
Targeting the Cholinergic System to Develop a Novel Therapy for Huntington’s Disease
Abstract
In this review, we outline the role of the cholinergic system in Huntington’s disease, and briefly describe the dysfunction of cholinergic transmission, cholinergic neurons, cholinergic receptors and cholinergic survival factors observed in post-mortem human brains and animal models of Huntington’s disease. We postulate how the dysfunctional cholinergic system can be targeted to develop novel therapies for Huntington’s disease, and discuss the beneficial effects of cholinergic therapies in pre-clinical and clinical studies.
INTRODUCTION
Huntington’s disease (HD) is an autosomal dominant, neurodegenerative disorder caused by an extended CAG repeat sequence in the HD gene [1]. Several mechanisms have been proposed to explain how an expanded CAG repeat sequence in the HD gene leads to the symptoms and neuropathology of HD [2–8]. We propose an updated review of the mechanisms through which the cholinergic system could be used to modify the functional deficits and neuropathology of HD.
The cholinergic hypothesis suggests that dysfunctional acetylcholine (ACh)-containing neurons and dysfunctional cholinergic transmission in the brain significantly contribute to the behavioural symptoms and neuropathology in disease, in this instance HD [9]. The link between the cholinergic hypothesis and neurodegenerative disease was proposed by Bartus and colleagues in a seminal review [10] which referred to findings from previous studies including the cognitive deficits produced by anticholinergic drugs in humans [11, 12] and primates [13], as well as reduced choline acetyltransferase (ChAT) activity, reduced ACh release and degeneration of cholinergic neurons in autopsied patients [14–20].
The electrophysiological properties of cholinergic neurons vary depending on their location in the brain. Cholinergic neurons in the ventral pallidum and magnocellular cholinergic neurons in other parts of the forebrain, identified by ChAT staining, displayed a large whole cell conductance, a hyperpolarized resting membrane potential, marked fast inward rectification, a prominent spike afterhyperpolarization (AHP), but did not fire spontaneously [21]. However, these findings were based on a single study in rat brain slices [21].
In addition cholinergic neurons in the basal forebrain of GFP-expressing transgenic mice can be either early-firing or late-firing neurons [22]. The early-firing neurons are more excitable and are more susceptible to depolarization blockade, while displaying prominent spike frequency adaptation. Conversely, late firing neurons are less excitable and maintain a tonic discharge at low frequencies. Early-firing neurons are thought to be involved in phasic changes in cortical ACh release associated with attention, while the late-firing neurons may support general arousal by maintaining tonic ACh levels [22].
The distinct electrophysiological properties of cholinergic neurons means that it would be possible to distinguish cholinergic neurons from non-cholinergic neurons in brain slices, which will simplify future physiological and pharmacological studies of these neurons. The existence of two distinct subtypes of basal forebrain cholinergic neurons and their electrophysiological properties suggests that each subtype has a different ACh release profile, which is supported by recent studies showing ACh release can be measured over seconds or minutes [23–25]. Further, it is thought that each subtype is involved in different aspects of synaptic plasticity [22].
The afferent inputs and efferent outputs of cholinergic neurons also vary according to their location. Muscarinic cholinoceptive neurons in the neocortex are directly innervated by the magnocellular basal nucleus, while basal forebrain cholinergic neurons receive afferent input from the prefrontal cortex [26]. Cholinergic neurons from the basal forebrain and upper brainstem project to several areas including the cerebral cortex, amygdala, hippocampus, olfactory bulb and the thalamic nuclei [27]. The amygdala and pyriform cortex also receives cholinergic projections from the substantia innominata [28]. In addition cholinergic neurons in and around the pedunculopontine nucleus were shown to send projections to the substantia nigra pars compacta (SNc) via nicotinic receptors [29]. The substantia nigra also receives innervation from cholinergic cells of the rostral pontine tegmentum [28].
Striatal cholinergic interneurons densely innervate the striatum resulting in the striatum having one of the highest levels of ACh in the brain. These neurons, known as tonically active neurons (TANs) of the striatum, function as pacemaker cells and exhibit single spiking or rhythmic discharges but fire autonomously at rest. They receive dopaminergic input from the SNc, glutamatergic input from the thalamus (from the intralaminar nuclei) and the cerebral cortex, as well as input from the brain stem. In addition they contact each other. TANS modulate other interneurons particularly the fast spiking parvalbumin positive GABAergic interneurons and most importantly the medium spiny neurons (MSNs) through complex synaptic interactions [30]. They project to virtually all MSN subtypes, including both D1- and D2-dopamine receptor expressing MSNs and directly act through M1 receptors located on MSNs [31, 32]. The combined activation of the A2A adenosine and D2 dopamine receptors on cholinergic interneurons decreases the release of ACh. This decrease reverses the blockade of L-type calcium channels, which are mediated by M1 muscarinic receptors located on MSNs, which triggers the entry of calcium into the MSN. This might in turn trigger endocannabinoid release at the postsynaptic sites of both D1- and D2R-expressing MSNs and thus depress the corticostriatal and thalamostriatal glutamatergic synaptic transmission of both the direct and indirect pathways [31, 32].
Mouse models of HD have shown anatomical changes of striatal cholinergic neurons and reduced thalamic input to striatal cholinergic interneurons [33]. Since cholinergic interneurons have different effects on the striatal spiny projection neurons of the direct and indirect pathways, the reduction of glutamatergic transmission from corticostriatal and thalamostriatal projections may be responsible for early motor impairments observed in HD [33].
EVIDENCE FOR THE CHOLINERGIC HYPOTHESIS IN HUNTINGTON’S DISEASE
Studies in post-mortem human HD brains and animal models have shown that dysfunctional cholinergic neurotransmission and cholinergic neurons may play a significant role in HD pathogenesis. Post-mortem brains of patients with HD have shown a reduction in the levels of ChAT, vesicular acetylcholine transporter (VAChT), vesicular monoamine transporter type-2 (VMAT2) and the pyruvate dehydrogenase complex, an essential enzyme for ACh synthesis [34–39]. Conversely, choline levels were down in the CSF of HD patients but there was no reduction in the levels of acetylcholinesterase (AChE), an enzyme which degrades ACh [40]. Impairments in cholinergic neurotransmission were also observed in animal models of HD. Young R6/1 transgenic mice exhibited reductions in AChE activity and ChAT and VAChT messenger ribonucleic acid (mRNA) and protein levels [41, 15]. Transgenic R6/2 mice also showed a significant reduction in the electrically-evoked release of ACh in striatal tissue, as well as a decrease in ChAT activity and synaptosomal high-affinity choline uptake with age [42, 43].
In HD, striatal GABAergic MSNs are preferentially degraded, while large aspiny cholinergic interneurons are relatively spared [44, 45]. However, the surviving striatal cholinergic neurons showed decreased ACh release, VAChT binding and significant reductions in ChAT activity, but persistent AChE activity [34, 45–47]. During the early and mid-stages of HD, these cholinergic neurons demonstrate neuronal dysfunction without cell death, which may be involved in the early symptomatology of HD [41, 48]. The dysfunction observed in striatal cholinergic neurons may impact ACh-dependent processes such as the induction of synaptic plasticity [43, 47], and may significantly affect the function of other neuronal populations, including MSNs [48–50]. However, in the later stages of HD, the loss of aspiny cholinergic neurons has been observed [51–53].
Studies carried out in post-mortem human HD brains have produced varying results. Some have shown a reduction in muscarinic acetylcholine receptor (mAChR) binding, and a decrease in the number of M2 mAChRs and total mAChRs [35, 37]. Conversely, early studies reported no change in the levels of nAChRs in post-mortem human HD brains [54, 55]. However, these studies also reported no alterations in the level of ChAT in human HD brains, which has since been refuted by subsequent studies, and further research is therefore needed to clarify the levels of nAChRs in the HD brain [34–37]. Muscarinic cholinergic receptors were also decreased in R6/2 mice [56].
Decreased levels of NGF, an essential regulator of cholinergic neuronal survival and cholinergic neurotransmission, were observed in HD patients and R6/1 mice [48, 57]. Altered levels of NGF receptors have also been reported in the hippocampus of HD patients and in R6/1 and HdhQ7/Q111 mice [58, 59].
CHOLINERGIC TREATMENTS FOR HUNTINGTON’S DISEASE
The first procholinergic treatment for HD was the ACh precursor, choline, which significantly reduced chorea in three out of eight patients after they were administered choline (Table 1) [60]. However, choline showed no beneficial effects in other clinical studies [61, 62].
Another therapeutic strategy was to increase ACh levels through the use of acetylcholinesterase inhibitors (AChEIs) which inhibited the breakdown of ACh by the enzyme AChE (Fig. 1). The AChEI, rivastigmine, enhanced cognitive performance and slowed motor deterioration in an open-label clinical trial of 21 HD patients [63]. In a follow-up study, patients treated with rivastigmine showed increased motor performance, reduced chorea, increased cognitive function and reduced functional disability, relative to controls, suggesting that rivastigmine may have long-term benefits as a potential treatment for HD [64]. A smaller clinical study on four HD patients reported that rivastigmine improved cognitive and behavioural function, but failed to improve motor function or functional performance (Table 1) [65]. The AChEI, donepezil, improved the cognitive function of R6/2 mice in a simple visual discrimination task (Table 2) [66]. However, the beneficial effects of donepezil did not translate to the clinic [67, 68]. Galantamine, another AChEI, attenuated neurological deficits, reduced striatal lesion volumes and decreased the number of apoptotic cells in the 3-nitropropionic acid rat model of HD [69]. Further, a 35 year old male patient treated with galantamine displayed an improvement of psychotic symptoms and chorea, but showed no change in cognitive function [70]. Interestingly, the beneficial effects of galantamine were attenuated by mecamylamine, a nAChR antagonist, which suggests that the beneficial effects of galantamine may be mediated through nAChR activation [69].
AChEIs can also activate the cholinergic anti-inflammatory pathway, which reduces the neuroinflammation associated with HD pathology [71, 72]. These anti-inflammatory effects were inhibited by the α7 nAChR antagonist MLA, but not by the α4β2 nAChR antagonist, dihydro-β-erythroidine, which suggests that the anti-inflammatory effects of the cholinergic anti-inflammatory pathway are mediated through α7 nAChRs [73–76]. Physostigmine, an AChEI which stimulates both nAChRs and mAChRs, was shown to reduce chorea in two studies [77, 78]. Muscarinic receptor activation was also shown to reverse impairments in spatial memory, enhance hippocampal cell proliferation in the dentate gyrus and the CA1 region of the hippocampus and reverse deficits in hippocampal neurogenesis (Fig. 1) [79].
An alternative therapeutic strategy was to use anticholinergic agents, such as muscarinic receptor antagonists, to reduce chorea since anticholinergics cause sedation. The mAChR antagonist, scopolamine, decreased chorea but increased lack of coordination, sedation and confusion [80]. Another mAChR antagonist, benztropine, produced similar but milder effects, while the peripheral anticholinergic agent, glycopyrrolate, had no effect on chorea [80]. Conversely, the M1 mAChR antagonist, olanzapine, improved motor function [81] and psychiatric function [82] in human HD patients. Other studies reported that olanzapine improved both motor and psychiatric function when administered alone [83] or together with valproate [84]. The M4 mAChR positive allosteric modulator VU0467154 was reported to improve motor function and reduce synaptic deficits in transgenic YAC128 mice [85]. Further, the depotentiation of spiny neurons was blocked by either scopolamine or hemicholinium in mouse and rat models, which suggests that the reversal of LTP requires the activation of muscarinic receptors [86].
The administration of NGF and NGF-promoting drugs, cells or microspheres, increased motor coordination [87], reduced clasping [87], increased spatial working memory [48, 88], showed neuroprotective effects [89–94], reversed cholinergic neuron atrophy [88], promoted hippocampal neurogenesis [48], and increased the expression of ChAT and VAChT [48, 95] in rodent models of HD (Table 2).
There are therefore several potential targets for therapeutic intervention in the cholinergic system including (1) increasing the amount of the cholinergic survival factor NGF (2) increasing the amount of the ACh precursor choline (3) using AChEIs which inhibit AChEs from breaking down ACh and (4) using mAChR/nAChR ligands to activate mAChRs and nAChRs. Of these, the most promising therapeutic intervention appears to be increasing NGF, because if NGF can restore cholinergic neurons to optimal function and enhance ACh release to enable proper MSN function, there is no need to artificially add choline, AChEIs or mAChR/nAChR ligands to the system.
CONCLUDING REMARKS
Studies showing the dysfunction of cholinergic transmission, cholinergic receptors, cholinergic neurons and their associated neurotrophic factors in human HD patients and animal models of HD, together with the close link between cholinergic neurons and MSNs, suggest that the dysfunctional cholinergic system plays a vital role in the symptoms and neuropathology of HD. This provides researchers with a rationale to develop potential therapies for HD by targeting the cholinergic system. Cholinergic treatments have shown beneficial effects in both pre-clinical and clinical studies, and have improved motor, cognitive and psychiatric function, as well as reducing striatal lesions, synaptic deficits, excitotoxicity, cell atrophy and cell death [63, 64, 69, 85, 88]. However, different studies often reported different efficacies for the same therapy, possibly due to differences in the model or criteria used to determine efficacy, dose administration technique and dosing regimen used [79]. In order to achieve optimal results, these factors need to be optimised. In addition, a potential cholinergic therapeutic should be administered during the stage of the disease where it can have the greatest effect, for example, a mAChR agonist should be administered before significant loss of mAChRs occurs. Nonetheless, we believe that the beneficial effects of cholinergic treatments in pre-clinical and clinical HD studies demonstrates that this is a promising avenue of HD research which requires further investigation, and that enhancing cholinergic function may lead to the development of a potential therapy for HD.
The interdependent nature of the cholinergic dysfunction and its associated neurotrophic factors means that a potential treatment targeted towards increasing cholinergic neurotrophic factors may also have beneficial effects on cholinergic neuronal survival and neurotransmission [48]. In addition, the widespread effects of the cholinergic system on brain function suggests that treatments targeted towards the normalization of cholinergic transmission in HD may have other unseen beneficial effects, such as improving the function of other neuronal populations, such as MSNs [48, 50, 96].
Based on the available data (see Table 1), one of the most-promising therapeutic avenues appears to be increasing the levels of NGF, either through direct administration, the administration of NGF-promoting drugs, such as riluzole, or NGF-secreting cells or microspheres. NGF is an important survival factor for cholinergic neurons, and thus mediates cholinergic transmission as well as the function of neurons innervated by cholinergic neurons, such as MSNs. To investigate the pre-clinical efficacy of NGF, an animal model that replicates the human condition as closely as possible such as the YAC128 transgenic mouse model of HD, could be used. This mouse contains the full-length human HD gene and exhibits the motor, cognitive and psychiatric symptoms of HD, as well as the neuropathological characteristics, such as striatal cell loss, huntingtin aggregates and altered synaptic currents [97, 98]. NGF does not cross the blood-brain-barrier [99], thus, we would deliver NGF by surgically implanting microspheres in the striatum of the mice to achieve a controlled and sustained delivery of NGF [91]. The mice would undergo behavioural testing, and various neuropathological endpoints including MSN and cholinergic cell survival as well as synaptic function [48, 85, 100, 101]. Since therapies geared towards a single target have failed to show beneficial effects in the clinic, a therapy, such as NGF, which can affect multiple targets, may have a higher chance of exhibiting beneficial effects in a clinical trial [102].
Recent studies have focused on specific cholinergic receptor subtype agonists and antagonists, in particular those targeting α4β2 and α7 nicotinic acetylcholine receptors (nAChRs). The selective α4β2 nAChR agonist SIB-1508Y exerted anti-Parkinsonian effects in monkeys [103]. The selective α4β2 agonists ZY-1 and TC-6683 increased cognitive function in preclinical animal models of AD, with TC-6683 having progressed to phase 2 clinical trials for AD [104, 105]. The selective α4β2 agonist altinicline, and partial α4β2 agonists cytisine and ispronicline displayed antidepressant-like effects [106, 107, 108]. The α7 nAChR agonist, PNU-282987, improved the motor coordination of rats [109]. Further, administration of EVP-6124, an α7 nAChR partial agonist, and TC-5619, a full α7 nAChR agonist produced improvements in the memory of rats [110, 111].
Given the beneficial effects of α4β2 and α7 nAChR agonists on motor, cognitive and psychiatric function, it may be beneficial to pursue a potential therapy which activates both α4β2 and α7 nAChRs. One such drug is varenicline, a potent, partial agonist at α4β2 nAChRs, and a potent, full agonist at α7 nAChRs [112, 113]. Varenicline increased basal locomotor activity in rats [114]. Varenicline also enhanced the cognitive function of mice [115, 116] and rats [117]. Further, varenicline showed antidepressant-like activity [118], and anxiolytic effects [119].
The potentially advantageous therapies described above which directly or indirectly target the cholinergic system may benefit HD patients, and should therefore be further investigated.
REFERENCES
[1] | Walker FO . Huntington’s disease. Lancet. (2007) ;369: (9557):218–28. |
[2] | Quintanilla RA , Johnson GVW . Role of mitochondrial dysfunction in the pathogenesis of Huntington’s disease. Brain Res Bull. (2009) ;80: (4-5):242–7. |
[3] | Estrada-Sánchez AM , Mejia-Toiber J , Massieu L . Excitotoxic neuronal death and the pathogenesis of Huntington’s disease. Arch Med Res. (2008) ;39: (3):265–76. |
[4] | Sanchez-Mejia RO , Friedlander RM . Caspases in Huntington’s disease. Neuroscientist. (2001) ;7: (6):480–9. |
[5] | Hoffner G , Djian P . Protein aggregation in Huntington’s disease. Biochimie. (2002) ;84: (4):273–8. |
[6] | Cha JH . Transcriptional dysregulation in Huntington’s disease. Trends Neurosci. (2000) ;23: (9):387–92. |
[7] | Smith R , Brundin P , Li JY . Synaptic dysfunction in Huntington’s disease: A new perspective. Cell Mol Life Sci. (2005) ;62: (17):1901–12. |
[8] | Martin DDO , Ladha S , Ehrnhoefer DE , Hayden MR . Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci. (2015) ;38: (1):26–35. |
[9] | Terry AV Jr , Buccafusco JJ . The cholinergic hypothesis of age and Alzheimer’s disease-related cognitive deficits: Recent challenges and their implications for novel drug development. J Pharmacol Exp Ther. (2003) ;306: (3):821–7. |
[10] | Bartus RT , Dean RL , Beer B , Lippa AS . The cholinergic hypothesis of geriatric memory dysfunction. Science. (1982) ;217: (4558):408–17. |
[11] | Longo VG . Behavioral and electroencephalographic effects of atropine and related compounds. Pharmacol Rev. (1966) ;18: (2):965–96. |
[12] | Drachman DA , Leavitt J . Human memory and the cholinergic system. A relationship to aging? Arch Neurol. (1974) ;30: (2):113–121. |
[13] | Bartus RT , Johnson HR . Short-term memory in the rhesus monkey: Disruption from the anti-cholinergic scopolamine. Pharmacol Biochem Behav. (1976) ;5: (1):39–46. |
[14] | Whitehouse PJ , Price DL , Struble RG , Clark AW , Coyle JT , Delon MR . Alzheimer’s disease and senile dementia: Loss of neurons in the basal forebrain. Science. (1982) ;215: (4537):1237–39. |
[15] | Eckenstein F , Sofroniew MV . Identification of central cholinergic neurons containing both choline acetyltransferase and acetylcholinesterase and of central neurons containing only acetylcholinesterase. J Neurosci. (1983) ;3: (11):2286–91. |
[16] | Perry EK , Gibson PH , Blessed G , Perry RH , Tomlinson BE . Neurotransmitter enzyme abnormalities in senile dementia. Choline acetyltransferase and glutamic acid decarboxylase activities in necropsy brain tissue. J Neurol Sci. (1977) ;34: (2):247–65. |
[17] | Nilsson L , Nordberg A , Hardy J , Wester P , Winblad B . Physostigmine restores 3H-acetylcholine efflux from Alzheimer brain slices to normal level. J Neural Transm. (1986) ;67: (3-4):275–85. |
[18] | Rylett RJ , Ball MJ , Colhoun EH . Evidence for high affinity choline transport in synaptosomes prepared from hippocampus and neocortex of patients with Alzheimer’s disease. Brain Res. (1983) ;289: (1-2):169–75. |
[19] | Bowen DM , Smith CB , White P , Davison AN . Neurotransmitter-related enzymes and indices of hypoxia in senile dementia and other abiotrophies. Brain. (1976) ;99: (3):459–96. |
[20] | Davies P , Maloney AJ . Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet. (1976) ;2: (8000):1403. |
[21] | Bengtson CP , Osborne PB . Electrophysiological properties of cholinergic and noncholinergic neurons in the ventral pallidal region of the nucleus basalis in rat brain slices. J Neurophysiol. (2000) ;83: (5):2649–60. |
[22] | Unal CT , Golowasch JP , Zaborszky L . Adult mouse basal forebrain harbors two distinct cholinergic populations defined by their electrophysiology. Front Behav Neurosci. (2012) ;6: (21):1–14. |
[23] | Hasselmo ME , Sarter M . Modes and models of forebrain cholinergic neuromodulation of cognition. Neuropsychopharmacology. (2011) ;36: :52–73. |
[24] | Parikh V , Kozak R , Martinez V , Sarter M . Prefrontal acetylcholine release controls cue detection on multiple timescales. Neuron. (2007) ;56: (1):141–54. |
[25] | Parikh V , Sarter M . Cholinergic mediation of attention: Contributions of phasic and tonic increases in prefrontal cholinergic activity. Ann N Y Acad Sci. (2008) ;1129: :225–35. |
[26] | Gaykema RP , Gaal G , Traber J , Hersh LB , Luiten PG . The basal forebrain cholinergic system: Efferent and afferent connectivity and long-term effects of lesions. Acta Psychiatr Scand Suppl. (1991) ;366: :14–26. |
[27] | Mesulam MM , Mufson EJ , Wainer BH , Levey AI . Central cholinergic pathways in the rat: An overview based on an alternative nomenclature (Ch1–Ch6). Neuroscience. (1983) ;10: (4):1185–201. |
[28] | McGeer E , McGeer PL . Possible destruction of cholinoceptive neurons by overstimulation of cholinergic tracts. Can J Physiol Pharmacol. (1986) ;64: (3):363–8. |
[29] | Clarke PB , Hommer DW , Pert A , Skirboll LR . Innervation of substantia nigra neurons by cholinergic afferents from pedunculopontine nucleus in the rat: Neuroanatomical and electrophysiological evidence. Neuroscience. (1987) ;23: (3):1011–9. |
[30] | Goldberg JA , Wilson CJ . The Cholinergic interneurons of the striatum: Intrinsic properties underlie multiple discharge patterns. In Steiner H, Tseng KY, editors. Handbook of Basal Ganglia Structure and Function; London, Academic Press; (2010) . pp. 133–146. |
[31] | Tozzi A , de Iure A , Di Filippo M , Tantucci M , Costa C , Borsini F , et al. The distinct role of medium spiny neurons and cholinergic interneurons in the D2/A2A receptor interaction in the striatum: Implications for Parkinson’s disease. J Neurosci. (2011) ;31: (5):1850–62. |
[32] | Calabresi P , Picconi B , Tozzi A , Ghiglieri V , DiFilippo M . Direct and indirect pathways of basal ganglia: A critical reappraisal. Nat Neurosci. (2014) ;17: (8):1022–30. |
[33] | Deng YP , Reiner A . Cholinergic interneurons in the Q140 knockin mouse model of Huntington’s disease: Reductions in dendritic branching and thalamostriatal input. J Comp Neurol. (2016) ;524: (17):3518–29. |
[34] | Suzuki M , Desmond TJ , Albin RL , Frey KA . Vesicular neurotransmitter transporters in Huntington’s disease: Initial observations and comparison with traditional synaptic markers. Synapse. (2001) ;41: (4):329–36. |
[35] | Wastek GJ , Stern LZ , Johnson PC , Yamamura HI . Huntington’s disease: Regional alteration in muscarinic cholinergic receptor binding in human brain. Life Sci. (1976) ;19: (7):1033–9. |
[36] | Spokes EG . Neurochemical alterations in Huntington’s chorea: A study of post-mortem brain tissue. Brain. (1980) ;103: (1):179–210. |
[37] | Lange KW , Javoy-Aqid F , Aqid Y , Jenner P , Marsden CD . Brain muscarinic cholinergic receptors in Huntington’s disease. J Neurol. (1992) ;239: (2):103–4. |
[38] | Butterworth J , Yates CM , Reynolds GP . Distribution of phosphate-activated glutaminase, succinic dehydrogenase, pyruvate dehydrogenase and gamma-glutamyl transpeptidase in post-mortem brain from Huntington’s disease and agonal cases. J Neurol Sci. (1985) ;67: (2):161–71. |
[39] | Sorbi S , Bird ED , Blass JP . Decreased pyruvate dehydrogenase complex activity in Huntington and Alzheimer brain. Ann Neurol. (1983) ;13: (1):72–8. |
[40] | Manyam BV , Giacobini E , Colliver JA . Cerebrospinal fluid acetylcholinesterase and choline measurements in Huntington’s disease. J Neurol. (1990) ;237: (5):281–284. |
[41] | Smith R , Chung H , Rundquist S , Maat-Schieman ML , Colgan L , Englund E , et al. Cholinergic neuronal defect without cell loss in Huntington’s disease. Hum Mol Genet. (2006) ;15: (21):3119–31. |
[42] | Vetter JM , Jehle T , Heinemeyer J , Franz P , Behrens PF , Jackisch R , et al. Mice transgenic for exon 1 of Huntington’s disease: Properties of cholinergic and dopaminergic pre-synaptic function in the striatum. J Neurochem. (2003) ;85: (4):1054–63. |
[43] | Farrar AM , Callahan JW , Abercrombie ED . Reduced striatal acetylcholine efflux in the R6/2 mouse model of Huntington’s disease: An examination of the role of altered inhibitory and excitatory mechanisms. Exp Neurol. (2011) ;232: (2):119–25. |
[44] | Waldvogel HJ , Kim EH , Tippett LJ , Vonsattel JP , Faull RL . The neuropathology of Huntington’s disease. Curr Top Behav Neurosci. (2015) ;22: :33–80. |
[45] | Holley SM , Joshi PR , Parievsky A , Galvan L , Chen JY , Fisher YE , et al. Enhanced GABAergic inputs contribute to functional alterations of cholinergic interneurons in the R6/2 mouse model of Huntington’s disease. eNeuro. (2015) ; e0008: :1–15. |
[46] | Ferrante RJ , Flint Beal M , Kowall NW , Richardson EP Jr , Martin JB . Sparing of acetylcholinesterase-containing striatal neurons in Huntington’s disease. Brain Res. (1987) ;411: (1):162–6. |
[47] | Pisani A , Bernardi G , Ding J , Surmeier DJ . Re-emergence of striatal cholinergic interneurons in movement disorders. Trends Neurosci. (2007) ;30: (10):545–53. |
[48] | Zhang H , Petit GH , Gaughwin PM , Hansen C , Ranganathan S , Zuo X , et al. NGF rescues hippocampal cholinergic neuronal markers, restores neurogenesis, and improves the spatial working memory in a mouse model of Huntington’s disease. J Huntingtons Dis. (2013) ;2: (1):69–82. |
[49] | Pawlik K , Rosenzweig MR . editors. The International Handbook of Psychology. London SAGE Publications Ltd; (2000) . |
[50] | Nithianantharajah J , Barkus C , Murphy M , Hannan AJ . Gene-environment interactions modulating cognitive function and molecular correlates of synaptic plasticity in Huntington’s disease transgenic mice. Neurobiol Dis. (2008) ;29: (3):490–504. |
[51] | Zuccato C , Ciammola A , Rigamonti D , Leavitt BR , Goffredo D , Conti L , et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science. (2010) ;293: (5529):493–8. |
[52] | Jenden DD , editor. Cholinergic Mechanisms and Psychopharmacology. New York: Plenum Press; (1978) . |
[53] | Alexi T , Hughes PE , van Roon-Mom WM , Faull RL , Williams CE , Gluckman PD . The IGF-I amino-terminal tripeptide glycine-proline-glutamate (GPE) is neuroprotective to striatum in the quinolinic acid lesion animal model of Huntington’s disease. Exp Neurol. (1999) ;159: (1):84–97. |
[54] | Whitehouse PJ , Kellar KJ . Nicotinic and muscarinic cholinergic receptors in Alzheimer’s disease and related disorders. J Neural Transm Suppl. (1987) ;24: :175–82. |
[55] | Perry EK , Perry RH , Smith CJ , Dick DJ , Candy JM , Edwardson JA , et al. Nicotinic receptor abnormalities in Alzheimer’s and Parkinson’s diseases. J Neurol Neurosurg Psychiatry. (1987) ;50: (6):806–9. |
[56] | Cha JH , Kosinski CM , Kerner JA , Alsdorf SA , Mangiarini L , Davies SW , et al. Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human huntington disease gene. Proc Natl Acad Sci U S A. (1998) ;95: (11):6480–5. |
[57] | Tasset I , Sanchez-Lopez F , Aguera E , Fernandez-Bolanos R , Sanchez FM, Cruz-Guerrero A. NGF and nitrosative stress in patients with Huntington’s disease. J Neurol Sci. (2012) ;315: (1-2):133–6. |
[58] | Brito V , Gines S . p75NTR in Huntington’s disease: Beyond the basal ganglia. Oncotarget. (2016) ;7: (1):1–2. |
[59] | Brito V , Giralt A , Enriquez-Barreto L , Puigdellivol M , Suelves N , Zamora-Moratalla A , et al. Neurotrophin receptor p75(NTR) mediates Huntington’s disease-associated synaptic and memory dysfunction. J Clin Invest. (2014) ;124: (10):4411–28. |
[60] | Davis KL , Mohs RC , Marin D , Purohit DP , Perl DP , Lantz M , et al. Cholinergic markers in elderly patients with early signs of Alzheimer disease. JAMA. (1999) ;281: (15):1401–6. |
[61] | Aquilonius SM , Eckernas SA . Choline therapy in Huntington chorea. Neurology. (1997) ;27: (9):887–9. |
[62] | Growdon JH , Wurtman RJ . Oral choline administration to patients with Huntington’s disease. Adv Neurol. (1979) ;23: :765–76. |
[63] | De Tommaso M , Specchio N , Sciruicchio V , Difruscolo O , Specchio LM . Effects of rivastigmine on motor and cognitive impairment in Huntington’s disease. Mov Disord. (2004) ;19: (12):1516–8. |
[64] | De Tommasso M , Difruscolo O , Sciruicchio V , Specchio N , Livrea P . Two years’ follow-up of rivastigmine treatment in Huntington disease. Clin Neuropharmacol. (2007) ;30: (1):43–6. |
[65] | Rot U , Kobal J , Sever A , Pirtosek Z , Mesec A . Rivastigmine in the treatment of Huntington’s disease. Eur J Neurol. (2002) ;9: (6):689–90. |
[66] | Murphy CA , Paterson NE , Chen A , Arias W , He D , Alosio W , et al. Cognitive deficits in the R6/2 mouse model of Huntington’s disease and their amelioration with Donepezil. Int J Comp Psychol. (2014) ;27: (3):397–407. |
[67] | Fernandez HH , Friedman JH , Grace J , Beason-Hazen S . Donepezil for Huntington’s disease. Mov Disorders. (2000) ;15: (1):173–6. |
[68] | Cubo E , Shannon KM , Tracy D , Jaglin JA , Bernard BA , Wuu J , et al. Effect of donepezil on motor and cognitive function in Huntington disease. Neurology. (2006) ;67: (7):1268–71. |
[69] | Park JE , Lee ST , Im WS , Chu K , Kim M . Galantamine reduces striatal degeneration in 3-nitropropionic acid model of Huntington’s disease. Neuroscience Lett. (2008) ;448: (1):143–7. |
[70] | Petrikis P , Andreou C , Piachas A , Bozikas VP , Karavatos A . Treatment of Huntington’s disease with galantamine. Int Clin Psychopharmacol. (2004) ;19: (1):49–50. |
[71] | Moller T . Neuroinflammation in Huntington’s disease. J Neural Transm. (2010) ;117: (8):1001–8. |
[72] | Crotti A , Glass CK . The choreography of neuroinflammation in Huntington’s disease. Trends Immunol. (2015) ;36: (6):364–73. |
[73] | Tyagi E , Agrawal R , Nath C , Shukla R . Inhibitory role of cholinergic system mediated via alpha7 nicotinic acetylcholine receptor in LPS-induced neuroinflammation. Innate Immun. (2010) ;16: (1):3–13. |
[74] | Han Z , Li L , Wang L , Degos V , Maze M , Hua S . Alpha-7 nicotinic acetylcholine receptor agonist treatment reduces neuroinflammation, oxidative stress, and brain injury in mice with ischemic stroke and bone fracture. J Neurochem. (2014) ;131: (4):498–508. |
[75] | Liu Y , Hu J , Wu J , Zhu C , Hui Y , Han Y , et al. α7 nicotinic acetylcholine receptor-mediated neuroprotection against dopaminergic neuron loss in an MPTP mouse model via inhibition of astrocyte activation. J Neuroinflammation. (2012) ;9: (98):1–15. |
[76] | Nizri E , Irony-Tur-Sinai M , Faranesh N , Lavon I , Lavi E , Weinstock M . Suppression of neuroinflammation and immunomodulation by the acetylcholinesterase inhibitor rivastigmine. J Neuroimmunol. (2008) ;203: (1):12–22. |
[77] | Aquilonius SM , Sjöström R . Cholinergic and dopaminergic mechanisms in Huntington’s chorea. Life Sci. (1971) ;10: (7):405–14. |
[78] | Fann WE , Gerber CJ , McKenzie GM . Physostigmine in rigid Huntington’s disease. Confin Neurol. (1973) ;35: (5):312–5. |
[79] | Van Kampen JM , Eckman CB . Agonist-induced restoration of hippocampal neurogenesis and cognitive improvement in a model of cholinergic denervation. Neuropharmacology. (2010) ;58: (6):921–9. |
[80] | Nutt JG . Effect of cholinergic agents in Huntington’s disease: A reappraisal. Neurology. (1983) ;33: (7):932–5. |
[81] | Bonelli RM , Mahnert FA , Niederwieser G . Olanzapine for Huntington’s disease: An open label study. Clin Neuropharmacol. (2002) ;25: (5):263–5. |
[82] | Squitieri F , Cannella M , Porcellini A , Brusa L , Simonelli M , Ruggieri S . Short-term effects of olanzapine in Huntington disease. Neuropsychiatry Neuropsychol Behav Neurol. (2001) ;14: (1):69–72. |
[83] | Laks J , Rocha M , Capitao C , Dominques RC , Ladeia G , Engelhardt E . Functional and motor response to low dose olanzapine in Huntington’s disease: Case report. Arq Neuropsiquiatr. (2004) ;62: (4):1092–94. |
[84] | Grove VE Jr , Quintanilla J , DeVaney GT . Improvement of Huntington’s disease with olanzapine and valproate. N Engl J Med. (2000) ;343: (13):973–4. |
[85] | Pancani T , Foster DJ , Moehle MS , Bichell TJ , Bradley E , Bridges TM , et al. Allosteric activation of M4 muscarinic receptors improve behavioral and physiological alterations in early symptomatic YAC128 mice. Proc Natl Acad Sci U S A. (2015) ;112: (45):14078–83. |
[86] | Picconi B , Passino E , Sgobio C , Bonsi P , Baroni I , Ghiglieri V , et al. Plastic and behavioral abnormalities in experimental Huntington’s disease: A crucial role for cholinergic interneurons. Neurobiol Dis. (2006) ;22: (1):143–52. |
[87] | Dey ND , Bombard MC , Roland BP , Davidson S , Lu M , Rossignol J , et al. Genetically engineered mesenchymal stem cells reduce behavioural deficits in the YAC128 mouse model of Huntington’s disease. Behav Brain Res. (2010) ;214: (2):193–200. |
[88] | Martinez-Serrano A , Fischer W , Bjorklund A . Reversal of age-dependent cognitive impairments and cholinergic neuron atrophy by NGF-secreting neural progenitors grafted to the basal forebrain. Neuron. (1995) ;15: (2):473–84. |
[89] | Mizuta I , Ohta M , Ohta K , Nishimura M , Mizuta E , Kuno S . Riluzole stimulates nerve growth factor, brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor synthesis in cultured mouse astrocytes. Neurosci Lett. (2001) ;310: (2-3):117–20. |
[90] | Cruz-Aquado R , Turner LF , Diaz CM , Pinero J . Nerve growth factor and striatal glutathione metabolism in a rat model of Huntington’s disease. Restor Neurol Neurosci. (2000) ;17: (4):217–21. |
[91] | Menei P , Pean JM , Nerriere-Daguin V , Jollivet C , Brachet P , Benoit JP . Intracerebral implantation of NGF-releasing biodegradable microspheres protects striatum against excitotoxic damage. Exp Neurol. (2000) ;161: (1):259–72. |
[92] | Perez-Navarro E , Alberch J , Arenas E , Calvo N , Marsal N . Nerve growth factor and basic fibroblast growth factor protect cholinergic neurons against quinolinic acid excitotoxicity in rat neostriatum. Eur J Neurosci. (1994) ;6: (5):706–11. |
[93] | Davies SW , Beardsall K . Nerve growth factor selectively prevents excitotoxin induced degeneration of striatal cholinergic neurones. Neurosci Lett. (1992) ;140: (2):161–4. |
[94] | Alberch J , Perez-Navarro E , Canals JM . Neurotrophic factors in Huntington’s disease. Prog Brain Res. (2004) ;146: :195–229. |
[95] | Venero JL , Beck KD , Hefti F . Intrastriatal infusion of nerve growth factor after quinolinic acid prevents reduction of cellular expression of choline acetyltransferase messenger RNA and trkA messenger RNA, but not glutamate decarboxylase messenger RNA. Neuroscience. (1994) ;61: (2):257–68. |
[96] | Deffains M , Bergman H . Striatal cholinergic interneurons and cortico-striatal synaptic plasticity in health and disease. Mov Disorders. (2015) ;30: (8):1014–25. |
[97] | Slow EJ , van Raamsdonk J , Rogers D , Coleman SH , Graham RK , Deng Y , et al. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Gen. (2003) ;12: (13):1555–67. |
[98] | Van Raamsdonk JM , Warby SC , Hayden MR . Selective degeneration in YAC mouse models of Huntington disease. Brain Res Bull. (2007) ;72: :124–31. |
[99] | Friden PM , Walus LR , Watson P , Doctrow SR , Kozarich JW , Backman C , et al. Blood-brain barrier penetration and in vivo activity of an NGF conjugate. Science. (1993) ;259: (5093):373–7. |
[100] | Precious SV , Kelly CM , Reddington AE , Vinh NN , Strickland RC , Pekarik V , et al. FoxP1 marks medium spiny neurons from precursors to maturity and is required for their differentiation. Exp Neurol. (2016) ;282: :9–18. |
[101] | Mandyam CD , Harburg GC , Eisch AJ . Determination of key aspects of precursor cell proliferation, cell cycle length and kinetics in the adult mouse subgranular zone. Neuroscience. (2008) ;146: (1):108–22. |
[102] | Rubinsztein DC , Carmichael J . Huntington’s disease: Molecular basis of neurodegeneration. Expert Rev Mol Med. (2003) ;5: (22):1–20. |
[103] | Schneider JS , Pope-Coleman A , Van Velson M , Menzaghi F , Lloyd GK . Effects of SIB-Y, a novel neuronal nicotinic acetylcholine receptor agonist, on motor behavior in parkinsonian monkeys. Mov Disorders. (1998) ;13: (4):637–42. |
[104] | Nie H , Wang Z , Zhao W , Lu J , Zhang C , Lok K , et al. New nicotinic analogue ZY-1 enhances cognitive functions in a transgenic mice model of Alzheimer’s disease. Neurosci Lett. (2013) ;537: :29–34. |
[105] | Mazurov AA , Miao L , Bhatti BS , Strachan JP , Akireddy S , Murthy S , et al. Discovery of 3-(5-chloro-2-furoyl)-3,7-diazabicyclo[3.3.0]octane (TC-AZDa novel highly selective α4β2 nicotinic acetylcholine receptor agonist for the treatment of cognitive disorders. J Med Chem. (2012) ;55: (21):9181–94. |
[106] | Ferguson SM , Brodkin JD , Lloyd GK , Menzaghi F . Antidepressant-like effects of the subtype-selective nicotinic acetylcholine receptor agonist, SIB-Y, in the learned helplessness rat model of depression. Psychopharmacology. (2000) ;152: (3):295–303. |
[107] | Mineur YS , Somenzi O , Picciotto MR . Cytisine, a partial agonist of high affinity nicotinic acetylcholine receptors, has antidepressant-like properties in male C57BL/6J mice. Neuropharmacology. (2007) ;52: (5):1256–62. |
[108] | Gatto GJ , Bohme GA , Caldwell WS , Letchworth SR , Traina1 VM , Obinu MC , et al. TC-An orally active neuronal nicotinic acetylcholine receptor modulator with antidepressant, neuroprotective and long-lasting cognitive effects. CNS Drug Rev. (2004) ;10: (2):147–66. |
[109] | Mannelli LDC , Bonaccini L , Zanardelli M , Mello T , Pacini A , Ghelardini C . Involvement of alpha7 nAChR downregulation in rat oxaliplatin-induced neuropathy. Ital J Anat Embryol. (2012) ;117: (2):57. |
[110] | Prickaerts J , Van Goethem NP , Chesworth R , Shapiro G , Boess FG , Methfessel C , et al. EVP-6124, a novel and selective α7 nicotinic acetylcholine receptor partial agonist, improves memory performance by potentiating the acetylcholine response of α7 nicotinic acetylcholine receptors. Neuropharmacology. (2012) ;62: (2):1099–110. |
[111] | Hauser TA , Kucinski A , Jordan KG , Gatto GJ , Wersinger SR , Hesse RA , et al. TC-An alpha7 neuronal nicotinic receptor-selective agonist that demonstrates efficacy in animal models of the positive and negative symptoms and cognitive dysfunction of schizophrenia. Biochem Pharmacol. (2009) ;78: (7):803–12. |
[112] | Rollema H , Russ C , Lee TC , Hurst RS , Bertrand D . Functional interactions of varenicline and nicotine with nAChR subtypes implicated in cardiovascular control. Nicotine Tob Res. (2014) ;16: (6):733–42. |
[113] | Mihalak KB , Carroll FI , Luetje CW . Varenicline is a partial agonist at α4β2 and a full agonist at α7 neuronal nicotinic receptors. Mol Pharmacol. (2006) ;70: :801–5. |
[114] | Zaniewska M , McCreary AC , Stefanski R , Przegalinski E , Filip M . Effect of varenicline on the acute and repeated locomotor responses to nicotine in rats. Synapse. (2008) ;62: :935–9. |
[115] | Gulick D , Gould TJ . Varenicline ameliorates ethanol-induced deficits in learning in C57BL/6 mice. Neurobiol Learn Mem. (2008) ;90: :230–6. |
[116] | Raybuck JD , Portugal GS , Lerman C , Gould TJ . Varenicline ameliorates nicotine withdrawal-induced learning deficits in C57BL/6 mice. Behav Neurosci. (2008) ;122: (5):1166–71. |
[117] | King J , Huang W , Chen W , Heffernan M , Shields J , Rane P , et al. A comparison of brain and behavioral effects of varenicline and nicotine in rats. Behav Brain Res. (2011) ;223: :42–7. |
[118] | Rollema H , Guanowsky V , Mineur YS , Shrikhande A , Coe JW , Seymour PA , et al. Varenicline has antidepressant-like activity in the forced swim test and augments sertraline’s effect. Eur J Pharmacol. (2009) ;605: :114–6. |
[119] | Turner JR , Castellano LM , Blendy JA . Nicotinic partial agonists Varenicline and Sazetidine-A have differential effects on affective behavior. J Pharm Exp Ther. (2010) ;334: (2):665–72. |
Figures and Tables
Fig.1
The cholinergic system in HD and targets for therapeutic intervention.
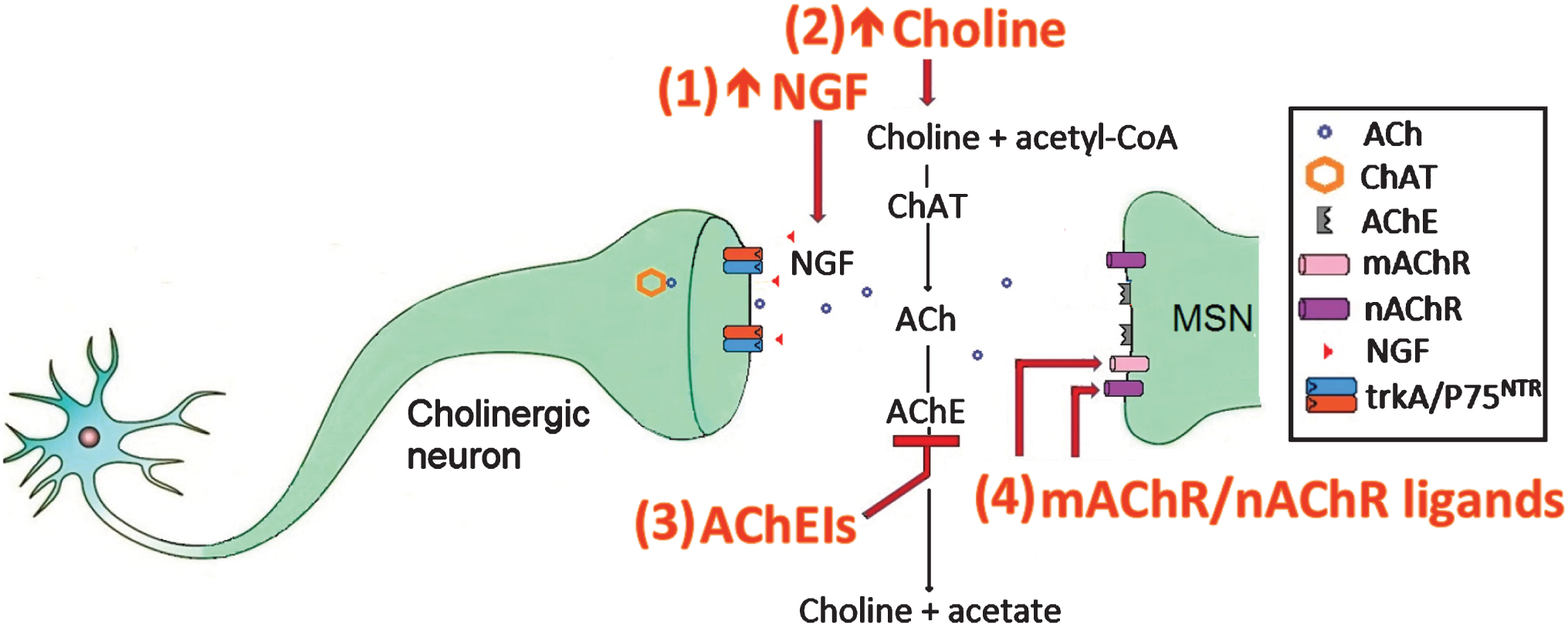
Table 1
Cholinergic-based treatments which have demonstrated beneficial effects in clinical HD studies. Strength of evidence criteria was based on proper study design, number of subjects improved, level of improvement, lack of side effects and replication by others
| Treatment | Beneficial effect(s) | Number of subjects studied | Reference(s) | Strength of | Comments |
| evidence | |||||
| Choline | reduces chorea | 8 | [60] | 3/10 | 3 out of 8 showed reduced chorea. |
| Rivastigmine | improves motor and cognitive function, reduces chorea | 21 patients, 17 patients, 4 patients | [63–65] | 8/10 | Beneficial cognitive and motor effects in a trial and then again in a follow-up study. Beneficial cognitive effects replicated in separate study. Study design could be improved. |
| Galantamine | improves psychotic symptoms and chorea | 1 patient | [70] | 3/10 | Positive effects, but only 1 patient. |
| Physostigmine | reduces chorea | 4 patients; 3 patients | [77, 78] | 4/10 | Reduction in chorea seen in both studies, but low number of patients studied. |
| Scopolamine | reduces chorea | 4 patients | [80] | 2/10 | Reduction in chorea, but side-effects include incoordination, sedation and confusion. |
| Benztropine | reduces chorea | 4 patients | [80] | 2/10 | Similar but milder effects compared to scopolamine |
| Olanzapine | improves motor and psychiatric function | 9 patients; 11 patients; 1 patient; 2 patients | [81–84] | 5/10 | Beneficial effects were replicated across studies. |
Table 2
Cholinergic-based treatments which have demonstrated beneficial effects in pre-clinical HD studies. Strength of evidence criteria was based on proper study design, number of subjects improved, level of improvement, lack of side effects and replication by others
| Treatment | Beneficial effect(s) | Animal model | Number of animals studied | Reference(s) | Strength of | Comments |
| evidence | ||||||
| Donepezil | improves cognitive function | R6/2 transgenic mice | First study: n = 73 (across 4 groups); second study: n = 55 (across 4 groups) | [66] | 8/10 | Appropriate study design, appropriate mouse model, high number of mice used, results replicated in individual studies. |
| Galantamine | reduces neurological deficits, striatal lesions and apoptosis | 3-nitropropionic acid rat model | n = 46 (across 5 groups) | [69] | 4/10 | Appropriate study design although some groups with low numbers of mice, animal model used not ideal. |
| VU0467154 | improves motor and synaptic function | YAC128 transgenic mice | Motor function: n = 41 (across 4 groups); synaptic function: n = 43 (across 4 groups) | [85] | 6/10 | Appropriate study design, highly valid animal model used. |
| Physostigmine | increases hippocampal cell proliferation | Wistar rats | n = 30 (across 5 groups) | [79] | 4/10 | Animal model not HD-specific, relatively low numbers of animals. |
| Oxotremorine | increases hippocampal cell proliferation, spatial working memory and reverses deficits in hippocampal neurogenesis | Wistar rats | n = 30 (across 5 groups) | [79] | 5/10 | Multiple beneficial effects but animal model not HD-specific. |
| NGF | improves motor and cognitive function; increases cholinergic cell survival, expression of ChAT, VAChT and trkA markers, hippocampal neurogenesis; reduces excitotoxicity and cholinergic cell atrophy | R6/1 mice, YAC128 mice, Sprague-Dawley rats, Wistar rats | R6/1 mice: n = 48 (across 4 groups); YAC128 mice: n = 64 (across 4 groups); SD rats study 1: n = 29 (across 4 groups); SD rats study 2: n = 27 (across 4 groups); SD rats study 3: n = 37 (across 4 groups); SD rats study 4: n = 18 (across 3 groups) | [48, 87, 88, 90–95] | 9/10 | Appropriate study designs, valid animal models used, numerous beneficial effects reported, beneficial effects found by multiple research groups. |
| Wistar rats: n = 8 (across 2 groups) |


