Neonatal Iron Supplementation Induces Striatal Atrophy in Female YAC128 Huntington’s Disease Mice
Abstract
Background: Dysregulation of iron homeostasis is implicated in the pathogenesis of Huntington’s disease. We have previously shown that increased iron intake in R6/2 HD neonatal mice, but not adult R6/2 HD mice potentiates disease outcomes at 12-weeks of age corresponding to advanced HD [Redox Biol. 2015;4 : 363–74]. However, whether these findings extend to other HD models is unknown. In particular, it is unclear if increased neonatal iron intake can promote neurodegeneration in mouse HD models where disease onset is delayed to mid-adult life.
Objective: To determine if increased dietary iron intake in neonatal and adult life-stages potentiates HD in the slowly progressive YAC128 HD mouse model.
Methods: Female neonatal mice were supplemented daily from days 10–17 with 120μg/g body weight of carbonyl iron. Adult mice were provided diets containing low (50 ppm), medium (150 ppm) and high (500 ppm) iron concentrations from 2-months of age. HD progression was determined using behavioral, brain morphometric and biochemical approaches.
Results: Neonatal-iron supplemented YAC128 HD mice had significantly lower striatal volumes and striatal neuronal cell body volumes as compared to control HD mice at 1-year of age. Neonatal-iron supplementation of HD mice had no effect on rota-rod motor endurance and brain iron or glutathione status. Adult iron intake level had no effect on HD progression. YAC128 HD mice had altered peripheral responses to iron intake compared to iron-matched wild-type controls.
Conclusions: Female YAC128 HD mice supplemented with nutritionally-relevant levels of iron as neonates demonstrate increased striatal degeneration 1-year later.
Introduction
Huntington’s disease (HD) is an ultimately fatal heritable neurodegenerative disorder caused by a CAG-repeat expansion in the polyglutamine-enco-ding tract within exon-1 of the huntingtin gene (HTT) [1]. Clinical signs are characterized by an involuntary movement disorder, weight loss and psychiatric disturbances that slowly progress to dementia. While HD has autosomal dominant inheritance with essentially 100% genetic penetrance there is significant variability in age of onset after correction for effects of the CAG expansion mutation size [2]. This variability can be explained by a combination of non-huntingtin genetic and environmental factors [3]. Identifying these factors may contribute to understanding of HD pathways and also suggest new approaches for slowing the progression of human HD.
Iron homeostasis is disrupted in mouse and human HD brain. Studies in R6/2 and N171–82Q HD mice which express N-terminal mutant HTT fragments (mHTT) have demonstrated increases in brain iron with disease progression [4, 5]. Further, iron-imaging and autopsy studies also support accumulation of iron in human HD brain [6, 7]. The mechanisms by which iron accumulates in HD brain are currently unclear. However, iron that is not chelated by protein or other molecules can undergo redox cycling and induce oxidative stress, which is a consistent feature of both cell and murine models as well as human HD [8–11]. While the extent to which iron homeostatic dysregulation may contribute to HD progression is unclear there is evidence to support protective effects of both weak and strong iron chelators in mouse HD [4, 12].
Iron is an essential dietary element. Intake levels in the human population vary widely during both development and adulthood due to natural or milk replacer feeding, variation in solid diet and also the level of iron supplementation [13–15]. To determine if such iron-intake variation may modify HD we previously tested the effect of elevated iron intake during neonatal and adult-life stages in the R6/2 mouse model of HD. We recently reported that iron supplementation in neonatal, but not adult, R6/2 mice potentiates established behavioral, biochemical and morphologic markers of HD [16]. R6/2 HD express the exon-1 fragment of mHTT with 150–180 CAG repeats. This model displays a particularly aggressive phenotype with behavioral deficits at 6 weeks of age and spontaneous death from advanced HD beginning at 12 weeks of age [17]. In the context of neonatal environmental influences on adult neurodegeneration in HD, a disadvantage of studying R6/2 mice is the short time from iron supplementation to advanced HD (10 weeks). Therefore, it is unclear how predictive these findings in R6/2 mice are to human HD where there is a several year delay from the neonatal period to time of disease onset.
YAC128 HD mice express the full-length human mutant huntingtin gene with 128 CAG repeats and are maintained on the FVB inbred rather than B6/CBA F1 genetic background of R6/2 mice [17, 18]. Importantly, they demonstrate delayed onset disease. Deficits in motor function and brain pathology develop around 9–12 months of age [19, 20]. Therefore, investigation of the effect of dietary iron in YAC128 mice would extend our previous findings by determining if results can be repeated in a mouse HD model that differs significantly from the R6/2 HD model. Here we report findings from such studies investigating the effects of elevated iron intake in neonatal and adult-life stage in YAC128 HD mice. Similar to our previous findings [16] we found that iron supplementation in neonatal YAC128 HD mice resulted in potentiation of striatal atrophy while there was no effect of supplementation in adult YAC128 mice.
Materials and Methods
Materials
Chemicals were from Sigma unless otherwise stated.
Mouse husbandry
YAC128 HD mice were maintained on the FVB-inbred background. Tails snips were obtained at 3 weeks of age and mice were genotyped by PCR as previously described [21]. Female mice only were used for experiments. Mice were maintained on a 12-hour light-dark cycle and were provided with access to food and water ad libitum. Unless otherwise state mice were fed cereal-based AIN76A rodent chow that contains 150 ppm iron. Each cage had a mouse igloo (Bio-Serv) and sani-chip bedding (Harlan Laboratories). All procedures were approved by the Institutional Animal Care and Use Committee and followed NIH guidelines.
Experimental design and iron supplementation
Neonatal iron study: Mice were supplemented daily on post-natal day (PND) 10–17 with 120μg/g carbonyl iron or vehicle (saline). This dose is estimated to provide a 40-fold increase in iron intake compared to mother’s milk [13]. Because the mice were genotyped after dosing, iron or vehicle treatments were randomly assigned to litters. At 4-weeks of age, after genotyping and weaning, treated mice were assigned to study cages such that each cage housed mice of the same genotype and treatment.
Adult iron study: Mice were genotyped then randomly assigned to iron-treatment groups at 4-weeks of age. Mice were caged by dietary-iron level and genotype. Base-line behavioral analyses were undertaken at 6 weeks. Iron diets were instituted at 8-weeks of age. Mice were fed one of three casein-based diets that contained 50 (low), 150 (medium) and 500 (high) ppm levels of iron added as ferric citrate (Harlan Laboratories).
Behavioral analyses
Accelerating Rota-rod: We used a rota-rod (IITC Life Science) set to accelerate from 5 to 25 rpm over 15 minutes. Mice were given one training day followed by three consecutive test days. For training, mice were placed on the rotating rod; each time the mouse fell off it was replaced on the rod up to 5 minutes. During testing, each mouse was placed on the rota-rod and the latency to fall was recorded. Trials terminated at 15 minutes of testing.
Spontaneous in-cage wheel running activity: Mice in the neonatal iron study were assessed for spontaneous motor activity via in-cage wheel analysis. Mice were placed individually in large cages with wireless running wheels (Med Associates, Inc.) for 4 days. Mice were then returned to their home cages. Data from days 2–4 were analyzed.
Neuropathology
To prepare tissues for stereological analyses, 12-month-old mice were sacrificed and perfused transcardially with 0.9% (w/v) heparinized (125 units/ml) saline for two minutes followed by 200 mls 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) at 12 mls / minute. Brains were extracted and post-fixed for 48 hours in the same solution. They were then weighed before transfer to cryoprotectant (10% glycerol, 2% DMSO, 0.1M phosphate buffer pH 7.4) solution for 5–10 days at 4°C. Serial 40μm sections representing the entirety of the striatum were obtained with a freezing microtome. Sections were mounted on glass slides such that every 12th section through the entire striatum were present on a single slide. Mounted sections were allowed to dry overnight, stained with a 0.5% (w/v) thionin solution then dehydrated and mounted in permount solution. Stereo Investigator Software (MBF Bioscience, Williston, VT) was used to quantify striatal volumes, cell body volumes and neuronal numbers as described previously [16, 22]. Briefly, in a single section at the level of the anterior commissure, the optical fractionator workflow and the nucleator method were used for estimation of neuronal cell body volume. For neuronal number estimates, each section on a slide that contained the striatum was analyzed using the optical fractionator workflow. Slides were coded such that the operator did not know the genotypes or iron treatments and were analyzed in a systematically randomized order.
Biochemical assays
Glutathione (GSH) was measured as previously described [9]. Tissue iron analysis was carried out essentially as previously described [23] but using an Agilent Technologies 7700 series inductively-coupled plasma mass spectrometer. This method measures total iron in the sample regardless of the form. Serum iron was measured using the ferrozine method (Pointe Scientific Inc.).
Statistical analysis
Analyses were performed using SAS software (version 9.4, Cary, NC). For behavioral analyses with repeated measures the MIXED procedure was used. For all other data, the general linear model (GLM) procedure was used. These methods are valid in the presence of unequal group sizes as was present in some of our experiments due to unequal genotype ratios and occasional losses during the year-long experiments. All main effects and interactions were investigated in the initial model. When significant main effects or interactions were present in the overall model, we performed pre-planned pairwise comparisons on all non-confounded pairs from the same time point. Wheel data was right skewed so are presented as means±95% confidence interval. All other results are reported as means±standard error of the mean (SEM). Unless otherwise stated in figure legends significance bars on graphs indicate pairwise comparisons. Significant differences between confounded comparisons (e.g. wild-type control and HD iron supplemented) are not shown. P-values less than 0.05 were considered significant.
Results
Effect of neonatal iron supplementation on motor test performance YAC128 HD mice. We evaluated forced (rota-rod) and spontaneous (wheel running) motor activity. Rota-rod testing revealed significant effects of age (F3,196 = 28.44, p < 0.001), genotype (F1,196 = 24.76, p < 0.0001) and an age x genotype interaction (F1,196 = 7.11, p = 0.0001) (Fig. 1A). However, there was no effect of neonatal iron supplementation (F1,69 = 0.88, p = 0.3521). Spontaneous wheel running activity was significantly altered by age (F4,155 = 9.82, p < 0.0001), and age x genotype (F4,155 = 2.57, p = 0.0400) and age x iron (F4,155 = 3.29, p = 0.0128) interactions (Fig. 1B). HD iron-supplemented mice had increased wheel running activity at 5 months compared to HD controls (p = 0.0086). Body-weight analysis demonstrated that iron supplementation had no effect on body weight. However, HD mice were significantly heavier than wild-type mice from 3 months of age (data not shown).
Neonatal iron supplementation potentiates neuropathological markers of HD. Brain weights were decreased by HD (F1,37 = 5.35, p = 0.0263). Iron-supplemented HD mice had significantly lower brain weights than wild-type iron-supplemented mice (p = 0.0083). Iron-supplemented HD mice had lower mean brain weights than HD non-supplemented mice; while the pair-wise p-value was 0.0214 the main effect of iron was not significant (F1,37 = 3.52, p = 0.0685) indicating a non-significant trend (Fig. 2A). To investigate possible effects of iron on brain mass in HD mice we quantified both unilateral striatal volume and striatal neuronal cell body volumes (Fig. 2B-D). Striatal volumes were decreased by HD (F1,37 = 6.49, p = 0.0152) and iron supplementation (F1,37 = 10.54, p = 0.0025). Striatal volume was significantly decreased in neonatal supplemented HD versus non-supplemented HD mice (p = 0.0018) (Fig. 2B). Striatal neuronal cell body volumes were significantly decreased in HD mice (F1,37 = 13.82, p = 0.0007). Iron supplementation further decreased neuronal volumes (F1,37 = 5.89, p = 0.0202) and there was a genotype x iron interaction (F1,37 = 4.79, p = 0.0351). HD iron-supplemented mice had significantly lower striatal neuronal cell body volumes as compared to wild-type iron-supplemented (p = 0.0001) and HD non-supplemented (p = 0.0016) mice (Fig. 2C-D). Analysis of total striatal neuron numbers unilaterally revealed that iron supplementation resulted in decreased neuronal estimates (F1,37 = 4.71, p = 0.0365). However, there was no effect of genotype (F1,37 = 0.03, p = 0.8630) and individual pair-wise comparisons were not significant.
Neonatal iron supplementation does not alter brain iron or glutathione levels. To investigate possible pathways by which iron may promote brain atrophy we determined both brain iron and glutathione levels in brain (Fig. 3A-C). While there was a trend for iron to be increased in HD cerebral cortex this was not significant (F1,24 = 2.51, p = 0.1557). Further, there was no effect of iron supplementation on brain iron in either genotype (F1,24 = 0.56, p = 0.4598) (Fig. 3A). Effects of iron supplementation on manganese, zinc and copper were not found (data not shown). Glutathione is an important aqueous phase antioxidant and changes in this system are present in both R6/2 and N171-82Q HD mice [9, 24]. We quantified glutathione (GSH) in brains of study mice. There were no significant effects of genotype or iron supplementation in cerebral cortex or striatum (Fig. 3B-C).
YAC128 HD mice have an altered peripheral response to neonatal iron supplementation. We investigated if peripheral markers of iron status are altered at 12-months of age following neonatal iron supplementation. We found a significant genotype x iron interaction (F1,33 = 5.47, p = 0.0260) on total serum iron levels. Iron-supplementation decreased total serum iron in wild-type mice (p = 0.0114) but not HD mice (p = 0.4596) (Fig. 4A). Liver is an important iron store in the body [25]; therefore we measured iron levels as an additional outcome of peripheral iron status. There was an effect of iron supplementation on liver iron (F1,45 = 4.14, p = 0.0478) but no effect of genotype (F1,45 = 1.84, p = 0.1812). HD iron-supplemented mice had significantly lower liver iron levels than HD non-supplemented (p = 0.0388) and wild-type iron supplemented (p = 0.0125) mice (Fig. 4B).
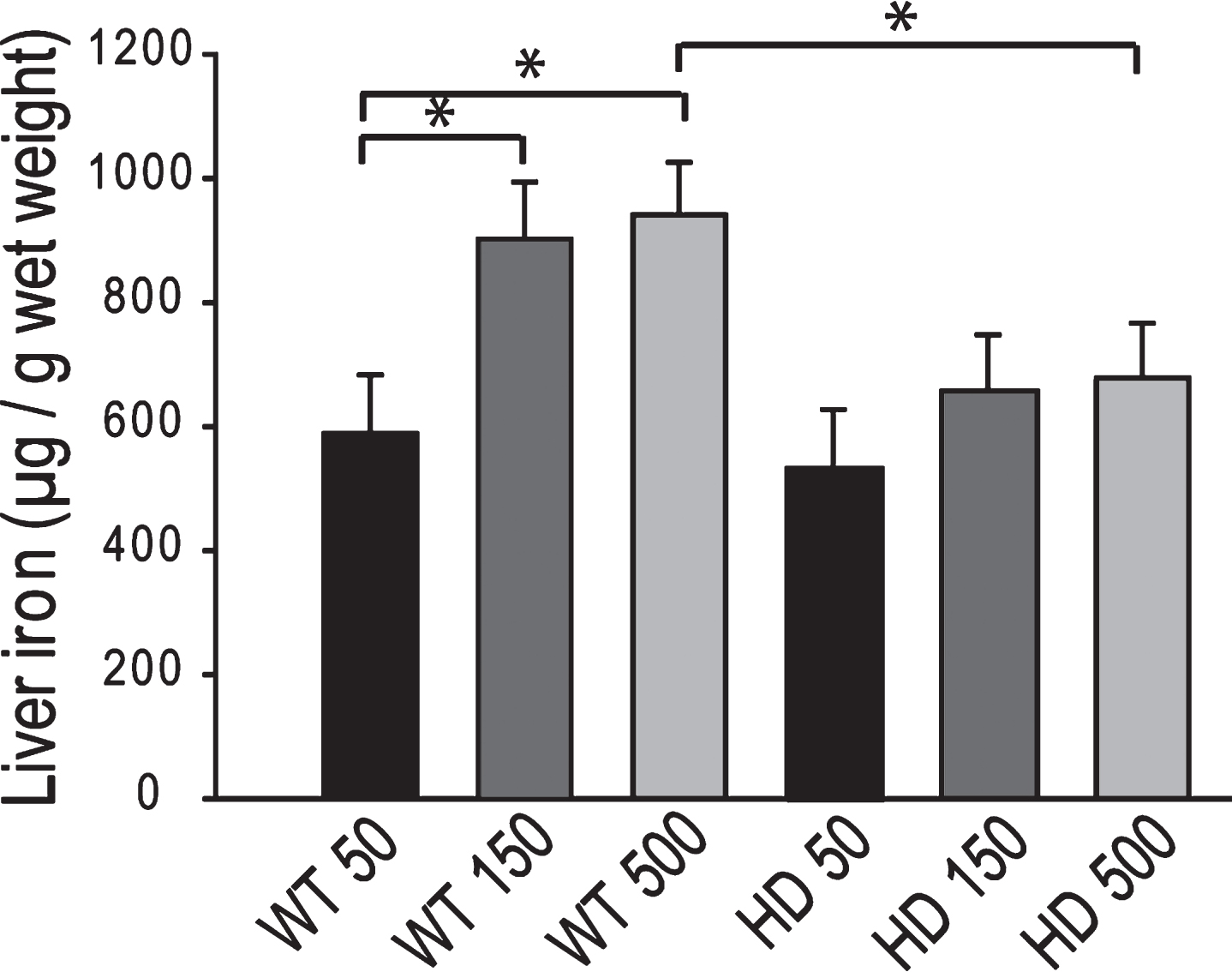
Iron intake level in adult YAC128 HD mice does not potentiate markers of HD progression. To determine if adult iron supplementation alters disease outcomes, we fed adult YAC128 HD and wild-type littermate mice diets containing 50 ppm, 150 ppm or 500 ppm from 2-12 months of age and determined disease outcomes. Rota-rod analysis revealed that HD mice performed significantly worse than wild-type mice (F1,389 = 103.02, p < 0.0001); however, dietary iron level had no effect on endurance (F2,116 = 1.41, p = 0.2482) (Fig. 5). Body weights were significantly increased by both HD (F1,389 = 132.42, p < 0.0001) and increasing dietary iron level (F2,116 = 4.81, p < 0.0099) (not shown). At 12-months of age HD mice had lower brain weights (F1,64 = 32.75, p < 0.0001), striatal volumes (F1,24 = 16.68, p = 0.0004) and striatal neuronal cell body volumes (F1,53 = 8.72, p = 0.0046); however, there were no significant effects of iron (Fig. 6A-C). Further, there were no effects of genotype or dietary iron on striatal neuronal number estimates (data not shown). Cerebro-cortical iron levels were increased in YAC128 HD compared to wild-type litter mates (F1,33 = 4.69, p = 0.0373); however, there was no effect of dietary iron supplementation (F2,33 = 1.05, p = 0.3591) (Fig. 7A). Cortical copper levels were increased in HD mice in this study (F2,32 = 12.36, p = 0.0077) but there was no effect of iron on copper. Manganese and zinc were not altered (data not shown). Genotype and dietary iron level resulted in an interaction for cortical glutathione (F2,52 = 3.96, p = 0.0254). Specifically, YAC128 HD mice on a low-iron diet had lower cortical glutathione than wild-type low-iron and HD high-iron groups (p-values: 0.0020 and 0.0196, respectively) (Fig. 7B). As for the neonatal study (Fig. 4B), we found that with the outcome of liver iron levels YAC128 HD mice responded differently than wild-type littermates at 12-months of age. HD mice had lower liver iron (F1,69 = 6.32, p = 0.0142) than wild-types; iron supplementation increased liver iron (F1,69 = 4.09, p = 0.0121). Medium and high iron intake in wild-type mice significantly increased liver iron (p-values 0.0228 and 0.0102); however, this did not occur in YAC128 HD mice (Fig. 8).
Discussion
The current study demonstrates that neonatal iron supplementation potentiates key morphologic markers of HD progression in female YAC128 HD mice (Fig. 2). This finding corroborates our prior study demonstrating that neonatal, but not adult, iron supplementation potentiates disease in the rapidly progressive R6/2 mouse HD model [16]. The studies support work investigating if iron supplementation in Htt mutation positive infants has a role in potentiating the human disease.
Human HD brains demonstrate degeneration and atrophy mainly involving the striatum and cerebral cortex [26]. Brain atrophy results from a combination of factors including neuronal loss but also neuronal atrophy [26]. Genetic mouse HD models largely recapitulate these changes. For example, both exon-1 and full-length human mHTT expressing R6/2 and YAC128 HD mice are reported to demonstrate reduced brain weights, striatal atrophy, and striatal neuronal atrophy and loss [21, 22]. In the neonatal study iron-supplemented HD mice had significantly lower striatal volumes and striatal neuronal volumes when compared to non-supplemented HD mice (Fig. 2B-C). Importantly, there were no effects of neonatal iron supplementation in wild-type mice on these outcomes (Fig. 2A-C). Therefore, YAC128 HD mice are specifically sensitive to the effects of increased neonatal iron intake as compared to wild-type litter-mate mice indicating an age-dependent genotype by environment interaction. Brain weights, striatal volumes and striatal neuron cell body volumes were not decreased in non-supplemented HD versus wild-type mice (Fig. 2A-C) even though they were in the adult iron study (Fig. 6A-C) suggesting that in the adult study there was greater HD progression at 12-months of age. The reasons for this are not fully clear. It is possible that the cereal versus casein-based diets used altered the course of HD in the two separate studies. Despite the brain morphometric changes strong behavioral correlates were not found. Neonatal supplementation in YAC128 mice had no effect on motor endurance even though activity was decreased in HD compared to wild-type mice (Fig. 1A). Spontaneous in-cage wheel running revealed decreased activity in non-supplemented YAC128 mice compared to wild-type controls that was not present in iron-supplemented mice at 5 months (Fig. 1B). The significance of this result is not fully clear as in early stages of mouse HD activity can be increased [27]. Significantly increased body weights in HD mice from 3 months of age (not shown) complicates interpretation of these data. In contrast to the neonatal iron study, and in agreement with our prior study in R6/2 HD mice [16], we found no evidence that increased iron intake in adult-life potentiates HD (Figs. 5–7). However, both adult and neonatal iron supplementation resulted in different peripheral iron responses at 1-year of age (Figs. 4, 8) indicating that YAC128 HD mice regulate systemic iron differently than wild-type littermates.
We found a trend or a significant increase in cortical iron in YAC128 HD mice (Figs. 3A, 7A) compared to wild-type litter-mates at 1-year of age. This result is consistent with findings in R6/2, N171–82Q and CAG140 HD mice all of which have elevated brain iron levels [5, 28]. Finding elevated cortical iron in YAC128 mice (Fig. 7A) is consistent with previous findings in slowly progressive HD in CAG140 HD mice [5] and demonstrates that crude elevations of brain iron are present early in the clinical course of mouse HD. Changes in brain iron as found in human HD [29] are therefore a consistent and early feature of murine HD. Amyloid precursor protein (APP) is necessary for iron export from neurons [30] and functions by stabilizing the iron-export protein ferroportin [31]. We have previously reported decreased brain APP by Western blot analysis in R6/2 HD mice [5, 16] providing a potential mechanism for elevated brain iron. However, we did not find decreases in brain APP in YAC128 mice at 1-year of age (not shown).
There are numerous mechanisms by which neonatal iron supplementation could promote brain atrophy. Significant elevations of brain iron and resulting oxidative stress is not an underlying pathway based on the absence of changes in cortical iron and brain glutathione (Fig. 3A-C). However, iron toxicity resulting from altered cellular distribution of iron within neurons and glial cells, which would not be detected by the ICP-MS methodology used, cannot be discounted by our data. Copper and manganese have both been implicated in HD pathogenesis [5, 32–34]. Copper and manganese can interact with iron during development [35, 36]. Despite this, we found no effect of iron supplementation status on brain copper or manganese levels suggesting that HD potentiation is not mediated through these elements. While we did not address effects of iron supplementation on absorption in this study we have previously reported that neonatal iron supplementation in R6/2 mice results in a smaller increase in liver iron than in supplemented wild-type littermates at 5 weeks of age [16]. Consistent with this, in both neonatal and adult iron studies supplemented YAC128 HD mice had lower liver iron levels than supplemented wild-type mice (Figs. 4B, 8). These findings show altered peripheral responses to iron between wild-type and HD mice and possibly greater homeostatic defenses against elevated iron uptake in HD mice. Epigenetic changes have been implicated in mediating the effects of diverse early-life environmental exposures on later life changes in brain [37]. Numerous epigenetic alterations have been described in HD mice [38]; therefore, this remains a plausible area of investigation.
Mouse post-natal days 10–17 of age corresponds approximately to birth through 20 weeks of age in a human infant [39]. We supplemented mice of this age with a level of iron as used in prior work [13, 16] that is estimated to provide an iron-intake level forty-fold greater than a naturally fed mice. Natural human milk contains 0.2–0.4 mg / L iron while human milk replacers contain 4–12 mg / L iron [40]. An infant fed a replacer containing 12 mg / L iron would therefore also have an intake level forty-fold over one fed a natural milk diet calculated on a body weight basis. Body surface area which correlates with metabolic rate is an alternative approach to translate dose across species [41]. Using this method the neonatal iron dose we used translates to less than a forty-fold increase in intake over maternal milk. While iron in natural milk is mainly present as highly bioavailable lactoferrin compared to lower available iron salts present in replacers [40], the level of neonatal iron supplementation used is comparable to that in a human infant receiving a fortified diet. Iron levels in the adult study were also chosen to be nutritionally relevant and to model widespread supplementation used in the USA [14]. In mice, 50 ppm is a low intake level, but sufficient to prevent iron deficiency anemia. The middle level, 150 ppm was chosen as this approximates the absolute level present in the cereal-based rodent chow. Five-hundred ppm, was chosen to model an iron-supplemented diet. While we cannot exclude the possibility that higher levels of adult iron intake potentiate mouse HD these levels would translate to intake above what is commonly used in the human population. Taken together, findings demonstrate that in YAC128 HD mice the neonatal period is a critical life-stage at which elevated iron intake can have deleterious effects on later disease progression and that the doses we used are relevant to infant nutrition.
In summary, we demonstrate that neonatal YAC128 HD mice when supplemented with iron demonstrate worsened brain morphologic markers at an early-stage of clinical HD. These findings largely corroborate our findings of a similar study in the rapidly progressive R6/2 model of HD [16]. However, the current findings also extend the earlier findings because they show that neonatal iron supplementation can alter HD outcomes up to a year after supplementation corresponding to middle-age in terms of biologic aging. Similar effects have also been reported in a mouse model of aging [13]. Human HD onset is typically in the third and fourth decades of life corresponding to early to mid-adult life. This and our earlier study [16] support studies investigating if iron supplementation in Htt mutation positive infants is an environmental modifier of HD age of onset or progression.
Conflict of Interest
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
Funding was provided by RO1 NS079450 from NINDS. We thank Jianfang Chen for assistance with mouse experiments and Merl Raisbeck and Roger Siemons for completion of ICP-MS analyses.
REFERENCES
[1] | A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell. (1993) ;72: (6:971–83. |
[2] | Andresen JM , Gayan J , Djousse L , Roberts S , Brocklebank D , Cherny SS , et al. The relationship between CAG repeat length and age of onset differs for Huntington’s disease patients with juvenile onset or adult onset. Ann Hum Genet. (2007) ;71: (Pt 3):295–301. |
[3] | Wexler NS , Lorimer J , Porter J , Gomez F , Moskowitz C , Shackell E , et al. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington’s disease age of onset. Proc Natl Acad Sci U S A. (2004) ;101: (10):3498–503. |
[4] | Chen J , Marks E , Lai B , Zhang Z , Duce JA , Lam LQ , et al. Correction: Iron accumulates in Huntington’s disease neurons: Protection by deferoxamine. PLoS One. (2013) ;8: (11):e77023. |
[5] | Fox JH , Kama JA , Lieberman G , Chopra R , Dorsey K , Chopra V , et al. Mechanisms of copper ion mediated Huntington’s disease progression. PLoS One. (2007) ;2: (3):e334. |
[6] | Rosas HD , Chen YI , Doros G , Salat DH , Chen NK , Kwong KK , et al. Alterations in brain transition metals in Huntington disease: An evolving and intricate story. Arch Neurol. (2012) ;69: (7):887–93. |
[7] | Dominguez DJ , Ng AC , Poudel G , Stout JC , Churchyard A , Chua P , et al. Iron accumulation in the basal ganglia in Huntington’s disease: Cross-sectional data from the IMAGE-HD study. J Neurol Neurosurg Psychiatry. (2015) . doi: 10.1136/jnnp-2014-310183 |
[8] | Firdaus WJ , Wyttenbach A , Giuliano P , Kretz-Remy C , Currie RW , Arrigo AP . Huntingtin inclusion bodies are iron-dependent centers of oxidative events. FEBS J. (2006) ;273: (23):5428–41. |
[9] | Fox JH , Barber DS , Singh B , Zucker B , Swindell MK , Norflus F , et al. Cystamine increases L-cysteine levels in Huntington’s disease transgenic mouse brain and in a PC12 model of polyglutamine aggregation. J Neurochem. (2004) ;91: (2):413–22. |
[10] | Browne SE , Bowling AC , MacGarvey U , Baik MJ , Berger SC , Muqit MM , et al. Oxidative damage and metabolic dysfunction in Huntington’s disease: Selective vulnerability of the basal ganglia. Ann Neurol. (1997) ;41: (5):646–53. |
[11] | Valko M , Jomova K , Rhodes CJ , Kuca K , Musilek K . Redox- and non-redox-metal-induced formation of free radicals and their role in human disease. Arch Toxicol. (2016) ;90: (1):1–37. |
[12] | Nguyen T , Hamby A , Massa SM . Clioquinol down-regulates mutant huntingtin expression in vitro and mitigates pathology in a Huntington’s disease mouse model. Proc Natl Acad Sci U S A. (2005) ;102: (33):11840–5. |
[13] | Kaur D , Peng J , Chinta SJ , Rajagopalan S , Di Monte DA , Cherny RA , et al. Increased murine neonatal iron intake results in Parkinson-like neurodegeneration with age. Neurobiol Aging. (2007) ;28: (6):907–13. |
[14] | Millen AE , Dodd KW , Subar AF . Use of vitamin, mineral, nonvitamin, and nonmineral supplements in the United States: The and National Health Interview Survey results. J Am Diet Assoc. (2004) ;104: (6):942–50. |
[15] | Institute of Medicine (US) Panel on Micronutrients. Dietary reference intakes for vitamin A, vitamin K, arsenic, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium and zinc. National Academy Press, Washington, DC; (2001) . |
[16] | Berggren KL , Chen J , Fox J , Miller J , Dodds L , Dugas B , et al. Neonatal iron supplementation potentiates oxidative stress, energetic dysfunction and neurodegeneration in the R6/2 mouse model of Huntington’s disease. Redox Biol. (2015) ;4: :363–74. |
[17] | Mangiarini L , Sathasivam K , Seller M , Cozens B , Harper A , Hetherington C , et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. (1996) ;87: (3):493–506. |
[18] | Hodgson JG , Agopyan N , Gutekunst CA , Leavitt BR , LePiane F , Singaraja R , et al. A YAC mouse model for Huntington’s disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron. (1999) ;23: (1):181–92. |
[19] | Van Raamsdonk JM , Pearson J , Slow EJ , Hossain SM , Leavitt BR , Hayden MR . Cognitive dysfunction precedes neuropathology and motor abnormalities in the YAC128 mouse model of Huntington’s disease. J Neurosci. (2005) ;25: (16):4169–80. |
[20] | Ferrante RJ Mouse models of Huntington’s disease and methodological considerations for therapeutic trials. Biochim Biophys Acta. (2009) ;1792: (6):506–20. |
[21] | Slow EJ , van Raamsdonk J , Rogers D , Coleman SH , Graham RK , Deng Y , et al. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet. (2003) ;12: (13):1555–67. |
[22] | Dodds L , Chen J , Berggren K , Fox J . Characterization of striatal neuronal loss and atrophy in the R6/2 mouse model of Huntington’s disease. PLoS Curr. (2014) ;6. |
[23] | Maynard CJ , Cappai R , Volitakis I , Cherny RA , Masters CL , Li QX , et al. Gender and genetic background effects on brain metal levels in APP transgenic and normal mice: Implications for Alzheimer beta-amyloid pathology. J Inorg Biochem. (2006) ;100: (5-6):952–62. |
[24] | Lu Z , Marks E , Chen J , Moline J , Barrows L , Raisbeck M , et al. Altered selenium status in Huntington’s disease: Neuroprotection by selenite in the N171-82Q mouse model. Neurobiol Dis. (2014) ;71: :34–42. |
[25] | Ganz T . Systemic iron homeostasis. Physiol Rev. (2013) ;93: (4):1721–41. |
[26] | Vonsattel JP , Keller C , Cortes Ramirez EP . Huntington’s disease - neuropathology. Handb Clin Neurol. (2011) ;100: :83–100. |
[27] | Menalled LB , Sison JD , Dragatsis I , Zeitlin S , Chesselet MF . Time course of early motor and neuropathological anomalies in a knock-in mouse model of Huntington’s disease with 140 CAG repeats. J Comp Neurol. (2003) ;465: (1):11–26. |
[28] | Chen J , Marks E , Lai B , Zhang Z , Duce JA , Lam LQ , et al. Iron accumulates in Huntington’s disease neurons: Protection by deferoxamine. PLoS One. (2013) ;8: (10):e77023. |
[29] | Rosas HD , Salat DH , Lee SY , Zaleta AK , Pappu V , Fischl B , et al. Cerebral cortex and the clinical expression of Huntington’s disease: Complexity and heterogeneity. Brain. (2008) ;131: (Pt 4):1057–68. |
[30] | Duce JA , Tsatsanis A , Cater MA , James SA , Robb E , Wikhe K , et al. Iron-export ferroxidase activity of beta-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell. (2010) ;142: (6):857–67. |
[31] | McCarthy RC , Park YH , Kosman DJ . sAPP modulates iron efflux from brain microvascular endothelial cells by stabilizing the ferrous iron exporter ferroportin. EMBO Rep. (2014) ;15: (7):809–15. |
[32] | Xiao G , Fan Q , Wang X , Zhou B . Huntington disease arises from a combinatory toxicity of polyglutamine and copper binding. Proc Natl Acad Sci U S A. (2013) ;110: (37):14995–5000. |
[33] | Stansfield KH , Bichell TJ , Bowman AB , Guilarte TR . BDNF and Huntingtin protein modifications by manganese: Implications for striatal medium spiny neuron pathology in manganese neurotoxicity. J Neurochem. (2014) ;131: (5):655–66. |
[34] | Kumar KK , Goodwin CR , Uhouse MA , Bornhorst J , Schwerdtle T , Aschner M , et al. Untargeted metabolic profiling identifies interactions between Huntington’s disease and neuronal manganese status. Metallomics. (2015) ;7: (2):363–70. |
[35] | Crowe A , Morgan EH . Iron and copper interact during their uptake and deposition in the brain and other organs of developing rats exposed to dietary excess of the two metals. J Nutr. (1996) ;126: (1):183–94. |
[36] | Chua AC , Morgan EH . Effects of iron deficiency and iron overload on manganese uptake and deposition in the brain and other organs of the rat. Biol Trace Elem Res. (1996) ;55: (1-2):39–54. |
[37] | Modgil S , Lahiri DK , Sharma VL , Anand A . Role of early life exposure and environment on neurodegeneration: Implications on brain disorders. Transl Neurodegener. (2014) ;3: :9. |
[38] | Valor LM , Guiretti D , Lopez-Atalaya JP , Barco A . Genomic landscape of transcriptional and epigenetic dysregulation in early onset polyglutamine disease. J Neurosci. (2013) ;33: (25):10471–82. |
[39] | Clancy B , Finlay BL , Darlington RB , Anand KJ . Extrapolating brain development from experimental species to humans. Neurotoxicology. (2007) ;28: (5):931–7. |
[40] | Institute of Medicine (US) Committee on the Evaluation of the Addition of Ingredients New to Infant Formula. Infant Formula Evaluating the Safety of New Ingredients.. The National Academic Press, Washington DC; (2004) . |
[41] | Reagan-Shaw S , Nihal M , Ahmad N . Dose translation from animal to human studies revisited. FASEB J. (2008) ;22: (3):659–61. |
Figures and Tables
Fig.1
Effect of neonatal iron supplementation on motor test performance in YAC128 HD mice. Mice were supplemented with iron or saline at 10–17 days. A. Rota-rod endurance in YAC128 HD and wild-type mice. YAC128 HD mice have significantly decreased rota-rod motor endurance from 6-months of age compared to wild-type mice. Iron supplementation does not alter rota-rod performance. n = 16–21, B. Spontaneous in-cage wheel running activity. Non-supplemented YAC128 HD mice have decreased wheel activity at 3, 5 and 12 months of age compared to wild-type controls. Iron-supplemented HD mice have increased wheel-running activity at 5-months of age compared to HD controls. P-values: *p < 0.05, **p < 0.01, ***p < 0.001, n = 12.
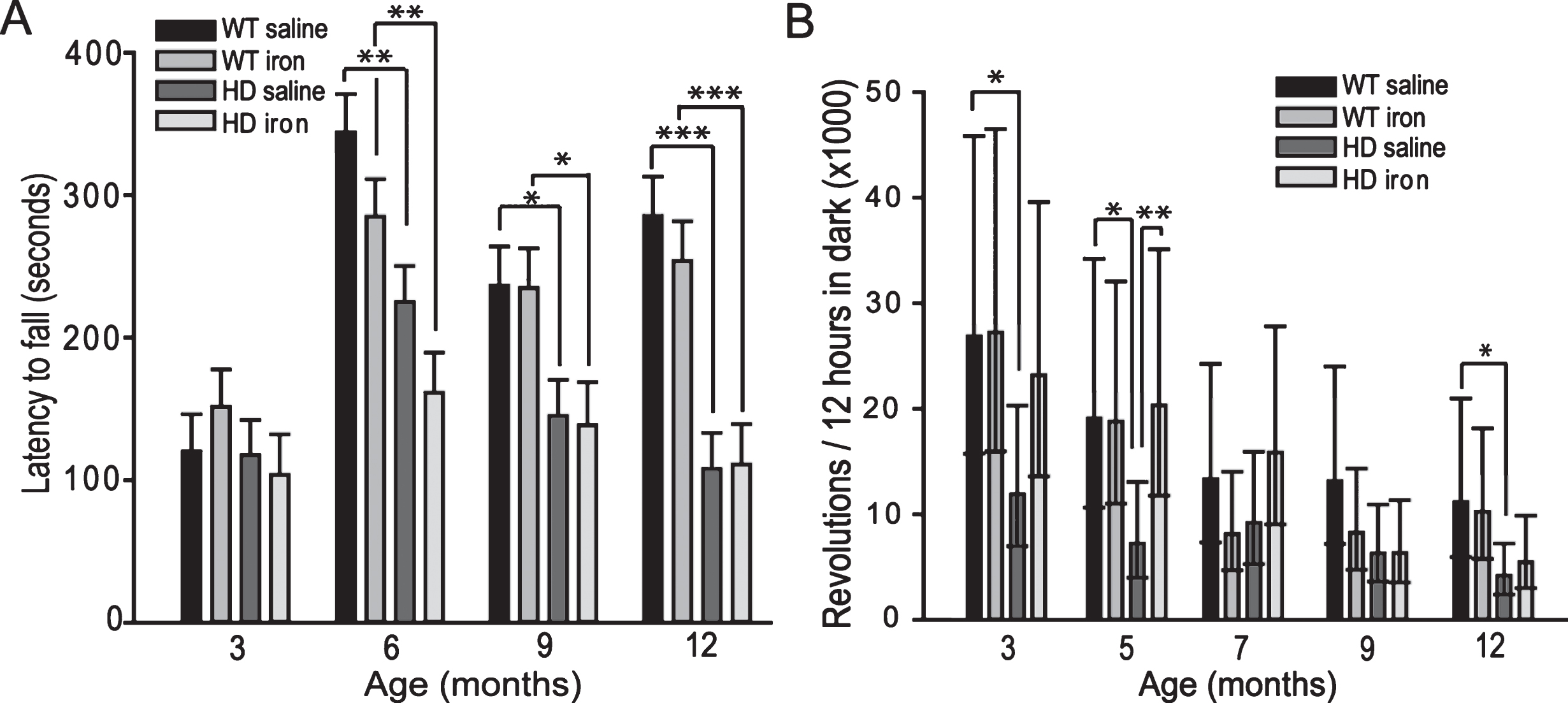
Fig.2
Neonatal iron supplementation results in brain and striatal atrophy in YAC128 HD mice. Mice were supplemented at days 10–17 with iron or saline and sacrificed at 1-year of age. A. Neonatal iron-supplementation decreases brain weights in YAC128 HD but not wild-type mice. B. Neonatal iron-supplementation decreases striatal volume in YAC128 HD mice but not wild-type mice. C-D. Striatal neuronal cell body volumes are decreased by neonatal iron supplementation in YAC128 HD but not wild-type mice (C). Representative images are shown in D. Arrows point to neurons. Scale bar = 25μm. P-values: *p < 0.05, **p < 0.01, ***p < 0.001, n = 8–12 mice.
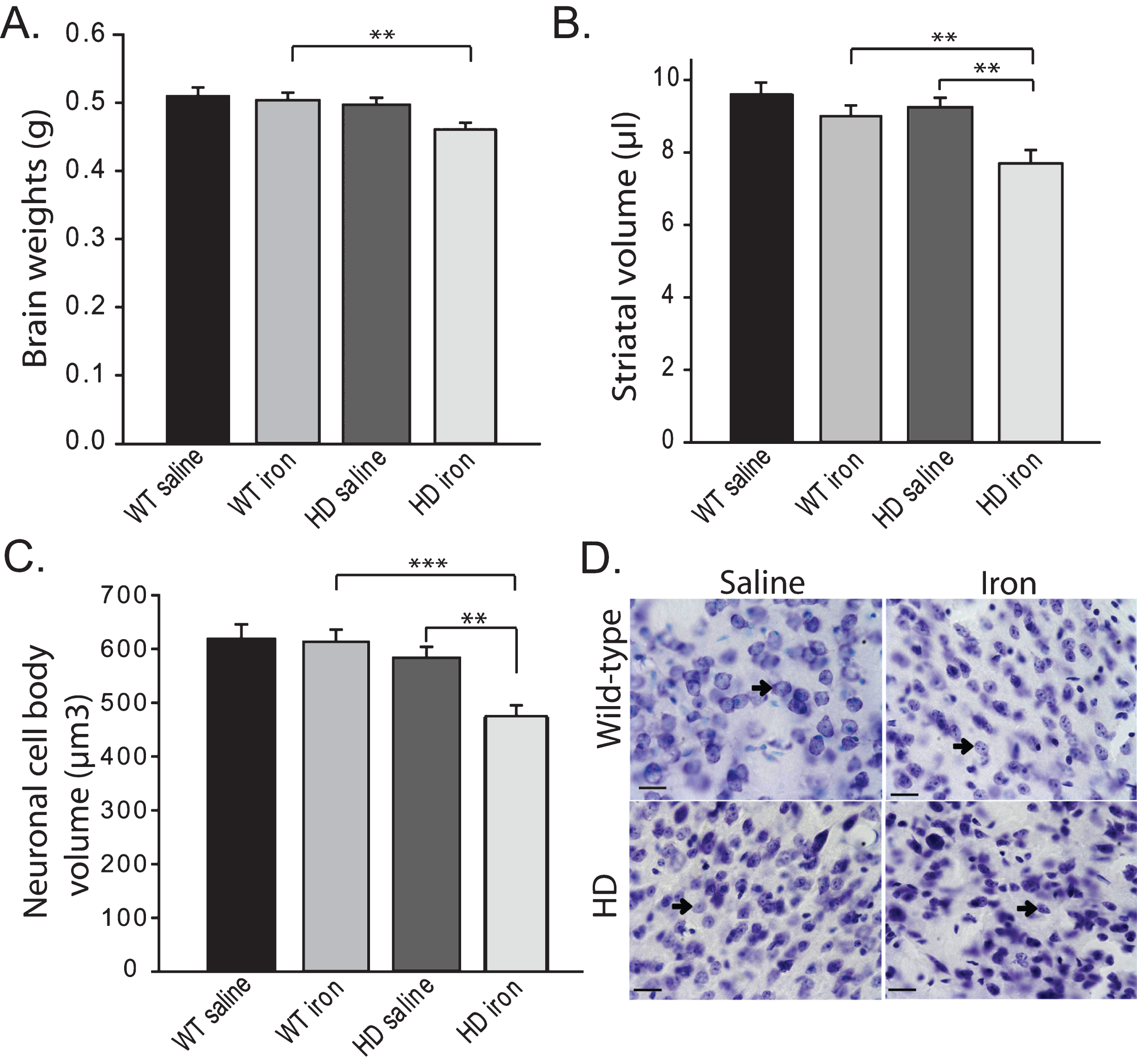
Fig.3
Neonatal iron supplementation does not alter brain iron or glutathione. Mice were supplemented at 10–17 days and sacrificed at 1-year of age. A. Cerebro-cortical iron is not altered by HD or the presence of neonatal iron supplementation. n = 6–7, B-C. Glutathione levels in cerebral cortex (B) and striatum (C) are not altered by HD or neonatal iron supplementation. n = 9–15 for cortex and 9–12 for striatum.
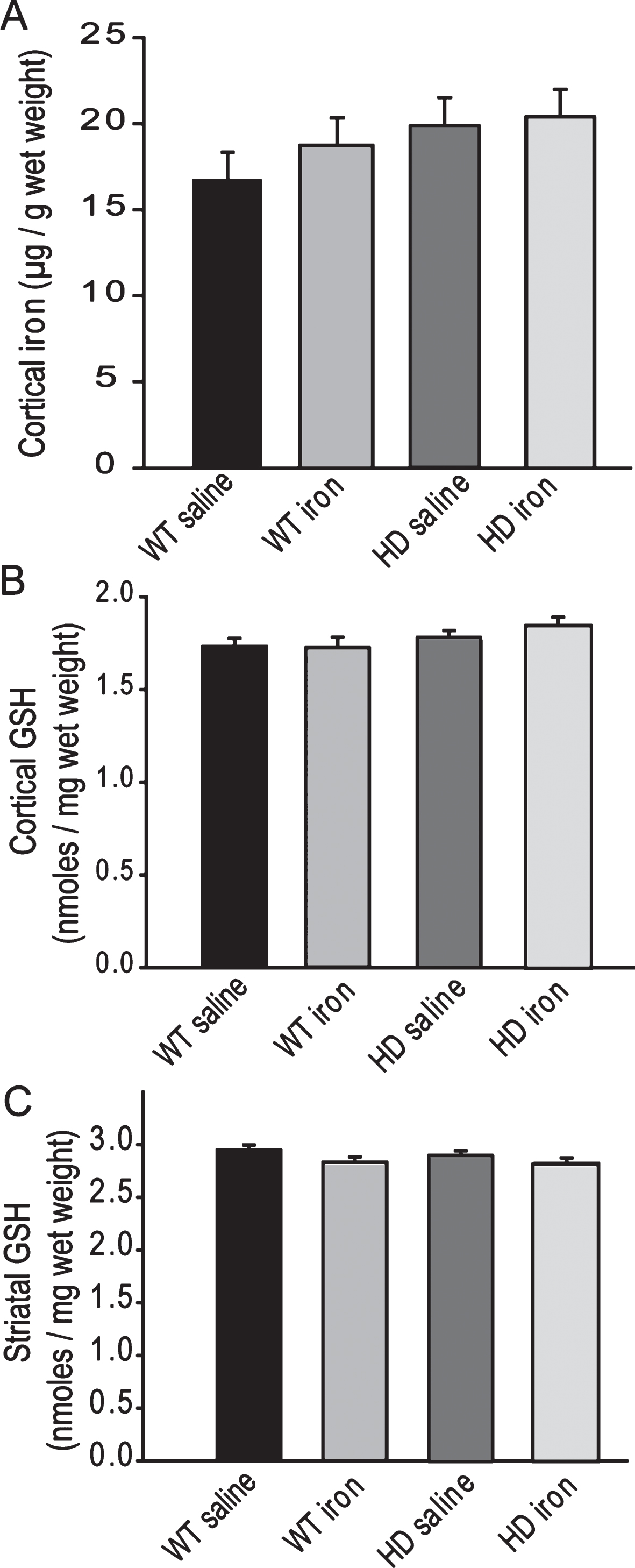
Fig.4
YAC128 HD mice have an altered peripheral response to neonatal iron supplementation. Mice were supplemented at 10–17 days and sacrificed at 1-year of age. A. Neonatal iron supplementation results in decreased serum iron at 1-year of age in wild-type but not YAC128 HD mice. n = 8–10, B. Iron-supplemented YAC128 HD, but not wild-type mice have significantly decreased liver iron at 1-year of age. P-value: *p < 0.05, n = 12–13.
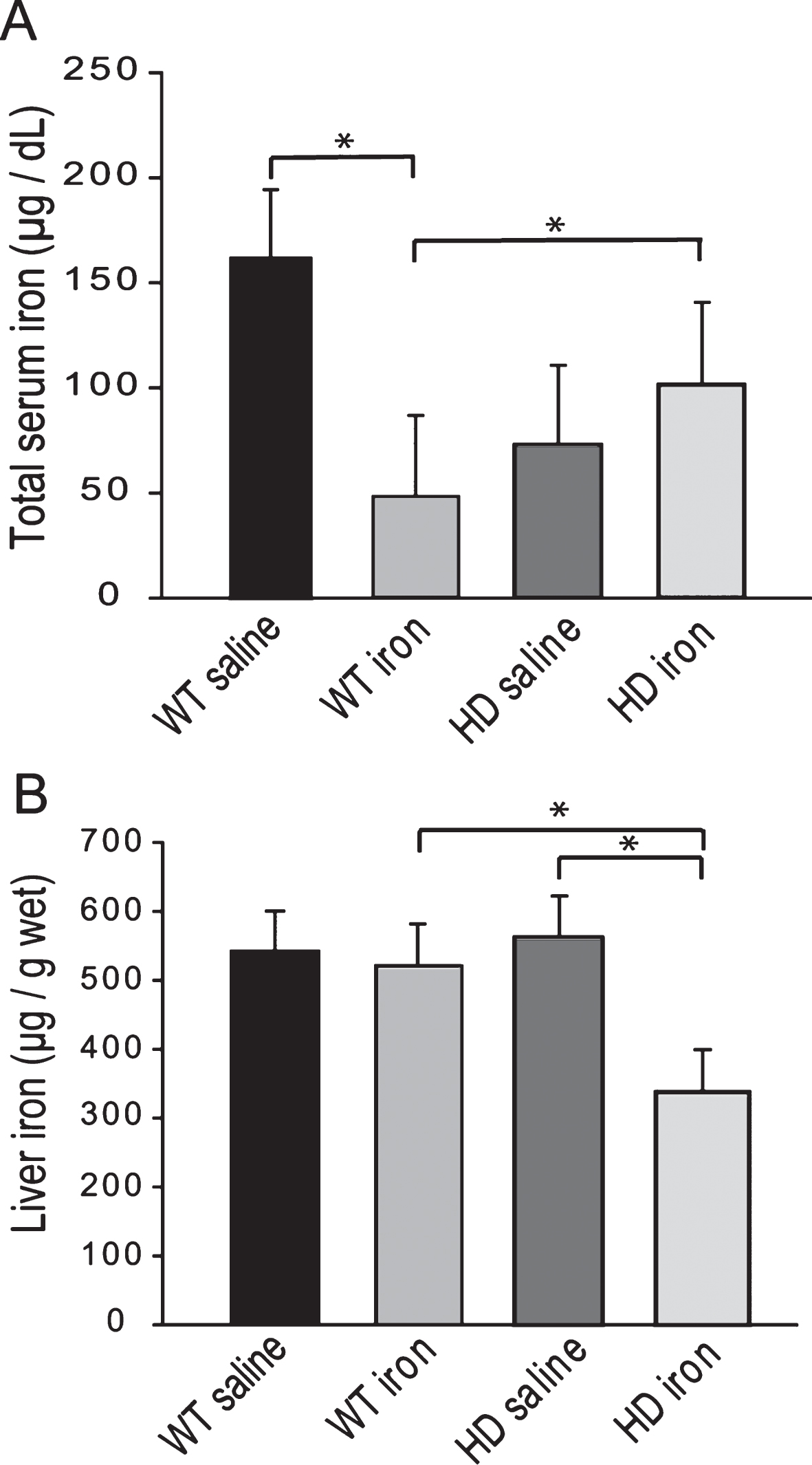
Fig.5
Adult iron supplementation does not alter rota-rod motor endurance in YAC128 HD mice. Mice were maintained on special diets from 2–12 months of age. YAC128 HD groups performed significantly worse on the rota-rod compared to respective iron-dose wild-type mouse groups. Iron supplementation had no effect on performance. P-values: *p < 0.05, **p < 0.01, ***p < 0.001, n = 19–24.
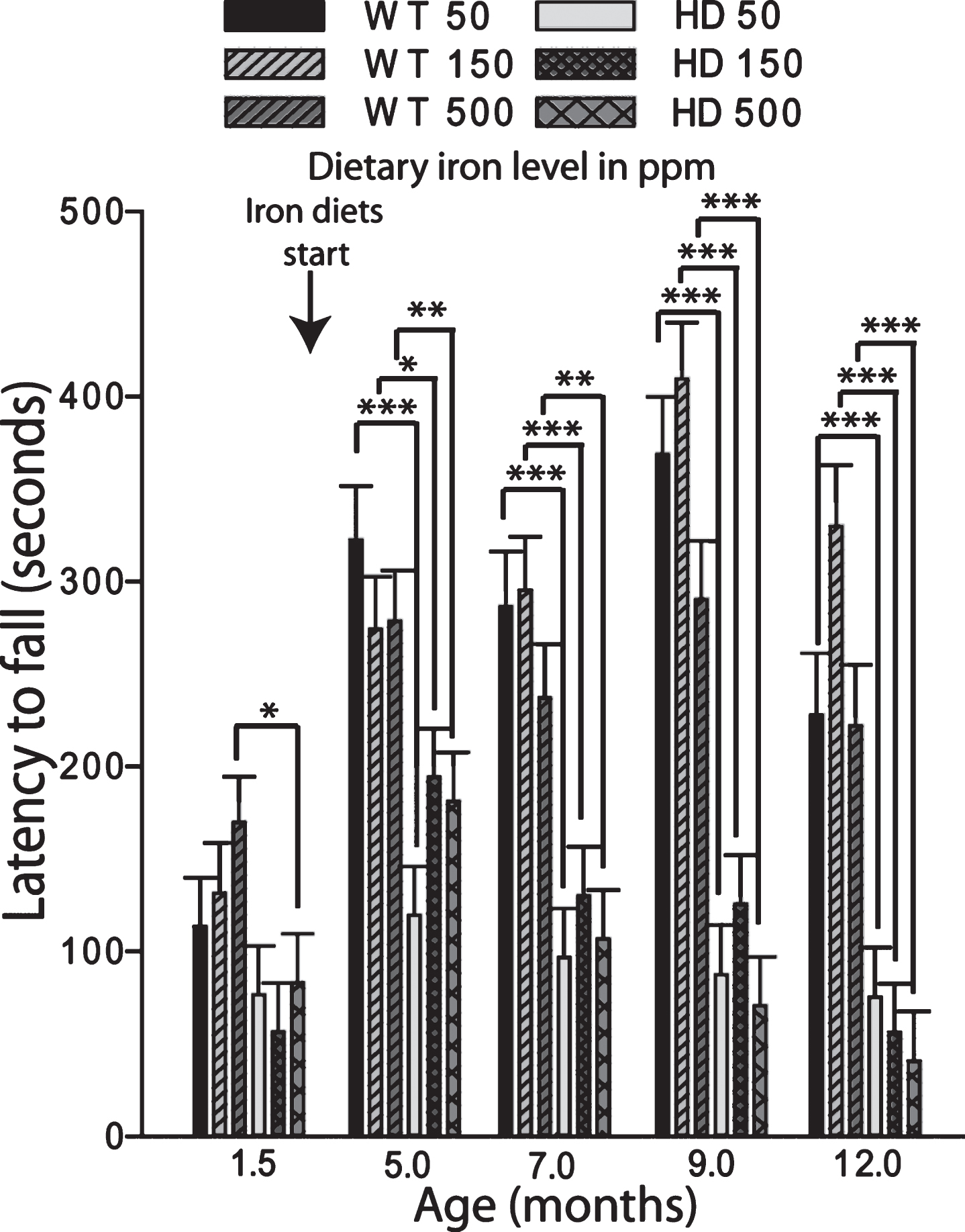
Fig.6
Adult iron supplementation does not exacerbate neurodegeneration markers in YAC128 HD mice. Mice were maintained on diets from 2–12 months of age. A. Brain weights of YAC128 HD and wild-type mice fed one of three custom iron diets. YAC128 mice have significantly decreased brain weights compared to wild-type litter-mates. Iron supplementation has no effect on brain weight in HD or wild-type mice. n = 12–15, B. Striatal volumes are decreased by HD but not by dietary iron level. There was a significant effect of genotype with YAC128 HD mice having lower striatal volumes across all iron groups, n = 4–6, C. Striatal cell body volumes are not altered by dietary iron intake level in YAC128 HD or wild-type mice. There was a significant effect of genotype with YAC128 HD mice having lower cell volumes across all iron groups. ME = main effect comparison. P-values: *p < 0.05, **p < 0.01, ***p < 0.001, n = 4–6.
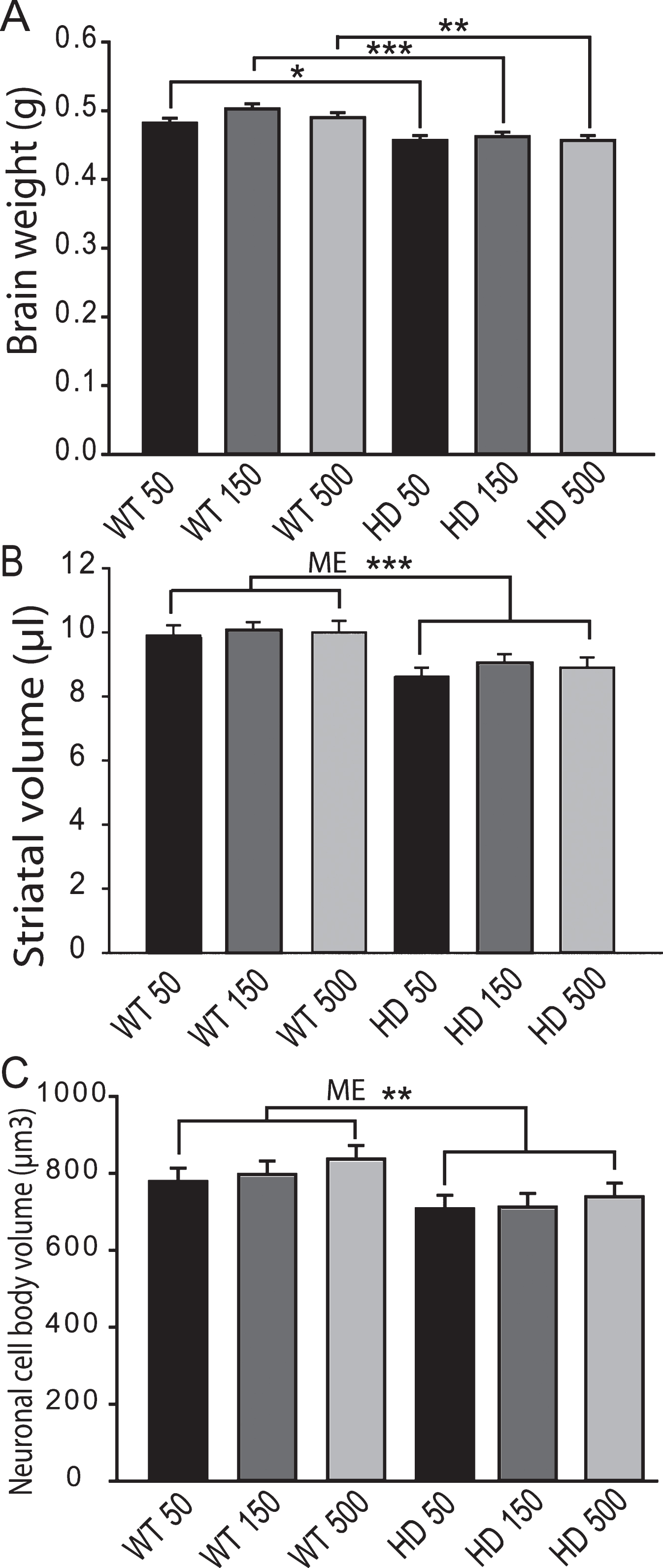
Fig.7
Brain iron and glutathione status in YAC128 HD mice supplemented with different levels of iron as adults. Mice were maintained on diets from 2–12 months of age. A. Cerebro-cortical iron levels are increased in HD mice but are not altered by iron-intake level. There was a significant effect of genotype (ME = main effect comparison). n = 5–9, B. Cortical GSH in wild-type is not altered by iron-intake level. Low versus high iron intake (150 and 500 ppm iron, respectively) results in decreased cortical glutathione in YAC128 mice. P-value: *p < 0.05, n = 9–11.
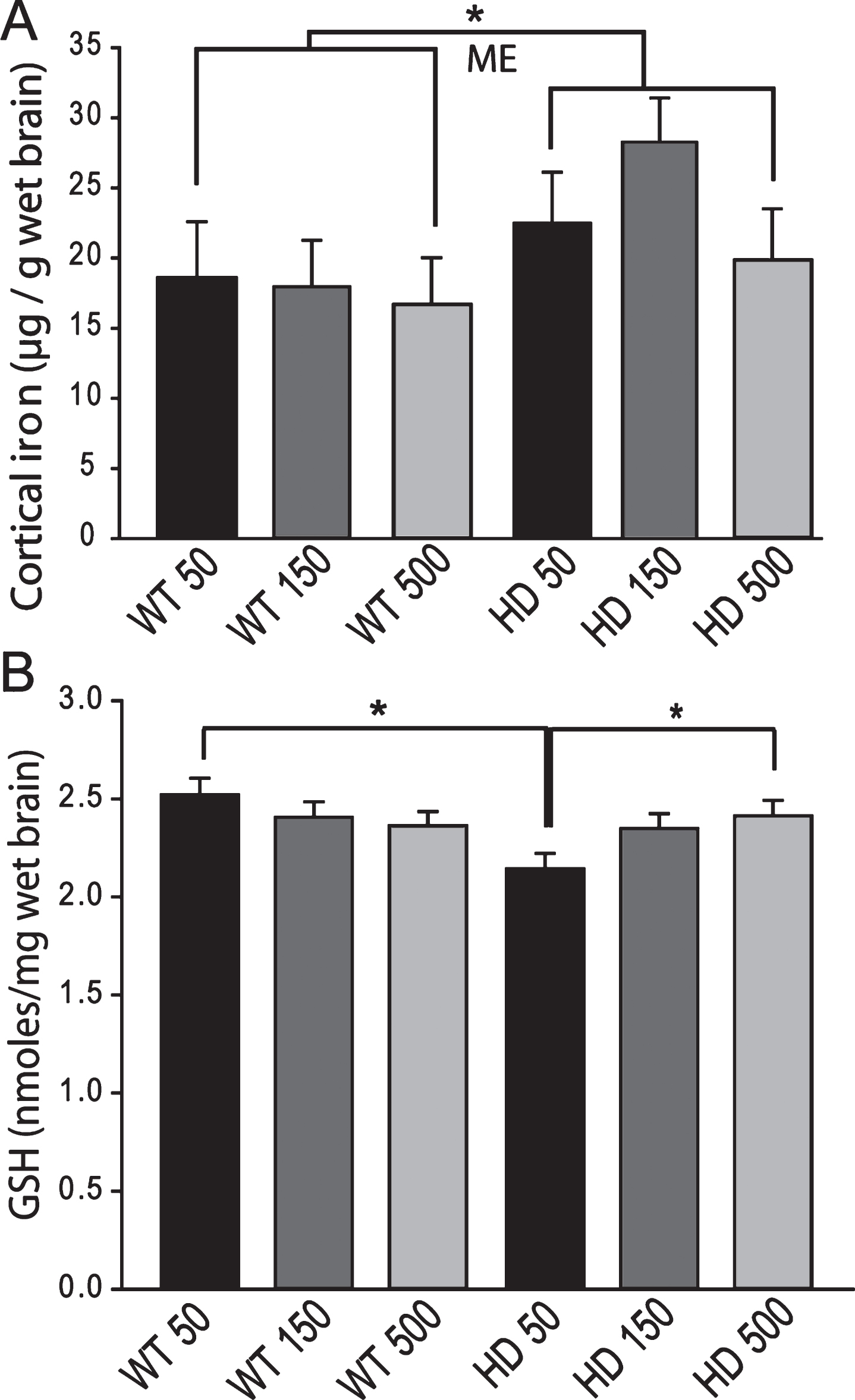
Fig.8
YAC128 HD mice have an altered peripheral response to adult iron intake level. Medium and high dietary iron intake levels in adult mice (150 and 500 ppm, respectively) results in increased liver iron in wild-type but not YAC128 HD mice at 1-year of age. P-value: *p < 0.05, n = 12–15.