
Dysregulation of Corticostriatal Connectivity in Huntington’s Disease: A Role for Dopamine Modulation
Abstract
Aberrant communication between striatum, the main information processing unit of the basal ganglia, and cerebral cortex plays a critical role in the emergence of Huntington’s disease (HD), a fatal monogenetic condition that typically strikes in the prime of life. Although both striatum and cortex undergo substantial cell loss over the course of HD, corticostriatal circuits become dysfunctional long before neurons die. Understanding the dysfunction is key to developing effective strategies for treating a progressively worsening triad of motor, cognitive, and psychiatric symptoms. Cortical output neurons drive striatal activity through the release of glutamate, an excitatory amino acid. Striatal outputs, in turn, release γ-amino butyric acid (GABA) and exert inhibitory control over downstream basal ganglia targets. Ample evidence from transgenic rodent models points to dysregulation of corticostriatal glutamate transmission along with corresponding changes in striatal GABA release as underlying factors in the HD behavioral phenotype. Another contributor is dysregulation of dopamine (DA), a modulator of both glutamate and GABA transmission. In fact, pharmacological manipulation of DA is the only currently available treatment for HD symptoms. Here, we review data from animal models and human patients to evaluate the role of DA in HD, including DA interactions with glutamate and GABA within the context of dysfunctional corticostriatal circuitry.
INTRODUCTION
Huntington’s disease (HD) is a fatal monogenetic neurodegenerative disorder that affects approximately 10–12 individuals per 100,000 of European ancestry [1]. Clinical manifestations include progressively deteriorating motor, cognitive, and psychiatric symptoms [2]. The motor phenotype normally begins with gait disturbances and a lack of coordination followed by chorea and jerky body movements [3, 4]. In late stages of HD, the motor alterations switch to bradykinesia and dystonia [5]. Although the motor signs are typically the trigger for seeking medical attention, cognitive symptoms often develop first. Early-stage cognitive decline is characterized by difficulties in decision-making, planning, and cognitive inflexibility [6, 7] followed in late stages by dementia [8]. Psychiatric signs also can emerge in HD and are frequently reported as “changes of personality” with symptoms such as impulsivity, irritability, aggression, and altered mood regulation, which lead progressively to apathy and depression [9]. Symptoms generally begin in the prime of life between 35–45 years of age and progressively worsen until death; survival after motor onset is approximately 15–20 years [10].
Although the etiology of HD has been broadly identified, its underlying neural mechanisms have been difficult to establish. The most prominent neural change is a massive loss of neurons in the striatum, which receives information from all areas of cerebral cortex and processes it for behavioral output via downstream loops that converge back onto cortex, especially motor regions [11, 12]. Cortical neurons also degenerate in HD, creating a widespread pattern of corticostriatal neuropathology [13, 14].
Long before cortical and striatal neurons die, however, they become dysfunctional, setting the stage for the emergence of the HD behavioral phenotype. In this review, we discuss corticostriatal connectivity and its dysfunction in HD, including alterations in downstream information flow through the circuitry of the basal ganglia (BG). The two most prominent transmitters in these circuits – glutamate, the excitatory amino acid released by corticostriatal neurons, and γ-amino butyric acid (GABA), the inhibitory amino acid released by striatal output neurons – are closely tied to HD neuropathology and have been discussed extensively [15–22]. We review both transmitters here to provide context for a more detailed discussion of dopamine (DA), a key modulator of corticostriatal information flow, along with evidence for its emerging role in HD.
Clinical overview
HD is an autosomal dominant neurodegenerative disorder caused by a trinucleotide repeat expansion within the huntingtin gene (HTT). The repeat involves the CAG codon, which translates into a long poly-glutamine (polyQ) sequence insertion in the mutant huntingtin protein (mHTT). Abnormal aggregates of mHTT have been proposed to cause cellular dysfunction and ultimately neuronal death [23–25]. Clinical manifestations are fully penetrant when the CAG repeat number exceeds 39. The number of repeats in the mutant gene (mHTT) roughly corresponds to an inverse association with HD onset such that the greater the number, the earlier the onset [26, 27]. A less common form of HD begins in childhood or adolescence and is known as juvenile HD. In this case, symptoms occur before the age of 20 years and usually include changes in handwriting along with learning disabilities, motor problems (such as slowness, rigidity, tremor, and muscular twitching), and, most commonly, epileptic seizures [28, 29].
Medium spiny neurons (MSNs), which account for more than 90% of the striatal neuronal population, are particularly vulnerable. Also known as striatal projection neurons (SPNs), their progressive degeneration is the main feature of HD [30]. Because mHTT is expressed throughout the body [31], the role of mHTT aggregates in causing the relatively selective loss of MSNs in striatum is unclear, but ample evidence shows that mHTT underlies MSN dysfunction as well as abnormal communication between cortical and striatal neurons [32–37]. In fact, data obtained from transgenic animal models of HD indicate that dysfunction of corticostriatal circuitry precedes frank degeneration and likely plays a key role in symptom onset [19, 38–40].
Rodent models
The generation of transgenic mice and rats has greatly advanced understanding of HD pathology by identifying potential mechanisms underlying HD progression [2]. Transgenic rodent models can be classified into three groups: truncated models (expressing only the first exon of the mutant gene), full-length models (expressing the complete mutant gene), and knock-in (KI) models (with direct insertion or “knock-in” of the CAG repeat expansion into the HTT gene).
Truncated models
Two of the most widely used truncated models belong to the R6 mouse line and are designated as R6/1 and R6/2. R6/1 mice contain about 115 CAG repeats, display symptoms from 4-5 months of age and live approximately 10–14 months. Robust motor symptoms do not occur until approximately 5–7 months of age [41]. In contrast, R6/2 mice, which can carry upwards of 150 CAG repeats, display a very early onset (4-5 weeks of age) and rapid progression of neurological signs leading to death by 15 weeks of age [41, 42]. Although the behavioral phenotype is robust and easy to characterize, the short lifespan limits the usefulness of this model for identifying the pre-symptomatic triggers of HD onset and the gradual progression of HD through adulthood. Like HD patients with a high number of CAG repeats, the R6/2 model appears to recapitulate juvenile HD [43, 44].
A truncated rat model, the tgHD51, carries a truncated mHTT cDNA fragment with 51 CAG repeats [45]. This model exhibits a high degree of similarity to the late-onset form of HD due to the relatively small number of CAG repeats. These animals survive up to 24 months and exhibit a slow cognitive decline and relatively mild motor deficits [46, 47].
Full-length models
Compared to truncated models, full-length models display a slower disease progression. In this group, two types of mice have been generated: the yeast artificial chromosome (YAC) mouse and the bacterial artificial chromosome (BAC) mouse. Three YAC models are available based on different CAG repeat lengths: YAC46, YAC72, and YAC128. The most commonly used is the YAC128 model. Although behavioral changes occur at approximately 7 months of age, YAC128 mice show some aspects of the human motor phenotype, such as hyperkinesia followed by bradykinesia [48, 49], but the overall symptom profile is relatively mild and the animals typically survive for more than 18 months. A BAC HD model carries 97 repeats and exhibits similarly late-onset motor deficits [50, 51]. Both YAC and BAC models display evidence of corticostriatal degeneration [49, 50].
KI models
Widely varying CAG repeat lengths, ranging from Q71 and Q94 to Q140 and Q175, characterize currently available KI models [52–54]. Although the inverse correlation between age of onset and number of CAG repeats (see above) would suggest a robust and early phenotype similar to the R6 line, KI mice more closely resemble the full-length rather than the truncated model. Heterozygous Q175 KIs, for example, exhibit first motor signs at 3-4 months of age and survive well past 18 months [55, 54]. An extensive evaluation of these mice reported subtle but significant cognitive and motor impairments along with alterations in the level of intracellular proteins involved in synaptic function and axonal transport [35]. These molecular changes may correspond to common HD neuropathological features such as striatal atrophy, cortical thinning, degeneration of MSNs, and dense mHTT inclusions [33].
Dysfunctional neuronal activity also has been found in KI models. The Q140 mouse shows many of the same neurological abnormalities in striatum and cortex as the R6/2 model [56, 57]. In addition, reduced synaptic transmission in the striatum has been correlated with hypokinetic symptoms in homozygous Q175 mice [58]. Although Q175s display slow disease progression and relatively subtle behavioral changes, this animal model contains the human mHTT allele with the expanded CAG repeat within the native mouse HTT gene.
CORTICOSTRIATAL CONNECTIVITY AND DYSFUNCTION
Alterations in the flow of information from cerebral cortex to striatum play a key role in the onset and progression of HD. Virtually all areas of cerebral cortex have the ability to activate striatal neurons through a massive topographically organized projection system. Cortical input to striatum is driven by glutamate. The corticostriatal system, however, is tightly regulated by dopamine (DA), a monoamine modulator, and γ-amino butyric acid (GABA), an amino acid synthesized from glutamate that exerts a strong inhibitory influence. Together, DA and GABA provide a critical counterbalance to the glutamate-induced excitation of striatal neurons, which in turn modulates the activity of downstream targets in the BG. Thus, DA- and GABA-mediated control of the activation of striatal neurons by glutamate allow for the motor and cognitive responses that define healthy BG function. HD disrupts all three transmitter systems, creating a slowly progressing motor and cognitive phenotype that ends in death. To gain some perspective on this disruption, it is important to understand the basic organization of cortico-BG circuits.
According to the classical model proposed more than 25 years ago [59, 60], cortical pyramidal neurons (CPNs) activate the striatum where they target interneurons as well as MSNs, which project to other BG targets. Without excitatory input, MSNs, because of a relatively high potassium conductance at rest, are in a hyperpolarized or “down” state. Glutamate input is required to move MSNs into a depolarized or “up” state where they are readily excitable by additional glutamate input. But even under excitatory drive from cortex, MSNs typically discharge at a relatively slow rate (<15 spikes/s). GABA may account for some of this inhibitory influence since MSNs receive input from GABA-releasing interneurons that typically discharge at a fast rate (see below). Moreover, MSNs, which also release GABA, send axon collaterals within the striatum. Thus, even though MSNs receive direct glutamate input from cortex, there are sufficient constraints on their activity to ensure that they are not chronically hyperactive.
MSNs send their GABA projections to two segments of the globus pallidus, forming the so-called direct and indirect pathways. Direct-pathway MSNs send axons to the internal segment of the globus pallidus (GPi) and the substantia nigra pars reticulata (SNpr). This pathway is considered direct because the GPi/SNpr is the output nuclei of the BG circuit, and thus these MSNs exert a direct influence on BG output. In contrast, indirect-pathway MSNs sends axons to the external segment of the globus pallidus (GPe). The GPe sends axons to the subthalamic nucleus (STN), which in turn projects to the GPi/SNpr. Thus, multiple synapses intervene before striatal information traveling via the indirect pathway reaches BG output.
Direct- and indirect-projecting MSNs can be distinguished by the peptides they contain. MSNs that stain positively for dynorphin (DYN) and substance-P (SP) project directly to the GPi/SNpr, whereas a positive stain for enkephalin (ENK) identifies indirect-projecting MSNs [61–63].
Interestingly, all these neuronal projections are GABAergic except for the STN projection to GPi/SNpr, which releases glutamate. As shown in Fig. 1, activation of the direct pathway inhibits BG output, and the opposite occurs when the indirect pathway is activated. Accordingly, these pathways can be considered to have opposite effects on behavior because the GPi/SNpr sends inhibitory GABA projections to thalamus, which in turn activates motor cortex via glutamate. Thus, based on the classical model of BG circuits, the direct pathway disinhibits movement by suppressing inhibitory input to thalamus, whereas the indirect pathway has the opposite effect on the GPi/SNpr and suppresses movement. Recent studies using optogenetic technology, however, have proposed that both pathways are necessary for the initiation of movement because both have been found to be simultaneously active during the performance of motor sequences [64, 65]. In addition, both pathways are required not only for the initiation of sequential movements, but also for the performance of learned action sequences [66]. Importantly, despite concurrent activation of the two pathways, their motor contribution is not identical. Direct pathway inactivation has been shown to slow initiation of movement but indirect pathway activation terminated it, whereas activation of the direct but not indirect pathway prolonged action sequences, suggesting that both pathways trigger movements by acting in concert rather than in opposition [66]. Together, this evidence suggests that BG circuitry is more complex than activation or inactivation of direct and indirect pathways in the control of movement. In fact, it has been reported that activation of the direct pathway can evoke both excitatory and inhibitory actions in different cell populations within the GPi/SNpr [67]. Thus, the BG output nuclei appear to integrate information from these two pathways by acting as a gate that can be opened or closed to release coordinated movement [67].
In HD, the indirect pathway seems to deteriorate first as denoted by the preferential loss of striatal ENK neurons [68], and decreased ENK immunoreactivity in GPe [69]. This loss, according to the canonical model of BG connectivity, would bias the system toward activation of the direct pathway and thus induce chorea [70]. The model is further supported by evidence that cortical input to the indirect pathway is higher than that for the direct pathway [71], which may contribute to the vulnerability of the indirect pathway in HD since glutamate in high concentrations can be neurotoxic [72]. But in addition to the neuropathology of the indirect pathway, it is likely that direct pathway MSNs can be dysfunctional even though they may not show early-stage deterioration. Recent in vitro evidence, for example, indicates that selective activation of the direct pathway in either YAC128 or R6/2 mice results in a decreased SNpr response relative to healthy wild-type controls [73]. Thus, both striatal projections are likely to make important contributions to the HD behavioral phenotype.
Glutamatergic cortical pyramidal neurons
Despite the topographic organization of cortical pyramidal neurons (CPNs) and their striatal projections, the potential influence of these projections on striatal activity is complex [71, 74–78]. This section highlights important characteristics of the control that corticostriatal connectivity exerts over the direct and indirect BG pathways.
The CPN projection to the striatum can be divided into two main types: the intra-encephalic tract (IT) and the pyramidal tract (PT). Each type has distinct connectivity with the striatum and involvement in motor control. For example, IT neurons from the motor cortex but not from sensorimotor cortex project both contralaterally and ipsilaterally, whereas PT neurons only project to the ipsilateral striatum [77]. The distribution of IT and PT neurons in cortical layer 5, which is highly involved in voluntary movement, is about 65% for IT and 90% for PT [74–77, 79]. In an elegant series of studies, Reiner and colleagues [77] demonstrated that these two types of neurons show very distinct and preferential synaptic contact onto striatal MSNs. These researchers found that 50.9% of IT-type corticostriatal neurons project to DYN/SP MSNs that form the direct pathway and only 12.6% to the indirect pathway. While 50.5% of PT-type corticostriatal neurons target ENK MSNs, which form the indirect pathway, only 21.3% project to the direct pathway [71, 77, 79]. In addition, PT neurons show complex firing activity during movement, while IT neurons fire during motor planning before movement occurs [80, 81]. These different electrophysiological properties also correspond to differences in glutamate release. For example, PT neurons that target the indirect pathway have a higher probability of glutamate release than IT neurons that target the direct pathway [82, 83]. This difference may make indirect pathway MSNs more susceptible to degeneration and death [68, 70, 84]. Nevertheless, dysregulated corticostriatal activity seems to be critical in the neuropathology of HD [40, 57, 85, 86].
Although the specific involvement of IT and PT corticostriatal connectivity has not been associated with the pathological features of HD, several studies have noted cortical alterations in HD patients and animal models. Both show evidence of cortical deterioration such as atrophy, demyelination, and decreased size of soma and dendritic field [35, 87–90]. Moreover, GABA transmission is decreased in HD patients [91] and in BACHD mice [92], suggesting a reduction of inhibitory tone. In HD mice, increased calcium currents were reported for CPNs [93], which also have hyperexcitable membranes [51]. Consistent with increased excitability of CPNs is evidence of altered firing patterns in vivo, including a decrease in synchronized activity among simultaneously recorded neurons [57]. Furthermore, preventing the expression of mHTT in cortical efferents improved striatal neuronal activity [36, 94], suggesting that mHTT alters striatal activity via dysregulated cortical input.
GABAergic medium spiny neurons
The neuronal population of the striatum is divided into two groups: projection neurons and interneurons. Projection neurons account for more than 90% of the neuronal population and include all MSNs [95, 96], while the remaining includes GABA- and acetylcholine (ACh)-containing interneurons [97, 98].
The projection system is an integral part of the cortico-BG-thalamo-cortical loop (see above). Striatal MSNs are activated by glutamate input from IT and PT cortical neurons, and information flows through the direct and indirect pathways to the BG output nuclei, the thalamus, and back to cortex (Fig. 1). Importantly, a separate region of thalamus, the intralaminar nuclei, also sends glutamate input to both direct and indirect MSNs as well as striatal interneurons [99–101]. Thalamostriatal axon terminals have a higher probability of glutamate release than corticostriatal axon terminals [102, 103]. Interestingly, the thalamic projection to striatum also shows signs of neuropathology in HD and is a likely contributor to the motor phenotype [104], much as the thalamostriatal projection also contributes to the motor signs of Parkinson’s disease [105]. In fact, using in vitro co-cultured thalamostriatal axons from YAC128 mice, investigators reported that functional synapses on MSNs are significantly altered. For example, thalamostriatal afferents showed decreased membrane capacitance and increased membrane resistance compared to corticostriatal afferents, suggesting that thalamostriatal axons are affected earlier [103]. Thus, thalamostriatal involvement in HD deserves further investigation.
Striatal MSN activity is often linked to movement. For instance, the motor-activating effects of psychomotor stimulants as well as spontaneous episodes of rearing or stepping excite many MSN populations, although some MSNs fail to respond to movement or are inhibited [106–109]. Moreover, after intra-striatal infusion of amphetamine, a widely studied stimulant, the activity of motor-related MSNs increases prior to behavioral activation, suggesting that MSN activity is not secondary to movement [110]. In addition, MSN motor-responsiveness is region-specific with dorsal and especially dorso-lateral striatum most sensitive to motor activation [106]. Thus, striatal activity can be key in cases of motor dysfunction.
In HD mouse models, one of the most consistent electrophysiological alterations in MSNs is a change in membrane properties. For example, in R6/2 mice, MSNs have a depolarized resting membrane potential and reduced membrane capacitance [88, 111], consistent with a significantly elevated firing rate during behavior relative to wild-type controls [112]. MSN calcium conductance is also increased, but at late stages, when R6/2s become hypokinetic, calcium conductance decreases [113]. Notably, these electrophysiological changes occur in conjunction with such morphological changes as decreased number of dendrites, loss of spines, decreased soma size, and decreased dendritic spread [22, 35, 88, 111]. Thus, MSNs undergo both functional and morphological changes over the course of HD.
MSN activity also is regulated by interneurons, and interestingly, the relationship is not reciprocal: interneurons innervate MSNs but not vice versa [20, 97, 114, 115]. Despite their small numbers, striatal interneurons exert a large influence on MSN activity owing to their extensive pattern of axonal branching. Thus, the interneuron population, which also receives glutamate input from cortex and thalamus, is another important factor in MSN dysfunction in HD.
Striatal interneurons
There are two main types of striatal interneurons: ACh and GABA. Early reports suggested that the interneuron population remained morphologically intact in HD [43, 116], but further assessments have found that degeneration of striatal GABA interneurons might be associated with dystonia [117].
GABA interneurons are classified according to the type of co-localized protein, peptide, or enzyme, which would include calcium-binding proteins (e.g., parvalbumin or calretinin), tyrosine hydroxylase (TH), neuropeptide Y, somatostatin, nitric oxide synthase and NADPH diaphorase [98, 118–120]. The best characterized is the parvalbumin-positive or fast-spiking interneuron (FSI), which accounts for approximately 20% of the total interneuron population [95]. FSIs show an increased expression gradient from medial to lateral dorsal striatum [121], which parallels input from motor and sensorimotor cortical regions [122], suggesting a selective role in motor integration. An important feature of FSIs is that they connect with other interneuron dendrites by gap junctions, which may provide strong control of striatal activity forming a large meshwork of interneuron-interneuron communication [123, 124]. In fact, data from electrophysiological studies suggest that FSIs participate in MSN spike initiation [125]. For instance, FSIs make synaptic contact with hundreds of MSNs, and many of these contacts are onto the somatic region. In contrast, MSN-to-MSN synaptic contacts occur distal to the cell body [126]. FSIs exhibit a brief action potential and a significantly higher firing rate than MSNs [115]. FSI activity, characterized by irregular, high frequency bursting, is believed to induce gamma oscillations in local field potentials (LFPs) [97, 127, 128], which sample pre- and postsynaptic activity within a relatively large area surrounding the recording electrode (see below). FSI burst firing may be important for the synchronous recruitment of MSNs throughout the sensoriomotor striatum causing LFP high-voltage spindles (HVSs), which have been reported to occur when there is no motivation to act or move [129].
Gap junctions seem to play an important role in spindle synchronization [130]. Because of the inhibitory influence of FSIs on MSNs [97, 115] hypofunction or loss of FSIs is associated dystonia [131, 132]. In fact, FSIs are lost in HD, suggesting a mechanism for dystonia onset [117].
Another striatal neuron type is the TH-positive interneuron. This cell type, named after TH, the rate-limiting step in DA synthesis, appears to play an important role in the pattern of striatal activity by making synaptic contact with MSNs [119, 120]. Although these interneurons are not DAergic, they have the unique capability of producing widespread and effective inhibition of both direct and indirect pathway MSNs [133]. Interestingly, it has been shown that DA enhances the ability of TH-positive interneurons to generate long-lasting depolarizing potentials [134]. Few studies have targeted TH-positive interneurons for analysis in HD, but available evidence from HD patients suggests that they are decreased in number [135]. As data accumulates on this particular class of interneurons, further studies of their role in HD, including the behavioral phenotype of HD mice, are needed.
ACh-containing interneurons appear to correspond to the group of tonically active neurons (TANs) identified in striatal electrophysiological recordings [136]. Unlike FSIs, TANs have a very long-duration action potential [137]. They receive prominent glutamate input from intralaminar thalamus [99, 101], and seem to control MSN activity through the release of ACh and consequent activation of postsynaptic muscarinic receptors, which increase MSN excitability [138, 139].
The activity of choline acetyl transferase, the enzyme that synthetizes ACh, is significantly decreased in HD patients [91] along with decreased binding and expression of the ACh vesicular transporter [140, 141]. In addition, HD patients treated with inhibitors of acetyl cholinesterase, to elevate Ach levels, showed some improvement in both cognitive function and motor performance [142]. Recent evidence also showed that deficient thalamic input to the dendrites of ACh interneurons occurs early in the lifespan of Q140 mice [143]. Collectively, these data suggest that altered ACh interneuron activity may play a role in the emergence of the HD behavioral phenotype.
DOPAMINE MODULATION
Because DA plays an important role in the modulation of excitatory and inhibitory signaling in both cortex and striatum to control the flow of information through cortico-BG circuits, DA dysfunction deserves attention as another potential mechanism underlying HD neuropathology. DA and its receptors have been broadly studied in the neuropathology of Parkinson’s disease, a progressive neurodegenerative disorder characterized by motor disturbances. Although DA system alterations in HD remain elusive, neurochemical findings have shown important changes in DA transmission that emerge at early stages of the disease and might be related to motor impairments, cognitive dysfunction, and psychiatric symptoms.
Dopamine overview
Two major DA pathways innervate cortical and striatal areas: nigrostriatal and mesocorticolimbic. The nigrostriatal pathway, which modulates the activity of striatal neurons in the control of movement [144], is defined by the projection of DA neurons from the substantia nigra pars compacta (SNpc) to dorsal striatum (Fig. 2). The degeneration of this pathway in Parkinson’s disease is the primary mechanism underlying the slowness of movement and shuffling gait of Parkinsonian patients. This pathway also has been implicated in cognitive function. Striatal regions with high DA innervation play an important role in cognitive tasks and cognitive flexibility [145, 146].
Psychiatric symptoms, which can also emerge in HD, are associated primarily with mesocorticolimbic dysfunction, although nigrostriatal abnormalities are likely to contribute [147–149]. The mesocorticolimbic pathway originates from DA cell bodies in the ventral tegmental area (VTA) with axons ascending to the nucleus accumbens or ventral striatum as well as large areas of frontal cortex, including the prelimbic and infralimbic areas of prefrontal cortex [144]. In the target areas of both pathways, DA modulates the influence of glutamate by adjusting the strength of the glutamate signal relative to background activity [150].
Several steps are involved in DA transmission as outlined in Fig. 2. Multiple pharmacological approaches have been developed to modulate each of these steps.
Dopamine in HD
In postmortem studies, HD patients lose more than 40% of DA neurons in SNpc relative to controls [151, 152] along with a significant decrease in DA terminals in the striatum [140]. This evidence is consistent with a significant decrease in immunoreactivity for TH in the SNpc and striatum [153, 154] and a significant decrease in striatal vesicular monoamine transporter-2 (VMAT-2), which transports newly synthesized DA into presynaptic vesicles [140]. Similarly, the DA transporter (DAT), located on DA axon terminals and removes extracellular DA after its release, shows a 50% reduction in HD postmortem specimens [140, 155] and in binding studies of HD striatum [156]. In line with this evidence, DA uptake is reduced in striatal tissue of HD patients relative to controls, and the volume of the HD substantia nigra is also decreased [157].
Evidence for altered DA transmission also appears in HD models. For example, decreased neuronal size in the SNpc of R6/1 mice correlates with an increase in mHTT aggregates [158]. In addition, an expansion of CAG repeats in SNpc is associated with decreased DA content [159]. Evoked DA release in brain slices also is decreased in R6/1 [160] and R6/2 mice relative to controls [161]. Microdialysis studies, moreover, indicate a 70% reduction in striatal extracellular DA content [158]. Interestingly, however, although decreased DA was found in symptomatic R6/1s relative to control mice, the opposite effect was reported for pre-symptomatic mice [159], suggesting that nigrostriatal DA transmission is differentially affected over the course of HD progression. In support of this view, TH activity relative to control was increased at 4 weeks of age in R6/2 mice with no changes in TH expression, but a decrease of both protein and activity occurred at 12 weeks of age [154]. In the same animal model, DAT was significantly reduced at late but not at early stages, although no DAT change was found in BACHD mice [162]. Interestingly, a decrease in striatal DA content was correlated with a reduction in total locomotor activity in both R6/2 and YAC128 mice [163]. Gait disturbances in tgHD rats also were also associated with decreased DA release from 12 to 15 months of age as measured by fast scan cyclic voltammetry [160].
Curiously, however, studies of the Q175 mouse model fail to present a consistent DA picture. In heterozygous Q175s, for example, hypokinesia is not accompanied by decreased DA content; in fact, the alteration in DA did not occur until the mice reached 12 months of age, and no changes in DA fibers were found [35]. In contrast, homozygous Q175s show decreased DA release in striatum accompanied by reduced TH immunoreactivity and VMAT-2 expression along with hypokinetic symptoms [58]. Furthermore, evidence of decreased DA content in the heterozygous nucleus accumbens is believed to contribute to cognitive neurological signs [164]. It appears that additional research is needed to clarify DA changes in the KI-Q175 model and to determine the extent to which the DA changes in this model parallel the changes in truncated and full-length HD models.
DA metabolism also is altered in animal models, but again with some inconsistency. For example, a significant decrease in the DA metabolite homovanillic acid was found in Q175, R6/2 and YAC128 mice [35, 165, 166], while DOPAC was found to be decreased in R6/2 and YAC128 mice [163] but increased in KI-Q175 at 6 months with no change at 12 or 16 months [35]. Collectively, these animal data suggest that DA transmission is altered in HD perhaps even at very early stages when no motor symptoms are apparent, but the lack of consistency across models suggests the data should be interpreted with caution.
Receptor expression and signaling
DA receptors belong to the large family of G-protein-coupled receptors. There are five types of mammalian DA receptors divided into two subfamilies according to their amino acid structure, homology, and biological response. The D1-like subfamily includes D1 and D5 receptors, while the D2-like subfamily consists of D2, D3 and D4 receptors. D1-like receptors are positively coupled to adenylyl cyclase (AC); their activation induces the intracellular accumulation of cyclic 3,5 adenosine-monophosphate (cAMP), which in turn, activates protein-kinase dependent cAMP (PKA) (Fig. 3). PKA is an important intracellular kinase directly involved in the phosphorylation of ion channels and activation of phosphatases such as DA regulated phosphatase-32 (DARPP-32) that play an important role in the electrophysiological properties of the neuronal membrane [167]. D2-like receptors, in contrast, are negatively coupled to AC. As a result, their activation decreases cAMP accumulation and PKA activation (Fig. 4) [168–170]. Growing evidence has shown, however, that activation of DA receptors is not only restricted to modulation of AC but also other complex transduction signaling pathways, depending on the brain area and any ongoing physiological and pathological conditions [171].
Striatal MSNs that comprise the direct pathway (SP/DYN), preferentially expresses D1-like receptors [62, 172, 173]. When activated, these receptors increase MSN activity by increasing time spent in the “up” or depolarized state [174]. Because these receptors are also located presynaptically on striatonigral terminals, they also increase GABA release in the GPi/SNpr [175, 176, 177]. Thus, the end result of D1-like activation of striatal MSNs is GABA-mediated inhibition of BG outputs, which in turn disinhibits the thalamic projection to motor cortical areas and promotes movement.
In contrast, D2-like receptors are found preferentially on striatal indirect pathway neurons [60], which project to the external segment of the globus pallidus (GPe). Activation of D2 receptors has been reported to have an inhibitory effect on MSNs by preventing the transition to an “up” state [62, 66, 172, 178]. In striatopallidal terminals, the presynaptic activation of D2 receptors will inhibit GABA release through the inhibition of PKA activity [179, 180, 181]. The inhibition of GABA release in GPe will activate the inhibitory GPe projection to the subthalamic nucleus (STN). Because the STN activates GPi/SNpr by releasing glutamate, inhibition of the STN ensures inhibition of BG outputs, and thus activates the thalamocortical system to promote movement. Without DA, however, cortical activation of indirect MSNs will tend to suppress movement by activating BG outputs.
Coordinated movement, therefore, depends on balanced DA transmission between D1- and D2-like receptors. In fact, recent evidence indicates that both pathways regulate movements by acting in a complementary manner [66]. In HD, a change in the D1- D2-receptor balance could trigger either excess of movement (chorea), an early-stage phenotype, or bradykinesia, which emerges later.
Transcriptional dysregulation of DA receptors has been found in HD patients. For instance, the mRNA for MSN D2 receptors is significantly decreased and this effect is exacerbated in advanced stages of HD, whereas the mRNA for D1 receptors showed an initial reduction and then a slight increase with HD progression [182]. Quantitative autoradiography has confirmed a loss of D1 and D2 receptors in striatum and also in corresponding terminal areas. A 55% loss of D1 receptors in the GPi/SNpr was found in pathological grade 1 HD, but no further loss in grade 3, while D2 receptors in GPe showed a 30% decrease in grade 1 that jumped to 55% in grade 3 [183]. Similar stage-dependent D2 receptor loss was found with positron emission tomography (PET) imaging such that the severity of neuropathological signs was strongly correlated with decreased expression of D2 receptors [155, 184–186]. Moreover, in R6/1, R6/2, and YAC128 [187–189] but not BACHD mice [162, 189], the binding and mRNA expression of D1 and D2 receptors were decreased, and again, the changes were correlated with disease progression.
DA signaling also changes in HD. In pre-symptomatic R6/2 mice, for example, striatal D1 receptor signaling is increased [113]; the same effect has been reported for pre-symptomatic Q175 mice [58]. In symptomatic R6/2 and YAC72 mice, D1 receptor expression is decreased [188, 190], but both cAMP and the level of phosphorylation of DARPP-32 are increased [113, 188].
Under normal conditions, the activity of DARPP-32 would be inhibited by calcineurin (phosphatase-2B), but the expression of calcineurin is decreased by about 30% in pre-sympomatic R6/2s and Q175s but further downregulated when motor signs appear [191], suggesting that D1 receptor signaling might be chronically activated. In support of this view, the expression of immediate early genes, which occurs after chronic activation of D1 receptors [192], is elevated in R6/2 mice [190], indicating augmented neuronal activity in this model and confirmed by recording of striatal MSNs in behaviorally active R6/2s [112]. Because D1 receptors are decreased but cAMP and DARPP-32 signaling are increased in HD models, one possibility is over-activation of AC (Fig. 3). Interestingly, increased AC activity has been found in other motor complications such as L-DOPA-induced dyskinesia [176, 177, 193, 194], autosomal dominant familiar dyskinesia, and myokymia [195].
Interestingly, D1 receptor agonists activate extracellular regulated kinase (ERK), and in HD, ERK signaling is consistently altered in striatum but not cortex [196, 197]. The increased phosphorylation of ERK can phosphorylate cAMP response-element binding protein (CREB), which is an important regulator of D1 receptor signaling [198, 199]. In fact, this pathway has been associated with movement disorders perhaps mediated by supersensitive D1 receptors [63, 200]. Increased CREB phosphorylation has been found in striatum but not cortex in R6/2 mice at late stages of disease [196], suggesting that striatal postsynaptic signaling transduction pathways might be involved in transcriptional alterations. In line with this point of view, massive transcriptional dysregulation has been reported in HD (see review [201]).
D1 receptors also could play a role in HD progression. Pharmacological activation of D1 receptors, for example, accelerates the formation of mHTT nuclear aggregates, and again the effect is mediated by the activation of transcription factors [202]. Furthermore, the same result was found with forskolin, which directly activates AC, further implicating the D1 receptor signal transduction pathway in the aggregation of mHTT (Fig. 3) [202, 203].
Altered D2 receptor signaling also is a likely contributor to the neuropathology of HD. In addition to modulation of cAMP signaling, D2 receptors can activate β-arrestin2, which recruits phosphatase-2A (PP2A), enhancing its interaction with thymoma viral protoncogen (Akt) (Fig. 4). This complex inactivates Akt, which in turn induces stimulation of the glycogen synthase kinase-3 β (GSK-3β) pathway [204–206]. Importantly, altered GSK-3β signaling is associated with HD neuropathology [90, 207]. Activation of GSK-3β induces phosphorylation of β-catenin, which can stimulate the ubiquitin-induced proteasomal degradation system [208]. Moreover, activation of this system induces disposal of mHTT, preventing its cellular aggregation (Fig. 4) [209, 210]. Interestingly, β-catenin phosphorylated levels were found to be decreased in HD cell lines, causing toxicity [211]. Recent studies using postmortem HD specimens found a 50% reduction of GSK-3β in frontal cortex [90, 207]. In R6/2 mice, decreased GSK-3β protein levels and activity were found in cortex and striatum but not hippocampus, and this corticostriatal change contributes to brain atrophy, behavioral alterations, and learning deficits in early symptomatic and symptomatic but not in pre-symptomatic HD mice [90]. Restoring expression of GSK-3β resulted in amelioration of the behavioral phenotype [90]. In primary striatal neurons cultured from the Q111 KI mouse, Akt signaling contributed to mHTT localization and this occurred prior to cell death [212].
In conditions of decreased DA transmission, the lack of D2 receptor activation would result in prolonged inactivation of GSK-3β and probably decreased ubiquitin-induced proteasomal degradation. To the best of our knowledge, the role of DA and its receptors in relation to GSK-3β signaling in HD has not been studied. Intriguingly, in conditions of DA depletion with subsequent chronic L-DOPA treatment, which is widely known to cause dyskinesia, constitutive Akt signaling was found in striatum [213]. Given the likely biphasic change in DA transmission observed in HD, this receptor pathway may play a role in the formation of mHTT aggregates.
Collectively, these data suggest a broad spectrum of changes in the DA system ranging from synthesis to release to receptors. But changes in DA are closely tied to changes in glutamate, and the interaction of these two systems has been associated with several aspects of HD neuropathology.
Interaction with glutamate
Dysregulation of DA and glutamate in HD has been widely proposed [17, 214–218]. In corticostriatal circuitry, DA modulates the glutamate system by controlling glutamate release or by the direct regulation of glutamate receptors [219–222]. For example, early studies showed that D1 receptors and N-methyl-D-aspartate (NMDA) receptors co-immunoprecipitate, suggesting a protein-protein interaction [223, 224]. This interaction prevents D1 receptor internalization [225]. Furthermore, activation of D1 receptors increased the expression of NMDA receptors at the membrane surface of corticostriatal axon terminals [226], as well as the membrane expression of both NMDA and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors in striatum. D1 receptors not only regulate NMDA surface expression, but the phosphorylated state of NMDA receptors through activation of PKA [222, 227], which can increase NMDA receptor ion currents [228]. In striatum, D1 receptor activation enhanced the NMDA response, indicating an important functional interaction [229–231].
In an interesting contrast, D2 receptor activation in striatum decreases the glutamate response evoked by AMPA [229]. D2 receptor activation also decreased the glutamate-evoked NMDA response by presynaptic modulation of glutamate release [232].
Abnormal DA and glutamate transmission has been associated with dysfunction and loss of MSNs in HD [214, 233]. It has been shown, for example, that increased NMDA activity could promote HD neurodegeneration [234, 235]. Interestingly, DA and glutamate act synergistically to induce apoptosis in YAC128 MSNs [214].
An important consideration regarding this interaction is the location of NMDA receptors. They are composed of different subunits that confer different properties on their activity [215, 236]. In the synapse, for example, NMDA receptors are mainly composed of NR1 and NR2B subunits, while extra-synaptic locations results in NR1, NR2B, and NR2A subunits [237]. The differential expression of these subunits has implications for the activation of specific intracellular signaling pathways. Synaptic NMDA receptors, for example, appear to activate anti-apoptotic pathways, whereas extra-synaptic receptors are associated with mitochondrial dysfunction and cell death [238]. Accordingly, YAC128 mice have increased expression of extra-synaptic NMDA receptors [239]. Blockade of these receptors, moreover, resulted in amelioration of striatal synaptic dysfunction and cell death [240]. Thus, it is possible to assume that the differential expression of synaptic and extra-synaptic NMDA receptors determines the extent of neurotoxicity, suggesting that a sustained increase in extracellular glutamate is the sole mechanism underlying cell death. DA, however, has been found to differentially modulate synaptic and extra-synaptic receptors. For instance, D1 receptor-mediated potentiation of NMDA responses is increased by genetic deletion of the NR2A subunit [236]. Moreover, NMDA receptors dominated by NR2A subunits are linked to glutamate responses in MSNs expressing D1 receptors, while NR2B-dominated NMDA receptors are associated with MSNs expressing D2 receptors [236]. This interplay is also important in neuronal morphological changes. Analysis of corticostriatal morphology after treatment with a D1 receptor agonist revealed an increase in the width of spine heads, and co-treatment with a NR2A antagonist, further enhanced this effect [241], suggesting that NMDA receptors counterbalance DA effects.
Another line of evidence indicates that in striatum proteins involved with postsynaptic receptor stabilization are dysregulated in HD [35]. NMDA and AMPA receptors interact with the postsynaptic density-95 (PSD-95) protein, a scaffolding protein that recruits receptors and signaling molecules. The interaction of glutamate receptors with PSD-95 has neuroprotective implications for HD [242]. PSD-95 contains the domain Src homology 3 (SH3), which in normal conditions interacts with HTT. An important feature of HD is that mHTT fails to bind to PSD-95 (Fig. 3) and also prevents the interaction with non-mutant HTT causing sensitization of NMDA and promoting apoptosis [242]. In both YAC128 and YAC72 mice, there is evidence of increased NMDA interaction with PSD-95 mediated by mHTT with implications for increased neurotoxicity [243]. PDS-95 is also decreased in Q175 mice [35].
PSD-95 also interacts with D1 receptors located in spines and prevents activation of D1 receptor signaling [244]. Moreover, PSD-95 forms ternary protein complexes with D1 and NMDA receptors [225]. Both receptors are expressed in MSN spine heads, where the majority of corticostriatal glutamatergic synapses are found [245]. Thus, failure of PSD-95 function would over-activate both NMDA and D1 receptors, resulting in dysfunctional corticostriatal communication. The modulation of DA by PSD-95 is still elusive, but evidence suggests that the genetic deletion of PSD-95 produces concomitant activation of D1 and NMDA receptors, resulting in motor impairments and striatal degeneration [246]. In addition, signaling pathways associated with D1 but not D2 receptors induce cell-death mediated by mHTT [202, 215]. It is likely, therefore, that increased expression of mHTT triggered by D1 receptors may prevent binding between NMDA-PSD-95 to promote sensitization of NMDA receptors.
In a further link to D1 receptors, glutamate release onto D1- but not D2-receptor-expressing striatal neurons is increased early in YAC128 mice, but later switches to a decrease [217]. Moreover, although D1 receptor expression may decrease, receptor signaling is increased as shown by enhanced cAMP and DARPP-32 phosphorylation [58, 113, 182, 183, 188], suggesting augmented PKA activation, which in turn will increase NMDA receptor phosphorylation [222, 227, 228, 247]. Increased NMDA receptor activity and the synergistic action of D1 receptors on intracellular calcium could activate apoptosis [214, 234, 235]. The over-activation of D1 receptor signaling may contribute to an increase in mHTT aggregates, which in turn could disrupt the binding of NMDA and PSD-95 causing further sensitization of NMDA receptors, increased calcium signaling, neurotoxicity, and overall dysregulation of neuronal activity [202, 214, 242, 243]. Apart from modulation of NMDA receptors, D1 receptor activation in normal conditions increases L-type calcium channel currents [248, 249], decreases somatic K+ currents [250], and decreases activation of currents evoked by N- and P/Q-type calcium channels [251]. These channels control the small conductance calcium-activated K+ channels, which play an important role in slowing MSN spiking activity [252]. The end result is increased neuronal activity in the direct pathway [253].
DA in motor alterations
Early evidence for the contribution of DA dysfunction to the motor alterations of HD emerged from a report that L-DOPA, widely used to treat Parkinson’s disease, caused dyskinesia in asymptomatic HD patients [254]. There also was evidence of extensive atrophy of the SNpc in HD patients [255] as much as a 40% decrease of DA neurons has been reported [151, 152]. Nevertheless, increased levels of DA were found in the nigrostriatal pathway of HD patients who showed chorea-like motor symptoms, but not rigidity [91]. Interestingly, YAC128 mice also display motor hyperactivity at early stages followed by motor deficits at late stages [49]. Similar effects are found in Q175 mice, including reduced DA release in the striatum consistent with late-stage hypokinetic symptoms [35, 58]. There is evidence that binding of tetrabenazine (TBZ), which decreases DA release by blocking VMAT-2, is significantly decreased in patients with akinesia and rigidity compared to patients with chorea [156]. Because motor symptoms vary with the stage of the disease, a thorough understanding of DA involvement is difficult to achieve when most postmortem studies are based on late-stage data.
In animal models, DA has been widely implicated in motor symptoms. For example, in hypokinetic Q175 homozygous mice, a loss of DA in the corticostriatal circuit impaired glutamate transmission and low gamma oscillations in striatal LFPs [58]. The change in gamma is interesting because in normal conditions an interaction between low and high gamma oscillations plays a role in motor states. Low gamma power in striatum, for example, occurs in response to movement initiation [256], while high gamma oscillations predominate during running [257]. In YAC128 mice, D1 receptor-expressing MSNs show an age-related increase in the frequency of excitatory postsynaptic currents [217]. Because of the location of D1 receptors on striatonigral axon terminals, D1 receptors are closely related to movement disorders. It seems likely, therefore, that increased activity in the direct pathway is in part responsible for chorea-like symptoms. In agreement with this view, targeted ablation of striatal D1 receptors in mice is sufficient to cause gait disturbances and involuntary movements [258]. In addition, optogenetic approaches have revealed that selective activation of the striatonigral pathway is directly involved not only in movement initiation but also in motor action sequences [66, 173]. This effect, moreover, seems to be mediated by the inhibition of subsets of SNpr neurons [67, 173]. In symptomatic Q140 KIs, when motor activation declines, burst firing increases in SNpr compared to wild-type controls [259]. More studies are needed to identify the role of D1 receptors in SNpr activity and its relation to movement.
It is widely accepted that alterations in DA transmission in HD are biphasic with an increase in release occurring at early stages [260, 261] corresponding to chorea and hyperkinesia, and a subsequent decrease leading to late-stage hypokinesia [163, 165]. Changes in DA receptors are likely to accompany changes in release. To be effective in ameliorating motor symptoms, therefore, opposing DA treatments may be required for early- versus late-stage HD (see below).
DA in cognition and behavioral inflexibility
Cognition includes a wide range of mental processing such as attention, memory, perception, learning, decision-making, and problem-solving. All these aspects of cognition involve corticostriatal circuits. Here, we focus on cognitive flexibility, which is routinely affected in HD.
Cognitive flexibility is the ability to switch strategies during learning and refers to how rapidly a subject is able to adapt to changing circumstances [262]. The lack of cognitive flexibility or behavioral inflexibility can be measured in learning tasks, for example, by assessing perseverative responses during reversal learning [262–265]. Repetitive sequential behaviors are another extreme form of behavioral inflexibility also associated with some psychiatric conditions such as Tourette’s syndrome and obsessive-compulsive disorder. Behavioral inflexibility has been observed in both HD patients and animal models [6, 7, 266].
Reversal learning impairments in HD were noted when HD patients performed significantly lower than controls in the Wisconsin Card Sort Task (WCST) [267], which measures abstract reasoning and the ability to change problem-solving strategies when needed. The impairment, moreover, was found in late but not early stages of HD [268, 269]. In another study, however, the only predictor of cognitive decline among nine neuropsychological tests was significantly poor WCST performance in asymptomatic patients at approximately 3.72 years before HD onset [270]. Reversal learning deficits also have been found in HD animal models. In R6/2 mice, reversal learning was altered along with presynaptic proteins associated with glutamate transmitter release [271]. Q175 mice also showed deficits in reversal learning [6]. In tgHD rats, both reversal learning and fear conditioning were impaired [272], similar results were found in YAC128 mice [273, 274]. Similarly, R6/2 mice have difficulty extinguishing conditioned fear, an effect that correlated with altered activity in prefrontal cortex [275]. In a two-choice swim test, Q175 mice showed an increased latency in decision making [55].
Motor activity also seems inflexible. Repetitive, inflexible or stereotypic behaviors have been observed in HD patients [276] and HD animal models [85, 86, 217, 277]. Interestingly, DA innervation of the striatum has long been known to play a critical role in the repetitive movements triggered by psychomotor stimulants [278], and D1 receptor agonists administered systemically or directly in the ventricle produce robust stereotypic grooming [279–281]. Whether a hyperdopaminergic state underlies the chorea and related-motor signs of HD remains to be established.
Flexibility in learning appears to involve the circuit from prefrontal cortex (PFC) to dorsomedial striatum [282–284], while the neural pathway from sensorimotor cortex (SMC) to dorsolateral striatum is associated with repetitive behaviors [285, 286] and decision-making [287]. Corticostriatal dysfunction has been correlated with cognitive impairment even at very early stages of HD [288, 289]. When striatal feedback to cortical areas via striato-BG-thalamo-cortical loops becomes dysfunctional, an inability to switch behavior is the result (for review [285]).
In pre-symptomatic and symptomatic HD patients, striatal D2 receptors are significantly decreased in patients with poor cognitive performance, but a D1 receptor decrease was associated only with altered sequential organization tasks [269]. Interestingly, increased activation of PKA was associated with disrupted recognition and spatial memory in HD [247], which could be explained by the lack of activation of D2 receptors or increased D1 receptor signaling [113]. Accordingly, both receptor types decrease gradually with progression of HD as the pre-symptomatic patients approached clinical onset [269, 290]. In addition, aberrant cortical synaptic plasticity and DA dysfunction has been reported for R6/1 mice; both D1 and D2 receptors are decreased in perirhinal cortex and the effects are reversed by quinpirole, a D2 receptor agonist [291]. Taken together, these data suggest that the ability of DA and its receptors to modulate corticostriatal circuitry contributes to the cognitive decline in HD.
DA in psychiatric symptoms
Although the diagnosis of HD is mostly based on motor and cognitive symptoms, psychiatric alterations also can be present, sometimes even years before the other symptoms become prominent [292, 293]. The prevalence of psychiatric symptoms in HD is variable, but mood disorders, irritability, aggression, impulsivity, psychotic episodes, rigidity, compulsive behavior and sexual disorders have been found in pre-symptomatic gene carriers [10, 293, 294]. Apathy is one of the most prevalent psychiatric symptoms in HD. A lack of interest in life activities and social interactions appears early and continues during HD progression, becoming one of the most disabling symptoms over time. Although 62% of early symptomatic patients show apathy, this trait is already evident in 32% of pre-symptomatic patients [2, 295–297].
Depression is the most common mood disorder in HD and has an early onset in about 50% of symptomatic patients [298, 299]. In fact, suicide among HD patients is 4 to 6 fold higher than in the general population [10, 300]. Irritability, impulsivity, and aggression can be additional reasons for hospitalization but are less prevalent than apathy and depression [301–303]. Psychotic episodes are even less common, but the data are difficult to interpret because antipsychotic medication, which blocks D2-like receptors, is sometimes used to ameliorate the chorea or motor hyperactivity that appears in early stages of HD. The frequency of paranoia, delusional states, anxiety, compulsive behavior, obsessive thoughts, and hallucinations has been estimated to range from 35–75% in HD patients [10, 302].
The collective group of psychiatric symptoms has been linked to dysfunctional activity in the PFC [149, 304–306]. For example, PET studies have reported evidence of hypometabolism in the PFC of depressed patients [307], and increased DA transmission has been found in the PFC during hallucinations and delusions [304, 305]. The PFC receives DA innervation from the ventral tegmental area (VTA) [170, 308]. This pathway also innervates the nucleus accumbens or ventral striatum, which plays a key role in reward [261]. DA in cortical areas can modulate synaptic responses controlling glutamate transmission [309, 310].
Neuroimaging studies of HD patients have implicated the PFC in these same psychiatric symptoms [298]. It already is clear from work on the R6/2 mouse model that information processing in the PFC is altered in HD mice [57]. In fact, abnormal neuronal activity in the prelimbic cortex, a region of the PFC with close ties to the amygdala, is correlated with reduced fear conditioning [275]. Although dysregulation of the corticostriatal circuit has been linked to motor and cognitive alterations in HD [36, 85, 86, 275, 311], very few studies have addressed the issue of DA system changes in HD associated with psychiatric disorders. Renoir and colleagues [312], using the forced swim test, found impaired DA transmission in R6/1 mice. This effect was reversed by bupropion, a weak DAT blocker, and a D1 receptor antagonist prevented the effect of bupropion, suggesting that D1 receptors might be involved in depression-like behaviors in HD. In fact, among the wide range of psychiatric symptoms identified in HD, depression is the only one associated with a decrease in DA transmission [306]. Interestingly, however, depression is not commonly associated with the progression of HD neuropathology, most likely because this symptom is sensitive to social and family environmental factors, which can vary widely among HD patients [297, 313].
DOPAMINE AS THERAPEUTIC TARGET
Because of DA involvement in motor control, cognition, and psychiatric symptoms, HD pharmacotherapy has targeted the DA system. To date, however, the overall success of this approach has been limited owing, in part, to side effects resulting from actions at non-DA receptors and, perhaps most importantly, from the increasing severity of HD progression, which limits long-term efficacy.
Inhibitors of vesicular monoamine transport
The VMAT-2 inhibitor, TBZ (Xenazine®), is the only federally approved drug in the United States used to treat HD, specifically chorea or episodes of uncontrollable movement [314–317]. TBZ inhibits VMAT-2 with a Ki ≈100 nmol/L [316, 318, 319]. By preventing the loading of DA into presynaptic vesicles, TBZ makes less DA available for release when DA neurons are activated [320].
Although VMAT-2 operates in norepinephrine and serotonin as well as DA neurons [315], TBZ has a higher affinity for striatal VMAT-2, making it possible to adjust the dose to limit the effects of this drug on monoamines in other brain regions [319]. The TBZ-induced reduction in chorea is evidence that this can be an effective strategy. Side effects, however, are still common. TBZ, for example, can exacerbate symptoms of depression and cognitive decline in HD patients [321]. Chronic treatment, moreover, results in the loss of efficacy and, more ominously, may cause cell death in the SNpc [322], suggesting that at least some of the side effect of TBZ could be caused by neurotoxicity. The pharmacogenetics of TBZ is another reason for concern. For instance, the activity of the cytochrome involved in TBZ metabolism, CY2D6, is genetically determined, making for fast or slow TBZ metabolizers. Thus, an effective dose with few side effects in one person may not apply to another [323].
Interestingly, a novel VMAT-2 inhibitor, NBI-641449, is undergoing phase III clinical trials for the treatment of hyperkinetic disorders. This compound has a higher affinity for VMAT-2 in DA neurons than in other monoamine neurons. Importantly for HD, NBI-641449 also decreases mHTT aggregates in cortex [324]. Further testing will determine if this compound is a viable alternative to TBZ.
DA antagonists
DA antagonists, including both typical and atypical antipsychotic drugs, have been used to block the hyper-DA state that contributes to the motor and psychiatric disorders in HD. Typical and atypical antipsychotics such as haloperidol, sulpiride, pimozidem, flufenazine, clozapine, olanzapine and risperidone have been used for some HD symptoms treatment. Haloperidol, a typical antipsychotic, is a potent antagonist of D2-like receptors with a slow kinetic dissociation and a high affinity for D3 receptors (0.74 nM) followed by D2 (1.55 nM) and D4 receptors (5–9 nM) [325–327]. Haloperidol is effective in reducing chorea [328–330] and also has been used for treating episodes of psychosis, aggression, and impulsivity [331]. Severe side effects, such as akathisia, dystonia, tardive dyskinesia, and neuroleptic malignant syndrome, have limited its use [331–333].
Sulpiride, another highly selective D2-like antagonist that has been used to treat dyskinesia in HD, has not been found to be very effective and induces side effects, such as drowsiness, in about 45% of patients [334]. Fluphenazine and pimozide are also typical antipsychotics that have been used to treat chorea [335]. The affinity of fluphenazine for DA receptors families is slightly higher for D1-like than D2-like receptors (KD≈0.7 and ≈3.2 nM, respectively) [336]. Pimozide has a higher selectivity for D2-like receptors [332]. Both of these antipsychotics have shown some efficacy in the treatment of chorea but side effects are common, and in the case of fluphenazine, low white blood cell levels limit its long-term use [337].
Clozapine and olanzapine belong to a group of atypical antipsychotics that show differential affinities for DA receptors. In fact, the affinity for the D2 receptor is lower (Ki = 157 nM) than for the D4 receptor (Ki = 25 nM). Clozapine also displays affinity for the D1-like receptor family [338]. Although the drug has shown some efficacy in reducing chorea [339], high doses were needed and the effect was seen only in antipsychotic naïve patients. No beneficial effect was reported for patients with a history of antipsychotic treatment. Clozapine also had several side effects, including drowsiness, fatigue, hyper-salivation, dizziness and walking difficulties [340]. Olanzapine, which also has an affinity for serotonin receptors, induces less akathisia and no dystonia or dyskinesia compared to haloperidol [333, 341], and is helpful in treating chorea and gait disturbances [342, 343]. Using the unified HD rating scale (UHDRS), olanzapine improved scores for chorea, depression, anxiety, irritability and obsessive thinking, but none reached statistical significance [344]. Increasing the dose appeared to have helped, but side effects also increased, resulting in a high risk of dyslipidemia, hyperglycemia and weight gain [345].
DA agonists
Unlike DA antagonists, which have been used to treat chorea, DA agonists, including L-DOPA, have been used as a treatment option for the hypokinetic symptoms or bradykinesia of HD. Aripiprazole (Abilify®) displays a very complex pharmacological profile. It is a partial agonist of D2-like receptors, with higher affinity for D2 and D3 receptors (Ki = 1.6 and 5.4 nM, respectively) than D4 receptors (Ki = 514 nM). Adding to the complexity, aripiprazole also displays high affinity for 5HT1A, 5HT2A, and 5HT2B receptors with Ki values ranging from 0.4 to 8.7 nM [346]. Aripiprazole also has been found to bind to adrenergic and muscarinic receptors but with lower affinities [347]. In clinical practice, treatment with aripiprazole showed improvement in motor disturbances and reduction of depression with fewer sedative effects than TBZ [348, 349]. Bromocriptine, another potent agonist of D2 receptors, was proposed as potential therapy for HD bradykinesia after the drug was shown to be effective in treating the bradykinesia in Parkinson’s patients, but despite some success in tests of animal models, no efficacy was found in HD patients [350].
L-DOPA, a DA precursor and hallmark treatment for Parkinson’s disease, has been found effective in treating gait disturbances and rigidity in HD, but L-DOPA-induced dyskinesia is a common side effect [351–353].
DA stabilizers
Two pharmacological properties play an important role in DA receptor operation: kinetic dissociation and intrinsic activity. DA receptors have dual intrinsic activity referred to as a high-affinity (high) state or low-affinity (low) state. The effect of DA greatly depends on this particular pharmacological property. For example, tonic firing of DA neurons would activate low-state affinity receptors, while burst firing would activate high-state affinity receptors [354]. The affinity of a compound for its receptors depends of the type of interaction between association and dissociation constants (Kon or Koff, respectively) [355]. This is an important characteristic because DA has a higher affinity for D1-like receptors than for D2-like receptors [355]. Evidence suggests, however, that D2 receptors located pre-synaptically to modulate the release of transmitters are in a high-affinity state (D2high) and display slower Koff, while postsynaptic receptors are equally distributed between high and low affinity states [356]. Thus, it may be possible to use the affinity state of DA receptors to develop more effective treatments. For example, we recently proposed that D3 receptor-selective compounds with bitropic activity may be effective antipsychotics with relatively few side effects [357, 358].
In this context, a novel class of compounds known as dopidines was found to be effective in differentially activating high or low state affinity receptors depending on DA concentration. Thus, these drugs became known as “DA stabilizers”. Pridopidine, the representative compound in this class, binds with different affinities to D2 receptors. It has a Ki = 7521 nM for D2 receptorshigh but a Ki = 17550 nM for D2 receptorslow. In addition, pridopidine rapidly dissociates from the receptor (Koff) and binds primarily to activated D2 receptors [356].
Pridopidine has been shown to modulate motor activity by a dual action, antagonizing D2 receptors and modulating glutamate [359]. In HD, pridopidine has shown promising results, although some individual differences and side effects have been reported [360, 361]. In R6/2 mice, pridopidine improved motor behavior, showed anti-apoptotic effects, promoted release of brain-derived neurotrophic factor, a neuroprotectant, and reduced mHTT aggregates. In HD clinical trials, pridopidine has been reported to cause small improvements in global motor scores, hand movements, and gait balance, but side effects such as falls, chorea, dizziness, and nausea also have been reported [360, 361].
Surprisingly, however, the effects of pridopidine are likely to include an action at sigma-1 receptors [362]. Several studies have reported potential benefits of selective sigma-1 receptor compounds in neurodegenerative diseases and stroke [363–365]. In fact, we have reported a highly selective sigma-1 receptor compound (Ki = 3.5 nM) that is neuroprotective, increases brain-derived trophic factor, reverses cognitive impairments, and decreases the behavioral effects of a potent hallucinogen [363, 366, 367]. Further confirmation of the pharmacodynamics of this compound is needed but seems a promising new treatment. Perhaps an action at both D2 and sigma-1 receptors could lead the way toward an effective treatment.
CONCLUSIONS
Corticostriatal dysfunction is an early contributor to the HD behavioral phenotype. Abnormalities in cortical glutamate input to striatal MSNs and interneurons disrupts the activity of the direct and indirect pathways, changing GABA transmission in the direct and indirect downstream pathways of the BG. DA, a key modulator of these pathways, also has a role to play in HD. Both D1- and D2-like receptor families have been implicated in the motor, cognitive, and psychiatric symptoms. Dysregulation of D1 receptor signaling, moreover, has been linked to the aggregation of mHTT and abnormal modulation of NMDA receptor activity. In fact, the majority of treatments for HD target DA even though the precise DA dysfunction has yet to be identified. DA, like glutamate and GABA, is intricately involved in striatal processing of cortical information and its progression through downstream BG circuits. Improved understanding of the mechanisms by which DA participates in the corticostriatal dysfunction underlying HD can have far-reaching effects in the ongoing search for an effective treatment.
CONFLICT OF INTERESTS
The authors have no conflicts to declare.
ACKNOWLEDGMENTS
Preparation of this review was supported by CHDI.
REFERENCES
[1] | Evans SJ , Douglas I , Rawlins MD , Wexler NS , Tabrizi SJ , Smeeth L . Prevalence of adult Huntington’s disease in the UK based on diagnoses recorded in general practice records. J Neurol Neurosurg Psychiatry. (2013) ;84: (10):1156–60. |
[2] | Bates GP , Dorsey R , Gusella JF , Hayden MR , Kay C , Leavitt BR , et al. Huntington disease. Nat Rev Dis Primers. (2015) ;1: :15005. |
[3] | Warby SC , Graham RK , Hayden MR . Huntington Disease. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al, editors. GeneReviews. Seattle;(1993) . pp. 1993–2016. |
[4] | Dayalu P , Albin RL . Huntington disease: Pathogenesis and treatment. Neurol Clin. (2015) ;33: (1):101–14. |
[5] | Novak MJ , Tabrizi SJ . Huntington’s disease. BMJ. (2010) ;340: :c3109. |
[6] | Farrar AM , Murphy CA , Paterson NE , Oakeshott S , He D , Alosio W , et al. Cognitive deficits in transgenic and knock-in HTT mice parallel those in Huntington’s disease. J Huntingtons Dis. (2014) ;3: (2):145–58. |
[7] | Curtin PC , Farrar AM , Oakeshott S , Sutphen J , Berger J , Mazzella M , et al. Cognitive training at a young age attenuates deficits in the zQ175 mouse model of HD. Front Behav Neurosci. (2016) ;9: :361. |
[8] | Frank S . Treatment of Huntington’s disease. Neurotherapeutics. (2014) ;11: (1):153–60. |
[9] | Paulsen JS , Conybeare RA . Cognitive changes in Huntington’s disease. Adv Neurol. (2005) ;96: :209–25. |
[10] | Roos RA . Huntington’s disease: A clinical review. Orphanet J Rare Dis. (2010) ;5: :40. doi: 10.1186/1750-1172-5-40 |
[11] | Parent A , Hazrati LN . Functional anatomy of the basal ganglia. I. The cortico-basal ganglia-thalamo-cortical loop. Brain Res Brain Res Rev. (1995) ;20: (1):91–127. |
[12] | Shipp S . The functional logic of corticostriatal connections. Brain Struct Funct. (2016) . doi: 10.1007/s00429-016-1250-9 |
[13] | Rüb U , Vonsattel JP , Heinsen H , Korf HW . The Neuropathology of Huntington s disease: Classical findings, recent developments and correlation to functional neuroanatomy. Adv Anat Embryol Cell Biol. (2015) ;217: :1–146. |
[14] | Mehrabi NF , Waldvogel HJ , Tippett LJ , Hogg VM , Synek BJ , Faull RL . Symptom heterogeneity in Huntington’s disease correlates with neuronal degeneration in the cerebral cortex. Neurobiol Dis. (2016) ;96: :67–74. |
[15] | Cepeda C , Itri JN , Flores-Hernandez J , Hurst RS , Calvert CR , Levine MS . Differential sensitivity of medium- and large-sized striatal neurons to NMDA but not kainate receptor activation in the rat. Eur J Neurosci. (2001) ;14: (10):1577–89. |
[16] | Cepeda C , Starling AJ , Wu N , Nguyen OK , Uzgil B , Soda T , et al. Increased GABAergic function in mouse models of Huntington’s disease: Reversal by BDNF. J Neurosci Res. (2004) ;78: (6):855–67. |
[17] | Andre VM , Cepeda C , Fisher YE , Huynh M , Bardakjian N , Singh S , et al. Differential electrophysiological changes in striatal output neurons in Huntington’s disease. J Neurosci. (2011) ;31: (4):1170–82. |
[18] | Raymond LA , Andre VM , Cepeda C , Gladding CM , Milnerwood AJ , Levine MS . Pathophysiology of Huntington’s disease: Time-dependent alterations in synaptic and receptor function. Neuroscience. (2011) ;198: :252–73. |
[19] | Estrada-Sanchez AM , Rebec GV . Corticostriatal dysfunction and glutamate transporter 1 (GLT1) in Huntington’s disease: Interactions between neurons and astrocytes. Basal Ganglia. (2012) ;2: (2):57–66. |
[20] | Ghiglieri V , Bagetta V , Calabresi P , Picconi B . Functional interactions within striatal microcircuit in animal models of Huntington’s disease. Neuroscience. (2012) ;211: :165–84. |
[21] | Valencia A , Sapp E , Kimm JS , McClory H , Ansong KA , Yohrling G , et al. Striatal synaptosomes from Hdh140Q/140Q knock-in mice have altered protein levels, novel sites of methionine oxidation, and excess glutamate release after stimulation. J Huntingtons Dis. (2013) ;2: (4):459–75. |
[22] | Indersmitten T , Tran CH , Cepeda C , Levine MS . Altered excitatory and inhibitory inputs to striatal medium-sized spiny neurons and cortical pyramidal neurons in the Q175 mouse model of Huntington’s disease. J Neurophysiol. (2015) ;113: (7):2953–66. |
[23] | Lu B , Palacino J . A novel human embryonic stem cell-derived Huntington’s disease neuronal model exhibits mutant huntingtin (mHTT) aggregates and soluble mHTT-dependent neurodegeneration. FASEB J. (2013) ;27: (5):1820–9. |
[24] | Watkin EE , Arbez N , Waldron-Roby E , O’Meally R , Ratovitski T , Cole RN , et al. Phosphorylation of mutant huntingtin at serine 116 modulates neuronal toxicity. PLoS One. (2014) ;9: (2):e88284. |
[25] | Hoffner G , Djian P . Polyglutamine aggregation in Huntington Disease: Does structure determine toxicity? Mol Neurobiol. (2015) ;52: (3):1297–314. |
[26] | Andrew SE , Goldberg YP , Kremer B , Telenius H , Theilmann J , Adam S , et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington’s disease. Nat Genet. (1993) ;4: (4):398–403. |
[27] | Lee JM , Gillis T , Mysore JS , Ramos EM , Myers RH , Hayden MR , et al. Common SNP-based haplotype analysis of the 4p16.3 Huntington disease gene region. Am J Hum Genet. (2012) ;90: (3):434–44. |
[28] | Rasmussen A , Macias R , Yescas P , Ochoa A , Davila G , Alonso E . Huntington disease in children: Genotype-phenotype correlation. Neuropediatrics. (2000) ;31: (4):190–4. |
[29] | Gencik M , Hammans C , Strehl H , Wagner N , Epplen JT . Chorea Huntington: A rare case with childhood onset. Neuropediatrics. (2002) ;33: (2):90–2. |
[30] | Vonsattel JP , Myers RH , Stevens TJ , Ferrante RJ , Bird ED , Richardson EP Jr . Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol. (1985) ;44: (6):559–77. |
[31] | van der Burg JM , Bjorkqvist M , Brundin P . Beyond the brain: Widespread pathology in Huntington’s disease. Lancet Neurol. (2009) ;8: (8):765–74. |
[32] | Lee FJ , Xue S , Pei L , Vukusic B , Chery N , Wang Y , et al. Dual regulation of NMDA receptor functions by direct protein-protein interactions with the dopamine D1 receptor. Cell. (2002) ;111: (2):219–30. |
[33] | DiFiglia M , Sapp E , Chase KO , Davies SW , Bates GP , Vonsattel JP , et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. (1997) ;277: (5334):1990–3. |
[34] | Smith R , Brundin P , Li JY . Synaptic dysfunction in Huntington’s disease: A new perspective. Cell Mol Life Sci. (2005) ;62: (17):1901–12. |
[35] | Smith GA , Rocha EM , McLean JR , Hayes MA , Izen SC , Isacson O , et al. Progressive axonal transport and synaptic protein changes correlate with behavioral and neuropathological abnormalities in the heterozygous Q175 KI mouse model of Huntington’s disease. Hum Mol Genet. (2014) ;23: (17):4510–27. |
[36] | Estrada-Sanchez AM , Burroughs CL , Cavaliere S , Barton SJ , Chen S , Yang XW , et al. Cortical efferents lacking mutant huntingtin improve striatal neuronal activity and behavior in a conditional mouse model of Huntington’s disease. J Neurosci. (2015) ;35: (10):4440–51. |
[37] | Pan Y , Daito T , Sasaki Y , Chung YH , Xing X , Pondugula S , et al. Inhibition of DNA methyltransferases blocks mutant huntingtin-induced neurotoxicity. Sci Rep. (2016) ;6: :31022. |
[38] | Cepeda C , Hurst RS , Calvert CR , Hernandez-Echeagaray E , Nguyen OK , Jocoy E , et al. Transient and progressive electrophysiological alterations in the corticostriatal pathway in a mouse model of Huntington’s disease. J Neurosci. (2003) ;23: (3):961–9. |
[39] | Levine MS , Cepeda C , Hickey MA , Fleming SM , Chesselet MF . Genetic mouse models of Huntington’s and Parkinson’s diseases: Illuminating but imperfect. Trends Neurosci. (2004) ;27: (11):691–7. |
[40] | Cepeda C , Wu N , Andre VM , Cummings DM , Levine MS . The corticostriatal pathway in Huntington’s disease. Prog Neurobiol. (2007) ;81: (5-6):253–71. |
[41] | Mangiarini L , Sathasivam K , Seller M , Cozens B , Harper A , Hetherington C , et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. (1996) ;87: (3):493–506. |
[42] | Schilling G , Becher MW , Sharp AH , Jinnah HA , Duan K , Kotzuk JA , et al. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum Mol Genet. (1999) ;8: (3):397–407. |
[43] | Ferrante RJ . Mouse models of Huntington’s disease and methodological considerations for therapeutic trials. Biochim Biophys Acta. (2009) ;1792: (6):506–20. |
[44] | Pollock K , Dahlenburg H , Nelson H , Fink KD , Cary W , Hendrix K , et al. Human mesenchymal stem cells genetically engineered to overexpress brain-derived neurotrophic factor improve outcomes in Huntington’s disease mouse models. Mol Ther. (2016) ;24: (5):965–77. |
[45] | von Hörsten S , Schmitt I , Nguyen HP , Holzmann C , Schmidt T , Walther T , et al. Transgenic rat model of Huntington’s disease. Hum Mol Genet. (2003) ;12: (6):617–24. |
[46] | Cao C , Temel Y , Blokland A , Ozen H , Steinbusch HW , Vlamings R , et al. Progressive deterioration of reaction time performance and choreiform symptoms in a new Huntington’s disease transgenic ratmodel. Behav Brain Res. (2006) ;170: (2):257–61. |
[47] | Fink KD , Rossignol J , Crane AT , Davis KK , Bavar AM , Dekorver NW , et al. Early cognitive dysfunction in the HD 51 CAG transgenic rat model of Huntington’s disease. Behav Neurosci. (2012) ;126: (3):479–87. |
[48] | Hodgson JG , Agopyan N , Gutekunst CA , Leavitt BR , LePiane F , Singaraja R , et al. A YAC mouse model for Huntington’s disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron. (1999) ;23: (1):181–92. |
[49] | Slow EJ , van Raamsdonk J , Rogers D , Coleman SH , Graham RK , Deng Y , et al. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet. (2003) ;12: (13):1555–67. |
[50] | Gray M , Shirasaki DI , Cepeda C , Andre VM , Wilburn B , Lu XH , et al. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J Neurosci. (2008) ;28: (24):6182–95. |
[51] | Spampanato J , Gu X , Yang XW , Mody I . Progressive synaptic pathology of motor cortical neurons in a BAC transgenic mouse model of Huntington’s disease. Neuroscience. (2008) ;157: (3):606–20. |
[52] | Menalled LB , Sison JD , Dragatsis I , Zeitlin S , Chesselet MF . Time course of early motor and neuropathological anomalies in a knock-in mouse model of Huntington’s disease with 140 CAG repeats. J Comp Neurol. (2003) ;465: (1):11–26. |
[53] | Menalled LB . Knock-in mouse models of Huntington’s disease. NeuroRx. (2005) ;2: (3):465–70. |
[54] | Menalled LB , Kudwa AE , Miller S , Fitzpatrick J , Watson-Johnson J , Keating N , et al. Comprehensive behavioral and molecular characterization of a new knock-in mouse model of Huntington’s disease: zQ175. PLoS One. (2012) ;7: (12):e49838. |
[55] | Heikkinen T , Lehtimaki K , Vartiainen N , Puolivali J , Hendricks SJ , Glaser JR , et al. Characterization of neurophysiological and behavioral changes, MRI brain volumetry and 1H MRS in zQ175 knock-in mouse model of Huntington’s disease. PLoS One. (2012) ;7: (12):e50717. |
[56] | Miller BR , Walker AG , Shah AS , Barton SJ , Rebec GV . Dysregulated information processing by medium spiny neurons in striatum of freely behaving mouse models of Huntington’s disease. J Neurophysiol. (2008) ;100: (4):2205–16. |
[57] | Walker AG , Miller BR , Fritsch JN , Barton SJ , Rebec GV . Altered information processing in the prefrontal cortex of Huntington’s disease mouse models. J Neurosci. (2008) ;28: (36):8973–82. |
[58] | Rothe T , Deliano M , Wojtowicz AM , Dvorzhak A , Harnack D , Paul S , et al. Pathological gamma oscillations, impaired dopamine release, synapse loss and reduced dynamic range of unitary glutamatergic synaptic transmission in the striatum of hypokinetic Q175 Huntington mice. Neuroscience. (2015) ;311: :519–38. |
[59] | Penney JB , Young AB . Speculations on the functional anatomy of basal ganglia disorders. Annu Rev Neurosci. (1983) ;6: :73–94. |
[60] | Albin RL , Young AB , Penney JB . The functional anatomy of basal ganglia disorders. Trends Neurosci. (1989) ;12: (10):366–75. |
[61] | Alexander GE , Crutcher MD . Functional architecture of basal ganglia circuits: Neural substrates of parallel processing. Trends Neurosci. (1990) ;13: (7):266–71. |
[62] | Gerfen CR , Engber TM , Mahan LC , Susel Z , Chase TN , Monsma FJ Jr , et al. D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science. (1990) ;250: (4986):1429–32. |
[63] | Gerfen CR , Miyachi S , Paletzki R , Brown P . D1 dopamine receptor supersensitivity in the dopamine-depleted striatum results from a switch in the regulation of ERK1/2/MAP kinase. J Neurosci. (2002) ;22: (12):5042–54. |
[64] | Jin X , Tecuapetla F , Costa RM . Basal ganglia subcircuits distinctively encode the parsing and concatenation of action sequences. Nat Neurosci. (2014) ;17: (3):423–30. |
[65] | Tecuapetla F , Matias S , Dugue GP , Mainen ZF , Costa RM . Balanced activity in basal ganglia projection pathways is critical for contraversive movements. Nat Commun. (2014) ;5: :4315. |
[66] | Tecuapetla F , Jin X , Lima SQ , Costa RM . Complementary contributions of striatal projection pathways to action initiation and execution. Cell. (2016) ;166: (3):703–15. |
[67] | Freeze BS , Kravitz AV , Hammack N , Berke JD , Kreitzer AC . Control of basal ganglia output by direct and indirect pathway projection neurons. J Neurosci. (2013) ;33: (47):18531–9. |
[68] | Richfield EK , Maguire-Zeiss KA , Vonkeman HE , Voorn P . Preferential loss of preproenkephalin versus preprotachykinin neurons from the striatum of Huntington’s disease patients. Ann Neurol. (1995) ;38: (6):852–61. |
[69] | Sapp E , Ge P , Aizawa H , Bird E , Penney J , Young AB , et al. Evidence for a preferential loss of enkephalin immunoreactivity in the external globus pallidus in low grade Huntington’s disease using high resolution image analysis. Neuroscience. (1995) ;64: (2):397–404. |
[70] | Albin RL , Reiner A , Anderson KD , Dure LS , Handelin B , Balfour R , et al. Preferential loss of striato-external pallidal projection neurons in presymptomatic Huntington’s disease. Ann Neurol. (1992) ;31: (4):425–30. |
[71] | Lei W , Jiao Y , Del Mar N , Reiner A . Evidence for differential cortical input to direct pathway versus indirect pathway striatal projection neurons in rats. J Neurosci. (2004) ;24: (38):8289–99. |
[72] | Deng YP , Albin RL , Penney JB , Young AB , Anderson KD , Reiner A . Differential loss of striatal projection systems in Huntington’s disease: A quantitative immunohistochemical study. J Chem Neuroanat. (2004) ;27: (3):143–64. |
[73] | Barry J , Cepeda C , Levine MS . Direct and Indirect striatal output pathways differentially affect their targets in mouse models of Huntongton’s disease. Poster Presentation. Hereditary Disease Foundation (HD2016). “The Milton Wexler Celebration of Life”. August 3-6, 2016. Cambridge, MA. |
[74] | Levesque M , Charara A , Gagnon S , Parent A , Deschenes M . Corticostriatal projections from layer V cells in rat are collaterals of long-range corticofugal axons. Brain Res. (1996) ;709: (2):311–5. |
[75] | Levesque M , Parent A . Axonal arborization of corticostriatal and corticothalamic fibers arising from prelimbic cortex in the rat. Cereb Cortex. (1998) ;8: (7):602–13. |
[76] | Reiner A , Dragatsis I , Zeitlin S , Goldowitz D . Wild-type huntingtin plays a role in brain development and neuronal survival. Mol Neurobiol. (2003) ;28: (3):259–76. |
[77] | Reiner A , Hart NM , Lei W , Deng Y . Corticostriatal projection neurons - dichotomous types and dichotomous functions. Front Neuroanat. (2010) ;25;4: :142. doi: 10.3389/fnana.2010.00142 |
[78] | Shepherd GM . Corticostriatal connectivity and its role in disease. Nat Rev Neurosci. (2013) ;14: (4):278–91. |
[79] | Reiner A . Organization of corticostriatal projection neuron types. In Steinerand H and Tseng KY, editors. Handbook of Basal Ganglia Structure and Function. New York: Academic Press; (2010) . pp. 323–340. |
[80] | Beloozerova IN , Sirota MG , Swadlow HA , Orlovsky GN , Popova LB , Deliagina TG . Activity of different classes of neurons of the motor cortex during postural corrections. J Neurosci. (2003) ;23: (21):7844–53. |
[81] | DeLong MR , Huang KT , Gallis J , Lokhnygina Y , Parente B , Hickey P , et al. Effect of advancing age on outcomes of deep brain stimulation for Parkinson disease. JAMA Neurol. (2014) ;71: (10):1290–5. |
[82] | Geinisman Y . Perforated axospinous synapses with multiple, completely partitioned transmission zones: Probable structural intermediates in synaptic plasticity. Hippocampus. (1993) ;3: (4):417–33. |
[83] | Sulzer D , Pothos EN . Regulation of quantal size by presynaptic mechanisms. Rev Neurosci. (2000) ;11: (2-3):159–212. |
[84] | Reiner A , Albin RL , Anderson KD , D’Amato CJ , Penney JB , Young AB . Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci U S A. (1988) ;85: (15):5733–7. |
[85] | Hong SL , Cossyleon D , Hussain WA , Walker LJ , Barton SJ , Rebec GV . Dysfunctional behavioral modulation of corticostriatal communication in the R6/2 mouse model of Huntington’s disease. PLoS One. (2012) ;7: (10):e47026. |
[86] | Miller BR , Walker AG , Barton SJ , Rebec GV . Dysregulated neuronal activity patterns implicate corticostriatal circuit dysfunction in multiple rodent models of Huntington’s disease. Front Syst Neurosci. (2011) ;5: :26. |
[87] | DiFiglia M . Excitotoxic injury of the neostriatum: A model for Huntington’s disease. Trends Neurosci. (1990) ;13: (7):286–9. |
[88] | Klapstein GJ , Fisher RS , Zanjani H , Cepeda C , Jokel ES , Chesselet MF , et al. Electrophysiological and morphological changes in striatal spiny neurons in R6/2 Huntington’s disease transgenic mice. J Neurophysiol. (2001) ;86: (6):2667–77. |
[89] | Spires TL , Grote HE , Garry S , Cordery PM , Van Dellen A , Blakemore C , et al. Dendritic spine pathology and deficits in experience-dependent dendritic plasticity in R6/1 Huntington’s disease transgenic mice. Eur J Neurosci. (2004) ;19: (10):2799–807. |
[90] | Fernandez-Nogales M , Hernandez F , Miguez A , Alberch J , Gines S , Perez-Navarro E , et al. Decreased glycogen synthase kinase-3 levels and activity contribute to Huntington’s disease. Hum Mol Genet. (2015) ;24: (17):5040–52. |
[91] | Spokes EG . Neurochemical alterations in Huntington’s chorea: A study of post-mortem brain tissue. Brain. (1980) ;103: (1):179–210. |
[92] | Gu X , Li C , Wei W , Lo V , Gong S , Li SH , et al. Pathological cell-cell interactions elicited by a neuropathogenic form of mutant Huntingtin contribute to cortical pathogenesis in HD mice. Neuron. (2005) ;46: (3):433–44. |
[93] | Andre VM , Cepeda C , Venegas A , Gomez Y , Levine MS . Altered cortical glutamate receptor function in the R6/2 model of Huntington’s disease. J Neurophysiol. (2006) ;95: (4):2108–19. |
[94] | Wang N , Gray M , Lu XH , Cantle JP , Holley SM , Greiner E , et al. Neuronal targets for reducing mutant huntingtin expression to ameliorate disease in a mouse model of Huntington’s disease. Nat Med. (2014) ;20: (5):536–41. |
[95] | Luk KC , Sadikot AF . GABA promotes survival but not proliferation of parvalbumin-immunoreactive interneurons in rodent neostriatum: An in vivo study with stereology. Neuroscience. (2001) ;104: (1):93–103. |
[96] | Tepper JM , Koos T , Wilson CJ . GABAergic microcircuits in the neostriatum. Trends Neurosci. (2004) ;27: (11):662–9. |
[97] | Kawaguchi Y . Physiological, morphological, and histochemical characterization of three classes of interneurons in rat neostriatum. J Neurosci. (1993) ;13: (11):4908–23. |
[98] | Kawaguchi Y , Wilson CJ , Augood SJ , Emson PC . Striatal interneurones: Chemical, physiological and morphological characterization. Trends Neurosci. (1995) ;18: (12):527–35. |
[99] | Lapper SR , Bolam JP . Input from the frontal cortex and the parafascicular nucleus to cholinergic interneurons in the dorsal striatum of the rat. Neuroscience. (1992) ;51: (3):533–45. |
[100] | Smith Y , Raju DV , Pare JF , Sidibe M . The thalamostriatal system: A highly specific network of the basal ganglia circuitry. Trends Neurosci. (2004) ;27: (9):520–7. |
[101] | Ding JB , Guzman JN , Peterson JD , Goldberg JA , Surmeier DJ . Thalamic gating of corticostriatal signaling by cholinergic interneurons. Neuron. (2010) ;67: (2):294–307. |
[102] | Ding J , Peterson JD , Surmeier DJ . Corticostriatal and thalamostriatal synapses have distinctive properties. J Neurosci. (2008) ;28: (25):6483–92. |
[103] | Kolodziejczyk K , Raymond LA . Differential changes in thalamic and cortical excitatory synapses onto striatal spiny projection neurons in a Huntington disease mouse model. Neurobiol Dis. (2016) ;86: :62–74. |
[104] | Deng YP , Wong T , Bricker-Anthony C , Deng B , Reiner A . Loss of corticostriatal and thalamostriatal synaptic terminals precedes striatal projection neuron pathology in heterozygous Q140 Huntington’s disease mice. Neurobiol Dis. (2013) ;60: :89–107. |
[105] | Parker PR , Lalive AL , Kreitzer AC . Pathway-specific remodeling of thalamostriatal synapses in parkinsonian mice. Neuron. (2016) ;89: (4):734–40. |
[106] | West MO , Michael AJ , Knowles SE , Chapin JK , Woodward DJ . Striatal unit activity and the linkage between sensory and motor events. In Schneider JS, Lidsky TI, editors. Basal Ganglia and Behavior: Sensory Aspects of Motor Functioning. Toronto: Hans Huber, (1987) . pp. 27–36 |
[107] | Gardiner TW , Iverson DA , Rebec GV . Heterogeneous responses of neostriatal neurons to amphetamine in freely moving rats. Brain Res. (1988) ;463: (2):268–74. |
[108] | Haracz JL , Tschanz JT , Wang Z , White IM , Rebec GV . Striatal single-unit responses to amphetamine and neuroleptics in freely moving rats. Neurosci Biobehav Rev. (1993) ;17: (1):1–12. |
[109] | White IM , Doubles L , Rebec GV . Cocaine-induced activation of striatal neurons during focused stereotypy in rats. Brain Res. (1998) ;810: (1-2):146–52. |
[110] | Wang Z , Rebec GV . Neuronal and behavioral correlates of intrastriatal infusions of amphetamine in freely moving rats. Brain Res. (1993) ;627: (1):79–88. |
[111] | Levine MS , Klapstein GJ , Koppel A , Gruen E , Cepeda C , Vargas ME , et al. Enhanced sensitivity to N-methyl-D-aspartate receptor activation in transgenic and knockin mouse models of Huntington’s disease. J Neurosci Res. (1999) ;58: (4):515–32. |
[112] | Rebec GV , Conroy SK , Barton SJ . Hyperactive striatal neurons in symptomatic Huntington R6/2 mice: Variations with behavioral state and repeated ascorbate treatment. Neuroscience. (2006) ;137: (1):327–36. |
[113] | Bibb JA , Yan Z , Svenningsson P , Snyder GL , Pieribone VA , Horiuchi A , et al. Severe deficiencies in dopamine signaling in presymptomatic Huntington’s disease mice. Proc Natl Acad Sci U S A. (2000) ;97: (12):6809–14. |
[114] | Tepper JM , Sharpe NA , Koos TZ , Trent F . Postnatal development of the rat neostriatum: Electrophysiological, light- and electron-microscopic studies. Dev Neurosci. (1998) ;20: (2-3):125–45. |
[115] | Koos T , Tepper JM . Inhibitory control of neostriatal projection neurons by GABAergic interneurons. Nat Neurosci. (1999) ;2: (5):467–72. |
[116] | Beal MF , Brouillet E , Jenkins BG , Ferrante RJ , Kowall NW , Miller JM , et al. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci. (1993) ;13: (10):4181–92. |
[117] | Reiner A , Shelby E , Wang H , Demarch Z , Deng Y , Guley NH , et al. Striatal parvalbuminergic neurons are lost in Huntington’s disease: Implications for dystonia. Mov Disord. (2013) ;28: (12):1691–9. |
[118] | Betarbet R , Turner R , Chockkan V , DeLong MR , Allers KA , Walters J , et al. Dopaminergic neurons intrinsic to the primate striatum. J Neurosci. (1997) ;17: (17):6761–8. |
[119] | Ibanez-Sandoval O , Tecuapetla F , Unal B , Shah F , Koos T , Tepper JM . Electrophysiological and morphological characteristics and synaptic connectivity of tyrosine hydroxylase-expressing neurons in adult mouse striatum. J Neurosci. (2010) ;30: (20):6999–7016. |
[120] | Tepper JM , Tecuapetla F , Koos T , Ibanez-Sandoval O . Heterogeneity and diversity of striatal GABAergic interneurons. Front Neuroanat. (2010) ;29;4: :150. doi: 10.3389/fnana.2010.00150 |
[121] | Gerfen CR , Baimbridge KG , Miller JJ . The neostriatal mosaic: Compartmental distribution of calcium-binding protein and parvalbumin in the basal ganglia of the rat and monkey. Proc Natl Acad Sci U S A. (1985) ;82: (24):8780–4. |
[122] | Ramanathan S , Hanley JJ , Deniau JM , Bolam JP . Synaptic convergence of motor and somatosensory cortical afferents onto GABAergic interneurons in the rat striatum. J Neurosci. (2002) ;22: (18):8158–69. |
[123] | Kita H , Kosaka T , Heizmann CW . Parvalbumin-immunoreactive neurons in the rat neostriatum: A light and electron microscopic study. Brain Res. (1990) ;17: (1-2):1–15. |
[124] | Fukuda T . Network architecture of gap junction-coupled neuronal linkage in the striatum. J Neurosci. (2009) ;29: (4):1235–43. |
[125] | Koos T , Tepper JM , Wilson CJ . Comparison of IPSCs evoked by spiny and fast-spiking neurons in the neostriatum. J Neurosci. (2004) ;24: (36):7916–22. |
[126] | Gustafson N , Gireesh-Dharmaraj E , Czubayko U , Blackwell KT , Plenz D . A comparative voltage and current-clamp analysis of feedback and feedforward synaptic transmission in the striatal microcircuit in vitro. J Neurophysiol. (2006) ;95: (2):737–52. |
[127] | Plenz D , Kitai ST . Up and down states in striatal medium spiny neurons simultaneously recorded with spontaneous activity in fast-spiking interneurons studied in cortex-striatum-substantia nigra organotypic cultures. J Neurosci. (1998) ;18: (1):266–83. |
[128] | Bracci E , Centonze D , Bernardi G , Calabresi P . Voltage-dependent membrane potential oscillations of rat striatal fast-spiking interneurons. J Physiol. (2003) ;549: (Pt 1):121–30. |
[129] | Berke JD , Okatan M , Skurski J , Eichenbaum HB . Oscillatory entrainment of striatal neurons in freely moving rats. Neuron. (2004) ;43: (6):883–96. |
[130] | Bennett MV , Zukin RS . Electrical coupling and neuronal synchronization in the Mammalian brain. Neuron. (2004) ;41: (4):495–511. |
[131] | Gernert M , Richter A , Loscher W . Subconvulsive dose of pentylenetetrazole increases the firing rate of substantia nigra pars reticulata neurons in dystonic but not in nondystonic hamsters. Synapse. (1999) ;33: (4):259–67. |
[132] | Gernert M , Hamann M , Bennay M , Loscher W , Richter A . Deficit of striatal parvalbumin-reactive GABAergic interneurons and decreased basal ganglia output in a genetic rodent model of idiopathic paroxysmal dystonia. J Neurosci. (2000) ;20: (18):7052–8. |
[133] | Xenias HS , Ibanez-Sandoval O , Koos T , Tepper JM . Are striatal tyrosine hydroxylase interneurons dopaminergic? J Neurosci. (2015) ;35: (16):6584–99. |
[134] | Ibanez-Sandoval O , Xenias HS , Tepper JM , Koos T . Dopaminergic and cholinergic modulation of striatal tyrosine hydroxylase interneurons. Neuropharmacology. (2015) ;95: :468–76. |
[135] | Huot P , Levesque M , Parent A . The fate of striatal dopaminergic neurons in Parkinson’s disease and Huntington’s chorea. Brain. (2007) ;130: (Pt 1):222–32. |
[136] | Aosaki T , Tsubokawa H , Ishida A , Watanabe K , Graybiel AM , Kimura M . Responses of tonically active neurons in the primate’s striatum undergo systematic changes during behavioral sensorimotor conditioning. J Neurosci. (1994) ;14: (6):3969–84. |
[137] | Kimura M , Rajkowski J , Evarts E . Tonically discharging putamen neurons exhibit set-dependent responses. Proc Natl Acad Sci U S A. (1984) ;81: (15):4998–5001. |
[138] | Perez-Rosello T , Figueroa A , Salgado H , Vilchis C , Tecuapetla F , Guzman JN , et al. Cholinergic control of firing pattern and neurotransmission in rat neostriatal projection neurons: Role of CaV2.1 and CaV2.2 Ca2+ channels. J Neurophysiol. (2005) ;93: (5):2507–19. |
[139] | Pisani A , Bernardi G , Ding J , Surmeier DJ . Re-emergence of striatal cholinergic interneurons in movement disorders. Trends Neurosci. (2007) ;30: (10):545–53. |
[140] | Suzuki M , Desmond TJ , Albin RL , Frey KA . Vesicular neurotransmitter transporters in Huntington’s disease: Initial observations and comparison with traditional synaptic markers. Synapse. (2001) ;41: (4):329–36. |
[141] | Smith R , Chung H , Rundquist S , Maat-Schieman ML , Colgan L , Englund E , et al. Cholinergic neuronal defect without cell loss in Huntington’s disease. Hum Mol Genet. (2006) ;15: (21):3119–31. |
[142] | Rot U , Kobal J , Sever A , Pirtosek Z , Mesec A . Rivastigmine in the treatment of Huntington’s disease. Eur J Neurol. (2002) ;9: (6):689–90. |
[143] | Deng YP , Reiner A . Cholinergic interneurons in the Q140 knock-in mouse model of Huntington’s disease: Reductions in dendritic branching and thalamostriatal input. J Comp Neurol. (2016) ;524: (17):3518–29. |
[144] | Groenewegen HJ . The basal ganglia and motor control. Neural Plast. (2003) ;10: (1-2):107–20. |
[145] | Backman L , Farde L . Dopamine and cognitive functioning: Brain imaging findings in Huntington’s disease and normal aging. Scand J Psychol. (2001) ;42: (3):287–96. |
[146] | Sleezer BJ , Hayden BY . Differential contributions of ventral and dorsal striatum to early and late phases of cognitive set reconfiguration. J Cogn Neurosci. (2016) ;15: :1–16. |
[147] | Rotaru DC , Lewis DA , Gonzalez-Burgos G . Dopamine D1 receptor activation regulates sodium channel-dependent EPSP amplification in rat prefrontal cortex pyramidal neurons. J Physiol. (2007) ;581: (3):981–1000. |
[148] | Scheggi S , Melis M , De Felice M , Aroni S , Muntoni AL , Pelliccia T , et al. PPARalpha modulation of mesolimbic dopamine transmission rescues depression-related behaviors. Neuropharmacology. (2016) ;110: (Pt A):251–9. |
[149] | Maia TV , Frank MJ . An integrative perspective on the role of dopamine in schizophrenia. Biol Psychiatry. (2016) ; doi: 10.1016/j.biopsych.2016.05.021 |
[150] | Kiyatkin EA , Rebec GV . Striatal neuronal activity and responsiveness to dopamine and glutamate after selective blockade of D1 and D2 dopamine receptors in freely moving rats. J Neurosci. (1999) ;19: (9):3594–609. |
[151] | Oyanagi K , Takeda S , Takahashi H , Ohama E , Ikuta F . A quantitative investigation of the substantia nigra in Huntington’s disease. Ann Neurol. (1989) ;26: (1):13–9. |
[152] | Gibb WR . Neuropathology of the substantia nigra. Eur Neurol. (1991) ;31: (Suppl 1):48–59. |
[153] | Bedard C , Wallman MJ , Pourcher E , Gould PV , Parent A , Parent M . Serotonin and dopamine striatal innervation in Parkinson’s disease and Huntington’s chorea. Parkinsonism Relat Disord. (2011) ;17: (8):593–8. |
[154] | Yohrling GJ 4th , Jiang GC , DeJohn MM , Miller DW , Young AB , Vrana KE , et al. Analysis of cellular, transgenic and human models of Huntington’s disease reveals tyrosine hydroxylase alterations and substantia nigra neuropathology. Brain Res Mol Brain Res. (2003) ;119: (1):28–36. |
[155] | Ginovart N , Lundin A , Farde L , Halldin C , Backman L , Swahn CG , et al. PET study of the pre- and post-synaptic dopaminergic markers for the neurodegenerative process in Huntington’s disease. Brain. (1997) ;120: (Pt 3):503–14. |
[156] | Bohnen NI , Koeppe RA , Meyer P , Ficaro E , Wernette K , Kilbourn MR , et al. Decreased striatal monoaminergic terminals in Huntington disease. Neurology. (2009) ;54: (9):1753–9. |
[157] | Kiferle L , Mazzucchi S , Unti E , Pesaresi I , Fabbri S , Nicoletti V , et al. Nigral involvement and nigrostriatal dysfunction in Huntington’s disease: Evidences from an MRI and SPECT study. Parkinsonism Relat Disord. (2013) ;19: (9):800–5. |
[158] | Petersen A , Puschban Z , Lotharius J , NicNiocaill B , Wiekop P , O’Connor WT , et al. Evidence for dysfunction of the nigrostriatal pathway in the R6/1 line of transgenic Huntington’s disease mice. Neurobiol Dis. (2002) ;11: (1):134–46. |
[159] | Dallerac GM , Levasseur G , Vatsavayai SC , Milnerwood AJ , Cummings DM , Kraev I , et al. Dysfunctional dopaminergic neurones in mouse models of Huntington’s disease: A role for SK3 channels. Neurodegener Dis. (2015) ;15: (2):93–108. |
[160] | Ortiz AN , Osterhaus GL , Lauderdale K , Mahoney L , Fowler SC , von Horsten S , et al. Motor function and dopamine release measurements in transgenic Huntington’s disease model rats. Brain Res. (2012) ;1450: :148–56. |
[161] | Johnson MA , Rajan V , Miller CE , Wightman RM . Dopamine release is severely compromised in the R6/2 mouse model of Huntington’s disease. J Neurochem. (2006) ;97: (3):737–46. |
[162] | Miller S , Hill Della Puppa G , Reidling J , Marcora E , Thompson LM , Treanor J . Comparison of phosphodiesterase 10A, dopamine receptors D1 and D2 and dopamine transporter ligand binding in the striatum of the R6/2 and BACHD mouse models of Huntington’s disease. J Huntingtons Dis. (2014) ;3: (4):333–41. |
[163] | Callahan JW , Abercrombie ED . In vivo dopamine efflux is decreased in striatum of both fragment (R6/2) and full-length (YAC128) transgenic mouse models of Huntington’s disease. Front Syst Neurosci. (2011) ;5: :61. |
[164] | Covey DP , Dantrassy HM , Zlebnik NE , Gildish I , Cheer JF . Compromised dopaminergic encoding of reward accompanying suppressed willingness to overcome high effort costs is a prominent prodromal characteristic of the Q175 mouse model of Huntington’s disease. J Neurosci. (2016) ;36: (18):4993–5002. |
[165] | Kish SJ , Shannak K , Hornykiewicz O . Elevated serotonin and reduced dopamine in subregionally divided Huntington’s disease striatum. Ann Neurol. (1987) ;22: (3):386–9. |
[166] | Mochel F , Durant B , Durr A , Schiffmann R . Altered dopamine and serotonin metabolism in motorically asymptomatic R6/2 mice. PLoS One. (2011) ;6: (3):e18336. |
[167] | Greengard P , Allen PB , Nairn AC . Beyond the dopamine receptor: The DARPP-32/protein phosphatase-1 cascade. Neuron. (1999) ;23: (3):435–47. |
[168] | Forn J , Krueger BK , Greengard P . Adenosine 3’,5’-monophosphate content in rat caudate nucleus: Demonstration of dopaminergic and adrenergic receptors. Science. (1974) ;186: (4169):1118–20. |
[169] | Arisawa M , Makino T , Izumi S , Iizuka R . Effect of prostaglandin D2 on gonadotropin release from rat anterior pituitary in vitro . Fertil Steril. (1983) ;39: (1):93–6. |
[170] | Missale C , Nash SR , Robinson SW , Jaber M , Caron MG . Dopamine receptors: From structure to function. Physiol Rev. (1998) ;78: (1):189–225. |
[171] | Rangel-Barajas C , Coronel I , Floran B . Dopamine receptors and neurodegeneration. Aging Dis. (2015) ;6: (5):349–68. |
[172] | Surmeier DJ , Yan Z , Song WJ . Coordinated expression of dopamine receptors in neostriatal medium spiny neurons. Adv Pharmacol. (1998) ;42: :1020–3. |
[173] | Chiken S , Sato A , Ohta C , Kurokawa M , Arai S , Maeshima J , et al. Dopamine D1 receptor-mediated transmission maintains information flow through the cortico-striato-entopeduncular direct pathway to release movements. Cereb Cortex. (2015) ;25: (12):4885–97. |
[174] | Surmeier DJ , Ding J , Day M , Wang Z , Shen W . D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. (2007) ;30: (5):228–35. |
[175] | Floran B , Aceves J , Sierra A , Martinez-Fong D . Activation of D1 dopamine receptors stimulates the release of GABA in the basal ganglia of the rat. Neurosci Lett. (1990) ;116: (1-2):136–40. |
[176] | Rangel-Barajas C , Silva I , Garcia-Ramirez M , Sanchez-Lemus E , Floran L , Aceves J , et al. 6-OHDA-induced hemiparkinsonism and chronic L-DOPA treatment increase dopamine D1-stimulated [(3)H]-GABA release and [(3)H]-cAMP production in substantia nigra pars reticulata of the rat. Neuropharmacology. (2008) ;55: (5):704–11. |
[177] | Rangel-Barajas C , Silva I , Lopez-Santiago LM , Aceves J , Erlij D , Floran B . L-DOPA-induced dyskinesia in hemiparkinsonian rats is associated with up-regulation of adenylyl cyclase type V/VI and increased GABA release in the substantia nigra reticulata. Neurobiol Dis. (2011) ;41: (1):51–61. |
[178] | Vicente AM , Galvao-Ferreira P , Tecuapetla F , Costa RM . Direct and indirect dorsolateral striatum pathways reinforce different action strategies. Curr Biol. (2016) ;26: (7):R267–9. |
[179] | Floran B , Floran L , Sierra A , Aceves J . D2 receptor-mediated inhibition of GABA release by endogenous dopamine in the rat globus pallidus. Neurosci Lett. (1997) ;237: (1):1–4. |
[180] | Gonzalez B , Paz F , Floran L , Aceves J , Erlij D , Floran B . Cannabinoid agonists stimulate [3H]GABA release in the globus pallidus of the rat when G(i) protein-receptor coupling is restricted: Role of dopamine D2 receptors. J Pharmacol Exp Ther. (2009) ;328: (3):822–8. |
[181] | Cooper AJ , Stanford IM . Dopamine D2 receptor mediated presynaptic inhibition of striatopallidal GABA(A) IPSCs in vitro. Neuropharmacology. (2001) ;41: (1):62–71. |
[182] | Augood SJ , Faull RL , Emson PC . Dopamine D1 and D2 receptor gene expression in the striatum in Huntington’s disease. Ann Neurol. (1997) ;42: (2):215–21. |
[183] | Richfield EK , O’Brien CF , Eskin T , Shoulson I . Heterogeneous dopamine receptor changes in early and late Huntington’s disease. Neurosci Lett. (1991) ;132: (1):121–6. |
[184] | Backman L , Robins-Wahlin TB , Lundin A , Ginovart N , Farde L . Cognitive deficits in Huntington’s disease are predicted by dopaminergic PET markers and brain volumes. Brain. (1997) ;120: (Pt 12):2207–17. |
[185] | Andrews TC , Weeks RA , Turjanski N , Gunn RN , Watkins LH , Sahakian B , et al. Huntington’s disease progression. PET and clinical observations. Brain. (1999) ;122: (Pt 12):2353–63. |
[186] | Glass M , van Dellen A , Blakemore C , Hannan AJ , Faull RL . Delayed onset of Huntington’s disease in mice in an enriched environment correlates with delayed loss of cannabinoid CB1 receptors. Neuroscience. (2004) ;123: (1):207–12. |
[187] | Cha JH , Kosinski CM , Kerner JA , Alsdorf SA , Mangiarini L , Davies SW , et al. Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human huntington disease gene. Proc Natl Acad Sci U S A. (1998) ;95: (11):6480–5. |
[188] | Ariano MA , Aronin N , Difiglia M , Tagle DA , Sibley DR , Leavitt BR , et al. Striatal neurochemical changes in transgenic models of Huntington’s disease. J Neurosci Res. (2002) ;68: (6):716–29. |
[189] | Pouladi MA , Stanek LM , Xie Y , Franciosi S , Southwell AL , Deng Y , et al. Marked differences in neurochemistry and aggregates despite similar behavioural and neuropathological features of Huntington disease in the full-length BACHD and YAC128 mice. Hum Mol Genet. (2012) ;21: (10):2219–32. |
[190] | Spektor BS , Miller DW , Hollingsworth ZR , Kaneko YA , Solano SM , Johnson JM , et al. Differential D1 and D2 receptor-mediated effects on immediate early gene induction in a transgenic mouse model of Huntington’s disease. Brain Res Mol Brain Res. (2002) ;102: (1-2):118–28. |
[191] | Gratuze M , Noel A , Julien C , Cisbani G , Milot-Rousseau P , Morin F , et al. Tau hyperphosphorylation and deregulation of calcineurin in mouse models of Huntington’s disease. Hum Mol Genet. (2015) ;24: (1):86–99. |
[192] | Berke JD , Paletzki RF , Aronson GJ , Hyman SE , Gerfen CR . A complex program of striatal gene expression induced by dopaminergic stimulation. J Neurosci. (1998) ;18: (14):5301–10. |
[193] | Feyder M , Bonito-Oliva A , Fisone G . L-DOPA-induced dyskinesia and abnormal signaling in striatal medium spiny neurons: Focus on dopamine D1 receptor-mediated transmission. Front Behav Neurosci. (2011) ;5: :71. |
[194] | Park HY , Kang YM , Kang Y , Park TS , Ryu YK , Hwang JH , et al. Inhibition of adenylyl cyclase type 5 prevents L-DOPA-induced dyskinesia in an animal model of Parkinson’s disease. J Neurosci. (2014) ;34: (35):11744–53. |
[195] | Chen YZ , Matsushita MM , Robertson P , Rieder M , Girirajan S , Antonacci F , et al. Autosomal dominant familial dyskinesia and facial myokymia: Single exome sequencing identifies a mutation in adenylyl cyclase 5. Arch Neurol. (2012) ;69: (5):630–5. |
[196] | Roze E , Betuing S , Deyts C , Marcon E , Brami-Cherrier K , Pages C , et al. Mitogen- and stress-activated protein kinase-1 deficiency is involved in expanded-huntingtin-induced transcriptional dysregulation and striatal death. FASEB J. (2008) ;22: (4):1083–93. |
[197] | Fusco FR , Anzilotti S , Giampa C , Dato C , Laurenti D , Leuti A , et al. Changes in the expression of extracellular regulated kinase (ERK 1/2) in the R6/2 mouse model of Huntington’s disease after phosphodiesterase IV inhibition. Neurobiol Dis. (2012) ;46: (1):225–33. |
[198] | Konradi C , Cole RL , Heckers S , Hyman SE . Amphetamine regulates gene expression in rat striatum via transcription factor CREB. J Neurosci. (1994) ;14: (9):5623–34. |
[199] | Sgambato V , Pages C , Rogard M , Besson MJ , Caboche J . Extracellular signal-regulated kinase (ERK) controls immediate early gene induction on corticostriatal stimulation. J Neurosci. (1998) ;18: (21):8814–25. |
[200] | Gerfen CR . D1 dopamine receptor supersensitivity in the dopamine-depleted striatum animal model of Parkinson’s disease. Neuroscientist. (2003) ;9: (6):455–62. |
[201] | Cha JH . Transcriptional signatures in Huntington’s disease. Prog Neurobiol. (2007) ;83: (4):228–48. |
[202] | Robinson P , Lebel M , Cyr M . Dopamine D1 receptor-mediated aggregation of N-terminal fragments of mutant huntingtin and cell death in a neuroblastoma cell line. Neuroscience. (2008) ;153: (3):762–72. |
[203] | Kim M , Velier J , Chase K , Laforet G , Kalchman MA , Hayden MR , et al. Forskolin and dopamine D1 receptor activation increase huntingtin’s association with endosomes in immortalized neuronal cells of striatal origin. Neuroscience. (1999) ;89: (4):1159–67. |
[204] | Beaulieu JM , Gainetdinov RR , Caron MG . The Akt-GSK-3 signaling cascade in the actions of dopamine. Trends Pharmacol Sci. (2007) ;28: (4):166–72. |
[205] | Mannoury la Cour C , Salles MJ , Pasteau V , Millan MJ . Signaling pathways leading to phosphorylation of Akt and GSK-3beta by activation of cloned human and rat cerebral D(2)and D(3) receptors. Mol Pharmacol. (2011) ;79: (1):91–105. |
[206] | Beaulieu JM , Del’guidice T , Sotnikova TD , Lemasson M , Gainetdinov RR . Beyond cAMP: The regulation of Akt and GSK3 by dopamine receptors. Front Mol Neurosci. (2011) ;4: :38. |
[207] | Lim NK , Hung LW , Pang TY , Mclean CA , Liddell JR , Hilton JB , et al. Localized changes to glycogen synthase kinase-3 and collapsin response mediator protein-2 in the Huntington’s disease affected brain. Hum Mol Genet. (2014) ;23: (15):4051–63. |
[208] | Doble BW , Woodgett JR . GSK- Tricks of the trade for a multi-tasking kinase. J Cell Sci. (2003) ;116: (Pt 7):1175–86. |
[209] | Hyrskyluoto A , Bruelle C , Lundh SH , Do HT , Kivinen J , Rappou E , et al. Ubiquitin-specific protease-14 reduces cellular aggregates and protects against mutant huntingtin-induced cell degeneration: Involvement of the proteasome and ER stress-activated kinase IRE1alpha. Hum Mol Genet. (2014) ;23: (22):5928–39. |
[210] | Chuang KH , Liang F , Higgins R , Wang Y . Ubiquilin/Dsk2 promotes inclusion body formation and vacuole (lysosome)-mediated disposal of mutated huntingtin. Mol Biol Cell. (2016) ;27: (13):2025–36. |
[211] | Carmichael J , Sugars KL , Bao YP , Rubinsztein DC . Glycogen synthase kinase-3beta inhibitors prevent cellular polyglutamine toxicity caused by the Huntington’s disease mutation. J Biol Chem. (2002) ;277: (37):33791-8(47). |
[212] | Bowles KR , Brooks SP , Dunnett SB , Jones L . Huntingtin subcellular localisation is regulated by kinase signalling activity in the StHdhQ111 model of HD. PLoS One. (2015) ;10: (12):e0144864. |
[213] | Bychkov E , Ahmed MR , Dalby KN , Gurevich EV . Dopamine depletion and subsequent treatment with L-DOPA, but not the long-lived dopamine agonist pergolide, enhances activity of the Akt pathway in the rat striatum. J Neurochem. (2007) ;102: (3):699–711. |
[214] | Tang TS , Chen X , Liu J , Bezprozvanny I . Dopaminergic signaling and striatal neurodegeneration in Huntington’s disease. J Neurosci. (2007) ;27: (30):7899–910. |
[215] | Paoletti P , Vila I , Rife M , Lizcano JM , Alberch J , Gines S . Dopaminergic and glutamatergic signaling crosstalk in Huntington’s disease neurodegeneration: The role of p25/cyclin-dependent kinase 5. J Neurosci. (2008) ;28: (40):10090–101. |
[216] | Andre VM , Cepeda C , Levine MS . Dopamine and glutamate in Huntington’s disease: A balancing act. CNS Neurosci Ther. (2010) ;16: (3):163–78. |
[217] | Andre VM , Fisher YE , Levine MS . Altered balance of activity in the striatal direct and indirect pathways in mouse models of Huntington’s disease. Front Syst Neurosci. (2011) ;5: :46. |
[218] | Gardoni F , Bellone C . Modulation of the glutamatergic transmission by dopamine: A focus on Parkinson, Huntington and addiction diseases. Front Cell Neurosci. (2015) ;9: :25. |
[219] | O’Donnell P , Grace AA . Tonic D2-mediated attenuation of cortical excitation in nucleus accumbens neurons recorded in vitro. Brain Res. (1994) ;634: (1):105–12. |
[220] | Levine MS , Altemus KL , Cepeda C , Cromwell HC , Crawford C , Ariano MA , et al. Modulatory actions of dopamine on NMDA receptor-mediated responses are reduced in D1A-deficient mutant mice. J Neurosci. (1996) ;16: (18):5870–82. |
[221] | Hernandez-Lopez S , Bargas J , Surmeier DJ , Reyes A , Galarraga E . D1 receptor activation enhances evoked discharge in neostriatal medium spiny neurons by modulating an L-type Ca2+ conductance. J Neurosci. (1997) ;17: (9):3334–42. |
[222] | Snyder GL , Fienberg AA , Huganir RL , Greengard P . A dopamine/D1 receptor/protein kinase A/dopamine- and cAMP-regulated phosphoprotein (Mr 32kDa)/protein phosphatase-1 pathway regulates dephosphorylation of the NMDA receptor. J Neurosci. (1998) ;18: (24):10297–303. |
[223] | Dunah AW , Wang Y , Yasuda RP , Kameyama K , Huganir RL , Wolfe BB , et al. Alterations in subunit expression, composition, and phosphorylation of striatal N-methyl-D-aspartate glutamate receptors in a rat 6-hydroxydopamine model of Parkinson’s disease. Mol Pharmacol. (2000) ;57: (2):342–52. |
[224] | Lee FJ , Xue S , Pei L , Vukusic B , Chery N , Wang Y , et al. Dual regulation of NMDA receptor functions by direct protein-protein interactions with the dopamine D1 receptor. Cell. (2002) ;111: (2):219–30. |
[225] | Fiorentini C , Gardoni F , Spano P , Di Luca M , Missale C . Regulation of dopamine D1 receptor trafficking and desensitization by oligomerization with glutamate N-methyl-D-aspartate receptors. J Biol Chem. (2003) ;278: (22):20196–202. |
[226] | Dunah AW , Sirianni AC , Fienberg AA , Bastia E , Schwarzschild MA , Standaert DG . Dopamine D1-dependent trafficking of striatal N-methyl-D-aspartate glutamate receptors requires Fyn protein tyrosine kinase but not DARPP-32. Mol Pharmacol. (2004) ;65: (1):121–9. |
[227] | Price CJ , Rintoul GL , Baimbridge KG , Raymond LA . Inhibition of calcium-dependent NMDA receptor current rundown by calbindin-D28k. J Neurochem. (1999) ;72: (2):634–42. |
[228] | Blank T , Nijholt I , Teichert U , Kugler H , Behrsing H , Fienberg A , et al. The phosphoprotein DARPP-32 mediates cAMP-dependent potentiation of striatal N-methyl-D-aspartate responses. Proc Natl Acad Sci U S A. (1997) ;94: (26):14859–864. |
[229] | Levine MS , Li Z , Cepeda C , Cromwell HC , Altemus KL . Neuromodulatory actions of dopamine on synaptically-evoked neostriatal responses in slices. Synapse. (1996) ;24: (1):65–78. |
[230] | Cepeda C , Levine MS . Dopamine and N-methyl-D-aspartate receptor interactions in the neostriatum. Dev Neurosci. (1998) ;20: (1):1–18. |
[231] | Flores-Hernandez J , Cepeda C , Hernandez-Echeagaray E , Calvert CR , Jokel ES , Fienberg AA , et al. Dopamine enhancement of NMDA currents in dissociated medium-sized striatal neurons: Role of D1 receptors and DARPP-32. J Neurophysiol. (2002) ;88: (6):3010–20. |
[232] | Higley MJ , Sabatini BL . Competitive regulation of synaptic Ca2+ influx by D2 dopamine and A2A adenosine receptors. Nat Neurosci. (2010) ;13: (8):958–66. |
[233] | Fan MM , Raymond LA . N-methyl-D-aspartate (NMDA) receptor function and excitotoxicity in Huntington’s disease. Prog Neurobiol. (2007) ;81: (5-6):272–93. |
[234] | Zeron MM , Hansson O , Chen N , Wellington CL , Leavitt BR , Brundin P , et al. Increased sensitivity to N-methyl-D-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington’s disease. Neuron. (2002) ;33: (6):849–60. |
[235] | Zeron MM , Fernandes HB , Krebs C , Shehadeh J , Wellington CL , Leavitt BR , et al. Potentiation of NMDA receptor-mediated excitotoxicity linked with intrinsic apoptotic pathway in YAC transgenic mouse model of Huntington’s disease. Mol Cell Neurosci. (2004) ;25: (3):469–79. |
[236] | Jocoy EL , Andre VM , Cummings DM , Rao SP , Wu N , Ramsey AJ , et al. Dissecting the contribution of individual receptor subunits to the enhancement of N-methyl-d-aspartate currents by dopamine D1 receptor activation in striatum. Front Syst Neurosci. (2011) ;5: :28. |
[237] | Tovar KR , Westbrook GL . The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci. (1999) ;19: (10):4180–8. |
[238] | Hardingham GE , Bading H . Coupling of extrasynaptic NMDA receptors to a CREB shut-off pathway is developmentally regulated. Biochim Biophys Acta. (2002) ;1600: (1-2):148–53. |
[239] | Milnerwood AJ , Gladding CM , Pouladi MA , Kaufman AM , Hines RM , Boyd JD , et al. Early increase in extrasynaptic NMDA receptor signaling and expression contributes to phenotype onset in Huntington’s disease mice. Neuron. (2010) ;65: (2):178–90. |
[240] | Dau A , Gladding CM , Sepers MD , Raymond LA . Chronic blockade of extrasynaptic NMDA receptors ameliorates synaptic dysfunction and pro-death signaling in Huntington disease transgenic mice. Neurobiol Dis. (2014) ;62: :533–42. |
[241] | Vastagh C , Gardoni F , Bagetta V , Stanic J , Zianni E , Giampa C , et al. N-methyl-D-aspartate (NMDA) receptor composition modulates dendritic spine morphology in striatal medium spiny neurons. J Biol Chem. (2012) ;287: (22):18103–14. |
[242] | Sun Y , Savanenin A , Reddy PH , Liu YF . Polyglutamine-expanded huntingtin promotes sensitization of N-methyl-D-aspartate receptors via post-synaptic density 95. J Biol Chem. (2001) ;276: (27):24713–8. |
[243] | Fan J , Cowan CM , Zhang LY , Hayden MR , Raymond LA . Interaction of postsynaptic density protein-95 with NMDA receptors influences excitotoxicity in the yeast artificial chromosome mouse model of Huntington’s disease. J Neurosci. (2009) ;29: (35):10928–38. |
[244] | Zhang J , Vinuela A , Neely MH , Hallett PJ , Grant SG , Miller GM , et al. Inhibition of the dopamine D1 receptor signaling by PSD-95. J Biol Chem. (2007) ;282: (21):15778–89. |
[245] | Hersch SM , Ciliax BJ , Gutekunst CA , Rees HD , Heilman CJ , Yung KK , et al. Electron microscopic analysis of D1 and D2 dopamine receptor proteins in the dorsal striatum and their synaptic relationships with motor corticostriatal afferents. J Neurosci. (1995) ;15: (7 Pt 2):5222–37. |
[246] | Zhang J , Saur T , Duke AN , Grant SG , Platt DM , Rowlett JK , et al. Motor impairments, striatal degeneration, and altered dopamine-glutamate interplay in mice lacking PSD-95. J Neurogenet. (2014) ;28: (1-2):98–111. |
[247] | Giralt A , Saavedra A , Carreton O , Xifro X , Alberch J , Perez-Navarro E . Increased PKA signaling disrupts recognition memory and spatial memory: Role in Huntington’s disease. Hum Mol Genet. (2011) ;20: (21):4232–47. |
[248] | Galarraga E , Hernandez-Lopez S , Reyes A , Barral J , Bargas J . Dopamine facilitates striatal EPSPs through an L-type Ca2+ conductance. Neuroreport. (1997) ;8: (9-10):2183–6. |
[249] | Gao T , Yatani A , Dell’Acqua ML , Sako H , Green SA , Dascal N , et al. cAMP-dependent regulation of cardiac L-type Ca2+ channels requires membrane targeting of PKA and phosphorylation of channel subunits. Neuron. (1997) ;19: (1):185–96. |
[250] | Kitai ST , Surmeier DJ . Cholinergic and dopaminergic modulation of potassium conductances in neostriatal neurons. Adv Neurol. (1993) ;60: :40–52. |
[251] | Surmeier DJ , Bargas J , Hemmings HC Jr , Nairn AC , Greengard P . Modulation of calcium currents by a D1 dopaminergic protein kinase/phosphatase cascade in rat neostriatal neurons. Neuron. (1995) ;14: (2):385–97. |
[252] | Vilchis C , Bargas J , Ayala GX , Galvan E , Galarraga E . Ca2+ channels that activate Ca2+-dependent K+ currents in neostriatal neurons. Neuroscience. (2000) ;95: (3):745–52. |
[253] | Surmeier DJ , Xu ZC , Wilson CJ , Stefani A , Kitai ST . Grafted neostriatal neurons express a late-developing transient potassium current. Neuroscience. (1992) ;48: (4):849–56. |
[254] | Klawans HC , Paulson GW , Barbeau A . Predictive test for Huntington’s chorea. Lancet. (1970) ;2: (7684):1185–6. |
[255] | Bernheimer H , Birkmayer W , Hornykiewicz O , Jellinger K , Seitelberger F . Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J Neurol Sci. (1973) ;20: (4):415–55. |
[256] | Masimore B , Schmitzer-Torbert NC , Kakalios J , Redish AD . Transient striatal gamma local field potentials signal movement initiation in rats. Neuroreport. (2005) ;16: (18):2021–4. |
[257] | von Nicolai C , Engler G , Sharott A , Engel AK , Moll CK , Siegel M . Corticostriatal coordination through coherent phase-amplitude coupling. J Neurosci. (2014) ;34: (17):5938–48. |
[258] | Kim HA , Jiang L , Madsen H , Parish CL , Massalas J , Smardencas A , et al. Resolving pathobiological mechanisms relating to Huntington disease: Gait, balance, and involuntary movements in mice with targeted ablation of striatal D1 dopamine receptor cells. Neurobiol Dis. (2014) ;62: :323–37. |
[259] | Murphy-Nakhnikian A , Dorner JL , Fischer BI , Bower-Bir ND , Rebec GV . Abnormal burst patterns of single neurons recorded in the substantia nigra reticulata of behaving 140 CAG Huntington’s disease mice. Neurosci Lett. (2012) ;512: (1):1–5. |
[260] | Garrett MC , Soares-da-Silva P . Increased cerebrospinal fluid dopamine and 3,4-dihydroxyphenylacetic acid levels in Huntington’s disease: Evidence for an overactive dopaminergic brain transmission. J Neurochem. (1992) ;58: (1):101–6. |
[261] | Jahanshahi A , Vlamings R , Kaya AH , Lim LW , Janssen ML , Tan S , et al. Hyperdopaminergic status in experimental Huntington disease. J Neuropathol Exp Neurol. (2010) ;69: (9):910–17. |
[262] | Izquierdo A , Brigman JL , Radke AK , Rudebeck PH , Holmes A . The neural basis of reversal learning: An updated perspective. Neuroscience. In Press (2016) . |
[263] | Rogers RD , Andrews TC , Grasby PM , Brooks DJ , Robbins TW . Contrasting cortical and subcortical activations produced by attentional-set shifting and reversal learning in humans. J Cogn Neurosci. (2000) ;12: (1):142–62. |
[264] | Cools R , Clark L , Owen AM , Robbins TW . Defining the neural mechanisms of probabilistic reversal learning using event-related functional magnetic resonance imaging. J Neurosci. (2002) ;22: (11):4563–7. |
[265] | Castañé A , Theobald DE , Robbins TW . Selective lesions of the dorsomedial striatum impair serial spatial reversal learning in rats. Behav Brain Res. (2010) ;210: (1):74–83. |
[266] | Lawrence AD , Sahakian BJ , Hodges JR , Rosser AE , Lange KW , Robbins TW . Executive and mnemonic functions in early Huntington’s disease. Brain. (1996) ;119: (5):1633–45. |
[267] | Josiassen RC , Curry LM , Mancall EL . Development of neuropsychological deficits in Huntington’s disease. Arch Neurol. (1983) ;40: (13):791–6. |
[268] | Lawrence AD , Hodges JR , Rosser AE , Kershaw A , Constant C , Rubinsztein DC , et al. Evidence for specific cognitive deficits in preclinical Huntington’s disease. Brain. (1998) ;121: (7):1329–41. |
[269] | Lawrence AD , Weeks RA , Brooks DJ , Andrews TC , Watkins LH , Harding AE , et al. The relationship between striatal dopamine receptor binding and cognitive performance in Huntington’s disease. Brain. (1998) ;121: (7):1343–55. |
[270] | Brandt J , Inscore AB , Ward J , Shpritz B , Rosenblatt A , Margolis RL , et al. Neuropsychological deficits in Huntington’s disease gene carriers and correlates of early “conversion”. J Neuropsychiatry Clin Neurosci. (2008) ;20: (4):466–72. |
[271] | Glynn D , Reim K , Brose N , Morton AJ . Depletion of Complexin II does not affect disease progression in a mouse model of Huntington’s disease (HD); support for role for complexin II in behavioural pathology in a mouse model of HD. Brain Res Bull. (2007) ;72: (2-3):108–20. |
[272] | Abada YS , Nguyen HP , Ellenbroek B , Schreiber R . Reversal learning and associative memory impairments in a BACHD rat model for Huntington disease. PLoS One. (2013) ;8: (11):e71633. |
[273] | Van Raamsdonk JM , Pearson J , Slow EJ , Hossain SM , Leavitt BR , Hayden MR . Cognitive dysfunction precedes neuropathology and motor abnormalities in the YAC128 mouse model of Huntington’s disease. J Neurosci. (2005) ;25: (16):4169–80. |
[274] | Brooks SP , Janghra N , Higgs GV , Bayram-Weston Z , Heuer A , Jones L , et al. Selective cognitive impairment in the YAC128 Huntington’s disease mouse. Brain Res Bull. (2012) ;88: (2-3):121–9. |
[275] | Walker AG , Ummel JR , Rebec GV . Reduced expression of conditioned fear in the R6/2 mouse model of Huntington’s disease is related to abnormal activity in prelimbic cortex. Neurobiol Dis. (2011) ;43: (2):379–87. |
[276] | Cummings JL , Cunningham K . Obsessive-compulsive disorder in Huntington’s disease. Biol Psychiatry. (1992) ;31: (3):263–70. |
[277] | Rangel-Barajas C , Bunner K , Barton SJ , Rebec GV . Striatal local field potential activity in the Q175 knock-in mouse model of Huntington’s disease during plus maze motor activity. Soc Neurosci Abstr. 2015;No. 303.10. |
[278] | Berridge KC , Aldridge JW , Houchard KR , Zhuang X . Sequential super-stereotypy of an instinctive fixed action pattern in hyper-dopaminergic mutant mice: A model of obsessive compulsive disorder and Tourette’s. BMC Biol. (2005) ;3: :4. |
[279] | Hartgraves SL , Randall PK . Dopamine agonist-induced stereotypic grooming and self-mutilation following striatal dopamine depletion. Psychopharmacology (Berl). (1986) ;90: (3):358–63. |
[280] | Berridge KC , Aldridge JW . Super-stereotypy II: Enhancement of a complex movement sequence by intraventricular dopamine D1 agonists. Synapse. (2000) ;37: (3):205–15. |
[281] | Taylor JL , Rajbhandari AK , Berridge KC , Aldridge JW . Dopamine receptor modulation of repetitive grooming actions in the rat: Potential relevance for Tourette syndrome. Brain Res. (2010) ;1322: :92–101. doi: 10.1016/j.brainres.2010.01.052 |
[282] | Levy R , Dubois B . Apathy and the functional anatomy of the prefrontal cortex-basal ganglia circuits. Cereb Cortex. (2006) ;16: (7):916–28. |
[283] | Haruno M , Kawato M . Heterarchical reinforcement-learning model for integration of multiple cortico-striatal loops: fMRI examination in stimulus-action-reward association learning. Neural Netw. (2006) ;19: (8):1242–54. |
[284] | Morris LS , Kundu P , Dowell N , Mechelmans DJ , Favre P , Irvine MA , et al. Fronto-striatal organization: Defining functional and microstructural substrates of behavioural flexibility. Cortex. (2016) ;74: :118–33. |
[285] | Langen M , Durston S , Kas MJ , van Engeland H , Staal WG . The neurobiology of repetitive behavior: … And men. Neurosci Biobehav Rev. (2011) ;35: (3):356–65. |
[286] | Langen M , Kas MJ , Staal WG , van Engeland H , Durston S . The neurobiology of repetitive behavior: Of mice … Neurosci Biobehav Rev. (2011) ;35: (3):345–55. |
[287] | Balleine BW , Delgado MR , Hikosaka O . The role of the dorsal striatum in reward and decision-making. J Neurosci. (2007) ;27: (31):8161–5. |
[288] | Liu W , Yang J , Chen K , Luo C , Burgunder J , Gong Q , et al. Resting-state fMRI reveals potential neural correlates of impaired cognition in Huntington’s disease. Parkinsonism Relat Disord. (2016) ;27: :41–6. |
[289] | Li W , Silva HB , Real J , Wang YM , Rial D , Li P , et al. Inactivation of adenosine A2A receptors reverses working memory deficits at early stages of Huntington’s disease models. Neurobiol Dis. (2015) ;79: :70–80. |
[290] | Weeks RA , Piccini P , Harding AE , Brooks DJ . Striatal D1 and D2 dopamine receptor loss in asymptomatic mutation carriers of Huntington’s disease. Ann Neurol. (1996) ;40: (1):49–54. |
[291] | Cummings DM , Milnerwood AJ , Dallerac GM , Waights V , Brown JY , Vatsavayai SC , et al. Aberrant cortical synaptic plasticity and dopaminergic dysfunction in a mouse model of Huntington’s disease. Hum Mol Genet. (2006) ;15: (19):2856–68. |
[292] | Craufurd D , Thompson JC , Snowden JS . Behavioral changes in Huntington disease. Neuropsychiatry Neuropsychol Behav Neurol. (2001) ;14: (4):219–26. |
[293] | Kingma EM , van Duijn E , Timman R , van der Mast RC , Roos RA . Behavioural problems in Huntington’s disease using the Problem Behaviours Assessment. Gen Hosp Psychiatry. (2008) ;30: (2):155–61. |
[294] | van Duijn E . Treatment of Irritability in Huntington’s disease. Curr Treat Options Neurol. (2010) ;12: (5):424–33. |
[295] | Tabrizi SJ , Scahill RI , Owen G , Durr A , Leavitt BR , Roos RA , et al. Predictors of phenotypic progression and disease onset in premanifest and early-stage Huntington’s disease in the TRACK-HD study: Analysis of 36-month observational data. Lancet Neurol. (2013) ;12: (7):637–49. |
[296] | Mason SL , Barker RA . Advancing pharmacotherapy for treating Huntington’s disease: A review of the existing literature. Expert Opin Pharmacother. (2016) ;17: (1):41–52. |
[297] | Martinez-Horta S , Perez-Perez J , van Duijn E , Fernandez-Bobadilla R , Carceller M , Pagonabarraga J , et al. Neuropsychiatric symptoms are very common in premanifest and early stage Huntington’s Disease. Parkinsonism Relat Disord. (2016) ;25: :58–64. |
[298] | Epping EA , Paulsen JS . Depression in the early stages of Huntington disease. Neurodegener Dis Manag. (2011) ;1: (5):407–14. |
[299] | Thompson JC , Harris J , Sollom AC , Stopford CL , Howard E , Snowden JS , et al. Longitudinal evaluation of neuropsychiatric symptoms in Huntington’s disease. J Neuropsychiatry Clin Neurosci. (2012) ;24: (1):53–60. |
[300] | Rosenblatt A , Leroi I . Neuropsychiatry of Huntington’s disease and other basal ganglia disorders. Psychosomatics. (2000) ;41: (1):24–30. |
[301] | Dewhurst K . Personality disorder in Huntington’s disease. Psychiatr Clin (Basel). (1970) ;3: (4):221–9. |
[302] | van Duijn E , Kingma EM , van der Mast RC . Psychopathology in verified Huntington’s disease gene carriers. J Neuropsychiatry Clin Neurosci. (2007) ;19: (4):441–8. |
[303] | van Duijn E , Reedeker N , Giltay EJ , Eindhoven D , Roos RA , van der Mast RC . Course of irritability, depression and apathy in Huntington’s disease in relation to motor symptoms during a two-year follow-up period. Neurodegener Dis. (2014) ;13: (1):9–16. |
[304] | Arnsten AF . Prefrontal cortical network connections: Key site of vulnerability in stress and schizophrenia. Int J Dev Neurosci. (2011) ;29: (3):215–23. |
[305] | Marek GJ . Serotonin and dopamine interactions in rodents and primates: Implications for psychosis and antipsychotic drug development. Int Rev Neurobiol. (2007) ;78: :165–92. |
[306] | Grace AA . Dysregulation of the dopamine system in the pathophysiology of schizophrenia and depression. Nat Rev Neurosci. (2016) ;17: (8):524–32. |
[307] | Baxter LR Jr . Neuroimaging studies of obsessive compulsive disorder. Psychiatr Clin North Am. (1992) ;15: (4):871–84. |
[308] | Weinberger DR , Berman KF , Chase TN . Mesocortical dopaminergic function and human cognition. Ann N Y Acad Sci. (1988) ;537: :330–8. |
[309] | Pennartz CM , Dolleman-Van der Weel MJ , Kitai ST , Lopes da Silva FH . Presynaptic dopamine D1 receptors attenuate excitatory and inhibitory limbic inputs to the shell region of the rat nucleus accumbens studied in vitro. J Neurophysiol. (1992) ;67: (5):1325–34. |
[310] | Nicola SM , Kombian SB , Malenka RC . Psychostimulants depress excitatory synaptic transmission in the nucleus accumbens via presynaptic D1-like dopamine receptors. J Neurosci. (1996) ;16: (5):1591–604. |
[311] | Dogan I , Eickhoff CR , Fox PT , Laird AR , Schulz JB , Eickhoff SB , et al. Functional connectivity modeling of consistent cortico-striatal degeneration in Huntington’s disease. Neuroimage Clin. (2015) ;7: :640–52. |
[312] | Renoir T , Argyropoulos A , Hannan AJ . Antidepressant-like effect of the norepinephrine-dopamine reuptake inhibitor bupropion in a mouse model of Huntington’s disease with dopaminergic dysfunction. J Huntingtons Dis. (2012) ;1: (2):261–6. |
[313] | Sprengelmeyer R , Orth M , Muller HP , Wolf RC , Gron G , Depping MS , et al. The neuroanatomy of subthreshold depressive symptoms in Huntington’s disease: A combined diffusion tensor imaging (DTI) and voxel-based morphometry (VBM) study. Psychol Med. (2014) ;44: (9):1867–78. |
[314] | Scott LJ . Tetrabenazine: For chorea associated with Huntington’s disease. CNS Drugs. (2011) ;25: (12):1073–85. |
[315] | Lane JD , Smith JE , Shea PA , McBride WJ . Neurochemical changes following the administration of depleters of biogenic monoamines. Life Sci. (1976) ;19: (11):1663–7. |
[316] | Pearson SJ , Reynolds GP . Depletion of monoamine transmitters by tetrabenazine in brain tissue in Huntington’s disease. Neuropharmacology. (1988) ;27: (7):717–9. |
[317] | Wang H , Chen X , Li Y , Tang TS , Bezprozvanny I . Tetrabenazine is neuroprotective in Huntington’s disease mice. Mol Neurodegener. (2010) ;5: :18-1326-5-18. |
[318] | Pettibone DJ , Totaro JA , Pflueger AB . Tetrabenazine-induced depletion of brain monoamines: Characterization and interaction with selected antidepressants. Eur J Pharmacol. (1984) ;102: (3-4):425–30. |
[319] | Mehvar R , Jamali F . Concentration-effect relationships of tetrabenazine and dihydrotetrabenazine in the rat. J Pharm Sci. (1987) ;76: (6):461–5. |
[320] | Chen JJ , Ondo WG , Dashtipour K , Swope DM . Tetrabenazine for the treatment of hyperkinetic movement disorders: A review of the literature. Clin Ther. (2012) ;34: (7):1487–504. |
[321] | Frank S , Ondo W , Fahn S , Hunter C , Oakes D , Plumb S , et al. A study of chorea after tetrabenazine withdrawal in patients with Huntington disease. Clin Neuropharmacol. (2008) ;31: (3):127–33. |
[322] | Fuentes P , Paris I , Nassif M , Caviedes P , Segura-Aguilar J . Inhibition of VMAT-2 and DT-diaphorase induce cell death in a substantia nigra-derived cell line–an experimental cell model for dopamine toxicity studies. Chem Res Toxicol. (2007) ;20: (5):776–83. |
[323] | Mehanna R , Hunter C , Davidson A , Jimenez-Shahed J , Jankovic J . Analysis of CYP2D6 genotype and response to tetrabenazine. Mov Disord. (2013) ;28: (2):210–5. |
[324] | Chen S , Zhang XJ , Xie WJ , Qiu HY , Liu H , Le WD . A new VMAT-2 inhibitor NBI-49 in the treatment of Huntington disease. CNS Neurosci Ther. (2015) ;21: (8):662–71. |
[325] | Leysen JE , Janssen PM , Gommeren W , Wynants J , Pauwels PJ , Janssen PA . In vitro and in vivo receptor binding and effects on monoamine turnover in rat brain regions of the novel antipsychotics risperidone and ocaperidone. Mol Pharmacol. (1992) ;41: (3):494–508. |
[326] | Leysen JE , Janssen PM , Megens AA , Schotte A . Risperidone: A novel antipsychotic with balanced serotonin-dopamine antagonism, receptor occupancy profile, and pharmacologic activity. J Clin Psychiatry. (1994) ;55: (Suppl):5–12. |
[327] | Malmberg , Mikaels , Mohell N . Agonist and inverse agonist activity at the dopamine D3 receptor measured by guanosine 5’–gamma-thio-triphosphate–35S- binding. J Pharmacol Exp Ther. (1998) ;285: (1):119–26. |
[328] | Leonard DP , Kidson MA , Brown JG , Shannon PJ , Taryan S . A double blind trial of lithium carbonate and haloperidol in Huntington’s chorea. Aust N Z J Psychiatry. (1975) ;9: (2):115–8. |
[329] | Barr AN , Fischer JH , Koller WC , Spunt AL , Singhal A . Serum haloperidol concentration and choreiform movements in Huntington’s disease. Neurology. (1988) ;38: (1):84–8. |
[330] | Gimenez-Roldan S , Mateo D . Huntington disease: Tetrabenazine compared to haloperidol in the reduction of involuntary movements. Neurologia. (1989) ;4: (8):282–7. |
[331] | Giannini AJ , Eighan MS , Loiselle RH , Giannini MC . Comparison of haloperidol and chlorpromazine in the treatment of phencyclidine psychosis. J Clin Pharmacol. (1984) ;24: (4):202–4. |
[332] | Giannini AJ , Price WA , Loiselle RH , Malone DW . Treatment of phenylcyclohexylpyrrolidine (PHP) psychosis with haloperidol. J Toxicol Clin Toxicol. (1985) ;23: (2-3):185–9. |
[333] | Haddad PM , Das A , Keyhani S , Chaudhry IB . Antipsychotic drugs and extrapyramidal side effects in first episode psychosis: A systematic review of head-head comparisons. J Psychopharmacol. (2012) ;26: (5 Suppl):15–26. |
[334] | Quinn N , Marsden CD . A double blind trial of sulpiride in Huntington’s disease and tardive dyskinesia. J Neurol Neurosurg Psychiatry. (1984) ;47: (8):844–7. |
[335] | Arena R , Iudice A , Virgili P , Moretti P , Menchetti G . Huntington’s disease: Clinical effects of a short-term treatment with pimozide. Adv Biochem Psychopharmacol. (1980) ;24: :573–5. |
[336] | Morgan DG , Finch CE . 3H]Fluphenazine binding to brain membranes: Simultaneous measurement of D-1 and D-2 receptor sites. J Neurochem. (1986) ;46: (5):1623–31. |
[337] | Terrence CF . Fluphenazine decanoate in the treatment of chorea: A double-blind study. Curr Ther Res Clin Exp. (1976) ;20: (2):177–83. |
[338] | Meltzer HY . An overview of the mechanism of action of clozapine. J Clin Psychiatry. (1994) ;55: :47–52. |
[339] | Bonuccelli U , Ceravolo R , Maremmani C , Nuti A , Rossi G , Muratorio A . Clozapine in Huntington’s chorea. Neurology. (1994) ;44: (5):821–3. |
[340] | van Vugt JP , Siesling S , Vergeer M , van der Velde EA , Roos RA . Clozapine versus placebo in Huntington’s disease: A double blind randomised comparative study. J Neurol Neurosurg Psychiatry. (1997) ;63: (1):35–9. |
[341] | Lieberman JA , Tollefson G , Tohen M , Green AI , Gur RE , Kahn R , et al. Comparative efficacy and safety of atypical and conventional antipsychotic drugs in first-episode psychosis: A randomized, double-blind trial of olanzapine versus haloperidol. Am J Psychiatry. (2003) ;160: (8):1396–404. |
[342] | Priller J , Ecker D , Landwehrmeyer B , Craufurd D . A Europe-wide assessment of current medication choices in Huntington’s disease. Mov Disord. (2008) ;23: (12):1788. |
[343] | Squitieri F , Cannella M , Giallonardo P , Maglione V , Mariotti C , Hayden MR . Onset and pre-onset studies to define the Huntington’s disease natural history. Brain Res Bull. (2001) ;56: (3-4):233–8. |
[344] | Paleacu D , Anca M , Giladi N . Olanzapine in Huntington’s disease. Acta Neurol Scand. (2002) ;105: (6):441–4. |
[345] | Bonelli RM , Niederwieser G , Tribl GG , Koltringer P . High-dose olanzapine in Huntington’s disease. Int Clin Psychopharmacol. (2002) ;17: (2):91–3. |
[346] | Nguyen CT , Rosen JA , Bota RG . Aripiprazole partial agonism at 5-HT2C: A comparison of weight gain associated with aripiprazole adjunctive to antidepressants with high versus low serotonergic activities. Prim Care Companion CNS Disord. (2012) ;14: (5): doi: 10.4088/PCC.12m01386 |
[347] | Davies MA , Sheffler DJ , Roth BL . Aripiprazole: A novel atypical antipsychotic drug with a uniquely robust pharmacology. CNS Drug Rev. (2004) ;10: (4):317–36. |
[348] | Brusa L , Orlacchio A , Moschella V , Iani C , Bernardi G , Mercuri NB . Treatment of the symptoms of Huntington’s disease: Preliminary results comparing aripiprazole and tetrabenazine. Mov Disord. (2009) ;24: (1):126–9. |
[349] | Ciammola A , Sassone J , Colciago C , Mencacci NE , Poletti B , Ciarmiello A , et al. Aripiprazole in the treatment of Huntington’s disease: A case series. Neuropsychiatr Dis Treat. (2009) ;5: :1–4. |
[350] | Caraceni TA , Girotti F , Giovannini P , Pederzoli M , Parati EA . Effects of DA agonist in Huntington disease hyperkinesia. Ital J Neurol Sci. (1980) ;1: (3):155–61. |
[351] | Loeb C , Roccatagliata G , La Medica G , Abbruzzese G , Albano C . Levodopa and Huntington’s chorea. J Neurol Neurosurg Psychiatry. (1976) ;39: (10):958–61. |
[352] | Reuter I , Hu MT , Andrews TC , Brooks DJ , Clough C , Chaudhuri KR . Late onset levodopa responsive Huntington’s disease with minimal chorea masquerading as Parkinson plus syndrome. J Neurol Neurosurg Psychiatry. (2000) ;68: (2):238–41. |
[353] | Mason SL , Barker RA . Emerging drug therapies in Huntington’s disease. Expert Opin Emerg Drugs. (2009) ;14: (2):273–97. |
[354] | Dreyer JK , Hounsgaard J . Mathematical model of dopamine autoreceptors and uptake inhibitors and their influence on tonic and phasic dopamine signaling. J Neurophysiol. (2013) ;109: (1):171–82. |
[355] | Seeman P , Van Tol HH . Dopamine receptor pharmacology. Curr Opin Neurol Neurosurg. (1993) ;6: (4):602–8. |
[356] | Pettersson F , Ponten H , Waters N , Waters S , Sonesson C . Synthesis and evaluation of a set of 4-phenylpiperidines and 4-phenylpiperazines as D2 receptor ligands and the discovery of the dopaminergic stabilizer 4-[3-(methylsulfonyl)phenyl]-1-propylpiperidine (huntexil, pridopidine, ACR16). J Med Chem. (2010) ;53: (6):2510–20. |
[357] | Luedtke RR , Rangel-Barajas C , Malik M , Reichert DE , Mach RH . Bitropic D3 dopamine receptor selective compounds as potential antipsychotics. Curr Pharm Des. (2015) ;21: (26):3700–24. |
[358] | Rangel-Barajas C , Malik M , Mach RH , Luedtke RR . Pharmacological modulation of abnormal involuntary DOI-induced head twitch response movements in male DBA/2J mice: II. Effects of D3 dopamine receptor selective compounds. Neuropharmacology. (2015) ;93: :179–90. |
[359] | Ponten H , Kullingsjo J , Lagerkvist S , Martin P , Pettersson F , Sonesson C , et al. In vivo pharmacology of the dopaminergic stabilizer pridopidine. Eur J Pharmacol. (2010) ;644: (1-3):88–95. |
[360] | Squitieri F , Landwehrmeyer B , Reilmann R , Rosser A , de Yebenes JG , Prang A , et al. One-year safety and tolerability profile of pridopidine in patients with Huntington disease. Neurology. (2013) ;80: (12):1086–94. |
[361] | de Yebenes JG , Landwehrmeyer B , Squitieri F , Reilmann R , Rosser A , Barker RA , et al. Pridopidine for the treatment of motor function in patients with Huntington’s disease (MermaiHD): A phase 3, randomised, double-blind, placebo-controlled trial. Lancet Neurol. (2011) ;10: (12):1049–57. |
[362] | Squitieri F , de Yebenes JG . Profile of pridopidine and its potential in the treatment of Huntington disease: The evidence to date. Drug Des Devel Ther. (2015) ;28: (9):5827–33. |
[363] | Luedtke RR , Perez E , Yang SH , Liu R , Vangveravong S , Tu Z , et al. Neuroprotective effects of high affinity Sigma1 receptor selective compounds. Brain Res. (2012) ;1441: :17–26. |
[364] | Francardo V , Bez F , Wieloch T , Nissbrandt H , Ruscher K , Cenci MA . Pharmacological stimulation of sigma-1 receptors has neurorestorative effects in experimental parkinsonism. Brain. (2014) ;137: (Pt 7):1998–2014. |
[365] | Hong J , Sha S , Zhou L , Wang C , Yin J , Chen L . Sigma-1 receptor deficiency reduces MPTP-induced parkinsonism and death of dopaminergic neurons. Cell Death Dis. (2015) ;6: :e1832. |
[366] | Malik M , Rangel-Barajas C , Sumien N , Su C , Singh M , Chen Z , et al. The effects of sigma (sigma1) receptor-selective ligands on muscarinic receptor antagonist-induced cognitive deficits in mice. Br J Pharmacol. (2015) ;172: (10):2519–31. |
[367] | Malik M , Rangel-Barajas C , Mach RH , Luedtke RR . The effect of the sigma-1 receptor selective compound LS-1-137 on the DOI-induced head twitch response in mice. Pharmacol Biochem Behav. (2016) ;148: :136–44. |
Figures and Tables
Fig.1
Schematic illustration of basal ganglia circuitry. Striatal medium spiny neurons (MSNs) receive excitatory corticostriatal input from IT and PT pyramidal neurons in cerebral cortex. Intra-telencephalic (IT) neurons preferentially activate substance P/D1 receptor (SP/D1R) MSNs that project to the internal globus pallidus and sustantia nigra pars reticulata (GPi/SNpr) forming the direct pathway. Pyramidal tract (PT) neurons activate enkephalin/D2 receptor (ENK/D2R) MSNs that form the indirect pathway and send projections to the external globus pallidus (GPe). GPe neurons in turn send axons to the subthalamic nucleus (STN), which projects to GPi/SNpr. GPi/SNpr is the basal ganglia output system that integrates direct and indirect pathway information and sends it back to cortex via thalamus.
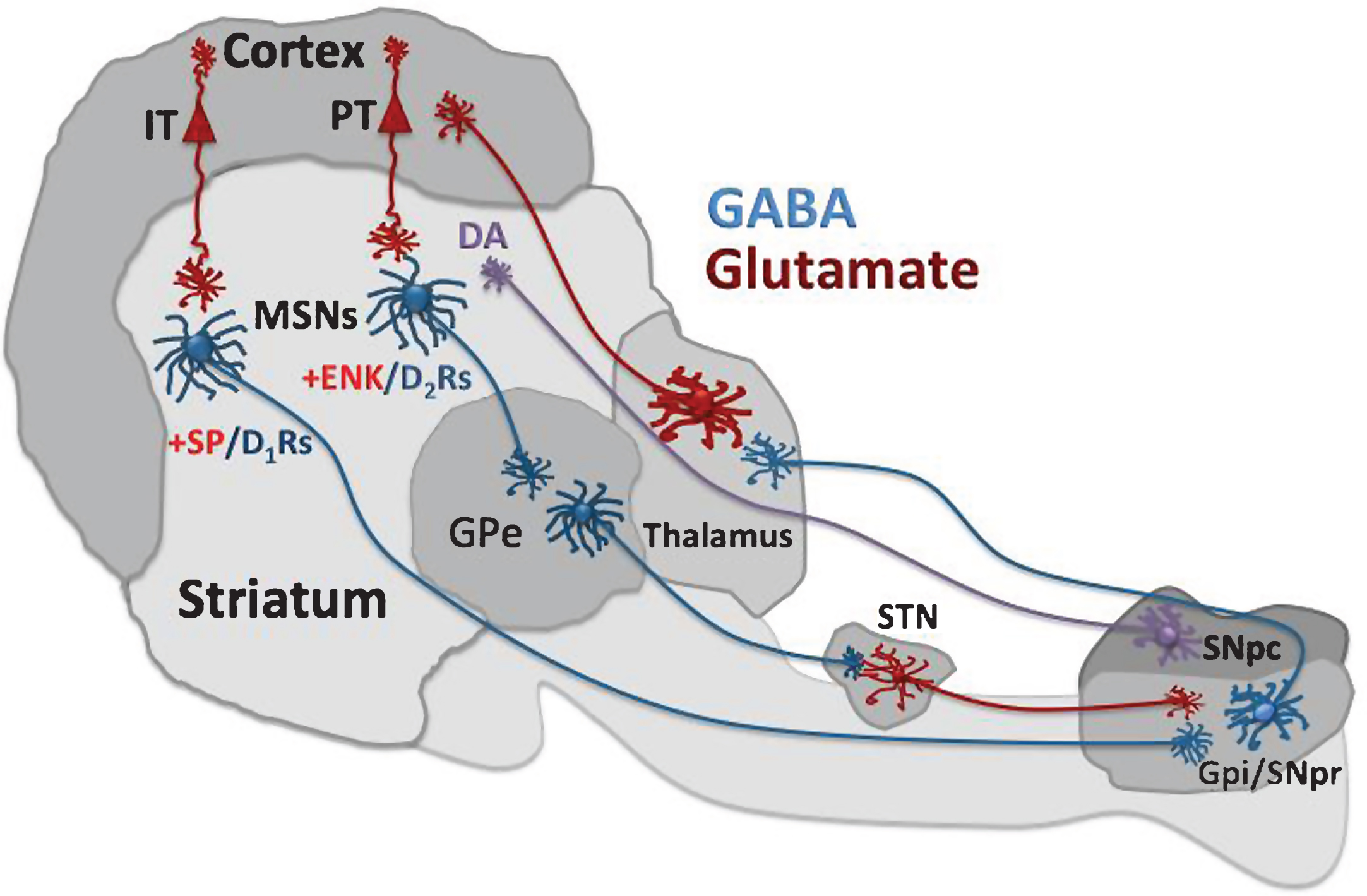
Fig.2
DA synthesis, vesicular loading, and release. DA is synthetized from tyrosine, which is converted to L-3,4-dihydroxyphenylalanine (L-DOPA) by tyrosine hydroxylase. Decarboxylation of L-DOPA by the L-amino acid decarboxylase (AADC) produces DA. Newly synthesized DA is incorporated into vesicles by vesicular monoamine transporter-2 (VMAT-2). Action potential activity allows the vesicles to fuse with the plasma membrane and release their contents into the synaptic cleft. Extracellular DA can activate postsynaptic receptors and be transported back to the terminal by the DA transporter (DAT). Remaining extracellular DA is metabolized by catechol-O-methyltransferase (COMT) or by monoamine oxidase (MAO). Presynaptic D2 receptors play an important role in the feedback regulation of DA release.
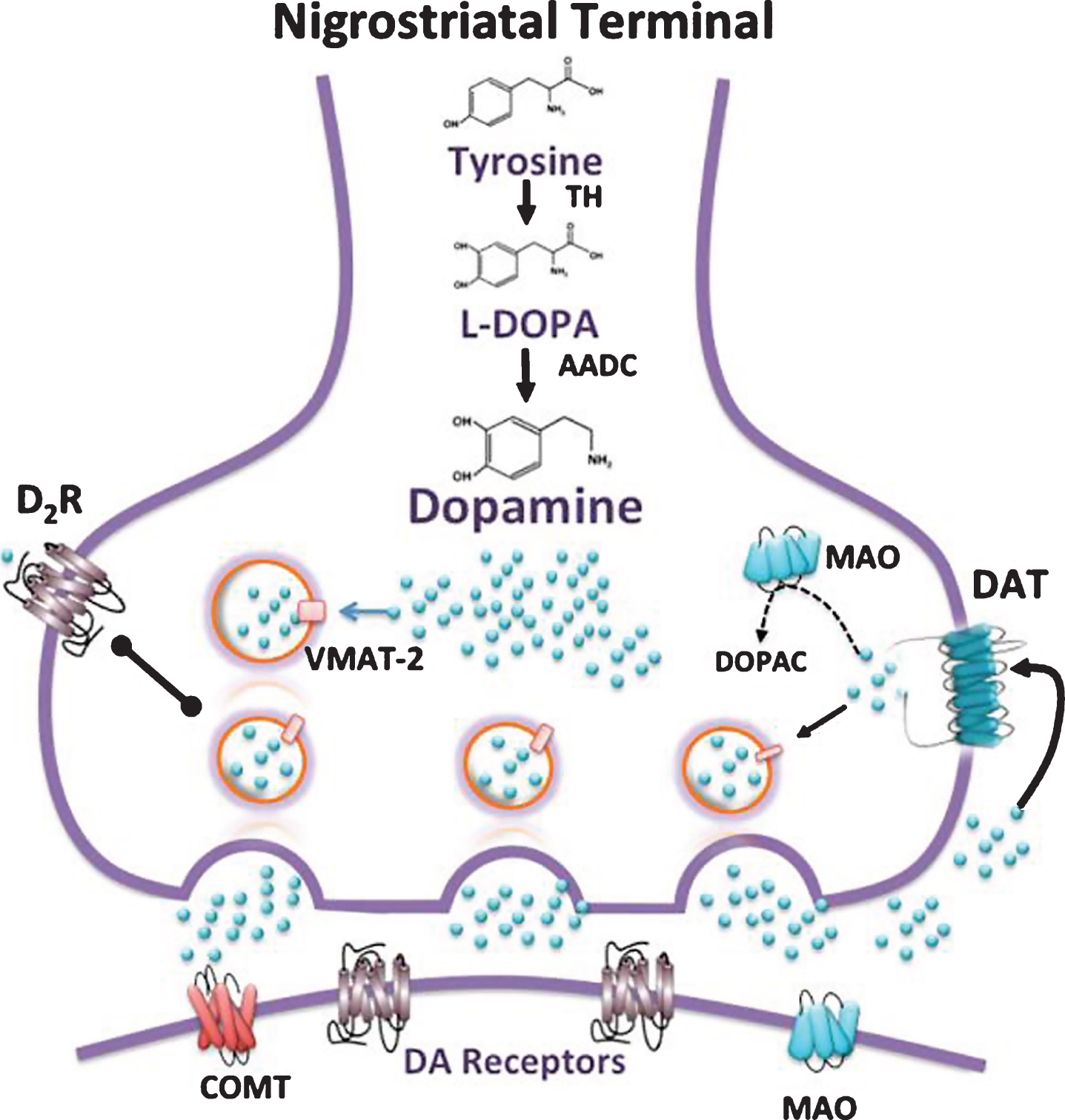
Fig.3
D1 and NMDA receptor signaling pathways and mHTT aggregates. The activation of D1 receptors results in G protein αs/olf coupling and the stimulation of adenylyl cyclase (AC), which transforms adenosine triphosphate (ATP) into 3′-5′-cyclic adenosine monophosphate (cAMP). This messenger activates protein kinase dependent cAMP (PKA), which phosphorylates cAMP-regulated phosphoprotein-32 (DARPP-32) leading to inactivation of phosphatase-1 (PP1). PKA can also phosphorylate NMDA receptors enhancing its activity. Through the phosphorylation of extracellular regulated kinase (ERK) and subsequent phosphorylation of mitogen- and stress- protein kinase-1 (MSK-1), PKA may activate the cAMP response element-binding protein (CREB), which is involved in gene transcription processes. Activation of the D1 receptor induces mHTT aggregations [202, 203], possibly by activating CREB. NMDA receptors interact with the postsynaptic density-95 (PSD-95) protein, which contains the domain Src homology 3 that interacts with HTT; mHTT prevents the interaction causing sensitization of NMDA receptors and promoting apoptosis. Stimulatory effects are indicated with arrows; inhibitory effects with a line ending in a circle. Broken lines indicate possible mechanism of action.
![D1 and NMDA receptor signaling pathways and mHTT aggregates. The activation of D1 receptors results in G protein αs/olf coupling and the stimulation of adenylyl cyclase (AC), which transforms adenosine triphosphate (ATP) into 3′-5′-cyclic adenosine monophosphate (cAMP). This messenger activates protein kinase dependent cAMP (PKA), which phosphorylates cAMP-regulated phosphoprotein-32 (DARPP-32) leading to inactivation of phosphatase-1 (PP1). PKA can also phosphorylate NMDA receptors enhancing its activity. Through the phosphorylation of extracellular regulated kinase (ERK) and subsequent phosphorylation of mitogen- and stress- protein kinase-1 (MSK-1), PKA may activate the cAMP response element-binding protein (CREB), which is involved in gene transcription processes. Activation of the D1 receptor induces mHTT aggregations [202, 203], possibly by activating CREB. NMDA receptors interact with the postsynaptic density-95 (PSD-95) protein, which contains the domain Src homology 3 that interacts with HTT; mHTT prevents the interaction causing sensitization of NMDA receptors and promoting apoptosis. Stimulatory effects are indicated with arrows; inhibitory effects with a line ending in a circle. Broken lines indicate possible mechanism of action.](https://content.iospress.com:443/media/jhd/2016/5-4/jhd-5-4-jhd160221/jhd-5-jhd160221-g003.jpg)
Fig.4
Alteration of the D2 receptor signaling pathway in HD. D2 receptor activation decreases cAMP signaling. D2 receptors can also activate β-arrestin2, which recruits phosphatase-2A (PP2A). PP2A in turn interacts with thymoma viral protoncogen (Akt). The PP2A-Akt complex promotes activation of glycogen synthase kinase-3 β (GSK-3β), which induces phosphorylation of β-catenin, a protein involved in the activation of the ubiquitin-induced proteasomal degradation system [209, 210]. Because GSK-3β expression is decreased in HD [90, 207] the decrease in phosphorylated β-catenin causes cellular toxicity [211]. Decreased activation of D2 receptors may be involved in the reduced degradation of mHTT. Stimulatory effects are indicated with arrows; inhibitory effects with a line ending in a circle. Broken lines indicate possible mechanism of action.
![Alteration of the D2 receptor signaling pathway in HD. D2 receptor activation decreases cAMP signaling. D2 receptors can also activate β-arrestin2, which recruits phosphatase-2A (PP2A). PP2A in turn interacts with thymoma viral protoncogen (Akt). The PP2A-Akt complex promotes activation of glycogen synthase kinase-3 β (GSK-3β), which induces phosphorylation of β-catenin, a protein involved in the activation of the ubiquitin-induced proteasomal degradation system [209, 210]. Because GSK-3β expression is decreased in HD [90, 207] the decrease in phosphorylated β-catenin causes cellular toxicity [211]. Decreased activation of D2 receptors may be involved in the reduced degradation of mHTT. Stimulatory effects are indicated with arrows; inhibitory effects with a line ending in a circle. Broken lines indicate possible mechanism of action.](https://content.iospress.com:443/media/jhd/2016/5-4/jhd-5-4-jhd160221/jhd-5-jhd160221-g004.jpg)


