
Indoleamine 2,3 Dioxygenase as a Potential Therapeutic Target in Huntington’s Disease
Abstract
Within the past decade, there has been increasing interest in the role of tryptophan (Trp) metabolites and the kynurenine pathway (KP) in diseases of the brain such as Huntington’s disease (HD). Evidence is accumulating to suggest that this pathway is imbalanced in neurologic disease states. The KP diverges into two branches that can lead to production of either neuroprotective or neurotoxic metabolites. In one branch, kynurenine (Kyn) produced as a result of tryptophan (Trp) catabolism is further metabolized to neurotoxic metabolites such as 3-hydroxykunurenine (3-HK) and quinolinic acid (QA). In the other branch, Kyn is converted to the neuroprotective metabolite kynurenic acid (KA). The enzyme Indoleamine 2,3 dioxygenase (IDO1) catalyzes the conversion of Trp into Kyn, the first and rate-limiting enzymatic step of the KP. This reaction takes place throughout the body in multiple cell types as a required step in the degradation of the essential amino acid Trp. Studies of IDO1 in brain have focused primarily on a potential role in depression, immune tolerance associated with brain tumours, and multiple sclerosis; however the role of this enzyme in neurodegenerative disease has garnered significant attention in recent years. This review will provide a summary of the current understanding of the role of IDO1 in Huntington’s disease and will assess this enzyme as a potential therapeutic target for HD.
ABBREVIATIONS USED IN TEXT
HD | Huntington’s disease |
Htt | Huntingtin |
Ido1 | Indoleamine 2,3 dioxygenase |
Tdo2 | Tryptophan 2,3 dioxygenase |
Ido2 | Indoleamine 2,3 dioxygenase 2 |
KP | Kynurenine pathway |
Kyn | Kynurenine |
LC/MS/MS | liquid chromatography tandem mass spectrometry |
Tryp | Tryptophan |
QA | Quinolinic acid |
KA | Kynurenic acid |
3-HAA | 3-hydroxyanthranilic acid |
3-HK | 3-hydroxykynurenine |
3-HAO | 3-hydroxyanthranilic acid oxidase |
MSN | medium spiny neurons |
mHtt | mutant huntingtin |
CNS | Central Nervous System |
BBB | Blood Brain Barrier |
NMDA | N-methyl-D-aspartate |
THE KYNURENINE PATHWAY IN THE BRAIN
Within the past decade, there have been significant advances in our understanding of the kynurenine pathway, both its role within the brain and its function within specific brain cell types. Furthermore, there is accumulating evidence suggesting perturbations of this pathway and imbalances in the levels of KP metabolites occur in many neurologic disease states. Tryptophan that enters the body as an essential amino acid in food can take two different routes; one is to enter the kynurenine pathway via the catalytic activity of IDO1 (along with two other enzymes, tryptophan 2,3 dioxygenase (TDO2) and IDO2) and the other is to enter the serotonergic pathway (SP) by the action of the enzyme tryptophan hydroxylase (Fig. 1). The Kynurenine pathway is the principle route of tryptophan (Trp) catabolism in the brain. The first step of this pathway is the conversion of L-tryptophan (L-Trp) to N-formyl-L-kynurenine, which is mainly catalyzed by the enzymes Indoleamine 2,3 dioxygenase (IDO1), IDO2 and TDO2. These are the known heme-containing oxido-reductase, which facilitate this reaction through the oxidative cleavage of the indole ring of Trp [1]. Therefore, the induction of this first enzymatic step is an important determinant of whether Trp is diverted from the SP and into the KP.
Downstream of IDO1 activity, kynurenine acts as a point of divergence of the KP, which splits into two branches that can lead to production of either neuroprotective or neurotoxic metabolites. In one branch, kynurenine produced as a result of Trp degradation is further catabolized to neurotoxic metabolites such as 3-hydroxykunurenine (3-HK) and quinolinic acid (QA). 3-HK generates hydrogen peroxide and other reactive oxygen species, and has been shown to result in neuronal cell death within the select brain region that has been implicated in the pathology of neurodegenerative disorders including HD [2].
In the other branch, kynurenine is converted to the neuroprotective metabolite kynurenic acid (KA), which is an endogenous glutamate antagonist. In addition to KA inhibitory action on NMDA receptors, it also acts on kainate- and AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid)-sensitive ionotropic glutamate receptors [3]. Another antiglutamatergic function of KA is through the inhibition of the α7 nicotinic acetylcholine receptors (nAChRs) known to enhance glutamate release [4]. Finally, KA acts as a potent free radical scavenger [5]. Metabolism of Trp through this pathway also results in production of nicotinamide adenine dinucleotide (NAD +) in mammalian tissues [6]. Figure 1 depicts the metabolites and enzymes of the kynurenine pathway (vertical) and parts of the serotonin pathway (horizontal).
THE ROLE OF THE KYNURENINE PATHWAY IN NEURODEGENERATION
Within the past two decades, there has been increasing evidence for the involvement of KP in neurodegenerative diseases such as amyotropic lateral sclerosis (ALS), Parkinson’s disease (PD), Alzheimer’s disease (AD) and Huntington’s disease (HD) [7–20]. This involvement generally manifests as an imbalance between the levels of neuroprotective and neurotoxic metabolites in the pathway.
In many neurologic disorders the ratio of QA/KA is modified in the plasma of patients or their susceptible brain regions. For example, an increased neostriatal QA/KA ratio, caused by a rise in QA levels has been observed in early grade HD brain at the first stages of the disease [21]. Similarly, elevated QA immunoreactivity has been observed in the hippocampus of AD patients in association with senile plaques [13].
Among KP neurotoxic metabolites, QA is an N-methyl-D-aspartate (NMDA) receptor agonist that has the potential to cause NMDA receptor-mediated neuronal damage and dysfunction, a mechanism thought to be involved in the pathogenesis of neurodegenerative diseases such as HD [22]. This effect of QA was initially inferred when intrastriatal injection of QA in rodents replicated HD-like striatal degeneration by binding to NMDA receptors and producing axon-sparing lesions in the striatum [23]. Currently, intrastriatal injection of QA is used in many laboratories around the world as a mouse model of acute excitotoxic neurodegeneration that may have relevance to the form of cell death that medium spiny neurons undergo in HD [22, 23].
A reduction in the levels of neuroprotective KA has been observed in the brain of HD patients as well as in several mouse models of HD [7, 24, 29]. Altered levels of KA in the CSF of patients with ALS have also been observed [18]. Mice deficient for kynurenine aminotransferase ii (KAT2), an enzyme in the KP that converts L-L-kyn into KA, have decreased levels of KA and are more susceptible to QA-induced toxicity [25].
Upstream of QA, 3-HK is an endogenous oxidative stress generator that is known to be elevated in the brains of HD and PD patients and HD mouse models [7, 26–28]. 3-HK causes neuronal cell death with apoptotic features and region selectivity to the striatum in both HD and PD [28, 29]. Although 3-HK is converted to QA through a few steps in the pathway, the toxic effect of 3-HK is at least partly independent of NMDA receptor mediated toxicity and is due to its ability to generate free radicals [61]. Intrastriatal co-application of 3-HK, at a concentration that did not produce neuronal damage on its own, increased the neurotoxicity of intrastriatal QA injections [30] demonstrating the ability of 3-HK to exacerbate QA-induced lesions in the striatum, despite its lack of known effects on NMDA receptors. The result of feeding neurotoxic metabolites such as KA and 3-HK to flies expressing mutant huntingtin (mHTT) suggested a causal involvement of kynurenine pathway metabolites in mHTT dependent neuron loss in this model system [31].
More recently, the enzyme kynurenine 3-monooxygenase (KMO) has attracted a lot of attention as a potential therapeutic target for HD and AD. This enzyme is known to be expressed primarily in microglia where it converts L-kyn to 3-HK [32, 33]. A trial of chronic oral administration of a KMO inhibitor extends lifespan, prevents synaptic loss and decreases brain microglial activation in the R6/2 model of HD [34]. This KMO inhibitor also prevented spatial memory deficits, anxiety-related behaviour, and synaptic loss in mouse model of AD. A more recent study of the KMO knock-out mice showed reduced amount of neurotoxic compounds in the brain of these animals [35].
This study by Giorgini et al. is a valuable example of how manipulating specific enzymes in the kynurenine pathway can divert the pathway from the neurotoxic branch to the neuroprotective branch and modify the net result of the pathway in terms of neurotoxicity. With regards to neuropathology and particularly neurodegeneration, the metabolites downstream of Kyn have been studied extensively compared to those upstream of Kyn. This review examines the potential role of the first step of the pathway with particular focus on IDO1 in neurologic disorders specifically in HD, and will assess its potential as a therapeutic target in HD.
ROLE OF IDO1 IN PERIPHERY
IDO1 is the first and the rate-limiting enzyme of the kynurenine pathway which is the principle route of tryptophan catabolism. IDO1 is highly expressed in mammalian organs such as lung, epididymis, small intestine and placenta [6]. In most other organs the expression is constitutively low but can be induced by the expression of cytokines such as interferon gamma (IFNy) [6]. IDO1 has also been implicated in immune tolerance and plays a role in maintaining the T cell homeostasis of self-antigen tolerance during inflammation [36–39]. Similarly, by increasing IDO1, cancer tumours suppress host T cells and enhance local Treg-mediated immunosuppression through Trp depletion [40]. During infection, IDO1 upregulation depletes Trp which in turn inhibits the proliferation of intracellular parasites [41, 42]. IDO1 is therefore a potential therapeutic target for multiple clinical settings such as cancer, chronic infections, autoimmunity, allergy, and potentially even transplantation [43].
During pregnancy, IDO1 induction prevents allogeneic fetal rejection by reducing the availability of Trp to maternal T cells and thus enhancing activation induced T cell death [44]. The induction of IDO1 shunts Trp into the KP and away from the SP. This induction generates a microenvironment in which maternal T cells become Trp-deprived and die. The IDO1-mediated suppression of T cell activity therefore prevents fetal rejection. This was discovered in 1998, when Munn et al. found that pharmacologic inhibition of IDO1 results in rapid T cell-induced rejection of all allogeneic embryos in pregnant mice [44]. Interestingly, immuno-tolerance as a result of Trp depletion during pregnancy has been linked to decreased serotonin levels and associated with post-partum depression [45].
ROLE OF IDO1 IN THE CNS
Previous work examining the role of IDO1 in the brain initially focused primarily on its role in immune tolerance associated with brain tumours [40, 46] and a role in infiltrating macrophages in autoimmune diseases such as multiple sclerosis (MS) [5, 37, 47]. Development of depression and depressive-like phenotypes as a result of IDO1-induced Trp degradation and reduced serotonin levels has been of more recent interest [19, 20, 48–50; 51]. A new study has been successful in showing that IDO1 contributes to the comorbidity of pain and depression [52].
The study of normal IDO1 function in brain has only recently been addressed in a small number of studies. In a recent study, we examined the brain response to an NMDA-mediated excitotoxic insult in mice lacking indoleamine 2,3 dioxygenase (Ido knockout or Ido–/– mice) [53]. The striatum of Ido–/– mice was less sensitive to NMDA receptor-mediated excitotoxic stress induced by QA compared to their wild-type littermates. Downstream metabolites of the KP were measured, and no changes in endogenous KA or QA levels were observed in the striatum of Ido–/– mice. However, 3-HK was significantly reduced in the striatum of Ido–/– mice. 3-HK is an endogenous oxidative stress generator, known to be increased in the brains of HD patients and mouse models of HD [3, 26]. Increased 3-HK causes neuronal cell death with apoptotic features and region selectivity to the striatum [29]. We proposed in this paper that the reduction in striatal 3-HK levels was neuroprotective in these ido–/– mice and that IDO1 inhibition may be a promising therapeutic strategy for HD [53].
The Ido1 transcript is highly enriched in mouse striatum relative to other brain regions [27, 54]. In addition, in silico analysis showed that Ido1 is co-expressed with many striatal-enriched genes found in previous studies [54]. Cell-type specificity of IDO1 expression has also been investigated in mouse brain [27]. Striatal neuronsand astrocytes express high levels of Ido1 mRNA at baseline, but ido1 transcript was not detectable in microglia.
IDO1 IN AD AND PD
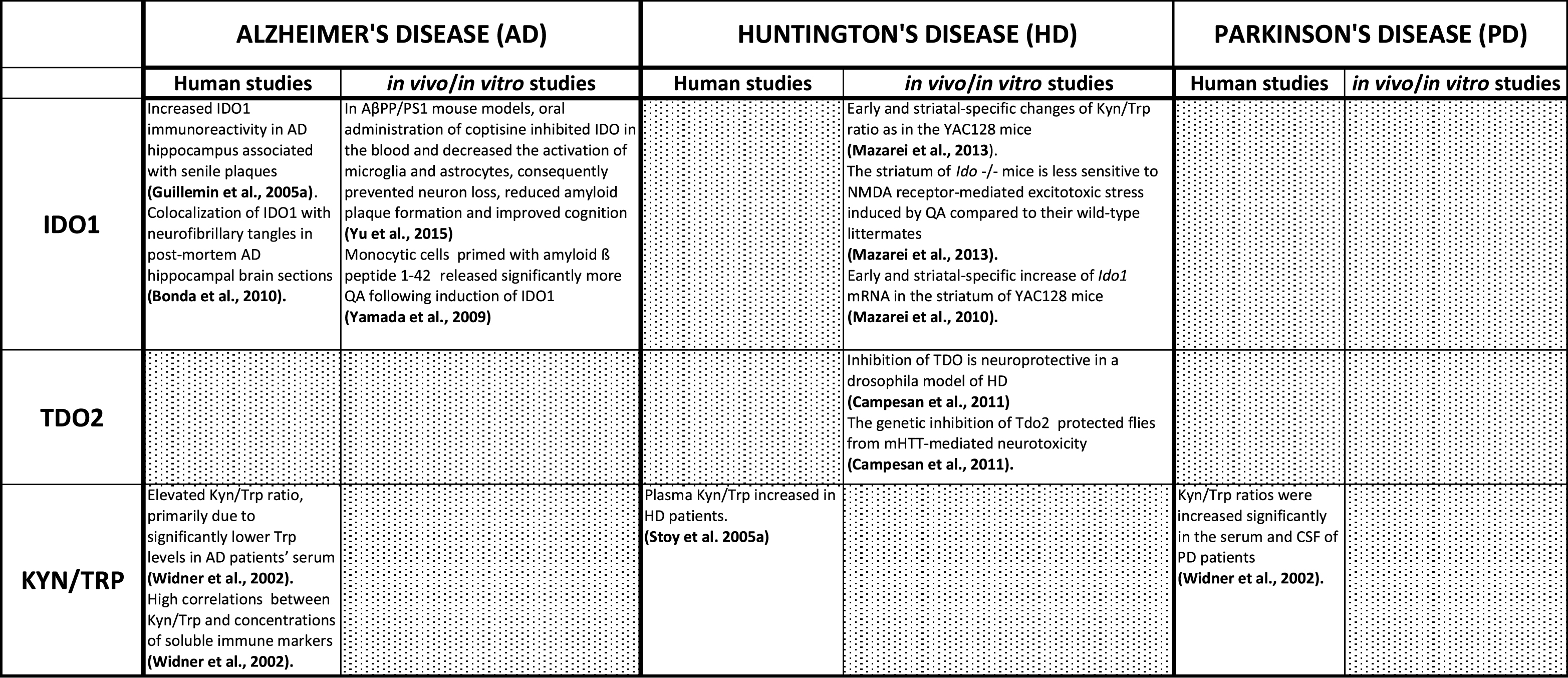
Since IDO1 facilitates the conversion of Tryp to Kyn, the ratio of Kyn/Trp is used as an estimate of IDO1 activity in many older studies. Widner and colleagues examined serum from 21 AD patients and 20 controls for Tryp and Kyn concentrations. Elevated Kyn/Trp ratio, primarily due to significantly lower Trp levels was observed in patients’ serum [11]. Interestingly, high correlations were found in patients between Kyn/Trp and concentrations of soluble immune markers such as neopterin, interleukin-2 receptor, and tumor necrosis factor receptor. Finally, this study demonstrated that increased IDO1 activity in patients’ serum correlated with cognitive impairment in AD [11]. These results suggest a possible link between increased KP activity and the pathogenic processes involved in AD.
Monocytic cells (macrophages and microglia) primed with amyloid ß peptide 1-42 [16] released significantly more QA following induction of IDO1. Interestingly, IDO1 induction did not occur when cells were primed with other peptides such as amyloid ß 1–40, or prion peptide 106–126 [55–57]. Moreover, analysis of human post-mortem brain samples has revealed increased IDO1 immunoreactivity in AD hippocampus, which was associated with senile plaques [13]. IDO1 is specifically co-localized with neurofibrillary tangles in post-mortem AD hippocampal brain sections [17]. IDO1 has also been implicated in PD, but has not been extensively investigated. Kyn and Trp levels in serum and CSF of 22 PD patients were compared to those of 11 controls and Kyn/Trp ratios were increased significantly in both the serum and CSF of PD patients [11]. Additionally, the serum and CSF Kyn/Trp ratios correlated with neopterin concentration reflecting immune activation, and also correlated with disease severity [11]. Inflammatory mechanisms, possibly linked to KP pathway activation, appear to be involved in the neuropathology of PD and AD (Table 1).
IDO1 IN HUNTINGTON’S DISEASE
Increased IDO1 activity in HD increases the production of the neurotoxic metabolites 3HK and QA, thus linking elevated susceptibility to NMDA-mediated neurotoxicity in HD with increased KP activity. Plasma Kyn to Trp ratio (a measure of IDO1 activity) is increased in HD [58]. While many studies have measured KP metabolites in the brain of HD patients and mouse models, IDO1 activity has not been investigated extensively. Our recent analysis of YAC128 mouse brains has revealed early and region-specific changes of Kyn/Trp ratio as a measure of IDO1 activity [27]. These studies revealed higher Kyn/Trp ratios in the striatum, indicating an important role for the first step of the kynurenine pathway in this model system
OTHER ENZYMES IN THE FIRST STEP OF TRYPTOPHAN DEGRADATION (TDO2 AND IDO2)
Despite both catalyzing the dioxygenation of tryptophan, the first step in the kynurenine pathway, the sequence similarity between TDO and IDO is low. Alignment of sequences across this family of enzymes is only possible on the basis of their structures. Human IDO shows activity toward a wider range of substrates than TDO, and is found throughout the body, while TDO is limited to the liver and epidermis. To date, no functional prokaryotic IDO has been identified, while TDO is found in many bacteria.
While IDO1 is expressed widely in most extra-hepatic tissues, TDO2 has been considered the main enzyme converting Trp to Kyn in the liver. Recently however, there has been increasing evidence for the role of TDO2 in the brain (59, 30). TDO2 and IDO1 are different in their regulation and are induced/inhibited through different physiological mechanisms. For example, the family of interferons (α, β, y), tumour necrosis factor α (TNFα), platelet activating factor, T-antigen 4, HIV1 proteins (Nef and Tat), Amyloid beta peptide 1–4, Trp, and Trp analogues are well-known inducers of IDO1 while TDO2 is mainly induced by Trp, Trp analogues, and glucocorticoids [60]. On the other hand, IL-4, nitric oxide, and COX-2 inhibitors are known to inhibit IDO1 expression whereas indoleamines, nicotinamide analogues, and several antidepressant drugs are effective inhibitors of TDO2 [60].
Interestingly, inhibition of TDO2-mediated Trp degradation has been shown to delay aging and aging-associated protein homeostasis in C. elegans [61]. IDO2 or IDO-like gene was first discovered in 2007 to be a paralog of IDO1. Both genes have very similar genomic structures and are located adjacent to each other on chromosome 8 in both mouse and human genomes. These genes are likely to be the result of gene duplication and seem to have similar enzymatic activity (ie. conversion of Trp to kyn) but are different in their expression patterns and their selectivity for some inhibitors [62]. Evolutionarily, the IDO ancestor gene was found to be more similar to IDO2 than IDO1 and while IDO1 is found in mammals and yeast, IDO2 is present only in mammals [6].
To examine the potential involvement of Ido2 and Tdo2 in facilitating Trp to Kyn conversion, the expression patterns of the transcripts corresponding to these enzymes was assessed in the brains of the YAC128 mouse model of HD and Ido–/– adult mice [27]. Ido1 and Ido2 proteins are encoded by genes adjacent to each other on human and mouse chromosome 8 and are 43% similar in amino acid sequence. These two genes are paralogs that have resulted from the duplication of an ancestral Ido gene. Tdo2 on the other hand is structurally unrelated to Ido1 and Ido2 but has similar functions [6]. While Tdo2 has mainly been postulated to function in the liver, it has a recently described function in adult neurogenesis and in anxiety-related behavior in mice [59]. This suggests a role of this enzyme in some higher brain functions. The Ido2 expression pattern is similar to that of Ido1 in that it is highly enriched in the striatum compared with other brain regions. Similar to Ido1, Ido2 mRNA was not expressed in the striatum of Ido–/– mice. This suggested that the knockout construct designed for the generation of Ido–/– mice also targeted Ido2, and that these animals are null for both Ido1 and Ido2. Unlike Ido1, Ido2 expression however, did not change significantly in the striatum of YAC128 mice. Tdo2 expression was not detectable in the striatum and was only present in the mouse cerebellum. Levels of cerebellar Tdo2 did not change in either of the YAC128 or Ido–/– mice [53]. These mouse data support a role for IDO1 as the primary KP enzyme involved in brain regions affected in HD.
The kinetic parameter Michaelis–Menten constant (Km) which is a reverse measure of enzymatic affinity for substrate, in this case Trp, was shown to be different for each of the three enzymes. The Km value is ∼20μM for human IDO1 and ∼100μM for human TDO2 [6]. The Km value of IDO2 is estimated to be 500–1000 fold higher than that of IDO1 [6]. These findings demonstrate that among the three enzymes; IDO1 has the highest affinity for substrate Trp, is highly expressed in striatum, and has elevated expression in HD brain and mouse models [27]. Increased peripheral Kyn levels were also found in the plasma of YAC128 mice. This Kyn may cross the blood brain barrier (BBB) and add to the pool of striatal Kyn. Plasma Kyn/Trp was also assessed in these mice, as the state of the KP in the periphery can affect the KP in the CNS and several KP metabolites can traverse the blood brain barrier (BBB) [3]. Increases in plasma Kyn and Kyn/Trp could potentially contribute to the increase in the pool of Kyn in the striatum where it is converted to 3-HK in a region-specific manner. Given the significant levels of transcript enrichment in the brain and much lower Km, it is very likely that IDO1 is the predominant enzyme responsible for the first step in the KP converting Trp to Kyn in the brain [53]. This study also showed that brain levels of IDO1 are further increased in HD, likely due to local inflammation.
IDO1 AS A THERAPEUTIC TARGET
Targeting the KP in HD has drawn a great deal of attention in the past few years. This involves strategies that shift the pathway toward generation of KA and away from production of 3-HK and QA. In other words, the therapeutic strategy is considered successful when the imbalance in KP resulted from increased neurotoxicity over neuroprotection is reversed. So far, KMO inhibition has been validated as a therapeutic strategy for HD. It has been shown that the peripheral inhibition of this enzyme by a novel orally bioavailable prodrug (JM6) decreased synaptic loss in brain and increased the lifespan of the R6/2 mouse model [34, 63].
While the first step of the KP is located upstream of both neuroprotective and neurotoxic branches, there is increasing evidence that inhibition of this step may push the balance towards the neuroprotective metabolites [61]. A study in Drosophila models of HD also showed that the reduction in the activity of the first step through genetic inhibition of Tdo2 (flies do not express Ido1 or Ido2) protected flies from mHTT-mediated neurotoxicity [31]. No therapeutic studies of IDO1 inhibitors have yet been published in mouse models of HD. The potential mode of administration of various IDO1 inhibitors, their specificity, and ability to cross the BBB are issues yet to be resolved in the development of compounds targeting IDO1 as effective therapies for HD.
SUMMARY
In summary, studies investigating IDO1 the first enzymatic step of the kynurenine pathway in neurodegeneration suggest that inhibition of this enzyme is likely neuroprotective in HD. The early andregion-specific induction of IDO1 in the brains of HD mouse models suggests that this enzyme plays a central role in the observed imbalance of the kynurenine pathway (summarized in fig. 2). The changes in IDO1 should be taken into account when assessing therapeutic targets that act downstream in this pathway. These findings open new doors toward understanding novel mechanisms involving tryptophan degradation and suggest potential future therapies for HD.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
We would like to thank A. Hill and Dr. T. Petkau for kind assistance and insightful comments on the manuscript. Funding support was provided by the Canadian Institutes of Health Research (BRL and GM).
REFERENCES
1 | Higuchi K, Hayaishi O(1967) Enzymatic formation of D-kynurenine from D-tryptophanArch Biochem Biophys120: 397403 |
2 | Okuda S, Nishiyama N, Saito H, Katsuki H(1998) 3-Hydroxykynurenine, an endogenous oxidative stress generator, causes neuronal cell death with apoptotic features and region selectivityJ Neurochem70: 299307 |
3 | Vécsei L, Szalárdy L, Fülöp F, Toldi J(2013) Kynurenines in the CNS: Recent advances and new questionsNat Rev Drug Discov12: 6482 |
4 | Marchi M, Risso F, Viola C, Cavazzani P, Raiteri M(2002) Direct evidence that release-stimulating alpha7 nicotinic cholinergic receptors are localized on human and rat brain glutamatergic axon terminalsJ Neurochem80: 10711078 |
5 | Lugo-Huitrón R, Blanco-Ayala T, Ugalde-Muñiz P, Carrillo-Mora P, Pedraza-Chaverrí J, Silva-Adaya D, Maldonado PD, Torres I, Pinzón E, Ortiz-Islas E, López T, García E, Pineda B, Torres-Ramos M, Santamaría A, La Cruz VP-D(2011) On the antioxidant properties of kynurenic acid: Free radical scavenging activity and inhibition of oxidative stressNeurotoxicol Teratol33: 538547 |
6 | Yuasa HJ, Ball HJ, Ho YF, Austin CJD, Whittington CM, Belov K, Maghzal GJ, Jermin LS, Hunt NH(2009) Characterization and evolution of vertebrate indoleamine 2, 3-dioxygenases IDOs from monotremes and marsupialsComp Biochem Physiol B Biochem Mol Biol153: 137144 |
7 | Sathyasaikumar KV, Stachowski EK, Wonodi I, Roberts RC, Rassoulpour A, McMahon RP, Schwarcz R(2011) Impaired kynurenine pathway metabolism in the prefrontal cortex of individuals with schizophreniaSchizophr Bull37: 11471156 |
8 | Sardar AM, Bell JE, Reynolds GP(1995) Increased concentrations of the neurotoxin 3-hydroxykynurenine in the frontal cortex of HIV-1-positive patientsJ Neurochem64: 932935 |
9 | Huengsberg M, Winer JB, Gompels M, Round R, Ross J, Shahmanesh M(1998) Serum kynurenine-to-tryptophan ratio increases with progressive disease in HIV-infected patientsClin Chem44: 858862 |
10 | Baran H, Hainfellner JA, Kepplinger B, Mazal PR, Schmid H, Budka H(2000) Kynurenic acid metabolism in the brain of HIV-1 infected patientsJ Neural Transm107: 11271138 |
11 | Widner B, Leblhuber F, Fuchs D(2002) Increased neopterin production and tryptophan degradation in advanced Parkinson’s diseaseJ Neural Transm109: 181189 |
12 | Clark CJ, Mackay GM, Smythe GA, Bustamante S, Stone TW, Phillips RS(2005) Prolonged survival of a murine model of cerebral malaria by kynurenine pathway inhibitionInfect Immun73: 52495251 |
13 | Guillemin GJ, Brew BJ, Noonan CE, Takikawa O, Cullen KM(2005) Indoleamine 2,3 dioxygenase and quinolinic acid immunoreactivity in Alzheimer’s disease hippocampusNeuropathol Appl Neurobiol31: 395404 |
14 | Walker DG, Link J, Lue L-F, Dalsing-Hernandez JE, Boyes BE(2006) Gene expression changes by amyloid beta peptide-stimulated human postmortem brain microglia identify activation of multiple inflammatory processesJ Leukoc Biol79: 596610 |
15 | Darlington LG, Mackay GM, Forrest CM, Stoy N, George C, Stone TW(2007) Altered kynurenine metabolism correlates with infarct volume in strokeEur J Neurosci26: 22112221 |
16 | Yamada A, Akimoto H, Kagawa S, Guillemin GJ(2009) Takikawa O, Proinflammatory cytokine interferon-gamma increases induction of indoleamine 2,3-dioxygenase in monocytic cells primed with amyloid beta peptide 1- Implications for the pathogenesis of Alzheimer’s diseaseJ Neurochem110: 791800 |
17 | Bonda DJ, Mailankot M, Stone JG, Garrett MR, Staniszewska M, Castellani RJ, Siedlak SL, Zhu X, Lee H-G, Perry G, Nagaraj RH, Smith MA(2010) Indoleamine 2,3-dioxygenase and 3-hydroxykynurenine modifications are found in the neuropathology of Alzheimer’s diseaseRedox Re15: 161168 |
18 | Chen Y, Stankovic R, Cullen KM, Meininger V, Garner B, Coggan S, Grant R, Brew BJ, Guillemin GJ(2010) The kynurenine pathway and inflammation in amyotrophic lateral sclerosisNeurotox Res18: 132142 |
19 | Fu X, Lawson MA, Kelley KW, Dantzer R(2011) HIV-1 Tat activates indoleamine 2,3 dioxygenase in murine organotypic hippocampal slice cultures in a p38 mitogen-activated protein kinase-dependent mannerJ Neuroinflammation8: 88 |
20 | Szabó N, Kincses ZT, Toldi J, Vécsei L(2011) Altered tryptophan metabolism in Parkinson’s disease: A possible novel therapeutic approachJ Neurol Sci15: 256260 |
21 | Guidetti P, Luthi-Carter RE, Augood SJ, Schwarcz R(2004) Neostriatal and cortical quinolinate levels are increased in early grade Huntington’s diseaseNeurobiol Dis17: 455461 |
22 | Beal MF, Kowall NW, Ellison DW, Mazurek MF, Swartz KJ, Martin JB(1986) Replication of the neurochemical characteristics of Huntington’s disease by quinolinic acidNature321: 168171 |
23 | Schwarcz R, Whetsell WO, Mangano RM(1983) Quinolinic acid: An endogenous metabolite that produces axon-sparing lesions in rat brainScience219: 316318 |
24 | Beal MF, Matson WR, Swartz KJ, Gamache PH, Bird ED(1990) Kynurenine pathway measurements in Huntington’s disease striatum: Evidence for reduced formation of kynurenic acidJ Neurochem55: 13271339 |
25 | Rossi F, Schwarcz R, Rizzi M(2008) Curiosity to kill the KAT (kynurenine aminotransferase): Structural insights into brain kynurenic acid synthesisCurr Opin Struct Biol18: 748755 |
26 | Guidetti P, Bates GP, Graham RK, Hayden MR, Leavitt BR, MacDonald ME, Slow EJ, Wheeler VC, Woodman B, Schwarcz R(2006) Elevated brain 3-hydroxykynurenine and quinolinate levels in Huntington disease miceNeurobiol Dis23: 190197 |
27 | Mazarei G, Budac DP, Lu G, Adomat H, Tomlinson Guns ES, Möller T, Leavitt BR(2013) Age-dependent alterations of the kynurenine pathway in the YAC128 mouse model of Huntington diseaseJ Neurochem127: 6852867 |
28 | Ogawa T, Matson WR, Beal MF, Myers RH, Bird ED, Milbury P, Saso S(1992) Kynurenine pathway abnormalities in Parkinson’s diseaseNeurology42: 17021706 |
29 | Sapko MT, Guidetti P, Yu P, Tagle DA, Pellicciari R, Schwarcz R(2006) Endogenous kynurenate controls the vulnerability of striatal neurons to quinolinate: Implications for Huntington’s diseaseExp Neurol197: 3140 |
30 | Laugeray A, Launay J-M, Callebert J, Surget A, Belzung C, Barone PR(2010) Peripheral and cerebral metabolic abnormalities of the tryptophan-kynurenine pathway in a murine model of major depressionBehav Brain Res210: 8491 |
31 | Schwarcz R, Guidetti P, Sathyasaikumar KV, Muchowski PJ(2010) Of mice, rats and men: Revisiting the quinolinic acid hypothesis of Huntington’s diseaseProg Neurobiol90: 230245 |
32 | Campesan S, Green EW, Breda C, Sathyasaikumar KV, Muchowski PJ, Schwarcz R, Kyriacou CP, Giorgini F(2011) The kynurenine pathway modulates neurodegeneration in a Drosophila model of Huntington’s diseaseCurr Biol1: 961966 |
33 | Foster AC, White RJ, Schwarcz R(1986) Synthesis of quinolinic acid by 3-hydroxyanthranilic acid oxygenase in rat brain tissue in vitro J Neurochem47: 2330 |
34 | Giorgini F, Möller T, Kwan W, Zwilling D, Wacker JL, Hong S, Tsai L-CL, Cheah CS, Schwarcz R, Guidetti P, Muchowski PJ(2008) Histone deacetylase inhibition modulates kynurenine pathway activation in yeast, microglia, and mice expressing a mutant huntingtin fragmentJ Biol Chem283: 73907400 |
35 | Zwilling D, Huang S-Y, Sathyasaikumar KV, Notarangelo FM, Guidetti P, Wu H-Q, Lee J, Truong J, Andrews-Zwilling Y, Hsieh EW, Louie JY, Wu T, Scearce-Levie K, Patrick C, Adame A, Giorgini F, Moussaoui S, Laue G, Rassoulpour A, Flik G, Huang Y, Muchowski JM, Masliah E, Schwarcz R, Muchowski PJ(2011) Kynurenine 3-monooxygenase inhibition in blood ameliorates neurodegenerationCell145: 863874 |
36 | Giorgini F, Huang S-Y, Sathyasaikumar KV, Notarangelo FM, Thomas MAR, Tararina M, Wu H-Q, Schwarcz R, Muchowski PJ(2013) Targeted deletion of kynurenine 3-monooxygenase (KMO) in mice: A new tool for studying kynurenine pathway metabolism in periphery and brainJ Biol Chem288: 3655436566 |
37 | Sakurai K, Zou J-P, Tschetter JR, Ward JM, Shearer GM(2002) Effect of 2 indoleamine 2,3-dioxygenase on induction of experimental autoimmune encephalomyelitisJ Neuroimmunol129: 186196 |
38 | Kwidzinski E, Bunse J, Aktas O, Richter D, Mutlu L, Zipp F, Nitsch R, Bechmann I(2005) Indolamine 2,3-dioxygenase is expressed in the CNS and down-regulates autoimmune inflammationFASEB J19: 13471349 |
39 | Platten M, Ho PP, Youssef S, Fontoura P, Garren H, Hur EM, Gupta R, Lee LY, Kidd BA, Robinson WH, Sobel RA, Selley ML, Steinman L(2005) Treatment of autoimmune neuroinflammation with a synthetic tryptophan metaboliteScience310: 850855 |
40 | Ravishankar B, Liu H, Shinde R, Chandler P, Baban B, Tanaka M, Munn DH, Mellor AL, Karlsson MCI, McGaha TL(2012) Tolerance to apoptotic cells is regulated by indoleamine 2,3-dioxygenaseProc Natl Acad Sci U S A109: 39093914 |
41 | Munn DH, Mellor AL(2007) Indoleamine 2,3-dioxygenase and tumor-induced toleranceJ of Clin Invest117: 11471154 |
42 | Pfefferkorn ER(1984) Interferon gamma blocks the growth of Toxoplasma gondii in human fibroblasts by inducing the host cells to degrade tryptophanProc Natl Acad Sci U S A81: 908912 |
43 | Nettelnbreker E, Zeidler H, Bartels H, Dreses-Werringloer U, Däubener W, Holtmann H, Köhler L(1998) Studies of persistent infection by Chlamydia trachomatis serovar K in TPA-differentiated U937 cells and the role of IFN-gammaJ Med Microbiol47: 141149 |
44 | Munn DH, Mellor AL(2013) Indoleamine 2,3 dioxygenase and metabolic control of immune responsesTrends Immunol34: 137143 |
45 | Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, Brown C, Mellor AL(1998) Prevention of allogeneic fetal rejection by tryptophan catabolismScience281: 11911193 |
46 | M’Baïlara K(2006) Decreased brain tryptophan availability as a partial determinant of post-partum bluesPsychoneuroendo31: 407413 |
47 | Adams S, Braidy N, Bessesde A, Brew BJ, Grant R, Teo C, Guillemin GJ(2012) The kynurenine pathway in brain tumor pathogenesisCancer Res72: 56495657 |
48 | Kwidzinski E, Bechmann I(2007) IDO expression in the brain: A double-edged swordJ Mol Med85: 13519 |
49 | Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW(2008) From inflammation to sickness and depression: When the immune system subjugates the brainNat Rev Neurosci9: 4656 |
50 | Moreau M, André C, O’Connor JC, Dumich SA, Woods JA, Kelley KW, Dantzer R, Lestage J, Castanon N(2008) Inoculation of Bacillus Calmette-Guerin to mice induces an acute episode of sickness behavior followed by chronic depressive-like behaviorBrain Behav Immun22: 10871095 |
51 | O’Connor JC, Lawson MA, André C, Briley EM, Szegedi SS, Lestage J, Castanon N, Herkenham M, Dantzer R, Kelley KW(2009) Induction of IDO by bacille Calmette-Guérin is responsible for development of murine depressive-like behaviorJ Immunol182: 32023212 |
52 | Leonard B, Maes M(2012) Mechanistic explanations how cell-mediated immune activation, inflammation and oxidative and nitrosative stress pathways and their sequels and concomitants play a role in the pathophysiology of unipolar depressionNeurosci Biobehav Rev36: 764785 |
53 | Kim H, Chen L, Lim G, Sung B, Wang S, McCabe MF, Rusanescu G, Yang L, Tian Y, Mao J(2012) Brain indoleamine 2,3-dioxygenase contributes to the comorbidity of pain and depressionThe J of Clin Invest122: 29402954 |
54 | Mazarei G, Budac DP, Lu G, Lee H, Möller T, Leavitt BR(2013) The absence of indoleamine 2,3-dioxygenase expression protects against NMDA receptor-mediated excitotoxicity in mouse brainExp Neurol249: 144148 |
55 | Mazarei G, Neal SJ, Becanovic K, Luthi-Carter R, Simpson EM, Leavitt BR(2010) Expression analysis of novel striatal-enriched genes in Huntington diseaseHum Mol Gen19: 609622 |
56 | Guillemin GJ, Smith DG, Smythe GA, Armati PJ, Brew BJ(2003) Expression of the kynurenine pathway enzymes in human microglia and macrophagesAdv Exp Med Biol527: 105112 |
57 | Guillemin GJ, Smythe GA, Veas LA, Takikawa O, Brew BJ(2003) A beta 1-42 induces production of quinolinic acid by human macrophages and microgliaNeuroreport14: 23112315 |
58 | Guillemin GJ, Williams KR, Smith DG, Smythe GA, Croitoru-Lamoury J, Brew BJ(2003) Quinolinic acid in the pathogenesis of Alzheimer’s diseaseAdv Exp Med Biol527: 167176 |
59 | Stoy N, Mackay GM, Forrest CM, Christofides J, Egerton M, Stone TW, Darlington LG(2005) Tryptophan metabolism and oxidative stress in patients with Huntington’s diseaseJ Neurochem93: 611623 |
60 | Kanai M, Funakoshi H, Takahashi H, Hayakawa T, Mizuno S, Matsumoto K, Nakamura T(2009) Tryptophan 2,3-dioxygenase is a key modulator of physiological neurogenesis and anxiety-related behavior in miceMol Brain2: 8 |
61 | Szalardy L, Klivenyi P, Zadori D, Fulop F, Toldi J, Vecsei L(2012) Mitochondrial disturbances, tryptophan metabolites and neurodegeneration: Medicinal chemistry aspectsCurr Med Chem12: 17971806 |
62 | Van der Goot AT, Zhu W, Vázquez-Manrique RP, Seinstra RI, Dettmer K, Michels H, Farina F, Krijnen J, Melki R, Buijsman RC, Ruiz Silva M, Thijssen KL, Kema IP, Neri C, Oefner PJ, Nollen EAA(2012) Delaying aging and the aging-associated decline in protein homeostasis by inhibition of tryptophan degradationProc Natl Acad Sci U S A109: 1491214917 |
63 | Ball HJ, Yuasa HJ, Austin CJD, Weiser S, Hunt NH(2009) Indoleamine 2,3-dioxygenase-2; a new enzyme in the kynurenine pathwayInt J Biochem Cell Biol41: 467471 |
64 | Thevandavakkam MA, Schwarcz R, Muchowski PJ, Giorgini F(2010) Targeting kynurenine 3-monooxygenase (KMO): Implications for therapy in Huntington’s diseaseCNS Neurol Disord Drug Targets9: 791800 |
Figures and Tables
Fig.1
Schematic of the KA pathway. Tryoptophan entering the CNS becomes a substrate for the kynurenine pathway (depicted vertically) or the serotonin pathway (depicted horizontally). The kynurenine pathway then diverges into two branches that can lead to production of either neuroprotective metabolites such as Kynurenic acid (KA) or neurotoxic metabolites such as 3-hydroxykunurenine (3-HK) and quinolinic acid (QA).
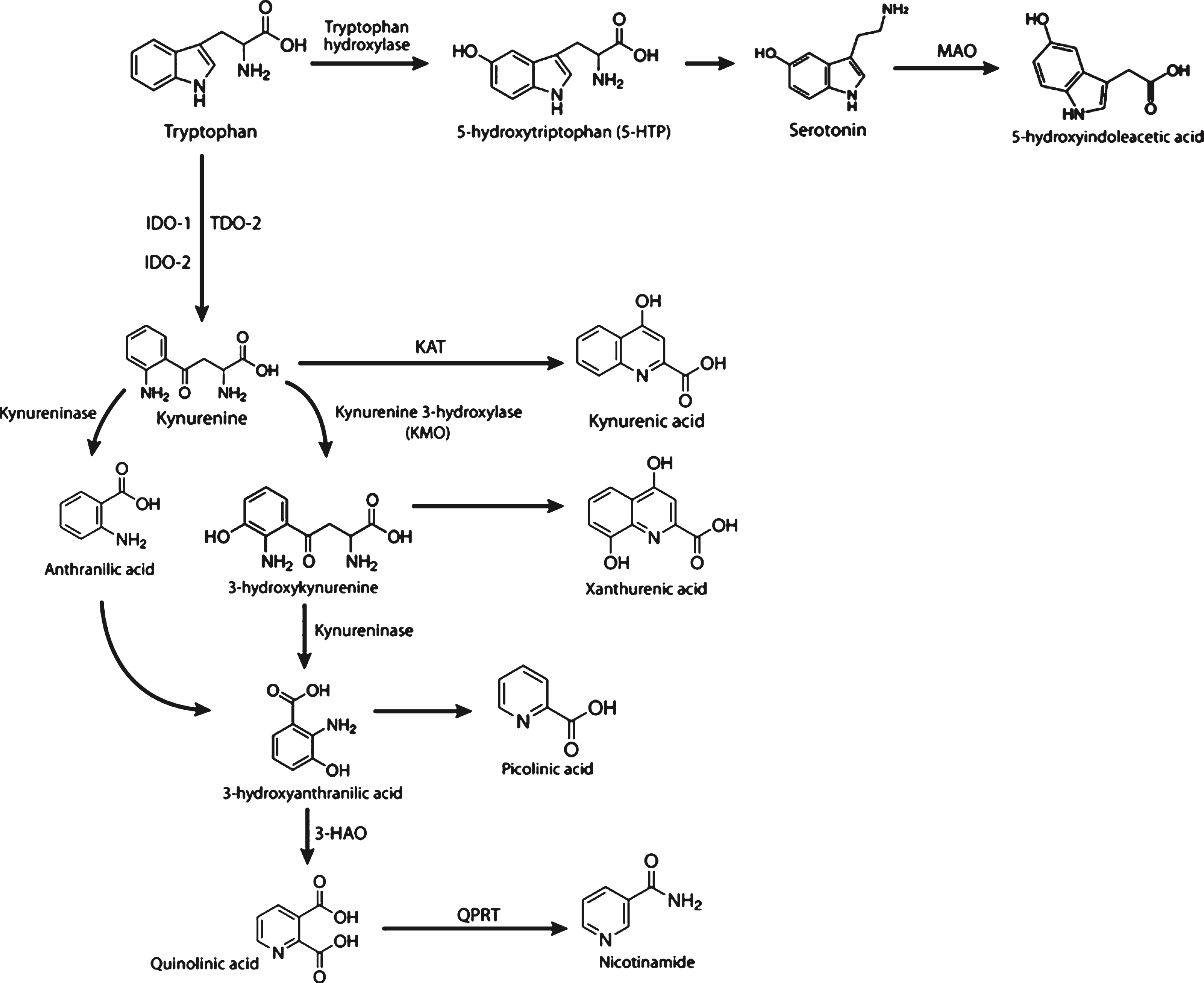
Fig.2
A model describing potential mechanism(s) involved in the selective vulnerability of the striatum in HD based on the YAC128 model. We propose that induction of Ido1 expression and activity in the striatum by mutant huntingtin (mHTT) and inflammation plays a central role in the observed imbalance of downstream kynurenine pathway metabolites. This imbalance, and altered transport of kynurenine pathway metabolites through the blood brain barrier (BBB) from blood to CNS, may result in increased sensitivity of striatal neurons to glutamate toxicity in HD. Dates (months) provided in the figure refer to findings at those ages of YAC128 mice, with 12 months representing an advanced stage with significant striatal cell loss.
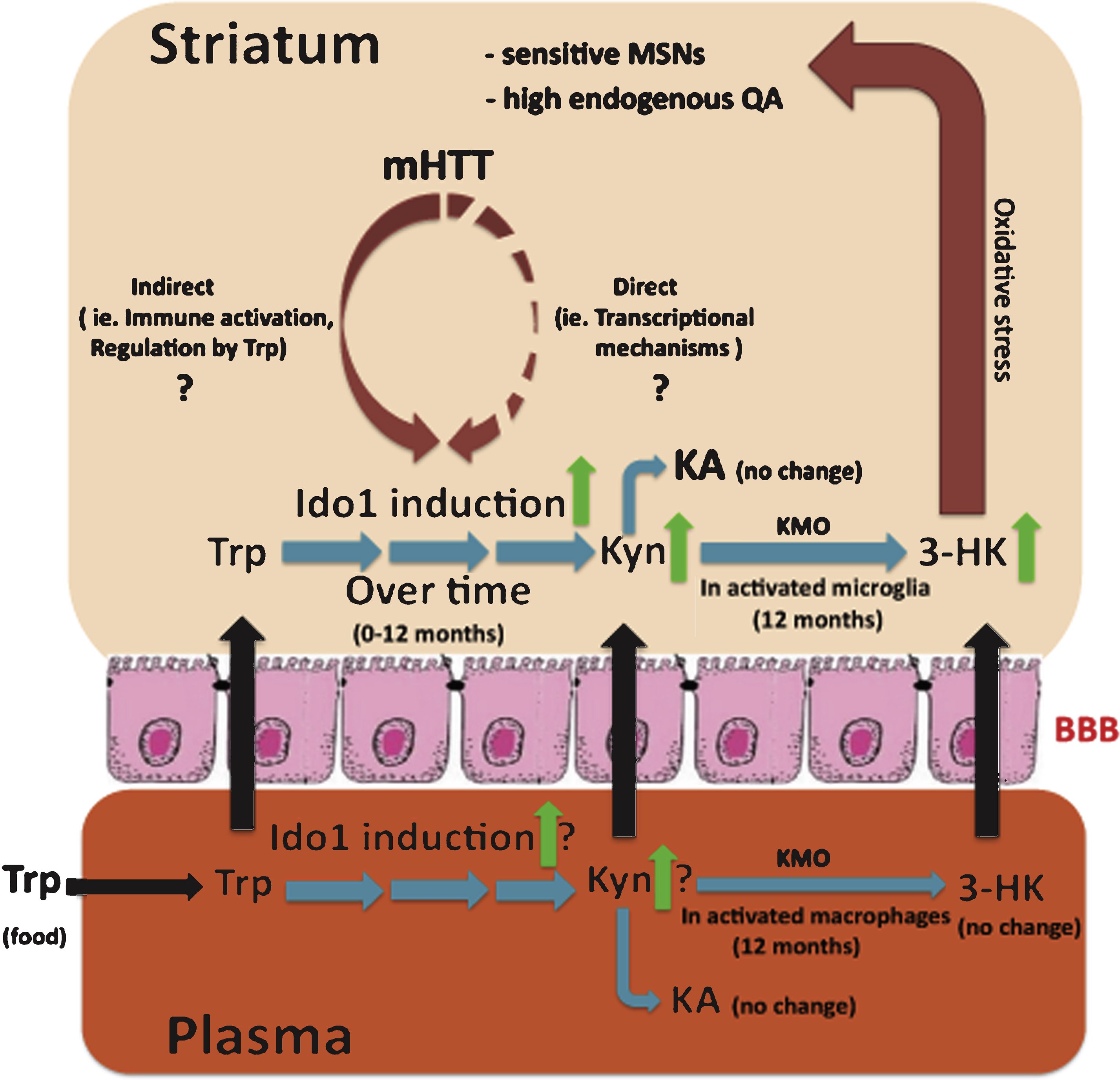
Table 1
Summary of IDO1, IDO2, and TDO changes reported in AD, HD, and PD model systems and patients
 |


