
Impaired Skeletal Development by Disruption of Presenilin-1 in Pigs and Generation of Novel Pig Models for Alzheimer’s Disease
Abstract
Background:
Presenilin 1 (PSEN1) is one of the genes linked to the prevalence of early onset Alzheimer’s disease. In mice, inactivation of Psen1 leads to developmental defects, including vertebral malformation and neural development. However, little is known about the role of PSEN1 during the development in other species.
Objective:
To investigate the role of PSEN1 in vertebral development and the pathogenic mechanism of neurodegeneration using a pig model.
Methods:
CRISPR/Cas9 system was used to generate pigs with different mutations flanking exon 9 of PSEN1, including those with a deleted exon 9 (Δexon9). Vertebral malformations in PSEN1 mutant pigs were examined by X-ray, micro-CT and micro-MRI. Neuronal cells from the brains of PSEN1 mutant pigs were analyzed by immunoflourescence, followed by image analysis including morphometric evaluation via image J and 3D reconstruction.
Results:
Pigs with a PSEN1 null mutation (Δexon9-12) died shortly after birth and had significant axial skeletal defects, whereas pigs carrying at least one Δexon9 allele developed normally and remained healthy. Effects of the null mutation on abnormal skeletal development were also observed in fetuses at day 40 of gestation. Abnormal distribution of astrocytes and microglia in the brain was detected in two PSEN1 mutant pigs examined compared to age-matched control pigs. The founder pigs were bred to establish and age PSEN1ΔE9/+ pigs to study their relevance to clinical Alzheimer’s diseases.
Conclusions:
PSEN1 has a critical role for normal vertebral development and PSEN1 mutant pigs serves as novel resources to study Alzheimer’s disease.
INTRODUCTION
PSEN1 is identified as one of the major causative genes for autosomal dominant early onset Alzheimer’s disease (AD).1 As a component of γ-secretase complex, PSEN1 processes the amyloid-β protein precursor (AβPP) and Notch family proteins.2–6 AβPP is processed by the γ-secretase complex, and abnormal activities of the γ-secretase complex have been suggested as a potential cause of AD. For example, mutations in PSEN1 are found in patients with familial AD (fAD).1,7 In addition, the deletion of exon 9 (Δexon9) in PSEN1 has been reported to cause early onset fAD.8–12 The mutations in PSEN1 alter enzymatic activity of the secretase and facilitate the accumulation of beta amyloids.13 Similarly, the deletion of exon9 results in the accumulation of the uncleaved PSEN1 proteins because the exon is responsible for its endoproteolytic cleavage function,14 thus leading to abnormal AβPP cleavage and abnormal amyloid peptide accumulation.15
As a key component of γ-secretase, PSEN1 also interacts with other key molecules during development and is essential for normal skeletal development. For example, PSEN1 induces proteolytic cleavage of the C-terminal fragment of Notch, and therefore, regulates nuclear translocation of the intracellular domain of Notch.16 Since Notch signaling is a key cascade pathway for normal somitogenesis, mutations in Notch signaling pathway genes lead to abnormal skeletal development in mice and humans.17–19 Specifically, the Psen1-null mouse dies shortly after birth and exhibits somite disorganization and rib cage abnormalities,20,21 and a mouse model carrying homozygous Δexon9 mutations presents neonatal mortality with skeletal defects.22 While the mouse models provide valuable information about the function of PSEN1 and its relevance to the pathogenesis of AD, limited alternative animal models are available to conduct comparative studies and effectively develop clinical solutions.
Pigs are a suitable preclinical large animal model to study human diseases, especially in the neuroscience field.23 Their gyrencephalic brain’s size and overall volume is about 10% of the size of a human brain and the gray-to-white matter ratio and the complexity of the distribution of crossing fibers in the pig brain are more similar to humans than rodent brain models.24,25 The lack of genetically engineered humanized models and assays discourage the use of pigs for biomedical applications; however, the development of genome editing tools has lowered the barrier for establishing additional clinically relevant pig models and more assays are now available including behavior and cognition assessments.26,27 Pig models carrying a pathogenic allele of AD, such as PSEN1 Δexon9, should present pathophysiology of clinical AD and improve our understanding of the disease beyond using currently available conventional laboratory animal models.
In this study, the function of PSEN1 was explored in pigs by using the CRISPR/Cas9 technology. Different types of mutations were introduced into the exon9 including deletion of the exon9 and impact of the mutations were investigated to understand physiological role of PSEN1 and its potential relevance to the AD.
MATERIALS AND METHODS
Animals
Use of animals was in accordance with approved protocol and standard operating procedures by the Animal Care and Use Committee of the University of Missouri (Protocol 13520).
In vitro embryo production
Porcine oocytes were collected from ovaries purchased from Desoto Biosciences LLC (Seymour, TN, USA) or Applied Reproductive Technology LLC (Madison, WI, USA). Cumulus oocyte complexes (COCs) were placed in 4-well dishes containing TCM-199 (Invitrogen) supplemented with 3.05-mM glucose, 0.91-mM sodium pyruvate, 0.57-mM cysteine, 10-ng/mL EGF, 0.5-mg/mL LH, 0.5-mg/mL FSH, 10-ng/mL gentamicin, and 0.1% polyvinyl alcohol (PVA) at 38.5°C, 5% CO2 in humidified air. After 42–44 h of maturation, cumulus cells were removed by vortexing in the presence of 0.03% hyaluronidase. Oocytes with a polar body were collected in manipulation medium (TCM-199 supplemented with 0.6 mM NaHCO3, 2.9 mM Hepes, 30 mM NaCl, 10 ng/ml gentamicin, and 3 mg/ml bovine serum albumin (BSA)) and placed in 50μl droplets of fertilization medium (modified Tris-buffered medium with 113. 1 mM NaCl, 3 mM KCl, 7.5 mM CaCl2, 11 mM glucose, 20 mM Tris, 2 mM caffeine, 5 mM sodium pyruvate, and 2 mg/ml BSA) in a group of 25–30 oocytes. Semen was prepared following the protocol described in our previous study,28 then sperm were added to the droplets with oocytes at a final concentration of 0.5×106/ml. Oocytes and sperm were co-incubated in fertilization medium at 38.5°C, 5% CO2 in humidified air for 4 h. After fertilization, embryos were cultured in PZM3 medium29 at 38.5°C, 5% CO2, 5% O2 in humidified air.
Microinjection
Three sgRNAs targeting different regions of PSEN1 gene were designed by using the CRISPR design tool, CRISPOR.30 To induce targeted deletion of exon 9 in PSEN1, two sgRNA combinations were used: sgRNA1&3 and sgRNA2&3 (Fig. 1A). The combinations are designed to introduce double strand breaks flanking exon 9 of PSEN1. Each sgRNA combination (10 ng/μl each) mixed with in vitro-synthesized Cas9 mRNA (20 ng/μl) was injected into the cytoplasm of presumptive zygotes after in vitro fertilization (IVF) by using a FemtoJet microinjector (Eppendorf) as previously described.31 Embryos were microinjected in manipulation medium on a heated stage of a Nikon inverted microscope. After the microinjection, the zygotes were washed and then cultured in PZM3 medium for 5-6 days.
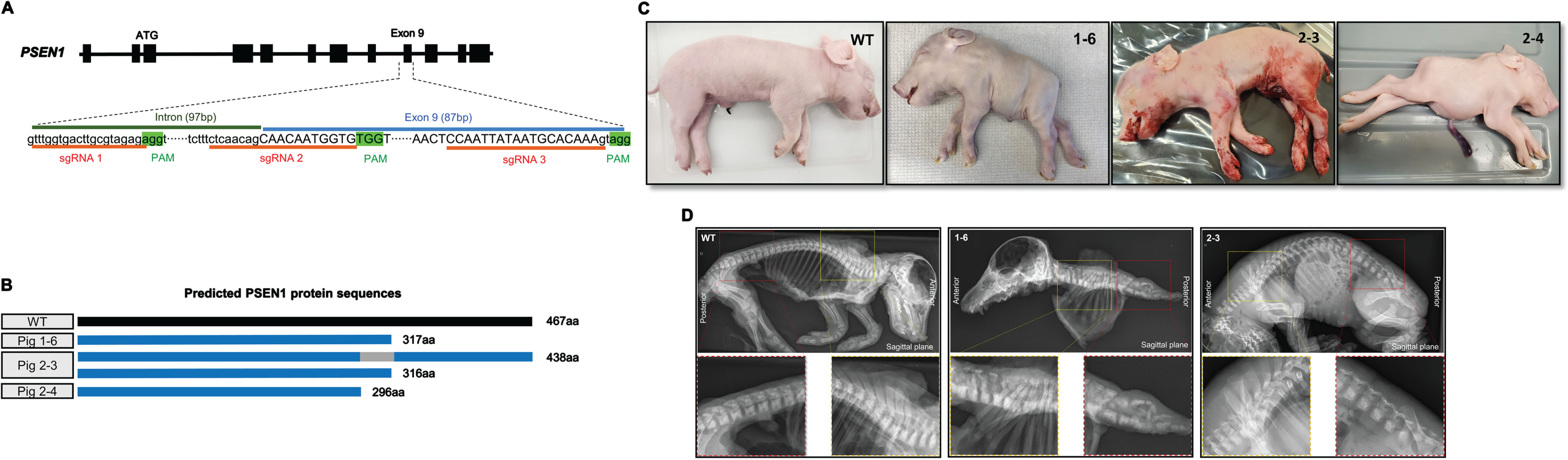
Fig. 1
Generation of PSEN1-targeted piglets and skeletal defects in the stillborn piglets. A) Strategies for editing the PSEN1 gene to induce a targeted deletion of exon 9 by using the CRISPR/Cas9 system. Two sgRNAs targeting near the 5’- and 3’ ends of exon 9 (sgRNA 1 and 3 or sgRNA 2 and 3) were simultaneously injected into porcine zygotes together with Cas9 mRNA. B) Predicted PSEN1 protein sequences expressed in the stillborn piglets based on their genotypes. Piglets 1–6 and 2–4 were expected to express truncated PSEN1 proteins, approximately 300aa long, while piglet 2-3 was expected to express a similar truncated protein as well as a 436 aa protein that was translated from its ΔE9 allele. C) External appearances of the malformations presented in stillborn PSEN1 null piglets compared to an age-matched wildtype (WT) pig. Among the three stillborn piglets, piglets 1–6 and 2–4 piglets exhibited a short axial body trunk whereas 2-3 exhibited a normal morphology except for a shortened tail. D) Radiographic images of PSEN1-targeted stillborn piglets. Compared to wildtype and 2-3 piglets, 1–6 had defects in the axial skeleton including vertebrae and ribs. The yellow rectangular box represents the thoracic vertebrae and the red box represents the lumbar and sacral vertebrae.
Surgical embryo transfer and fetal collection
Day 6 blastocysts (n = 30–50) developed from zygotes injected with Cas9 mRNA and sgRNAs targeting PSEN1 were surgically transferred into oviducts of surrogate gilts near the ampullary-isthmic junction on days 4 or 5 after exhibiting standing estrus as previously described.32 Pregnancies were carried to term or a pregnant surrogate was sacrificed on gestation day 40 and fetuses were collected from the uterus to detect the impact of PSEN1 disruption during fetal development. A total of four transfers were performed and three resulted insuccessful pregnancy.
Genotyping
To genotype PSEN1-targeted pigs and fetuses, gDNA was isolated from ear tips of piglets and fetal skin tissue, respectively, by using the GeneJET Genomic DNA Purification Kit (Thermo Fisher Scientific) following the manufacturer’s instructions. Primers flanking projected double strand break sites were designed to amplify target region of PSEN1 genes: forward primer 5’- CATTGTTGCTGGCGTGAACAC-3’ and reverse primer 5’-CTTGGCTGTACGAACTGGGT-3’. The target regions were PCR amplified by using Dream Taq DNA Polymerase (Thermo Fisher Scientific). PCR conditions were as follows, initial denature at 95°C for 2 min, denature at 95°C for 30 s, annealing at 60°C for 30 s and extension at 72°C for 30 s for 34 cycles, 72°C for 5 min and holding at 4°C. To identify sequences of PSEN1 mRNA after the targeted modifications, total RNA was isolated from the brain tissue of the stillborn piglets by using the RNeasy Mini Kits (Qiagen), then cDNA was synthesized by using the Maxima H Minus First Strand cDNA Synthesis Kit (Thermo Fisher Scientific). The synthesized cDNA was used as a template for RT-PCR and primers were designed to amplify the transcript of the PSEN1 gene flanking the CRISPR/Cas9 target region: forward primer 5’-TTGAAACAGCTCAGGAGAGAAA-3’; reverse primer 5’- CTTGGCTGTACGAACTGGGT-3’. RT-PCR was performed by using the Dream Taq DNA Polymerase (Thermo Fisher Scientific). PCR conditions were as follows, initial denature at 95°C for 2 min, denature at 95°C for 30 s, annealing at 60°C for 30 s and extension at 72°C for 30 s for 38 cycles, 72°C for 5 min and holding at 4°C. The PCR products were purified by using a GeneJet PCR Purification Kit (Thermo Fisher Scientific) and were then ligated into pCR 2.1 TA cloning vector (Thermo Fisher Scientific). Ten colonies of each cloned sample were Sanger sequenced.
RT-qPCR
To assess the transcript abundance of GFAP and AIF1 in the brain, tissue homogenization of cortex samples was performed followed by mRNA isolation using the Dynabeads™ mRNA DIRECT™ Purification Kit (Invitrogen). The Maxima H Minus First Strand cDNA Synthesis Kit (Thermo Fisher Scientific) was used to synthesize cDNA and RT-qPCR amplification was performed using the QIAquant 96 5plex (Qiagen) with Fast SYBR™ Green Master Mix (Applied Biosystems) and appropriate primers (Supplementary Table 1). The porcine GAPDH gene was used as an internal control to normalize transcript level of the target genes as relative quantification. Each sample was run independently and in quadruplicate at the following PCR conditions: initial denature at 95°C for 2 min, denature at 95°C for 5 s and annealing at 60°C for 30 s for 35 cycles.
X-ray, micro-CT, and micro-MRI imaging
X-ray radiography imaging of neonatal piglets was performed by using an OTC18M Radiographic System (Del Medical, Bloomingdale, IL). Micro-CT and -MRI imaging of the fetal skeleton was performed by using a VECTor6CTUHROI Micro-CT system (MILabs, Houten, The Netherlands) and a 7.0 Tesla MRI system (Bruker Biospin, Billerica, MA), respectively. Dorsal and sagittal images were taken from day 40 fetuses stored in 70% ethanol at 4°C. All the images were measured and processed by using the MicroDicom software.
Tissue treatment
Brains of euthanized wildtype and PSEN1 pigs were collected, and cerebral cortex sections were trimmed into regions of interest and fixed in 4% paraformaldehyde. After four days, the fixed tissue was washed with 1X PBS and transferred into 30% sucrose in PBS until the tissues sank. Tissue blocks were frozen in Tissue-Tek™ CRYO-OCT Compound (Andwin Scientific) and maintained at –80°C. Tissue samples were cryo-sectioned at 20μm thickness and placed in 50/50 Glycerol-PBS + 0.1% NaN3 (Sodium azide) and stored at 4°C.
Immunofluorescence
Using the floating method, tissue samples were removed from the storage solution and were washed three times with 1X PBS followed by incubation in blocking solution (5% NGS in PBS + 0.3% Triton X-100) for 1 h at room temperature. Primary antibodies, GFAP (Novus, NBP1-05198), NeuN (Millipore, MAB377B), and Iba1 (FUJIFILM Wako Shibayagi, 019-19741) were prepared by using 5% NGS in PBST solution with the following concentrations GFAP (1:2000), NeuN and Iba1 (1:1000) and incubated at 4°C overnight. Primary antibodies were visualized by using the secondary antibodies, Gt anti-chicken Alexa-flour 488 (Thermo Fisher, A32931), Gt anti-mouse Alexa-Flour 568 (Thermo Fisher, A-21124), and Gt anti-rabbit Alexa-flour 647 (Thermo Fisher, A-21245), which were prepared in 5% NGS in PBST solution with a concentration of 1:1000 and incubated for 2 h at room temperature. The tissues were washed three times with 1X PBS and mounted using Flouromount- G with DAPI (Invitrogen). Slides were left to dry at room temperature in the dark and moved to 4°C until imaging.
Image analysis to identify neural cells
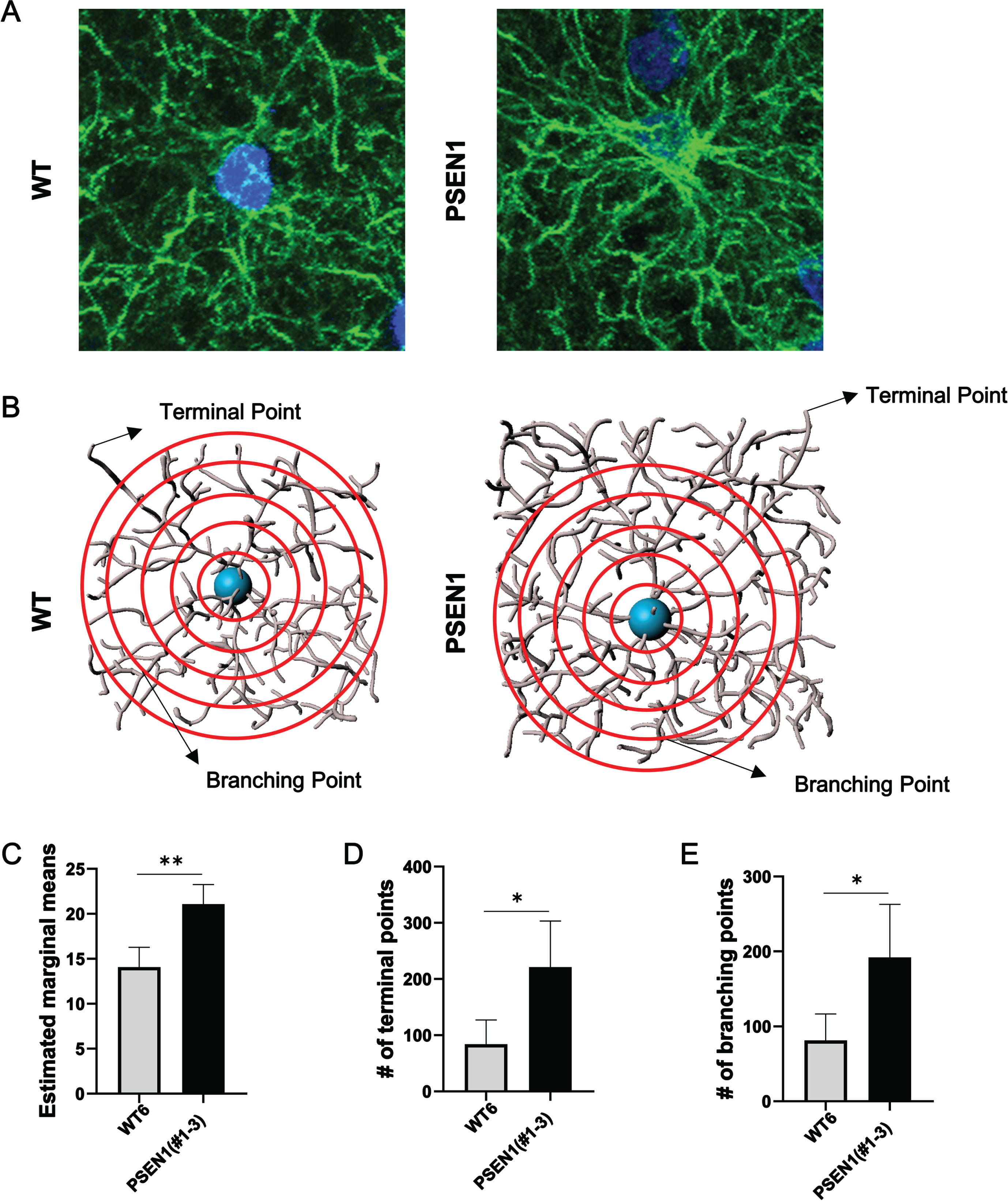
The number of cells and intensity of fluorescent signals was quantified using Image J (NIH). To quantify Iba1 positive areas to reflect microglia, first images were exported to Image J and a region of interest (ROI) was selected around Iba1 positive regions. Using the same ROI, 10 random microglia from each animal were selected and duplicated. Then, Z-projections were generated, and areas of signal were selected using color thresholding and the signal was quantified. Student’s T-test was used to analyze the cell number and intensity of fluorescent signals (p < 0.05). For 3-D reconstruction of astrocytes, images were taken using a Leica SP8 Lightning confocal microscope and image stacks were imported into Imaris 10.1.1. Five random astrocytes were selected per animal for reconstruction, and their processes were traced using the filament trace tool. Morphometric data such as the total number of terminal points, total number of branching points, and Sholl analyses were conducted using the images. Following 3D reconstruction of astrocytes, concentric spheres around the filament originating from the soma with an incremental radius of 1μm were used to assess the complexity of the astrocytes via Sholl intersections. Statistical comparisons were performed via Student’s T-test for the number of terminal and branching points and ANCOVA for Sholl analysis. p value less than 0.05 was considered significant.
RESULTS
Generation of pigs carrying modified PSEN1 gene
Ten piglets were farrowed from two surrogates carrying PSEN1 edited embryos (Table 1). As expected, all the piglets carried mutations adjacent to exon 9 of PSEN1 at the genomic DNA level without wildtype sequences (Table 1). Five (ID: 1-2, 1-3, 1-4, 2-1, and 2-2) out of the 10 piglets carried at least one allele with the intended exon 9 deletion. Four piglets (ID: 1-1, 1-3, 2-1, and 2-3) were predicted to retain a functional PSEN1 allele because changes in the nucleotide sequences did not lead to a premature stop codon. To determine how the changes in the PSEN1 gene altered the mRNA sequence, brain tissue was collected from the stillborn piglets and subsequent PSEN1 mRNA sequences were captured by RT-PCR followed by Sanger sequencing. Only a single mRNA type was detected from piglet (1-6) while the other two piglets (2-3 and 2-4) possessed two different mRNAs; the mRNA sequences matched changes to their genomic DNA (Table 1). Based on the mRNA sequences, the expected PSEN1 protein sequences of the stillborn piglets were compared to the wildtype PSEN1 sequences (Table 1 and Fig. 1B). All the stillborn piglets had truncated PSEN1 proteins as compared to the wildtype protein (467aa). Two stillborn piglets (1-6 and 2-4) were expected to express a single truncated PSEN1 protein (317aa and 296aa, respectively). Piglet 2-3 was expected to express two types of truncated PSEN1 (438aa and 316aa). Piglet 2-3 had an allele lacking the exon 9 sequence at the genomic level, and the translated protein from the allele (438aa) was projected to be partially functional - the exon 9 deletion results in the loss of 30aa from the wildtype protein. Other than the three pigs, the remaining pigs thrived and remained healthy. These healthy pigs carried at least one allele of wildtype or PSEN1 lacking exon 9. Because the pigs were raised as breeders, no mRNA was extracted from their brains to identify changes at the mRNA level.
Table 1
Genotyping of PSEN1 targeted pigs
| Pigs | Phenotype | Sex | sgRNA combinations | Mutations in gDNA* | Mutations in mRNA | Predicted protein | |
| 1-1 | Normal | Female | sgRNA 1&3 | Mosaic w/o wild | (IF/IF/IF/IF) | N/A | N/A |
| 1-2 | Normal | Male | sgRNA 1&3 | Biallelic | (–/ΔE9) | N/A | N/A |
| 1-3 | Normal | Male | sgRNA 1&3 | Biallelic | (IF/ΔE9) | N/A | N/A |
| 1-4 | Normal | Female | sgRNA 1&3 | Mosaic w/o wild | (–/–/ΔE9) | N/A | N/A |
| 1-5 | Normal | Female | sgRNA 1&3 | Mosaic w/o wild | (–/–/–/–) | N/A | N/A |
| 1-6 | Malformation (stillborn) | Female | sgRNA 1&3 | Biallelic | (–/–) | Single mRNA transcript | Single protein |
| - 31bp deletion in exon9 | - Truncated protein (317aa) (V310A, S311Q, K312V, S314H, N315K, Y316T, N317L, Δ318-467) | ||||||
| 2-1 | Normal | Female | sgRNA 2&3 | Mosaic w/wild | (+/ΔE9/IF) | N/A | N/A |
| 2-2 | Normal | Male | sgRNA 2&3 | Mosaic w/o wild | (ΔE9/–/–) | N/A | N/A |
| 2-3 | Normal (stillborn) | Female | sgRNA 2&3 | Mosaic w/o wild | (–/–/–/–/IF) | Two mRNA transcripts | Two proteins |
| - Exon 9 (87bp) deletion | - Truncated protein (438aa) (S291C, Δ292-320) | ||||||
| - 1bp insertion in exon 9 | - Truncated protein (316aa) (V294G, W295V, L296V, V297G, N298E, M299Y, A300G, E301R, G302R, D303R, E305R, A306S, Q307P, R308K, K309E, V310G, S311I, K312Q, N313K, S314L, N315Q, Y316L, Δ317-467) | ||||||
| 2-4 | Malformation (stillborn) | Male | sgRNA 2&3 | Biallelic | (–/–) | Two mRNA transcripts | Single protein |
| - 1bp deletion in exon9 and 19bp insertion between exon9 and exon10 | - Truncated protein (296aa) (V294C, W295G, L296W, Δ297-467) | ||||||
| - 1bp deletion in exon9 and 1bp insertion between exon9 and exon10 |
*IF, in-frame mutation including mutations in intron sequences; –, frame shift mutation; ΔE9, deletion of exon9; +, wildtype.
Skeletal defects in PSEN1 knockout newborn piglets
Two of the stillborn piglets (1-6 and 2-4) that carried PSEN1 alleles with premature stop codons demonstrated a severely truncated body axis after birth (Fig. 1C). To correlate the effect of PSEN1 mutations to their skeletal malformation, bone structures of PSEN1-targeted piglets were visualized by X-ray imaging and clearing tissues. Consistent with the external phenotype, normal skeletal development including vertebrae, ribs, and sternum was observed in piglet 2-3 (Fig. 1D). The bone structure of piglet 2-3 was similar to that of the neonatal wildtype piglet. On the other hand, significant skeletal defects were observed in piglet 1-6 (Fig. 1D, and Supplementary Figure 1). Compared to wildtype and 2-3, morphology of all vertebrae was abnormal, and the number of vertebrae in the spinal column was decreased in piglet 1-6. In addition, the number of ribs was decreased and the connections between ribs and vertebrae were irregular. The types of mutations in PSEN1 and the radiology analysis link the role of PSEN1 to vertebrate development.
Deficiency of PSEN1 causes vertebral malformation during fetal development in pigs
To verify whether the mutated PSEN1 is responsible for abnormal axial skeleton formation during early development, PSEN1 mutant fetuses were generated and collected at day 40 of gestation. The fetuses also carried mutations in APP (Supplementary Figure 2); however, changes in APP were considered to have no impact on vertebrate development as APP null pigs have normal spine development (Supplementary Figure 3). Eight fetuses were recovered, and four fetuses (1 through 4) displayed normal external appearance, but the other four fetuses (5 through 8) showed abnormal features in their shape and size (Fig. 2A). For example, the body axial length (from head to tail) of the four fetuses (5 through 8) was shorter (<4 cm) than that of the other fetuses (1 through 4) (>4.5 cm). Among the 4 fetuses with abnormal appearance, fetuses 5 and 8 presented a short body trunk and upward fixed hind legs, clearly resembling the abnormal phenotype of the stillborn neonatal piglets (1-6 and 2-4). Sequences of PSEN1 mRNA revealed that fetus 5 and 8 only carried truncated PSEN1 (290∼325aa), further verifying the impact of the truncated PSEN1 on vertebrate development (Fig. 2B and Table 2). Consistent with the radiological findings in the neonatal piglets, defects in the axial skeleton were identified in the fetuses after micro-CT imaging analysis; compared with fetus 1, fetus 5 had an unclear vertebral column and fewer ribs (Fig. 2C, D). Similarly, micro-MRI imaging revealed underdeveloped vertebrae in fetus 5, whereas fetus 1 had well-developed vertebrae and intervertebral discs (Fig. 2C, D). Thus, it appears that disruption of PSEN1 can induce defective vertebral formation during early fetal development in pigs and specific types of PSEN1 mutations are responsible for abnormal vertebrate development.
Fig. 2
Defective vertebral development in PSEN1 targeted fetuses. A) Images of PSEN1 targeted fetuses collected at day 40. While 4 fetuses (1∼4) had a normal appearance (top 4 panels), other 4 fetuses (5∼8) showed abnormalities in size and shape. B) Predicted PSEN1 protein sequences expressed in the PSEN1 targeted piglets. Fetuses 1∼4 were expected to express a single protein translated from an exon 9 deleted allele, whereas fetuses 5∼8 were expected to express smaller truncated protein(s). C, D) Abnormal skeletal development in PSEN1-targeted fetuses. In contrast to the normal axial skeleton in fetus 1 (left panel), defective development of vertebrae and ribs was detected in fetus 5 (right panel) by micro-CT and MRI imaging and analysis. In the micro-MRI images, the yellow rectangular box represents the thoracic vertebrae and the red box represents the lumbar and sacral vertebrae. Red arrows indicate vertebrae and intervertebral discs and yellow arrow indicates the spinal canal.
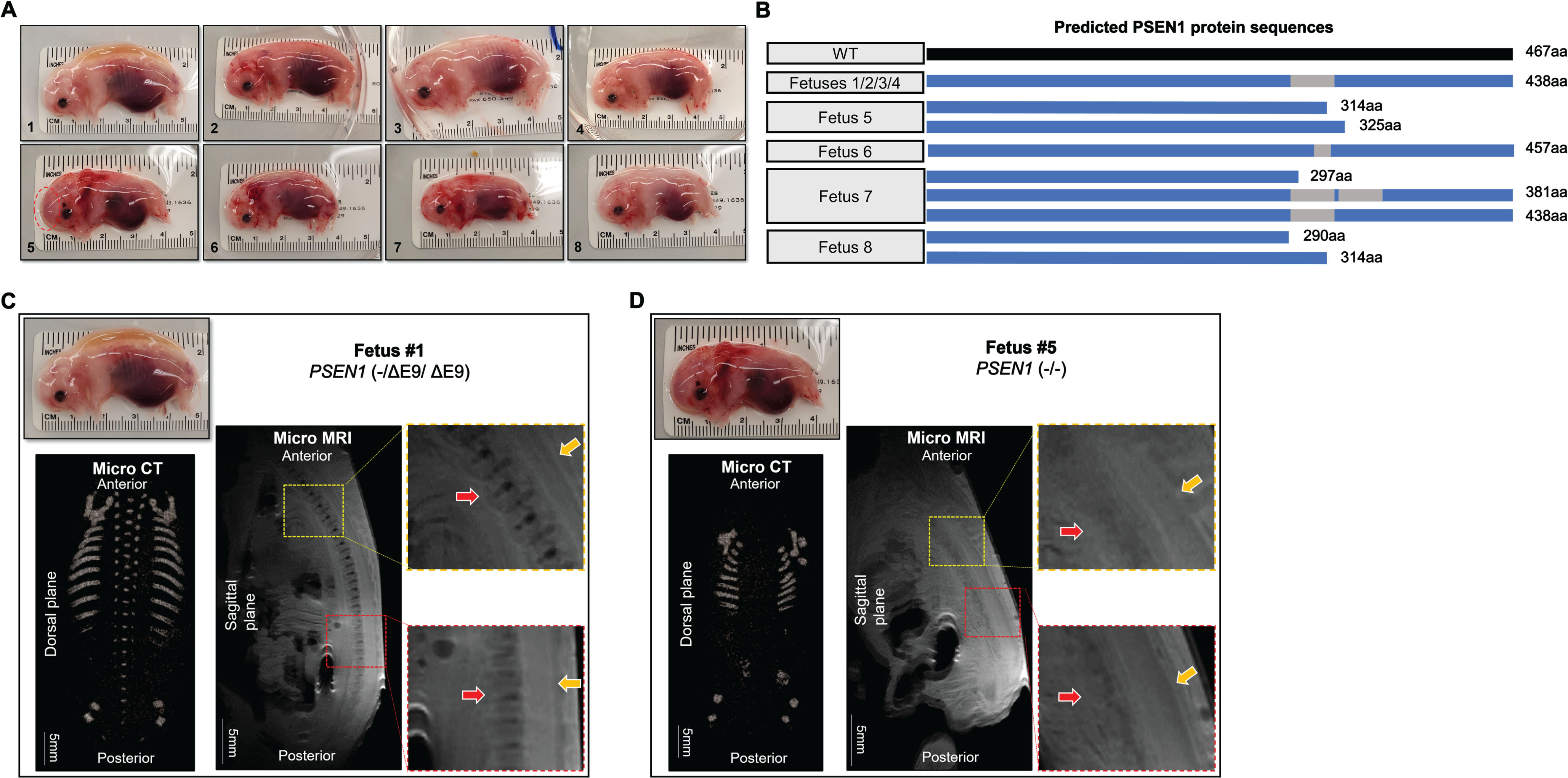
Table 2
Genotyping of PSEN1 targeted fetuses
| Fetus | Phenotype | Sex | sgRNA combinations | Mutations in gDNA* | Mutations in mRNA | Predicted protein | |
| 1 | Normal | Male | sgRNA 2&3 | Mosaic w/o wild | (–/ΔE9/ΔE9) | Single mRNA transcript | Single protein |
| - Exon 9 (87bp) deletion | - Truncated protein (438aa) (S291C, Δ292-320) | ||||||
| 2 | Normal | Female | sgRNA 2&3 | Biallelic | (–/ΔE9) | Single mRNA transcript | Single protein |
| - Exon 9 (87bp) deletion | - Truncated protein (438aa) (S291C, Δ292-320) | ||||||
| 3 | Normal | Female | sgRNA 2&3 | Biallelic | (–/ΔE9) | Single mRNA transcript | Single protein |
| - Exon 9 (87bp) deletion | - Truncated protein (438aa) (S291C, Δ292-320) | ||||||
| 4 | Normal | Female | sgRNA 2&3 | Homozygous | (ΔE9/ΔE9) | Single mRNA transcript | Single protein |
| - Exon 9 (87bp) deletion | - Truncated protein (438aa) (S291C, Δ292-320) | ||||||
| 5 | Malformation | Female | sgRNA 2&3 | Biallelic | (–/–) | Two mRNA transcripts | Two proteins |
| - 53bp insertion between intron 8 and exon 9- 7bp deletion in exon 9 | - Truncated protein (314aa) (Δ291-294, L296T, M299D, A300N, E301G, G302Q, D303F, P304Y, E305N, A306S, Q307S, R308 C , K309 S , V310 L , S311 E , K312 A , N313 K , S314 A , N315 K , Y316 S , N317 D , A318 W, Δ319-467) | ||||||
| - Truncated protein (325aa) (Δ294-295, S 320A, T 321Q, G 322V, E 323N, S 324H, Q 325K, D 326T, S 327L, Δ328-467) | |||||||
| 6 | Normal | Male | sgRNA 2&3 | Biallelic | (–/–) | Single mRNA transcript | Single protein |
| - 1bp insertion and 31bp deletion in exon 9 | - Truncated protein (457aa) (V294G, W295V, L296V, V297G, N298E, M299Y, A300G, E301R, G302R, D303R, E305R, A306S, Q307P, R308K, Δ309-318, Q319E, S320G) | ||||||
| 7 | Normal | Female | sgRNA 2&3 | Mosaic w/o wild | (–/–/ΔE9) | Three mRNA transcripts | Three proteins |
| - 2bp and 1bp insertion in exon 9 | - Truncated protein (297aa) (V294W, W295C, L296G, V297W, Δ298-467) | ||||||
| - Exon 9 and 10 (258bp) deletion | - Truncated protein (381aa) (Δ291-294, Δ296-377) | ||||||
| - Exon 9 (87bp) deletion | - Truncated protein (438aa) (S291C, Δ292-320) | ||||||
| 8 | Malformation | Female | sgRNA 2&3 | Biallelic | (–/–) | Two mRNA transcripts | Two proteins |
| - 2bp insertion and 4bp deletion in exon 9 | - Truncated protein (290aa) (Δ291-467) | ||||||
| - 5bp deletion in exon 9 | - Truncated protein (314aa) (W295G, L296E, V297Y, N298G, M299R, A300R, E301R, G302P, D303R, P304S, E305P, A306K, Q307E, R308G, K309I, V310Q, S311K, K312L, N313Q, S314L, Δ315-467) |
* -, frame shift mutation; ΔE9, deletion of exon9.
Abnormal neural cells in the absence of functional PSEN1
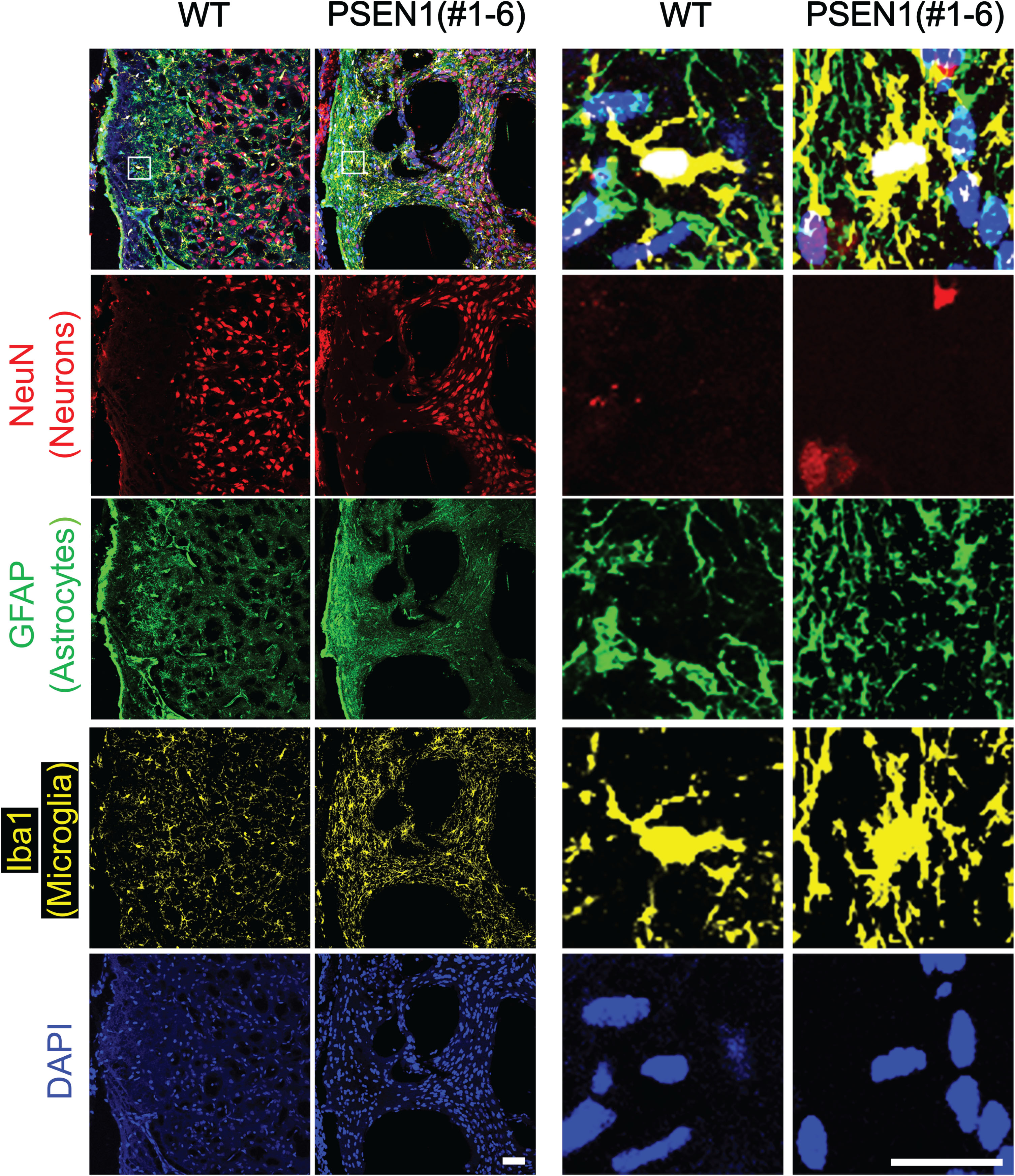
Brain from PSEN1 mutants were examined to investigate the role of PSEN1 on brain development and function. When cranial bones were dissected, hemorrhages were observed in all stillborn piglets (data not shown). PSEN1 mutant fetuses also had visible intracranial hemorrhages in the brain; the most severe hemorrhage was visible on the forebrain region of fetus 5 (Fig. 2A, red dotted oval). Immunofluorescence was performed on the brain of PSEN1 null newborn piglets to further investigate cellular distribution due to the lack of functional PSEN1 (Fig. 3). The density of astrocytes and microglia in the brain of PSEN1 null pigs appeared to be higher (n = 2) compared to the wildtype counterpart (n = 1) (Fig. 3). In PSEN1 null pigs, the density of microglial cells tended to be higher compared to the wildtype (p = 0.06) (Supplementary Figure 4B). In addition, Iba1 positive areas reflecting microglial activity was significantly increased in PSEN1 null pigs (Supplementary Figure 4A, B). The GFAP signal appear to be brighter in PSEN1 null pigs, but no morphological differences in GFAP+ cells were noticed. Clustering of GFAP+ and Iba1+ cells was observed from immunofluorescence, indicating potential activation of astrocytes and microglia, respectively. No difference in the distribution of neurons, i.e., NeuN+ cells, was observed (Supplementary Figure 4B).
Fig. 3
Abnormal neural cells in PSEN1 knockout newborn pigs. Immunohistochemistry was used to identify the distribution of neuronal cells in the frontal cerebral cortex of PSEN1 knockout pigs. Cells positive for markers representing astrocytes (GFAP) and microglia (Iba1) were clustered in the brain of PSEN1 knockout pigs compared to an age-matched wildtype control. Scale bars indicate 100μm.
One aged founder pig (#1–3) carrying a PSEN1ΔE9 allele and an in-frame allele (Table 1) was sacrificed at two years old to investigate if the abnormal distribution of neural cells could also be found in an adult PSEN1 mutant pig. Similar to the histological findings in the neonatal PSEN1 null pig (#1–6), the number of Iba1 positive cells and Iba1 positive area were greater in the PSEN1 mutant pig’s brain than in adult (1 year old) wildtype control pigs (Fig. 4 and Supplementary Figure 4C). The number of neurons was also higher in the PSEN1 mutant pig (Fig. 4 and Supplementary Figure 4C). The Sholl analysis was performed to evaluate the complexity of astrocytes in the PSEN1 mutant pig (Fig. 5). Specifically, Sholl intersections, defined as the number of branching points on a concentric sphere, were used to assess the spatial distribution of the astrocytes (Fig. 5A, B). Using this analysis, astrocytes in the PSEN1 mutant pig (#1–3) were found to be more complex than astrocytes in the wildtype control pig (Fig. 5B-D), indicating that the PSEN1 founder’s astrocytes were in a more reactive state. Due to the neonatal lethality, only a few animals were available for the histological analysis and no statistical analysis was conducted using a sufficient number of biological samples.
Fig. 4
Abnormal neural cells in PSEN1 adult pigs. Immunohistochemistry was used to identify the distribution of neuronal cells in the frontal cerebral cortex of PSEN1 mutant pigs. Cells positive for markers representing astrocytes (GFAP) and microglia (Iba1) were clustered in the brain of PSEN1 knockout pigs compared to an age-matched wildtype control. Scale bars indicate 100μm.
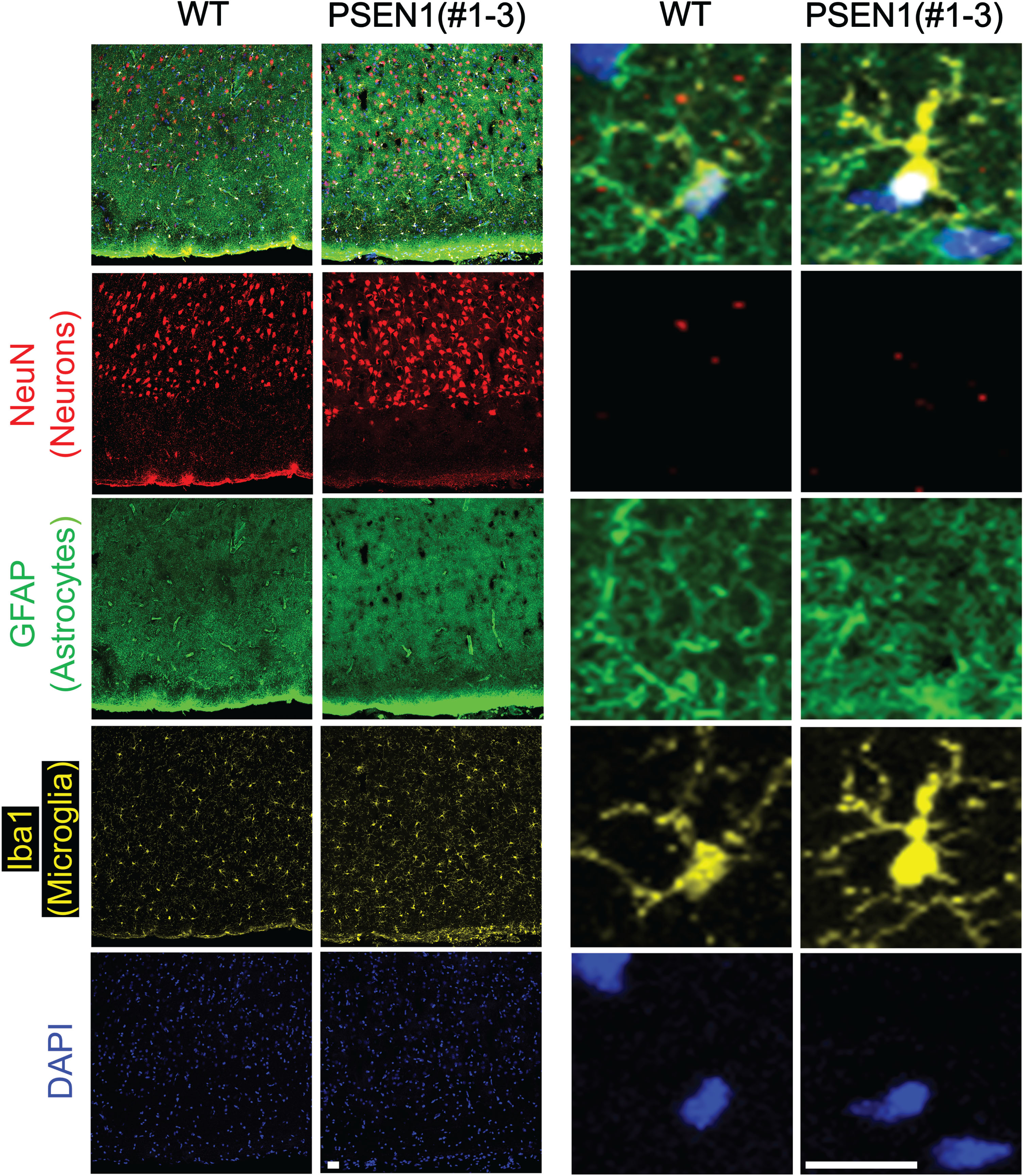
Fig. 5
Astrocyte morphology in an adult PSEN1 mutant pig. A) Representative images of GFAP positive astrocytes in a 1-year-old wildtype (WT) pig and a 2-year-old PSEN1 mutant pig (#1–3). B) 3D reconstructions of astrocytes from the PSEN1 (#1–3) and wildtype pig for Sholl analysis. Results of the Sholl analysis revealed more complex astrocyte morphology in pig #1–3 through (C) increased marginal means, (D) number of terminal points, and (E) number of branching points in the astrocytes of the PSEN1 mutant pig than in astrocytes from the wildtype pig. *p < 0.05, **p < 0.01, respectively.
To investigate the abnormal distribution of astrocytes and microglia in PSEN1 mutant pigs at the molecular level, RT-qPCR was performed to measure the level of GFAP and AIF1 (Iba1) transcripts in the brain of the pigs. Transcript abundance of GFAP and AIF1 were increased in the cortex of PSEN1 mutant pigs (#1–6 and #1–3) compared to wildtype pigs (Supplementary Figure 5), indicating the levels of GFAP and AIF1 may remain abnormal as the PSEN1 mutant pigs age. Because of the limited PSEN1 founder pigs available for the analysis, we could not secure a sufficient number of biological replications. However, the levels of GFAP and AIF1 were higher in PSEN1 mutant pigs compared to two individual wildtype pigs in both age groups.
Establishment of PSEN1 mutant pigs to study Alzheimer’s disease
Founder pigs carrying mutated PSEN1 were bred to establish pig models representing AD. Due to the phenotypes expressed by null PSEN1 piglets, founder PSEN1 pigs (RRID: NSRRC_0094) have been outcrossed into domestic pig lines, NIH mini pig lines (RRID: NSRRC_0014), and Minnesota Mini pig lines (RRID: NSRRC_0005) to generate heterozygous animals with reduced adult size. To date, 39 domestic and 29 half-mini F1 piglets have been produced. The oldest F1 pigs have reached puberty (Supplementary Figure 6) and are being outcrossed into Minnesota Mini lines (RRID: NSRRC_0005), again to reduce their size. Both male and female pigs have been produced, and they are being aged to validate their relevance to clinical AD as PSEN1ΔE9/+ pigs are genetically disposed to mimic clinical phenotypes in AD patients.
DISCUSSION
As a causative gene linked to fAD, the function of PSEN1 has centered around its involvement in the pathophysiology of AD.33 Beyond its involvement in AD, previous studies using genetically engineered mouse models have elucidated its role in vertebrate development. Targeted disruption of PSEN1 resulted in neonatal lethality in Psen1 null mice.20–22,34 Specifically, Psen1 null mice presented skeletal malformations, such as a truncated body axis and deformed ribcage. Elucidating the global function of PSEN1 in other mammals has been a challenge because of the difficulty in introducing targeted modifications in nonconventional laboratory animals. Application of the CRISPR/Cas9 system allowed for rapid inactivation of PSEN1 to study its function and establish pig models carrying a clinically relevant allele of AD.
We have generated 10 piglets and 16 fetuses carrying modified PSEN1, and all pig/fetuses lacking functional PSEN1 resulted in abnormal spine development with the piglets dying soon after birth. These findings are similar to previous reports in mice,20 illustrating the conserved role of PSEN1 between different mammalian species. On the other hand, piglets carrying different PSEN1 mutations did not have any developmental abnormalities and were able to thrive. In contrast to the neonatal lethality of Δexon9 mutations in the mouse.22 piglets carrying at least one Δexon9 allele appeared normal at birth. In a non-human primate study using marmosets, an introduction of the Δexon9 mutation resulted in an increased ratio of Aβ42/Aβ40 in primary fibroblast cells and ability to thrive.35 Similarly, another marmoset model carrying C410Y or A426P mutations in PSEN1 had elevated plasma Aβ42 levels, but no developmental defects were reported.36,37 Founder PSEN1 pigs carrying at least one Δexon 9 allele or in-frame mutations developed normally and were fertile.
The abnormal spine development in PSEN1–/– pigs could be due to the involvement of PSEN1 on the Notch signaling pathway, which plays a key role in vertebrate body axis formation by regulating somitogenesis during early embryonic development. As a unit of γ-secretase, PSEN1 is involved in the cleavage of the Notch receptor and release of the active Notch intracellular domain (NICD) into the nucleus.38 If NICD is not appropriately released by abnormal PSEN1 function, it may lead to defective spine development. In mice, mutations in Notch receptors, ligands, and effectors result in defective somitogenesis.39–43 Similarly, in humans, mutations in Notch ligands and effectors also cause axial skeletal abnormalities.19,44,45 Consistent with the previous mouse study,20 all the stillborn PSEN1 null piglets presented intracranial hemorrhages (data not shown). The hemorrhage was not detected in pigs carrying at least one Δexon9 PSEN1 allele, or in-frame mutations; indicating the importance of PSEN1 coded by exons 10–12 for normal brain structure. Since PSEN1 deficiency is known to induce the upregulation of PSEN2, an isoform of PSEN1, through a compensatory mechanism,46,47 we cannot exclude the possible influence of PSEN2 on normal vertebral development in pigs with Δexon9 mutations. Further analysis of the expression of PSEN2 in pigs carrying PSEN1 mutations will provide a more comprehensive picture of the impact of the deletion of exon 9 in PSEN1 on the development of vertebrae in the pig.
Signs of neuroinflammation were detected in the brain of PSEN1 null pigs. Markers representing astrocytes (GFAP) and microglia (Iba1) were clustered in the PSEN1 null brain samples. Specifically, increased cell density of microglia and a larger Iba1 positive area were observed under the absence of functional PSEN1, which indicates possible neuroinflammation in the PSEN1 brain compared to the wildtype control. Analysis of astrocytes in an aged PSEN1 mutant pig (#1–3) also suggested a possible hyperactivation of astrocytes in the presence of abnormal PSEN1. Due to the availability of samples, brains from adult pigs at different ages were used (1- versus 2-years). However, as the brain reaches maturity around six months of age and brain cell density does not increase any further in pigs,48,49 the age difference should not have affected the outcome of these analyses. Astrocytes, microglia, and their communication are critical for brain function, and the activity of astrocytes50 and microglia51 is known to hyperactivate in AD patients52–55 and rodent AD models.56–59 As a large animal species, it is challenging to obtain a sufficient number of samples to fully characterize the signs of neuroinflammation in pigs. A recent study using marmosets reported increased pathological amyloid beta and neuroinflammatory biomarkers in the brains of PSEN1 mutants.37 Additionally, gliosis was reported in the PSEN1 marmosets due to elevated populations of Iba1 positive phagocytic microglia in the brain, which is similar to the abnormal distribution of Iba1 positive cells observed in the PSEN1 pigs. Clinical relavance of the neuroinflammation will be compared after establishing a line of PSEN1ΔE9/+ pigs and aging them until their brain is fully matured.48,60
Little success has been reported on developing large animal models that can recapitulate the clinical signs of fAD.61–63 To the best of our knowledge, this is the first report of PSEN1ΔE9/+ pigs, and the pig models are both being expanded by breeding and being aged to validate clinical relevance of these mutations. As a large animal model, expanding and aging the F1 generation of PSEN1ΔE9/+ pigs takes over one year. Propagation of the pig models is underway and considering high similarities between pig and human brain,64 we expect to see similar clinical signs of AD in these pigs once they are aged. Removal of PSEN1 exon9 had a distinctive effect in pigs compared to a previous report in the mouse.22 The unique phenotype indicates the need to study the PSEN1ΔE9/+ phenotype in pigs to further understand the role of PSEN1 beyond rodent models. While no phenotyping efforts are included in this study, we have also generated APP knockout pig models. The pigs are growing and do not appear to have any developmental defects. APP and PSEN1 are both genetically linked to the prevalence of AD65 and their specific interactions lead to amyloid beta production, which is at the center of AD pathophysiology.13 Pigs with these mutations will greatly facilitate functional studies to understand the pathogenic mechanism of fAD and the development of appropriate treatments.
Using the CRISPR/Cas9 system, two different genetically engineered pig models have been rapidly established and the role of PSEN1 in piglet development was explored. These pig models will serve as novel resources to expand our understanding of AD and allow us to follow the progression of the disease throughout the lifespan of the pigs.
AUTHOR CONTRIBUTIONS
Kyungjun Uh (Conceptualization; Methodology; Writing – original draft); Kaylynn Monarch (Data curation; Formal analysis; Methodology; Writing – review & editing); Emily D. Reese (Data curation; Methodology); Katherine Rodriguez (Data curation; Methodology); Junchul Yoon (Formal analysis; Methodology; Validation); Lee D. Spate (Formal analysis; Methodology); Melissa S. Samuel (Methodology; Resources); Sehwon Koh (Data curation; Formal analysis; Methodology; Writing – review & editing); Paula R. Chen (Formal analysis; Methodology); Timothy J. Jarome (Data curation; Formal analysis; Funding acquisition); Timothy A. Allen (Formal analysis; Funding acquisition; Investigation); Randall S. Prather (Funding acquisition; Project administration; Resources; Supervision; Writing – original draft); Kiho Lee (Conceptualization; Funding acquisition; Investigation; Project administration; Resources; Supervision; Writing – original draft; Writing – review & editing).
ACKNOWLEDGMENTS
We thank all current and former members of Lee’s laboratory at the University of Missouri. We also appreciate the efforts of the National Swine Resource and Research Center (NSRRC) staff in animal production and husbandry.
FUNDING
Funding for the National Swine Resource and Research Center is from the Office Of The Director, National Institutes Of Health (OD), the National Institute Of Allergy And Infectious Diseases (NIAID), and National Heart, Lung, And Blood Institute (NHLBI) grant U42OD011140. The research was also in part funded by the National Institute on Aging (R21AG079292).
CONFLICT OF INTEREST
The Curators of the University of Missouri have filed intellectual property protection for the genetically engineered pig models.
DATA AVAILABILITY
The data supporting the findings of this study are available within the article and/or its supplementary material, or upon request from the corresponding author.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-231297.
REFERENCES
1. | Sherrington R , Rogaev EI , Liang Y , et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature (1995) ; 375: : 754–760. |
2. | De Strooper B , Saftig P , Craessaerts K , et al. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature (1998) ; 391: : 387–390. |
3. | De Strooper B , Annaert W , Cupers P , et al. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature (1999) ; 398: : 518–522. |
4. | Wolfe MS , Xia W , Ostaszewski BL , et al. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature (1999) ; 398: : 513–517. |
5. | Struhl G and Greenwald I. Presenilin is required for activity and nuclear access of Notch in Drosophila. Nature (1999) ; 398: : 522–525. |
6. | Ye Y , Lukinova N and Fortini ME. Neurogenic phenotypes and altered Notch processing in Drosophila Presenilin mutants. Nature (1999) ; 398: : 525–529. |
7. | Levy-Lahad E , Wasco W , Poorkaj P , et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science (1995) ; 269: : 973–977. |
8. | Crook R , Verkkoniemi A , Perez-Tur J , et al. A variant of Alzheimer’s disease with spastic paraparesis and unusual plaques due to deletion of exon 9 of presenilin 1. Nat Med (1998) ; 4: : 452–455. |
9. | Prihar G , Verkkoniem A , Perez-Tur J , et al. Alzheimer disease PS-1 exon 9 deletion defined. Nat Med (1999) ; 5: : 1090. |
10. | Smith MJ , Kwok JB , McLean CA , et al. Variable phenotype of Alzheimer’s disease with spastic paraparesis. Ann Neurol (2001) ; 49: : 125–129. |
11. | Dumanchin C , Tournier I , Martin C , et al. Biological effects of four PSEN1 gene mutations causing Alzheimer disease with spastic paraparesis and cotton wool plaques. Hum Mutat (2006) ; 27: : 1063. |
12. | Le Guennec K , Veugelen S , Quenez O , et al. Deletion of exons 9 and 10 of the Presenilin 1 gene in a patient with Early-onset Alzheimer disease generates longer amyloid seeds. Neurobiol Dis (2017) ; 104: : 97–103. |
13. | Hampel H , Hardy J , Blennow K , et al. The amyloid-β pathway in Alzheimer’s disease. Mol Psychiatry (2021) ; 26: : 5481–5503. |
14. | Lee MK , Borchelt DR , Kim G , et al. Hyperaccumulation of FAD-linked presenilin 1 variants in vivo. Nat Med (1997) ; 3: : 756–760. |
15. | Thinakaran G , Borchelt DR , Lee MK , et al. Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron (1996) ; 17: : 181–190. |
16. | Andersson ER , Sandberg R and Lendahl U. Notch signaling: simplicity in design, versatility in function. Development (2011) ; 138: : 3593–3612. |
17. | Zanotti S and Canalis E. Notch signaling and the skeleton. Endocr Rev (2016) ; 37: : 223–253. |
18. | McDaniell R , Warthen DM , Sanchez-Lara PA , et al. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. Am J Hum Genet (2006) ; 79: : 169–173. |
19. | Bulman MP , Kusumi K , Frayling TM , et al. Mutations in the human delta homologue, DLL3, cause axial skeletal defects in spondylocostal dysostosis. Nat Genet (2000) ; 24: : 438–441. |
20. | Shen J , Bronson RT , Chen DF , et al. Skeletal and CNS defects in Presenilin-1-deficient mice. Cell (1997) ; 89: : 629–639. |
21. | Donoviel DB , Hadjantonakis AK , Ikeda M , et al. Mice lacking both presenilin genes exhibit early embryonic patterning defects. Genes Dev (1999) ; 13: : 2801–2810. |
22. | Koizumi K , Nakajima M , Yuasa S , et al. The role of presenilin 1 during somite segmentation. Development (2001) ; 128: : 1391–1402. |
23. | Whyte JJ and Prather RS. Genetic modifications of pigs for medicine and agriculture. Mol Reprod Dev (2011) ; 78: : 879–891. |
24. | Saikali S , Meurice P , Sauleau P , et al. A three-dimensional digital segmented and deformable brain atlas of the domestic pig. J Neurosci Methods (2010) ; 192: : 102–109. |
25. | Musigazi GU , De Vleeschauwer S , Sciot R , et al. Brain perfusion fixation in male pigs using a safer closed system. Lab Anim (2018) ; 52: : 413–417. |
26. | Kornum BR and Knudsen GM. Cognitive testing of pigs (Sus scrofa) in translational biobehavioral research. Neurosci Biobehav Rev (2011) ; 35: : 437–451. |
27. | Allen LM , Murphy DA , Roldan V , et al. Testing spatial working memory in pigs using an automated T-maze. Oxf Open Neurosci (2023) ; 2: : kvad010. |
28. | Lei S , Ryu J , Wen K , et al. Increased and prolonged human norovirus infection in RAG2/IL2RG deficient gnotobiotic pigs with severe combined immunodeficiency. Sci Rep (2016) ; 6: : 25222. |
29. | Yoshioka K , Suzuki C , Tanaka A , et al. Birth of piglets derived from porcine zygotes cultured in a chemically defined medium. Biol Reprod (2002) ; 66: : 112–119. |
30. | Concordet JP and Haeussler M. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res (2018) ; 46: : W242–W245. |
31. | Uh K , Ryu J , Farrell K , et al. TET family regulates the embryonic pluripotency of porcine preimplantation embryos by maintaining the DNA methylation level of NANOG. Epigenetics (2020) ; 15: : 1228–1242. 2. |
32. | Lee K , Redel BK , Spate L , et al. Piglets produced from cloned blastocysts cultured in vitro with GM-CSF. Mol Reprod Dev (2013) ; 80: : 145–154. |
33. | Lanoiselée HM , Nicolas G , Wallon D , et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med (2017) ; 14: : e1002270. |
34. | Wong PC , Zheng H , Chen H , et al. Presenilin 1 is required for Notch1 and DII1 expression in the paraxial mesoderm. Nature (1997) ; 387: : 288–292. |
35. | Sasaguri H , Sato K , Kumita W , et al. Generation of non-human primate models of Alzheimer’s disease. Alzheimers Dement (2021) ; 17: : e052561. |
36. | Rizzo SJS , Homanics GE , Park JE , et al. Establishing the marmoset as a non-human primate model of Alzheimer’s disease. Alzheimers Dement (2021) ; 17: : e049952. |
37. | Homanics GE , Park JE , Bailey L , et al. Early molecular events of autosomal-dominant Alzheimer’s disease in marmosets with PSEN1 mutations. Alzheimers Dement (2024) ; 20: : 3455–3471. |
38. | Bray SJ . Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol (2006) ; 7: : 678–689. |
39. | Conlon RA , Reaume AG and Rossant J. Notch1 is required for the coordinate segmentation of somites. Development (1995) ; 121: : 1533–1545. |
40. | Bessho Y , Sakata R , Komatsu S , et al. Dynamic expression and essential functions of Hes7 in somite segmentation. Genes Dev (2001) ; 15: : 2642–2647. |
41. | Evrard YA , Lun Y , Aulehla A , et al. lunatic fringe is an essential mediator of somite segmentation and patterning. Nature (1998) ; 394: : 377–381. |
42. | Kusumi K , Sun ES , Kerrebrock AW , et al. The mouse pudgy mutation disrupts Delta homologue Dll3 and initiation of early somite boundaries. Nature genetics (1998) ; 19: : 274–278. |
43. | Shifley ET , Vanhorn KM , Perez-Balaguer A , et al. Oscillatory lunatic fringe activity is crucial for segmentation of the anterior but not posterior skeleton. Development (2008) ; 135: : 899–908. |
44. | Sparrow DB , Chapman G , Wouters MA , et al. Mutation of the LUNATIC FRINGE gene in humans causes spondylocostal dysostosis with a severe vertebral phenotype. Am J Hum Genet (2006) ; 78: : 28–37. |
45. | Sparrow DB , Guillén-Navarro E , Fatkin D , et al. Mutation of Hairy-and-Enhancer-of-Split-7 in humans causes spondylocostal dysostosis. Hum Mol Genet (2008) ; 17: : 3761–3766. |
46. | Watanabe H , Iqbal M , Zheng J , et al. Partial loss of presenilin impairs age-dependent neuronal survival in the cerebral cortex. J Neurosci (2014) ; 34: : 15912–15922. |
47. | Placanica L , Tarassishin L , Yang G , et al. Pen2 and presenilin-1 modulate the dynamic equilibrium of presenilin-1 and presenilin-2 gamma-secretase complexes. J Biol Chem (2009) ; 284: : 2967–2977. |
48. | Conrad MS , Dilger RN and Johnson RW. Brain growth of the domestic pig (Sus scrofa) from 2 to 24 weeks of age: a longitudinal MRI study. Dev Neurosci (2012) ; 34: : 291–298. |
49. | Fang M , Lorke DE , Li J , et al. Postnatal changes in functional activities of the pig’s brain: a combined functional magnetic resonance imaging and immunohistochemical study. Neurosignals (2005) ; 14: : 222–233. |
50. | Verkhratsky A , Olabarria M , Noristani HN , et al. Astrocytes in Alzheimer’s disease. Neurotherapeutics (2010) ; 7: : 399–412. |
51. | Hansen DV , Hanson JE and Sheng M. Microglia in Alzheimer’s disease. J Cell Biol (2018) ; 217: : 459–472. 20171201. |
52. | Beach TG and McGeer EG. Lamina-specific arrangement of astrocytic gliosis and senile plaques in Alzheimer’s disease visual cortex. Brain Res (1988) ; 463: : 357–361. |
53. | Griffin WS , Stanley LC , Ling C , et al. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci U S A (1989) ; 86: : 7611–7615. |
54. | McGeer PL , Itagaki S , Tago H , et al. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci Lett (1987) ; 79: : 195–200. |
55. | Tooyama I , Kimura H , Akiyama H , et al. Reactive microglia express class I and class II major histocompatibility complex antigens in Alzheimer’s disease. Brain Res (1990) ; 523: : 273–280. |
56. | Verkhratsky A , Zorec R , Rodríguez JJ , et al. Astroglia dynamics in ageing and Alzheimer’s disease. Curr Opin Pharmacol (2016) ; 26: : 74–79. |
57. | Rodríguez JJ , Olabarria M , Chvatal A , et al. Astroglia in dementia and Alzheimer’s disease. Cell Death Differ (2009) ; 16: : 378–385. |
58. | Verkhratsky A , Zorec R and Parpura V. Stratification of astrocytes in healthy and diseased brain. Brain Pathol (2017) ; 27: : 629–644. |
59. | Frautschy SA , Yang F , Irrizarry M , et al. Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol (1998) ; 152: : 307–317. |
60. | Winter JD , Dorner S , Lukovic J , et al. Noninvasive MRI measures of microstructural and cerebrovascular changes during normal swine brain development. Ped Res (2011) ; 69: : 418–424. |
61. | Kragh PM , Nielsen AL , Li J , et al. Hemizygous minipigs produced by random gene insertion and handmade cloning express the Alzheimer’s disease-causing dominant mutation APPsw. Transgenic Res (2009) ; 18: : 545–558. |
62. | Søndergaard LV , Ladewig J , Dagnæs-Hansen F , et al. Object recognition as a measure of memory in 1-2 years old transgenic minipigs carrying the APPsw mutation for Alzheimer’s disease. Transgenic Res (2012) ; 21: : 1341–1348. |
63. | Jakobsen JE , Johansen MG , Schmidt M , et al. Expression of the Alzheimer’s disease mutations AβPP695sw and PSEN1M146I in double-transgenic göttingen minipigs. J Alzheimers Dis (2016) ; 53: : 1617–1630. |
64. | Hoffe B and Holahan MR. The use of pigs as a translational model for studying neurodegenerative diseases. Front Physiol (2019) ; 10: : 838. |
65. | Bagyinszky E , Youn YC , An SS , et al. The genetics of Alzheimer’s disease. Clin Interv Aging (2014) ; 9: : 535–551. |

