
Cerebrospinal Fluid Biomarkers for Huntington’s Disease
Abstract
Cerebrospinal fluid (CSF) is enriched in brain-derived components and represents an accessible and appealing means of interrogating the CNS milieu to study neurodegenerative diseases and identify biomarkers to facilitate the development of novel therapeutics. Many such CSF biomarkers have been proposed for Huntington’s disease (HD) but none has been validated for clinical trial use. Across many studies proposing dozens of biomarker candidates, there is a notable lack of statistical power, consistency, rigor and validation. Here we review proposed CSF biomarkers including neurotransmitters, transglutaminase activity, kynurenine pathway metabolites, oxidative stress markers, inflammatory markers, neuroendocrine markers, protein markers of neuronal death, proteomic approaches and mutant huntingtin protein itself. We reflect on the need for large-scale, standardized CSF collections with detailed phenotypic data to validate and qualify much-needed CSF biomarkers for clinical trial use in HD.
INTRODUCTION
Despite over two decades of research since the identification of the specific cause of Huntington’s disease (HD), there remains no treatment that can prevent the onset or slow down the progression of this devastating autosomal dominant neurodegenerative condition [1].
The cellular and molecular processes underlying the natural history of the disease have been well characterized since the causative gene mutation in HTT was discovered in 1993 [2]. A CAG trinucleotide repeat expansion leads to a mutant, polyglutamine-expanded form of the HTT protein (mHTT) which aggregates and is toxic to neuronal cells, particularly to vulnerable striatal cells. This gives rise to neuronal dysfunction and death and ultimately the signs and symptoms of HD [3].
However, several novel therapeutic candidates are in ongoing or planned clinical trials, perhaps most noteworthy being the various ‘gene silencing’ or huntingtin-lowering approaches that use targeted nucleotide-based therapeutics to reduce expression of mHTT. The first clinical trial of such a therapy – an antisense oligonucleotide therapeutic called Ionis-HTTRx – is now underway [4].
Current clinical outcome measures such as the unified Huntington’s disease rating scale (UHDRS) [5] are well-established and will remain important for the conduct of trials and licensing of novel drugs. But they are limited by lack of ability to distinguish between symptom relief and amelioration of the underlying pathological process, and are by nature subjective and subject to inter- and intra-rater variability and the influence of confounders such as the placebo effect [6]. In addition, by definition they offer no ability to detect preclinical changes that occur in HTT gene mutation carriers who are yet to develop signs of symptoms of HD. With the ultimate goal of treating individuals to prevent neurodegeneration and disease onset, this shortfall presents a huge obstacle in the design of future trials of prospective therapies.
The development of disease-specific biomarkers has never been more important. In order for these treatments to be validated it will be essential to have objective and reliable measures.
A biomarker may serve one or more specific functions: diagnostic, aiding more precise definition of onset; prognostic, giving an indication of how the disease will develop in an individual; natural history, to more accurately determine disease stage; and finally pharmacokinetic, pharmacodynamic and efficacy biomarkers, that may independently assess therapeutic response to and the effectiveness of a potential intervention. Biomarkers need to be quantitative and ideally, quantification should be easy and cheap; certainly, it should be reliable. Some particularly robust biomarkers may, after an extensive, formal process of evaluation and validation [7, 8], be qualified for use as surrogate endpoints for clinical trials leading to regulatory approval, but many possible functions of biomarkers such as elucidating pathobiology and informing go/no-go decisions can be fulfilled with less stringent levels ofscrutiny.
Much progress has been made in recent years in developing and evaluating biomarkers for HD, most notably in the fields of quantitative clinical measures and cognitive measures where studies such as TRACK-HD have, through head-to-head comparison of candidate biomarkers, effectively defined a toolkit of robust biomarkers for use as outcome measures in clinical trials [9, 10]. Numerous trials now in progress employ these biomarkers as secondary or exploratory endpoints (regulatory approval on the basis of a surrogate primary endpoint is a milestone yet to be attained by any biomarker in neurodegenerative disease) [11–13].
Modifying the course of a progressive disease requires an understanding of the chemistry and biology of health, their derangement by the disease, and their modulation by an intervention. This is difficult to do without tools to interrogate directly the biochemical milieu of the affected system – in the case of HD, the human central nervous system. Direct sampling of the target organ by brain biopsy is possible but carries high risk. Conventional imaging methods can tell us about brain structure but are indirect and cannot report on the biochemical changes underlying structural pathology. Molecular imaging techniques, such as magnetic resonance spectroscopy or positron emission tomography, can only report on a limited repertoire of chemical substances or changes. Biofluid markers that reflect neuropathology, ideally by virtue of being centrally involved in it, are therefore highly desirable – but so far notably lacking in HD [3, 6, 14].
It is theoretically possible to identify and quantify substances in more accessible biofluids, such as blood and urine, that are derived from the CNS and altered by the progression of a neurodegenerative disease, and can be distinguished from peripherally-derived substances. The quest for robust biofluid biomarkers in HD has hitherto yielded no such substances. Changes have been reported that are detectable in peripheral blood and that reflect matched peripheral and central processes, including putative neuroinflammatory [15] and metabolic markers [16]. In most cases the overwhelming mass of the peripheral tissues inevitably dwarfs the contribution of substances that are released from dying neurons and can penetrate the blood-brain barrier, especially given the very slow rate of CNS cell death in HD. Even seemingly ‘CNS-specific’ entities that can be detected in peripheral blood, such as brain-derived neurotrophic factor (BDNF) [17], may have peripheral sources that confound their interpretation [18].
Changes in peripheral biofluids that nonetheless mirror neuropathology could have some function as biomarkers of early efficacy, but only in the very defined situation of a therapeutic confidently expected to act in the same manner centrally and peripherally; or as an indication of ‘overflow’ into the peripheral circulation of a CNS-administered drug. TR-FRET detection of mHTT in blood-derived leukocytes might, for instance, be used to assess target engagement by a peripherally-administered huntingtin-lowering compound [19] that was known to be brain-penetrant.
There remains a clear need for biochemical markers that can reliably inform us about neuropathology. Cerebrospinal fluid (CSF), is produced within the CNS and, while it begins as an ultrafiltrate of blood plasma, it receives substantive contributions from the brain. That 20% of its protein content is brain-derived [20] gives reason to hope that relevant CSF biomarkers can be identified. Experience from other neurodegenerative diseases suggests that such markers can be found and can be useful – e.g. β-amyloid and tau protein in Alzheimer’s disease (AD) [21] or α-synuclein and phosphorylated tau in Parkinson’s disease (PD) [22] and that the lumbar puncture (LP) or ‘spinal tap’ procedure by which CSF is routinely obtained is safe and well-tolerated even for observational studies [23]. Despite our certainty of the cause of the disease, the field of CSF biomarkers for HD lags behind the more common neurodegenerativediseases.
Studying CSF is not a novel idea in HD research. Molecules have been quantified in HD CSF since the early 1970s but progress has been hampered by low sample numbers, inconsistent procedures, relatively unreliable quantification techniques and a lack of targets investigated with a plausible mechanistic link to HD pathology. Few if any results have been replicated and we have no validated CSF biomarkers for any aspect of HD.
Nonetheless there has been much recent progress. Here we review previous CSF studies in HD and consider what robust biomarker leads have been identified. Figure 1 summarizes HD CSF biomarker research over the past four decades. We conclude with a focus on the most promising targets and work underway to prosecute them efficiently.
NEUROTRANSMITTERS
The earliest studies examining CSF in HD took a particular interest in neurotransmitters as a quantifiable surrogate for neuronal function and dysfunction. GABAergic medium spiny neurons were known to degenerate early in HD, triggering studies of CSF GABA. The first study successfully to detect GABA in HD CSF, in 1977, used a radioreceptor assay [24]. Lower GABA levels were observed in 19 HD patient CSF samples compared to a control group of 26 subjects with other neurological and psychiatric disorders [25]. Manyam and colleagues included individuals at-risk for developing HD in their investigation of CSF GABA in HD [26] (it would be 5 further years before a genetic marker was found and 15 before the HTT gene was identified). A subset of the at-risk individuals had lower than normal CSF GABA closer to the patients with HD, suggesting there could be early changes in GABA before symptoms manifest. Homocarnosine, a dipeptide containing GABA, was later found to be significantly lower in HD patient CSF [27]. In contrast Bonnet et al. found total GABA and homocarnosine to be significantly higher in HD CSF, in conflict with previous results [28]. The promise of GABA-related markers was further dampened after results of a randomized, double-blind, placebo-controlled crossover, clinical trial in 4 HD patients using Isoniazid (with pyridoxine), a treatment which increased GABA [29]. This failed to produce significant improvement in clinical measurements [30] but did produce side effects. A later study analyzing the samples obtained from this trial revealed that, in addition to GABA elevation, isoniazid treatment increased levels of aspartate, asparagine, homocarnosine, ornithine, histidine, a-aminobutyric acid, isoleucine, leucine and alanine in CSF [31]. Manyam et al. concluded that the therapy’s effect on amino acids in general likely contributed to the undesired clinical response observed as a result of the treatment. GABA has not been re-studied since 1987 but, on the evidence to date, it seems unlikely to be a clinically relevant marker ofdisease [32].
Similarly to GABA, choline-related proteins were measured in HD CSF because of loss of neurons using acetylcholine and the caudate, affected disproportionately in HD, having the highest acetylcholinesterase (AChE) activity in the brain. After having shown the broad effects of isoniazid, Manyam et al. extended their study to include choline [33]. They measured levels of choline and AChE activity in CSF samples from the 6 placebo-treated HD patients, and 9 controls who did not take part in the trial. Choline was significantly lower after age adjustment in HD patients compared to controls, but AChE showed no difference. There was no effect on choline levels and AChE activity after treatment with isoniazid.
Other amino acids were investigated using high pressure ion-exchange chromatography in a study involving 12 HD patients, 8 at-risk offspring and 16 non-choreic controls, each having morning CSF collection after fasting – a study design ahead of its time for minimizing potential confounders [34]. Out of the 27 measured, lower levels in HD patients were seen in asparagine, isoleucine, leucine, phenylalanine, histidine, arginine, alpha-aminoadipic acid and homocarnosine compared to non-choreic controls, with tyrosine the only to increase. At-risk offspring were split into two groups with 5 stated to be “neurophysiologically conspicuous offspring” (based on eye movement disturbance in electronystagmographic examinations) and displaying a similar trend of amino acid imbalance in the CSF. This may indicate early disturbance of metabolism in HD but because of low sample number, lack of a genetic test to identify gene carriers in the at-risk offspring, and no replication of these results since the genetic test was established, it remains unclear whether any neurotransmitter amino acids could provide early markers of disease onset.
The striatum receives dopaminergic input from the substantia nigra. Neuronal loss in the striatum – and therefore loss of postsynaptic striatal dopamine receptors – during the course of HD creates an imbalance due to a reduction in dopamine uptake (DA). This disproportion was thought to potentially contribute to chorea on the basis of an overactive effect of dopamine on the striatum [35]. Dopamine (DA) metabolites were therefore a source of early interest as possible CSF markers.
Homovanillic acid (HVA) a CSF metabolite of DA and indicator of dopaminergic activity, has been measured in HD on several occasions, generating contradictory findings. Where some have found no difference in CSF HVA in HD [36, 37] others reported a reduction [38] – neither case supporting the original theory of overactive dopamine. DA metabolites studied by Garrett and colleagues in HD CSF included DA itself, 3,4-dihydroxyphenylacetic acid (DOPAC), homovanillic acid (HVA), noradrenaline (NA), 3-methoxy-4-hydroxyphenylglycol (MHPG), and 5-hydroxyindoleacetic acid (5-HIAA). Increased CSF levels of DA and DOPAC were taken to reflect increased dopaminergic activity in HD yet levels of HVA were not significantly different [39].
García Ruiz and colleagues again measured CSF levels of HVA, 5-HIAA, and tryptophan in 20 HD patients against four control groups: 15 healthy controls, 23 untreated PD patients, 38 patients with dystonia and 61 patients with other neurological disorders, using high-performance liquid chromatography (HPLC) with electrochemical detection [40]. HVA was significantly lower in HD compared to controls and other neurological patients adding more evidence that HVA is not a reliable marker of dopaminergic activity and the true role of the dopaminergic pathway in HD is poorly understood.
TRANSGLUTAMINASE ACTIVITY
Polyglutamine – the expanded amino acid sequence that confers toxicity upon mHTT – has been shown to be a good substrate for transglutaminases, enzymes that catalyze linkage reactions between substances containing glutaminyl and lysyl residues or polyamines. Polyglutamine expansion in HD increases its effectiveness as a substrate for transglutaminases in vitro. Transglutaminases have been implicated in the aggregation of mHTT, and cystamine (among other actions, an inhibitor of transglutaminase) ameliorated symptoms in transgenic HD mice, supporting the notion that transglutaminase activity may contribute to the toxic gain of function of mHTT [41]. Nɛ-(γ-l-Glutamyl)-l-lysine (GGEL) is one substance produced from a transglutaminase reaction and therefore used as a marker for transglutaminase activity. Jeitner et al. found significantly higher levels of GGEL in CSF of 36 HD patients compared to 27 controls (being treated for various spinal injuries) using liquid chromatography with electrochemical detection (LCEC) [42]. Building on the previous evidence from GGEL levels in HD CSF, Jeitner and colleagues went on to investigate whether other glutamylpolyamines would also reflect alterations of TGase activity in HD. They used HPLC-EC to quantify GGEL, γ-glutamylspermidine, γ-glutamylputrescine, and bis-γ-glutamylputrescine simultaneously in CSF [43]. These were all found to be elevated in manifest HD compared to controls. Cysteamine, a reduced form of cystamine, was recently tested in a clinical trial in HD patients. The results have only so far been released in unreviewed summary form; the drug did not meet its primary endpoint and CSF was not collected so pharmacodynamic data on the compound’s putative transglutaminase inhibitor mechanism will not be forthcoming [44].
KYNURENINE PATHWAY METABOLITES
The NMDA receptor agonist quinolinic acid (QA), and the concept of excitotoxicity as a contributor to neuropathology, became of huge interest in HD CSF after it was discovered that direct intra-striatal administration is selectively toxic to medium spiny neurons and produces deficits that mimic some aspects of HD. Before the identification of the causative gene, QA-lesioned rodents were the main experimental model for HD.
QA is a downstream product of the kynurenine pathway, by which the neurotransmitter amino acid tryptophan is degraded in mammals [45]. As well as its possible role in HD neuropathology, affirmed by subsequent work in animal models and human post-mortem samples [46, 47], the pathway and in particular the enzyme kynurenine mono-oxygenase, is a high-priority target for therapeutic development [1]. Its study in CSF also serves to illustrate the historical shortcomings in the field and the urgent need for greater rigor.
From the observations in animal and human brains, it was initially hypothesized that an increase would be seen in QA in HD patient CSF but in one study using a radioenzymatic assay, no significant difference was observed [48]. This study included just 10 patients and 7 controls and was most likely underpowered to detect an effect, especially given its controls were patients with schizophrenia and there was no standardization of dietary conditions (known to affect KP metabolite levels) or the time of day when samples were collected, and no information recorded about medications or the processing of CSF samples.
In a second study three years later, a similar result was found but this was a faithful replication inasmuch as it suffered the same shortcomings as the original work: Small subject numbers (9 patients and 9 ‘hospital patient’ controls), no standardization of sampling or processing conditions and insufficient information about possible confounds such as medication [49].
In a broad-ranging study in 1992, Heyes and colleagues set out to quantify several KP metabolites in CSF from HD patients and a broad range of other neurological and physical diseases. The total number of subjects was large but the HD sample relatively small and inconsistently reported (the number of patients was reported as both 30 and 13). Using HPLC they found CSF levels of the neuroprotective metabolite kynurenic acid (KA) were lower in HD than in controls, and a similar pattern in Alzheimer’s disease; CSF QA was elevated in inflammatory disease but not altered in neurodegenerative diseases [50]. The decreased KA finding echoed an earlier report from 23 HD patients and 50 controls “undergoing myelography or being evaluated for fever or headache” by Beal and colleagues [51].
This cluster of publications on CSF kynurenine pathway metabolites is edifying. There were – and still are – strong reasons to suspect that relevant, disease-related alterations in metabolites ought to be detectable in CSF; but the only studies available to us in 2016 were conducted using inconsistent methods over two decades ago – before we could even be certain that the patient volunteers had HD rather than a phenocopy syndrome – and yielded mixed or negative results we cannot interpret because they likely lacked the power to test their hypotheses definitively.
OXIDATIVE STRESS
Oxidative damage has been associated with many neurodegenerative diseases including HD [52] and through early QA-lesioned animal models, their study overlaps with the kynurenine pathway. Markers of oxidative stress have been investigated in HD blood plasma and brain tissue of disease models but few have been quantified in CSF. F2-isoprostanes are a marker for lipid peroxidation. Using gas chromatography/negative ion chemical ionization mass spectrometry (GC/NICIMS) these were found to be significantly elevated in 20 HD patients but not in 7 patients with multiple system atrophy or in 23 controls (who underwent CSF collection for diagnostic purposes) [53]. Montine and colleagues had previously shown increased levels of F2-isoprostanes in AD compared to controls [54] and used these results for direct comparison to HD. It is important to note that there was large overlap in disease groups and controls, so the utility of F2-isoprostanes as markers of disease state appears low. Several clinical trials of purportedly antioxidant compounds such as ethyl-EPA [55] and coenzyme Q10 [56] have produced negative clinical outcomes and the lack of suitable pharmacodynamic CSF biomarkers precludes the interpretation of possible subclinical effects.
INFLAMMATORY MARKERS
Overactivation of myeloid cells, including microglia, and subtle abnormalities of the innate immune system, are among the earliest biochemical changes that have so far been detected in HD patients [15, 57]. Most of this work has been carried out in peripheral blood or ex vivo cells [58] but in 2007 Dalrymple and colleagues found that plasma elevations of clusterin were mirrored in CSF from 20 patients and 10 controls in the first report from a CSF collection with dedicated matched contemporaneous healthy controls from the general population, rather than from patients under investigation for other conditions [12]. Their finding of elevated IL-6 and IL-8 in blood plasma was reproduced in patient CSF in 2008 by Björkqvist, et al. [15].
More recently, YKL-40, a member of the glycosyl hydrolase family 18 and a marker of microglial activation [59], was investigated as a CSF marker in 54 HD mutation carriers (27 manifest and 27 premanifest) after reported elevation in other diseases associated with neuroinflammation [60]. YKL-40 was noted to increase with age in healthy controls but, after age-correction only a trend of elevated YKL-40 was seen in manifest HD that did not reach statistical significance. Novel therapeutic approaches include compounds aimed at modulating microglial activity (e.g. the current Legato-HD trial of laquinimod [11]) so YKL-40 and other microglial and inflammatory markers remain of interest. It may be that an emphasis on standardization of CSF collection and processing methods, to eliminate variation from time of day, medication or diet, can reveal these substances to be useful markers reflecting relevant and tractable disease-related changes in HD.
Similar to other neurodegenerative diseases, there is an increase in clusterin levels as well as cytokines IL-6 and IL-8 reported in HD [57]. A small sample proteomic study detected significantly higher levels of prothrombin and haptoglobin which both are proteins associated with inflammatory response [61]. Like clusterin, IL-6 and IL-8, upregulation of haptoglobin has been seen in other diseases including AD and traumatic brain injury [62, 63]. Prothrombin correlated with disease severity and has yet to be tested in other diseases. Further investigation will be needed to ascertain its effectiveness as a CSF marker for disease progression specific to HD.
NEUROENDOCRINE MARKERS
Hypothalamic neuroendocrine abnormalities have been suggested to contribute to many symptoms seen in HD such as weight loss, depression and disruption of sleep [64], supported by evidence of hypothalamic cell loss. Neuroendocrine markers have been targets for several CSF studies in HD. Corticotropin releasing factor (CRF) and 5-HIAA were investigated in HD after higher and lower levels, respectively, were observed in the CSF of patients suffering from major depression [65, 66]. CRF and 5-HIAA were measured in 56 early stage HD patients (drug-free) using a radioimmunoassay (for CRF) and HPLC (for 5-HIAA) [67]. Aliquots were taken from 21 subjects with no known neurological disease and undergoing spinal anesthesia for control comparison of CRF, while a control group of 4 individuals was used to compare levels of 5-HIAA.Mean CRF was higher in HD patients (with or without depression) than controls, and correlated with severity of depression whereas 5-HIAA showed no significant difference. Again, low sample numbers are limiting and no demographic information was presented for the control group. Although a biochemical underpinning for depressive symptoms in HD seems plausible, little can be concluded with confidence here.
Ghrelin, the ‘hunger’ hormone which also regulates the reward mechanism in dopamine neurons and effects sleep and depression, was quantified in plasma and CSF (15 HD patients and 20 healthy controls) alongside leptin, a hormone associated with satiety [68]. Ghrelin was increased and leptin decreased in plasma of the HD patients but neither was altered in CSF. Orexin A (hypocretin) levels were investigated by Björkqvist and colleagues in 37 HD patients and 30 controls, but no significant differences were found and this was replicated in two separate studies [69–71]. Cocaine and amphetamine regulated transcript (CART), a neuropeptide associated in increased anxiety and lower food intake was significantly higher in HD CSF (n = 39) compared to control CSF (n = 29), after quantification using a radioimmunoassay [72]. Björkqvist and colleagues investigated the relationship between anxiety symptoms and levels of CSF CART but found no significant correlation. Evidence for elevated CART in HD CSF was replicated by Gaberyet al. [73].
PROTEIN MARKERS OF NEURONAL DEATH
Proteins released by dying neurons that can be quantified in CSF have been studied much more comprehensively in other neurological diseases than in HD. Tau protein (a component of microtubules) and the light subunit of neurofilament triple protein (NFL, a component of the neuronal cytoskeleton) are reasonably well-established as markers of neuronal death that appear to be widely applicable across neurodegenerative diseases [74]. Tau in particular is already in clinical use in the diagnostic process of Alzheimer’s disease alongside with levels of beta-amyloid species. Tau and NFL have both been examined in small sample sets in HD. NFL levels were higher in the CSF of 35 HD patients compared to 35 matched controls, and correlated with UHDRS Total Functional Capacity [75], a clinical measure of disease progression. The same group measured CSF levels of tau and found a significant elevation in HD though, due to overlap, tau could not provide categorical distinction between groups and showed no correlations with clinical measures [76]. As with many initial findings, these results have yet to be replicated and the effect of inter-subject variability in sampling conditions remainsto be seen.
A further complication in the investigation of low-abundance proteins in CSF is the effect of different materials used in sample handling. Polystyrene and polyethylene tubes have been shown to artefactually diminish CSF levels of amyloid beta compared with polypropylene tubes, through adsorption of the protein to the plastic surface [77]. Similar effects may well be seen with any other protein; most have not been investigated for such technical effects. Such observations call for absolutely strict standardization of all equipment within and between studies. In our experience this requires great effort and close attention, since we are aware of at least two cases where plasticware described by the manufacturer as polypropylene has later emerged as being made from another plastic. Other granular technical considerations such as aliquot size can have a significant effect on the measured levels of proteins [78]. Careful planning and due diligence around all aspects of CSF studies are paramount.
PROTEOMIC APPROACHES
In contrast to hypothesis-driven approaches, proteomic discovery has the potential to identify novel biomarker candidates and pathogenic pathways through the analysis of many proteins from the same sample. In HD CSF, proteomic work has so far been limited by small sample sizes and lack of necessary analytic technology. Fang and colleagues integrated 5 sets of proteomic profiles, obtained by different laboratories, from a single HD sample set. They incorporated gene expression profiling data from a study by Ge et al. [79] to identify the most probable origin of the detected proteins. As expected, they showed that brain-specific proteins were more likely to be found in CSF than in plasma. Inevitably they identified many proteins that were altered in HD CSF, but the majority of these disease-associated changes were in proteins which were said to be liver-specific. There was a rather surprising tendency for brain-derived proteins to decline in HD CSF, with the exception of neuroinflammatory markers [80]. These findings highlight the inconsistencies between proteomic techniques and the nature of such studies as hypothesis-generating engines for future targeted work in properly powered sample sets. This replication work has yet to be carriedout in HD.
A small sample proteomic study of 9 HD patients detected significantly higher levels of the immune-associated proteins prothrombin and haptoglobin (see Inflammatory Markers, above) as well as Apolipoprotein A-IV (ApoA). ApoA is a glycoprotein associated with inhibition of food intake and regulation of body weight. All three were higher in HD patients compared to matched controls [61] but these changes were not independently replicated using a more quantitative assay – though Fang and colleagues had also found robust elevations of ApoA in HD CSF in their previous proteomic work [80].
Another proteomic study used surface-enhanced laser desorption/ionization time-of-flight (SELDI-TOF) mass spectrometry to analyze the proteomic profile of CSF from 39 preHD gene carriers, 55 manifest HD and 27 HD gene negative controls. 10 out of the 85 peaks detected showed significant difference between manifest HD and controls. After age-adjustment and Bonferroni correction, only ubiquitin remained significantly higher in manifest HD patients, with a trend in preHD compared to controls. The correlation between ubiquitin and CAG-age product (CAP) and other clinical measures was statistically significant [81]. Ubiquitin is added post-transcriptionally to flag proteins for degradation by the ubitiquitin-proteaseome system (UPS), and its elevation in CSF may reflect the gradual accumulation of unwanted proteins, perhaps including mHTT, in cells of the CNS. Failure of the UPS to cope with the buildup of abnormal proteins is suggested to be involved in the pathogenesis of HD and other neurodegenerative diseases [82]. This finding warrants further investigation and its longitudinal predictive value needs to be determined in a suitable patient cohort.
MUTANT HUNTINGTIN
Understanding the function of the huntingtin protein and its mutant counterpart has been a major focus of HD research since the gene was discovered in 1993. With many approaches aimed at reducing the expression of mHTT, its quantification in CSF has been highly desirable but proved extremely challenging. mHTT is a large, aggregation-prone protein expressed mostly intracellularly, and since HD generally progresses slowly, mHTT would be expected to be released very gradually into the CSF from dying neurons. Consequently its CSF concentration is certainly extremely low and several generations of improvement in antibodies and mHTT-quantifying assays failed to yield a method sensitive enough to quantify it [19].
Successful quantification of soluble CSF mutant huntingtin was only achieved in early 2015 [83]. We used a single molecule counting (SMC) immunoassay, and a combination of HTT N-terminal-detecting 2B7 antibody and polyglutamine-binding MW1 antibody, to quantify mHTT for the first time in CSF at femtomolar sensitivity (Fig. 2). mHTT was significantly higher in manifest HD and preHD compared to controls with a roughly threefold difference seen between preHD and manifest. mHTT also correlated with clinical phenotype as measured by motor and cognitive scores. Crucially, these associations remained statistically significant after adjustment for disease burden score (a product of age and CAG repeat length), indicating that CSF mHTT level has independent power to predict clinical phenotype in HD – the first molecular measure reported to show this ability. Moreover, mHTT level was seen to correlate significantly with CSF concentrations of NFL and Tau, suggesting a neuronal origin for the mHTT detected. These findings were shown in two independent cohorts of 12 participants in London and 40 in Vancouver. Work is now underway to validate this assay according to the Food and Drug Administration (FDA) and European Medicines Agency (EMA) guidelines for biomarker validation for clinical trial use. Assays for total huntingtin level are also under development.
Shortly afterwards, Southwell and colleagues reported mHTT quantification in CSF with immunoprecipitation and flow cytometry (IP-FCM) using a combination of MW1 and HDB4E10 antibodies, the latter recognizing a C-terminal binding site [84]. In a single cohort, overlapping with the Vancouver dataset from our work, they confirmed the presence of mHTT in mutation carriers but not controls and a tendency to rise with onset and clinical manifestations. They went on to show that the assay was capable of detecting mHTT in CSF from Hu97/18 mice expressing human mHTT, and that the signal in CSF was correlated significantly with brain mHTT level after intracerebroventricular administration of an HTT-lowering antisense oligonucleotide. These findings (Fig. 3) confirm a likely neuronal origin for mHTT detected in CSF and affirm its promise as a highly pathogenically relevant biomarker for HTT-lowering trials.
Both these assays and their successors under development are likely to be valuable in the investigation of HTT in CSF in humans and model animals. While the SMC immunoassay appears highly robust and reproducible, and gives a more quantitative indication of concentration, perhaps making it the more likely candidate for clinical trial use, the IP-FCM assay uses more widely available technology and may therefore find a more widespread role in the preclinicalsetting.
All forms of mutant huntingtin are not alike, and a means of quantifying the most pathogenic forms in CSF would be valuable. Tan and colleagues developed a cell-based aggregation assay to quantify the proportion of cells with aggregates, and amount of aggregates in lysate, and demonstrated that seeding of aggregation could be triggered by synthetic polyglutamine oligomers and by CSF from transgenic rats and human HD patients [85]. The study provides the first evidence that CSF could be used to study not only the quantity of mHTT but its pathogenic properties, offering potential as a diagnostic and prognostic marker.
CONCLUSION
It is ever more likely that robust, useful CSF biomarkers of HD will be developed. The current challenge is not a numerical shortage of hypotheses or of small studies proposing potential CSF biomarkers for HD. The investigators in the early studies established the field using the methods and standards of the day, and it would be unfair to judge them by standards that have changed dramatically in the intervening decades. We now have at our disposal advanced methodologies for sensitively, accurately and reproducibly quantifying metabolites in biofluids. Moreover we are now definitively in a new era where targeted HD therapeutics are entering clinical trials, giving more pressing reasons than ever to wish to know what HD-related changes can be found, or what drug-induced changes can be detected.
Experience from other neurodegenerative diseases suggests that the greatest prima facie utility for neuronal proteins in CSF is as natural history biomarkers. A small cadre of such candidates can be proposed based on findings in HD to date: Tau and NFL to measure neuronal death and mHTT as a ‘smoking gun’ – the disease agent itself, released from the neurons it is killing. Even these require urgent replication and longitudinal study to examine their prognostic powers and response to therapeutic intervention.
Other classes of biomarker may be able to shed light on specific aspects of HD natural history or progression, and in carefully chosen contexts may serve as targeted pharmacodynamic markers for clinical trials of specific therapeutics. But each of these dozens of proposed markers and its assay requires full replication and validation according to stringent criteria if they are to meet their potential to advance therapeutics.
The greatest challenge is to replicate previous promising findings in large adequately powered cohorts using the latest assay techniques, and the unglamorous but crucial work of standardizing methods across sites and between studies. CSF collection efforts need to move from ad hoc to concerted, with optimized protocols and equipment and with controls recruited contemporaneously from an age-matched, genuinely healthy population. Clinical phenotyping needs to be comprehensive and standardized based on the best available evidence. With these aims in mind, we have embarked on HDClarity (http://hdclarity.net), a major multi-site study of CSF in HD, which aims to recruit 600 participants at up to 30 sites globally using optimized techniques with rigorous quality control and assurance. These samples and data will be subject to multiple analyses to identify head-to-head those biomarkers with the most power to detect disease-related changes in HD or to act as pharmacodynamic tools to facilitate the rapid testing of a new generation of promising therapeutic candidates.
ACKNOWLEDGMENTS INCLUDING SOURCES OF SUPPORT
EJW and LMB are supported by the Medical Research Council. This work was supported in part by the National Institute for Health Research University College London Hospitals Biomedical Research Centre and the UCL Leonard Wolfson Experimental Neurology Centre. HDClarity is supported by the CHDI Foundation, Inc. and uses the ENROLL-HD platform (http://enroll-hd.org).
CONFLICT OF INTEREST
EJW has undertaken consultancy work for Ionis, Roche and Shire Pharmaceuticals, from which all fees were paid to his employer UCL to support his research. LMB has no conflict of interest to declare.
REFERENCES
[1] | Wild EJ , Tabrizi SJ . Targets for future clinical trials in Huntington’s disease: What’s in the pipeline? Mov Disord. (2014) ;29: :1434–45. |
[2] | The Huntington’s Disease Collaborative Research GrouA novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. (1993) ;72: :971–83. |
[3] | Ross CA , Aylward EH , Wild EJ , Langbehn DR , Long JD , Warner JH , et al. Huntington disease: Natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol. (2014) ;10: :204–16. |
[4] | Gallagher J . Landmark Huntington’s trial starts - BBC News. BBC News Website 2015. http://www.bbc.co.uk/news/health-34552041 (accessed March 1, 2016). |
[5] | Huntington Study GrouUnified Huntington’s disease rating scale: Reliability and consistency. Mov Disord. (1996) ;11: (2):136–42. |
[6] | Scahill RI , Wild EJ , Tabrizi SJ . Biomarkers for Huntington’s disease: An update. Expert Opin Med Diagn. (2012) ;6: :371–5. |
[7] | Andreasson U , Perret-Liaudet A , van Waalwijk van Doorn LJC , Blennow K , Chiasserini D , Engelborghs S , et al. A Practical Guide to Immunoassay Method Validation. Front Neurol. (2015) ;6: :179. |
[8] | EMA. Guideline on bioanalytical method validation. EMEA, Comm Med Prod Hum Use. (2012) ;44: :1–23. |
[9] | Tabrizi SJ , Reilmann R , Roos RAC , Durr A , Leavitt B , Owen G , et al. Potential endpoints for clinical trials in premanifest and early Huntington’s disease in the TRACK-HD study: Analysis of 24-month observational data. Lancet Neurol. (2012) ;11: :42–53. |
[10] | Tabrizi SJ , Scahill RI , Owen G , Durr A , Leavitt BR , Roos RA , et al. Predictors of phenotypic progression and disease onset in premanifest and early-stage Huntington’s disease in the TRACK-HD study: Analysis of 36-month observational data. Lancet Neurol. (2013) ;12: :637–49. |
[11] | ClinicalTrials.gov. A Clinical Study in Subjects With Huntington’s Disease to Assess the Efficacy and Safety of Three Oral Doses of Laquinimod. ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT02215616 (accessed March 1, 2016). |
[12] | ClinicalTrials.gov. Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of ISIS-HTTRx in Patients With Early Manifest Huntington’s Disease. ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT02519036 (accessed March 1, 2016). |
[13] | ClinicalTrials.gov. Randomized, Placebo Controlled Study Of The Efficacy And Safety Of PF-02545920 In Subjects With Huntington’s Disease. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT02197130 (accessed March 1, 2016). |
[14] | Wild EJ , Tabrizi SJ . Biomarkers for Huntington’s disease. Expert Opin Med Diagn. (2008) ;2: :47–62. |
[15] | Björkqvist M , Wild EJ , Thiele J , Silvestroni A , Andre R , Lahiri N , et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J Exp Med. (2008) ;205: :1869–77. |
[16] | Mochel F , Charles P , Seguin F , Barritault J , Coussieu C , Perin L , et al. Early energy deficit in Huntington disease: Identification of a plasma biomarker traceable during disease progression. PLoS One. (2007) ;2: :e647. |
[17] | Ciammola A , Sassone J , Cannella M , Calza S , Poletti B , Frati L , et al. Low brain-derived neurotrophic factor (BDNF) levels in serum of Huntington’s disease patients. Am J Med Genet Part B Neuropsychiatr Genet. (2007) ;144: :574–7. |
[18] | Fujimura H , Altar CA , Chen R , Nakamura T , Nakahashi T , Kambayashi JI , et al. Brain-derived neurotrophic factor is stored in human platelets and released by agonist stimulation. Thromb Haemost. (2002) ;87: :728–34. |
[19] | Weiss A , Träger U , Wild EJ , Grueninger S , Farmer R , Landles C , et al. Mutant huntingtin fragmentation in immune cells tracks Huntington’s disease progression. J Clin Invest. (2012) ;122: :3731–6. |
[20] | Reiber H . Proteins in cerebrospinal fluid and blood: Barriers, CSF flow rate and source-related dynamics. Restor Neurol Neurosci. (2003) ;21: :79–96. |
[21] | Rosén C , Zetterberg H . Cerebrospinal fluid biomarkers for pathological processes in Alzheimer’s disease. Curr Opin Psychiatry. (2013) ;26: :276–82. |
[22] | Hall S , Surova Y , Öhrfelt A , Zetterberg H , Lindqvist D , Hansson O . CSF biomarkers and clinical progression of Parkinson disease. Neurology. (2015) ;84: :57–63. |
[23] | Peskind ER , Riekse R , Quinn JF , Kaye J , Clark CM , Farlow MR , et al. Safety and acceptability of the research lumbar puncture. Alzheimer Dis Assoc Disord. (2005) ;19: :220–5. |
[24] | Enna SJ , Wood JH , Snyder SH . γ-Aminobutyric acid (GABA) in human cerebrospinal fluid: Radioreceptor assay. J Neurochem. (1977) ;28: :1121–4. |
[25] | Enna SJ , Stern LZ , Wastek GJ , Yamamura HI . Cerebrospinal fluid gamma-aminobutyric acid variations in neurological disorders. Arch Neurol. (1977) ;34: :683–5. |
[26] | Manyam NVB , Hare TA , Katz L , Glaeser BS . Huntington’s disease: Cerebrospinal fluid GABA levels in at-risk individuals. Arch Neurol. (1978) ;35: :728. |
[27] | Böhlen P , Tell G , Schechter PJ , Koch-Weser J , Agid Y , Coquillat G , et al. Cerebrospinal fluid homocarnosine in Huntington’s disease. Life Sci. (1980) ;26: :1009–12. |
[28] | Bonnet AM , Tell G , Schechter PJ , Grove J , Saint-Hilaire MH , De Smet Y , et al. Cerebrospinal fluid GABA and homocarnosine concentrations in patients with Friedreich’s ataxia, Parkinson’s disease, and Huntington’s chorea. Mov Disord. (1987) ;2: :117–23. |
[29] | Manyam NVB , Hare TA , Katz L . Effect of isoniazid on cerebrospinal fluid and plasma GABA levels in Huntington’s disease. Life Sci. (1980) ;26: :1303–8. |
[30] | Tell G , Böhlen P , Schechter PJ , Koch-Weser J , Agid Y , Bonnet AM , et al. Treatment of Huntington disease with gamma-acetylenic GABA, an irreversible inhibitor of GABA-transaminase: Increased CSF GABA and homocarnosine without clinical amelioration. Neurology. (1981) ;31: :207–11. |
[31] | Manyam BV , Ferraro TN , Hare TA . Isoniazid-induced alteration of CSF neurotransmitter amino acids in Huntington’s disease. Brain Res. (1987) ;408: :125–30. |
[32] | Manyam BV , Katz L , Hare TA , Kaniefski K , Tremblay RD . Isoniazid-induced elevation of CSF GABA levels and effects on chorea in Huntington’s disease. Ann Neurol. (1981) ;10: :35–7. |
[33] | Manyam BV , Giacobini E , Colliver JA . Cerebrospinal fluid acetylcholinesterase and choline measurements in Huntington’s disease. J Neurol. (1990) ;237: :281–4. |
[34] | Oepen G , Cramer H , Bernasconi R , Martin P . Huntington’s disease - imbalance of free amino acids in the cerebrospinal fluid of patients and offspring at-risk. Arch Psychiatr Nervenkr. (1982) ;231: :131–40. |
[35] | Klawans HL Jr . A pharmacologic analysis of Huntington’s chorea. Eur Neurol. (1970) ;4: :148–63. |
[36] | Klawans HL Jr . Cerebrospinal fluid homovanillic acid in Huntington’s chorea. J Neurol Sci. (1971) ;13: :277–9. |
[37] | Mattsson B , Persson S -å . Cerebrospinal homovanillic acid and 5-hydroxy-indoleacetic acid in huntington’s chorea. Acta Psychiatr Scand. (1974) ;50: :245–59. |
[38] | Curzon G , Gumpert J , Sharpe D . Amine metabolites in the cerebrospinal fluid in Huntington’s chorea. J Neurol Neurosurg Psychiatry. (1972) ;35: :514–9. |
[39] | Garrett MC , Soares-da-Silva P . Increased cerebrospinal fluid dopamine and 3,4-dihydroxyphenylacetic acid levels in Huntington’s disease: Evidence for an overactive dopaminergic brain transmission. J Neurochem. (1992) ;58: :101–6. |
[40] | García Ruiz PJ , Mena MA , Sanchez Bernardos V , Díaz Neira W , Gimenez Roldan S , Benitez J , et al. Cerebrospinal fluid homovanillic acid is reduced in untreated Huntington’s disease. Clin Neuropharmacol. (1995) ;18: :58–63. |
[41] | Cooper AJL , Jeitner TM , Gentile V , Blass JP . Cross linking of polyglutamine domains catalyzed by tissue transglutaminase is greatly favored with pathological-length repeats: Does transglutaminase activity play a role in (CAG)n/Qn-expansion diseases? Neurochem Int. (2002) ;40: :53–67. |
[42] | Jeitner TM , Bogdanov MB , Matson WR , Daikhin Y , Yudkoff M , Folk JE , et al. Nɛ-(γ-l-Glutamyl)-l-lysine (GGEL) is increased in cerebrospinal fluid of patients with Huntington’s disease. J Neurochem. (2001) ;79: :1109–12. |
[43] | Jeitner TM , Matson WR , Folk JE , Blass JP , Cooper AJL . Increased levels of gamma-glutamylamines in Huntington disease CSF. J Neurochem. (2008) ;106: :37–44. |
[44] | Raptor Pharmaceutical Corp. Raptor Plans to Advance RP103 in a Registration Study in Huntington’s Disease Based on Favorable Treatment Effects at 36 Months in CYST-HD Trial 2015. http://ir.raptorpharma.com/releasedetail.cfm?ReleaseID=946463 (accessed March 15, 2016). |
[45] | Vécsei L , Szalárdy L , Fülöp F , Toldi J . Kynurenines in the CNS: Recent advances and new questions. Nat Rev Drug Discov. (2013) ;12: :64–82. |
[46] | Sathyasaikumar KV , Stachowski EK , Amori L , Guidetti P , Muchowski PJ , Schwarcz R . Dysfunctional kynurenine pathway metabolism in the R6/2 mouse model of Huntington’s disease. J Neurochem. (2010) ;113: :1416–25. |
[47] | Bohár Z , Toldi J , Fülöp F , Vécsei L . Changing the face of kynurenines and neurotoxicity: Therapeutic considerations. Int J Mol Sci. (2015) ;16: :9772–93. |
[48] | Schwarcz R , Tamminga CA , Kurlan R , Shoulson I . Cerebrospinal fluid levels of quinolinic acid in Huntington’s disease and schizophrenia. Ann Neurol. (1988) ;24: :580–2. |
[49] | Heyes MP , Swartz KJ , Markey SP , Beal MF . Regional brain and cerebrospinal fluid quinolinic acid concentrations in Huntington’s disease. Neurosci Lett. (1991) ;122: :265–9. |
[50] | Heyes MP , Saito K , Crowley JS , Davis LE , Demitrack MA , Der M , et al. Quinolinic acid and kynurenine pathway metabolism in inflammatory and non-inflammatory neurological disease. Brain. (1992) ;115: :1249–73. |
[51] | Beal MF , Matson WR , Swartz KJ , Gamache PH , Bird ED . Kynurenine pathway measurements in Huntington’s disease striatum: Evidence for reduced formation of kynurenic acid. J Neurochem. (1990) ;55: (4):1327–39. |
[52] | Chen X , Guo C , Kong J . Oxidative stress in neurodegenerative diseases. Neural Regen Res. (2012) ;7: :376–85. |
[53] | Montine TJ , Beal MF , Robertson D , Cudkowicz ME , Biaggioni I , O’Donnell H , et al. Cerebrospinal fluid F2-isoprostanes are elevated in Huntington’s disease. Neurology. (1999) ;52: :1104. |
[54] | Montine TJ , Markesbery WR , Morrow JD , Roberts LJ . Cerebrospinal fluid F2-isoprostane levels are increased in Alzheimer’s disease. Ann Neurol. (1998) ;44: :410–3. |
[55] | Huntington Study Group TREND-HD Investigators. Randomized controlled trial of ethyl-eicosapentaenoic acid in Huntington disease: The TREND-HD study. Arch Neurol. (2008) ;65: :1582–9. |
[56] | National Institute of Neurological Disorders and Stroke. Announcement of 2CARE Early Study Closure 2014. http://www.ninds.nih.gov/disorders/clinical_trials/2CARE-Early-Study-Closure.htm (accessed March 10, 2016). |
[57] | Dalrymple A , Wild EJ , Joubert R , Sathasivam K , Björkqvist M , Petersén A , et al. Proteomic profiling of plasma in Huntington’s disease reveals neuroinflammatory activation and biomarker candidates. J Proteome Res. (2007) ;6: :2833–40. |
[58] | Träger U , Andre R , Lahiri N , Magnusson-Lind A , Weiss A , Grueninger S , et al. HTT-lowering reverses Huntington’s disease immune dysfunction caused by NFκB pathway dysregulation. Brain. (2014) ;137: :819–33. |
[59] | Aronson NN , Blanchard CJ , Madura JD . Homology modeling of glycosyl hydrolase family 18 enzymes and proteins. J Chem Inf Comput Sci. (1997) ;37: :999–1005. |
[60] | Vinther-Jensen T , Budtz-Jørgensen E , Simonsen AH , Nielsen JE , Hjermind LE . YKL-40 in cerebrospinal fluid in Huntington’s disease–a role in pathology or a nonspecific response to inflammation? Parkinsonism Relat Disord. (2014) ;20: :1301–3. |
[61] | Huang Y-C , Wu Y-R , Tseng M-Y , Chen Y-C , Hsieh S-Y , Chen C-M . Increased prothrombin, apolipoprotein A-IV, and haptoglobin in the cerebrospinal fluid of patients with Huntington’s disease. PLoS One. (2011) ;6: :e15809. |
[62] | Pantoni L , Inzitari D . Abnormal penetration of haptoglobin through the blood-brain-barrier (BBB) into the cerebro-spinal fluid (CSF) in Alzheimer’s disease patients. Acta Neurol Scand. (1995) ;81: :225. |
[63] | Conti A , Sanchez-Ruiz Y , Bachi A , Beretta L , Grandi E , Beltramo M , et al. Proteome study of human cerebrospinal fluid following traumatic brain injury indicates fibrin(ogen) degradation products as trauma-associated markers. J Neurotrauma. (2004) ;21: :854–63. |
[64] | Petersén Å . The role of hypothalamic and neuroendocrine changes in the pathogenesis of huntington’s disease – Current understanding and implications for future treatments. Eur Neurol Rev. (2010) ;5: :49–53. |
[65] | Nemeroff CB , Widerlöv E , Bissette G , Walléus H , Karlsson I , Eklund K , et al. Elevated concentrations of CSF corticotropin-releasing factor-like immunoreactivity in depressed patients. Science. (1984) ;226: :1342–4. |
[66] | Post RM , Ballenger JC , Goodwin FK . Cerebrospinal Fluid Studies of Neurotransmitter Function in Manic and Depressive Illness. In: Wood JH, editor. Neurobiol. Cerebrospinal Fluid, New York: Springer US. (1980) ;685–717. |
[67] | Kurlan R , Caine E , Rubin A , Nemeroff CB , Bissette G , Zaczek R , et al. Cerebrospinal fluid correlates of depression in Huntington’s disease. Arch Neurol. (1988) ;45: :881–3. |
[68] | Popovic V , Svetel M , Djurovic M , Petrovic S , Doknic M , Pekic S , et al. Circulating and cerebrospinal fluid ghrelin and leptin: Potential role in altered body weight in Huntington’s disease. Eur J Endocrinol. (2004) ;151: :451–5. |
[69] | Meier A , Mollenhauer B , Cohrs S , Rodenbeck A , Jordan W , Meller J , et al. Normal hypocretin-1 (orexin-A) levels in the cerebrospinal fluid of patients with Huntington’s disease. Brain Res. (2005) ;1063: :201–3. |
[70] | Björkqvist M , Petersén Å , Carroll J , Nielsen J , Ecker D , Mulder H , et al. Cerebrospinal fluid levels of orexin-A are not a clinically useful biomarker for Huntington disease. Clin Genet. (2006) ;70: :78–9. |
[71] | Gaus SE , Lin L , Mignot E . CSF hypocretin levels are normal in Huntington’s disease patients. Slee. (2005) ;28: :1607–8. |
[72] | Björkqvist M , Leavitt BR , Nielsen JE , Landwehrmeyer B , Ecker D , Mulder H , et al. Cocaine- and amphetamine-regulated transcript is increased in Huntington disease. Mov Disord. (2007) ;22: :1952–4. |
[73] | Gabery S , Murphy K , Schultz K , Loy CT , McCusker E , Kirik D , et al. Changes in key hypothalamic neuropeptide populations in Huntington disease revealed by neuropathological analyses. Acta Neuropathol. (2010) ;120: :777–88. |
[74] | Blennow K , Hampel H , Weiner M , Zetterberg H . Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol. (2010) ;6: :131–44. |
[75] | Constantinescu R , Romer M , Oakes D , Rosengren L , Kieburtz K . Levels of the light subunit of neurofilament triplet protein in cerebrospinal fluid in Huntington’s disease. Parkinsonism Relat Disord. (2009) ;15: :245–8. |
[76] | Constantinescu R , Romer M , Zetterberg H , Rosengren L , Kieburtz K . Increased levels of total tau protein in the cerebrospinal fluid in Huntington’s disease. Parkinsonism Relat Disord. (2011) ;17: :714–5. |
[77] | Lewczuk P , Beck G , Esselmann H , Bruckmoser R , Zimmermann R , Fiszer M , et al. Effect of sample collection tubes on cerebrospinal fluid concentrations of tau proteins and amyloid β peptides. Clin Chem. (2005) ;52: :331–2. |
[78] | Toombs J , Paterson RW , Lunn MP , Nicholas JM , Fox NC , Chapman MD , et al. Identification of an important potential confound in CSF AD studies: Aliquot volume. Clin Chem Lab Med. (2013) ;51: :2311–7. |
[79] | Ge X , Yamamoto S , Tsutsumi S , Midorikawa Y , Ihara S , Wang SM , et al. Interpreting expression profiles of cancers by genome-wide survey of breadth of expression in normal tissues. Genomics. (2005) ;86: :127–41. |
[80] | Fang Q , Strand A , Law W , Faca VM , Fitzgibbon MP , Hamel N , et al. Brain-specific proteins decline in the cerebrospinal fluid of humans with Huntington disease. Mol Cell Proteomics. (2009) ;8: :451–66. |
[81] | Vinther-Jensen T , Simonsen AH , Budtz-Jørgensen E , Hjermind LE , Nielsen JE . Ubiquitin: A potential cerebrospinal fluid progression marker in Huntington’s disease. Eur J Neurol. (2015) ;22: (10):1378–84. |
[82] | Dantuma NP , Bott LC . The ubiquitin-proteasome system in neurodegenerative diseases: Precipitating factor, yet part of the solution. Front Mol Neurosci. (2014) ;7: :70. |
[83] | Wild EJ , Boggio R , Langbehn D , Robertson N , Haider S , Miller JRC , et al. Quantification of mutant huntingtin protein in cerebrospinal fluid from Huntington’s disease patients. J Clin Invest. (2015) ;125: :1–8. |
[84] | Southwell AL , Smith SEP , Davis TR , Caron NS , Villanueva EB , Xie Y , et al. Ultrasensitive measurement of huntingtin protein in cerebrospinal fluid demonstrates increase with Huntington disease stage and decrease following brain huntingtin suppression. Sci Rep. (2015) ;5: :12166. |
[85] | Tan Z , Dai W , van Erp TGM , Overman J , Demuro A , Digman MA , et al. Huntington’s disease cerebrospinal fluid seeds aggregation of mutant huntingtin. Mol Psychiatry. (2015) :1–8. |
Figures and Tables
Fig.1
A timeline of CSF biomarker studies in HD. Substances previously reported to change significantly in HD compared to controls are marked by the year of first published finding. Triangular markers indicate the identification of the HTT gene as the cause of HD.
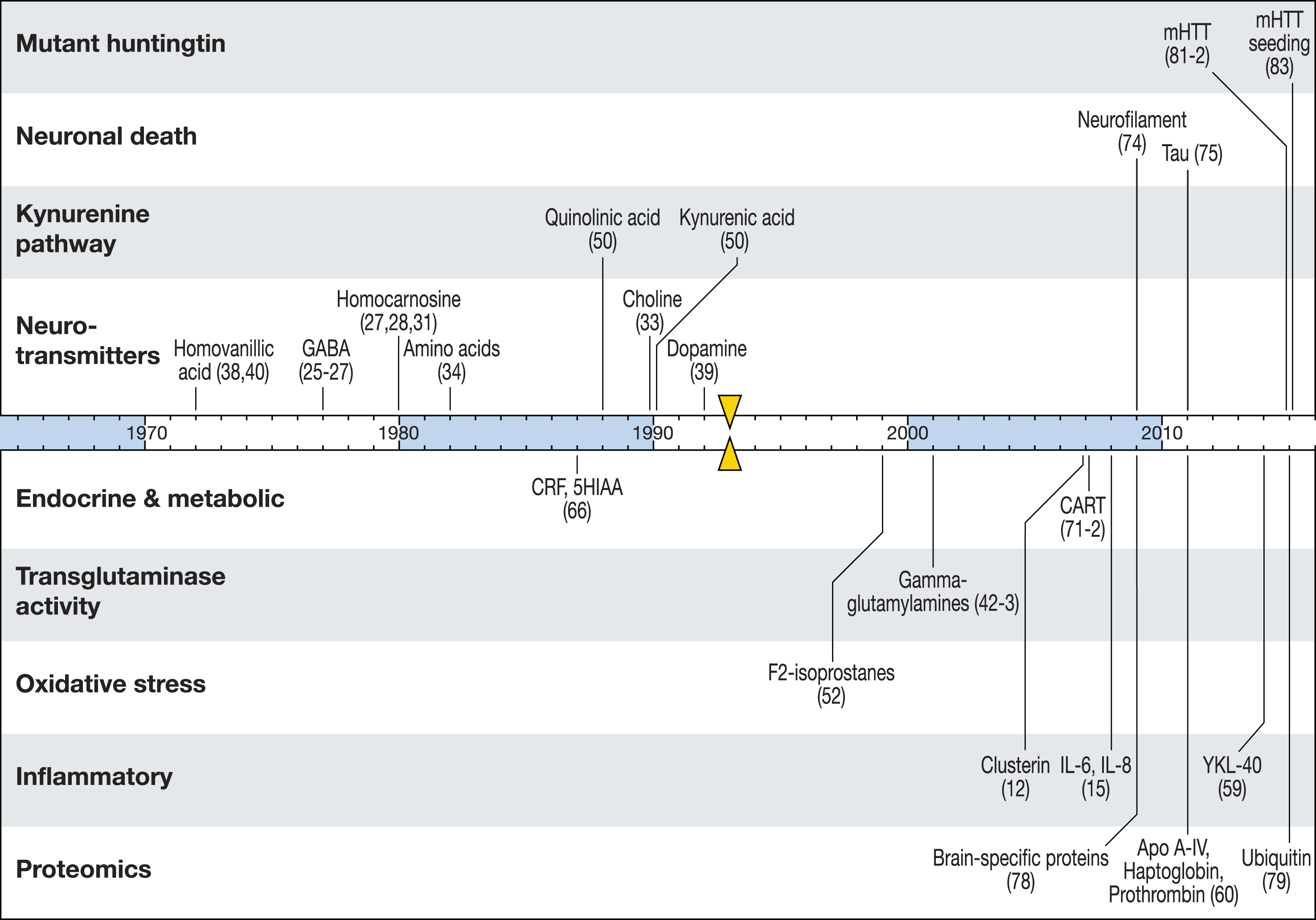
Fig.2
First successful quantification of mHTT in CSF using the Singulex SMC immunoassay. mHTT was significantly higher in premanifest than controls and in manifest HD than premanifest in two cohorts from (A) London cohort and (B) Vancouver. mHTT correlated with (C) disease burden score and (D) Unified Huntington’s Disease Rating Scale motor score. Adapted from Wild et al. [83] with the author’s and publisher’s permission.
![First successful quantification of mHTT
in CSF using the Singulex SMC immunoassay. mHTT was significantly higher in premanifest than controls and in
manifest HD than premanifest in two cohorts from (A) London cohort and (B) Vancouver. mHTT correlated with (C)
disease burden score and (D) Unified Huntington’s Disease Rating Scale motor score. Adapted from Wild et al. [83]
with the author’s
and publisher’s permission.](https://content.iospress.com:443/media/jhd/2016/5-1/jhd-5-1-jhd160196/jhd-5-jhd160196-g002.jpg)
Fig.3
CSF mHTT positively correlates with brain levels after treatment of HTT-lowering anti-sense oligonucleotide. The IP-FCM mHTT assay confirmed (A) mHTT can distinguish between controls and gene carriers; (B) mHTT levels in brain lysate and CSF strongly correlate after HTT-lowering treatment; and reduction in mHTT levels increases with ASO dosage in (C) brain and (D) CSF. Adapted from Southwell et al. [83] with the author’s and publisher’s permission.
![CSF mHTT positively correlates with brain levels after treatment of
HTT-lowering anti-sense oligonucleotide. The IP-FCM mHTT assay confirmed (A) mHTT
can distinguish between controls and gene carriers; (B) mHTT levels in brain lysate
and CSF strongly correlate after HTT-lowering treatment; and reduction in mHTT levels increases with ASO dosage in (C) brain and (D) CSF. Adapted from Southwell et al. [83] with the author’s and publisher’s permission.](https://content.iospress.com:443/media/jhd/2016/5-1/jhd-5-1-jhd160196/jhd-5-jhd160196-g003.jpg)


