s00109-013-1092-3 |
[55] | Kloster E , Saft C , Epplen JT , Arning L . CNR1 variation is associated with the age at onset in Huntington disease. Eur J Med Genet. (2013) ;56: :416–9. doi: 10.1016/j.ejmg.2013.05.007 |
[56] | Garriga-Canut M , Agustín-Pavón C , Herrmann F , Sánchez A , Dierssen M , Fillat C , et al. Synthetic zinc finger repressors reduce mutant huntingtin expression in the brain of R6/2 mice. Proc Natl Acad Sci U S A. (2012) ;109: :E3136–45. doi: 10.1073/pnas.1206506109 |
[57] | Nemudryi AA , Valetdinova KR , Medvedev SP , Zakian SM . TALEN and CRISPR/Cas genome editing systems: Tools of discovery. Acta Naturae. (2014) ;6: :19–40. |
[58] | Cermak T , Doyle EL , Christian M , Wang L , Zhang Y , Schmidt C , et al. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. (2011) ;39: :e82. doi: 10.1093/nar/gkr218 |
[59] | Zhang Z , Wu E , Qian Z , Wu W-S . A multicolor panel of TALE-KRAB based transcriptional repressor vectors enabling knockdown of multiple gene targets. Sci Re. (2014) ;4: :7338. doi: 10.1038/srep07338 |
[60] | Adachi H , Katsuno M , Waza M , Minamiyama M , Tanaka F , Sobue G . Heat shock proteins in neurodegenerative diseases: Pathogenic roles and therapeutic implications. Int J Hyperthermia. (2009) ;25: (8):647–54. doi: 10.3109/02656730903315823 |
[61] | Konermann S , Brigham MD , Trevino AE , Joung J , Abudayyeh OO , Barcena C , et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. (2014) ;517: :583–88. doi: 10.1038/nature14136 |
[62] | Gilbert LA , Larson MH , Morsut L , Liu Z , Brar GA , Torres SE , et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. (2013) ;154: :442–51. doi: 10.1016/j.cell.2013.06.044 |
[63] | Day JJ . New approaches to manipulating the epigenome. Dialogues Clin Neurosci. (2014) ;16: :345–57. |
[64] | Meister GE , Chandrasegaran S , Ostermeier M . An engineered split M. HhaI-zinc finger fusion lacks the intended methyltransferase specificity. Biochem Biophys Res Commun. (2008) ;377: :226–30. doi: 10.1016/j.bbrc.2008.09.099 |
[65] | Nomura W , Barbas CF . In vivo site-specific DNA methylation with a designed sequence-enabled DNA methylase. J Am Chem Soc. (2007) ;129: :8676–7. doi: 10.1021/ja0705588 |
[66] | Meister GE , Chandrasegaran S , Ostermeier M . Heterodimeric DNA methyltransferases as a platform for creating designer zinc finger methyltransferases for targeted DNA methylation in cells. Nucleic Acids Res. (2010) ;38: :1749–59. doi: 10.1093/nar/gkp1126 |
[67] | Chaikind B , Kilambi KP , Gray JJ , Ostermeier M . Targeted DNA methylation using an artificially bisected M.HhaI fused to zinc fingers. PLoS One. (2012) ;7: :e44852. doi: 1008 10.1371/journal.pone.0044852 |
[68] | Chaikind B , Ostermeier M . Directed evolution of improved zinc finger methyltransferases. PLoS One. (2014) ;9: :e96931. doi: 10.1371/journal.pone.0096931 |
[69] | Li F , Papworth M , Minczuk M , Rohde C , Zhang Y , Ragozin S , et al. Chimeric DNA methyltransferases target DNA methylation to specific DNA sequences and repress expression of target genes. Nucleic Acids Res. (2007) ;35: :100–12. doi: 10.1093/nar/gkl1035 |
[70] | Nunna S , Reinhardt R , Ragozin S , Jeltsch A , Cannistra S , Rescigno P , et al. Targeted methylation of the epithelial cell adhesion molecule (EpCAM) promoter to silence its expression in ovarian cancer cells. PLoS One. (2014) ;9: :e87703. doi: 10.1371/journal.pone.0087703 |
[71] | Chen H , Kazemier HG , de Groote ML , Ruiters MHJ , Xu G-L , Rots MG . Induced DNA demethylation by targeting Ten-Eleven Translocation 2 to the human ICAM-1 promoter. Nucleic Acids Res. (2014) ;42: :1563–74. doi: 10.1093/nar/gkt1019 |
[72] | Bernstein DL , Le Lay JE , Ruano EG , Kaestner KH . TALE-mediated epigenetic suppression of CDKN2A increases replication in human fibroblasts. J Clin Invest. (2015) ;125: :1998–2006. doi: 10.1172/JCI77321 |
[73] | Li K , Pang J , Cheng H , Liu W-P , Di J-M , Xiao H-J , et al. Manipulation of prostate cancer metastasis by locus-specific modification of the CRMP4 promoter region using chimeric TALE DNA methyltransferase and demethylase. Oncotarget. (2015) ;6: :10030–44. doi: 10.18632/oncotarget.3192 |
[74] | Maeder ML , Angstman JF , Richardson ME , Linder SJ , Cascio VM , Tsai SQ , et al. Targeted DNA demethylation and activation of endogenous genes using programmable TALE-TET1 fusion proteins. Nat Biotechnol. (2013) ;31: :1137–42. doi: 10.1038/nbt.2726 |
[75] | McDonald JI , Celik H , Rois LE , Fishberger G , Fowler T , Rees R , et al. Reprogrammable CRISPR/Cas9-based system for inducing site-specific DNA methylation. Biol Open. (2016) ;5: :866–74. doi: 10.1242/bio.019067 |
[76] | Xu X , Tao Y , Gao X , Zhang L , Li X , Zou W , et al. A CRISPR-based approach for targeted DNA demethylation. Cell Discov. (2016) ;2: :16009. doi: 10.1038/celldisc.2016.9 |
[77] | Choudhury SR , Cui Y , Lubecka K , Stefanska B , Irudayaraj J . CRISPR-dCas9 mediated TET1 targeting for selective DNA demethylation at BRCA1 promoter. Oncotarget. (2014) ;7: :46545–56. doi: 10.18632/oncotarget.10234 |
[78] | Konermann S , Brigham MD , Trevino AE , Hsu PD , Heidenreich M , Cong L , et al. Optical control of mammalian endogenous transcription and epigenetic states. Nature. (2013) ;500: :472–6. doi: 10.1038/nature12466 |
[79] | Mendenhall EM , Williamson KE , Reyon D , Zou JY , Ram O , Joung JK , et al. Locus-specific editing of histone modifications at endogenous enhancers. Nat Biotechnol. (2013) ;31: :1133–6. doi: 10.1038/nbt.2701 |
[80] | Hilton IB , D’Ippolito AM , Vockley CM , Thakore PI , Crawford GE , Reddy TE , et al. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol. (2015) ;33: :510–7. doi: 10.1038/nbt.3199 |
[81] | Valton J , Dupuy A , Daboussi F , Thomas S , Maréchal A , Macmaster R , et al. Overcoming transcription activator-like effector (TALE) DNA binding domain sensitivity to cytosine methylation. J Biol Chem. (2012) ;287: :38427–32. doi: 10.1074/jbc.C112.408864 |
[82] | Vojta A , Dobrinić P , Tadić V , Bočkor L , Korać P , Julg B , et al. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. (2016) ;44: :5615–28. doi: 10.1093/nar/gkw159 |
[83] | Lee J , Hwang YJ , Kim KY , Kowall NW , Ryu H . Epigenetic mechanisms of neurodegeneration in Huntington’s disease. Neurotherapeutics. (2013) ;10664–76. doi: 10.1007/s13311-013-0206-5 |
[84] | Coppieters N , Dieriks BV , Lill C , Faull RLM , Curtis MA , Dragunow M . Global changes in DNA methylation and hydroxymethylation in Alzheimer’s disease human brain. Neurobiol Aging. (2014) ;35: :1334–44. doi: 10.1016/j.neurobiolaging.2013.11.031 |
[85] | Sanchez-Mut JV , Aso E , Panayotis N , Lott I , Dierssen M , Rabano A , et al. DNA methylation map of mouse and human brain identifies target genes in Alzheimer’s disease. Brain. (2013) ;136: :3018–27. doi: 10.1093/brain/awt237 |
[86] | Grayson DR , Jia X , Chen Y , Sharma RP , Mitchell CP , Guidotti A , et al. Reelin promoter hypermethylation in schizophrenia. Proc Natl Acad Sci. (2005) ;102: :9341–6. doi: 10.1073/pnas.0503736102 |
[87] | Robertson KD . DNA methylation and human disease. Nat Rev Genet. (2005) ;6: :597–610. doi: 10.1038/nrg1655 |
[88] | Park C-Y , Halevy T , Lee DR , Sung JJ , Lee JS , Yanuka O , et al. Reversion of FMR1 methylation and silencing by editing the triplet repeats in Fragile X iPSC-derived neurons. Cell Re. (2015) ;13: :234–41. doi: 10.1016/j.celrep.2015.08.084 |
[89] | Kooistra SM , Helin K . Molecular mechanisms and potential functions of histone demethylases. Nat Rev Mol Cell Biol. (2012) ;13: :297. doi: 10.1038/nrm3327 |
[90] | Portela A , Esteller M . Epigenetic modifications and human disease. Nat Biotechnol. (2010) ;28: :1057–68. doi: 10.1038/nbt.1685 |
Figures and Tables
Fig.1
Table of gene editing tools survey for disease modeling. Advantages of the tools are marked in green frames.
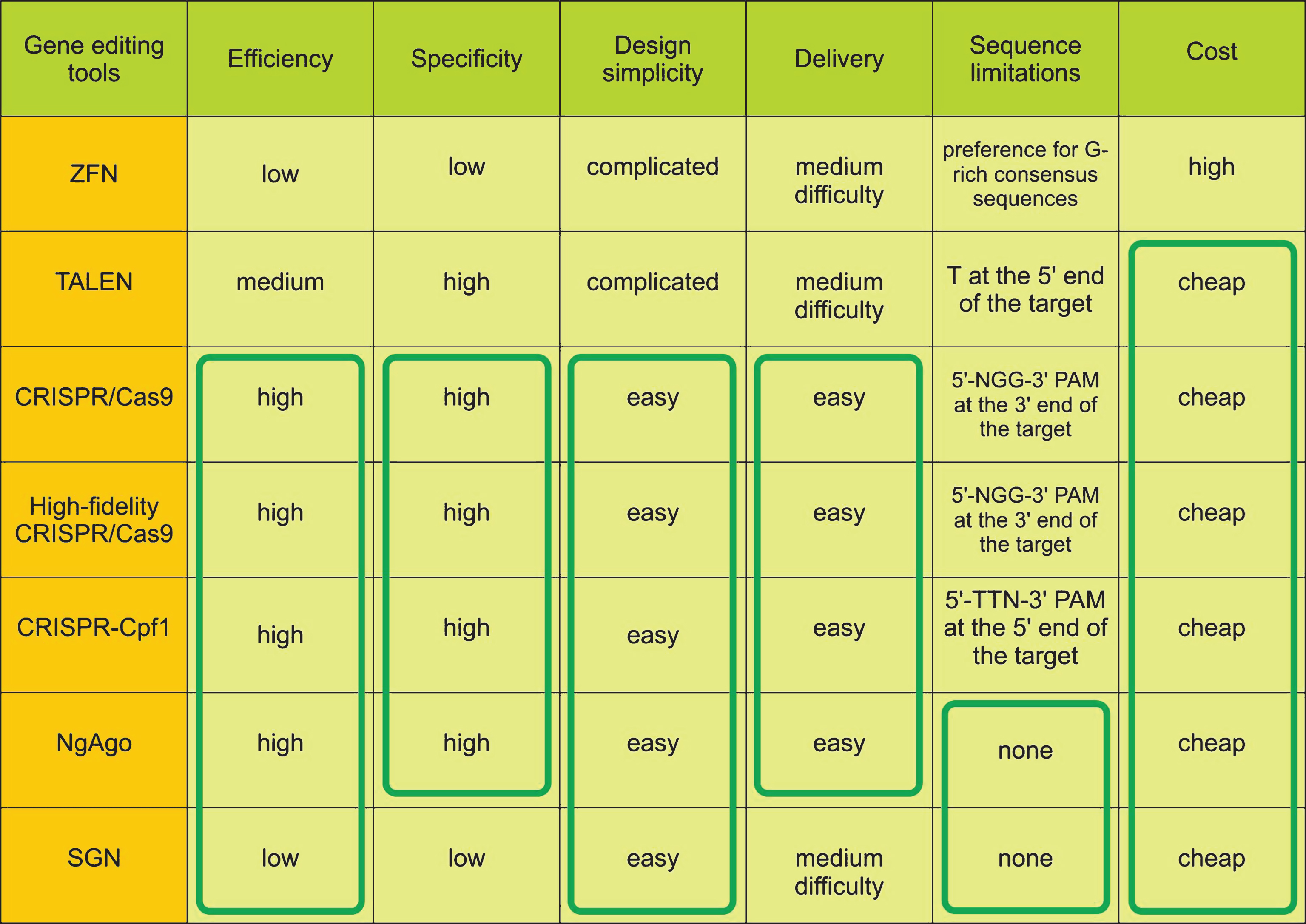
Fig.2
Generation of isogenic cell line panel with various numbers of CAG repeats using gene editing tools.
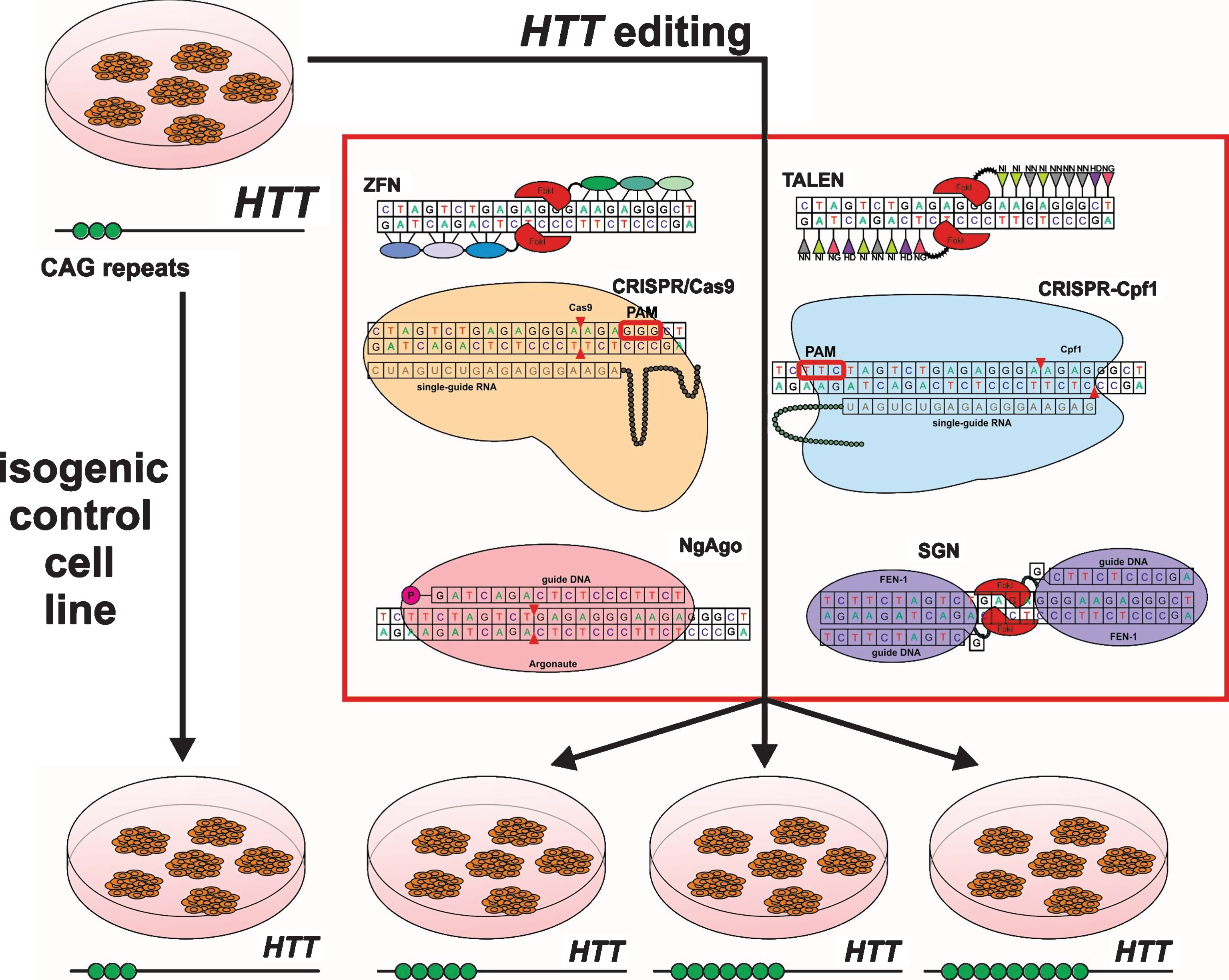
Table 1
Gene editing tools for targeted epigenome modifications
| Alteration in DNA methylation | |||
| ZFN-based DNA methylases and demethylases | M.HhaI+ZFN | No evidence of specificity | [64, 65] |
| M.EcoHK31I+ZFN | Low specificity at higher expression levels of M.EcoHK31I+ZFN | [66] | |
| M.HhaI+ZFN | Upgraded tool with high efficiency (∼60%) and undetectable levels of methylation at a non-target sites | [67] | |
| M.SssI+ZFN | High levels of CpG methylation at the target site (up to ∼80%) and nearly unobservable levels of methylation at off-target sites (<1%) | [68] | |
| Dnmt3a+ZFN | Targeted methylation of HSV-1 (herpes simplex virus-1) IE175k promoter and reduction of HSV-1 titer during HSV-1 lytic infection | [69] | |
| Dnmt3a+ZFN | Specifically increased methylation of the EpCAM promoter in SKOV3 cells from 4–8% in untreated cells to 30%. Specific for the target region. Hypermethylated EpCAM promoter showed a 60–80% reduction of EpCAM expression (mRNA level and decreased proliferation rate) | [70] | |
| TET1+ZFN | Reactivation of originally epigenetically silenced ECAM1 and EpCAM | [71] | |
| TALE-based DNA methylases and demethylases | DNMT3A+TALE | Specific methylation of the targeted gene locus in human cells. Methylation of CDKN2A that inhibits the proliferation of primary human cells leading to increased replication in these cells | [72] |
| Methylation of CRMP4 resulted in increasing invasion and migration of cancer cells in vitro and metastasis in vivo | [73] | ||
| TET1+TALE | Increased expression of the KLF4, RHOXF2, HBB genes | [74] | |
| Demethylation of CRMP4 resulted in decreasing invasion and migration of cancer cells in vitro and no metastasis in vivo | [73] | ||
| CRISPR-based DNA methylases and demethylases | DNMT3A+CRISPR | Decrease in CDKN2A, ARF, and Cdkn1a expression | [75] |
| TET1+CRISPR | Significant upregulation of RANKL, MAGEB2 or MMP2 transcription in a close correlation to DNA demethylation of their neighboring CpGs in the promoters with tenuous off-target effects | [76] | |
| Upregulation of BRCA1 that suppressed in tumors | [77] | ||
| Histone modifications | |||
| TALE-based histone effectors | epiTALEs carrying histone methyltransferase and histone deacetylase domains | In primary neurons and Neuro 2a cells, levels of H3K9me1, H4K20me3, H3K27me3, H3K9ac, and H4K8ac were altered | [78] |
| LSD1+TALE | Efficient demethylaton of a candidate enhancer in the stem cell leukemia locus in K562 erythroleukemia cells, without affecting control regions. Inactivation of enhancer chromatin by TALE-LSD1 frequently results in down-regulation of proximal genes, indicating target genes of enhancer | [79] | |
| CRISPR-based histone effectors | p300+CRISPR | Highly specific acetylation of histone H3 lysine 27 at target sites and following transcriptional activation of target genes from promoters and both proximal and distal enhancers | [80] |


