
Induced Pluripotent Stem Cells in Huntington’s Disease Research: Progress and Opportunity
Abstract
Induced pluripotent stem cells (iPSCs) derived from controls and patients can act as a starting point for in vitro differentiation into human brain cells for discovery of novel targets and treatments for human disease without the same ethical limitations posed by embryonic stem cells. Numerous groups have successfully produced and characterized Huntington’s disease (HD) iPSCs with different CAG repeat lengths, including cells from patients with one or two HD alleles. HD iPSCs and the neural cell types derived from them recapitulate some disease phenotypes found in both human patients and animal models. Although these discoveries are encouraging, the use of iPSCs for cutting edge and reproducible research has been limited due to some of the inherent problems with cell lines and the technological differences in the way laboratories use them. The goal of this review is to summarize the current state of the HD iPSC field, and to highlight some of the issues that need to be addressed to maximize their potential as research tools.
INTRODUCTION
Since their development in 2006, induced pluripotent stem cells (iPSCs) have generated excitement due to their potential to produce appropriate cell models to facilitate discovery of novel targets and treatments for human disease [1]. iPSCs derived from controls and patients can act as a starting point for in vitro differentiation into human brain cells without the same ethical limitations posed by embryonic stem cells. The first human Huntington’s disease (HD) iPSCs with mutations of 54 CAG and 72 CAG repeats in the huntingtin gene were created from HD patient fibroblasts in 2008 by Park et al. using retroviral induction of four pluripotency factors: cMyc, Klf4, Oct4, and Sox2 [2]. Since then, numerous groups have successfully produced and characterized iPSCs with different CAG repeat lengths, including cells from patients with one or two HD alleles. HD iPSCs and the neural cell types derived from them recapitulate some disease phenotypes found in both human patients and animal models. Although these discoveries are encouraging, the use of iPSCs for cutting edge and reproducible research has been limited due to some of the inherent problems with cell lines and the technological differences in the way laboratories use them. The goal of this review is to summarize the current state of the HD iPSC field, and to highlight some of the issues that need to be addressed to maximize their potential as research tools (Fig. 1).
The current inventory of HD iPSCs
Early efforts at reprogramming adult cells to iPSCs relied upon lentiviral [3] or retroviral [1] delivery of cDNAs encoding pluripotency factors [4]. iPSCs have been successfully created from a variety of somatic cell types including fibroblasts [1, 3], blood cells [5, 6], renal epithelial cells [7], and keratinocytes [8, 9]. Several reviews discuss the history and recent advances in reprogramming methods used to produce human iPSCs [10–13]. At present, the best characterized HD iPSC lines have been produced from patient fibroblasts using lentivirus [14–16] or retrovirus [2, 17–28] to express a combination of pluripotency factors, including: Oct3/4, Klf-4, Sox2, c-Myc, SSEA4, LIN-28, NANOG, and p53 shRNA (to increase efficiency). However, the potential for off-target effects due to random viral insertions motivated scientists to develop novel non-integrating approaches for delivery including Sendai-virus [29], adenovirus [30] and episomal vectors [6, 31], as well as RNA transfection [32], protein [33], and small molecule [34, 35] based methods [4]. More recently, HD researchers have created and begun to characterize cells produced using non-integrating, episomal vectors for induction of pluripotency [36–42].
Numerous iPSC lines exist with CAG repeats in the range of wild-type to that of HD in the Huntingtin gene (HTT) (from 17 CAGs to 180 CAGs) [2, 14–28, 36–43]. Many of these cell lines were created by the HD iPSC Consortium and are available through the newly established NINDS Human Cell and Data Repository (NHCPR) including 8 unaffected and 18 HD iPCS lines (the catalog for cell lines can be viewed at the following website: https://stemcells.nindsgenetics.org/). The best described HD iPSC line called HD4 was produced by Park et al. (2008) and contains 72 CAGs [2, 17–20, 23–25, 28]. HD4 was used by the Ellerby laboratory to create two cell lines corrected at the HD locus to 21 CAGs by homozygous recombination [17]. HD4 was also used to create an HD allelic series with 21, 72 and 97 CAGs using gene editing employing the CRISPR/Cas9 system along with an antibody screen to confirm the presence of an expanded polyglutamine region in cell lines [44]. These cell lines together are very useful because they offer an isogenic background on which to delineate effects of the HD mutation. Although isogenic lines are a “gold standard” for a well-controlled iPSC experiment, the inherent variability that has been found among control iPSCs substantiates a need for more isogenic lines from additional HD iPSCs.
iPSCs derived from individuals with juvenile onset HD (>60 CAGs) have been used more frequently for genomic and proteomic studies than iPSCs from individuals with adult onset HD (39–60 CAG) [2, 14–28, 36–43]. However, only about 5% of HD patients have a juvenile onset (prior to age 20) form of the disease, with associated CAG repeat lengths greater than 60 [45–47]. Tables 1 & 2 show summaries of gene expression and protein changes based upon available data sets. The majority of reported work with iPSCs from those with adult onset HD has been with a single cell line derived by the HD iPSC Consortium in 2012 [15, 36]. This line has 60 CAG repeats which is at the high end of the CAG spectrum for adult onset HD [46, 47]. Additional cell lines with repeats ranging from 43–60 CAGs have been described, they are less well characterized [22, 26, 39, 40, 42, 43]. This is likely in part due to the greater ease of detection of mutant huntingtin protein with longer polyglutamine repeats using antibodies that preferentially recognize extended polyglutamine stretches, and the possibility that longer CAG expansions may give more robust changes, as they do in animal models. At present no iPSCs have been produced from patients bearing “intermediate” CAG repeat lengths (27–35 CAGs) or patients with 36–39 CAGs and reduced disease penetrance. Future work with iPSC derived neuronal cells could provide insight into the molecular changes underlying the behavioral changes identified in intermediate repeat length patients [48], and the differences in disease penetrance with low repeat lengths.
Search for phenotypes unveils numerous CAG repeat length dependent and independent properties
Several common phenotypes exist for neural stem cells (NSCs) (also called neural precursor cells (NPCs)) and neuronal cultures differentiated from adult and juvenile HD iPSCs. NSCs from both adult and juvenile-onset patients have decreased metabolic rate (respiratory capacity or ATP/ADP ratio) [15, 17, 19], reduced filamentous actin protein and cell-cell adhesion [15], as well as parallel changes in gene expression compared to NSCs from controls of genes related to nervous system development, cell assembly and organization [15]. HD neuronal cultures derived from both adult and juvenile-onset iPSCs likewise have increased levels of cell death, caspase 3/7 expression in response to BDNF withdrawal, and altered gene expression in pathways related to cell function/signaling and tissue development [15]. One caveat, however, is that differentiated neuronal cultures are by nature composed of a heterogeneous mixture of cell types and the cell death discovered in this study was not demonstrated to be specific to neurons.
Some unique differences were identified when NSCs from adult onset HD iPSCs were compared to those from juvenile onset HD (Tables 1 & 2). Pathway analysis of gene array data showed alterations in axonal guidance in NSCs from juvenile HD (72, 109 and 180 CAGs) that were not present in NSCs from those with adult onset (60 CAGs) [15, 19]. The axon guidance changes were further associated with altered netrin and netrin receptor expression in 72 CAG NSCs [19]. Analysis of gene array data also indicated the presence of alterations in calcium signaling in an HD NSC line with 60 CAGs, but not in ones with 109 or 180 CAGs [15]. These changes in gene expression may have functional relevance in corresponding iPSC induced neuronal cultures: increasing glutamate levels was associated with increased calcium dyshomeostasis and cell death in neuronal cultures with 60 CAGs but not in those with 180 CAGs [15]. As mentioned above, neuronal cultures were heterogeneous and these changes also were not demonstrated to be specific to neurons. But, altered calcium signaling after glutamate treatment was also observed in low repeat (37 and 51 CAGs) embryonic stem cell (ESC)-derived forebrain neuronal cultures [49]. Finally, Mattis et al. (2015) identified a Nestin-positive cell population sensitive to BDNF-withdrawal induced cell death that was present only in neuronal cultures from juvenile-onset cell lines (109 and 180 CAGs) but not adult onset (60 CAGs) [36].
Phenotypes that may be relevant to disease have been identified in naïve and differentiated HD iPSCs. These include conditions related to altered cell growth [18, 24], adhesion [15, 17], differentiation [18, 36], protein clearance (proteasomal, autophagic) [15, 16, 25, 40], survival [15, 17–19, 36, 38], oxidative stress/antioxidant response [18, 37, 41], metabolism [14, 15, 17, 19], growth factor signaling [15, 17, 20, 28, 36, 41] mitochondrial fragmentation [14], and electrophysiological properties [15]. Tables 3 & 4 list known phenotypes of HD cell lines. Conclusions about HD iPSC phenotypes have been drawn from a maximum of three HD cell lines compared to one or two control lines in a given publication [15, 16, 19, 36, 41, 42], with many groups using a single cell line or pair of cell lines compared to controls [14, 17, 18, 20–22, 24, 24–28, 37–40, 43] (see also Tables 1–4 for number of cell lines used in specific studies with their corresponding references). A major problem therefore in phenotype characterization has been the limited efforts to validate results in multiple iPSCs with similar CAG repeats or a range of CAG repeat lengths.
In future studies, one solution to this problem is to use multiple control and HD cell lines. The number of cell lines used should be determined using a power analysis based on data from individual phenotypes, just as in animal studies. Robust phenotypes may require testing far fewer iPSC lines compared to that necessary for more subtle phenotypes. We recommend comparing at least three control and three HD lines lines established from different individuals as an absolute minimum even for robust phenotypes. Subtle phenotypes may require as many as 10–12 cell lines per group and may require HD lines to have very similar CAG repeats. Ideally at least one genetically corrected iPSC paired with its isogenic HD iPSC should also be included, but two corrected clones are preferred to control for off target effects of genome editing. Alternatively, a huntingtin lowering strategy could be used (discussed later in “Maximizing the potential for research with HD iPSCs”). It would also be helpful if researchers currently growing other iPSCs could test published phenotypes in their cells and report conformational studies. Although these types of studies may not be novel and thus may seem of low importance, reproducible HD phenotypes in human cells validated across laboratories is of high value. Several of the current phenotypes have also been shown to occur in animal HD models, increasing confidence that they are real changes that may be important for HD; however, even for changes already shown in other HD models, demonstrating these changes in just one human HD iPSC line is of limited use.
Varied methods create mixed results: comparing apples to apples or apples to oranges?
Considerable variability exists in the culture methods used to reprogram somatic cells, and to derive NSCs and neurons from HD iPSCs. This limits the ability to draw broad conclusions from the existing published data. Though reprogramming techniques have been consistent: 17 of 24 publications HD cell lines were produced by retroviral or lentiviral reprogramming, the terms ‘NSC/NPC’ and ‘neuron’ were used to describe cell populations that varied in a multitude of ways including: protein markers of differentiation, growth factor dependence, and surface matrix [50–54]. The marked differences that exist in culture protocols can be explained to some extent by the ongoing progress made in the iPSC field to improve differentiation efficiency and specificity. Nonetheless, as with iPSCs from patients with other neurodegenerative diseases, the relative homogeneity of cell lines (i.e. few patient donors or few cell lines used during comparisons), when combined with variability in reprogramming and differentiation methods, may disguise subtle but important phenotypes, and make it challenging to determine which phenotypes are disease-relevant rather than due to culture conditions, clonal variation, or genetic background of the patient [55].
There are several protocols for differentiation of iPSCs favoring certain neuronal subtypes including medium spiny neurons (MSNs), the GABA-ergic projection neurons of the striatum that is particularly vulnerable in HD [15, 24, 36, 43, 56–64]. Table 5 summarizes salient features of MSN differentiation protocols. The source of MSNs in the developing human striatum is the lateral geniculate eminence (LGE) of the ventral telencephalon [65, 66]. LGE precursors are characterized by expression of several transcription factors including: GSX2, DLX2, ISL1, BCL11B(CTIP2), FOXP1, and FOXP2 [67]. Mature MSNs are characterized by the co-expression of several protein markers including: DARPP32, BCL11B, FOXP1 or FOXP2 [68, 69].
Driving the differentiation of MSNs from human pluripotent stem cells requires the manipulation of several signaling pathways, including Wnt/β-catenin, and Sonic Hedghog (Shh), and bone morphogenic protein (BMP) well described in a recent review by Fjodorova et al. (2015) [70]. The inconsistent and frequently limited efficiency of existing MSN differentiation protocols, ranging from (5–80% MSN yield, see Table 5) is in part due to the complexity of factors that combine to specify MSN fate. Differentiation of MSNs from iPSC is a lengthy process, with protocols ranging from 3–16 weeks. Production of MSNs from iPSCs requires the derivation of LGE-like neural precursor cells, rather than those approximating the medial or caudal geniculate eminence, the maturation of the neurons rather than maintenance of multipotent neural precursor cells or development of glia, and finally the designation of MSNs rather than the olfactory bulb interneurons [65].
The general pattern used in the currently available MSN differentiation protocols includes neural induction using stromal cell co-culture [24, 56], exposure to growth factors to induce embryoid body/neural rosette formation [15, 36, 43, 58, 60, 71], or dual-SMAD/BMP inhibition [62]. Neural induction is followed by specification of LGE precuror/MSN fate using a combination of factors including Shh and DKK1 (a WNT-inhibitor) [15, 36, 56–58, 71, 72], or more recently Activin A, a TGFβ signaling protein [62]. Alternatively, a recent protocol describes the efficient direct induction of adult human fibroblasts to DARPP32+ neurons using lentiviral infection with miRNAs: MiR-9/9* and miR-124 as described by Yoo et al. [73] to encourage neuronal differentiation, and expression transcription factors: CTIP2(BCL11B), DLX1, DLX2, and MYT1L (CDM) to specifically encourage MSN fate [61] (Table 5).
MSN differentiation protocols have largely been described in control cell lines [57–64], with only a few attempted using HD iPSC lines [14, 15, 27, 28, 37]. Further study is necessary to understand how individual cell lines, HD or control, will respond to each differentiation protocol.
Drug discovery and the use of stress induced phenotypes
Drug screens using animal cell models of HD that express endogenous mutant huntingtin (knock-in) or overexpress mutant huntingtin exogenously, as well as those using human cells overexpressing huntingtin, have failed to identify a compound that prevents, slows or reverses disease onset. A well-characterized human neuronal cell model with huntingtin expression from the endogenous gene locus could be a valuable asset in developing a therapeutic. Numerous screens have already been performed using HD iPSC derived cells [14, 19, 20, 25–27, 36–39, 43]. Table 6 describes initial efforts using HD iPSCs and derived neuronal cells as models for drug screening. Interestingly, in contrast with the analysis of gene/protein changes and characterization of disease relevant phenotypes which were mostly performed in NSCs bearing juvenile repeat lengths (>60 CAGs), experimental screens have largely focused on neuronal cultures from HD iPSCs, and have included low repeat cell lines (43–47 CAGs) [14, 19, 20, 25–27, 36–39, 43]. However for each study, testing is often limited to one or two HD iPSC lines, often without a control or corrected cell line for comparison.
Most phenotypic readouts used to test the effectiveness of novel therapies in HD iPSC derived neuronal cells include a stressor such as growth factor withdrawal, oxidative stress, DNA damage, or glutamate toxicity [14, 19, 20, 25–27, 36–39, 43] (Table 6). The effect of a drug on cell response to cytokine treatment [26], induced DNA breakage [37], proteasome inhibition [25], H2O2 or Mn2 + treatment [37, 43] and growth factor withdrawal [27, 36, 38, 39], have been investigated. The TNFα inhibitor XPro-1595 lowered cytokine (TNFα and ILβ) induced apoptosis activation and iNOS levels in astrocytes and neurons, respectively, differentiated from iPSCs with 43 CAGs [26]. Adenosine receptor 2A agonists CGS-21680 and APEC produced a dose dependent reduction of oxidative stress toxicity induced by exposure to H2O2 in 43 CAG neuronal cultures, as measured by decreased γH2AX induction and caspase3 cleavage [43]. The microRNA 196a (miR196a) was identified as a candidate that might impact HD using microarray data from transgenic HD monkey tissue; miR196a was found to decrease MG-132 induced EM-48 positive huntingtin aggregates in neuronal cultures differentiated from HD iPSCs with 72 CAGs through an unknown mechanism [25]. An ATM (ataxia-telangiectasia mutated) protein inhibitor KU55933 reversed both neocarzinostatin (a DNA damaging agent) induced increases in phosphorylation of p53, CHK2 and γH2AX, and Mn2 + decreases in p53 phosphorylation in 70 and 180 CAG “striatal-like” NPCs [37].
Several groups have tested drug effects of growth factor (BDNF or FGF/LIF) withdrawal on cell survival [19, 27, 36, 38, 39]. Although cells were grown in medium containing essential nutrients, acute withdrawal of neurotrophins might also be considered a stress once cells have become dependent upon them. Lu et al. (2014) showed that an alternate ATM protein inhibitor, KU60019, reduced BDNF withdrawal-induced increases in TUNEL-positive nuclei and caspase 3 activation in 109 and 180 CAG mixed neuronal cell cultures [38]. Furthermore, to elucidate mechanisms of BDNF-withdrawal induced cell death in neuronal cultures differentiated from HD iPSCs with 109 and 180 CAG, Mattis et al. (2015) showed reversal of increased TUNEL+nuclei after BDNF withdrawal using several compounds including a calcium chelator (BAPTA), TRKB antibody agonist (αTrkB), MAPK signaling inhibitor (SB2390463), NMDAR antagonist (memantine) and AMPA/Kainate antagonist (CNQX), and allele specific antisense-oligonucleotides of mutant huntingtin (ASO1, ASO2, ASO3) [36]. Importantly, the susceptible population in the neuronal culture was Nestin+ radial glia-like cells, not neurons.
Likewise, Yao et al. (2015) explored the potential role of G-protein coupled receptor 52 (GPR52) in mediating mutant huntingtin protein toxicity. They found that siRNA knockdown of GPR52 both reduced mutant huntingtin protein levels, and BDNF withdrawal induced caspase 3 activation in HD iPSC derived neurons with 47 and 70 CAG repeats [27]. In addition, Dickey et al. (2016) described the ability of a PPARδ activator (KD3010) to reduce numbers of condensed nuclei after BDNF withdrawal from 60 CAG derived neuronal cultures [39]. Finally, Ring et al. (2015) assessed the ability of recombinant TGFβ1/TGFβ2 and netrin-1 to reverse increases in caspase3/7 activity and decreases in maximum respiratory capacity that occur after FGF/LIF withdrawal in NSCs with 72 CAGs; TGFβ1/TGFβ2 treatment was likewise found to be beneficial to HD NSCs under unstressed conditions [19].
Aside from the TGFβ experiment by Ring et al. [19], only two other groups have tested the effects of a compound on iPSC phenotypes without the addition of a stressor. Charbord et al. (2013) demonstrated that treatment of 72 CAG NSCs with a Repressor element-1 silencing transcription factor (REST) inhibitor X5050 produced increased RE1 gene expression and reduced REST activity when compared to control ESC derived NSCs [20]. Guo et al. (2013) showed that a Drp1-selective peptide inhibitor, P110-TAT, reversed: neurite shortening, decreases in mitochondrial membrane potential and ATP level, increases in cell death, mitochondrial fragmentation, and mitochondrial reactive oxygen species (ROS) in mixed neuronal cultures derived from iPSCs derived from juvenile HD patient fibroblasts of unreported CAG length (GM05539, Coriell Cell Repository) [14].
In other disease fields, additional phenotypes have been identified using alternative methods of aging iPSC derived neurons well described in a recent review by Studer et al. (2015) [74]. Recently, exogenous expression of progerin, a mutated form of the nuclear envelope protein lamin A associated with disorder of premature aging, has been used to mimic aging [75]. Overexpression of progerin in iPSC derived dopaminergic neurons resulted in elevated levels of reactive oxygen species (ROS) and DNA damage consistent with the neuronal aging [75]. The same experimental procedure was also used in iPSC derived dopaminergic neurons from Parkinson disease (PD) patients bearing PINK1 and Parkin mutations, and expression of progerin unveiled a decrease in dendrite length specific to PD patient neurons, not identified in progerin-negative cells [75]. The introduction of progerin, along with continued work to understand the molecular mechanisms underlying normal aging could lead to an improved understanding of HD neurodegeneration and future cell models for drug screening. Care should be used with interpretation of results identified using progerin overexpression, however, since cells will be burdened with two mutant proteins, one of which is overexpressed. Furthermore, iPSCs from patients with Hutchinson-Gilford progeria syndrome (HGPS) and atypical Werner syndrome (AWS) which contain mutations in their endogenous LMA gene, have dysmorphic nuclei and premature senescence [76].
Phenotypes relating to disease mechanism
It is possible that aging and stress might compound or enhance secondary effects of mutant huntingtin but not affect primary drivers of HD progression. If the underlying primary problem causing disease in humans HD is not related to stress or aging, then screening for compounds that reverse stress induced cell death or age related morphology changes may be insufficient to identify novel therapeutics for HD that target crucial changes. Assessing the ability of a given therapeutic to reverse additional alterations such as cell morphology, cell signaling activity or gene expression in the absence of stress may allow for more confident conclusions about drug efficacy [77]. Additional phenotypes useful for screening could include any of the number of disease relevant phenotypes already identified in HD iPSC derived NSC/NPCs and neurons, though further work is necessary to determine the extent to which these phenotypes can be generalized using HD iPSC lines derived from a larger patient population (Tables 3 & 4).
Utilizing HD iPSCs as a model for early CNS development in HD patients, and determining phenotypes that precede neuronal death may be an equally important and a more readily attainable goal. Study of rodent and human cell models indicate the influence of mutant huntingtin on neural development in general, and specifically related to: premature onset of neural differentiation [78], altered Notch signaling [78] and aberrant mitotic spindle orientation [79, 80]. Recent work has indicated alterations to HD patient brains years before symptom onset, including: basal ganglia cell death [81], altered striatal/cortical volume and morphology [81, 82–84], cortico-striatal connectivity [85, 86], decreased cortical inhibition [87], and increased oligodendrocyte number [88, 89]. Early cognitive changes have likewise been identified in HD patients related to altered cortico-striatal circuitry [90]. Behavioral changes and altered synaptic connectivity, preceding the onset of motor symptoms, have also been identified in transgenic mouse models of HD [91–94].These findings have encouraged the hypothesis that the huntingtin mutation may alter molecular pathways, cell phenotypes and activity during neural development, and the cumulative effects of these changes over time eventually lead to disease onset well described in several reviews: [50, 88, 95, 96].
Genomic and proteomic analyses of HD iPSC and derived neuronal cells support the importance of continued exploration of the role of mutant huntingtin in disrupting the normal differentiation and maturation of human cells. Differentiation of HD iPSCs to NSCs and neurons has uncovered differences in the efficiency [18] and composition of cultures [36], when compared to iPSCs from healthy controls. Furthermore, comparisons of gene microarray and RNAseq data between HD4 (72 CAGs) HD iPSCs and a cell line corrected to 21 CAGs identified axonal guidance and cadherin, TGF β and SMAD3 signaling [17, 19] as pathways significantly altered in HD cells. Altered expression of genes specifically related to striatal development were also identified in the same cells [19].
Other brain cell types may contribute to HD pathology. Changes to astrocytes, microglia, and oligodendrocytes have been observed in both HD patients [89, 97–101], and transgenic animal models of HD [101–108]. Several protocols have been shown to produce astrocytes [109–114] and oligodendrocytes [115–118] from human iPSCs (45). Two groups have used HD iPSCs to produce GFAP+astrocytes [26, 40] (Tables 3 & 6). Juopperi et al. (2012) reported a CAG-repeat dependent increase in cytoplasmic vacuolization in HD astrocytes derived from 50 CAG and 109 CAG iPSC lines, and related the change to a potential alteration in autophagy as overnight treatment with chloroquine, an autophagy inhibitor, increased LC3+ positive vacuoles in HD cells [40]. Hsiao et al. (2014) showed that a TNF-α inhibitors, XPro-1595, lowers cytokine (TNF-α & IL-β) induced iNOS production in astrocytes derived from iPSCs with 43 CAGs [26]. So far, no work has been published describing HD iPSC derived oligodendrocytes. Further exploration of HD iPSC derived glia, along with co-culture of iPSC derived neurons and glia, may shed light on sources of non-cell autonomous toxicity in HD pathogenesis.
Technical caveats that may undermine phenotype characterization
Despite their advantages, unanswered questions about iPSC as a model remain. One area of particular interest for investigating age-related diseases such as HD is obliteration of age from the patient sample. Several studies indicate that induced pluripotency reverses typical age-related phenotypes, such as telomere shortening [119–121] and mitochondrial dysfunction [74, 121, 122]. A total reversal of age related changes such as alterations in levels of oxidative stress, DNA packaging and damage, nuclear morphology and related gene expression changes occurred in fibroblasts differentiated from iPSCs that had been collected from donor patients in different age ranges [74]. Transdifferentiation or directed differentiation to neurons from aged fibroblasts may be one approach to maintaining age [4, 61, 77]. Another issue is the potential for reprogrammed cells to bear an ‘epigenetic memory’ of their somatic cell source [4, 74, 123–125]. A recent study by Kim et al. (2011) indicated that iPSCs retain some residual DNA methylation from their parental cell type (fibroblast or blood cell), rending them more readily differentiated to their parent cell’s fate [124]. However, recent work also suggests that repeated passaging [126] or chromatin modifying agents [127] can diminish or fully reset this ‘epigenetic memory’ [124].
The extent to which the phenomenon of epigenetic memory and limited pluripotency may affect neuronal cells derived from of HD iPSCs is as yet, uncertain. Reprogramming, continuous passage (cell dissociation and re-culturing) to high passage number, and differentiation of iPSCs have been shown to lead to genetic instability [128–132]. Investigators using iPSCs are thus cautioned when using highly passage cells. Encouraging results showed few changes in human iPSCs due to mutant huntingtin. The HD iPSC Consortium (2013) reported an expansion from 110 to 118 CAGs in one HD iPSC derived NSC line, but no changes in iPSC karyotype [15]. Camnasio et al. (2012) reported occasional changes in karyotype in both control and HD iPSCs but stable CAG repeat lengths [16]. This is in contrast to NSCs established from embryonic brain tissue from homozygous CAG140 knock-in mice which became tetraploid after very few passages in culture [133]. However, a recent finding by Tidball et al. (2016) showed increased somatic instability in HD iPSCs using siRNA targeting p53 to increase the efficiency of reprogramming [42]. Further study is necessary to determine the extent to which additional changes occur in human HD iPSC lines and differentiated cells, but so far the CAG repeat expansion and presence of mutant huntingtin protein do not seem to impose genetic instability.
While progress towards understanding HD mechanisms using iPSC has been unquestionably substantial, there are challenges in drawing broad conclusions about disease pathogenesis from these studies. Limitations on the number of HD and control cell lines used are understandable considering the cost and effort in maintaining human stem cells. However, given the relatively small number of cell lines studied, care should be taken when interpreting results. Although HD is caused by just one gene, the patient donors have diverse genetic backgrounds that could account for many of the changes observed. As with any experiment, sample size increases confidence in results. Efforts should be supported for additional testing of existing cell lines and for continued creation of new cell lines to overcome this limitation. Furthermore, determining the presence of certain phenotypes and susceptibility to specific treatments one day may be possible using iPSC derived neurons allowing for tailored treatments (so called “personalized medicine”) [134]. The availability of cell lines from diverse genetic backgrounds is critical for these efforts.
Maximizing the potential for research with HD iPSCs
Genetic correction to create non-disease CAG repeat lengths in iPSCs with the same genetic background as diseased cells in theory removes at least one confounding variable thereby diminishing barriers to identifying less subtle phenotypes. An et al. (2012) reported the successful use of homologous recombination to correct the 72 CAG allele of an HD4 iPSC line to 21 CAG [17]. Recent advances in gene editing technology should make genetic correction of iPSC easier [50–52, 77, 135–138]. The potential for zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENS), and clustered regularly-interspaced short palindromic repeats/Cas 9 (CRISPR/Cas9) to modify human stem cells has been well described [4, 55, 138]. Each method bears limitations in the targeting of guiding DNA/RNA sequences, as well as differing propensities for off-target effects [55, 138], although off target effects are reduced in pluripotent stem cells in contrast to other cells [139]. Current methods of gene editing rely on endogenous machinery to repair DNA breaks produced by an exogenous nuclease (i.e., CRISPR or Fok1) [4, 135, 138]. Several recent findings indicate that the molecular pathways underlying DNA repair may be altered or aberrantly active in HD iPSCs [18, 37, 38, 42, 43], consistent with somatic CAG repeat expansions in post-mortem brain tissue from HD patients, and oxidative DNA damage in HD animal models [140]. These existing changes in HD cells could have adverse effects on the efficiency of genetic corrections, and susceptibility of cells to off-target effects. There are ongoing efforts to target single nucleotide polymorphisms (SNPs) specific to the mutated HD allele and to contract the expanded CAG repeat in heterozygous cell lines [53] in order to create a series of iPSC lines with different CAG expansions lengths on the same genetic background to determine CAG repeat length dependent phenotypes. An HD allelic series 21, 72 and 97 CAGs, was successfully created by An et al., (2014) using the CRISPR/Cas9 gene editing system [44]. One practical problem is that unfortunately many of the existing iPSC lines are not heterozygous for SNPs that could be targeted by CRISPR/Cas9 making allele-specific changes difficult in these lines (our unpublished observations).
Just as an ‘epigenetic memory’ of the somatic cell source may persist in iPSCs after reprogramming, it is as yet unknown if contracting expanded CAG repeats in HD iPSCs will fully reverse the effects of the HD mutation on derived neuronal cells. Using a genetic correction as the control will obscure phenotypes that cannot be reversed since both the mutant and the isogenic corrected line will continue to share the condition; thus they will not be observed as “different”. Analysis of gene and protein expression and epigenetic modifications, comparing both HD iPSC line and iPSC lines derived from healthy patient controls is necessary to understand the extent to which genetic correction in HD iPSC lines can reverse HD phenotypes; it is possible that some phenotypes can be reversed in one cell line with a particular genetic background, but not in another ([for in-depth coverage of this topic please see [141]). For HD, only one iPSC (HD4) has been subjected to correction. Correction by homologous recombination of the iPSC line HD4 from 72 CAGs to 21 CAGs reversed phenotypes of altered TGF-beta and cadherin gene expression [17] and changes to gene ontology categories including synaptic assembly, axonal guidance and SMAD3 signaling [19]. Far fewer changes were found between HD4 and the corrected line, versus HD4 and an unrelated control iPSC line, supporting the notion that an isogenic background is a good control. Studies comparing iPSC derived neurons from healthy controls to Parkinson patients bearing mutations in LRRK2 gene demonstrated that gene expression cluster profiles did not partition with the mutation, indicating the normal genomic variability had a stronger effect than the mutation [142]; meaningful changes were only found using an isogenic corrected control. Additional studies comparing phenotypes of HD4 and the corrected cell line with numerous control cell lines will be informative. These issues are particularly important in considering the potential of genetically corrected HD iPSCs as a tissue source for cell replacement therapy [45, 52, 53, 123].
As an alternative to genetic editing, levels of mutant protein can be lowered using methods that specifically target mutant huntingtin RNA stability and protein expression including: anti-sense oligonucleotides (ASOs), small interfering RNA (siRNA), short hairpin RNA (shRNA) [53], and microRNAs (miRs). Mattis et al. 2015 reported the ability of allele-specific ASOs to lower levels of mutant huntingtin expression and reverse BDNF-withdrawal induced toxicity in 109 and 180 CAG HD iPSC derived neurons [36]. As mentioned above, many existing iPSC lines are homozygous at SNPs that could be used for allele-specific targeting by known useful siRNAs, shRNAs, and miRs making allele specific knockdown of mutant huntingtin difficult or impossible using these methods. Zinc finger proteins (ZFPs) are also being developed (by Sangamo) to repress transcription of the HTT allele bearing expanded CAGs and thus specifically reduced mutant huntingtin protein levels.
CONCLUSIONS
Significant progress has been made in the HD iPSC field. Now that numerous HD iPSCs are available to researchers, individual studies should be performed using as many cell lines as appropriate based on a power analysis for individual phenotypes. The limited number of studies on iPSCs and their differentiated progeny using lines with adult onset CAG lengths is concerning since adult onset HD represents the vast majority of HD cases. Work with HD embryonic stem (ES) cells has focused on lower repeat lengths (37–51 CAG) [49, 143–145]. However, use of HD ES cells abrogates the ability to correlate cellular phenotypes with patient symptoms, a major effort underway in other neurodegenerative diseases using iPSC models [134]. Renewed efforts should be undertaken to continue to increase the repertoire of available stem cell lines as existing lines age with passage. Further investigations should be directed toward identifying phenotypes in HD iPSCs bearing an expanded allele in the adult onset range and investigating changes that occur in the absence of exogenous stress. New methods using 3-dimensional culture or co-culture systems may be key to unveiling phenotypes in the absence of stress. Finally, it would be helpful if the field as a whole would settle on one or two differentiation schemes to improve comparison among studies. Thanks to the pioneering work of numerous HD investigators, HD iPSCs are just beginning to show their promise and may be the key to finally identifying treatments useful in HD patients.
CONFLICT OF INTERESTS
The authors have no conflicts to declare.
ACKNOWLEDGMENTS
We thank Marian DiFiglia for invaluable help editing the manuscript. KKG is funded by CHDI.
REFERENCES
[1] | Takahashi K , Yamanaka S . Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. (2006) ;126: :663–76. doi: 10.1016/j.cell.2006.07.024 |
[2] | Park I-H , Arora N , Huo H , Maherali N , Ahfeldt T , Shimamura A , et al. Disease-specific induced pluripotent stem (iPS) cells. Cell. (2008) ;134: :877–86. doi: 10.1016/j.cell.2008.07.041 |
[3] | Yu J , Vodyanik MA , Smuga-Otto K , Antosiewicz-Bourget J , Frane JL , Tian S , et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. (2007) ;318: :1917–20. doi: 10.1126/science.1151526 |
[4] | Qiang L , Fujita R , Abeliovich A . Remodeling neurodegeneration: Somatic cell reprogramming-based models of adult neurological disorders. Neuron. (2013) ;78: :957–69. doi: 10.1016/j.neuron.2013.06.002 |
[5] | Loh Y-H , Agarwal S , Park I-H , Urbach A , Huo H , Heffner GC , et al. Generation of induced pluripotent stem cells from human blood. Blood. (2009) ;113: :5476–9. doi: 10.1182/blood-2009-02-204800 |
[6] | Chou B-K , Mali P , Huang X , Ye Z , Dowey SN , Resar LM , et al. Efficient human iPS cell derivation by a non-integrating plasmid from blood cells with unique epigenetic and gene expression signatures. Cell Res. (2011) ;21: :518–29. doi: 10.1038/cr.2011.12 |
[7] | Aasen T , Raya A , Barrero MJ , Garreta E , Consiglio A , Gonzalez F , et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat Biotechnol. (2008) ;26: :1276–84. doi: 10.1038/nbt.1503 |
[8] | Raab S , Klingenstein M , Liebau S , Linta L . A comparative view on human somatic cell sources for iPSC generation. Stem Cells Int ((2014) )768391. doi: 10.1155/2014/768391 |
[9] | Zhou T , Benda C , Duzinger S , Huang Y , Li X , Li Y , et al. Generation of induced pluripotent stem cells from urine. J Am Soc Nephrol JASN. (2011) ;22: :1221–8. doi: 10.1681/ASN.2011010106 |
[10] | Schlaeger TM , Daheron L , Brickler TR , Entwisle S , Chan K , Cianci A , et al. A comparison of non-integrating reprogramming methods. Nat Biotechnol. (2015) ;33: :58–63. doi: 10.1038/nbt.3070 |
[11] | González F , Boué S , Izpisúa Belmonte JC . Methods for making induced pluripotent stem cells: Reprogramming á la carte. Nat Rev Genet. (2011) ;12: :231–42. doi: 10.1038/nrg2937 |
[12] | Hu K . All roads lead to induced pluripotent stem cells: The technologies of iPSC generation. Stem Cells Dev. (2014) ;23: :1285–300. doi: XXXXX |
[13] | Brouwer M , Zhou H , Kasri NN . Choices for Induction of Pluripotency: Recent Developments in Human Induced Pluripotent Stem Cell Reprogramming Strategies. Stem Cell Rev Re. (2015) ;12: :54–72. doi: 10.1007/s12015-015-9622-8 |
[14] | Guo X , Disatnik M-H , Monbureau M , Shamloo M , Mochly-Rosen D , Qi X . Inhibition of mitochondrial fragmentation diminishes Huntington’s disease-associated neurodegeneration. J Clin Invest. (2013) ;123: :5371–88. doi: 10.1172/JCI70911 |
[15] | The HD iPSC Consortium. Induced pluripotent stem cells from patients with Huntington’s disease show CAG-repeat-expansion-associated phenotypes. Cell Stem Cell. (2012) ;11: :264–78. doi: 10.1016/j.stem.2012.04.027 |
[16] | Camnasio S , Carri AD , Lombardo A , Grad I , Mariotti C , Castucci A , et al. The first reported generation of several induced pluripotent stem cell lines from homozygous and heterozygous Huntington’s disease patients demonstrates mutation related enhanced lysosomal activity. Neurobiol Dis. (2012) ;46: :41–51. doi: 10.1016/j.nbd.2011.12.042 |
[17] | An MC , Zhang N , Scott G , Montoro D , Wittkop T , Mooney S , et al. Genetic correction of Huntington’s disease phenotypes in induced pluripotent stem cells. Cell Stem Cell. (2012) ;11: :253–63. doi: 10.1016/j.stem.2012.04.026 |
[18] | Chae J , Kim D , Lee N , Jeon Y , Jeon I , Kwon J , et al. Quantitative proteomic analysis of induced pluripotent stem cells derived from a human Huntington’s disease patient. Biochem J. (2012) ;446: :359–71. doi: 10.1042/BJ20111495 |
[19] | Ring KL , An MC , Zhang N , O’Brien RN , Ramos EM , Gao F , et al. Genomic analysis reveals disruption of striatal neuronal development and therapeutic targets in human Huntington’s disease neural stem cells. Stem Cell Re. (2015) ;5: :1023–38. doi: 10.1016/j.stemcr.2015.11.005 |
[20] | Charbord J , Poydenot P , Bonnefond C , Feyeux M , Casagrande F , Brinon B , et al. High throughput screening for inhibitors of REST in neural derivatives of human embryonic stem cells reveals a chemical compound that promotes expression of neuronal genes. Stem Cells Dayt Ohio. (2013) ;31: :1816–28. doi: 10.1002/stem.1430 |
[21] | Ooi J , Hayden MR , Pouladi MA . Inhibition of excessive monoamine oxidase A/B activity protects against stress-induced neuronal death in Huntington disease. Mol Neurobiol. (2015) ;52: :1850–61. doi: 10.1007/s12035014-8974-4 |
[22] | Martín-Flores N , Romaní-Aumedes J , Rué L , Canal M , Sanders P , Straccia M , et al. RTP801 is involved in mutant huntingtin-induced cell death. Mol Neurobiol ((2015) )1–12. doi: 10.1007/s12035-015-9166-6 |
[23] | Jeon I , Choi C , Lee N , Im W , Kim M , Oh S-H , et al. In vivo roles of a patient-derived induced pluripotent stem cell line (HD72-iPSC) in the YAC128 model of Huntington’s disease. Int J Stem Cells. (2014) ;7: :43–7. doi: 10.15283/ijsc.2014.7.1.43 |
[24] | Jeon I , Lee N , Li J-Y , Park I-H , Park KS , Moon J , et al. Neuronal Properties, in vivo effects, and pathology of a Huntington’s disease patient-derived induced pluripotent stem cells. Stem Cells. (2012) ;30: :2054–62. doi: 10.1002/stem.1135 |
[25] | Cheng P-H , Li C-L , Chang Y-F , Tsai S-J , Lai Y-Y , Chan AWS , et al. miR-196a ameliorates phenotypes of Huntington disease in cell, transgenic mouse, and induced pluripotent stem cell models. Am J Hum Genet. (2013) ;93: :306–12. doi: 10.1016/j.ajhg.2013.05.025 |
[26] | Hsiao H-Y , Chiu F-L , Chen C-M , Wu Y-R , Chen H-M , Chen Y-C , et al. Inhibition of soluble tumor necrosis factor is therapeutic in Huntington’s disease. Hum Mol Genet. (2014) :ddu151. doi: 10.1093/hmg/ddu151 |
[27] | Yao Y , Cui X , Al-Ramahi I , Sun X , Li B , Hou J , et al. A striatal-enriched intronic GPCR modulates huntingtin levels and toxicity. eLife. (2015) ;4. doi: 10.7554/eLife.05449 |
[28] | Zhang N , An MC , Montoro D , Ellerby LM . Characterization of human Huntington’s disease cell model from induced pluripotent stem cells. PLoS Curr. (2010) ;2: :RRN1193. doi: 10.1371/currents.RRN1193 |
[29] | Ban H , Nishishita N , Fusaki N , Tabata T , Saeki K , Shikamura M , et al. Efficient generation of transgene-free human induced pluripotent stem cells (iPSCs) by temperature-sensitive Sendai virus vectors. Proc Natl Acad Sci U S A. (2011) ;108: :14234–9. doi: 10.1073/pnas.1103509108 |
[30] | Stadtfeld M , Nagaya M , Utikal J , Weir G , Hochedlinger K . Induced pluripotent stem cells generated without viral integration. Science. (2008) ;322: :945–9. doi: 10.1126/science.1162494 |
[31] | Okita K , Matsumura Y , Sato Y , Okada A , Morizane A , Okamoto S , et al. A more efficient method to generate integration-free human iPS cells. Nat Methods. (2011) ;8: :409–12. doi: 10.1038/nmeth.1591 |
[32] | Warren L , Manos PD , Ahfeldt T , Loh Y-H , Li H , Lau F , et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell. (2010) ;7: :618–30. doi: 10.1016/j.stem.2010.08.012 |
[33] | Kim D , Kim C-H , Moon J-I , Chung Y-G , Chang M-Y , Han B-S , et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell. (2009) ;4: :472–6. doi: 10.1016/j.stem.2009.05.005 |
[34] | Hou P , Li Y , Zhang X , Liu C , Guan J , Li H , et al. Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science. (2013) ;341: :651–4. doi: 10.1126/science.1239278 |
[35] | Yu J , Chau KF , Vodyanik MA , Jiang J , Jiang Y . Efficient feeder-free episomal reprogramming with small molecules. PloS One. (2011) ;6: :e17557. doi: 10.1371/journal.pone.0017557 |
[36] | Mattis VB , Tom C , Akimov S , Saeedian J , Østergaard ME , Southwell AL , et al. HD iPSC-derived neural progenitors accumulate in culture and are susceptible to BDNF withdrawal due to glutamate toxicity. Hum Mol Genet. (2015) ;24: :3257–71. doi: 10.1093/hmg/ddv080 |
[37] | Tidball AM , Bryan MR , Uhouse MA , Kumar KK , Aboud AA , Feist JE , et al. A novel manganese-dependent ATM-p53 signaling pathway is selectively impaired in patient-based neuroprogenitor and murine striatal models of Huntington’s disease. Hum Mol Genet. (2014) :ddu609. doi: 10.1093/hmg/ddu609 |
[38] | Lu X-H , Mattis VB , Wang N , Al-Ramahi I , van den Berg N , Fratantoni SA , et al. Targeting ATM ameliorates mutant Huntingtin toxicity in cell and animal models of Huntington’s disease. Sci Transl Med. (2014) ;6: :268ra178–268ra178. doi: 10.1126/scitranslmed.3010523 |
[39] | Dickey AS , Pineda VV , Tsunemi T , Liu PP , Miranda HC , Gilmore-Hall SK , et al. PPAR-δ is repressed in Huntington’s disease, is required for normal neuronal function and can be targeted therapeutically. Nat Med. (2016) ;22: :37–45. doi: 10.1038/nm.4003 |
[40] | Juopperi TA , Kim WR , Chiang C-H , Yu H , Margolis RL , Ross CA , et al. Astrocytes generated from patient induced pluripotent stem cells recapitulate features of Huntington’s disease patient cells. Mol Brain. (2012) ;5: :17. doi: 10.1186/1756-6606-5-17 |
[41] | Szlachcic WJ , Switonski PM , Krzyzosiak WJ , Figlerowicz M , Figiel M . Huntington disease iPSCs show early molecular changes in intracellular signaling, the expression of oxidative stress proteins and the p53 pathway. Dis Model Mech. (2015) ;8: :1047–57. doi: 10.1242/dmm.019406 |
[42] | Tidball AM , Neely MD , Chamberlin R , Aboud AA , Kumar KK , Han B , et al. Genomic instability associated with p53 knockdown in the generation of Huntington’s disease human induced pluripotent stem cells. PloS One. (2016) ;11: :e0150372. doi: 10.1371/journal.pone.0150372 |
[43] | Chiu F-L , Lin J-T , Chuang C-Y , Chien T , Chen C-M , Chen K-H , et al. Elucidating the role of the A2A adenosine receptor in neurodegeneration using neurons derived from Huntington’s disease iPSCs. Hum Mol Genet. (2015) ;24: :6066–79. doi: 10.1093/hmg/ddv318 |
[44] | An MC , O’Brien RN , Zhang N , Patra BN , De La Cruz M , Ray A , et al. Polyglutamine disease modeling: Epitope based screen for homologous recombination using CRISPR/Cas9 system. PLoS Curr. (2014) ;6. doi: 10.1371/currents.hd.0242d2e7ad72225efa72f6964589369a |
[45] | Fink KD , Deng P , Torrest A , Stewart H , Pollock K , Gruenloh W , et al. Developing stem cell therapies for juvenile and adult-onset Huntington’s disease. Regen Med. (2015) ;10: :623–46. doi: 10.2217/rme.15.25 |
[46] | Douglas I , Evans S , Rawlins MD , Smeeth L , Tabrizi SJ , Wexler NS . Juvenile Huntington’s disease: A population-based study using the General Practice Research Database. BMJ Open. (2013) ;3: :e002085. doi: 10.1136/bmjopen-2012-002085 |
[47] | Quarrell O , O’Donovan KL , Bandmann O , Strong M . The prevalence of juvenile Huntington’s disease: A review of the literature and meta-analysis. PLoS Curr. (2012) ;4. doi: 10.1371/4f8606b742ef3 |
[48] | Killoran A , Biglan KM , Jankovic J , Eberly S , Kayson E , Oakes D , et al. Characterization of the Huntington intermediate CAG repeat expansion phenotype in PHAROS. Neurology. (2013) ;80: :2022–>7. doi: 10.1212/WNL.0b013e318294b304 |
[49] | Niclis JC , Pinar A , Haynes JM , Alsanie W , Jenny R , Dottori M , et al. Characterization of forebrain neurons derived from late-onset Huntington’s disease human embryonic stem cell lines. Front Cell Neurosci. (2013) ;7. doi: 10.3389/fncel.2013.00037 |
[50] | Zhang N , Bailus BJ , Ring KL , Ellerby LM . iPSC-based drug screening for Huntington’s disease. Brain Res. (2015) . doi: 10.1016/j.brainres.2015.09.020 |
[51] | Mattis VB , Svendsen CN . Modeling Huntington’s disease with patient-derived neurons. Brain Res (2015) . doi: 10.1016/j.brainres.2015.10.001 |
[52] | Liu L , Huang J-S , Han C , Zhang G-X , Xu X-Y , Shen Y , et al. Induced pluripotent stem cells in Huntington’s disease: Disease modeling and the potential for cell-based therapy. Mol Neurobiol. (2015) :1–11. doi: 10.1007/s12035-015-9601-8 |
[53] | Golas MM , Sander B . Use of human stem cells in Huntington disease modeling and translational research. Exp Neurol. (2016) . doi: 10.1016/j.expneurol.2016.01.021 |
[54] | Kaye JA , Finkbeiner S . Modeling Huntington’s disease with induced pluripotent stem cells. Mol Cell Neurosci. (2013) ;56: :50–64. doi: 10.1016/j.mcn.2013.02.005 |
[55] | Hendriks WT , Warren CR , Cowan CA . Genome editing in human pluripotent stem cells: Approaches, pitfalls, and solutions. Cell Stem Cell. (2016) ;18: :53–65. doi: 10.1016/j.stem.2015.12.002 |
[56] | Aubry L , Bugi A , Lefort N , Rousseau F , Peschanski M , Perrier AL . Striatal progenitors derived from human ES cells mature into DARPP32 neurons in vitro and in quinolinic acid-lesioned rats. Proc Natl Acad Sci. (2008) ;105: :16707–12. doi: 10.1073/pnas.0808488105 |
[57] | Nicoleau C , Varela C , Bonnefond C , Maury Y , Bugi A , Aubry L , et al. Embryonic stem cells neural differentiation qualifies the role of Wnt/β-Catenin signals in human telencephalic specification and regionalization. Stem Cells. (2013) ;31: :1763–74. doi: 10.1002/stem.1462 |
[58] | Li X-J , Zhang X , Johnson MA , Wang Z-B , Lavaute T , Zhang S-C . Coordination of sonic hedgehog and Wnt signaling determines ventral and dorsal telencephalic neuron types from human embryonic stem cells. Dev Camb Engl. (2009) ;136: :4055–63. doi: 10.1242/dev.036624 |
[59] | El-Akabawy G , Medina LM , Jeffries A , Price J , Modo M . Purmorphamine increases DARPP-32 differentiation in human striatal neural stem cells through the Hedgehog pathway. Stem Cells Dev. (2011) ;20: :1873–87. doi: 10.1089/scd.2010.0282 |
[60] | Ma L , Hu B , Liu Y , Vermilyea SC , Liu H , Gao L , et al. Human embryonic stem cell-derived GABA neurons correct locomotion deficits in quinolinic acid-lesioned mice. Cell Stem Cell. (2012) ;10: :455–64. doi: 10.1016/j.stem.2012.01.021 |
[61] | Victor MB , Richner M , Hermanstyne TO , Ransdell JL , Sobieski C , Deng P-Y , et al. Generation of human striatal neurons by MicroRNA-dependent direct conversion of fibroblasts. Neuron. (2014) ;84: :311–23. doi: 10.1016/j.neuron.2014.10.016 |
[62] | Arber C , Precious SV , Cambray S , Risner-Janiczek JR , Kelly C , Noakes Z , et al. Activin A directs striatal projection neuron differentiation of human pluripotent stem cells. Dev Camb Engl. (2015) ;142: :1375–86. doi: 10.1242/dev.117093 |
[63] | Graveland GA , Williams RS , DiFiglia M . Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington’s disease. Science. (1985) ;227: :770–3. doi: 10.1126/science.3155875 |
[64] | Reiner A , Albin RL , Anderson KD , D’Amato CJ , Penney JB , Young AB . Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci. (1988) ;85: :5733–7. |
[65] | Waclaw RR , Wang B , Pei Z , Ehrman LA , Campbell K . Distinct temporal requirements for the homeobox gene Gsx2 in specifying striatal and olfactory bulb neuronal fates. Neuron. (2009) ;63: :451–65. doi: 10.1016/j.neuron.2009.07.015 |
[66] | Olsson M , Björklund A , Campbell K . Early specification of striatal projection neurons and interneuronal subtypes in the lateral and medial ganglionic eminence. Neuroscience. (1998) ;84: :867–76. |
[67] | Onorati M , Castiglioni V , Biasci D , Cesana E , Menon R , Vuono R , et al. Molecular and functional definition of the developing human striatum. Nat Neurosci. (2014) ;17: :1804–15. doi: 10.1038/nn.3860 |
[68] | Takahashi K , Liu F-C , Hirokawa K , Takahashi H . Expression of Foxp2, a gene involved in speech and language, in the developing and adult striatum. J Neurosci Res. (2003) ;73: :61–72. doi: 10.1002/jnr.10638 |
[69] | Tamura S , Morikawa Y , Iwanishi H , Hisaoka T , Senba E . Foxp1 gene expression in projection neurons of the mouse striatum. Neuroscience. (2004) ;124: :261–7. doi: 10.1016/j.neuroscience.2003.11.036 |
[70] | Fjodorova M , Noakes Z , Li M . How to make striatal projection neurons. Neurogenesis. (2015) ;2: :e1100227. doi: 10.1080/23262133.2015.1100227 |
[71] | Lin L , Yuan J , Sander B , Golas MM . In vitro differentiation of human neural progenitor cells into striatal GABAergic neurons. Stem Cells Transl Med. (2015) ;4: :775–88. doi: 10.5966/sctm.2014-0083 |
[72] | Delli Carri A , Onorati M , Lelos MJ , Castiglioni V , Faedo A , Menon R , et al. Developmentally coordinated extrinsic signals drive human pluripotent stem cell differentiation toward authentic DARPP-32+medium-sized spiny neurons. Dev Camb Engl. (2013) ;140: :301–12. doi: 10.1242/dev.084608 |
[73] | Yoo AS . MicroRNA-mediated conversion of human fibroblasts to neurons. Nature. (2011) ;476: :228–31. doi: 10.1038/nature10323 |
[74] | Studer L , Vera E , Cornacchia D . Programming and reprogramming cellular age in the era of induced pluripotency. Cell Stem Cell. (2015) ;16: :591–600. doi: 10.1016/j.stem.2015.05.004 |
[75] | Miller JD , Ganat YM , Kishinevsky S , Bowman RL , Liu B , Tu EY , et al. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell. (2013) ;13: :691–705. doi: 10.1016/j.stem.2013.11.006 |
[76] | Liu G-H , Suzuki K , Qu J , Sancho-Martinez I , Yi F , Li M , et al. Targeted gene correction of laminopathy-associated LMNA mutations in patient-specific iPSCs. Cell Stem Cell. (2011) ;8: :688–94. doi: 10.1016/j.stem.2011.04.019 |
[77] | Khurana V , Tardiff DF , Chung CY , Lindquist S . Toward stem cell-based phenotypic screens for neurodegenerative diseases. Nat Rev Neurol. (2015) ;11: :339–50. doi: 10.1038/nrneurol.2015.79 |
[78] | Nguyen GD , Gokhan S , Molero AE , Mehler MF . Selective roles of normal and mutant huntingtin in neural induction and early neurogenesis. PLoS One. (2013) ;8: :e64368. doi: 10.1371/journal.pone.0064368 |
[79] | Molina-Calavita M , Barnat M , Elias S , Aparicio E , Piel M , Humbert S . Mutant huntingtin affects cortical progenitor cell division and development of the mouse neocortex. J Neurosci. (2014) ;34: :10034–40. doi: 10.1523/JNEUROSCI.0715-14.2014 |
[80] | Lopes C , Aubert S , Bourgois-Rocha F , Barnat M , Rego AC , Déglon N , et al. Dominant-negative effects of adult-onset huntingtin mutations alter the division of human embryonic stem cells-derived neural cells. PloS One. (2016) ;11: :e0148680. doi: 10.1371/journal.pone.0148680 |
[81] | Nopoulos P , Magnotta VA , Mikos A , Paulson H , Andreasen NC , Paulsen JS . Morphology of the cerebral cortex in preclinical Huntington’s disease. Am J Psychiatry. (2007) ;164: :1428–34. doi: 10.1176/appi.ajp.2007.06081266 |
[82] | Paulsen JS , Nopoulos PC , Aylward E , Ross CA , Johnson H , Magnotta VA , et al. Striatal and white matter predictors of estimated diagnosis for Huntington disease. Brain Res Bull. (2010) ;82: :201–7. doi: 10.1016/j.brainresbull.2010.04.003 |
[83] | Aylward EH . Change in MRI striatal volumes as a biomarker in preclinical Huntington’s disease. Brain Res Bull. (2007) ;72: :152–8. doi: 10.1016/j.brainresbull.2006.10.028 |
[84] | Wolf RC , Thomann PA , Thomann AK , Vasic N , Wolf ND , Landwehrmeyer GB , et al. Brain structure in preclinical Huntington’s disease: A multi-method approach. Neurodegener Dis. (2013) ;12: :13–22. doi: 10.1159/000338635 |
[85] | Unschuld PG , Joel SE , Liu X , Shanahan M , Margolis RL , Biglan KM , et al. Impaired cortico-striatal functional connectivity in prodromal Huntington’s disease. Neurosci Lett. (2012) ;514: :204–9. doi: 10.1016/j.neulet.2012.02.095 |
[86] | Harrington DL , Rubinov M , Durgerian S , Mourany L , Reece C , Koenig K , et al. Network topology and functional connectivity disturbances precede the onset of Huntington’s disease. Brain J Neurol. (2015) ;138: :2332–46. doi: 10.1093/brain/awv145 |
[87] | Philpott AL , Cummins TDR , Bailey NW , Churchyard A , Fitzgerald PB , Georgiou-Karistianis N . Cortical inhibitory deficits in premanifest and early Huntington’s disease. Behav Brain Res. (2016) ;296: :311–7. doi: 10.1016/j.bbr.2015.09.030 |
[88] | Kerschbamer E , Biagioli M . Huntington’s disease as neurodevelopmental disorder: Altered chromatin regulation, coding, and non-coding RNA Transcription. Front Neurosci. (2016) ;9. doi: 10.3389/fnins.2015.00509 |
[89] | Gómez-Tortosa E , MacDonald ME , Friend JC , Taylor SAM , Weiler LJ , Cupples LA , et al. Quantitative neuropathological changes in presymptomatic Huntington’s disease. Ann Neurol. (2001) ;49: :29–34. |
[90] | Unschuld PG , Joel SE , Pekar JJ , Reading SA , Oishi K , McEntee J , et al. Depressive symptoms in prodromal Huntington’s disease correlate with Stroop-interference related functional connectivity in the ventromedial prefrontal cortex. Psychiatry Res Neuroimaging. (2012) ;203: :166–74. doi: 10.1016/j.pscychresns.2012.01.002 |
[91] | Puigdellívol M , Cherubini M , Brito V , Giralt A , Suelves N , Ballesteros J , et al. A role for Kalirin-7 in corticostriatal synaptic dysfunction in Huntington’s disease. Hum Mol Genet. (2015) ;24: :7265–85. doi: 10.1093/hmg/ddv426 |
[92] | Deng YP , Wong T , Bricker-Anthony C , Deng B , Reiner A . Loss of corticostriatal and thalamostriatal synaptic terminals precedes striatal projection neuron pathology in heterozygous Q140 Huntington’s disease mice. Neurobiol Dis. (2013) ;60: :89–107. doi: 10.1016/j.nbd.2013.08.009 |
[93] | McKinstry SU , Karadeniz YB , Worthington AK , Hayrapetyan VY , Ozlu MI , Serafin-Molina K , et al. Huntingtin is required for normal excitatory synapse development in cortical and striatal circuits. J Neurosci. (2014) ;34: :9455–72. doi: 10.1523/JNEUROSCI.4699-13.2014 |
[94] | Hickey MA , Kosmalska A , Enayati J , Cohen R , Zeitlin S , Levine MS , et al. Extensive early motor and non-motor behavioral deficits are followed by striatal neuronal loss in knock-in Huntington’s disease mice. Neuroscience. (2008) ;157: :280–95. doi: 10.1016/j.neuroscience.2008.08.041 |
[95] | Brennand K . Is Huntington’s disease a neurodevelopmental disorder? Sci Transl Med. (2016) ;8: :320ec1–320ec1. doi: 10.1126/scitranslmed.aad9760 |
[96] | Humbert S . Is Huntington disease a developmental disorder? EMBO Rep. (2010) ;11: :899. doi: 10.1038/embor.2010.182 |
[97] | Sapp E , Kegel KB , Aronin N , Hashikawa T , Uchiyama Y , Tohyama K , et al. Early and progressive accumulation of reactive microglia in the Huntington disease brain. J Neuropathol Exp Neurol. (2001) ;60: :161–72. |
[98] | Pavese N , Gerhard A , Tai YF , Ho AK , Turkheimer F , Barker RA , et al. Microglial activation correlates with severity in Huntington disease A clinical and PET study. Neurology. (2006) ;66: :1638–43. doi: 10.1212/01.wnl.0000222734.56412.17 |
[99] | Singhrao SK , Thomas P , Wood JD , MacMillan JC , Neal JW , Harper PS , et al. Huntingtin protein colocalizes with lesions of neurodegenerative diseases: An investigation in Huntington’s, Alzheimer’s, and Pick’s diseases. Exp Neurol. (1998) ;150: :213–22. doi: 10.1006/exnr.1998.6778 |
[100] | Myers RH , Vonsattel JP , Paskevich PA , Kiely DK , Stevens TJ , Cupples LA , et al. Decreased neuronal and increased oligodendroglial densities in Huntington’s disease caudate nucleus. J Neuropathol Exp Neurol.. (1991) ;50: :729–42. |
[101] | Faideau M , Kim J , Cormier K , Gilmore R , Welch M , Auregan G , et al. In vivo expression of polyglutamine-expanded huntingtin by mouse striatal astrocytes impairs glutamate transport: A correlation with Huntington’s disease subjects. Hum Mol Genet. (2010) ;19: :3053–67. doi: 10.1093/hmg/ddq212 |
[102] | Bradford J , Shin J-Y , Roberts M , Wang C-E , Li X-J , Li S . Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms. Proc Natl Acad Sci U S A. (2009) ;106: :22480–5. doi: 10.1073/pnas.0911503106 |
[103] | Bradford J , Shin J-Y , Roberts M , Wang C-E , Sheng G , Li S , et al. Mutant huntingtin in glial cells exacerbates neurological symptoms of Huntington disease mice. J Biol Chem. (2010) ;285: :10653–61. doi: 10.1074/jbc.M109.083287 |
[104] | Shin J-Y , Fang Z-H , Yu Z-X , Wang C-E , Li S-H , Li X-J . Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J Cell Biol. (2005) ;171: :1001–12. doi: 10.1083/jcb.200508072 |
[105] | Jiang R , Diaz-Castro B , Looger LL , Khakh BS . Dysfunctional calcium and glutamate signaling in striatal astrocytes from Huntington’s disease model mice. J Neurosci. (2016) ;36: :3453–70. doi: 10.1523/JNEUROSCI.3693-15.2016 |
[106] | Tong X , Ao Y , Faas GC , Nwaobi SE , Xu J , Haustein MD , et al. Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nat Neurosci. (2014) ;17: :694–703. doi: 10.1038/nn.3691 |
[107] | Boussicault L , Hérard A-S , Calingasan N , Petit F , Malgorn C , Merienne N , et al. Impaired brain energy metabolism in the BACHD mouse model of Huntington’s disease: Critical role of astrocyte-neuron interactions. J Cereb Blood Flow Metab. (2014) ;34: :1500–10. doi: 10.1038/jcbfm.2014.110 |
[108] | Hsiao H-Y , Chen Y-C , Huang C-H , Chen C-C , Hsu Y-H , Chen H-M , et al. Aberrant astrocytes impair vascular reactivity in Huntington disease. Ann Neurol. (2015) ;78: :178–92. doi: 10.1002/ana.24428 |
[109] | Zhou S , Szczesna K , Ochalek A , Kobolák J , Varga E , Nemes C , et al. Neurosphere based differentiation of human iPSC improves astrocyte differentiation. Stem Cells Int. (2016) ;2016. doi: 10.1155/2016/4937689 |
[110] | Krencik R , Weick JP , Liu Y , Zhang Z-J , Zhang S-C . Specification of transplantable astroglial subtypes from human pluripotent stem cells. Nat Biotechnol. (2011) ;29: :528–34. doi: 10.1038/nbt.1877 |
[111] | Emdad L , D’Souza SL , Kothari HP , Qadeer ZA , Germano IM . Efficient differentiation of human embryonic and induced pluripotent stem cells into functional astrocytes. Stem Cells Dev. (2012) ;21: :404–10. doi: 10.1089/scd.2010.0560 |
[112] | Roybon L , Lamas NJ , Garcia AD , Yang EJ , Sattler R , Lewis VJ , et al. Human stem cell-derived spinal cord astrocytes with defined mature or reactive phenotypes. Cell Re. (2013) ;4: :1035–48. doi: 10.1016/j.celrep.2013.06.021 |
[113] | Zhang P-W , Haidet-Phillips AM , Pham JT , Lee Y , Huo Y , Tienari PJ , et al. Generation of GFAP::GFP astrocyte reporter lines from human adult fibroblast-derived iPS cells using zinc-finger nuclease technology. Glia. (2016) ;64: :63–75. doi: 10.1002/glia.22903 |
[114] | Shaltouki A , Peng J , Liu Q , Rao MS , Zeng X . Efficient generation of astrocytes from human pluripotent stem cells in defined conditions. Stem Cells Dayt Ohio. (2013) ;31: :941–52. doi: 10.1002/stem.1334 |
[115] | Wang S , Bates J , Li X , Schanz S , Chandler-Militello D , Levine C , et al. Human iPSC-derived oligodendrocyte progenitor cells can myelinate and rescue a mouse model of congenital hypomyelination. Cell Stem Cell. (2013) ;12: :252–64. doi: 10.1016/j.stem.2012.12.002 |
[116] | Hu B-Y , Du Z-W , Zhang S-C . Differentiation of human oligodendrocytes from pluripotent stem cells. Nat Protoc. (2009) ;4: :1614–22. doi: 10.1038/nprot.2009.186 |
[117] | Izrael M , Zhang P , Kaufman R , Shinder V , Ella R , Amit M , et al. Human oligodendrocytes derived from embryonic stem cells: Effect of noggin on phenotypic differentiation in vitro and on myelination in vivo. Mol Cell Neurosci. (2007) ;34: :310–23. doi: 10.1016/j.mcn.2006.11.008 |
[118] | Czepiel M , Balasubramaniyan V , Schaafsma W , Stancic M , Mikkers H , Huisman C , et al. Differentiation of induced pluripotent stem cells into functional oligodendrocytes. Glia. (2011) ;59: :882–92. doi: 10.1002/glia.21159 |
[119] | Marion RM , Strati K , Li H , Tejera A , Schoeftner S , Ortega S , et al. Telomeres acquire embryonic stem cell characteristics in induced pluripotent stem cells. Cell Stem Cell. (2009) ;4: :141–54. doi: 10.1016/j.stem.2008.12.010 |
[120] | Suhr ST , Chang EA , Rodriguez RM , Wang K , Ross PJ , Beyhan Z , et al. Telomere dynamics in human cells reprogrammed to pluripotency. PloS One. (2009) ;4: :e8124. doi: 10.1371/journal.pone.0008124 |
[121] | Lapasset L , Milhavet O , Prieur A , Besnard E , Babled A , Aït-Hamou N , et al. Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev. (2011) ;25: :2248–53. doi: 10.1101/gad.173922.111 |
[122] | Prigione A , Hossini AM , Lichtner B , Serin A , Fauler B , Megges M , et al. Mitochondrial-associated cell death mechanisms are reset to an embryonic-like state in aged donor-derived iPS cells harboring chromosomal aberrations. PloS One. (2011) ;6: :e27352. doi: 10.1371/journal.pone.0027352 |
[123] | Goldman SA ((2016) ) Stem and progenitor cell-based therapy of the central nervous system: Hopes, hype, and wishful thinking. Cell Stem Cell 18: :174–88. doi: 10.1016/j.stem.2016.01.012 |
[124] | Kim K , Doi A , Wen B , Ng K , Zhao R , Cahan P , et al. Epigenetic memory in induced pluripotent stem cells. Nature. (2010) ;467: :285–90. doi: 10.1038/nature09342 |
[125] | Srikanth P , Young-Pearse TL . Stem cells on the brain: Modeling neurodevelopmental and neurodegenerative diseases using human induced pluripotent stem cells. J Neurogenet. (2014) ;28: :5–29. doi: 10.3109/01677063.2014.881358 |
[126] | Polo JM , Liu S , Figueroa ME , Kulalert W , Eminli S , Tan KY , et al. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nat Biotechnol. (2010) ;28: :848–55. doi: 10.1038/nbt.1667 |
[127] | Mikkelsen TS , Hanna J , Zhang X , Ku M , Wernig M , Schorderet P , et al. Dissecting direct reprogramming through integrative genomic analysis. Nature. (2008) ;454: :49–55. doi: 10.1038/nature07056 |
[128] | Kang X , Yu Q , Huang Y , Song B , Chen Y , Gao X , et al. Effects of integrating and non-integrating reprogramming methods on copy number variation and genomic stability of human induced pluripotent stem cells. PLoS One. (2015) ;10: :e0131128. doi: 10.1371/journal.pone.0131128 |
[129] | Laurent LC , Ulitsky I , Slavin I , Tran H , Schork A , Morey R , et al. Dynamic changes in the copy number of pluripotency and cell proliferation genes in human ESCs and iPSCs during reprogramming and time in culture. Cell Stem Cell. (2011) ;8: :106–18. doi: 10.1016/j.stem.2010.12.003 |
[130] | Lister R , Pelizzola M , Kida YS , Hawkins RD , Nery JR , Hon G , et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. (2011) ;471: :68–73. doi: 10.1038/nature09798 |
[131] | Ruiz S , Lopez-Contreras AJ , Gabut M , Marion RM , Gutierrez-Martinez P , Bua S , et al. Limiting replication stress during somatic cell reprogramming reduces genomic instability in induced pluripotent stem cells. Nat Commun. (2015) ;6: :8036. doi: 10.1038/ncomms9036 |
[132] | Hussein SM , Batada NN , Vuoristo S , Ching RW , Autio R , Närvä E , et al. Copy number variation and selection during reprogramming to pluripotency. Nature. (2011) ;471: :58–62. doi: 10.1038/nature09871 |
[133] | Ritch JJ , Valencia A , Alexander J , Sapp E , Gatune L , Sangrey GR , et al. Multiple phenotypes in Huntington disease mouse neural stem cells. Mol Cell Neurosci. (2012) ;50: :70–81. doi: 10.1016/j.mcn.2012.03.011 |
[134] | McNeish J , Gardner JP , Wainger BJ , Woolf CJ , Eggan K . From dish to bedside: Lessons learned while translating findings from a stem cell model of disease to a clinical trial. Cell Stem Cell. (2015) ;17: :8–10. doi: 10.1016/j.stem.2015.06.013 |
[135] | Hargus G , Ehrlich M , Hallmann A-L , Kuhlmann T . Human stem cell models of neurodegeneration: A novel approach to study mechanisms of disease development. Acta Neuropathol (Berl). (2014) ;127: :151–73. doi: 10.1007/s00401-013-1222-6 |
[136] | Imaizumi Y , Okano H . Modeling human neurological disorders with induced pluripotent stem cells. J Neurochem. (2014) ;129: :388–99. doi: 10.1111/jnc.12625 |
[137] | Vasileva EA , Shuvalov OU , Garabadgiu AV , Melino G , Barlev NA . Genome-editing tools for stem cell biology. Cell Death Dis. (2015) ;6: :e1831. doi: 10.1038/cddis.2015.167 |
[138] | Li M , Suzuki K , Kim NY , Liu G-H , Izpisua Belmonte JC . A cut above the rest: Targeted genome editing technologies in human pluripotent stem cells. J Biol Chem. (2014) ;289: :4594–9. doi: 10.1074/jbc.R113.488247 |
[139] | Veres A , Gosis BS , Ding Q , Collins R , Ragavendran A , Brand H , et al. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell. (2014) ;15: :27–30. doi: 10.1016/j.stem.2014.04.020 |
[140] | Jonson I , Ougland R , Larsen E . DNA repair mechanisms in Huntington’s disease. Mol Neurobiol. (2013) ;47: :1093–102. doi: 10.1007/s12035-013-8409-7 |
[141] | Musunuru K . Genome editing of human pluripotent stem cells to generate human cellular disease models. Dis Model Mech. (2013) ;6: :896–904. doi: 10.1242/dmm.012054 |
[142] | Reinhardt P , Schmid B , Burbulla LF , Schöndorf DC , Wagner L , Glatza M , et al. Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell. (2013) ;12: :354–67. doi: 10.1016/j.stem.2013.01.008 |
[143] | Bradley CK , Scott HA , Chami O , Peura TT , Dumevska B , Schmidt U , et al. Derivation of Huntington’s disease-affected human embryonic stem cell lines. Stem Cells Dev. (2010) ;20: :495–502. doi: 10.1089/scd.2010.0120 |
[144] | Feyeux M , Bourgois-Rocha F , Redfern A , Giles P , Lefort N , Aubert S , et al. Early transcriptional changes linked to naturally occurring Huntington’s disease mutations in neural derivatives of human embryonic stem cells. Hum Mol Genet. (2012) ;21: :3883–95. doi: 10.1093/hmg/dds216 |
[145] | Jacquet L , Neueder A , Földes G , Karagiannis P , Hobbs C , Jolinon N , et al. Three Huntington’s disease specific mutation-carrying human embryonic stem cell lines have stable number of CAG repeats upon in vitro differentiation into cardiomyocytes. PLoS One. (2015) ;10. doi: 10.1371/journal.pone.0126860 |
Figures and Tables
Fig.1
Induced pluripotent stem cells (iPSC) in Huntington’s disease research: progress and opportunity. Schematic shows cell types of the neural lineage that can be differentiated from iPSCs. The relative purity attainable of differentiated cultures is indicated in parenthesis (homogeneous or heterogeneous). Differentiated cultures are rarely 100% pure but may reach 95% homogeneity depending on cell type. Neuronal cultures often contain significant numbers of glia including Nestin-positive neural stem cells (NSCs), astrocytes and oligodendrocytes (heterogeneous). Dotted arrows highlight areas where more studies are needed.
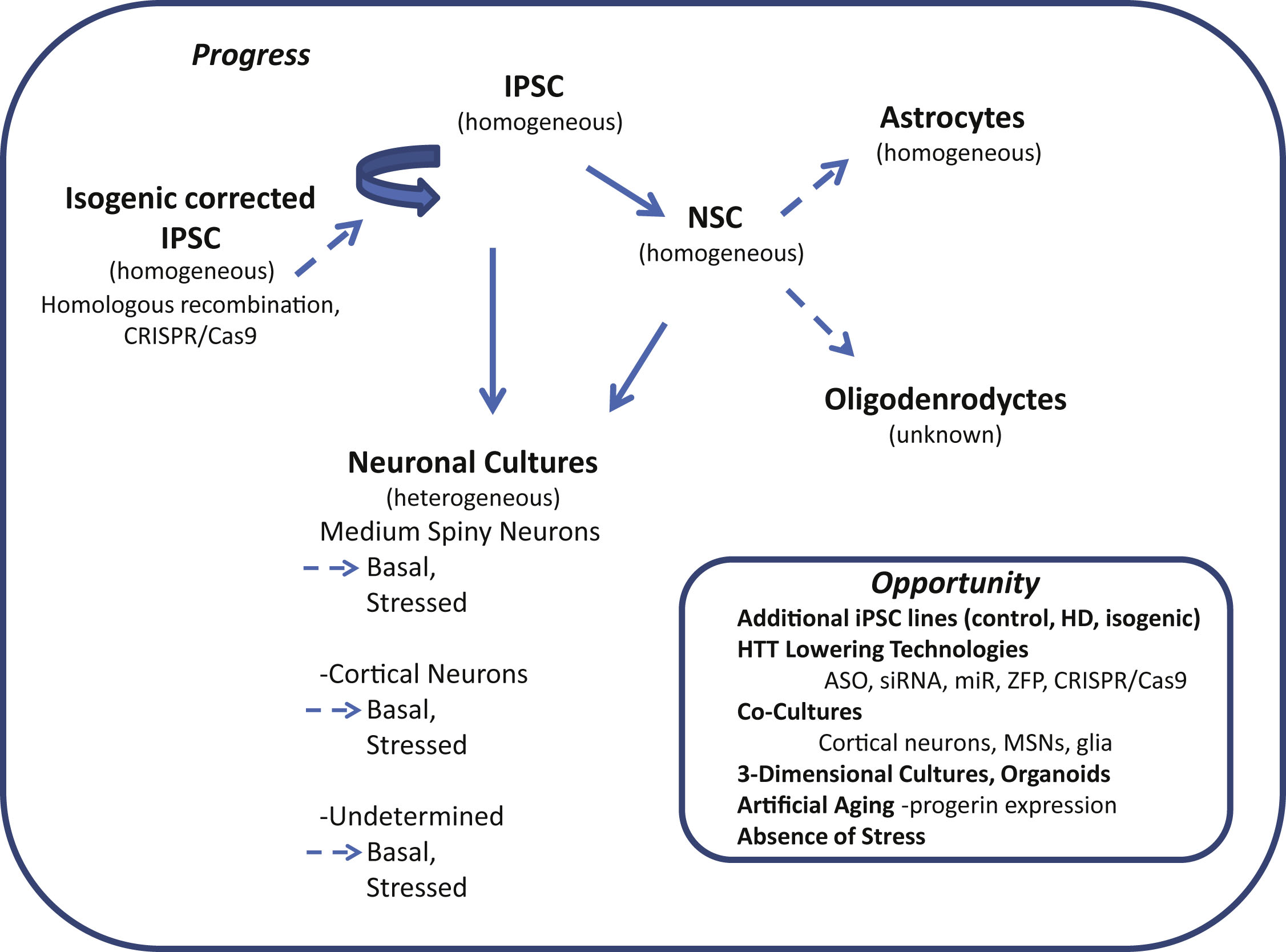
Table 1
Gene/Protein pathways changes in HD iPSCs and derived cells
| Initial cell Type | Differentiated cell type (Reference) | Cell markers | Clone name (CAG Repeat length) | Gene or protein assay type | Identified gene or protein change in HD cells | Reference |
| Retroviral iPSCs (OCT4, SOX2, KLF4, c-MYC))&H9 Embryonic Stem Cells | Undifferentiated iPSCs | Not described. | HD(72CAG), H9 ES (control Embryonic Stem Cells) | High resolution 2 dimensional electrophoresis | Increase in: SOD1, GST, PRX1, PRX2,PRX6, BFT;Decrease in: GPX1, CB1, STMN-1 | [Chae et al., 2012] |
| Retroviral iPSCs [OCT4, SOX2, KLF4, c-MYC))&H9 Embryonic Stem Cells | Undifferentiated iPSCs | Not described. | HD(72CAG), H9 ES (control Embryonic Stem Cells) | Biological Network Analysis-MetaCoreTM | Protein changes identified by 2-dimensional electrophoresis play a role in regulation of transcription factors: p53, c-Myc, E2F1, YY1 and NF-κB | [Chae et al., 2012] |
| Retroviral iPSCs (OCT4, SOX2, KLF4, c-MYC) | Undifferentiated iPSCs | Nanog, Sox2, Oct4, SSEA4, TRA-1-60 | HDc116(21CAG), HD4(72CAG), HFib2-IPS5 (control) | Gene Microarray | Pathway analysis following gene microarray indicates increased caspase-related signaling molecules/TGFβ family genes and decreased cadherin family genes | [An et al., 2012] |
| Lentiviral iPSCs (OCT4, SOX2, KLF4, c-MYC, LIN28, NANOG) | FGF/EGF dependent neural stem cell monolayer derived from dissociated neurospheres | Nestin, PAX6, SOX1, SOX2, Musashi | HD33i.8(33CAG), HD28i.2(28CAG), HD21i.5(21CAG), HD60i.3/HD60i.4(60CAG), HD109i.1 (109CAG)HD180i.5(180CAG) | Whole Transcript Expression Profiling/Ingenuity Pathway Tools | HD vs. Control NSCs differed in: Cell Signaling, Organismal Development, Genetic Disorder; Cell Cycle, Cancer, Cellular Assemby and Organization; Connnective Tissue Disorders, Genetic Disorder, Dermatological Diseases and Conditions; Tissue Development, Embryonic Development, Cellular Development; Gene Expression, Cellular Movement, Nervous System Development and Function | [The HD iPSC Consortium, 2012] |
| Lentiviral iPSCs (OCT4, SOX2, KLF4, c-MYC, LIN28, NANOG) | FGF/EGF dependent neural stem cell monolayer derived from dissociated neurospheres | Nestin, PAX6, SOX1, SOX2, Musashi | HD33i.8(33CAG), HD28i.2(28CAG), HD21i.5(21CAG), HD60i.3/HD60i.4(60CAG), HD109i.1 (109CAG)HD180i.5(180CAG) | Whole Transcript Expression Profiling/Ingenuity Pathway Tools | (109/180CAG) vs. Control NSCs differed in: Neurological Disease; Axonal Guidance Signaling | [The HD iPSC Consortium, 2012] |
| Lentiviral iPSCs (OCT4, SOX2, KLF4, c-MYC, LIN28, NANOG) | FGF/EGF dependent neural stem cell monolayer derived from dissociated neurospheres | Nestin, PAX6, SOX1, SOX2, Musashi | HD33i.8(33CAG), HD28i.2(28CAG), HD21i.5(21CAG), HD60i.3/HD60i.4(60CAG), HD109i.1 (109CAG)HD180i.5(180CAG) | Whole Transcript Expression Profiling/Ingenuity Pathway Tools | (60CAG) vs. Control NSCs differed in: Calcium Signaling; Skeletal and Muscular System Development and Function | [The HD iPSC Consortium, 2012] |
| Lentiviral iPSCs (OCT4, SOX2, KLF4, c-MYC, LIN28, NANOG) | FGF/EGF dependent neural stem cell monolayer derived from dissociated neurospheres | Nestin, PAX6, SOX1, SOX2, Musashi | HD33i.8(33CAG), HD28i.2(28CAG), HD21i.5(21CAG), HD60i.3/HD60i.4(60CAG), HD109i.1 (109CAG)HD180i.5(180CAG) | Whole Transcript Expression Profiling/Ingenuity Pathway Tools | (180CAG) vs. (60CAG) NSCs differed in: Calcium Signaling; Genetic Disorder | [The HD iPSC Consortium, 2012] |
| Retroviral iPSCs (OCT4, SOX2, KLF4, c-MYC) | FGF/LIF dependent neural stem cells derived from neural rosettes induced by STEM-LIF neural induction media | Nestin | HDc116(21CAG corrected), HD4(72CAG) | Weighted Gene Coexpression Network Analysis (RNA-seq) | Altered gene ontology categories: ECM organization, regulation of synapse assembly, mammalian phenotype-abnormal survival or perinatal lethality, axonal guidance, targets of transcription factor SMAD3; Upstream regulators of altered genes: TGF-β, β-estradiol, TNF-α | [Ring et al., 2015] |
| Lentiviral iPSCs (OCT4, SOX2, KLF4, c-MYC, LIN28, NANOG) | Neurons differentiated from dissociated FGF/EGF neurospheres induced by FGF/EGF withdrawal and addition of BDNF, cAMP, Shh, Dkk1, and valproic acid [Aubry et al.,2008] | DARPP32, MAP1, MAP2a/b, Mash1, Bcl-1B, BIII-Tubulin | HD33i.8(33CAG), HD28i.2(28CAG), HD21i.5(21CAG), HD60i.3/HD60i.4(60CAG), HD109(109CAG), HD180i.5(180CAG) | Whole Transcript Expression Profiling/Ingenuity Pathway Tools | Cellular growth and proliferation, cellular assembly and organization, cellular function and maintenance; cell-to-cell signaling and interaction, connective tissue development and function, lipid metabolism; tissue development, embryonic development, organ development, connective tissue disorders, genetic disorder, dermatological diseases and conditions. cellular assembly and organization, cellular function and maintenance, cellular movement | [The HD iPSC Consortium, 2012] |
| Episomal iPSCs (OCT4, SOX2, KLF4, c-MYC, NANOG) | “Striatal” neuronal cells differentiated from IPS derived neurospheres (iPSCs->EBs->neuronal rosettes->neurospheres) by FGF withdrawal in B27 media [Chiu et al., 2015] | TUJ1,MAP2, GABA, GAD65, Calbindin, or DARPP-32 | HD-IPS-A1(43CAG), HD-IPS-B4(43CAG), control IPS 1 &2 | Gene Microarray | Decrease in RAD51/52 (double stranded break repair), ADORA2A (adenosine 2A receptor), PENK (proenkephalin), ARPP21 (cAMP regulated phosphoprotein), GPR88 (G-protein coupled receptor 88), RGS4 (regulator of G-protein signaling 4), GSST1/GSST2 (oxidative stress repair), (glutathione-S transferase) Increase in:, DRD1/2 (dopamine receptor 1/2), GAD65 (glutamic acid decarboxylase 2), COMT (Catechol-O-methyl transferase) | [Chiu et al., 2015] |
Table 2
Individual Gene/Protein changes in HD iPSCs and derived cells
| Initial cell type | Cell type differentiation (Reference) | Cell markers | Clone name (CAG Repeat length) | Identified gene or protein change in HD cells | Reference |
| Retroviral iPSCs (OCT4, SOX2, KLF4, c-MYC) | Undifferentiated iPSCs | Nanog, Sox2, Oct4,SSEA4, TRA-1-60 | HDc116(21CAG), HD4(72CAG), HFib2-IPS5 (control) | Increase in mRNA levels of cadherin family genes: CDK inhibitor 2B, ID2, ID4, PITX2, THBS1 and LEFTY2 | [An et al., 2012] |
| Retroviral iPSCs (OCT4, SOX2, KLF4, c-MYC) | Undifferentiated iPSCs | Nanog, Sox2, Oct4,SSEA4, TRA-1-60 | HDc116(21CAG), HD4(72CAG), HFib2-IPS5 (control) | Decreased cadherin family mRNA levels: protocadherin 11, protocadherin beta 13, protocadherin gamma subfamily A10 and A2 | [An et al., 2012] |
| Episomal iPSCs (Oct4, Sox2, KLF4, LMYC, LIN28 and shRNA to p53) | Undifferentiated iPSCs | Oct-4, SOX2, NANOG, GDF2, REX01 | ND42228, ND42230 (71CAG);ND42223, ND42224 (109CAG); ND41658 (17/18CAG), ND42245 (21CAG) | Decreased ERK phosphorylation (Thr202/Tyr204), no change in β-catenin levels or phosphorylation (Ser33/37), increased SOD1 expression | [Szlachcic et al., 2015] |
| Retroviral iPSCs (OCT4, SOX2, KLF4, c-MYC)&H9 Embryonic Stem Cells | Undifferentiated iPSCs/ES Cells | Not described | HD(72CAG), HD2(72CAG), 551-8 IPS(control iPSCs), H9 ES (control Embryonic Stem Cells) | Decrease in oxidative stress response related proteins: SOD1, GST, Gpx1; increase in anti-oxidant response proteins: Prx1,Prx2, Prx6 | [Chae et al., 2012] |
| Retroviral iPSCs (OCT4, SOX2, KLF4, c-MYC)&H9 Embryonic Stem Cells | Undifferentiated iPSCs/ES Cells | Not described | HD(72CAG), HD2(72CAG), 551-8 IPS(control iPSCs), H9 ES (control Embryonic Stem Cells) | Decrease in cytoplasmic localization, increase in nuclear localization of Prx1 | [Chae et al., 2012] |
| Episomal iPSCs | Undifferentiated iPSCs | Not described | HD70-2(70CAG), HD180-4(180CAG); CC-1, CE-6 (controls) | Elevated p53, p-p53, p-ATM protein levels | [Tidball et al., 2014] |
| Episomal iPSCs (OCT4, SOX2, KLF4, L-MYC, LIN28, p53 shRNA) | Striatal-like neurons derived from FGF/EGF dependent neurospheres after FGF withdrawal/induction with: Shh, Dkk1, BDNF, cAMP, and valproic acid | TUJ1, DARPP32 | HD60(60CAG), HD109(109CAG), HD180(180CAG), Controls(28CAG, 33W) | Increased GRIN2B (NMDA receptor subunit) mRNA in 109CAG and 180CAG, NOT 60CAG | [Mattis et al., 2015] |
| Retroviral iPSCs (OCT4, SOX2, KLF4, c-MYC) | FGF/LIF dependent neural stem cells derived from neural rosettes induced by STEM-LIF neural induction media | Nestin | HDc116(21CAG corrected), HD4(72CAG) | Decrease in mRNA specific to striatal development: DARPP32, CTIP2, FOXP1, ISL1,TBR1, PAX6, increase in FOXP2 mRNA | [Ring et al., 2015] |
| Retroviral iPSCs (OCT4, SOX2, KLF4, c-MYC) | FGF and LIF dependent NSC produced from selection of neuronal rosettes apparent after plating of EBs (serum dep.) [Zhang et al., 2010] | Nestin | HDc116(21CAG), HD4(72CAG), HFib2-IPS5 (control) | Decreased TGF β1 mRNA | [An et al., 2012] |
| Retroviral iPSCs (OCT4, SOX2, KLF4, c-MYC) | FGF and LIF dependent NSC produced from selection of neuronal rosettes apparent after plating of EBs (serum dep.) [Zhang et al., 2010] | Nestin | HDc116(21CAG), HD4(72CAG), HFib2-IPS5 (control) | Decrease in BDNF mRNA | [An et al., 2012] |
| Retroviral iPSCs (OCT4, SOX2, Klf4 and cMyc) | FGF/EGF dependent neural stem cells grown as a monolayer after neuroepithelial induction with SB431542 and Noggin [Feyeux et al., 2012] | Not described | HD1-IPS4(72CAG), RC9 and SA-01(WT Embryonic Stem Cells) | Decrease in BDNFII and BDNFIV mRNA | [Charbord et al., 2013] |
| Retroviral iPSCs (OCT4, SOX2, KLF4, c-MYC) | LIF dependent neural precursor cells induced by small molecule inhibitors: SB431542 and CHIR99021 [Li et al., 2011] | Nestin | CAG33(33CAG) and CAG180(180CAG) | Decrease in MAO-A and MAO-B mRNA levels, increase in MAO-A and MAO-B activity | [Ooi et al., 2015] |
| Retroviral iPSCs (OCT4, SOX2, KLF4, c-MYC) | FGF/LIF dependent neural stem cells derived from neural rosettes induced by STEM-LIF neural induction media | Nestin | HDc116(21CAG corrected), HD4(72CAG) | Increase in TGF-β precursor, dimer and monomer protein levels; increased SMAD-2 phosphorylation, increased SMAD-2 phosphorylation after human recombinant TGF-β stimulation | [Ring et al., 2015] |
| Retroviral iPSCs (OCT4, SOX2, KLF4, c-MYC) | FGF and LIF dependent NSC produced from selection of neuronal rosettes apparent after plating of EBs (serum dep.) [Zhang et al., 2010] | Nestin | HDc116(21CAG), HD4(72CAG), HFib2-IPS5 (control) | Decreased n-cadherin mRNA, and n-cadherin protein | [An et al., 2012] |
| Lentiviral iPSCs (OCT4, SOX2, KLF4, c-MYC, LIN28, NANOG) | FGF/EGF dependent neural stem cell monolayer derived from dissociated neurospheres | Nestin, PAX6, SOX1, SOX2, Musashi | HD33i.8(33CAG), HD28i.2(28CAG), HD60i.3/HD60i.4(60CAG), HD180i.5/HD180i.7(180CAG) | Decreased actin protein as measured by a phalloidin binding assay | [The HD iPSC Consortium, 2012] |
| Retroviral iPSCs (OCT4, SOX2, KLF4, c-MYC) | FGF/LIF dependent neural stem cells derived from neural rosettes induced by STEM-LIF neural induction media | Nestin | HDc116(21CAG corrected), HD4(72CAG) | Increased netrin mRNA, increase in netrin receptor: DCC, UNC5D mRNA, decrease in netrin receptor mRNA: UNC5CL, UNC5C, UNC5B | [Ring et al., 2015] |
| Retroviral iPSCs (OCT4, SOX2, KLF4, c-MYC) | Telencephalic progenitors induced by small molecule inhibitors: SB431542, LDN193189, IWR1 | Not described | CSD83iCTR33n1(33CAG) and CS21HD60n5(60CAG) | Increase in Akt/mTOR signlaing effector RTP801(REDD1) protein levels | [Martín-Flores et al., 2015] |
| Episomal iPSCs (OCT4, SOX2, KLF4, c-MYC, NANOG) | “Striatal” neuronal cells differentiated from IPS derived neurospheres (iPSCs->embryoid bodies->neuronal rosettes->neurospheres) by FGF withdrawal in B27 media [Chiu et al., 2015] | TUJ1,MAP2, GABA, GAD65,Calbindin,or DARPP-32 | HD-IPS-A1(43CAG), HD-IPS-B4(43CAG), control IPS 1 &2 | Decrease in A2AR mRNA expression, decrease in A2AR protein levels | [Chiu et al., 2015] |
| Retroviral iPSCs (OCT4, SOX2, KLF4, c-MYC)&H9 Embryonic Stem Cells | Neuronal cells derived after FGF withdrawal from neurospheres formed by selection of PA6 cell co-culture induced neural rosettes [Kawasaki et al., 2000] | MAP2 | HD(72CAG), HD2(72CAG), 551-8 IPS(control iPSCs), H9 ES (control Embryonic Stem Cells) | Decrease in western blot detected cytoskeleton associated protein expression: Cfl-1, Stmn-1, Facn-1 and Sept-2, confirmed by immunocytochemistry | [Chae et al., 2012] |
| Retroviral iPSCs (OCT4, SOX2, KLF4, c-MYC)&H9 Embryonic Stem Cells | Neuronal cells derived after FGF withdrawal from neurospheres formed by selection of PA6 cell co-culture induced neural rosettes [Kawasaki et al., 2000] | MAP2 | HD(72CAG), HD2(72CAG), 551-8 IPS(control iPSCs), H9 ES (control Embryonic Stem Cells) | Decrease in oxidative stress response related proteins: SOD1, GST, Gpx1; increase in anti-oxidant response proteins: Prx1,Prx2, Prx6 | [Chae et al., 2012] |
| Retroviral iPSCs (OCT4, SOX2, KLF4, c-MYC)&H9 Embryonic Stem Cells | Neuronal cells derived after FGF withdrawal from neurospheres formed by selection of PA6 cell co-culture induced neural rosettes [Kawasaki et al., 2000] | MAP2 | HD(72CAG), HD2(72CAG), 551-8 IPS(control iPSCs), H9 ES (control Embryonic Stem Cells) | Increase in double strand DNA damage related mRNA: ATM, BFT3; Increase in BFT3 protein and phosphorylation of ATM, p53 and H2AX; Increase in apoptosis related protein cleavage of: Bid, caspase-3, caspase-7 and caspase-9 | [Chae et al., 2012] |
Table 3
Functional phenotypes in HD iPSCs and derived cells
| Initial cell type | Cell type differentiation (Reference) | Cell markers | Clone name (CAG repeat length) | HD cell phenotype | reference |
| Lentiviral (OCT4, SOX2, KLF4, c-MYC) iPSCs | Undifferentiated iPSCs | NANOG, TRA1-81, OCT4 | HD-iPShom4F1/2 and 3F1 (42/44CAG),HD-iPShom4F1/2 and 3F1 (42/44CAG),HD-iPShom4F3 (39/43CAG), HD-iPShet3F1 (17/45CAG), WT-iPShom3F1 (15/17CAG)WT-iPShom4F1 (15/18CAG) | No difference number of mitoticcells as measured by PH3 phospho-histone expression (ICC) | [Camnasio et al., 2012] |
| Retroviral iPSCs (OCT4, SOX2, KLF4, c-MYC) | Undifferentiated iPSCs | Nanog, Sox2, Oct4,SSEA4, TRA-1-60 | HDc116(21CAG), HD4(72CAG), HFib2-IPS5 (control) | No difference in proliferation rate (BRDU assay) | [An et al., 2012] |
| Retroviral iPSCs (OCT4, SOX2, KLF4, c-MYC) | Undifferentiated iPSCs | Nanog, Sox2, Oct4,SSEA4, TRA-1-60 | HDc116(21CAG), HD4(72CAG), HFib2-IPS5 (control) | Decreased ERK phosphorylation in response to FGF | [Zhang et al., 2010] |
| Lentiviral (OCT4, SOX2, KLF4, c-MYC) iPSCs | Undifferentiated iPSCs | NANOG, TRA1-81, OCT4 | HD-iPShom4F1/2 and 3F1 (42/44CAG),HD-iPShom4F1/2 and 3F1 (42/44CAG),HD-iPShom4F3 (39/43CAG), HD-iPShet3F1 (17/45CAG), WT-iPShom3F1 (15/17CAG)WT-iPShom4F1 (15/18CAG) | Increased lysosomal activity as measured by LysoTracker® Red DND-99 fluorescence, increased in response to 48 hrs sucrose treament | [Camnasio et al., 2012] |
| Lentiviral iPSCs (OCT4, SOX2, KLF4, c-MYC) | Undifferentiated iPSCs | NANOG, TRA1-81, OCT4 | HD-iPShom4F1/2 and 3F1 (42/44CAG),HD-iPShom4F3 (39/43CAG), HD-iPShet3F1 (17/45CAG),WT-iPShom3F1 (15/17CAG) | No difference in caspase 3/7 (*** also no unique difference in homozygote vs. heterozygote cells) | [Camnasio et al., 2012] |
| Episomal iPSCs | Undifferentiated iPSCs | Not described. | HD35(5,7,9) (35CAG), HD57(1,4,6,7,15) (57CAG), HD58(1,3,13,19,29,21,31,34) (58CAG), HD70(2,5,11) (70CAG), HD180(1,3,4,6,10,14,16) (180CAG); CA(11,24,26,30), CC(1,2,3,5) CD(2,3,10,12), CE-(2,3,4,6), C-ESS(6,10) (controls) | Increase chromosomal abnormalities after reprogramming based upon karyotype data, no significant differences in number of chromosomal abnormalities accumulated after continuous passaging | [Tidball et al., 2016] |
| Lentiviral iPSCs (OCT4, SOX2, KLF4, c-MYC) | Undifferentiated iPSCs | NANOG, TRA1-81, OCT4 | HD-iPShom4F1/2 and 3F1 (42/44CAG),HD-iPShom4F3 (39/43CAG), HD-iPShet3F1 (17/45CAG),WT-iPShom3F1 (15/17CAG) | No difference in caspase 3/7 (*** also no unique difference in homozygote vs. heterozygote cells) | [Camnasio et al., 2012] |
| Episomal iPSCs | Undifferentiated iPSCs | Not described. | HD35(5,7,9) (35CAG), HD57(1,4,6,7,15) (57CAG), HD58(1,3,13,19,29,21,31,34) (58CAG), HD70(2,5,11) (70CAG), HD180(1,3,4,6,10,14,16) (180CAG); CA(11,24,26,30), CC(1,2,3,5) CD(2,3,10,12), CE-(2,3,4,6), C-ESS(6,10) (controls) | Increase chromosomal abnormalities after reprogramming based upon karyotype data, no significant differences in number of chromosomal abnormalities accumulated after continuous passaging | [Tidball et al., 2016] |
| Retroviral iPSCs (OCT4, SOX2, KLF4, c-MYC) | FGF and LIF dependent NSC produced from selection of neuronal rosettes apparent after plating of EBs (serum dep.) [Zhang et al., 2010] | Nestin | HDc116(21CAG), HD4(72CAG), HFib2-IPS5 (control) | Decreased maximum respiratory rate as measured by oxygen consumption rate after uncoupling via FCCP treatment | [An et al., 2012] |
| Lentiviral iPSCs (OCT4, SOX2, KLF4, c-MYC, LIN28, NANOG) | FGF/EGF dependent neural stem cell monolayer derived from dissociated neurospheres | Nestin, PAX6, SOX1, SOX2, Musashi | HD33i.8(33CAG), HD28i.2(28CAG), HD60i.3/HD60i.4(60CAG), HD180i.5/HD180i.7(180CAG) | Decreased intracellular ATP, decreased ATP/ADP ratio | [The HD iPSC Consortium, 2012] |
| iPSCs (OCT4, SOX2, KLF4, c-MYC, NANOG) | FGF dependent neurospheres (iPSCs->EBs->neuronal rosettes->neurospheres) | TUJ1,MAP2, GABA, GAD65,Calbindin,or DARPP-32 | HD-IPS-A1 (43CAG),HD-IPS-B4 (43CAG) | No difference in expansion rate | [Chiu et al., 2015] |
| Lentiviral iPSCs (OCT4, SOX2, KLF4, c-MYC, LIN28, NANOG) | FGF/EGF dependent neural stem cell monolayer derived from dissociated neurospheres | Nestin, PAX6, SOX1, SOX2, Musashi | HD33i.8(33CAG), HD28i.2(28CAG), HD60i.3/HD60i.4(60CAG), HD180i.5/HD180i.7(180CAG) | Decreased sphere formation from single cell suspension after 12 hours | [The HD iPSC Consortium, 2012] |
| Retroviral iPSCs (OCT4, SOX2, KLF4, c-MYC) | FGF/LIF dependent neural stem cells derived from neural rosettes induced by STEM-LIF neural induction media | Nestin | HDc116(21CAG corrected), HD4(72CAG) | Increase in TGF-β precursor, dimer and monomer protein levels; increased SMAD-2 phosphorylation, increased SMAD-2 phosphorylation after human recombinant TGF-β stimulation | [Ring et al., 2015] |
| Retroviral iPSCs (OCT4, SOX2, KLF4, c-MYC)&H9 Embryonic Stem Cells | FGF-dependent neurospheres derived from PA6 cell co-culture induced neural rosettes, transplanted into QA-lesioned Sprague-Dawley rats [Kawasaki et al., 2000] | Nestin, DARPP32, GABA, MAP2, GAD65/67,SVP38 | HD(72CAG),HD2(72CAG), 551-8 IPS(control iPSCs), H9 ES (control Embryonic Stem Cells) | Form EM-48 positive aggregates, 40 weeks after transplantation | [Jeon et al., 2012] |
| Lentiviral (OCT4, SOX2, KLF4, c-MYC) iPSCs | Neural Cells, Day 5 of neuronal induction using small molecule SB431542 and Noggin | Not described | HD-iPShom4F1/2 and 3F1 (42/44CAG), HD-iPShom4F3 (39/43CAG), HD-iPShet3F1 (17/45CAG), WT-iPShom3F1 (15/17CAG), WT-iPShom4F1 (15/18CAG) | Increased lysosomal activity as measured by LysoTracker® Red DND-99 fluorescence | [Camnasio et al., 2012] |
| Lentiviral iPSCs (OCT4, SOX2, KLF4, c-MYC, LIN28, NANOG) | FGF/EGF dependent neural stem cell monolayer derived from dissociated neurospheres | Nestin, PAX6, SOX1, SOX2, Musashi | HD33i.8(33CAG), HD180i.5/HD180i.7(180CAG) | Increased cleaved caspase-3 | [The HD iPSC Consortium, 2012] |
| Episomal iPSCs | Astrocytes | GFAP, s100β | F-HD-IPS (50CAG), D-HD-IPS (109CAG) | CAG repeat dependent increase in cytoplasmic vacuolization; further increase in vacuolization and LC3(+) vacuoles following chloroquine treatment | [Juopperi et al., 2012] |
| Retroviral iPSCs (OCT4, SOX2, KLF4, c-MYC)&H9 Embryonic Stem Cells | Neuronal cells derived after FGF withdrawal from neurospheres formed by selection of PA6 cell co-culture induced neural rosettes [Kawasaki et al., 2000] | MAP2 | HD(72CAG),HD2(72CAG), 551-8 IPS(control iPSCs), H9 ES (control Embryonic Stem Cells) | Form less MAP2+ neurons, with shorter neurite length | [Chae et al., 2012] |
| Lentiviral iPSCs (OCT4, SOX2, KLF4, c-MYC, LIN28, NANOG) | Neuronal cells differentiated from dissociated FGF/EGF dependent neuropheres after FGF/EGF withdrawal and addition of ascorbic acid and cAMP | MAP2a/b, GABA | HD33i.8(33CAG), HD60i.3/HD60i.4(60CAG), HD180i.5/HD180i.7(180CAG) | 180CAG cells fail to produce inward/outward currents and fire action potentials during whole cell patch clamp recordings | [The HD iPSC Consortium, 2012] |
| Lentiviral iPSCs (OCT4, SOX2, KLF4, c-MYC, LIN28, NANOG) | Neuronal cultures differentiated from dissociated FGF/EGF neurospheres induced by FGF/EGF withdrawal and addition of BDNF, cAMP, Shh, Dkk1, and valproic acid [Aubry et al.,2008] | DARPP32, MAP1, MAP2a/b, Mash1, Bcl-1B, BIII-Tubulin | HD33i.8(33CAG), HD180i.5(180CAG) | Increase in cell death as measured by number of condensed nuclei | [The HD iPSC Consortium, 2012] |
| Episomal iPSCs (OCT4, SOX2, KLF4, L-MYC, LIN28, p53 shRNA) | Striatal-like neurons derived from FGF/EGF dependent neurospheres after FGF withdrawal/induction with: Shh, Dkk1, BDNF, cAMP, and valproic acid | TUJ1, DARPP32 | HD60(60CAG), HD109(109CAG), HD180(180CAG), Controls(28CAG, 33W) | Increase in persistent Nestin+cells after 42 days of differentiation; Nestin+cells are more susceptible to BDNF-withdrawal induced increased cell death as measured by number of TUNEL+nuclei | [Mattis et al., 2015] |
| Lentiviral (OCT4, SOX2, KLF4, c-MYC) iPSCs | Differentiated Neurons, Day 30 of neuronal induction using small molecule SB431542 and Noggin | BIII Tubulin, MAP2, PAX6, Nestin, GABA | HD-iPShom4F1/2 and 3F1 (42/44CAG),HD-iPShet3F1 (17/45CAG),WT-iPShom3F1 (15/17CAG) | Increased macroautophagy as measured by increased LC3 BII/LC3 B1 protein ratio | [Camnasio et al., 2012] |
| Retroviral iPSCs (OCT4, SOX2, KLF4, c-MYC)&H9 Embryonic Stem Cells | Neuronal cells derived after FGF withdrawal from neurospheres formed by selection of PA6 cell co-culture induced neural rosettes [Kawasaki et al., 2000] | MAP2 | HD(72CAG),HD2(72CAG), 551-8 IPS(control iPSCs), H9 ES (control Embryonic Stem Cells) | Increase in cell death as measured by number of TUNEL+nuclei | [Chae et al., 2012] |
| iPSCs (OCT4, SOX2, KLF4, c-MYC, NANOG) | “Striatal” neuronal cells differentiated from IPS derived neurospheres (iPSCs->Ebs->neuronal rosettes->neurospheres) by FGF withdrawal in B27 media [Chiu et al., 2015] | TUJ1,MAP2, GABA, GAD65,Calbindin,or DARPP-32 | HD-IPS-A1 (43CAG),HD-IPS-B4 (43CAG), control IPS 1 &2 | No difference in caspase 3 cleavage or H2AX phosphorylation levels | [Chiu et al. 2015] |
| Lentiviral iPSCs (OCT4, SOX2, KLF4, c-MYC, LIN28, NANOG) | Neurons differentiated from dissociated FGF/EGF neurospheres induced by FGF/EGF withdrawal and addition of BDNF, cAMP, Shh, Dkk1, and valproic acid [Aubry et al.,2008] | DARPP32, MAP1, MAP2a/b, Mash1, Bcl-1B, BIII-Tubulin | HD33i.8(33CAG), HD60i.3/HD60i.4(60CAG), HD180i.5/HD180i.7(180CAG) | Increased risk of cell death as determined by Kaplan-Meier analysis (reversed by addition of 4x concentration of BDNF) | [The HD iPSC Consortium, 2012] |
| Lentiviral (OCT4, SOX2, KLF4, c-MYC) iPSCs | Neurons differentiated after neuroepithelial induction with small molecules: SB431542 and Noggin, followed by striatal specification with Shh, Dkk, BDNF, ascorbic acid and cAMP [[HD iPSC Consortium, 2012]] | BIII Tubulin (TUJ1), MAP2, GAD67, DARPP32 | GM05539 (Juvenile onset- no CAG repeat provided), Contro1 (Control iPSCs) | Increased mitochondrial fragmentation in neurites of GAD67+ neurons, shorter neurite length | [Guo et al., 2013] |
| Lentiviral (OCT4, SOX2, KLF4, c-MYC) iPSCs | Neurons differentiated after neuroepithelial induction with small molecules: SB431542 and Noggin, followed by striatal specification with Shh, Dkk, BDNF, ascorbic acid and cAMP [HD iPSC Consortium, 2012] | BIII Tubulin (TUJ1), MAP2, GAD67, DARPP32 | GM05539 (Juvenile onset- no CAG repeat provided), Control1 (Control iPSCs) | Decreased neurite length in DARPP32+ neurons | [Guo et al., 2013] |
| Lentiviral (OCT4, SOX2, KLF4, c-MYC) iPSCs | Neurons differentiated after neuroepithelial induction with small molecules: SB431542 and Noggin, followed by striatal specification with Shh, Dkk, BDNF, ascorbic acid and cAMP [HD iPSC Consortium, 2012] | BIII Tubulin (TUJ1), MAP2, GAD67, DARPP32 | GM05539 (Juvenile onset- no CAG repeat provided), Control1 (Control iPSCs) | Decreased mitochondrial membrane potential (MMP), increased mitochondrial ROS production, decreased ATP, increased cell death (lactate dehydrogenase level) | [Guo et al., 2013] |
Table 4
Cell response to stress in HD iPSC and derived cells
| Initial cell type | Cell type differentiation (Reference) | Cell markers | Clone name (CAG repeat length) | Stressor | HD cell phenotype | Reference |
| Retroviral iPSCs (OCT4, SOX2, KLF4, c-MYC) | Undifferentiated iPSCs | Nanog, Sox2, Oct4,SSEA4, TRA-1-60 | HDc116(21CAG), HD4(72CAG), HFib2-IPS5 (control) | Growth factor withdrawal | No difference in caspase3/7 activation following serum/growth factor withdrawal | [An et al., 2012] |
| Episomal iPSCs | Undifferentiated iPSCs | Not described | HD58-19(58CAG), HD70-2(70CAG), HD180-4(180CAG); CC-1, CD-2, CE-6 (controls) | Induced DNA breakage | Significantly less of a loss of cell viability in response to neocarzinostatin exposure at multiple concentrations, compared to control cell lines | [Tidball et al., 2016] |
| Episomal iPSCs | Undifferentiated iPSCs | Not described | HD70-2(70CAG), HD180-4(180CAG); CC-1, CE-6 (controls) | Induced DNA breakage | Significantly increased p-p53 and total p53 protein, and p-ATM after neocarzinostatin treatment at multiple concentrations, similar magnitude of change after neocarzinostatin induction as control cells, but HD cells had higher baseline p-p53, p53 and ATM expression | [Tidball et al., 2016] |
| Episomal iPSCs | Astrocytes | GFAP, s100β | F-HD-IPS (50CAG), D-HD-IPS (109CAG) | Inhibition of Protein Clearance | Increase in autophagy; increase in LC3+ cytoplasmic vacuoles after chloroquine treatment | [Juopperi et al., 2012] |
| Episomal iPSCs | Striatal-like neural progenitors induced by small molecules SB31542 and purmorphamine [Delli Carri et al., 2013] | ISLET1, PAX6, FOXG1 | HD70-1(70CAG), HD180-4(180CAG) | Altered Ion Concentration | Decreased p-p53(Ser15), p-CHK2(Thr68) and p-Akt (Ser473) after treatment with Mn2 +; Decreased Mn2 + uptake after 24 hrs; No effect upon caspase3 activation or PARP phosphorylation | [Tidball et al., 2014] |
| Retroviral iPSCs (OCT4, SOX2, KLF4, c-MYC) | FGF and LIF dependent NSC produced from selection of neuronal rosettes apparent after plating of EBs (serum dep.) [Zhang et al., 2010] | Nestin | HDc116(21CAG), HD4(72CAG), HFib2-IPS5 (control) | Growth factor withdrawal | Increase in cell death as indicated by increase in TUNEL+cells and elevated caspase 3/7 activation in response to FGF/LIF withdrawal | [An et al., 2012] |
| Retroviral iPSCs (OCT4, SOX2, KLF4, c-MYC) | FGF/LIF dependent neural stem cells derived from neural rosettes induced by STEM-LIF neural induction media | Nestin | HDc116(21CAG corrected), HD4 (72CAG) | Growth factor withdrawal | Increased caspase3/7 activity and decreased maximal respiratory capacity after FGF/LIF withdrawal | [Ring et al, 2015] |
| Retroviral iPSCs (OCT4, SOX2, KLF4, c-MYC)&H9 Embryonic Stem Cells | FGF dependent NSC produced from selection of neuronal rosettes after plating of EBs (serum dep.) | Nestin, SOX1, PAX6 | HD IPS(72CAG), WT IPS, H9 Embryonic Stem Cells | Growth factor withdrawal | Elevated caspase 3/7 activity in response to EGF withdrawal | [Zhang et al., 2010] |
| Episomal iPSCs | Striatal-like neural progenitors induced by small molecules SB31542 and purmorphamine [Delli Carri et al., 2013] | ISLET1, PAX6, FOXG1 | HD70-1(70CAG), HD180-4(180CAG) | Induced DNA breakage | Elevated p-p53 (Ser15), p-CHK2(Thr68) and p-H2AX(Ser139) in response to double stranded breaks after neocarzinostatin treatment | [Tidball et al., 2014] |
| Lentiviral iPSCs (OCT4, SOX2, KLF4, c-MYC, LIN28, NANOG) | Neurons differentiated from dissociated FGF/EGF neurospheres induced by FGF/EGF withdrawal and addition of BDNF, cAMP, Shh, Dkk1, and valproic acid [Aubry et al.,2008] | DARPP32, MAP1, MAP2a/b, Mash1, Bcl-1B, BIII-Tubulin | HD33i.8(33CAG), HD180i.5(180CAG) | Inhibition of Protein Clearance | Increase in cell death as measured by number of condensed nuclei in response to autophagy inhibition (3-MA) but not proteasome inhibition (lactacystin) | [The HD iPSC Consortium, 2012] |
| Lentiviral iPSCs (OCT4, SOX2, KLF4, c-MYC, LIN28, NANOG) | Neuronal cells differentiated from dissociated FGF/EGF dependent neuropheres after FGF/EGF withdrawal and addition of ascorbic acid and cAMP | MAP2a/b, GABA | HD33i.8(33CAG), HD60i.4(60CAG), HD180i.5/HD180i.7(180CAG) | Glutamate Pulse | Increased calcium dyshomeostasis, further increased in 60CAG in response to 14 days glutamate pretreatment; increased cell death following glutamate pulses as measured by increased in TUNEL+nuclei | [The HD iPSC Consortium, 2012] |
| Episomal iPSCs (OCT4, SOX2, KLF4, L-MYC, LIN28, p53 shRNA) | Striatal-like neurons derived from FGF/EGF dependent neurospheres after FGF withdrawal/induction with: Shh, Dkk1, BDNF, cAMP, and valproic acid | TUJ1, DARPP32 | HD60(60CAG), HD109(109CAG), HD180(180CAG), Controls(28CAG, 33W) | Growth factor withdrawal | BDNF withdrawal induces increased cell death as measured by number of TUNEL+cells | [Mattis et al., 2015] |
| Lentiviral iPSCs (OCT4, SOX2, KLF4, c-MYC, LIN28, NANOG) | Neurons differentiated from dissociated FGF/EGF neurospheres induced by FGF/EGF withdrawal and addition of BDNF, cAMP, Shh, Dkk1, and valproic acid [Aubry et al., 2008] | DARPP32, MAP1, MAP2a/b, Mash1, Bcl-1B, BIII-Tubulin | HD33i.8(33CAG), HD109i.1(109CAG), HD180i.5(180CAG) | Growth factor withdrawal | Increased cell death after 48 hrs. BDNF withdrawal as measured by condensed nuclei | [The HD iPSC Consortium, 2012] |
| Lentiviral iPSCs (OCT4, SOX2, KLF4, c-MYC, LIN28, NANOG) | Neurons differentiated from dissociated FGF/EGF neurospheres induced by FGF/EGF withdrawal and addition of BDNF, cAMP, Shh, Dkk1, and valproic acid [Aubry et al., 2008] | DARPP32, MAP1, MAP2a/b, Mash1, Bcl-1B, BIII-Tubulin | HD33i.8(33CAG), HD28i.2(28CAG), HD60i.3/HD60i.4(60CAG), HD180i.5/HD180i.7(180CAG) | Growth factor withdrawal | Increased caspase-3/7 activity after 24 hrs BDNF withdrawal | [The HD iPSC Consortium, 2012] |
| Episomal iPSCs | Mixed neuronal cells derived from neurospheres [The HD iPSC Consortium, 2012] | GFAP, TUJ1, DARPP32 | GM09197 (180/18CAG), ND39258 (109/19CAG), GM05400 (21/18CAG) | Growth factor withdrawal | Increased cell death as measured by number of TUNEL+nuclei after BDNF withdrawal | [Lu et al., 2014] |
| iPSCs (OCT4, SOX2, KLF4, c-MYC, NANOG) | “Striatal” neuronal cells differentiated from IPS derived neurospheres (iPSCs->Embryoid bodies->neuronal rosettes->neurospheres) by FGF withdrawal in B27 media [Chiu et al., 2015] | TUJ1,MAP2, GABA, GAD65,Calbindin,or DARPP-32 | HD-IPS-A1 (43CAG), HD-IPS-B4 (43CAG), control IPS 1 &2 | H202 Treatment | Dose dependent increase in γH2AX levels after H202 treatment | [Chiu et al., 2015] |
Table 5
Differentiation protocols of medium spiny neurons (MSNs) from human stem cells
| Initial cell Type | Method | Differentiation length | Differentiation efficiency | Transplantation study | Reference |
| Control human ESC | Stromal cell co-culture to produce neural precursor cells, then treated by BDNF, Shh and DKK1, and allowed to mature in the prescence of BDNF but without Shh and DKK1 | 62 days | 22% MAP2+ of which 53% also DARPP32+ | Cells were transplanted at various points(23–72 days) of differentiation into quinolinic (QA)-lesioned rats to determine optimal differentiation stage (eg.neuroepithlial to mature neuron). Tissue was likewise analyzed at multiple timepoints post-graft. Cells transplanted from early differentiation stages produced “teratoma-like regions” in host brains, not present in later stages. Grafts of differentiation Day 45 cells produced DARRP32+ neuron-like cells which co-expressed MAP2 and NeuN. Furthermore, 2 months post-graft animals suffered from weight loss, decreased activity and hemiparesis found to coincide with grafts that expanded outside of the striatum largely due to the proliferation of Nestin+neural precuror cells | [Aubry et al., 2008] Used with HD iPSC by: [Zhang et al., 201]); [HD IPSC Consortium, 2013] |
| Control human ESC | Neuroepithelial cells grown as spheres were induced by treatment with media containing N2 supplement, then supplemented with DKK1/Shh to induce ventral telencephalic precursor fate, and terminal differentiation produced in Neurobasal media, containing N2/B27 supplements, along with trophic factors: BDNF, GDNF and IGF1 | 30 days | 40% ISLET1+ cells, with a population of cells co-expressing GAD65/67, MAP2 cells and BIII-Tubulin, and a further sub-population observed to be DARPP32+ | Not reported | [Li et al., 2009] |
| Control immortalized human striatal NSCs (FGF/EGF dependent) | Neuronal differention was induced in neural stem cells by growth factor withdrawal (FGF/EGF) in B27-supplemented media. Purmorphamine (a Shh agonist) was added for either 7 or 21 days of differentiation (with longer time period yielding higher percentages of DARPP32+ cells) | 3 weeks | ∼30% MAP2+ cells of which 20–25% co-expressed DARPP32 | Not reported | [El-Akabawy et al., 2011] |
| Control human ESC | Pax-6+ neuroepithelial cells grown as spheres were induced by treatment with media containing N2 supplement, then disrupted and treated with Shh *or small molecule agonist purmorphamine) to induce LGE-like progenitors, which were then directed towards a neuronal fate using culture conditions containing BDNF, GDNF, cAMP, IGF and valproic acid | 47 days | 93.2% of cells were BIII-Tubulin+, of which 90.2% were GABA+, while 89.7% of GABA+neurons were also DARPP32+ | Transplanted LGE-like progenitors into striatum of QA-lesioned immunodeficient (SCID) mice, tissue analyzed 4 months post-graft showed the development of human GABA+neurons in both forebrain and spinal tissue. An estimated 58.6% cells of grafted cells were GABA+forebrain cells expressing DARPP32+, co-stained with CTIP+and Meis2+, while remaining cells were largely Nestin+/Pax6- progenitors along with a small population of neuronal cells positive for calretinin, calbindin or parvalbumin. GABA+cells formed connections with host neurons (dopaminergic and glutamatergic). Grafted forebrain LGE-like progenitors also reversed motor deficits in QA-lesioned mice. | [Ma et al., 2012] Used with HD iPSC by: [Yao et al., 2015] |
| Human iPSC and ESC: HD(72CAG), HD2(72CAG), 551-8 IPS(control iPSC), H9 ES (control ESC) | FGF-dependent neurospheres were derived from iPSC using PA6 (stromal) cell co-culture induced neural rosettes, with neuronal maturation encouraged by FGF withdrawal and addition of BDNF (Kawasaki et al., 2000) | Not reported | 27% DARPP32+, and co-expressing: GSH-2 or DLX2 | Neural precursor cells were transplanted into striatum of QA-lesioned Sprague Dawley rats, tissue analyzed 12 weeks post-graft. Both HD and WT transplanted cells formed MAP2+ cells, of which some co-expressed DARPP32 and GAD65/67. Both HD and WT cells likewise led to improvements in several behavioral measures 4–12 weeks post graft. Animals receiving HD iPSC did form EM-48 positive aggregates, 40 weeks after transplantation in a later experiment | [Jeon et al., 2012] |
| Human iPSC: HD33i.8(33CAG), HD28i.2(28CAG), HD60i.3/HD60i.4(60CAG), HD180i.5/HD180i.7 (180CAG) | Based upon Aubry et al. (2008) protocol, neurons differentiated from dissociated FGF/EGF neurospheres after FGF/EGF withdrawal and addition of BDNF, cAMP, Shh, Dkk1, and valproic acid | 72 days | <10% BIII-Tubulin+or MAP2a/b+,<5% were DARPP-32/Bcl11B positive in all cell lines, but significant variability was observed between differentiations | Not reported | [HD iPSC Consortium, 2012] Used with HD iPSC by: [Guo et al., 2013] |
| Control human ESC and iPSC | Dual SMAD inhibition using SB431542/Noggin (or dorsomorphin/LDN-193189) per Chambers et al. (2009), with ventral telencephalic fate specified by Shh treatment | 60 days | 51% MAP2ab+, of which 20% are also DARPP32+ /CTIP2 (BCl11B)+ | Transplanted human ESC derived cells after 38 days of differentiation into striatum of QA-lesioned rats, tissue analyzed at 3, 6 and 9 weeks post-graft. After 3 weeks, some cells maintained proliferative potential (Ki67+), while after 6 weeks the graft contained a large population of MAP2ab+/BIII-Tubulin+neurons, and after 9 weeks cells expressing telecephalic markers: FOXP1/FOXP2, and MSN marker: DARPP32 were observed. Grafts also improved a behavioral phenotype in lesioned rats: apomorphine-induced turns were decrease in animals receiving grafts | [Delli Carri et al., 2013] Used with HD iPSC by: [Tidball et al., 2014] |
| Control human ESC and iPSC | Dual SMAD inhibition using SB431542/LDN-193189 based upon Chambers et al. (2009), with ventral telencephalic fate specified via Shh and DKK1 treatment | 45 days | ∼40% MAP2+, of which< 5% also DARPP32+ | Transplanted human ESC derived cells after 25 days of differentiation into striatum of quinolinic acid lesioned adult nude rats, tissue analyzed 5 months post-graft, showed significant DARPP32+ cells, co-stained with CTIP2 and FOXP1 | [Nicoleau et al., 2013] |
| Control human fibroblasts (post-natal and adult) | Neural induction of human fibroblasts directed by lentiviral infection with miRNAs: MiR-9/9* and miR-124 as described by Yoo et al. (2011), and transcription factors: CTIP2(BCL11B), DLX1, DLX2, and MYT1L (CDM) to further specify DARPP32+ neuronal cell fate | 35 days | 90% MAP2+ neurons, of which 80% were GABA+, while 70% were DARPP32+ | Transplanted EGFP expressing human fibroblasts into striatum of immunodeficient mice, two weeks after neural induction via lentiviral infection, tissue analyzed Day 50 post-graft. 91% of MAP2+ cells co-expressed DARPP32. Cells were observed to have formed a high density of dendritic spines, to be electrophysiologally active, and developed axonal projections to the substantia nigra and globus pallidus. However, a small number of cells were observed to have migrated outside of the striatal boundary, particulalry those failing to express striatal specific (CTIP2) after lentiviral infection | [Victor et al., 2014] |
| Control human iPSC | Two separate differentiation protocols were used to drive a neuronal fate from human iPSC derived neural precursor cells in N2/B27 supplemented media | (two step): 21 days (three step): 60 days | (two step): 51% of MAP2+ cells co-expressed DARPP32 | Not reported | [Lin et al., 2015] |
| (two step): induction by treatment with valproic acid, followed by maturation in media containing BDNF, GDNF, IGF1 and dibutyryl-cAMP | (three step): 86% of MAP2+ cells co-expressed DARPP32 | ||||
| (three step):induction using BDNF, Shh, DKK-1 and rock inhibitor Y-27632, maintenance in BDNF, Shh and DKK-1 containing media, and maturation in media containing only BDNF | |||||
| Human iPSC: HD-IPS-B4 (43CAG) | Neuroepithelial cells were grown from of iPSC after formation of embryoid bodies, and selection for neural rosettes in N2/B27 supplemented media. Neuronal fate was further encouraged by growth factor withdrawal (FGF) | 16 weeks | 60–80% DARPP32+ /GABA+cells | Not reported | [Chiu et al., 2015] |
| Control human | Dual SMAD inhibition using SB431542/dorsomorphin/LDN with ventral telencephalic fate specified by Activin A treatment | 36 days | 20–50% DARPP32+, which coexpress: BCl11B(CTIP2)+,GABA+, GAD65/67+ and PSD95+ | Transplanted LGE-like neural precursor cells into the striatum of a rat model of HD, and tissue analyzed 4,8, and 16 weeks post-graft. DARPP32+ cells were apparent after 8 weeks, and comprised of 49% of graft cells present at 16 weeks. Nestin+cells were observed at 4 and 8 weeks, but had disappeared by 16 weeks. Graft cells received mid-brain dopaminergic and cortical glutamatergic inputs. No behavioral improvement was observed in grafted HD rats | [Arber et al.,2015] |
| Human iPSC: HD109(109CAG), HD180(180CAG), Controls(28CAG, 33W) | After FGF/EGF dependent NSCs grown as spheres were derived from human iPSC, neuronal differentiation was induced by FGF withdrawal and culture with: Shh, Dkk1, BDNF, cAMP, and valproic acid | 42 days | DARPP32+ /TUJ1+ neuronal cells described, but efficiency not reported | Not reported | [Mattis et al., 2015] |
Table 6
Testing novel compounds in HD iPSCs and derived cells
| Initial cell type | Differentiated cell type (Reference) | Cell markers | Clone name (CAG repeat length) | Treatment type | Effect on HD cell phenotype | Reference |
| Retroviral (OCT4, SOX2, Klf4 and c-MYC) iPSCs | Neuronal cells differentiated in N3 media [Wernig et al., 2002] | BIII-Tubulin | GM23225 (72CAG) | miR-196a delivered by lentiviral (FUW) infection | Decrease in EM-48 positive huntingtin MG-132 induced aggregates, and lowered levels of MAB 2166 detected mutant Htt protein | [Cheng et al., 2013] |
| iPSCs (OCT4, SOX2, KLF4, c-MYC, NANOG) | “Striatal” neuronal cells differentiated from IPS derived neurospheres (iPSCs⟶embryoid bodies⟶neuronal rosettes⟶neurospheres) by FGF withdrawal in B27 media [Chiuet al., 2015] | TUJ1,MAP2, GABA, GAD65, Calbindin,or DARPP-32 | HD-IPS-A1 (43CAG) HD-IPS-B4 (43CAG) | A2AR agonists: CGS-21680 &APEC | Dose dependent reduction in H2O2 induced activation of γH2AX, active/pro-caspase3 ratio and TUNEL+nuclei; effect reversed by A2AR shRNA, A2AR antagonism (SCH5826)or selective PKA inhibition (H-89) | [Chiu et al., 2015] |
| Retroviral (Oct3/4, NANOG, SOX2, KLF4, c-MYC) iPSCs | Astrocytes derived from iPSCs in B27 media+ciliary neurotrophic factor[Hsiaoet al., 2014] | GFAP | HD IPS- Patient A(43CAG) | TNF-α inhibitor: XPro-1595 | Decrease in cytokine: TNF- α+IL-1β induced iNOS production | [Hsiao et al., 2014] |
| Neuronal cells derived from NSCs (EBs⟶NSCs) by FGF withdrawal in B27 media [Hsiao et al., 2014] | BIII-Tubulin | HD IPS- Patient A(43CAG) | TNF-α inhibitor: XPro-1595 | Decrease in cytokine: TNF- α+IL-1β induced caspase3 cleavage | ||
| Retroviral (OCT4, KLF4, SOX2 and c-MYC) iPSCs | Neurons derived from ventral progenitors (induced with Shh and purmorphamine) [Ma et al., 2012] | TUJ1, GABA, DARPP32 | HD IPS (47CAG) HD IPS (70CAG) | GPR52 siRNA | Reduces mHtt protein levels (2B7 and HTRF assay) | [Yao et al., 2015] |
| HD IPS (47CAG) | GPR52 siRNA | Reduces BDNF-withdrawal induced caspase 3 activation, neuronal loss as measured by density of TUJ1/DAPI (+) cells and increases neuronal process complexity (Sholl Analysis) | ||||
| Episomal iPSCs | Mixed neuronal cells derived from neurospheres [The HD iPSC Consortium, 2012] | GFAP, TUJ1, DARPP32 | GM09197 (180/18CAG) ND39258 (109/19CAG) GM05400 (21/18CAG) | ATM inhibitor: KU60019 | Reduces BDNF-withdrawal induced TUNEL(+) nuclei in HD iPSCs derived cells with no effect on control cells | [Lu et al., 2014] |
| Episomal iPSCs | Medium spiny neurons derivedusing MS5-cell co-culture [Aubry et al., 2008] | GFAP, TUJ1, DARPP32 | HD IPS 60i4 (60CAG) | PPARδ activator: KD3010 | Reduces BDNF-withdrawal induced cell death asmeasured by the degree of hoesct-detected nuclear condensation | [Dickey et al., 2015] |
| Episomal iPSCs | Striatal-like neural progenitors induced by small molecules SB31542 and purmorphamine [Delli Carri et al., 2013] | ISLET1, PAX6, FOXG1 | HD70-1(70CAG), HD180-4(180CAG) | ATM inhibitor: KU55933 | Reverses decreased phosphorylation of p53 in response Mn2 +concentration increase, and increased phosphorylation of p53, CHK2 and H2AX in response to neocarzinostatin treatment | [Tidball et al., 2014] |
| Episomal iPSCs (OCT4, SOX2, KLF4, c-MYC, LIN28, p53 shRNA) | Striatal-like neurons derived from FGF/EGF dependent neurospheres after FGF withdrawal/induction with: Shh, Dkk1, BDNF, cAMP, and valproic acid | TUJ1, DARPP32 | HD109(109CAG), HD180(180CAG), Controls(28CAG, 33W) | Calcium Chelator: BAPTA TRKB antibody agonist:αTrkB MAPK signaling (p38) inhibitor: SB239063 allele specific antisense oligonucleotides: ASO-1, 2 &3 NMDAR antagonism:memantine; AMPA/Kainate antagonism: CNQX; mGlu I/IIglutamate receptor antagonism:S-MCPG | Reverses BDNF withdrawal induced cell death as measured by number of TUNEL+nuclei | [Mattis et al., 2015] |
| Retroviral iPSCs (OCT4, SOX2, KLF4, c-MYC) | FGF/LIF dependent neural stem cells derived from neural rosettes induced by STEM-LIF neural induction media | Nestin | HDc116(21CAG corrected), HD4 (72CAG) | Human Recombinant TGFβ1/2 Netrin | Reverses increase in caspase3/7 activity and decrease in maximal respiratory capacity after FGF/LIF withdrawal | [Ring et al., 2015] |
| Lentiviral (OCT4, SOX2, KLF4, c-MYC) iPSCs | Neurons differentiated after neuroepithelial induction with small molecules: SB431542 and Noggin, followed by striatal specification with Shh, Dkk, BDNF, ascorbic acid and cAMP [HD iPSC Consortium, 2013] | BIII Tubulin (TUJ1), MAP2, GAD67, DARPP32 | GM05539 (Juvenile onset- no CAG repeat provided), Control-1 (Control iPSCs) | Drp1-selective peptide inhibitor: P110-TAT | Reverses mitochondrial fragmentation and shortened neurites in GAD67+ neurons, shortened neurites in DARPP32+ neurons, and decreased mitochondrial membrane potential (MMP), increased mitochondrial ROS production, decreased ATP, and increased cell death (lactate dehydrogenase level) in mixed neuronal culture; shRNA to p53 likewise reversed decreases in neurite length in GAD67+ neurons and MMP in mixed neuronal culture, as well as increases in mitochondrial ROS production and cell death | [Guo et al., 2013] |
| Retroviral (OCT4, SOX2, Klf4 and c-MYC) | FGF/EGF dependent neural stem cells grown as a monolayer after neuroepithelial induction with SB431542 and Noggin [Feyeux et al., 2012] | Not described | HD1-IPS4 (72CAG), RC9 and SA-01(WT Embryonic Stem Cells) | Repressor element-1 silencing transcription factor (REST) inhibitor: X5050 | Dose dependent reduction in REST activity (luciferase activity) and increase in RE1 gene mRNA: SNAP25, BDNF, and Syp; similar levels of activity and transcriptional increases to NSC derived from WT Embryonic Stem Cells | [Charbordet al., 2013] |


