
Oxidative Stress and Huntington’s Disease: The Good, The Bad, and The Ugly
Abstract
Redox homeostasis is crucial for proper cellular functions, including receptor tyrosine kinase signaling, protein folding, and xenobiotic detoxification. Under basal conditions, there is a balance between oxidants and antioxidants. This balance facilitates the ability of oxidants, such as reactive oxygen species, to play critical regulatory functions through a direct modification of a small number of amino acids (e.g. cysteine) on signaling proteins. These signaling functions leverage tight spatial, amplitude, and temporal control of oxidant concentrations. However, when oxidants overwhelm the antioxidant capacity, they lead to a harmful condition of oxidative stress. Oxidative stress has long been held to be one of the key players in disease progression for Huntington’s disease (HD). In this review, we will critically review this evidence, drawing some intermediate conclusions, and ultimately provide a framework for thinking about the role of oxidative stress in the pathophysiology of HD.
INTRODUCTION
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disorder resulting in a neuronal degeneration in the striatum, followed by degeneration of the cerebral cortex and thalamus during later stages of the disease. The initial clinical manifestations include personality and mood changes which are sometimes followed by a cognitive decline, and then involuntary choreiform movements, bradykinesia, dystonia (in some patients), rigidity, and dementia ultimately leading to death approximately 15–20 years from the age at onset [1, 2]. The genetic abnormality in HD involves an expansion of unstable CAG repeats in exon 1 of the huntingtin gene [3, 4]. The mutation results in a polyglutamine tract at the N’ terminal of the mutant huntingtin (mHtt) protein. The pathology of the disease has been attributed to toxic gain of functions for the mutant huntingtin protein, such as protein aggregation, transcriptional dysregulation, defective energy metabolism, oxidative stress, excitotoxicity, and inflammation [5–17], as well as to the loss of beneficial functions of wild type huntingtin protein (Htt), which includes BDNF translation, vesicle transport and as scaffold for autophagic machinery [18–21]. Despite remarkable progress in our understanding of this disease, the molecular logic connecting mHtt aggregates with cell dysfunction and pathological symptoms remains unclear. Transcriptionaldysregulation, mitochondrial dysfunction, oxidative stress and neuronal excitotoxicity are some of the key pathways consistently abnormal in cellular and mouse models of HD, as well as in autopsy tissue from HD patients. Herein, we will review the evidence regarding oxidative stress as a primary mediator of HD pathogenesis.
OXIDATIVE STRESS AND HD
A number of laboratories have provided evidence supporting the hypothesis that oxidative stress is a primary event in HD neuropathology [22–26]. Nevertheless, trials involving classical “antioxidants” in human HD patients have largely been unsuccessful [27–32]. It is important to emphasize that while molecular events, such as transcriptional dysregulation, protein aggregation, and mitochondrial dysfunction, have been linked to HD pathogenesis, it is still not clear whether oxidative stress causes HD, or is a consequence of more primary events [7, 33–35]. This uncertainty provides a compelling reason to review the putative molecular regulatory connections between redox changes and the established early events, such as mHtt aggregate formation and transcriptional dysregulation. This review will also hopefully stimulate efforts to discover novel strategies for reducing oxidative stress not just through the use of non-specific antioxidants, but also by targeting redox modulatory agents to these established early and causal events. Additionally, we will critically analyze the current support for a role for oxidative stress in HD, the shortcomings that have hindered progress and alternative redox therapeutic options that should dominate attention of the field to potentially achieve better therapeutic success in Huntington’s disease.
OXIDATIVE STRESS
Oxidative stress can be operationally defined as an imbalance between oxidants and anti-oxidants, in favor of oxidants, sufficient to result in cell damage, dysfunction or death. Oxidants are short-lived molecules with an unpaired electron in one or more of their outer orbitals and, therefore, they have the theoretical capacity to steal electrons from any cellular constituent, including proteins, lipids, and DNA. Cells produce these oxidants either in the form of reactive oxygen species (ROS), reactive nitrogen species (RNS), or reactive lipid species (RLS).
SOURCES OF ROS, RNS AND RLS
In mammalian cells, mitochondria are considered major sources of ROS production. Mitochondriause the electron transport chain to create a proton gradient and produce ATP. In this bioenergetic process, a small percentage of the electrons transferred down the electron transport chain for energy production inappropriately interact with oxygen molecules to produce superoxide free radicals (O2-). The resultant superoxide free radicals can readily react with other biomolecules to form other reactive molecules, including hydrogen peroxide, peroxyl (ROO.), hydroperoxyl (HOO.), and peroxinitrite (ONOO.) radicals [36, 37]. In this way, ROS or RNS are produced as normal byproducts of mitochondrial metabolism. Additionally, there are other sources of intracellular ROS production, such as cytochrome P450 enzymes of the endoplasmic reticulum, peroxisomal flavin oxidases, xanthine oxidase, and plasma membrane NADPH oxidases [38]. Other mediators of electrophilic stress are derived from lipids and are called RLS. RLS are defined as the oxidized lipid products including aldehydes such as HNE, malondialdehyde and acrolein as well as the A- and J- series isoprostanes etc. Their electrophilic nature allows them to covalently modify nucleic acids, lipids, and proteins [39, 40]. RLS can be produced by enzymatic (via the actions of lipoxygenase and cyclooxygenase enzymes) as well as non-enzymatic oxidation of polyunsaturated fatty acids (PUFAs) [39]. To limit damage and optimize local concentrations of ROS, RNS, and RLS for signaling, mammalian cells have a number of antioxidant molecules and enzymes, including but not limited to superoxide dismutases (SODs), peroxiredoxins (PRDXs), glutathione peroxidases (GPXs), glutathione reductases (GRs), glutathione transferases (GSTs), thioredoxins (TRXs), thioredoxin reductases (TRXRs), sulfiredoxins (SRXs), catalase, NADPH:qinone oxidoreductases (NQs), glutaredoxins (GRXs), glutathione (GSH), γ-tocopherol (vitamin E), ascorbic acid (vitamin C), carotenoids, and flavonoids [36, 41].
ROS AS SIGNALING MOLECULES
While better known as mediators of injury, ROS primarily evolved as signaling molecules in prokaryotes to facilitate adaptation to the changes in their immediate environment. Specifically, in prokaryotes ROS such as peroxide serve as important transcriptional regulators (Table 1). In eukaryotes, ROS evolved with a variety of diversified signaling functions. Amino acid residues such as cysteine and selenocysteine are particularly sensitive to the redox changes as they have the ability to not only donate electrons, but also to be reduced by cellular antioxidant enzymes. It is important to note that not all cysteine residues within a protein are equally sensitive to redox changes. Only cysteine residues with sufhydryl groups having low pKa (pKa 4-5) are sensitive to oxidation [42]. Within the physiological pH range, cysteine residues can exist as either thiolate anion (Cys-S-) or protonated cysteine thiol (Cys-SH) depending upon the local redox environment. Hydrogen peroxide is a key regulatory factor in dictating the redox state of cysteine residues within a protein [43]. Additionally, other molecules, such as NO and H2S, can also regulate the redox state of cysteine residues [44]. Under normal physiological conditions, for ROS to work as a signaling molecule, the ROS must generate reversible oxidation and exhibit substrate specificity [45]. The cysteine residues within a protein can be further oxidized into sulfenic (Cys-SOH), sulfinic (Cys-SO2H), or sulfonic forms (Cys-SO3H). The sulfenic form can be reduced back to a thiolate anion by different disulfide reductases, such as glutaredoxin and thioredoxin, and the sulfinic form can be reduced by sulfiredoxin, while the sulfonic forms are irreversible oxidation states. In addition, the cysteine residues can also be modified in other ways, including S-nitrosylation and S-glutathionylation, which are also reversiblemodifications.
ROS as molecules also exhibit substrate specificity. For instance, ROS very specifically regulate the catalytic activity of protein tyrosine phosphatases (PTPs). PTPs regulate a cascade of signaling molecules involved in growth factor signaling bycatalyzing the dephosphorylation of tyrosine residues in target proteins. PTPs, however, depend upon a redox sensitive cysteine for their enzymatic activity. ROS, by catalyzing the oxidation of this reactive cysteine, lead to inactivation of the PTPs, which, in turn, results in increased tyrosine phosphorylation of proteins involved in growth factor signaling. Moreover, the oxidative inactivation of PTPs is reversible[46, 47].
Spatially localized ROS production has been shown to be another important contributory factor in inducing effective redox signaling. For instance, ROS produced by NADPH oxidases present at the plasma membrane, which catalyzes redox changes mainly in membrane proteins, foster activation of kinase signaling and second messengers leading to modulation of nuclear proteins including transcription factors. While many nuclear transcription factors (TFs) have been shown to be redox-modulated, surprisingly, little is known whether redox changes occur in the cytoplasm or within the nucleus. [48, 49]. If they occur primarily in the nucleus, little is known about the factors in that subcellular compartment that govern its redox state, redox signaling, or redox related damage. Future studies that focus on the source of oxidants in the nuclear compartment may enhance our understanding of nuclear redox regulation.
HUNTINGTON’S DISEASE AND ABERRANT REDOX SIGNALING - AN UNEXPLORED CONNECTION
Currently, ROS are well established as signaling molecules involved in a variety of signal transduction pathways (Fig. 1), but very few of these signaling pathways have been examined in the context of oxidants and HD. Rather, most of the information regarding the redox regulation of signaling is currentlyemerging from the cancer field. For instance, cell survival pathway, such as the Erk pathway, is regulated in a redox associated manner by mitogen activated protein kinase phosphatase-3 (MKP-3). MKP-3 has a reactive cysteine whose oxidation results in inactivation of the phosphatase activity, leading to activation of the ERK pathway [50, 51]. Besides this, caspases contain a reactive cysteine, which regulates their activity. Under physiological conditions, mitochondrial caspase 3 and 9 are nitrosylated and, therefore, inactive while in the presence of pro-apoptotic signals, caspases get activated through the denitrosylation of their reactive cysteines [52]. Likewise, the activity of chaperones involved in proper protein folding, such as Hsp70, Hsp90 and PDI, are also redox-regulated through the reactive cysteine residues present in these proteins [53–55].
ROS also play important signaling functions in regulation of inflammatory responses. Immune cells express a number receptors, including Toll-like receptors (TLRs), NOD-like receptors (NLRs), and Rig-like receptors (RLRs), which, when bound to either microorganism-derived pathogen-associated molecular patterns (PAMPs) or endogenous cell-derived damage-associated molecular patterns (DAMPs), elicit the secretion of cytokines in order to fight pathogens or repair damaged tissue. TLR ligand lipopolysaccharides elicit inflammatory cytokines by stimulating ROS production [56]. Moreover, T cell and B cell activation has also been shown to require ROS signaling [57, 58]. Now, it is a well-established notion that the amount of ROS production dictates the degree of pro-inflammatory response. However, it is important to emphasize here that macrophages differentiate into at least two different phenotypes, called M1 and M2 macrophages, which have different consequential fates [59, 60]. Ligands, such as LPS and/or IFN-γ, activate macrophage differentiation into type 1 (M1 phenotype) through the pathway called the classical activation pathway. M1 macrophages produce pro-inflammatory cytokines, such as IL-1β, and TNF-α making it more vulnerable to cell death. By contrast, M2 macrophages differentiate in response to IL-4 or IL-13 and promote neuroprotective effects by producing anti-inflammatory cytokines, such as IL-6, IL-10, TGF-β, and neurotrophins. It is, therefore, very important to undertake studies focusing on understanding how ROS differentially regulate these two macrophage phenotypes or which of the ROS species are important for regulation of one phenotype, but not the other one. This information will be helpful in targeting only the ROS species/the pathways involved in activation of the M1 phenotype while sparing or even promoting the ROS species/pathway involved in the activation of the M2 phenotype. ROS as signaling molecules play important regulatory functions in a number of other pathways described in later sections of this review. Studies from our own lab have elucidated methods for spatially, temporally and quantitatively manipulating ROS such as peroxide. Specifically, we have utilized a cDNA of D-amino acid oxidase from red yeast as a strategy to tune the levels of peroxide in a cells. These studies have confirmed prior notions that low levels of peroxide can act as a second messenger, while higher levels can induce non cell-autonomous toxicity [61]. These methods could be applied to the study of Huntington’sdisease to facilitate understanding of cell type and compartments where ROS dysregulation may be greatest [38].
REDOX ACTIVE METALS IN HUNTINGTON’S DISEASE
Because of unpaired electrons in their outermost orbital shell, transitional metals exhibit the dynamic property of losing or gaining electrons easily. Their chemical nature enables some transitional metals (eg. Fe, Cu, Zn) to take part in a number of physiological redox reactions [62]. A growing number of studies suggest metal dyshomeostasis may be a part of HD pathogenesis [63]. In particular, iron (Fe) and copper (Cu) have been implicated as mediators of pathology. Iron and Copper have been shown to accumulate in post-mortem brain tissues of HD patients; in brain tissues from R6/2 mice; and in a Drosophila model of HD [64–66]. A recent MRI study showed enhanced accumulation of iron in basal ganglia and cortex of human HD patients and found its correlation with the CAG repeat number and severity of the disease pathogenesis [65]. Interestingly, few groups have attempted to understand the molecular basis of metal dyshomeostasis. Specifically, changes in expression of specific receptors and transporters of these metals such as transferrin receptors, ferroportin and metallothioneins have not been systematically examined in HD [67, 68].
Iron is classically believed to mediate oxidative damage via Fenton chemistry [62], although more recent studies suggest that iron may permit toxicity via its ability to activate iron containing proteins such as the hypoxia inducible factor prolyl hydroxylases (HIF PHDs) [69]. By contrast, copper has been shown to directly interact with N-terminal end of huntingtin to catalyze cysteine oxidation, cross-linking at itsN-terminal end and consequent mHtt oligomerization. Incomplete autophagic clearance of mHtt leads to increases in huntingtin aggregates [70–72]. Besides this, the excessive copper can also increase ROS production because of its capability to participate in a number of electron-transfer reactions.
In order to deal with the metal overload, a host of metal chelators have been tried as therapeutic options in HD. However, the problem associated with most of high affinity metal chelators is that in addition to having high metal binding capacity, they are very poor in their ability to cross blood brain barrier because of their molecular weight (>500) or hydrophilicnature [73]. For instance, the intra-ventricular delivery of deferoxamine (DFO), a canonical iron chelator, was shown to improve motor behavior in R6/2 mice [67], but systemic DFO (MW = 657) would not be expected to cross the BBB [69], thus limiting its potential clinical application.
In order to overcome the problem of CNS penetration, moderate affinity hydrophobic metal chelators such as 8-hydroxyquinoline (8-HQ) and its derivatives such as clioquinol and PBT2 have been developed and examined not only in AD but HD. The oral administration of clioquinol, a Cu/Fe chelator, was shown to delay the neuropathology significantly in R6/2 mice [74]. Recently, an improved clioquinol analog, PBT2, with better capability to cross BBB, came into the market. PBT2 has been shown to improve motor phenotype in R6/2 mice and to diminish the disease pathogenesis in C. elegans model of HD [75]. Very recently, PBT2 was found to be safe and well tolerable in early-stage to mid-stage HD patients [76]. Although the sample size was smaller and the clinical benefits were not very clear in this study, the outcome looks promising. Non-selective metal chelation thus looks promising, but may risk the unintended consequence of stealing metals from physiological metalloenzymes. This may necessitate an alternative approach of identifying the key molecular players perturbed by the excessive accumulation or altered distribution of these redox active metals and focusing on these players as therapeutic targets. For instance, we have shown Hif-prolyl hydroxylases (Hif-PHDs) as key targets for neuroprotection from oxidative death in vitro or brain hemorrhage in vivo. In this context, inhibiting Hif-PHDs provides neuroprotection independent of global iron chelation and suppression of Fenton chemistry [69, 77]. The model advanced in these studies is that metal chelators, including oxyquinolines optimally abrogate cell death by inhibiting the activity of pro-death transcription factors such as ATF4, thus leading to repression of death gene expression. This model has yet to be explored in the contextof HD.
BIOMARKERS OF OXIDATIVE DAMAGE
While small, transient discrepancies in oxidant levels and antioxidant defense are critical for physiological signaling by ROS, a sustained imbalance between ROS and antioxidants in favor of excessiveROS is believed to lead to irreversible cell damage, dysfunction, and death. A holy grail for the oxidative stress field has been identification of biomarkers for redox dyshomeostasis that suggest that this damage will or has occurred. Importantly, an oxidative stress product must be sensitive to oxidative stress changes, show specificity towards a particular oxidative pathway and must also be chemically stable in order to qualify as a good oxidative stress biomarker [78]. Practical and non-invasive methods of sample collection and detection are important criteria for identification of a good oxidative stress biomarker [78]. Unfortunately, none of the currently available tools appropriately meet criteria for an ideal oxidative biomarker, in part because increases in many currently used biomarkers could occur because of increased production or decreased turnover. Commonly accepted biomarkers of oxidative damage include oxidative modifications to DNA, proteins, lipids, and these have all been examined in the context of Huntington’s disease [79–81]. 8-hdroxydeoxyguanosine (8-OHdg), an oxidation product of DNA, is one of the most commonly used markers of oxidative damage to nucleic acid. Of note, leukocyte 8OHdg was shown as a very sensitive biomarker of Huntington’s disease in the PREDICT-HD study [79]. While this may reflect something about disease presence or course, it is unclear whether it reflects increased oxidative stress, or diminished turnover or repair of oxidized bases in HD.
The amino acids, cysteine and methionine, which are present in proteins, are very sensitive to oxidation [45]. Oxidation of protein side chains leads to carbonylation, which is viewed as an important biomarker of protein oxidation [82]. The oxidative attack on proteins can also lead to the formation of nitrated products such as 3-nitrotyrosine (3-NO2-Tyr) or halogenated products such as 3-chlorotyrosine (Cl-Tyr) and 3-bromotyrosine [83]. These oxidation products are also some of the commonly used markers of protein oxidation. Recently, other redox dependent post-translational modifications of proteins such as S-nitosylation, S-sulfenylation, S-glutathionylation, and S-sulfhydration have also been used as markers of protein oxidation [84].
Lipid peroxidation is a potential consequence of a specific type of oxidative stress. Oxidative damage to membrane lipids leads to changes in properties of cell membranes such as the fluidity, and inactivation of membrane associated enzymes or receptors. ROS oxidizes polyunsaturated fatty acids (PUFA) of phospholipid membranes, leading to the formationof oxidized products, such as malonaldehyde, 4-hydroxy-2-nonenal (4-HNE) and isoprostanes [39]. Some of these biomarkers and their methods of detections have wider applications while others have narrower applications with respect to in-vitro system versus in-vivo system or usage of samples such as brain tissue, CSF, blood, plasma, serum or urine. Additionally, the usage of these biomarkers and their methods of detection vary very much among different studies which complicates the comparison of these studies.
Despite these limitations, a host of research groupshave leveraged currently available redox biomarkers of DNA, proteins and lipids such as 8-hydroxy-2-deoxyguanosine, protein carbonyls, 3-nitrotyrosine, malondialdehyde, 3-hydroxykynurenine, 3-hydroxy-anthranilic acid, and TBARS etc. to investigate a role for oxidative stress in different models of HD [22, 23, 25, 80, 81, 85, 86]. Of note, the changes in these oxidative biomarkers should be interpreted very cautiously as redox modifications might have causal role in disease pathogenesis or might be consequence of cell death. Accordingly, careful evaluation of redox biomarkers through different stages of disease pathogenesis in mouse model of HD by Gil-Mohapel’s group, although correlative, provides potentially important information about the involvement of oxidative stress in disease pathogenesis [80].
OXIDATIVE DNA DAMAGE
Oxidative damage to DNA is believed, under basal conditions, to be a very common event and over 100 oxidative modifications to DNA have already been identified. These modifications can induce cell death or dysfunction via mutagenesis, replication blocks, or transcriptional blocks [87]. Damage to DNA is believed to be mediated via reactive oxygen species produced by the mitochondria, although it is unclear how ROS from mitochondria diffuse into the nucleus to damage DNA without interacting with the proteins or lipids in the nuclear envelope. NADPH oxidase 4 has been found to be localized in the nucleus of endothelial cells [48], but its role in neuronal nuclear redox homeostasis has not yet been explored. Whatever the source is, there is a multimodal antioxidant detoxification system that exists in the nucleus to detoxify hydrophilic and lipophilic oxidants [88]. Several groups have investigated whether oxidative damage to DNA occurs in HD, however, a major limitation of these studies is that they often include analysis of tissue where death or the commitment to die may have already occurred [23, 89, 90]. Accordingly, it is again not clear whether the changes reflect oxidative events that are causal to death, or those that are consequential to death. Increases in 8-hydroxydeoxyguanosine (a biomarker of oxidative damage to DNA) in the nucleus [89] or mitochondria [91] are seen in animal models of HD, as well as in blood [92] and serum [29] from humans. In some studies, these biomarkers have been used to identify the dose of a putative antioxidant, but, again, there is no evidence that directly correlates or associates these changes to cell death; thus, they could simply be tombstones of cells already committed to die. Indeed, it is possible anything that prevents cell death will prevent the appearance of oxidative biomarkers. For instance, Difiglia’s group has elegantly shown that the drugs which protect neurons also decrease the level of an oxidative biomarker [93]. Likewise, very recently, McMurray’s group has beautifully shown that treating HdhQ150 mice at different stages of disease progression with a synthetic antioxidant, XJB-5-131 not only improves motor function, and prevents neuronal death but also diminishes the level of oxidative biomarker 8-OHDG in the nucleus as well as mitochondria in striatal cells [94]. Similarly, future studies should identify the latest time point a drug can be given in the disease process, and then, in parallel experiments, in vivo and in vitro assays will examine whether changes in oxidative biomarkers can be observed before cells are ‘committed to die’. Such studies would provide more confidence as to whether oxidative DNA damage events are causally related to death in HD.
DNA OXIDATION, DNA REPAIR, AND EXPANDED POLYGLUTAMINE REPEATS
Oxidative damage to DNA induces DNA repair pathways that function in order to remove oxidized bases and restore the normal structure and function of DNA. Base excision repair (BER) plays a key role in repairing most oxidative damage. Oxidized bases, such as 8-oxo-guanosine (8-oxoG), 5-hydroxyuracil (5-OH-uracil), and 5-hydroxycytosine (5-OHC), are removed by BER enzymes that include OGG1, NEIL1, NEIL2, NEIL3, and APEX1. However, the removal of oxidized bases may trigger CAGexpansion [95].
The level of accumulation of oxidative DNA damage has been shown to correlate tightly with the degree of CAG repeats in R6/1 mice [96]. Interestingly, the somatic expansion has been shown to be length and sequence dependent. Expansions were found not to occur with short CAG repeats or in sequences which lacked the ability of forming specific secondary structures [96]. Specifically, the hairpin loop and other secondary structures formed during nucleotide repeat expansion were shown to make it more susceptible to oxidative damage, leading to the accumulation of more oxidative bases [97] which, in turn, could enhance the incidence of aberrant repair and increase the instability of trinucleotide repeats [98]. These findings suggest that the repair of damaged DNA may lead to the expansion and instability of CAG repeats in mutant huntingtin (Fig. 2). It is formally possible that mechanisms involved in repair of oxidized bases might promote tissue specific expansion of trinucleotide repeats and lead to selective vulnerability of some neurons in HD, as opposed to others.
Interestingly, the findings that R6/1 mice deficient in the BER enzymes OGG-1 or NEIL1 or mismatch repair (MMR) enzyme Msh2 showing a remarkable decrease in somatic CAG repeat instability [96, 99, 100] support the idea that the amount of DNA damage in a particular tissue dictates the rate of instability of trinucleotide repeats. However, Goula et al. [101] have emphasized that FEN1, a BER enzyme, and HMGB1, a BER co-factor, are potential contributing factors in determining the tissue specific variability to repeat instability. More recently, it has been shown that pools of oxidized bases, along with defective BER processing, were the combined factors involved in CAG expansion in R6/2 mice [102]. Defective BER processing in the striatum of R6/2 mice may indeed contribute to CAG expansion and region specific differences in accumulation of oxidized bases or capacities for BER processing. Variability in BER processing in distinct tissues of the CNS provides a testable model of selective vulnerability in HD. In fact, recently, McMurray’s group has shown that suppressing somatic expansion delays the onset of pathophysiology in HD mice, HdhQ150 [103, 104]. These studies raise the intriguing possibility that cell loss in HD is intimately coupled to somatic expansion of trinucleotide repeats during the lifetime of an afflictedmouse.
Of note, data has also been presented supporting the notion that increased oxidative DNA damage in HD may result not from increased oxidation of DNA, but from diminished repair. Mutant huntingtin has been shown to impair the DNA repair machinery by directly interacting with the non-homologous end joining repair protein ku70 and, thus, increasing the double strand breaks. Indeed, overexpression of ku70 was shown to diminish HD neuropathology [105]. Moreover, mutant huntingtin has also been shown to increase the level of ROS in neuronal and non-neuronal cells [106, 107]. These studies lead to a model in which increased oxidative damageto DNA combined with inefficient repair of that damaged DNA forms the basis for somatic expansion of trinucleotide repeats and ultimate neuronalloss.
IS OXIDATIVE STRESS A CAUSE OR A CONSEQUENCE IN HD PATHOLOGY?
Despite the numerous caveats mentioned above, several converging lines of inquiry in different HD models implicate oxidative stress as a key player in the pathogenesis of HD. Classical studies from postmortem brains of HD patients have demonstrated a significant increase in the level of oxidative damage. For instance, groups have examined the striatum and cortex from post-mortem brain samples and found a substantial increase in DNA double strand breaks (a potential result of free radical damage) [22, 24, 108]; a significant increase in the level of 8-OHDG (a biomarker of DNA oxidation) [23, 89]; a significant increase in the level of cytoplasmic lipofuscin (a peroxidation product of polyunstaturated fatty acids (PUFA)) [22, 109]; an increase in 3-nitrotyrosine (3-NT, a biomarker for peroxynitrite-induced oxidative damage of proteins) and malonadialdehyde (MDA, an oxidized product of PUFA used as a biomarker of lipid peroxidation) [22]. Interestingly, the oxidative marker lipofuscin was reported to be increased in a very selective manner only in susceptible neurons and not in spared neurons in the caudatenucleus [22].
Mitochondrial dysfunction is considered to be one of the key defects in HD pathogenesis. Recently, the mitochondrial creatine kinase, along with other mitochondrial proteins involved in oxidative phosphorylation (Cytochrome b-c1 complex subunit 2 and alpha subunit of ATPase) and Krebs cycle (citrate synthase and aconitase), were shown to exhibit significant increase in carbonylation (a type of protein oxidation catalyzed by reactive oxygen species) in the striatum of HD postmortem brain [25, 110]. Moreover, the protein levels of antioxidant enzymes, including peroxiredoxin 1, 2 and 6 (Prx 1, 2 and 6), glutathione peroxidases 1 and 6 (GPX1 and 6), and the activities of mitochondrial superoxide dismutase (MnSOD) and catalase, were also found to be remarkably elevated in the striatum and cortex of autopsied brain samples of HD patients [25]. In other studies, activities of complexes II, III, and IV were also reported to be significantly reduced in the striatum of HD patients [89, 111–113].
In a separate study, two key enzymes, pyridoxal kinase and antiquitin 1, which are involved in pyridoxal-5-phosphate (the active form of vitamin B6) metabolism were also found to be carbonylated in the striatum of HD patients [110]. Pyridoxal-5-phosphate plays an important role in the synthesis of neurotransmitters (serotonin, dopamine, and GABA) and the versatile antioxidant, glutathione [114].
Together, these studies from post-mortem brain specimens suggest that oxidative damage of biomolecules may be increased and may be related to neuronal loss in HD. However, again, these studies do not address whether oxidative stress is caused directly by mutant huntingtin or is simply a manifestation of dying neurons. In this regard, a recent study showed a significant increase in lipid peroxidation and decrease in glutathione levels in the plasma of asymptomatic HD gene carriers compared with age and sex matched healthy subjects [115]. Other similar studies have shown enhanced level of lipid peroxidation in the plasma of symptomatic patients compared to that of healthy subjects [85, 116]. Recently, the expression of genes such as AHCY1, ACO-2, OXCT-1 encoding S-adenosyl L-homocysteine hydrolase 1, aconitase 2 and enzymes involved in 3-oxoacid CoA transferase 1 proteins, were found to be involved in oxidative stress response and mitochondrial energy metabolism and were shown to be downregulated in the peripheral leukocytes of HD gene carriers, as well as in symptomatic HD patients compared to that of age and sex matched healthy controls [117]. In another recent study of HD patients, the levels of different oxidative markers, including leukocyte 8-OHDG and plasma MDA levels, were higher, but the activities of antioxidants, such as Cu/Zn-SOD and GPX1 in erythrocytes, were much lower in HD patients compared to that of healthy controls [92]. Very recently, oxidative markers, such as MDA and advanced oxidation protein products (AOPP), were shown to be significantly higher in the serum of symptomatic HD patients compared to that of age matched healthy controls. AOPP demonstrated a high correlation with motor severity of patients [118].
These crucial findings of changes in peripheral oxidative biomarkers in asymptomatic as well as symptomatic HD patients compared to that of healthy controls are consistent with the notion that oxidative stress is a dominant event in HD pathogenesis. However, there are many conflicting reports regarding changes in activities of antioxidants, such as Cu/Zn SOD [92, 119], GPX1 [92, 118] and mitochondrial complexes [120–123], in peripheral tissues which suggest that changes in oxidative biomarkers could be the result of diminished turnover of oxidative biomarkers rather than their increased generation. Ciancarelli et al. (2014) [119] recently demonstrated a lack of correlation between most of these peripheral oxidative biomarkers and clinical outcome measures in HD patients, which indicates that although there are higher levels of oxidative biomarkers in the blood of HD patients compared to the healthy controls, the correlation between the oxidative stress and selective neuronal vulnerability in the striatum and cortex or the psycho-motor defects in HD patients are high. It is also unclear to what extent changes in distinct populations reflect dietary differences or extent or choice of drug regimens for HD patients. Together, along with the failure of classical antioxidants in human clinical trials, these studies create an unclear picture of the role of oxidative stress in HD.
USE OF ANIMAL STUDIES TO ENHANCE OUR UNDERSTANDING OF OXIDATIVE STRESS IN HD
While human studies are useful to define an association, animal studies can be useful to establish or infer causality. Several transgenic mice models of HD have been created that mimic the neuropathology of human HD patients and express either the N-terminal fragment (R6/1 and R6/2 mouse) or full length mHtt (YAC128 and BACHD mice). Moreover, based on the mitochondrial hypothesis of HD pathogenesis, some chemically-induced mice models of HD, such as malonate (A reversible inhibitor of mitochondrial complex II) and 3-nitropropionic acid (3-NP, an irreversible inhibitor of mitochondrial complex II) injected mice, have also been produced that lead to oxidative stress and show selective striatal neuronal atrophy and defective neuro-motor function very similar to that seen in human HD patients [124–129]. Many studies in mouse and fly models of HD have provided support that oxidative damage to DNA, proteins, and lipids is a common association of Huntington’s disease related neurodegeneration. In one study using 7–12 week old R6/2 mice, a progressive increase in the level of mitochondrial oxidative DNA damage was shown with age [130]. In a separate study, BACHD mice expressing full length mHtt protein were shown to exhibit a significant increase in the level of Ataxia-telangiectasia mutated (ATM) protein [131], which is known to become activated in response to DNA damage [132] and oxidative stress [133]. Furthermore, increased levels of oxidative DNA damage (8-OHDG level) have been found in R6/2 at 12–14 weeks of age [134].
In a recent proteomic study, a number of proteins, such as α-enolase, γ-enolase aconitase (neuron specific enolase), voltage dependent anion channel 1 (VDAC1), heat shock protein 90 (hsp90), and creatine kinase, were found to show significant oxidative damage in 10 week R6/2 mice compared to that of either 4 week old R6/2 mice or with age matched wild type mice [26]. In other studies, increased levels of different oxidative markers were seen in the striatum of BACHD mice, N171-82Q mice, and the 3-NP model of HD [130, 135, 136]. Recently, a significant increase in the S-glutathionylation of TRPC5 (The Ca2 +-permeable transient receptor potential cation channel 5) was shown in the striatum of 12 month old YAC128 HD mice as an evidence that oxidative stress is a dominant player in HD neuropathology [137]. Collectively, these findings are correlative and are consistent with the notion that mitochondrial dysfunction and/or oxidative stress occurs at relatively late stages of the HD phenotype based on the timing in these HD models. Many of these studies suffer in not manipulating antioxidant defenses or repair enzymes related to oxidant damage. Accordingly, they provide tantalizing support for the oxidative stress model of HD pathogenesis, but they do not go far enough. A major concern when one observes multiple indices of oxidative damage is that the cell has made a commitment to die and accordingly it has disabled homeostatic mechanisms that prevent buildup of oxidative damage in proteins. It is essential moving forward that the HD community be vigilant about performing studies in which redox balance can be modulated via specific molecular or pharmacological tools. Otherwise, the relationship between oxidative damage and primary pathogenesis will remainelusive.
Indeed, there are limited number of studies that indicate that oxidative stress might play an important causal function. These studies should be used as models on which to base further study. One study employed both CAG140 knock-in mice and mHtt transfected COS-1 cells and showed that the N- terminal fragment of mHtt was shown to be sensitive to oxidation at specific cysteine residues. Oxidative modification of these cysteines promoted oligomerization and delayed the clearance of soluble fraction of mHtt [70]. Recently, double strand breaks and DNA damage responses were found in PC12 cells expressing mHtt, as well as in the striatum of R6/2 transgenic HD mouse even before aggregate formation of mHtt [138]. Similarly, in in vitro models of HD16Q and HD150Q cells, oxidative stress has been shown to enhance the aggregation of mHtt and cell death. These studies highlight the varied role that redox modifications can play in promoting HD pathogenesis. They could, on one hand, modify mutant htt directly to promote protein aggregation; alternatively, redox modification could activate Erk and transglutaminase to foster crosslinking of mutant htt with other proteins in the cytoplasm or nucleus. Other, but not exclusive models involve disruption of redox homeostasis downstream of aggregated htt. The numerous potential nodes of redox modulation highlight an additional complexity to study the role of oxidative stress in HD.
CAN ONE MEASURE AN INCREASE IN ROS DIRECTLY IN HUNTINGTON’S DISEASE?
Another holy grail for the field of redox biology has been the development of techniques to monitor local concentrations of distinct oxidant species dynamically in living cells. It is clear that our ability to understand calcium’s role as a second messenger and toxin was greatly facilitated by Roger Tsien’s discovery of quin-2 and fura-2-first generation calcium sensitive reporters that allowed new insights in calcium gradients and micro-domains in intact cells [139, 140]. Despite intensive effort, attempts to measure increases in ROS concentrations dynamically in living cells have been fraught with problems. Indeed, many of the dyes used to track changes in concentrations of reactive oxygen and reactive nitrogen species are not ratiometric (and thus are sensitive to changes in cell geometry); are difficult or impossible to calibrate to a single oxidant species; and do not enjoy selectivity for a particular oxidant species [141, 142]. The recent development of HyPer, a fusion protein of the prokaryotic transcription factor, OxyR and Yellow fluorescent protein has given rise to significant optimism that these technical challenges are being solved [143]. However, HyPer and other new probes have yet to be applied to HD models in vitro or in vivo using 2-photon microscopy to minimize photo-bleaching and maximize signal.
Additionally, only a few studies have examined redox changes during the asymptomatic and prodromal stages of HD pathogenesis [138, 144–146]. Tunable models of HD, in which mutant huntingtin can be turned off at a particular age, allow a rigorous dissection of when cells are committed to die due to HD. Examining oxidative biomarkers before any evidence of irreversible cell death appears is essential to make the case for oxidative damage as a primary event, rather than as a modulator or tombstone.In addition, the cell type specific manifestation (MSNs, interneuron, astrocytes, oligodendrocytes or microglia) of oxidative damage to the reportedproteins is also unclear.
BRIDGING TRANSCRIPTIONAL DYSREGULATION WITH OXIDATIVE STRESS OPENS NEW ROADS OF THERAPEUTIC OPPORTUNITIES
A major thrust of longstanding interest to both the redox biology and the Huntington’s disease community has been epigenetic and transcriptional homeostatic mechanisms to oxidant and metabolic stress. A dominant theme in these investigations has been the notion that if adaptive transcriptional responses to stresses such as oxidant stress are inhibited by mutant huntingtin, then this will set up a condition where stress is persistent rather than compensated. Precise understanding of the dominant mechanisms of adaptation to oxidative stress is necessary to fully vet this model.
The functional association of redox processes with adaptive transcriptional processes are conserved in all aerobic organisms throughout evolution, from bacteria to mammals. ROS are necessary byproducts of oxygen metabolism in aerobic organisms. There are a host of transcriptional proteins that are redox-regulated (Table 1). Moreover, the mammalian transcriptional machinery responds to redox changes via multiple mechanisms (Fig. 3). For example, cysteine oxidation regulates transcription in a host of ways; and transcription factors known to be regulated via cysteine oxidation, such as NF-κB, p53, and CREB, have been shown to be dysregulated in HD models [147–149]. AP-1 family proteins such as FRA-2 and JUND have been shown to be associated with mHtt led extensive DNA methylation in an in vitro model of HD employing STHdhQ111 striatal cells [150]. Ref-1 (APE-1), a protein that regulates the oxidation state of cysteines important in DNA binding of specific transcription factors (Table 1), was recently shown to be disrupted in the mitochondria of an in vitro model of HD using STHdhQ111 cells [90]. However, Ref-1 also has a very important role in base excision repair of oxidative damage in DNA [151, 152]. Therefore, it would interesting to understand which of these two functions of Ref-1 is more important in terms of Huntington’s disease. Interestingly, it is as yet unknown whether redox changes really play a critical regulatory role in mediating thedysregulation of these transcription players in the case of HD.
Importantly, reactive lipid species (RLS) are the key sources of oxidative stress [39]. In order to counteract the RLS mediated oxidativc stress, cells have evolved a transcriptional pathway modified andactivated by electrophilic stress, called nuclear factor E2-related factor-2 (Nrf-2). It is a basic leucine zipper (bZIP) TF that can activate a host of genes involved in electrophile counter-attack and xenobiotic defense. Under basal conditions, Nrf-2 binds to the Kelch like ECH associated protein-1 (Keap-1) [153, 154], a cysteine rich protein [155] in the cytosol, which facilitates its ubiquitination and proteasomal degradation [156, 157]. Under electrophilic stress, critical cysteines on Keap-1 are alkylated. Accordingly, Keap-1 changes its conformation [155], which causes release of Nrf-2 followed by its nuclear translocation [158].Once within the nucleus, Nrf-2 forms heterodimers with other transcriptional regulators, such as Maf family proteins, and binds to the antioxidant response element (ARE, 5’-TGAG/CnnnGC-3’). The ARE is present in the promoter regions of established Nrf-2 target genes involved in cellular defense and metabolism. [159]. Nrf-2 has been shown to be a critical transcriptional player in neuroprotection in different HD models. Indeed, Nrf2-/- mice when exposed to 3-NP or malonate at the doses not having any effect in wild type mice have been found to show more prominent lesion volumes [160–162]. Moreover, Jeffrey Johnson’s group has elegantly shown that astrocytic overexpression of Nrf-2 protects striatal neurons from mitochondrial complex II inhibition in malonate model of HD [163]. Triterpenoids such as CDDO-EA (2-Cyano-3, 12-Dioxooleana-1,9-Dien-28-Oic acid-ethyl amide) and CDDO-TFEA (CDDO-trifluoroethyl amide) when fed to N171-82Q HD mice were shown to upregulate Nrf-2 target genes in brain and peripheral tissues, decrease oxidative stress, improve motor behavior and enhance longevity [136]. Similarly, Dimethyl fumarate (DMF), a clinically approved Nrf-2 activator, when given orally to the HD mice models such as R6/2 and YAC128, was shown to result in increased Nrf2 neuronal immunoreactivity, improvement of motor impairment, preservation of neuronal subpopulations in striatum and cortex as well as enhanced survival [164]. It is still unclear whether DMF actually gets to the brain to activate Nrf-2 dependent genes. Rather, recent studies indicate that DMF can also activate BDNF expression, although this appears to be negatively regulated by Nrf-2 [165]. A high priority for HD therapeutics is the development of Nrf-2 activators that penetrate through the BBB but are not electrophiles and thus do not have toxicity.
As mentioned above, physiological levels of ROS serve as very important signaling molecules [61, 166]. By affecting signaling pathways, ROS can, indirectly, affect a whole array of transcriptional processes. There are many other TFs which are regulated by changes in the redox states of NAD, NADP, or glutathione. For instance, the transcriptional activities of SIRT1, a deacetylase which interacts with PGC-1α, a well-established regulator ofmitochondrial function and energy metabolism [167, 168], and CTBP-1, a co-repressor regulating the expression of BDNF by binding with transcription factor NRSF [169], depend upon the NAD-NADH ratio [170, 171]. Additionally, the class III histonedeacetylases (class III HDACs; also known as sirtuins) are NAD+ dependent epigenetic modulators. Prior studies from our laboratory have shown that glutathione depletion can induce acetylation of Sp1 and Sp3 to induce a frustrated compensatory response to oxidative stress. Indeed, modulation of class I HDACs which enhance Sp1/Sp3 acetylation, leads to increased Sp1/Sp3 DNA binding and resistance to glutathione depletion, 3-Nitroproprionic acid induced striatal damage; and transgenic overexpression of mutant huntingtin [172]. These findings raise several interesting possibilities. First, they raise the possibility that class I HDAC inhibitors could modify HD pathogenesis by augmenting compensatory responses to oxidative stress rather than by effecting mHtt mediated transcriptional suppression. Alternatively, mHtt may induce toxicity by repressingtranscription factors such as Sp1/Sp3 and, in turn, repress genes involved in compensation for oxidative stress. Furthermore, we have shown that Sp1 and Sp3 DNA binding activities are dramatically induced by oxidative stress in cortical neurons, (Fig. 4) [173]. Increased DNA binding of Sp1 and Sp3, in part, reflects the enhanced level of these proteins in nuclei of cortical neurons. Moreover, we have shown that protein levels of Sp1 and Sp3 are induced in the neostriatum in 3-NP model, as well as the R6/2 model of HD mice [36]. A consistent higher expression of full-length Sp1 or Sp3 has been shown to protect neurons from oxidative stress, DNA damage, or both [173]. Interestingly, Sp1 is a coactivator of cystathionine-γ-lyase, the biosynthetic enzyme for cysteine, a major target of redox regulation. In a recent study, a significant depletion of cystathionine-γ-lyase was found in an in vitro model of HD, STHdhQ111 cells, as well as in different brain regions of R6/2 and in the striatum of Q175 mice. The defect was correlated with the transcriptional dysfunction of Sp1, leading to the defective activity of cystathionine-γ-lyase. Cysteine supplementation was shown to reverse the abnormalities both in in vitro as well as in vivo models of HD [174]. Together, these studies suggest that mhtt via its ability to directly modify Sp1 and Sp3 [175] may repress genes involved in redox homeostasis. Strategies to augment Sp1 and Sp3 function, including HDAC inhibition, represent an opportunity to restore homeostasis via augmentation of genes involved in antioxidant defense. Future studies will clarify whether gene repression in HD extends to many homeostatic pathways (ER stress, mitochondrial stress) or whether oxidative stress is a dominant mediator of cell death and injury. As distinct transcription factors including ATF-4, Nrf-2, NF-κ-B and Sp-1 are activated via distinct oxidant stimuli in distinct CNS cell types, future studies will need to explore whether genes activated by other redox regulated transcription factors are repressed by mHtt to gain a more complete picture of transcriptional redox signaling in HD and their unique and redundant roles in oxidative stress homeostasis. Indeed, other coactivators or transcription factors including the PGC1alpha family may influence redox homeostasis indirectly via their ability to induce mitochondrial biogenesis [176]. Recent studies suggest that mitochondria not only act as ROS producers, but surprisingly ROS scavengers as well [177, 178]. Accordingly, as techniques for measuring ROS specifically in mitochondria become available, experiments designed to understand the role of mitochondrial biogenesis in increasing or decreasing ROS production can be clarified.
CONCLUDING REMARKS
A number of laboratories have garnered evidence supporting the notion that oxidative stress is one of dominant events in HD pathogenesis. However, the failure of antioxidants to modify disease progression in human HD patients has led to growing impatience over the oxidative stress hypothesis in HD neurodegeneration. The reasons for therapeutic failure are multifactorial and include poor understanding of the precise targets of antioxidants related in part to a lack of specific oxidative biomarkers; the possibility that more than one oxidant is mediating damage and therefore antioxidants with specificity for one species over another might only do part of the job; the possibility that the therapeutic effects of antioxidant supplementation are counterbalanced by negative effects of these agents on physiological redox signaling in the brain; and the possibility that stoichiometric antioxidant simply do not get to the brain in concentrations adequate to neutralize toxic ROS, if theyexist.
Another rational view of oxidative stress in HD advanced by our group and others (Sol Snyder at Hopkins; and Jeff Johnson at Wisconsin) is that it emerges due to a failure of compensatory transcriptional responses. This failure is mediated in part due to the direct repression of adaptive gene expression by mhtt. As Leslie Thompson’s group has shown, this could be due to direct interactions of mhtt with coactivators, or due to direct interaction of mhtt with redox-regulated transcription factors [e.g. Sp1; see [175]]. Repression of adaptive responses to oxidative stress leads to persistent stress at a cellular, local and systemic level and ultimately cell demise. According to this model, understanding how to derepress these homeostatic responses will result in the augmentation of many genes that ultimately will serve to restore redox homeostasis in HD. The tools for measuring ROS, and the genes that compensate for ROS are now becoming available to address this intriguing model experimentally. Such studies will likely involve small molecule enhancers of adaptive transcription and will give rise to novel and more robust approaches to treat oxidative stress in HD and other diseases compared to traditional stoichiometricantioxidants.
CONFLICT OF INTEREST
The authors declare that there is not conflict of interest in publishing the paper.
ACKNOWLEDGMENTS
The author’s own publications cited here in this review were supported by the National Institute on Aging of the National Institutes of Health under award number P01 AG14930-15A1 and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation.
REFERENCES
[1] | Vonsattel JP , DiFiglia M . Huntington disease. J Neuropathol Exp Neurol. (1998) ;57: (5):369–84. PubMed PMID: 9596408. |
[2] | Ross CA , Tabrizi SJ . Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. (2011) ;10: (1):83–98. PubMed PMID: 21163446. |
[3] | A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell. (1993) ;72: (6):971–83. PubMed PMID: 8458085. |
[4] | Langbehn DR , Brinkman RR , Falush D , Paulsen JS , Hayden MR , International Huntington’s Disease Collaborative G. A new model for prediction of the age of onset and penetrance for Huntington’s disease based on CAG length. Clin Genet. (2004) ;65: (4):267–77. PubMed PMID: 15025718. |
[5] | Gipson TA , Neueder A , Wexler NS , Bates GP , Housman D . Aberrantly spliced HTT, a new player in Huntington’s disease pathogenesis. RNA Biol. (2013) ;10: (11):1647–52. Pubmed Central PMCID: 3907474. |
[6] | Hatters DM . Putting huntingtin “aggregation” in view with windows into the cellular milieu. Curr Top Med Chem. (2012) ;12: (22):2611–22. PubMed PMID: 23339311. |
[7] | Kumar A , Vaish M , Ratan RR . Transcriptional dysregulation in Huntington’s disease: A failure of adaptive transcriptional homeostasis. Drug Discov Today. (2014) ;19: (7):956–62. PubMed PMID: 24662036. Pubmed Central PMCID: 4082751. |
[8] | Mochel F , Haller RG . Energy deficit in Huntington disease: Why it matters. J Clin Invest. (2011) ;121: (2):493–9. PubMed PMID: 21285522. Pubmed Central PMCID: 3026743. |
[9] | Yano H , Baranov SV , Baranova OV , Kim J , Pan Y , Yablonska S , et al. Inhibition of mitochondrial protein import by mutant huntingtin. Nat Neurosci. (2014) ;17: (6):822–31. PubMed PMID: 24836077. Pubmed Central PMCID: 4174557. |
[10] | Ayala-Pena S . Role of oxidative DNA damage in mitochondrial dysfunction and Huntington’s disease pathogenesis. Free Radic Biol Med. (2013) ;62: :102–10. PubMed PMID: 23602907. Pubmed Central PMCID: 3722255. |
[11] | Pitts A , Dailey K , Newington JT , Chien A , Arseneault R , Cann T , et al. Dithiol-based compounds maintain expression of antioxidant protein peroxiredoxin 1 that counteracts toxicity of mutant huntingtin. J Biol Chem. (2012) ;287: (27):22717–29. PubMed PMID: 22577145. Pubmed Central PMCID: 3391089. |
[12] | Rotblat B , Southwell AL , Ehrnhoefer DE , Skotte NH , Metzler M , Franciosi S , et al. HACE1 reduces oxidative stress and mutant Huntingtin toxicity by promoting the NRF2 response. Proc Natl Acad Sci U S A. (2014) ;111: (8):3032–7. PubMed PMID: 24516159. Pubmed Central PMCID: 3939919. |
[13] | Reijonen S , Kukkonen JP , Hyrskyluoto A , Kivinen J , Kairisalo M , Takei N , et al. Downregulation of NF-kappaB signaling by mutant huntingtin proteins induces oxidative stress and cell death. Cell Mol Life Sci. (2010) ;67: (11):1929–41. PubMed PMID: 20232225. |
[14] | Molero AE , Arteaga-Bracho EE , Chen CH , Gulinello M , Winchester ML , Pichamoorthy N , et al. Selective expression of mutant huntingtin during development recapitulates characteristic features of Huntington’s disease. Proc Natl Acad Sci U S A. (2016) ;113: (20):5736–41. PubMed PMID: 27140644. Pubmed Central PMCID: 4878495. |
[15] | Sepers MD , Raymond LA . Mechanisms of synaptic dysfunction and excitotoxicity in Huntington’s disease. Drug Discov Today. (2014) ;19: (7):990–6. PubMed PMID: 24603212. |
[16] | Crotti A , Benner C , Kerman BE , Gosselin D , Lagier-Tourenne C , Zuccato C , et al. Mutant Huntingtin promotes autonomous microglia activation via myeloid lineage-determining factors. Nat Neurosci. (2014) ;17: (4):513–21. PubMed PMID: 24584051. Pubmed Central PMCID: 4113004. |
[17] | Andre R , Carty L , Tabrizi SJ . Disruption of immune cell function by mutant huntingtin in Huntington’s disease pathogenesis. Curr Opin Pharmacol. (2016) ;26: :33–8. PubMed PMID: 26461267. |
[18] | Zuccato C , Tartari M , Crotti A , Goffredo D , Valenza M , Conti L , et al. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet. (2003) ;35: (1):76–83. PubMed PMID: 12881722. |
[19] | Twelvetrees AE , Yuen EY , Arancibia-Carcamo IL , MacAskill AF , Rostaing P , Lumb MJ , et al. Delivery of GABAARs to synapses is mediated by HAP1-KIF5 and disrupted by mutant huntingtin. Neuron. (2010) ;65: (1):53–65. PubMed PMID: 20152113. |
[20] | Ochaba J , Lukacsovich T , Csikos G , Zheng S , Margulis J , Salazar L , et al. Potential function for the Huntingtin protein as a scaffold for selective autophagy. Proc Natl Acad Sci U S A. (2014) ;111: (47):16889–94. PubMed PMID: 25385587. PubMed Central PMCID: 4250109. |
[21] | Rui YN , Xu Z , Patel B , Chen Z , Chen D , Tito A , et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat Cell Biol. (2015) ;17: (3):262–75. PubMed PMID: 25686248. PubMed Central PMCID: 4344873. |
[22] | Browne SE , Ferrante RJ , Beal MF . Oxidative stress in Huntington’s disease. Brain Pathol. (1999) ;9: (1):147–63. PubMed PMID: 9989457. |
[23] | Polidori MC , Mecocci P , Browne SE , Senin U , Beal MF . Oxidative damage to mitochondrial DNA in Huntington’s disease parietal cortex. Neurosci Lett. (1999) ;272: (1):53–6. PubMed PMID: 10507541. |
[24] | Stack EC , Matson WR , Ferrante RJ . Evidence of oxidant damage in Huntington’s disease: Translational strategies using antioxidants. Ann N Y Acad Sci. (2008) ;1147: :79–92. PubMed PMID: 19076433. |
[25] | Sorolla MA , Reverter-Branchat G , Tamarit J , Ferrer I , Ros J , Cabiscol E . Proteomic and oxidative stress analysis in human brain samples of Huntington disease. Free Radic Biol Med. (2008) ;45: (5):667–78. PubMed PMID: 18588971. |
[26] | Perluigi M , Poon HF , Maragos W , Pierce WM , Klein JB , Calabrese V , et al. Proteomic analysis of protein expression and oxidative modification in r6/2 transgenic mice: A model of Huntington disease. Mol Cell Proteomics. (2005) ;4: (12):1849–61. PubMed PMID: 15968004. |
[27] | Feigin A , Kieburtz K , Como P , Hickey C , Claude K , Abwender D , et al. Assessment of coenzyme Q10 tolerability in Huntington’s disease. Mov Disord. (1996) ;11: (3):321–3. PubMed PMID: 8723151. |
[28] | Huntington Study G. A randomized, placebo-controlled trial of coenzyme Q10 and remacemide in Huntington’s disease. Neurology. (2001) ;57: (3):397–404. PubMed PMID: 11502903. |
[29] | Hersch SM , Gevorkian S , Marder K , Moskowitz C , Feigin A , Cox M , et al. Creatine in Huntington disease is safe, tolerable, bioavailable in brain and reduces serum 8OH2’dG. Neurology. (2006) ;66: (2):250–2. PubMed PMID: 16434666. |
[30] | Huntington Study Group T-HDI. Randomized controlled trial of ethyl-eicosapentaenoic acid in Huntington disease: The TREND-HD study. Arch Neurol. (2008) ;65: (12):1582–9. PubMed PMID: 19064745. |
[31] | Ranen NG , Peyser CE , Coyle JT , Bylsma FW , Sherr M , Day L , et al. A controlled trial of idebenone in Huntington’s disease. Mov Disord. (1996) ;11: (5):549–54. PubMed PMID: 8866496. |
[32] | Safety and tolerability of the free-radical scavenger OPC-14117 in Huntington’s disease. The Huntington Study Group. Neurology. (1998) ;50: (5):1366–73. PubMed PMID: 9595988. |
[33] | Luthi-Carter R , Strand A , Peters NL , Solano SM , Hollingsworth ZR , Menon AS , et al. Decreased expression of striatal signaling genes in a mouse model of Huntington’s disease. Hum Mol Genet. (2000) ;9: (9):1259–71. PubMed PMID: 10814708. |
[34] | Achour M , Le Gras S , Keime C , Parmentier F , Lejeune FX , Boutillier AL , et al. Neuronal identity genes regulated by super-enhancers are preferentially down-regulated in the striatum of Huntington’s disease mice. Hum Mol Genet. (2015) ;24: (12):3481–96. PubMed PMID: 25784504. |
[35] | Iannicola C , Moreno S , Oliverio S , Nardacci R , Ciofi-Luzzatto A , Piacentini M . Early alterations in gene expression and cell morphology in a mouse model of Huntington’s disease. J Neurochem. (2000) ;75: (2):830–9. PubMed PMID: 10899961. |
[36] | Pisoschi AM , Pop A . The role of antioxidants in the chemistry of oxidative stress: A review. Eur J Med Chem. (2015) ;97: :55–74. PubMed PMID: 25942353. |
[37] | Weidinger A , Kozlov AV . Biological Activities of Reactive Oxygen and Nitrogen Species: Oxidative Stress versus Signal Transduction. Biomolecules. (2015) ;5: (2):472–84. PubMed PMID: 25884116. PubMed Central PMCID: 4496681. |
[38] | Kim GH , Kim JE , Rhie SJ , Yoon S . The role of oxidative stress in neurodegenerative diseases. Exp Neurobiol. (2015) ;24: (4):325–40. PubMed PMID: 26713080. PubMed Central PMCID: 4688332. |
[39] | Higdon A , Diers AR , Oh JY , Landar A , Darley-Usmar VM . Cell signalling by reactive lipid species: New concepts and molecular mechanisms. Biochem J. (2012) ;442: (3):453–64. PubMed PMID: 22364280. PubMed Central PMCID: 3286857. |
[40] | Hauck AK , Bernlohr DA . Oxidative stress and lipotoxicity. J Lipid Res. (2016) . PubMed PMID: 27009116. |
[41] | Birben E , Sahiner UM , Sackesen C , Erzurum S , Kalayci O . Oxidative stress and antioxidant defense. World Allergy Organ J. (2012) ;5: (1):9–19. PubMed PMID: 23268465. PubMed Central PMCID: 3488923. |
[42] | Rhee SG , Bae YS , Lee SR , Kwon J . Hydrogen peroxide: A key messenger that modulates protein phosphorylation through cysteine oxidation. Sci STKE. (2000) ;2000: (53):pe 1–. PubMed PMID: 11752613. |
[43] | Rhee SG . Cell signaling. H2O2, a necessary evil for cell signaling. Science. (2006) ;312: (5782):1882–3. PubMed PMID: 16809515. |
[44] | Finkel T . From sulfenylation to sulfhydration: What a thiolate needs to tolerate. Sci Signal. (2012) ;5: (215):pe 10. PubMed PMID: 22416275. |
[45] | Janssen-Heininger YM , Mossman BT , Heintz NH , Forman HJ , Kalyanaraman B , Finkel T , et al. Redox-based regulation of signal transduction: Principles, pitfalls, and promises. Free Radic Biol Med. (2008) ;45: (1):1–17. PubMed PMID: 18423411. PubMed Central PMCID: 2453533. |
[46] | Lee SR , Kwon KS , Kim SR , Rhee SG . Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J Biol Chem. (1998) ;273: (25):15366–72. PubMed PMID: 9624118. |
[47] | Kwon J , Lee SR , Yang KS , Ahn Y , Kim YJ , Stadtman ER , et al. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc Natl Acad Sci U S A. (2004) ;101: (47):16419–24. PubMed PMID: 15534200. PubMed Central PMCID: 534546. |
[48] | Matsushima S , Kuroda J , Ago T , Zhai P , Park JY , Xie LH , et al. Increased oxidative stress in the nucleus caused by Nox4 mediates oxidation of HDAC4 and cardiac hypertrophy. Circ Res. (2013) ;112: (4):651–63. PubMed PMID: 23271793. PubMed Central PMCID: 3574183. |
[49] | Weyemi U , Dupuy C . The emerging role of ROS-generating NADPH oxidase NOX4 in DNA-damage responses. Mutat Res. (2012) ;751: (2):77–81. PubMed PMID: 22580379. |
[50] | Seth D , Rudolph J . Redox regulation of MAP kinase phosphatase 3. Biochemistry. (2006) ;45: (28):8476–87. PubMed PMID: 16834321. |
[51] | Levinthal DJ , Defranco DB . Reversible oxidation of ERK-directed protein phosphatases drives oxidative toxicity in neurons. J Biol Chem. (2005) ;280: (7):5875–83. PubMed PMID: 15579467. |
[52] | Mannick JB , Schonhoff C , Papeta N , Ghafourifar P , Szibor M , Fang K , et al. S-Nitrosylation of mitochondrial caspases. J Cell Biol. (2001) ;154: (6):1111–6. PubMed PMID: 11551979. PubMed Central PMCID: 2150810. |
[53] | Hoppe G , Chai YC , Crabb JW , Sears J . Protein s-glutathionylation in retinal pigment epithelium converts heat shock protein 70 to an active chaperone. Exp Eye Res. (2004) ;78: (6):1085–92. PubMed PMID: 15109915. |
[54] | Nardai G , Sass B , Eber J , Orosz G , Csermely P . Reactive cysteines of the 90-kDa heat shock protein, Hsp90. Arch Biochem Biophys. (2000) ;384: (1):59–67. PubMed PMID: 11147836. |
[55] | Tsai B , Rodighiero C , Lencer WI , Rapoport TA . Protein disulfide isomerase acts as a redox-dependent chaperone to unfold cholera toxin. Cell. (2001) ;104: (6):937–48. PubMed PMID: 11290330. |
[56] | West AP , Brodsky IE , Rahner C , Woo DK , Erdjument-Bromage H , Tempst P , et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature. (2011) ;472: (7344):476–80. PubMed PMID: 21525932. PubMed Central PMCID: 3460538. |
[57] | Sena LA , Li S , Jairaman A , Prakriya M , Ezponda T , Hildeman DA , et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity. (2013) ;38: (2):225–36. PubMed PMID: 23415911. PubMed Central PMCID: 3582741. |
[58] | Wheeler ML , Defranco AL . Prolonged production of reactive oxygen species in response to B cell receptor stimulation promotes B cell activation and proliferation. J Immunol. (2012) ;189: (9):4405–16. PubMed PMID: 23024271. PubMed Central PMCID: 3515638. |
[59] | Kigerl KA , Gensel JC , Ankeny DP , Alexander JK , Donnelly DJ , Popovich PG . Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. (2009) ;29: (43):13435–44. PubMed PMID: 19864556. PubMed Central PMCID: 2788152. |
[60] | Mills CD . M1 and M2 macrophages: Oracles of health and disease. Crit Rev Immunol. (2012) ;32: (6):463–88. PubMed PMID: 23428224. |
[61] | Haskew-Layton RE , Payappilly JB , Smirnova NA , Ma TC , Chan KK , Murphy TH , et al. Controlled enzymatic production of astrocytic hydrogen peroxide protects neurons from oxidative stress via an Nrf2-independent pathway. Proc Natl Acad Sci U S A. (2010) ;107: (40):17385–90. PubMed PMID: 20855618. PubMed Central PMCID: 2951414. |
[62] | Brazier MW , Wedd AG , Collins SJ . Antioxidant and metal chelation-based therapies in the treatment of prion disease. Antioxidants (Basel). (2014) ;3: (2):288–308. PubMed PMID: 26784872. PubMed Central PMCID: 4665489. |
[63] | Muller M , Leavitt BR . Iron dysregulation in Huntington’s disease. J Neurochem. (2014) ;130: (3):328–50. PubMed PMID: 24717009. |
[64] | Bartzokis G , Lu PH , Geschwind DH , Tingus K , Huang D , Mendez MF , et al. Apolipoprotein E affects both myelin breakdown and cognition: Implications for age-related trajectories of decline into dementia. Biol Psychiatry. (2007) ;62: (12):1380–7. PubMed PMID: 17659264. |
[65] | Rosas HD , Chen YI , Doros G , Salat DH , Chen NK , Kwong KK , et al. Alterations in brain transition metals in Huntington disease: An evolving and intricate story. Arch Neurol. (2012) ;69: (7):887–93. PubMed PMID: 22393169. PubMed Central PMCID: 3652228. |
[66] | Fox JH , Kama JA , Lieberman G , Chopra R , Dorsey K , Chopra V , et al. Mechanisms of copper ion mediated Huntington’s disease progression. PLoS One. (2007) ;2: (3):e334. PubMed PMID: 17396163. PubMed Central PMCID: 1828629. |
[67] | Chen J , Marks E , Lai B , Zhang Z , Duce JA , Lam LQ , et al. Iron accumulates in Huntington’s disease neurons: Protection by deferoxamine. PLoS One. (2013) ;8: (10):e77023. PubMed PMID: 24146952. PubMed Central PMCID: 3795666. |
[68] | Hands SL , Mason R , Sajjad MU , Giorgini F , Wyttenbach A . Metallothioneins and copper metabolism are candidate therapeutic targets in Huntington’s disease. Biochem Soc Trans. (2010) ;38: (2):552–8. PubMed PMID: 20298220. |
[69] | Karuppagounder SS , Alim I , Khim SJ , Bourassa MW , Sleiman SF , John R , et al. Therapeutic targeting of oxygen-sensing prolyl hydroxylases abrogates ATF4-dependent neuronal death and improves outcomes after brain hemorrhage in several rodent models. Sci Transl Med. (2016) ;8: (328):328ra29. PubMed PMID: 26936506. |
[70] | Fox JH , Connor T , Stiles M , Kama J , Lu Z , Dorsey K , et al. Cysteine oxidation within N-terminal mutant huntingtin promotes oligomerization and delays clearance of soluble protein. J Biol Chem. (2011) ;286: (20):18320–30. PubMed PMID: 21454633. PubMed Central PMCID: 3093904. |
[71] | Xiao G , Fan Q , Wang X , Zhou B . Huntington disease arises from a combinatory toxicity of polyglutamine and copper binding. Proc Natl Acad Sci U S A. (2013) ;110: (37):14995–5000. PubMed PMID: 23980182. PubMed Central PMCID: 3773747. |
[72] | Mitomi Y , Nomura T , Kurosawa M , Nukina N , Furukawa Y . Post-aggregation oxidation of mutant huntingtin controls the interactions between aggregates. J Biol Chem. (2012) ;287: (41):34764–75. PubMed PMID: 22891249. PubMed Central PMCID: 3464579. |
[73] | Barnham KJ , Bush AI . Biological metals and metal-targeting compounds in major neurodegenerative diseases. Chem Soc Rev. (2014) ;43: (19):6727–49. PubMed PMID: 25099276. |
[74] | Nguyen T , Hamby A , Massa SM . Clioquinol down-regulates mutant huntingtin expression in vitro and mitigates pathology in a Huntington’s disease mouse model. Proc Natl Acad Sci U S A. (2005) ;102: (33):11840–5. PubMed PMID: 16087879. PubMed Central PMCID: 1187967. |
[75] | Cherny RA , Ayton S , Finkelstein DI , Bush AI , McColl G , Massa SM . PBT2 reduces toxicity in a C. elegans model of polyQ aggregation and extends lifespan, reduces striatal atrophy and improves motor performance in the R6/2 mouse model of Huntington’s disease. J Huntingtons Dis. (2012) ;1: (2):211–9. PubMed PMID: 25063332. |
[76] | Huntington Study Group Reach HDI. Safety, tolerability, and efficacy of PBT2 in Huntington’s disease: A phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. (2015) ;14: (1):39–47. PubMed PMID: 25467848. |
[77] | Siddiq A , Ayoub IA , Chavez JC , Aminova L , Shah S , LaManna JC , et al. Hypoxia-inducible factor prolyl 4-hydroxylase inhibition. A target for neuroprotection in the central nervous system. J Biol Chem. (2005) ;280: (50):41732–43. PubMed PMID: 16227210. PubMed Central PMCID: 2586128. |
[78] | Frijhoff J , Winyard PG , Zarkovic N , Davies SS , Stocker R , Cheng D , et al. Clinical relevance of biomarkers of oxidative stress. Antioxid Redox Signal. (2015) ;23: (14):1144–70. PubMed PMID: 26415143. PubMed Central PMCID: 4657513. |
[79] | Long JD , Matson WR , Juhl AR , Leavitt BR , Paulsen JS , PREDICT-HD Investigators and Coordinators of the Huntington Study Group. 8OHdG as a marker for Huntington disease progression. Neurobiol Dis. (2012) ;46: (3):625–34. PubMed PMID: 22414782. PubMed Central PMCID: 3784019. |
[80] | Brocardo PS , McGinnis E , Christie BR , Gil-Mohapel J . Time-course analysis of protein and lipid oxidation in the brains of Yac128 Huntington’s disease transgenic mice. Rejuvenation Res. (2016) ;19: (2):140–8. PubMed PMID: 26371883. |
[81] | Tunez I , Sanchez-Lopez F , Aguera E , Fernandez-Bolanos R , Sanchez FM , Tasset-Cuevas I . Important role of oxidative stress biomarkers in Huntington’s disease. J Med Chem. (2011) ;54: (15):5602–6. PubMed PMID: 21678912. |
[82] | Thanan R , Oikawa S , Hiraku Y , Ohnishi S , Ma N , Pinlaor S , et al. Oxidative stress and its significant roles in neurodegenerative diseases and cancer. Int J Mol Sci. (2015) ;16: (1):193–217. PubMed PMID: 25547488. PubMed Central PMCID: 4307243. |
[83] | Dalle-Donne I , Rossi R , Colombo R , Giustarini D , Milzani A . Biomarkers of oxidative damage in human disease. Clin Chem. (2006) ;52: (4):601–23. PubMed PMID: 16484333. |
[84] | Murray CI , Van Eyk JE . Chasing cysteine oxidative modifications: Proteomic tools for characterizing cysteine redox status. Circ Cardiovasc Genet. (2012) ;5: (5):591. PubMed PMID: 23074338. PubMed Central PMCID: 3500588. |
[85] | Stoy N , Mackay GM , Forrest CM , Christofides J , Egerton M , Stone TW , et al. Tryptophan metabolism and oxidative stress in patients with Huntington’s disease. J Neurochem. (2005) ;93: (3):611–23. PubMed PMID: 15836620. |
[86] | Browne SE , Beal MF . Oxidative damage in Huntington’s disease pathogenesis. Antioxid Redox Signal. (2006) ;8: (11-12):2061–73. PubMed PMID: 17034350. |
[87] | Andrade-Lima LC , Veloso A , Ljungman M . Transcription blockage leads to new beginnings. Biomolecules. (2015) ;5: (3):1600–17. PubMed PMID: 26197343. PubMed Central PMCID: 4598766. |
[88] | Li J , O W , Li W , Jiang ZG , Ghanbari HA . Oxidative stress and neurodegenerative disorders. Int J Mol Sci. (2013) ;14: (12):24438–75. PubMed PMID: 24351827. PubMed Central PMCID: 3876121. |
[89] | Browne SE , Bowling AC , MacGarvey U , Baik MJ , Berger SC , Muqit MM , et al. Oxidative damage and metabolic dysfunction in Huntington’s disease: Selective vulnerability of the basal ganglia. Ann Neurol. (1997) ;41: (5):646–53. PubMed PMID: 9153527. |
[90] | Siddiqui A , Rivera-Sanchez S , Castro Mdel R , Acevedo-Torres K , Rane A , Torres-Ramos CA , et al. Mitochondrial DNA damage is associated with reduced mitochondrial bioenergetics in Huntington’s disease. Free Radic Biol Med. (2012) ;53: (7):1478–88. PubMed PMID: 22709585. PubMed Central PMCID: 3846402. |
[91] | Polyzos A , Holt A , Brown C , Cosme C , Wipf P , Gomez-Marin A , et al Mitochondrial targeting of XJB-5-131 attenuates or improves pathophysiology in HdhQ150 animals with well-developed disease phenotypes. Hum Mol Genet. (2016) . PubMed PMID: 26908614. |
[92] | Chen CM , Wu YR , Cheng ML , Liu JL , Lee YM , Lee PW , et al. Increased oxidative damage and mitochondrial abnormalities in the peripheral blood of Huntington’s disease patients. Biochem Biophys Res Commun. (2007) ;359: (2):335–40. PubMed PMID: 17543886. |
[93] | Valencia A , Sapp E , Reeves PB , Alexander J , Masso N , Li X , et al. Reagents that block neuronal death from Huntington’s disease also curb oxidative stress. Neuroreport. (2012) ;23: (1):10–5. PubMed PMID: 22045254. |
[94] | Polyzos A , Holt A , Brown C , Cosme C , Wipf P , Gomez-Marin A , et al. Mitochondrial targeting of XJB-5-131 attenuates or improves pathophysiology in HdhQ150 animals with well-developed disease phenotypes. Hum Mol Genet. (2016) ;25: (9):1792–802. PubMed PMID: 26908614. |
[95] | Entezam A , Lokanga AR , Le W , Hoffman G , Usdin K . Potassium bromate, a potent DNA oxidizing agent, exacerbates germline repeat expansion in a fragile X premutation mouse model. Hum Mutat. (2010) ;31: (5):611–6. PubMed PMID: 20213777. PubMed Central PMCID: 2951473. |
[96] | Kovtun IV , Liu Y , Bjoras M , Klungland A , Wilson SH , McMurray CT . OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature. (2007) ;447: (7143):447–52. PubMed PMID: 17450122. PubMed Central PMCID: 2681094. |
[97] | Jarem DA , Wilson NR , Delaney S . Structure-dependent DNA damage and repair in a trinucleotide repeat sequence. Biochemistry. (2009) ;48: (28):6655–63. PubMed PMID: 19527055. |
[98] | Pearson CE , Nichol Edamura K , Cleary JD . Repeat instability: Mechanisms of dynamic mutations. Nat Rev Genet. (2005) ;6: (10):729–42. PubMed PMID: 16205713. |
[99] | Mollersen L , Rowe AD , Illuzzi JL , Hildrestrand GA , Gerhold KJ , Tveteras L , et al. Neil1 is a genetic modifier of somatic and germline CAG trinucleotide repeat instability in R6/1 mice. Hum Mol Genet. (2012) ;21: (22):4939–47. PubMed PMID: 22914735. PubMed Central PMCID: 3607484. |
[100] | Manley K , Shirley TL , Flaherty L , Messer A . Msh2 deficiency prevents in vivo somatic instability of the CAG repeat in Huntington disease transgenic mice. Nat Genet. (1999) ;23: (4):471–3. PubMed PMID: 10581038. |
[101] | Goula AV , Berquist BR , Wilson DM , Wheeler VC , Trottier Y , Merienne K . Stoichiometry of base excision repair proteins correlates with increased somatic CAG instability in striatum over cerebellum in Huntington’s disease transgenic mice. PLoS Genet. (2009) ;5: (12):e1000749. PubMed PMID: 19997493. PubMed Central PMCID: 2778875. |
[102] | Cilli P , Ventura I , Minoprio A , Meccia E , Martire A , Wilson SH , et al. Oxidized dNTPs and the OGG1 and MUTYH DNA glycosylases combine to induce CAG/CTG repeat instability. Nucleic Acids Res. (2016) . PubMed PMID: 26980281. |
[103] | Budworth H , Harris FR , Williams P , Lee do Y , Holt A , Pahnke J , et al. Suppression of somatic expansion delays the onset of pathophysiology in a mouse model of Huntington’s disease. PLoS Genet. (2015) ;11: (8):e1005267. PubMed PMID: 26247199. PubMed Central PMCID: 4527696. |
[104] | Budworth H , McMurray CT . Problems and solutions for the analysis of somatic CAG repeat expansion and their relationship to Huntington’s disease toxicity. Rare Dis. (2016) ;4: (1):e1131885. PubMed PMID: 27141411. PubMed Central PMCID: 4838321. |
[105] | Enokido Y , Tamura T , Ito H , Arumughan A , Komuro A , Shiwaku H , et al. Mutant huntingtin impairs Ku70-mediated DNA repair. J Cell Biol. (2010) ;189: (3):425–43. PubMed PMID: 20439996. PubMed Central PMCID: 2867301. |
[106] | Wyttenbach A , Sauvageot O , Carmichael J , Diaz-Latoud C , Arrigo AP , Rubinsztein DC . Heat shock protein 27 prevents cellular polyglutamine toxicity and suppresses the increase of reactive oxygen species caused by huntingtin. Hum Mol Genet. (2002) ;11: (9):1137–51. PubMed PMID: 11978772. |
[107] | Hands S , Sajjad MU , Newton MJ , Wyttenbach A . In vitro and in vivo aggregation of a fragment of huntingtin protein directly causes free radical production. J Biol Chem. (2011) ;286: (52):44512–20. PubMed PMID: 21984825. PubMed Central PMCID: 3247975. |
[108] | Dragunow M , Faull RL , Lawlor P , Beilharz EJ , Singleton K , Walker EB , et al. In situ evidence for DNA fragmentation in Huntington’s disease striatum and Alzheimer’s disease temporal lobes. Neuroreport. (1995) ;6: (7):1053–7. PubMed PMID: 7632894. |
[109] | Braak H , Braak E . Allocortical involvement in Huntington’s disease. Neuropathol Appl Neurobiol. (1992) ;18: (6):539–47. PubMed PMID: 1488086. |
[110] | Sorolla MA , Rodriguez-Colman MJ , Tamarit J , Ortega Z , Lucas JJ , Ferrer I , et al. Protein oxidation in Huntington disease affects energy production and vitamin B6 metabolism. Free Radic Biol Med. (2010) ;49: (4):612–21. PubMed PMID: 20639122. |
[111] | Brennan WA Jr , Bird ED , Aprille JR . Regional mitochondrial respiratory activity in Huntington’s disease brain. J Neurochem. (1985) ;44: (6):1948–50. PubMed PMID: 2985766. |
[112] | Stahl WL , Swanson PD . Biochemical abnormalities in Huntington’s chorea brains. Neurology. (1974) ;24: (9):813–9. PubMed PMID: 4277376. |
[113] | Gu M , Gash MT , Mann VM , Javoy-Agid F , Cooper JM , Schapira AH . Mitochondrial defect in Huntington’s disease caudate nucleus. Ann Neurol. (1996) ;39: (3):385–9. PubMed PMID: 8602759. |
[114] | Sorolla MA , Rodriguez-Colman MJ , Vall-llaura N , Tamarit J , Ros J , Cabiscol E . Protein oxidation in Huntington disease. Biofactors. (2012) ;38: (3):173–85. PubMed PMID: 22473822. |
[115] | Klepac N , Relja M , Klepac R , Hecimovic S , Babic T , Trkulja V . Oxidative stress parameters in plasma of Huntington’s disease patients, asymptomatic Huntington’s disease gene carriers and healthy subjects: A cross-sectional study. J Neurol. (2007) ;254: (12):1676–83. PubMed PMID: 17990062. |
[116] | Christofides J , Bridel M , Egerton M , Mackay GM , Forrest CM , Stoy N , et al. Blood 5-hydroxytryptamine, 5-hydroxyindoleacetic acid and melatonin levels in patients with either Huntington’s disease or chronic brain injury. J Neurochem. (2006) ;97: (4):1078–88. PubMed PMID: 16573644. |
[117] | Chang KH , Chen YC , Wu YR , Lee WF , Chen CM . Downregulation of genes involved in metabolism and oxidative stress in the peripheral leukocytes of Huntington’s disease patients. PLoS One. (2012) ;7: (9):e46492. PubMed PMID: 23029535. PubMed Central PMCID: 3459918. |
[118] | Pena-Sanchez M , Riveron-Forment G , Zaldivar-Vaillant T , Soto-Lavastida A , Borrero-Sanchez J , Lara-Fernandez G , et al. Association of status redox with demographic, clinical and imaging parameters in patients with Huntington’s disease. Clin Biochem. (2015) ;48: (18):1258–63. PubMed PMID: 26210848. |
[119] | Ciancarelli I , De Amicis D , Di Massimo C , Di Scanno C , Pistarini C , D’Orazio N , et al. Peripheral biomarkers of oxidative stress and their limited potential in evaluation of clinical features of Huntington’s patients. Biomarkers. (2014) ;19: (6):452–6. PubMed PMID: 24980251. |
[120] | Arenas J , Campos Y , Ribacoba R , Martin MA , Rubio JC , Ablanedo P , et al. Complex I defect in muscle from patients with Huntington’s disease. Ann Neurol. (1998) ;43: (3):397–400. PubMed PMID: 9506560. |
[121] | Parker WD Jr , Boyson SJ, Luder AS, Parks JK. Evidence for a defect in NADH: Ubiquinone oxidoreductase (complex I) in Huntington’s disease. Neurology. (1990) ;40: (8):1231–4. PubMed PMID: 2143271. |
[122] | Powers WJ , Haas RH , Le T , Videen TO , Hershey T , McGee-Minnich L , et al. Normal platelet mitochondrial complex I activity in Huntington’s disease. Neurobiol Dis. (2007) ;27: (1):99–101. PubMed PMID: 17543533. PubMed Central PMCID: 2140002. |
[123] | Turner C , Cooper JM , Schapira AH . Clinical correlates of mitochondrial function in Huntington’s disease muscle. Mov Disord. (2007) ;22: (12):1715–21. PubMed PMID: 17557337. |
[124] | Beal MF , Brouillet E , Jenkins B , Henshaw R , Rosen B , Hyman BT . Age-dependent striatal excitotoxic lesions produced by the endogenous mitochondrial inhibitor malonate. J Neurochem. (1993) ;61: (3):1147–50. PubMed PMID: 7689641. |
[125] | Brouillet E , Jenkins BG , Hyman BT , Ferrante RJ , Kowall NW , Srivastava R , et al. Age-dependent vulnerability of the striatum to the mitochondrial toxin 3-nitropropionic acid. J Neurochem. (1993) ;60: (1):356–9. PubMed PMID: 8417157. |
[126] | Brouillet E , Guyot MC , Mittoux V , Altairac S , Conde F , Palfi S , et al. Partial inhibition of brain succinate dehydrogenase by 3-nitropropionic acid is sufficient to initiate striatal degeneration in rat. J Neurochem. (1998) ;70: (2):794–805. PubMed PMID: 9453576. |
[127] | Beal MF , Brouillet E , Jenkins BG , Ferrante RJ , Kowall NW , Miller JM , et al. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci. (1993) ;13: (10):4181–92. PubMed PMID: 7692009. |
[128] | Borlongan CV , Koutouzis TK , Randall TS , Freeman TB , Cahill DW , Sanberg PR . Systemic 3-nitropropionic acid: Behavioral deficits and striatal damage in adult rats. Brain Res Bull. (1995) ;36: (6):549–56. PubMed PMID: 7538873. |
[129] | Guyot MC , Hantraye P , Dolan R , Palfi S , Maziere M , Brouillet E . Quantifiable bradykinesia, gait abnormalities and Huntington’s disease-like striatal lesions in rats chronically treated with 3-nitropropionic acid. Neuroscience. (1997) ;79: (1):45–56. PubMed PMID: 9178864. |
[130] | Acevedo-Torres K , Berrios L , Rosario N , Dufault V , Skatchkov S , Eaton MJ , et al. Mitochondrial DNA damage is a hallmark of chemically induced and the R6/2 transgenic model of Huntington’s disease. DNA Repair (Amst). (2009) ;8: (1):126–36. PubMed PMID: 18935984. PubMed Central PMCID: 3268004. |
[131] | Lu XH , Mattis VB , Wang N , Al-Ramahi I , van den Berg N , Fratantoni SA , et al. Targeting ATM ameliorates mutant Huntingtin toxicity in cell and animal models of Huntington’s disease. Sci Transl Med. (2014) ;6: (268):268ra178. PubMed PMID: 25540325. |
[132] | Shiloh Y , Ziv Y . The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol. (2013) ;14: (4):197–210. PubMed PMID: 23847781. |
[133] | Ditch S , Paull TT . The ATM protein kinase and cellular redox signaling: Beyond the DNA damage response. Trends Biochem Sci. (2012) ;37: (1):15–22. PubMed PMID: 22079189. PubMed Central PMCID: 3259275. |
[134] | Bogdanov MB , Andreassen OA , Dedeoglu A , Ferrante RJ , Beal MF . Increased oxidative damage to DNA in a transgenic mouse model of Huntington’s disease. J Neurochem. (2001) ;79: (6):1246–9. PubMed PMID: 11752065. |
[135] | Gray M , Shirasaki DI , Cepeda C , Andre VM , Wilburn B , Lu XH , et al. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J Neurosci. (2008) ;28: (24):6182–95. PubMed PMID: 18550760. PubMed Central PMCID: 2630800. |
[136] | Stack C , Ho D , Wille E , Calingasan NY , Williams C , Liby K , et al. Triterpenoids CDDO-ethyl amide and CDDO-trifluoroethyl amide improve the behavioral phenotype and brain pathology in a transgenic mouse model of Huntington’s disease. Free Radic Biol Med. (2010) ;49: (2):147–58. PubMed PMID: 20338236. PubMed Central PMCID: 2916021. |
[137] | Hong C , Seo H , Kwak M , Jeon J , Jang J , Jeong EM , et al. Increased TRPC5 glutathionylation contributes to striatal neuron loss in Huntington’s disease. Brain. (2015) ;138: (Pt 10):3030–47. PubMed PMID: 26133660. PubMed Central PMCID: 4643628. |
[138] | Illuzzi J , Yerkes S , Parekh-Olmedo H , Kmiec EB . DNA breakage and induction of DNA damage response proteins precede the appearance of visible mutant huntingtin aggregates. J Neurosci Res. (2009) ;87: (3):733–47. PubMed PMID: 18831068. |
[139] | Tsien RY , Pozzan T , Rink TJ . Calcium homeostasis in intact lymphocytes: Cytoplasmic free calcium monitored with a new, intracellularly trapped fluorescent indicator. J Cell Biol. (1982) ;94: (2):325–34. PubMed PMID: 6980885. PubMed Central PMCID: 2112871. |
[140] | Grynkiewicz G , Poenie M , Tsien RY . A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. (1985) ;260: (6):3440–50. PubMed PMID: 3838314. |
[141] | Winterbourn CC . The challenges of using fluorescent probes to detect and quantify specific reactive oxygen species in living cells. Biochim Biophys Acta. (2014) ;1840: (2):730–8. PubMed PMID: 23665586. |
[142] | Kalyanaraman B , Darley-Usmar V , Davies KJ , Dennery PA , Forman HJ , Grisham MB , et al. Measuring reactive oxygen and nitrogen species with fluorescent probes: Challenges and limitations. Free Radic Biol Med. (2012) ;52: (1):1–6. PubMed PMID: 22027063. PubMed Central PMCID: 3911769. |
[143] | Belousov VV , Fradkov AF , Lukyanov KA , Staroverov DB , Shakhbazov KS , Terskikh AV , et al. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nat Methods. (2006) ;3: (4):281–6. PubMed PMID: 16554833. |
[144] | Perez-Severiano F , Rios C , Segovia J . Striatal oxidative damage parallels the expression of a neurological phenotype in mice transgenic for the mutation of Huntington’s disease. Brain Res. (2000) ;862: (1-2):234–7. PubMed PMID: 10799690. |
[145] | Perez-Severiano F , Escalante B , Vergara P , Rios C , Segovia J . Age-dependent changes in nitric oxide synthase activity and protein expression in striata of mice transgenic for the Huntington’s disease mutation. Brain Res. (2002) ;951: (1):36–42. PubMed PMID: 12231454. |
[146] | Brocardo PS , McGinnis E , Christie BR , Gil-Mohapel J . Time-course analysis of protein and lipid oxidation in the brains of Yac128 Huntington’s disease transgenic mice. Rejuvenation Res 2016–. PubMed PMID: 26371883. |
[147] | Khoshnan A , Patterson PH . The role of IkappaB kinase complex in the neurobiology of Huntington’s disease. Neurobiol Dis. (2011) ;43: (2):305–11. PubMed PMID: 21554955. PubMed Central PMCID: 3124142. |
[148] | Bae BI , Xu H , Igarashi S , Fujimuro M , Agrawal N , Taya Y , et al. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington’s disease. Neuron. (2005) ;47: (1):29–41. PubMed PMID: 15996546. |
[149] | Shimohata T , Nakajima T , Yamada M , Uchida C , Onodera O , Naruse S , et al. Expanded polyglutamine stretches interact with TAFII130, interfering with CREB-dependent transcription. Nat Genet. (2000) ;26: (1):29–36. PubMed PMID: 10973244. |
[150] | Ng CW , Yildirim F , Yap YS , Dalin S , Matthews BJ , Velez PJ , et al. Extensive changes in DNA methylation are associated with expression of mutant huntingtin. Proc Natl Acad Sci U S A. (2013) ;110: (6):2354–9. PubMed PMID: 23341638. PubMed Central PMCID: 3568325. |
[151] | Demple B , Harrison L . Repair of oxidative damage to DNA: Enzymology and biology. Annu Rev Biochem. (1994) ;63: :915–48. PubMed PMID: 7979257. |
[152] | Luo M , He H , Kelley MR , Georgiadis MM . Redox regulation of DNA repair: Implications for human health and cancer therapeutic development. Antioxid Redox Signal. (2010) ;12: (11):1247–69. PubMed PMID: 19764832. PubMed Central PMCID: 2864659. |
[153] | Motohashi H , Yamamoto M . Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol Med. (2004) ;10: (11):549–57. PubMed PMID: 15519281. |
[154] | Itoh K , Wakabayashi N , Katoh Y , Ishii T , Igarashi K , Engel JD , et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. (1999) ;13: (1):76–86. PubMed PMID: 9887101. PubMed Central PMCID: 316370. |
[155] | Dinkova-Kostova AT , Holtzclaw WD , Cole RN , Itoh K , Wakabayashi N , Katoh Y , et al. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci U S A. (2002) ;99: (18):11908–13. PubMed PMID: 12193649. PubMed Central PMCID: 129367. |
[156] | Kobayashi A , Kang MI , Okawa H , Ohtsuji M , Zenke Y , Chiba T , et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. (2004) ;24: (16):7130–9. PubMed PMID: 15282312. PubMed Central PMCID: 479737. |
[157] | Cullinan SB , Gordan JD , Jin J , Harper JW , Diehl JA . The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: Oxidative stress sensing by a Cul3-Keap1 ligase. Mol Cell Biol. (2004) ;24: (19):8477–86. PubMed PMID: 15367669. PubMed Central PMCID: 516753. |
[158] | Smirnova NA , Haskew-Layton RE , Basso M , Hushpulian DM , Payappilly JB , Speer RE , et al. Development of Neh2-luciferase reporter and its application for high throughput screening and real-time monitoring of Nrf2 activators. Chem Biol. (2011) ;18: (6):752–65. PubMed PMID: 21700211. PubMed Central PMCID: 3251032. |
[159] | Kaspar JW , Niture SK , Jaiswal AK . NrfINrf2 (Keap1) signaling in oxidative stress. Free Radic Biol Med. (2009) ;47: (9):1304–9. PubMed PMID: 19666107. PubMed Central PMCID: 2763938. |
[160] | Calkins MJ , Jakel RJ , Johnson DA , Chan K , Kan YW , Johnson JA . Protection from mitochondrial complex II inhibition in vitro and in vivo by Nrf2-mediated transcription. Proc Natl Acad Sci U S A. (2005) ;102: (1):244–9. PubMed PMID: 15611470. PubMed Central PMCID: 538748. |
[161] | Shih AY , Imbeault S , Barakauskas V , Erb H , Jiang L , Li P , et al. Induction of the Nrf2-driven antioxidant response confers neuroprotection during mitochondrial stress in vivo. J Biol Chem. (2005) ;280: (24):22925–36. PubMed PMID: 15840590. |
[162] | Calkins MJ , Townsend JA , Johnson DA , Johnson JA . Cystamine protects from 3-nitropropionic acid lesioning via induction of nf-e2 related factor 2 mediated transcription. Exp Neurol. (2010) ;224: (1):307–17. PubMed PMID: 20406637. PubMed Central PMCID: 2885467. |
[163] | Calkins MJ , Vargas MR , Johnson DA , Johnson JA . Astrocyte-specific overexpression of Nrf2 protects striatal neurons from mitochondrial complex II inhibition. Toxicol Sci. (2010) ;115: (2):557–68. PubMed PMID: 20211941. PubMed Central PMCID: 2871759. |
[164] | Ellrichmann G , Petrasch-Parwez E , Lee DH , Reick C , Arning L , Saft C , et al. Efficacy of fumaric acid esters in the R6/2 and YAC128 models of Huntington’s disease. PLoS One. (2011) ;6: (1):e16172. PubMed PMID: 21297955. PubMed Central PMCID: 3031519. |
[165] | Brennan MS , Patel H , Allaire N , Thai A , Cullen P , Ryan S , et al. Pharmacodynamics of dimethyl fumarate are tissue specific and involve NRF2-dependent and -independent mechanisms. Antioxid Redox Signal. (2016) ;24: (18):1058–71. PubMed PMID: 26980071. |
[166] | D’Autreaux B , Toledano MB . ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. (2007) ;8: (10):813–24. PubMed PMID: 17848967. |
[167] | Rodgers JT , Lerin C , Haas W , Gygi SP , Spiegelman BM , Puigserver P . Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. (2005) ;434: (7029):113–8. PubMed PMID: 15744310. |
[168] | Nemoto S , Fergusson MM , Finkel T . SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1alpha. J Biol Chem. (2005) ;280: (16):16456–60. PubMed PMID: 15716268. |
[169] | Garriga-Canut M , Schoenike B , Qazi R , Bergendahl K , Daley TJ , Pfender RM , et al. 2-Deoxy-D-glucose reduces epilepsy progression by NRSF-CtBP-dependent metabolic regulation of chromatin structure. Nat Neurosci. (2006) ;9: (11):1382–7. PubMed PMID: 17041593. |
[170] | Fulco M , Schiltz RL , Iezzi S , King MT , Zhao P , Kashiwaya Y , et al. Sir2 regulates skeletal muscle differentiation as a potential sensor of the redox state. Mol Cell. (2003) ;12: (1):51–62. PubMed PMID: 12887892. |
[171] | Zhang Q , Piston DW , Goodman RH . Regulation of corepressor function by nuclear NADH. Science. (2002) ;295: (5561):1895–7. PubMed PMID: 11847309. |
[172] | Ryu H , Lee J , Olofsson BA , Mwidau A , Dedeoglu A , Escudero M , et al. Histone deacetylase inhibitors prevent oxidative neuronal death independent of expanded polyglutamine repeats via an Sp1-dependent pathway. Proc Natl Acad Sci U S A. (2003) ;100: (7):4281–6. PubMed PMID: 12640146. PubMed Central PMCID: 153084. |
[173] | Ryu H , Lee J , Zaman K , Kubilis J , Ferrante RJ , Ross BD , et al. Sp1 and Sp3 are oxidative stress-inducible, antideath transcription factors in cortical neurons. J Neurosci. (2003) ;23: (9):3597–606. PubMed PMID: 12736330. |
[174] | Paul BD , Sbodio JI , Xu R , Vandiver MS , Cha JY , Snowman AM , et al. Cystathionine gamma-lyase deficiency mediates neurodegeneration in Huntington’s disease. Nature. (2014) ;509: (7498):96–100. PubMed PMID: 24670645. PubMed Central PMCID: 4349202. |
[175] | Dunah AW , Jeong H , Griffin A , Kim YM , Standaert DG , Hersch SM , et al. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington’s disease. Science. (2002) ;296: (5576):2238–43. PubMed PMID: 11988536. |
[176] | Lin J , Handschin C , Spiegelman BM . Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. (2005) ;1: (6):361–70. PubMed PMID: 16054085. |
[177] | Starkov AA . The role of mitochondria in reactive oxygen species metabolism and signaling. Ann N Y Acad Sci. (2008) ;1147: :37–52. PubMed PMID: 19076429. PubMed Central PMCID: 2869479. |
[178] | Starkov AA , Andreyev AY , Zhang SF , Starkova NN , Korneeva M , Syromyatnikov M , et al. Scavenging of H2O2 by mouse brain mitochondria. J Bioenerg Biomembr. (2014) ;46: (6):471–7. PubMed PMID: 25248416. PubMed Central PMCID: 4634880. |
[179] | Song B , Scheuner D , Ron D , Pennathur S , Kaufman RJ . Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J Clin Invest. (2008) ;118: (10):3378–89. PubMed PMID: 18776938. PubMed Central PMCID: 2528909. |
[180] | Freeman TC , Wood IS , Sirinathsinghji DJ , Beechey RB , Dyer J , Shirazi-Beechey SP . The expression of the Na+/glucose cotransporter (SGLT1) gene in lamb small intestine during postnatal development. Biochim Biophys Acta. (1993) ;1146: (2):203–12. PubMed PMID: 8452856. |
[181] | Tell G , Zecca A , Pellizzari L , Spessotto P , Colombatti A , Kelley MR , et al. An environment to nucleus signaling system operates in B lymphocytes: Redox status modulates BSAP/Pax-5 activation through Ref-1 nuclear translocation. Nucleic Acids Res. (2000) ;28: (5):1099–105. PubMed PMID: 10666449. PubMed Central PMCID: 102597. |
Figures and Tables
Fig.1
A brief overview of involvement of reactive oxygen species in multiple physiological and pathological functions as signaling molecules. Recent findings have placed ROS as very critical signaling factors which are involved in regulating not only pathological functions but also in a host of functions necessary for normal cellular function. Therefore, a very careful attention is needed before considering antioxidants as therapeutic options for Huntington’s disease.
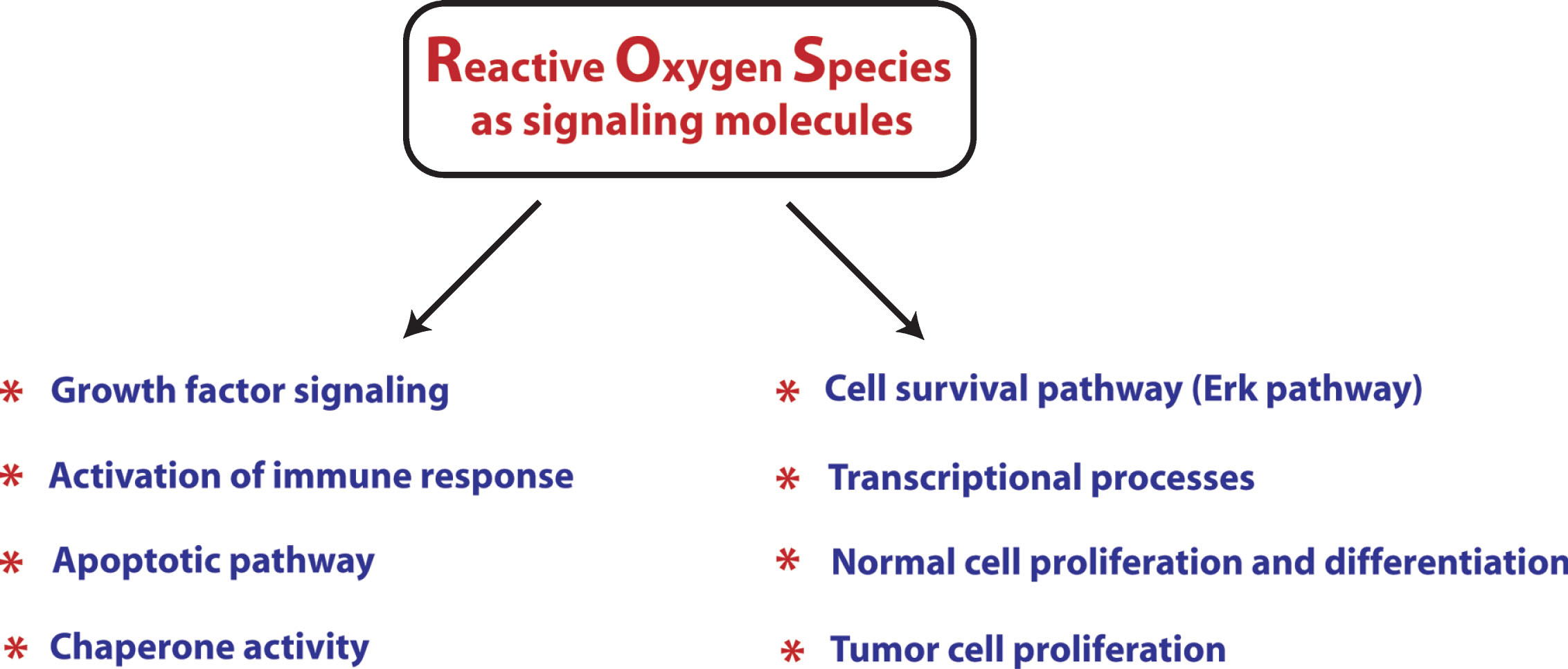
Fig.2
A coupled chain of oxidation-repair cycle leads to expansion and instability of huntingtin polyglutamine repeats. DNA oxidation induces an adaptive response in the form of recruitment of DNA repair enzymes. These enzymes are recruited in response to either hairpin loop structure or other secondary structures formed either because of oxidized bases and/or base mismatches leading to strand slippage and instability. Moreover, a complex cyclic interplay between oxidized bases and DNA repair enzymes leads to further expansion of trinucleotide repeats and its instability.
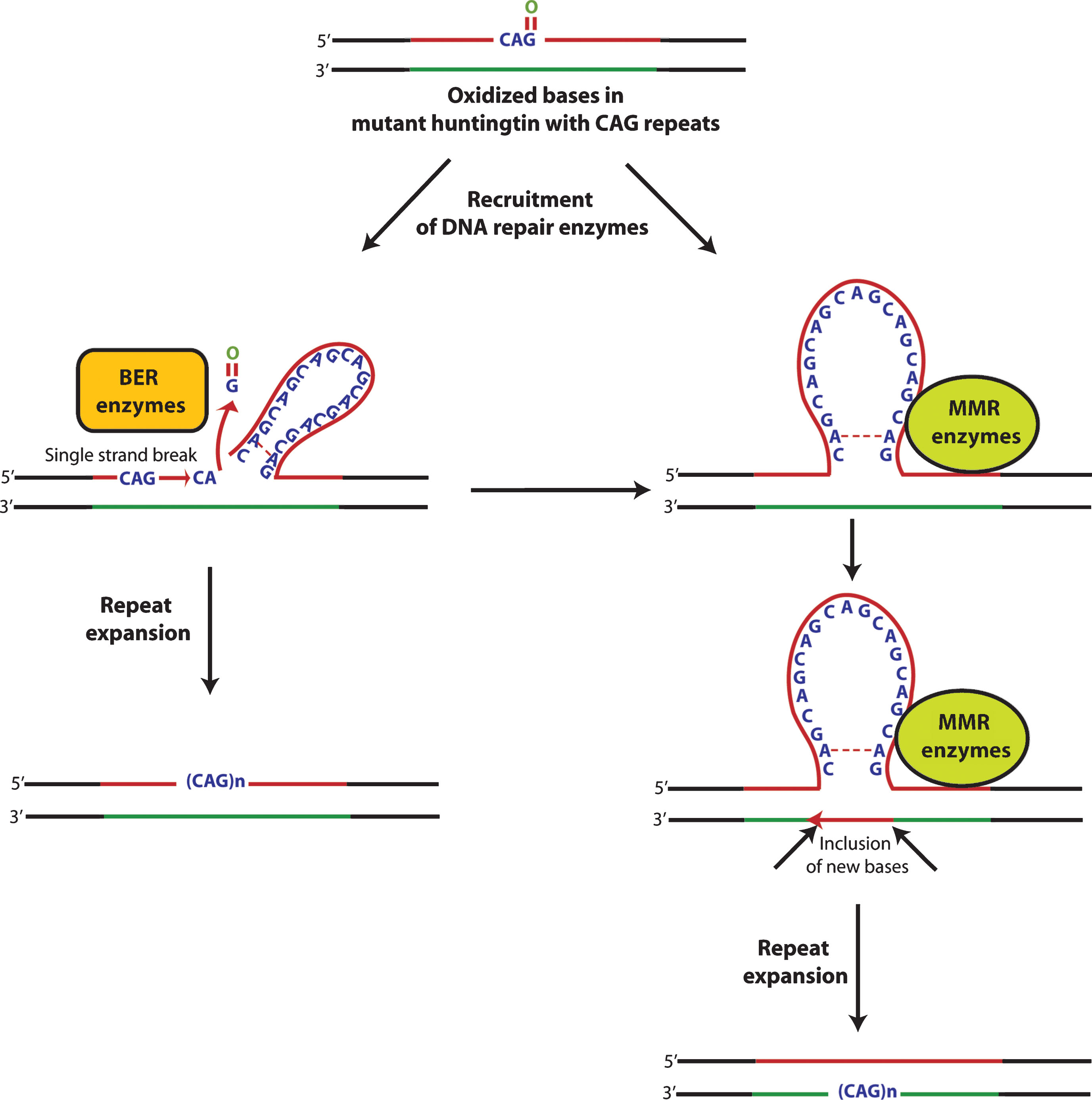
Fig.3
A schematic representation of redox regulation of different kinds of transcriptional processes. A. An example of the redox regulation of the TF binding to the promoters of its target genes through an upstream regulator, Ref-1. A number of TFs have one or more reactive cysteines in their DNA binding domain, which are redox regulated by Ref-1 (APE-1) protein, which, in turn, is also redox regulated through its reactive cysteine. B. An example of the redox regulation of the TF binding to the promoters of its target genes under oxidative stress. The binding of Sp-1/Sp-3 to the promoters of its target genes is also redox regulated. Under basal condition, the binding of Sp-1/Sp-3 is very weak while oxidative stress induces their strong binding with the promoters of their target genes, which has been shown to be neuroprotective by our group. C. An example of indirect redox regulation of TFs by the change in redox states of co-factors such as NAD and NADH. The binding of CTBP-1, a co-repressor regulating the expression of BDNF with transcription factor NRSF depends upon NAD/NADH ratio. D. An example of the regulation of transcriptional processes through redox associated post-translation modifications. Redox regulation of HDAC2 through S-nitrosylation leads to its release, which, in turn, increases acetylation and enhances CREB dependent transcription.
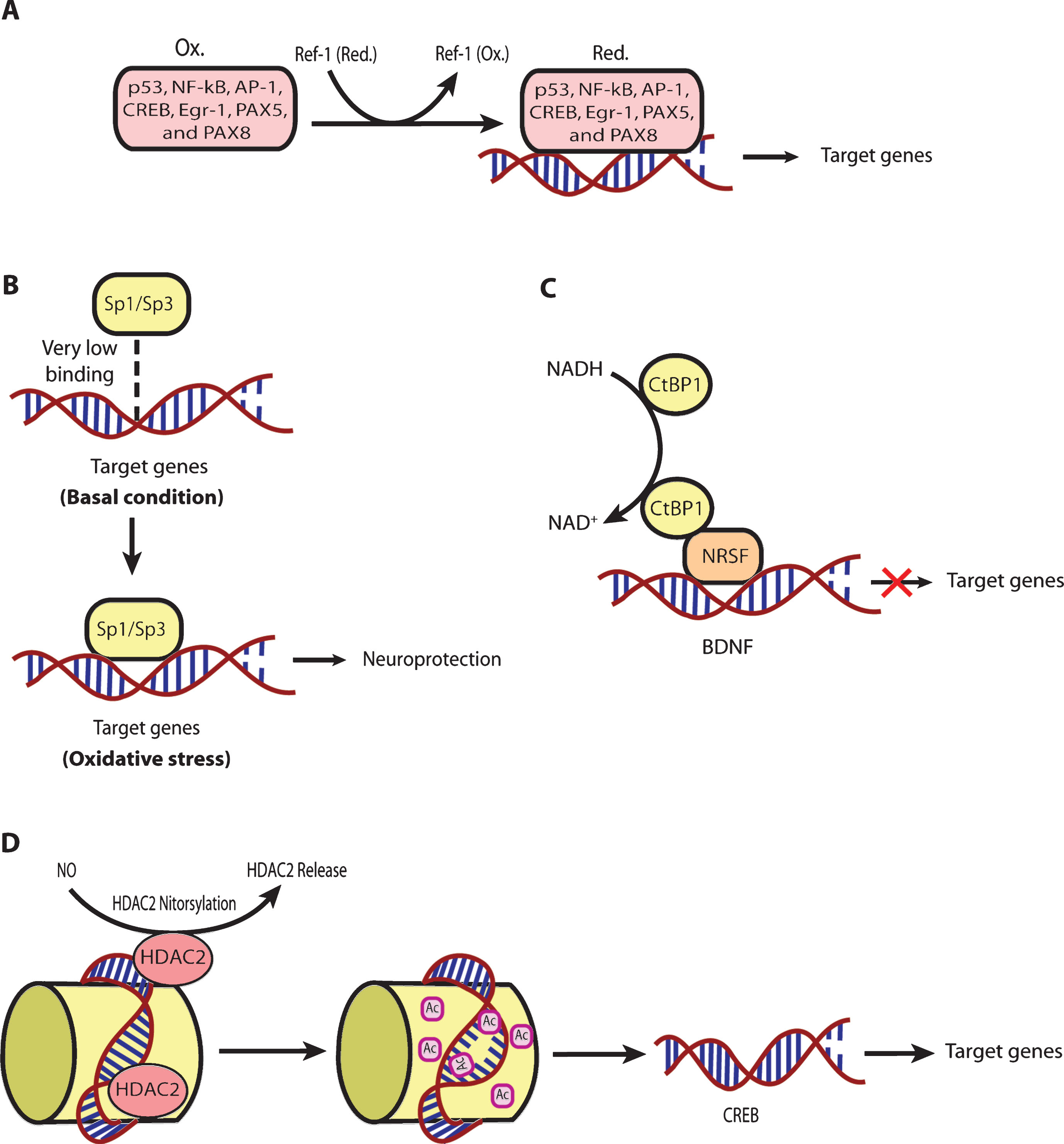
Fig.4
Increases in Sp1 and Sp3 DNA binding induced by the glutamate analog HCA are inhibited by antioxidants. Sp1 and Sp3 DNA binding in cortical neurons are activated by hydrogen peroxide. Induction of Sp1 and Sp3 DNA binding by HCA-induced glutathione depletion (4 hr) is decreased by the antioxidant iron chelator DFO (100 μm; A) and the lipid peroxidation inhibitor BHA (10 μm; B). C, Addition of exogenous peroxide, generated by the enzyme DAAO and its substrate d-ala (20 mm) for 4 hr increases Sp1 and Sp3 DNA binding in a concentration-dependent manner in cortical neurons. The induction is observed despite no morphological or biochemical evidence of cell death in cortical neurons. D, Addition of catalase abrogates Sp1 and Sp3 DNA binding induced by d-ala (20 mm) and DAAO (5 mU). Examples are representative of three to five independent experiments. (With permission from J. Neurosci.).
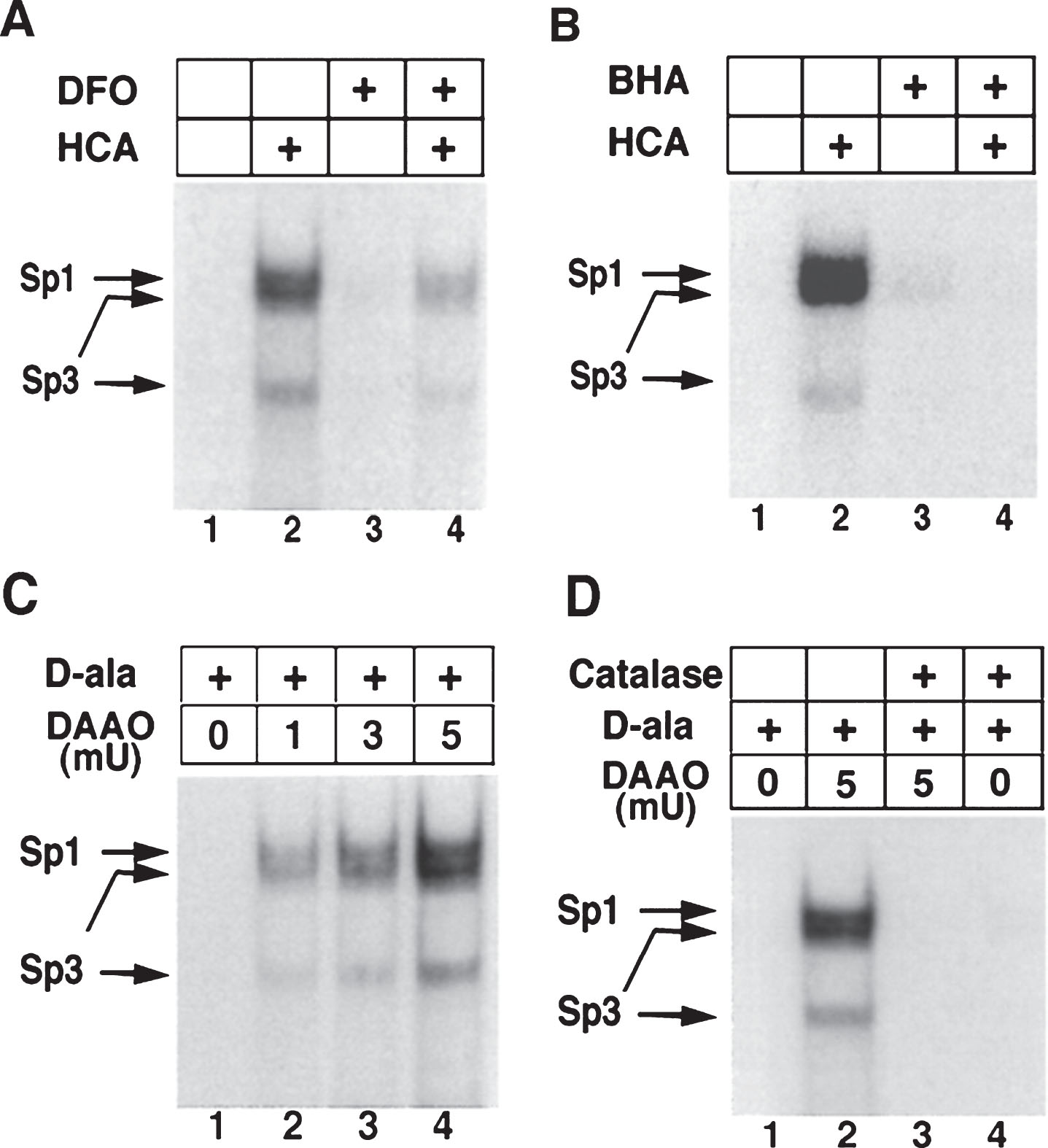
Table 1
Redox modifications of transcriptional players
| Transcriptional players | Redox targets | Consequences | References |
| SoxS (Bacteria) | Fe-S cluster | Changes in the activity of SoxS | [70] |
| OxyR (Bacteria) | Cys199 | Changes in the activity of OxyR | [160] |
| YAP1 (Yeast) | Cys 598 and Csy620 in its NES terminal end | Regulation of nuclear export | [161, 174] |
| Ref-1 (Mammals) | Cys 65 and Cys 93 in its N-terminal end | Regulation of the redox states of TFs such as | [162] |
| AP-1, NF-κB, p53, PAX5, PAX8, and Egr-1 | |||
| AP-1 (Mammals) | Cys residuse in basic region of Fos and Jun | Changes in its DNA binding activity | [179, 180] |
| NF-κB (Mammals) | Cys62 of p50 sub-unit | Changes in its DNA binding activity | [138, 141] |
| p53 (Mammals) | Multiple Cys residues | Changes in its DNA binding activity | [80] |
| PAX5 and PAX8 (Mammals) | Cyst37 and Cys49 in Prd domain | Changes in its DNA binding activity | [40, 181] |
| Egr-1 (Mammals) | Cys residues in zinc finger domain | Changes in its DNA binding activity | [41] |


