
Polyamine Dysregulation and Nucleolar Disruption in Alzheimer’s Disease
Abstract
A hypothesis of Alzheimer’s disease etiology is proposed describing how cellular stress induces excessive polyamine synthesis and recycling which can disrupt nucleoli. Polyamines are essential in nucleolar functions, such as RNA folding and ribonucleoprotein assembly. Changes in the nucleolar pool of anionic RNA and cationic polyamines acting as counterions can cause significant nucleolar dynamics. Polyamine synthesis reduces S-adenosylmethionine which, at low levels, triggers tau phosphorylation. Also, polyamine recycling reduces acetyl-CoA needed for acetylcholine, which is low in Alzheimer’s disease. Extraordinary nucleolar expansion and/or contraction can disrupt epigenetic control in peri-nucleolar chromatin, such as chromosome 14 with the presenilin-1 gene; chromosome 21 with the amyloid precursor protein gene; chromosome 17 with the tau gene; chromosome 19 with the APOE4 gene; and the inactive X chromosome (Xi; aka “nucleolar satellite”) with normally silent spermine synthase (polyamine synthesis) and spermidine/spermine-N1-acetyltransferase (polyamine recycling) alleles. Chromosomes 17, 19 and the Xi have high concentrations of Alu elements which can be transcribed by RNA polymerase III if positioned nucleosomes are displaced from the Alu elements. A sudden flood of Alu RNA transcripts can competitively bind nucleolin which is usually bound to Alu sequences in structural RNAs that stabilize the nucleolar heterochromatic shell. This Alu competition leads to loss of nucleolar integrity with leaking of nucleolar polyamines that cause aggregation of phosphorylated tau. The hypothesis was developed with key word searches (e.g., PubMed) using relevant terms (e.g., Alzheimer’s, lupus, nucleolin) based on a systems biology approach and exploring autoimmune disease tautology, gaining synergistic insights from other diseases.
INTRODUCTION
The “polyamine” hypothesis
Development of a hypothesis for Alzheimer’s disease (AD) can begin by exploring systems biology and autoimmune disease tautology to identify synergistic concepts that improve our perceptions of AD etiology. Using this approach, and the concept of polyamine dysregulation and nucleolar disruption proposed previously for lupus, we can develop a scenario for AD, suggesting new areas of research.
Hypothesis: “Polyamine dysregulation and detrimental Alu element expression can disrupt nucleoli and peri-nucleolar chromatin leading to tau hyperphosphorylation and aggregation, reduced acetylcholine, and other features of Alzheimer’s disease”.
Alzheimer’s disease complexity
AD is an extremely complex neurodegenerative and autoimmune disease involving an estimated 49% of cellular pathways in one way or another (instigation, progression or simply consequential) [1]. The many intersections and shared resources among pathways suggest key factors may be involved, such as altered levels of the methyl donor S-adenosylmethionine (SAM) needed for intracellular localization of proteins and RNAs, synthesis of polyamines, and epigenetic control by methylation of DNA and histones. Another key factor to consider is altered levels of the metabolic regulator acetyl-CoA involved in polyamine recycling, metabolism of proteins, carbohydrates, lipids, and synthesis of the neurotransmitter acetylcholine, which is low in AD [2]. AD may also involve loss of integrity in major components such as nucleoli, mitochondria, proteasomes, membranes, and chromosomes. Since AD complexity crosses multiple areas of research (genetics, epigenetics, immunology, neurology, and more), we need a systems biology approach to further our understanding, rather than a narrow reductionist approach focused on one or two suspected proteins, such as tau or amyloid-β (Aβ). In addition, we need to explore autoimmune disease tautology to gain insights from synergy of concepts across different diseases.
Autoimmune disease tautology
Autoimmune disease tautology refers to common characteristics often found among the more than 100 known autoimmune diseases and variants. Among these diseases we typically find female predominance, initial onset in mid-to-late adulthood, greater severity with earlier disease onset, and polyautoimmunity (i.e., multiple autoimmune diseases in a patient) [3]. With regards to polyautoimmunity, 25% of autoimmune patients present more than one autoimmune disease [4]. AD is an autoimmune disease displaying many characteristics of autoimmune disease tautology. In addition, patients with other autoimmune disorders have an increased risk for AD [5], such as AD and rheumatoid arthritis (RA) [6], AD and multiple sclerosis (MS) [7], while there are reports regarding associations between dementia and systemic lupus erythematosus (SLE) [8, 9]. As examples of common aspects in AD and other autoimmune diseases, Sjögren’s syndrome (SjS) can be a primary disease, or it can be a secondary disease in AD[10], SLE[11], RA [12], MS [13], systemic sclerosis (SSc) [14], and Parkinson’s disease (PD) [15]. Also, acrolein-conjugated proteins can appear in AD [16] and SjS [17] with appearance of acrolein-conjugated proteins in SjS closely corresponding to disease intensity. And there is suspected aberrant activity of Alu elements in SLE [18] and AD [19]. These observations suggest the possibility of similarities in underlying mechanisms in AD and other autoimmune diseases, but differing in cell types involved, immune system accessibility (e.g., behind the blood-brain barrier in AD versus systemic in SLE) and triggering factors (e.g., viruses, physical trauma, metals, UV light, and more). With regards to other diseases, there may be parallels between AD and cognitive difficulties (aka “brain fog”) in SLE and long COVID. Furthermore, survivors of human immunodeficiency virus (HIV) infection show an increased risk of AD as they age, suggesting a possible common adverse residual effect from prior cellular stresses, such as viral infections [20]. This residual effect may also help explain the appearance of autoimmunity in some other immunodeficiency diseases[21]. The potential residual effect may forewarn us of a future increase in AD cases, such as in long COVID patients. Therefore, common aspects of AD and other autoimmune diseases suggest we may gain insights into AD from research results obtained from other autoimmune diseases.
Higher-order epigenetics
Much of autoimmune disease research has been genetics-based, with a reductionist approach focusing on a few genes with suspected key roles. Often varied interpretations arise as to the importance of different proteins in the disease process, such as tau and Aβ in AD, whereas they may simply be involved as a consequence of earlier events. Modeling usually involves human gene sequences inserted in mice with the assumption that it represents a useful disease-like scenario, but this disregards potentially important differences, such as the fact that mice lack Alu elements found in abundance in humans and disregards differences in chromosome structures. For example, the human X chromosome is submetacentric whereas the mouse X is telocentric. This can result in greater difficulty in human cells in the establishment and/or maintenance of X inactivation in humans compared to mice [22].
Beyond genetics, mid-to-late adulthood onset of many autoimmune diseases suggests a major role for accumulated epigenetic damage and dysregulated gene control. However, most epigenetics-based research focuses on lower levels of epigenetics at the base pair, nucleosome, or gene levels with the aim of identifying specific genes with altered epigenetic markers. Genome-wide and epigenome-wide association studies can point to candidate sequences, but analysis of such data can be problematic since significant genetic mutations, insertions, deletions, or the epigenetic context may be missed if they require involvement of multiple genes or events, occur in only a few cells, and/or require years of accumulating damage to reach an observable effect. In addition, potentially hampering understanding of the female bias of many autoimmune diseases, X-linked genes are sometimes down-weighted or ignored since the number of X chromosomes and the extent of X chromosome inactivation can vary from cell to cell in an individual.
There is need for understanding involvement in diseases of higher levels of epigenetic control, such as the chromosome level, including both protein coding and non-coding sequences, heterochromatin versus euchromatin, gene clusters, and domains, including topologically associated domains (TADs), such as nucleolar associated domains. Recent advances in chromosome conformation capture techniques and organoid development are promising steps, but too many variables remain currently to accurately analyze chromatin in normal, stressed, and disease states.
KEY PLAYERS IN THE HYPOTHESIS
Polyamines
The polyamines (spermidine, spermine, and the precursor putrescine) (Fig. 1A),
Fig. 1
Polyamines. A) The main polyamines of concern in this discussion are putrescine, spermidine, and spermine. Their flexibility (all single bonds), high cationic charges at physiological pH (+2,+3,+4, respectively), and length (∼8 Å, ∼12 Å, ∼16 Å, respectively) give the polyamines unique characteristics beneficial in important interactions with larger anions, such as in folding of RNA transcripts, stabilizing chromatin, and modulating ion channels. (Darker spheres are nitrogen atoms, white spheres are carbon atoms, and hydrogens are not shown. Also, the acetylated forms of spermidine and spermine are not shown.) B) Depiction of a small nuclear aggregate of polyamines (NAP). Note that the NAP has an overall cationic charge which could associate it with anions (e.g., DNA). (Medium and large NAPs are not shown.) Putrescine (typically at only trace amounts) is a limiting factor in appearance of NAPs.
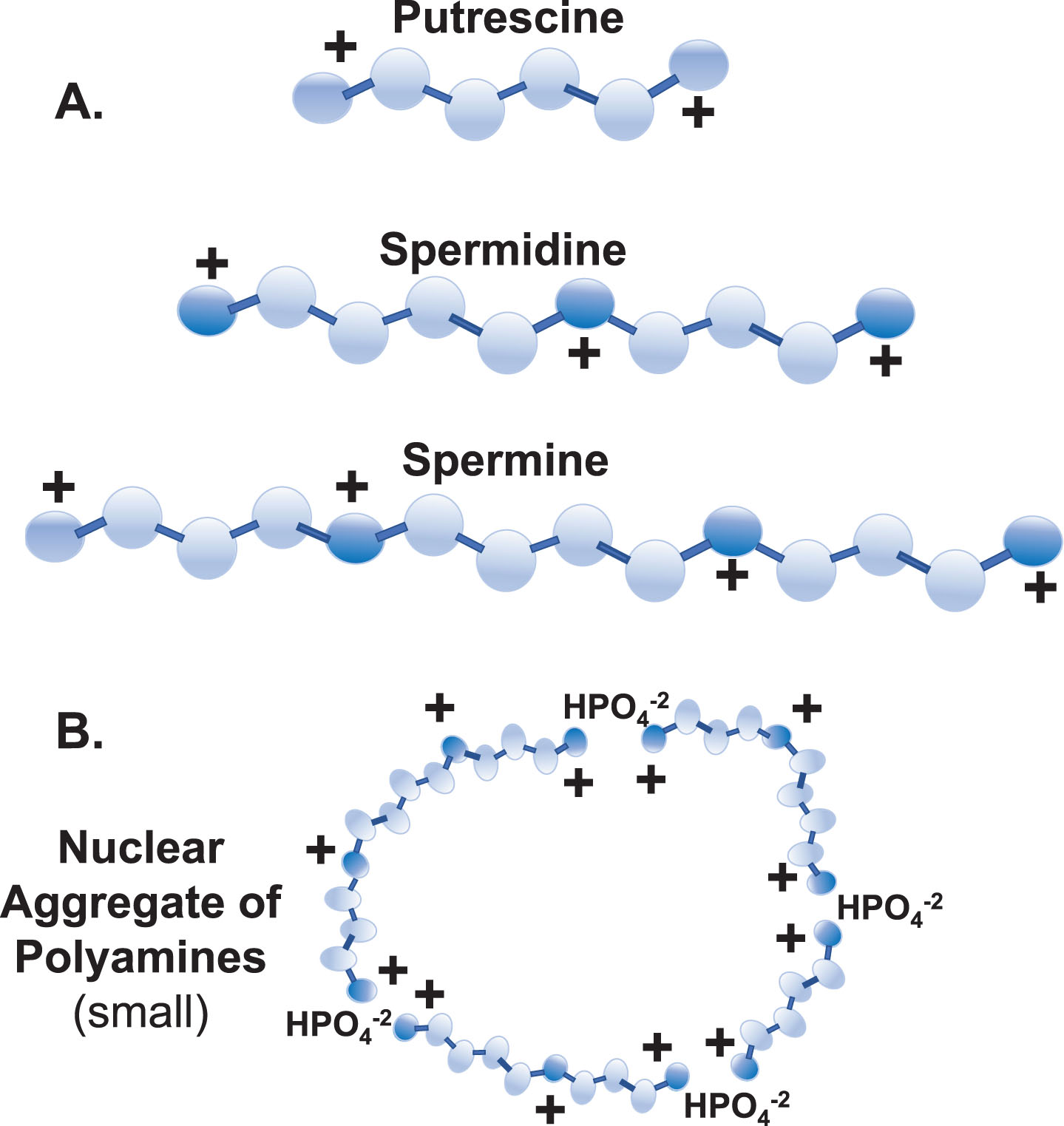
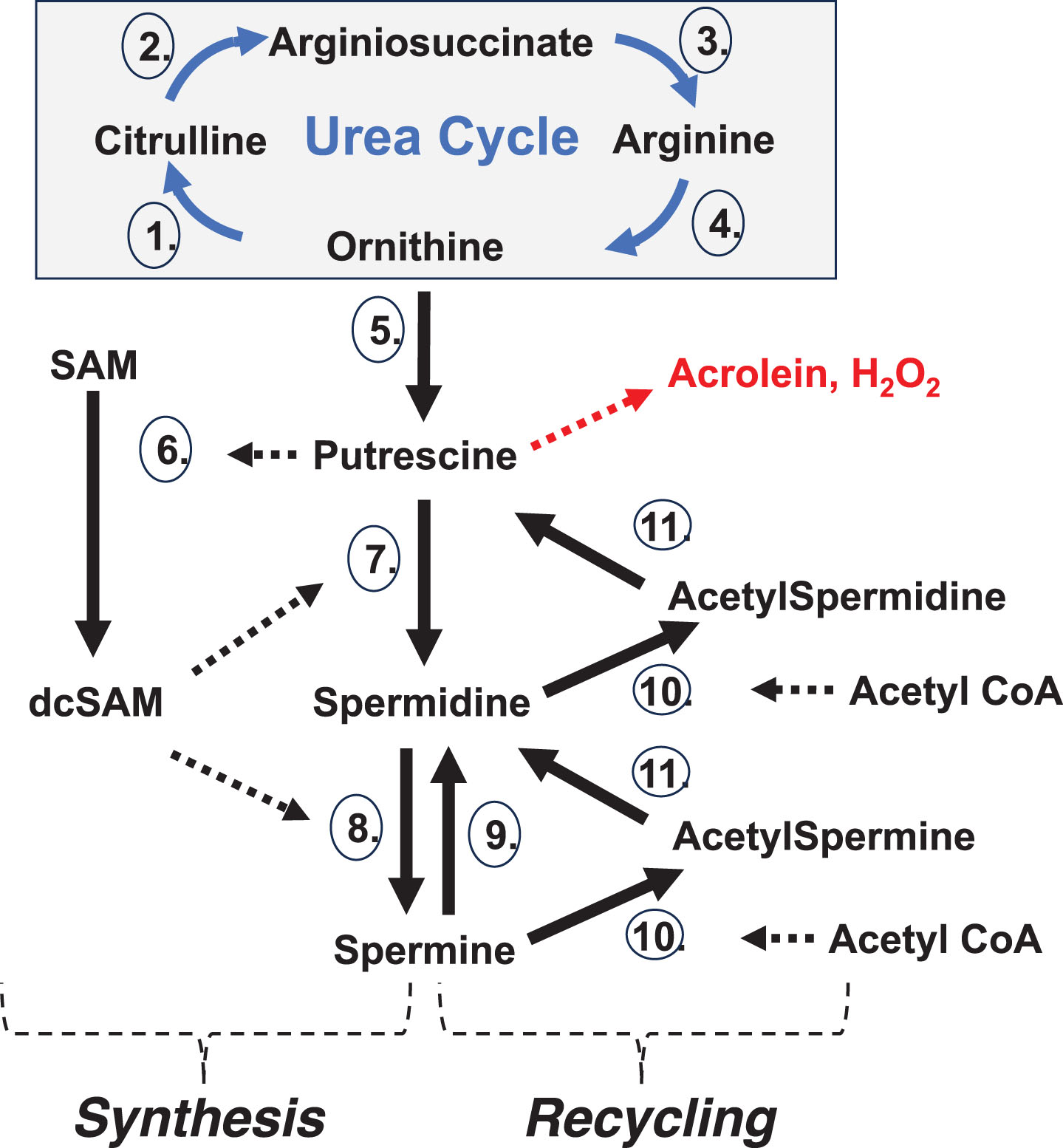
Fig. 2
Polyamine Synthesis and Recycling. Synthesis: De novo polyamine synthesis begins with decarboxylation of ornithine (from the Urea Cycle) by ornithine decarboxylase (5.), which produces putrescine. Putrescine allosterically stimulates decarboxylation of S-adenosylmethionine (SAM) by SAM decarboxylase (6.) to create decarboxylated SAM (dcSAM). dcSAM then provides an aminopropyl group for the conversion of putrescine to spermidine by spermidine synthase (7.) and again for conversion of spermidine to spermine by spermine synthase (8.). ODC is tightly controlled with an ODC antizyme and rapid ODC RNA and protein turnover to control polyamine levels. Recycling: Further control of polyamine levels is exerted by acetylation of spermine and spermidine by spermidine/spermine N1 acetyltransferase (10.) using acetyl CoA and then polyamine oxidase (11.) converts the acetylated polyamine to the next lower form. Putrescine created by polyamine recycling can trigger further polyamine synthesis without using ODC since putrescine can allosterically stimulate AMD1 and serve as input to SRM. Spermine oxidase (9.) can provide rapid reduction of spermine levels. In addition, polyamine recycling can generate harmful hydrogen peroxide and acrolein. Enzymes:1. Ornithine Transcarbamylase (OTC); 2. Argininosuccinate Synthetase 1 (ASS1); 3. Argininosuccinate Lyase (ASL); 4. Arginase 1 (ARG1); 5. Ornithine Decarboxylase (ODC); 6. S-Adenosylmethionine Decarboxylase 1 (AMD1); 7. Spermidine Synthase (SRM); 8. Spermine Synthase (SMS); 9. Spermine Oxidase (SMOX); 10. Spermidine/Spermine N1 Acetyltransferase 1 (SAT1); 11. Polyamine Oxidase (PAO).
Polyamines are intimately involved in folding of RNAs in the nucleolus and this is especially important with regards to nucleolar dynamics and functioning during a cellular stress response. In fact, changes in polyamine levels drive nucleolar morphological changes [30, 31]. Overall, spermidine and spermine are typically at sub-millimolar to millimolar concentrations in cells whereas putrescine, the precursor, is usually kept at only trace amounts to control initiation of polyamine synthesis and to protect SAM levels since putrescine can allosterically increase SAM decarboxylase (AMD1) activity, converting SAM to decarboxylated SAM (dcSAM), as an initial step in polyamine synthesis. SAM provides a methyl group for epigenetic control whereas dcSAM, which cannot serve as a methyl donor, provides an amino-propyl group in polyamine synthesis. Keeping putrescine levels low also prevents unwanted appearance of nuclear aggregates of polyamines (NAPs) (Fig. 1B), which are rings of putrescine, spermidine, spermine and phosphate moieties [32]. NAPs can provide coordinated binding of multiple polyamines that could cause persistence of normally transient DNA conformations (e.g., Z-DNA and cruciforms), compared to the binding of individual polyamines [33]. NAPs could also interfere with proper RNA folding in nucleoli and stabilize autoantigenic complexes including in neutrophil extracellular traps.
Nucleolar polyamines make many transient interactions with RNA transcripts, temporarily neutralizing some of the RNA’s self-repulsion to allow RNA folding or binding of proteins. Therefore, we can think of nucleolar polyamines as abundant and mobile whereas the vast majority of nuclear and cytoplasmic polyamines are at lower concentrations and primarily bound to DNA, RNA, proteins, phospholipids, or other anionic structures. An increase in polyamines can cause changes in the nucleoli but also can lead to increased polyamine recycling as a means of recovery with the potential to alter the balance of polyamine and acetylated polyamine levels. Thus, under extraordinary stress a cell may lose control of polyamines with wasteful use of SAM and acetyl-CoA, even generation of harmful acrolein and hydrogen peroxide, and the nucleolus may not be able to respond properly to subsequent stresses. One can imagine an initial change in nucleolar volume due to synthesis of cationic polyamines that counters increases in anionic RNA content, but then further variations in nucleolar size and efficiency could occur as increased polyamine recycling converts polyamines to acetylated polyamines with reduced cationic character and weaker service as counter ions making the nucleolus less productive, even counterproductive. Stressful events, such as chemically induced oxidative stress, can cause altered polyamine levels by activating transcription of ornithine decarboxylase (ODC) for polyamine synthesis and spermidine/spermine N1 acetyl transferase (SAT1) for polyamine recycling [34]. Wasteful synthesis and recycling could continue until lower SAM and/or acetyl-CoA levels slow the process. It has been reported that SAM levels in AD brains are greatly decreased [35]. This could be due to allosteric over stimulation of AMD1 by putrescine which is appearing from increased ODC activity and/or polyamine recycling by SAT1. As a result of low levels of SAM, there can be induction of p38 mitogen-activated protein kinase [36] and possibly other kinases that lead to phosphorylation of tau and possible cell cycle arrest. Increased SAT1 activity could cause lower acetyl-CoA levels which could interfere with acetylation of tau, a step that is thought to precede phosphorylation of tau. This could allow more phosphorylation of sites on tau yielding hyperphosphorylated tau and interfering with tau’s moderation of Aβ fibrillation [37]. Acetylation of tau has been shown to inhibit tau degradation and tau function [38, 39]. On the other hand, tau hyperphosphorylation could increase polyamine binding and shift the hyperphosphorylated tau from a soluble state towards an aggregated and less soluble state. Acetylation of tau at lysine 280 has been reported as a pathological modification that alters tau solubility [40]. Some of the connections between polyamine metabolism abnormalities and induction of tauopathies have been reported previously [41].
Translation of SAT1 RNA is suppressed by nucleolin, an important nucleolar protein, which keeps the SAT1 RNA sequestered [42]. However, in AD there is decreased nucleolin in the nucleolus and nucleus [43] which could allow for release of pre-existing SAT1 RNA with a sudden great increase in polyamine recycling. The lower nucleolin levels could result from leakage of nucleolin following nucleolar disruption, or binding of nucleolin by an abundance of RNA pol III transcribed Alu transcripts, or a purported role of nucleolin in moving pathogenic material to the cell surface [44]. Without sufficient nucleolin, products of polyamine catabolism, such as acrolein and hydrogen peroxide, could lead to oxidative damage to mitochondria [45]. Increased putrescine from SAT1 activity can allosterically increase AMD1 activity reducing SAM while supporting more polyamine synthesis. Putrescine originating from polyamine recycling can even exceed the de novo synthesis of putrescine from ornithine (originating from the Urea cycle) by the tightly controlled ODC enzyme. Regarding the Urea cycle, we should note that increased arginase activity has been reported in brains of AD patients [46]. Meanwhile, SAT1 activity reduces available acetyl-CoA needed for acetylcholine synthesis, acetylation of tau, histone acetylation and other metabolic uses of acetyl-CoA.
The resulting mayhem from abnormal stress and wasteful polyamine metabolism could include: lowered SAM and acetyl-CoA levels; great swings in polyamine and acetylated polyamine levels; appearance of cytotoxic acrolein and hydrogen peroxide; expression of RNA pol III Alu transcripts; nucleolar leaking and fragmentation; reduced nucleolar assembly of ribonucleoproteins; release of high concentrations of polyamines into the nucleus proper and the cytoplasm; release of autoantigens, some possibly containing viral components (e.g., EBNA1); appearance of NAPs; damage to mitochondria; polyamine interference with channels and receptors; altered synapse activity including reduced plasticity; and increased polyamine involvement in coordination of transmembrane proteins that leads to clustering and abnormal deposition (e.g., altered plaque formation).
Polyamines and AD
Many aspects of polyamines in relation to AD have emerged over the past three decades. Polyamines are important in neurons with involvement in microtubule assembly [47], amyloid deposition [48], countering oxidative stress, and ion channel regulation to mention a few routine functions. Also, additional polyamines are stored nearby in astrocytes and glial cells if needed. However, altered polyamine levels in the brain of AD patients have been reported, including increased levels of ODC, the initial enzyme in polyamine synthesis [49], with partial localization of ODC from the nucleus to the cytoplasm [50], and increased accumulation of the ODC antizyme inhibitor 2 (AZIN2) in AD brains [51]. In murine models of tauopathies, over expression of AZIN2 can cause an abnormal polyamine response with increase in acetylated polyamines and abnormal tau deposition [41]. Altered polyamine levels impact multiple aspects of AD, such as condensing hyperphosphorylated tau which is an adverse effect in AD [52]. The concept of an abnormal polyamine stress response in AD and in aging in general has been proposed recently [53]. An accumulation of Aβ associated with spermine, rising to levels of toxicity, in AD brain neurons could result from dysregulation of polyamine enzymes and polyamine intake [54]. Recent studies suggest that polyamines in the brain are locally synthesized as opposed to uptake from the blood [55]. With regards to neurons and neurodegenerative diseases, polyamine homeostasis is important for synapse functioning and plasticity by, for example, altering channel conductance [56]. Consider the fact that Ca+2 ions flowing through calcium permeable channels step from one anionic residue to the next. However, the ∼16 Å long spermine (+4) in the channel can mask multiple anionic residues preventing calcium movement. Polyamines can also mask receptors in neurons and alter the receptor’s signaling activity.
The nucleolus
With regards to higher order epigenetics, a major heterochromatic structure in the cell is the nucleolus. Unlike the cell membrane and nuclear envelope, the nucleolus does not have a delimiting lipid-like membrane, but instead maintains a heterochromatic shell. The lumen of a nucleolus has relatively low DNA content since the space is primarily dedicated to RNA folding and ribonucleoprotein assembly. However, the nucleolar lumen can contain some DNA, such as: 1) ribosomal genes in nucleolar organizing regions (NORs), 2) DNA being repaired, 3) DNA incorporating into centromeres, and 4) Xi DNA during maintenance of X chromosome inactivation. Within the nucleolus are fibrillar centers in which RNA is transcribed. Surrounding the fibrillar center is a layer called the dense fibrillar component for pre-processing of RNA transcripts, and the rest of the volume is the granular component (GC) for ribonucleoprotein assembly. Regarding nucleolar dynamics, there can be more than one nucleolus in a cell and nucleolar size, number, and proteome can change as needs arise. The nucleolar proteome, estimated to be 4,500 different proteins, has ongoing exchange between the nucleolus, nucleus and cytoplasm depending on requirements for different nucleolar activities [57]. Basic functions of the nucleoli are folding of nascent RNAs and assembly of ribonucleoprotein complexes, such as generation of ribosomes and tRNAs for translation. These functions require sufficient open volume for folding linear RNAs and high levels of the cationic polyamines to act as counter ions to neutralize the anionic self-repulsion in RNAs. In recent years specific nucleolar functions have emerged including assembly of signal recognition particles (SRP), spliceosome components, ribosomal components, and centromeres [58, 59]. Additional functions are DNA repair, processing of utility RNA transcripts (e.g., structural RNAs), refolding of RNAs as needed, and segregation of heterochromatin and euchromatin [60]. This makes the nucleolus a valuable target for viruses as they use the nucleoli for viral replication and assembly, but this misappropriation can lead to a nucleolar stress response. Besides viral activation, other triggers of nucleolar stress response (e.g., chemicals) and the subsequent effects have been reported [61].
Among chromosomes associated with nucleoli are acrocentric chromosomes with NORs in their short arms: chromosomes 13, 14, 15, 21, and 22. A primary function of the nucleolus is creating ribosomes for which the NORs contain key ribosomal genes. Epigenetic control of genes in the heterochromatic shell and in peri-nucleolar chromosomes could be adversely affected by stress-driven nucleolar morphological changes, particularly expansion, that require remodeling of the nucleolar shell. Although the nucleolus is one of the most dynamic, multi-functional and active components of the cell, and organization of the overall genome is centered around the nucleolus [62], integrity of the nucleolus is dependent on the stability of its heterochromatic shell and control of changes in polyamine levels since polyamines in nucleoli, have important dynamic roles in nucleolar functions and drive nucleolar changes in response to stress [31]. Nucleoli disappear during mitosis but reconstitute around NORs after cell division. Reformation of the nucleolar heterochromatic shell involves stabilization of the shell by nucleolin bound to Alu element sequences in structural RNA transcripts, i.e., nucleolar “duct tape”. However, in AD there is decreased nucleolin in the nucleolus and nucleus, potentially leaving the nucleolar shell vulnerable to loss of integrity [43].
Peri-nucleolar chromosomes
The inactive X chromosome (Xi; aka, the “Barr body”) is a major heterochromatic structure (i.e., higher order epigenetics) in cells of most females (46,XX; 47,XXX; etc.) and Klinefelter males (47,XXY). In humans, most X chromosome genes are not related to sex differentiation. Therefore, female cells (46,XX) need only one active X per cell to attain gene expression equivalent to male cells (46,XY). X chromosome inactivation begins early in embryogenesis when each female cell will randomly select one X (maternal X or paternal X) to remain active (Xa) and the other X becomes the inactive X (Xi). Any additional X chromosomes (e.g., 47,XXX females) will also be inactivated. Subsequent daughter cells will maintain the same inactivation pattern. As a result, based on the original embryonic choices, the adult female will have tissue sections in which all cells have a maternally derived Xi next to sections in which all cells have a paternally derived Xi. Inactivation initiates from the long arm (Xq) at Xq13 with expression of the X inactivation specific transcript (XIST) RNA transcripts which spread over and bind contiguous chromatin, first over the Xq and then the short arm (Xp), anchoring the XIST RNA at LINE-1 sites and, as the XIST RNA binds, it recruits epigenetic silencing factors (e.g., DNA methyl transferases) [63]. As a result, the Xi forms a dense heterochromatic body that localizes at the nuclear periphery against the nuclear envelope and is frequently next to nucleoli which assist in maintaining X inactivation. Xq (primarily the Xi core) will have approximately 95% of genes suppressed and Xp (primarily at the Xi surface) with approximately 65% of genes suppressed resulting in 75–85% of Xi genes suppressed. Several X-linked genes have purported significance in autoimmune diseases, but of particular importance in this discussion are, at Xp22, spermine synthetase (SMS), spermidine/spermine N1 acetyltransferase (SAT1) and a “hot” LINE-1 (i.e., fully functional reverse transcriptase activity). In addition, in the Pseudo-Autosomal Region 1 (PAR1) towards the terminus of Xp, there is a high concentration of Alu elements (28.8% versus genome average of 11%) [64]. Normally these genes are suppressed on the Xi but, with Xi disruption, these genes could be expressed with significant detrimental effects. Disruption of normal XIST RNA functioning in controlling the Xi is suspected of contributing to the female bias in SLE and RA [65].
The Xi is particularly vulnerable to disruption due to its heavy demand for methylation and repackaging following replication and due to the Xi’s peripheral location near the nuclear envelope that, compared to other chromosomes, delays the Xi’s completion of replication (late S phase or even early Gap 2) (Fig. 3A). A stress-related decrease in available SAM could hamper methylation of the Xi and lead to the opening of sequestered endogenous gene alleles (including X-linked polyamine gene alleles), Alu elements, and latent viral genes. In addition, the Xi is sandwiched between the nuclear envelope and a nucleolus making the Xi (aka, the “nucleolar satellite”) vulnerable to epigenetic disruption and spatially limited in responding intact to occasional stress-driven nucleolar dynamics (Fig. 3B).
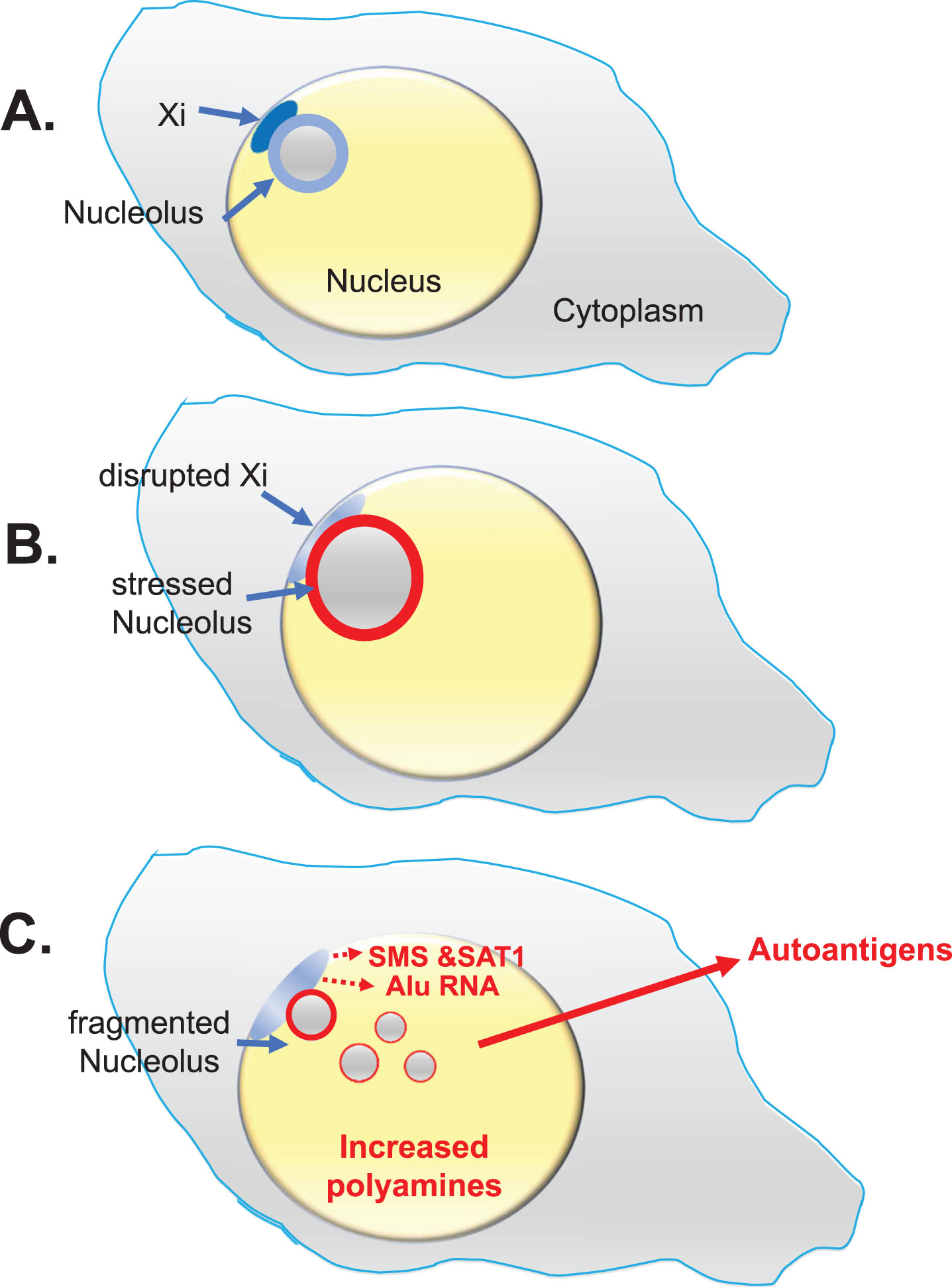
Fig. 3
Lupus Scenario. This scenario from the “X chromosome-nucleolus nexus” hypothesis was proposed for autoimmune diseases, especially SLE, to explain the female bias of autoimmune diseases based on involvement and vulnerability of the inactive X chromosome (Xi) with its normally epigenetically silenced polyamine gene alleles and a high concentration of Alu elements in the X’s pseudo-autosomal region 1 (PAR1). A) The Xi, a dense heterochromatic structure, localizes to the nuclear periphery (at the nuclear envelope) with approximately 75-85% of Xi genes epigenetically suppressed. The Xi (aka, the “nucleolar satellite” or “Barr body”) is typically next to a nucleolus where the Xi can be impacted by nucleolar dynamics during a cellular stress response. B) Stress can increase polyamine levels which causes expansion of the nucleolus, disrupting the Xi and opening previously sequestered gene alleles. C) Previously silent Xi genes at Xp22, spermine synthase (SMS) and spermidine/spermine-1-acetyltransferase (SAT1), could be expressed, further increasing polyamine synthesis and recycling. In addition, an abundance of Alu elements in PAR1 can be opened for expression by RNA pol III. RNA pol III Alu RNA transcripts compete with structural RNAs for binding nucleolin, thereby disrupting nucleolar integrity and releasing high levels of nucleolar polyamines and formation of autoantigens such as Z-DNA. Expression of a “hot” LINE-1, also at Xp22, could lead to reverse transcription.
Other chromosomes that could be disrupted by nucleolar stress are the aforementioned NOR-containing chromosomes around which the nucleolus forms following mitosis. This intimate involvement of NOR-containing chromosomes is understandable since ribosome formation is a major function of nucleoli. Of particular interest with regards to AD is chromosome 14 with the presenilin-1 (PSEN1) gene and chromosome 21 with the amyloid β precursor protein (A βPP) gene. PSEN1 and AβPP are both involved in the generation of Aβ, a protein of significant interest in AD due to adverse amyloid plaque formation. It should be noted that Down’s syndrome patients, with an extra chromosome 21, have an increased risk for AD with amyloid plaque buildup and earlier onset of AD, possibly due to the extra copy of A βPP [66].
Chromosomes 17 and 19 also closely associate and/or coordinate with the nucleolus, (i.e., possible TAD-like interactions) [62] and may suffer nucleolar driven disruption of their established epigenetic control. Chromosomes 17 and 19 have genes involved in DNA repair, which is a major function of the nucleolus. In addition, chromosome 17 contains the tau gene involved in hyperphosphorylated tau aggregates in AD and chromosome 17 contains the myeloperoxidase (MPO) gene which, with over expression, can lead to increased memory difficulties in AD [67]. Also, chromosome 19 can contain the apolipoprotein E4 (APOE4) allele that, compared to APOE2 and APOE3, accelerates deposition of amyloid [68]. Both chromosomes 17 and 19 contain more than double the average genomic gene density making any recovery of epigenetic control following disruption more difficult due to competition between neighboring genes for DNA binding proteins, negative DNA super coiling stress needed for nucleosome formation, and reestablishment of proper DNA and protein epigenetic modifications.
Alu elements and nucleolar disruption
Alu elements are short, interspersed elements, GC-rich, and average 300 bases in length. There are more than 106 Alu elements in the human genome comprising 11% of the genomic DNA, but expression of most Alu elements, which contain an internal RNA polymerase III (RNA pol III) transcription start site, is suppressed by a positioned nucleosome over the RNA pol III transcription start site. In effect, Alu expression by RNA pol III generates Alu retrotransposons which could potentially place demands on SAM levels for methylation, especially if reverse transcribed. Thirty-seven neurological and neurodegenerative diseases have been identified that are believed to involve harmful Alu activity [19]. An accumulation of Alu RNA, as observed in some neurological diseases (e.g., dementia and Creutzfeldt-Jakob disease), has been proposed as having a possible cytotoxic role in AD[69]. With regards to AD, there is importance of Alu elements in neurological network formation in development of human cognition, but Alu expression could also disrupt epigenetic control of genes involved in mitochondrial functioning, potentially leading to mitochondrial-driven apoptosis.
Alu elements have proliferated as retro elements in the human genome facilitated by reverse transcriptases, such as LINE-1 [70]. Although LINE-1 elements account for 17% of the human genome and 34% of the X chromosome, most are considered non-functional for reverse transcription with only an estimated 90 still viable, which are referred to as “hot” LINE-1s, but those are typically epigenetically suppressed [71]. One “hot” LINE-1 on the X chromosome short arm (Xp) at Xp22 is of particular interest with the possibility that it could become active from one or both X chromosomes in stressed cells. Reverse transcription activity has been demonstrated to be biased towards LINE-1 RNA at 3,000-fold greater compared to reverse transcription of other RNAs since the nascent functional LINE-1 protein is formed near the LINE-1 RNA currently being translated by the ribosome[72]. It was also found that reverse transcription of Alu RNA was 300-fold greater compared to other RNAs since Alu RNA localizes to ribosomes due to the Alu’s similarity to the SRP’s Alu domain which localizes the SRP to the ribosome where the SRP monitors emerging nascent polypeptides for extracellular protein signals [73].
Alu element sequences are often located within genes such as the structural RNAs that bind nucleolin in stabilizing the nucleolar shell, the “duct tape” mentioned above. However, Alu elements can be independently transcribed by RNA pol III using the Alu’s internal RNA pol III transcription start site if the positioned nucleosome over the start site is displaced or disrupted. Extensive disruption of chromatin could lead to a flood of Alu transcripts created by an abundance of RNA pol III typically present near the nucleolus, similar to an Alu “heat shock” response [74]. Many RNA transcripts processed in the nucleolus are created by RNA pol III, thus explaining the local availability of significant amounts of RNA pol III enzyme near nucleoli. Since RNA pol III does not require energy and is quite processive, there can be a rapid accumulation of RNA pol III Alu transcripts and even “hot” LINE-1 reverse transcribed hypomethylated Alu DNA. Re-establishment of epigenetic control of Alu elements in stressed chromatin would be difficult since Alu elements can form alternate conformations (e.g., cruciforms)from fluxing negative (underwinding) DNA supercoiling stress released from disrupted nucleosomes. Alternate DNA conformations can obstruct reformation of the positioned nucleosomes. Each nucleosome stores one negative supercoil (underwinding stress)wrapped around the histone core while the DNA strands are in B-DNA (right hand coiling) form. Reformation of disrupted nucleosomes with bound histones must compete with cationic polyamines, other DNA-binding proteins, and alternate transient conformations of negative supercoiling stress storage such as Z-DNA (left hand coiling), cruciforms, and strand separation. Also, Alu elements in chromatin, being very GC-rich, require extensive methylation which could be hampered by low levels of SAM. Especially problematic in re-establishing control are sites with a high density of Alu elements and other active genes, such as the pseudo-autosomal region 1 (PAR1) on the X chromosome (28.8% Alu) [64], chromosome 17 (18% Alu overall and 30% Alu at 17p13.3) [75], and chromosome 19 (25.8% Alu overall) [76]. Other chromosomes may also harbor Alu-rich content, such as NOR-containing chromosome 22’s long arm, 22q, which is 19% Alu [77]. Disruption of high concentrations of Alu elements in chromatin could lead to Alu cruciforms and/or strand separation followed by hybridization of strands from different Alu elements (inter-Alu hybridization) making it even more difficult to recover the original DNA strand pairings and heterochromatic structure, i.e., creating a tangle of disorganized chromatin with loss of epigenetic control, degrading cell viability.
Opening of chromatin could expose sites of DNA damage needing repair, a function involving the nucleolus and genes from chromosomes 17 and 19. This may explain the UV light sensitivity in SLE since opening DNA repair genes on 17 and/or 19 could also open the high concentrations of Alu elements on these chromosomes. The fact that nucleolar levels of the DNA repair scaffold protein, poly [ADP-ribose] polymerase 1(PARP1), are reduced in AD suggests PARP1 may be leaking from the nucleolus and is being recruited to sites of DNA damage or is involved with increased RNA transcripts. PARP1 is a key, multi-functional protein involved in DNA repair of single- and double-strand breaks, stabilizing replication forks, and facilitating chromatin remodeling [78]. In addition, PARP1 is involved in RNA stability and turnover [79]. Viral RNA, excessive Alu RNA, newly exposed DNA damage in the nucleus, and/or nucleolar disruption may explain reduced levels of PARP1 in the nucleolus reported in AD [80].
A danger from expression of a large number of RNA pol III Alu transcripts is that there could be competition between RNA pol III Alu transcripts and the Alu sequences in the structural RNA in the complexes of nucleolin/structural RNAs that stabilize the nucleolar shell. This competition could lead to compromise of the shell resulting in nucleolar inefficiency, leaking, fragmentation and even dissolution of nucleoli as the RNA pol III Alu transcripts usurp the supply of available nucleolin without contributing to maintenance of nucleolar structural integrity[81]. This insult to nucleolar integrity could release the high concentrations of free polyamines from the nucleolus into the nucleus proper and the cytoplasm where the preexisting polyamines, which are at lower concentrations and are relatively less mobile, are bound to DNA, RNA, proteins, and phospholipids. The sudden flood of free polyamines from the nucleolus could cause aggregation of anionic moieties such as hyperphosphorylated tau and stabilization of transient DNA and RNA conformations (e.g., Z-DNA and cruciforms). This flood of polyamines could also accelerate amyloid deposition [48], interfere with neuron signal transduction, interfere with receptors, interfere with mitochondrial calcium channels, and stabilize autoantigenic complexes. Other neurological diseases also involve problems from increased polyamines, potentially involving a flood of released nucleolar polyamines. Higher levels of polyamines are seen in red blood cells in amyotrophic lateral sclerosis (ALS) and PD patients [82]. This could involve difficulties with polyamine levels and nucleolar integrity prior to or concurrent with the extrusion of nuclei from the red blood cells, or it may involve greater uptake of polyamines arising from other cells, bacteria, or other triggers. Polyamines stimulate misfolding and abnormal aggregation of α-synuclein in PD [83] and are involved in Lewy body formation in PD [84], while excess polyamines in ALS can create receptor and channel blocking issues [85].
Another problem that could arise with excess RNA pol III Alu transcripts is that the transcripts could compete with the 7SL RNA for binding of SRP9/14 protein dimers in the nucleolus leading to lower levels of functional SRP, a nucleoprotein complex that identifies nascent polypeptides that code for proteins which will be released extracellularly via the endoplasmic reticulum. Incomplete SRPs localizing at ribosomes could still bind nascent polypeptides of extracellular proteins and escort the ribosome/mRNA/SRP/polypeptide complex to an SRP receptor in the endoplasmic reticulum, but incomplete SRPs would not be able to halt translation during the translocation. Also, lower levels of normal SRP could miss some extracellular polypeptides and fail to temporarily halt translation of those emerging nascent extracellular polypeptides. These errors would allow exposure of the nascent extracellular polypeptide currently undergoing translation to intracellular enzymes (e.g., transglutaminases) [86]. In this scenario, polyamines could be conjugated to proteins (aka, polyamination) by transglutaminases and be oxidized down to the acrolein-conjugated proteins previously mentioned in AD and SjS. This could be particularly problematic with a sudden flood of polyamines, such as from the nucleolus. Acrolein, being a cytotoxic agent, is suspected of involvement in neurodegenerative diseases.
Here importance has been placed on Alu elements due to their abundance (>106 copies) and their demonstrated effect on nucleolar integrity, but there are many other RNA sequences that could provide disruptive effects, such as long non-coding RNA genes (lncRNAs;>200 bases) that can affect expression of other genes cis (≤10K bp)or trans (>10K bp) away from the lncRNA gene.
Triggers
A variety of triggers have been associated with AD either as a single event or as multiple events with an accumulation of adverse effects. Some triggering factors that can lead to AD are metals [87], bacteria [88], viruses [89], and physical trauma [90]. Abnormal nucleolar stress can be a common component in AD onset and progression. Metals (e.g., aluminum, copper, iron, zinc) in increased levels can accumulate; alter blood-brain barrier permeability; impact oxidative stress; and adversely substitute for each other as co-factors in enzymatic activity causing cellular stress [91]. We can also consider the concept of the autoimmune/inflammatory syndrome induced by adjuvants (ASIA; aka, Shoenfeld’s syndrome) [92], in which immunogenic components added to vaccines or drug formulations to stimulate a focused immune reaction to pathogenic material may actually provoke an autoimmune disease due to a moregeneralized reaction to anomalous material of endogenous origin, such as from a disrupted nucleolus, or material from reactivated latent viral genes.
Physical trauma can contribute to AD since damaged proteins need to be replaced. More ribosomes and tRNA than normally available are needed for this recovery and therefore the nucleolar volume and activity must increase, driven by polyamines. Intracellular and extracellular structural tension when disrupted could expose damaged proteins and DNA damage that need repair; open unwanted gene expression; disrupt nutrient flow (e.g., damaged channels); interfere with cell-to-cell contacts; and interfere with cell-matrix adhesion.
In the case of stress triggered by bacteria (e.g., gut microbiota), bacteria do not control their production of putrescine and spermidine. This can raise circulating polyamine levels during infection and those excess polyamines can enter cells via polyamine transporters potentially causing an abnormal polyamine stress response [93]. With regards to cellular stress caused by viral activity, it is to the virus’s benefit to increase nucleolar capacity, creating more ribosomes and tRNAs to facilitate viral replication and assembly. Since polyamines induce increased nucleolar capacity and activity, stimulating polyamine synthesis is a common early step in viral activation [94]. One of the best examples of viral impact on nucleoli via increased polyamines is Epstein-Barr virus (EBV; aka, “human herpes virus 4”) which is suspected of playing a role in many autoimmune diseases, including in SLE [95]. Approximately 85 viral proteins and 50 non-coding RNAs that originate from the EBV genome have been identified for which EBV needs host cell assistance for reproduction [96]. Upon activation, EBV induces the host cell’s c-Myc proto-oncogene protein to increase gene expression of up to 15% of genes in the human genome [97]. There are more than 70 genes induced by c-Myc that generate proteins which localize to the nucleolus, such as nucleolin and nucleophosmin [98]. Among the genes targeted by c-Myc is ODC, which initiates polyamine synthesis by generating putrescine [99]. Activity of spermidine synthase (SRM) and spermine synthase (SMS) are also induced by c-Myc, thereby increasing polyamine synthesis[100]. And EBV upregulates RNA pol III transcription which can generate structural RNAs, ribosomal RNAs, tRNAs, Alu RNAs and many other non-coding RNAs, as well as for expression of viral RNAs [101]. Thus, the virus can hijack much of the host cell’s nucleolar functioning for viral use by increasing polyamine levels and RNA pol III activity, making abnormal RNA pol III transcription a potential factor in diseases [102]. Another example of a virus known to impact the nucleolar structure upon activation is adeno-associated virus type 2 (AAV2) [103].
Whatever triggering event occurs, subsequent chromatin disruption can induce the cGAS/STING (cyclic GMP-AMP synthase/stimulator of interferon genes) pathway that monitors chromatin and the cytoplasm to alert the immune system of DNA and/or RNA anomalies arising due to acute insults or age-related degeneration. Most cGAS is tightly bound to chromatin in the nucleus [104]. When cGAS senses anomalies, it can dimerize and become active, generating cGMP that activates STING. The cGAS/STING pathway can induce ODC which will increase polyamine synthesis and further deplete SAM [105]. When SAM is extensively reduced, it can induce p38 kinase that then phosphorylates tau, leading to AD issues with hyperphosphorylated tau.
The “X chromosome-nucleolus nexus” hypothesis
The “X chromosome-nucleolus nexus” hypothesis, originally proposed in 2015, describes a scenario in which stress-driven nucleolar changes disrupt peri-nucleolar chromatin, which can lead to onset of SLE with possible parallels in other autoimmune diseases [22]. Due to the female bias of many autoimmune diseases, the hypothesis was primarily focused on the Xi’s vulnerability and proximity to the nucleolus (Fig. 3A) during a nucleolar stress response with expansion of the nucleolus that could potentially disrupt the Xi (Fig. 3B) and open sequestered gene alleles, leading to loss of dosage compensation. X-linked gene alleles of particular interest are SMS involved in polyamine synthesis, and SAT1 involved in polyamine recycling. Both SMS and SAT1 are located at Xp22, and both are normally silent on the Xi, but expressed from the active X (Xa) [106]. Over expression of SMS and/or SAT1, possibly from reactivation on a disrupted Xi, could lead to futile rounds of polyamine synthesis and recycling that draw down SAM levels and acetyl-CoA. Additional polyamines created could further impact nucleolar dynamics.
Besides the X-linked polyamine genes, another problem is the opening of Alu elements, such as the high concentration of Alu elements in the Xi’s PAR1. RNA pol III enzyme near the nucleoli could rapidly generate an abundance of RNA pol III Alu transcripts using the internal transcription start site within Alu elements. The Alu transcripts could flood the nucleolus, form abnormal ribonucleoprotein complexes, and interfere with SRP assembly and functioning. There may even be reverse transcription of Alu elements by fully functional LINE1 proteins, such as a “hot” LINE1 gene at Xp22 on the Xi. High levels of Alu DNA have been observed in the free DNA in SLE patients’ sera which may arise from reverse transcription [18]. In addition, since many autoantigens in SLE are, at least transiently, resident in the nucleolus, it suggests that there is extraordinary stress occurring in nucleoli in SLE that can lead to improper conformations, modifications and/or assemblies of ribonucleoprotein complexes, such as assemblies containing viral proteins. Nucleolar-associated autoantigens in SLE can include Ku, Ro, La, nucleolin, and more [107]. These entities, in some autoantigenic state or complex, could trigger an autoimmune reaction which then expands via epitope spreading to the more abundant normal forms.
Also in 2015, a report was published describing how segments of RNA pol III Alu transcripts, even as short as 20 bases, can disrupt the integrity of nucleoli by competing with the Alu sequence in the structural RNA normally bound to nucleolin, leading to nucleolar inefficiencies, leaking, and even fragmenting [81]. This provided a possible connection between disruption of the Xi with its abundance of Alu elements in PAR1 and nucleolar disruption with release of autoantigenic material, leading to development of autoimmune diseases like SLE (Fig. 3C). Nucleolar fragmentation could also release high levels of nucleolar polyamines which could bind material in the general nucleus disrupting transcription and replication and stabilizing normally transient conformations, like cruciforms (e.g., in Alu sequences) and Z-DNA in autoantigenic forms. Z-DNA is a frequent target for autoantibodies in SLE and RA [108], and the anti-DNA autoantibodies show greater avidity in the presence of spermine as if spermine was involved in the initial provocation [109]. This suggests there may be epitope spreading from the initial autoantigenic anomalous entity. Subsequently, updates to the original “X chromosome-nucleolus nexus” hypothesis were published incorporating nucleolar disruption by RNA pol III Alu transcripts [110–112].
There is relevance for the “X chromosome-nucleolus nexus” hypothesis in AD since, for example, in AD there are alterations in nucleolar output affecting protein synthesis [113, 114] and there are increased polyamine levels in the brains of AD patients including increased levels of ODC, the initial enzyme involved in polyamine synthesis [115, 116].
THE POLYAMINE HYPOTHESIS: ALZHEIMER’S DISEASE SCENARIO
Originating from the earlier “X chromosome-nucleolus nexus” hypothesis, which primarily focused on SLE and the Xi, the “polyamine” hypothesis was developed using a broader scope based on systems biology and autoimmune disease tautology. In regard to AD, the “polyamine” hypothesis suggests events that may precede abnormal aggregation of tau and Aβ deposition difficulties, thereby allowing us to move past the debates on “tau versus Aβ” as the primary instigators.
Polyamine dysregulation in AD can include altered polyamine metabolism, intake, interactions, and localization. Interactions of polyamines with ion channels, receptors, synapses, nucleic acids, and phospholipids are frequently discussed activities, but polyamines can also participate in amyloid deposition, microtubule assembly and in condensing hyperphosphorylated tau. Thus, polyamine dysregulation is a key aspect of tau neuropathies and amyloid problems. Others have recently discussed polyamine functions and abnormalities in AD and dementia with regards to Aβ plaque formation [48], tau neuropathies [117], metabolic dysregulation including polyamine storage and stress responses[52], autophagy control [118], protection against lipid peroxidation [119], channel and receptor control [85], and general involvement in age-related disease [120]. Those works point to importance of polyamines in AD and neurological disorders in general.
A simplified neuron in its normal state is depicted with NOR containing chromosomes (13, 14, 15, 21, 22) in the nucleolar heterochromatic shell and other relevant chromosomes (Xi, 17, 19) in close proximity to the nucleolus (Fig. 4A). The “polyamine” hypothesis could occur in other cell types and lead to other related autoimmune diseases, but neurons with polyamine dysregulation and tau and Aβ aggregation are of significant importance in AD. Therefore, a neuron will be the focus for further discussion of the hypothesis (Figs. 4 and 5).
Fig. 4
Alzheimer’s Scenario. This scenario improves on the “X chromosome-nucleolus nexus” hypothesis by providing additional insights based on Alzheimer’s research. It could also be called the “polyamine dysregulation and nucleolar disruption” hypothesis, or simply the “polyamine” hypothesis. This hypothesis potentially has relevance in multiple autoimmune diseases. A) Chromosomes (13, 14, 15, 21, 22) containing nucleolar organizing regions (NORs) are part of the nucleolar heterochromatic shell and contain genes of importance in AD: presenilin-1 (PSEN1) on chromosome 14 and amyloid precursor protein (APP) on chromosome 21. Other chromosomes of interest are chromosome 17 with the tau (TAU) gene and the myoperoxidase (MPO) gene, and chromosome 19 with the ApoE4 allele. Chromosomes 17 and 19 can localize near the nucleolus since they contain genes involved in DNA repair, a function of the nucleolus. B) Stress can increase polyamine levels causing nucleolar expansion, potentially disrupting neighboring chromosomes (e.g., Xi, 17, 19). Chromatin disruption can open previously sequestered sequences, such as Alu elements, which comprise 11% of the genome. Xi PAR1 is ∼29% Alu, 17p13.3 in chromosome 17 is 30% Alu, and all of chromosome 19 is 25.8% Alu. C) An abundance of RNA pol III enzyme typically found near nucleoli can express “free” Alu transcripts that compete with RNA pol II transcribed structural RNAs (containing intronic Alu sequences) bound to nucleolin, thereby disrupting the nucleolar heterochromatic shell. As a result, nucleoli lose their integrity with leaking and even fragmentation. SAT1 transcripts, previously sequestered by nucleolin, increase polyamine recycling which reduces acetyl-CoA (impacting acetylcholine levels) and generates putrescine further increasing polyamine synthesis (impacting SAM levels). Low SAM levels can induce p38 kinase phosphorylation of Tau, which can aggregate when bound by released nucleolar polyamines. Nucleolar poly [ADP-ribose] polymerase 1 (PARP1) levels drop as PARP1 binds exposed DNA damage and an abundance of RNA.
![Alzheimer’s Scenario. This scenario improves on the “X chromosome-nucleolus nexus” hypothesis by providing additional insights based on Alzheimer’s research. It could also be called the “polyamine dysregulation and nucleolar disruption” hypothesis, or simply the “polyamine” hypothesis. This hypothesis potentially has relevance in multiple autoimmune diseases. A) Chromosomes (13, 14, 15, 21, 22) containing nucleolar organizing regions (NORs) are part of the nucleolar heterochromatic shell and contain genes of importance in AD: presenilin-1 (PSEN1) on chromosome 14 and amyloid precursor protein (APP) on chromosome 21. Other chromosomes of interest are chromosome 17 with the tau (TAU) gene and the myoperoxidase (MPO) gene, and chromosome 19 with the ApoE4 allele. Chromosomes 17 and 19 can localize near the nucleolus since they contain genes involved in DNA repair, a function of the nucleolus. B) Stress can increase polyamine levels causing nucleolar expansion, potentially disrupting neighboring chromosomes (e.g., Xi, 17, 19). Chromatin disruption can open previously sequestered sequences, such as Alu elements, which comprise 11% of the genome. Xi PAR1 is ∼29% Alu, 17p13.3 in chromosome 17 is 30% Alu, and all of chromosome 19 is 25.8% Alu. C) An abundance of RNA pol III enzyme typically found near nucleoli can express “free” Alu transcripts that compete with RNA pol II transcribed structural RNAs (containing intronic Alu sequences) bound to nucleolin, thereby disrupting the nucleolar heterochromatic shell. As a result, nucleoli lose their integrity with leaking and even fragmentation. SAT1 transcripts, previously sequestered by nucleolin, increase polyamine recycling which reduces acetyl-CoA (impacting acetylcholine levels) and generates putrescine further increasing polyamine synthesis (impacting SAM levels). Low SAM levels can induce p38 kinase phosphorylation of Tau, which can aggregate when bound by released nucleolar polyamines. Nucleolar poly [ADP-ribose] polymerase 1 (PARP1) levels drop as PARP1 binds exposed DNA damage and an abundance of RNA.](https://content.iospress.com:443/media/jad/2024/98-3/jad-98-4-jad231184/jad-98-jad231184-g004.jpg)
Fig. 5
Stress Impact on Polyamines. Stress can lead to wasteful rounds of polyamine synthesis and recycling. A) Increased polyamine synthesis occurs from stress as the cell needs to increase nucleolar activity for recovery. B) Extensive polyamine synthesis reduces SAM which can trigger p38 kinase phosphorylation of tau. Subsequent abnormal release of nucleolar polyamines can aggregate phosphorylated tau. C) “Free” Alu transcribed by RNA pol III can compete with structural RNAs (stRNA) bound to nucleolin. This weakens the nucleolar heterochromatic shell. D) “Free” Alu transcripts can also compete nucleolin off sequestered SAT1 transcripts. The SAT1 protein can cause super induction of polyamine recycling that draws down acetyl-CoA, impacting acetylcholine levels, and creates putrescine which induces further polyamine synthesis. Also, there is potential creation of cytotoxic acrolein and hydrogen peroxide. Polyamine recycling by acetylation and oxidation works to reestablish polyamine levels as the cell recovers from stress. Enzymes:1. Ornithine Transcarbamylase (OTC);2. Argininosuccinate Synthetase 1 (ASS1); 3. Argininosuccinate Lyase (ASL);4. Arginase 1 (ARG1);5. Ornithine Decarboxylase (ODC); 6. S-Adenosylmethionine Decarboxylase 1 (AMD1);7. Spermidine Synthase (SRM); 8. Spermine Synthase (SMS);9. Spermine Oxidase (SMOX);10. Spermidine/Spermine N1 Acetyltransferase 1 (SAT1);11. Polyamine Oxidase (PAO).
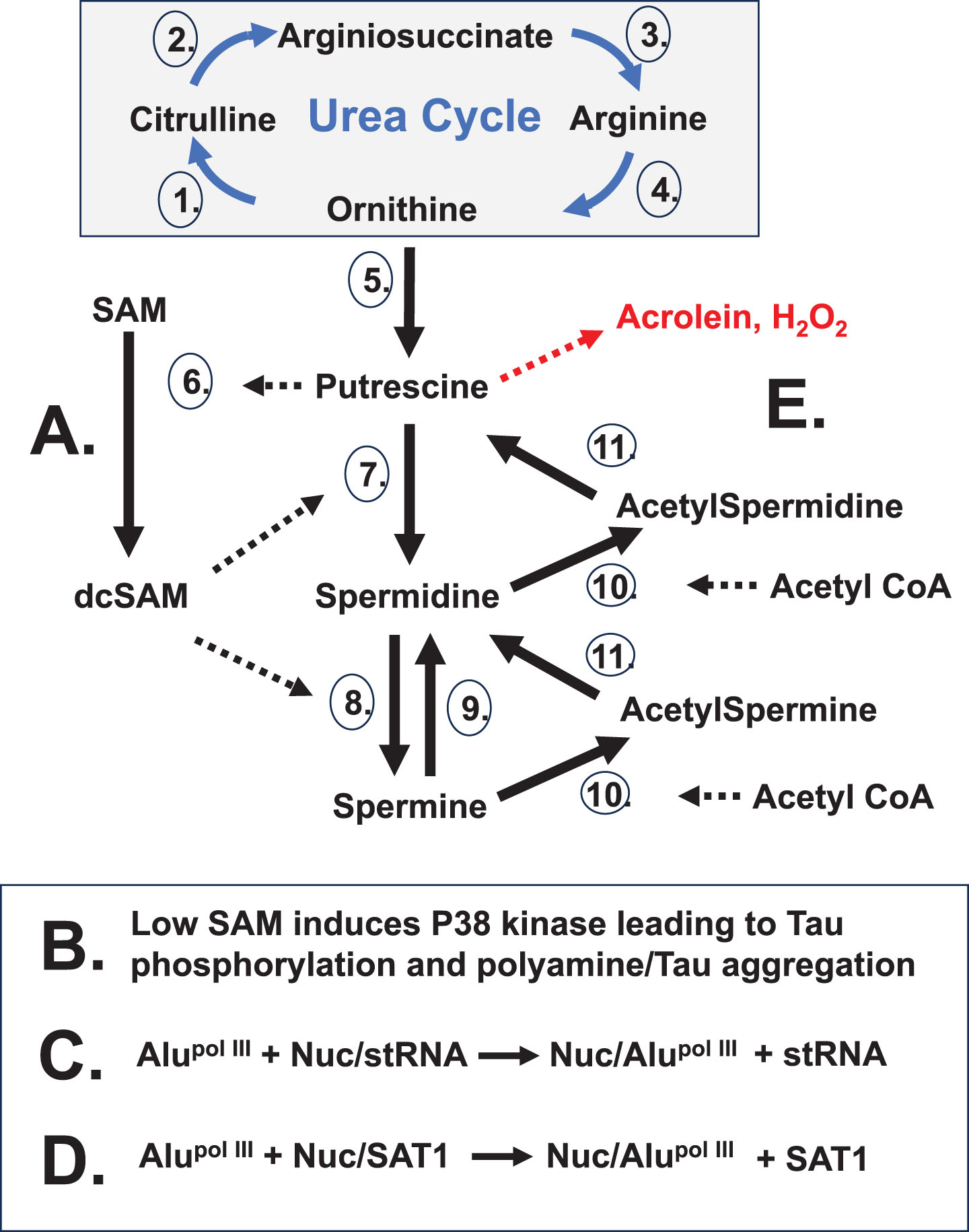
Cellular stress can arise from a number of different triggers creating a need for increased nucleolar activity for cell recovery. Since polyamines are at their highest intracellular levels in the nucleolus where they act as counter ions in RNA folding and ribonucleoprotein assembly and since polyamines drive increased nucleolar volume and activity, there is increased polyamine synthesis to stimulate the nucleolus following insult from the various triggers. Among the triggers, the hypothesis is best exemplified by EBV, and possibly other herpes viruses which can cause infections with suspected roles in autoimmune diseases. With regards to AD, some EBV proteins are suspected of a direct role in initiation of amyloid aggregate formation[121]. EBV involvement is suspected in at least seven major diseases [122], including AD and PD. Latency of the viral genes can explain why these diseases typically do not have initial onset until adulthood when sufficient disruption has accumulated. Other triggers that disrupt chromatin could open latent viral genes which then add to the stress.
Viruses can persist by insertion of their genes in chromatin, especially in fragile sites, which are slow to replicate, and are particularly vulnerable to DNA damage. A recent study of EBV insertions (in fragile sites and elsewhere) in Burkitt lymphoma cell lines shows a non-random pattern of potential EBV insertion sites in every chromosome with chromosomes 1–8, 10, and 12 having abundant sites of integration [123]. Susceptibility of some individuals to adverse viral activity may depend on the cell type, the means of viral entry, the viral load established, and the vulnerability of sites in which the viral genes may be inserted. Fragile site B (FRAXB) on the Xi short arm is a particularly interesting possibility since the Xi lags in replication and repackaging and the Xi is vulnerable to disruption by an expanding nucleolus. Viral activation could also entail replication and release of virions that infect other brain cells as AD progresses.
Upon activation, EBV induces the host cell’s c-Myc protein to increase gene expression of up to 15% of genes in the human genome, including genes that localize to the nucleolus. Among the genes targeted by c-Myc is ODC, which initiates polyamine synthesis (Fig. 5A) by generating putrescine. Putrescine (from ODC or SAT1) allosterically induces an increase in decarboxylation of SAM by adenosylmethionine decarboxylase (AMD1), thereby reducing SAM needed for cellular methylation. Also, c-Myc induces increased polyamine synthesis and upregulates RNA pol III transcription to generate RNA for ribosomes and tRNAs as well as for expression of viral RNAs. Thus, the virus can hijack much of the host cell’s nucleolar functioning for viral use by increasing polyamine and RNA pol III activity. Latent EBV genes can express microRNAs, non-coding RNAs, and a variety of proteins that are nuclear antigens (EBNAs 1, 2, 3A, 3B, 3C, EBNA-LP) or membrane proteins (LMPs 1, 2A, 2B) [124].
The increase in polyamine synthesis will reduce available SAM which can lead to induction of tau phosphorylation by kinases (e.g., p38 kinase), even to the extent of creating hyperphosphorylated tau (Fig. 5B) [125]. If acetyl-CoA is low, which could occur with extensive polyamine recycling, acetylation of residues on tau could be faulty and possibly leave more sites open to phosphorylation. This gives tau more anionic character, which can lead to aggregation by cationic polyamines, including an abundance of free nucleolar polyamines released when nucleolar integrity is compromised. The initial trigger-induced increase in polyamine levels will cause expansion of the nucleolus, potentially disrupting epigenetic control in peri-nucleolar chromatin (Fig. 4B). Disruption of the Xi can include opening of the SMS and SAT1 alleles that are normally silent but now lead to further increase in polyamine synthesis and recycling.
As the nucleolus expands with increased polyamines, there is need for more nucleolin in order to stabilize the nucleolar heterochromatic shell using nucleolin bound to an Alu sequence in a structural RNA transcript. However, as chromatin is disrupted by the expanding nucleolus, it can open Alu elements to RNA pol III transcription (Fig. 4C). Some chromosomes (e.g., Xi, 17, 19, 22) near or in the nucleolus have exceptionally high concentrations of Alu elements, as mentioned earlier. A flood of RNA pol III Alu transcripts could bind nucleolin but not contribute to stabilizing the nucleolar heterochromatic shell, leading to leaking of nucleolar polyamines and even fragmentation of the nucleolus (Fig. 5C). In addition, nucleolin sequesters SAT1 RNA but release of SAT1 RNA when available nucleolin in the nucleolus drops or is sequestered by RNA pol III Alu transcripts, could trigger sudden intense polyamine recycling by SAT1 which reduces acetyl-CoA levels (Fig. 5D, E). Lower acetyl-CoA would hamper acetylcholine synthesis, histone acetylation, and other uses of acetyl-CoA, including acetylation of some residues in tau, which appears to be a step that normally precedes tau phosphorylation. Eventually reduction of acetyl-CoA can also hamper polyamine recycling. However, while there is acetyl-CoA available, the increased SAT1 activity will create putrescine that allosterically increasesd decarboxylation of SAM by AMD1 leading to further production of polyamines. Increased SAT1 activity could also generate acrolein and acrolein-conjugated proteins. Fluctuations in the ratios and levels of polyamines and acetylated polyamines and decreased nucleolar integrity would affect nucleolar efficiency in RNA folding and ribonucleoprotein assembly. The resulting dynamics of the nucleolus could disrupt neighboring chromatin and hinder re-establishment of epigenetic control, leading to dysregulation in other pathways. There could also be mitochondrial damage since mitochondrial-related genes in the nucleus may be disrupted and polyamines could block channels supplying calcium to mitochondria [126]. We should consider the possibility that, with their strong cationic charges, polyamines can induce phase shifts moving some materials from their normal solubility to less soluble states. Since polyamines are involved in amyloid plaque deposition, this process could be altered with abnormal release of nucleolar polyamines, along with disruption of epigenetic control of many AD-related genes, such as presenilin-1, tau, APOE4, the amyloid precursor protein on chromosomes 14, 17, 19, and 21, respectively. Other consequences would follow (e.g., plaques, tangles, lower acetylcholine, low SAM, low acetyl-CoA, low levels of structural RNA/nucleolin complexes, calcium channel disruption, acrolein-conjugated proteins, altered synaptic activity, and more).
THERAPEUTIC POSSIBILITIES
The “polyamine” hypothesis provides many potential therapeutic targets. Since the targets are important in normal cellular activity, it may require ongoing administration of a cocktail of low doses of a variety of drugs to slow or prevent AD without destroying cells.
Research targeting polyamine enzymes for cancer therapeutics has been explored for many years, but typically the approach towards cancers is to use acute, high dose treatment in an attempt to eradicate a patient’s cancer based on the cancer cells’ heavy dependence on polyamines. This body of knowledge may provide potential drug candidates that could control or suppress some autoimmune diseases if administered at lower doses over a longer term. For example, diflouromethylornithine (DFMO; aka, eflornithine), an inhibitor of ODC, gave promising results in a mouse model of SLE but, when tested in humans, it had side-effects, such as tinnitus and hearing loss which, in rare cases, was irreversible. Another polyamine-related target, AMD1 could be targeted, at its active site and/or allosteric site, to slow its conversion of SAM to decarboxylated SAM. Acetyl-CoA levels may be preserved by inhibition of SAT1. To counter activation of p38 MAPK kinase and other kinases that may be activated by low SAM, kinase inhibitors are being explored to reduce hyperphosphorylation of tau [127].
Recent research on positive effects of spermidine in dementia patients [128] and age-related diseases in general [129] suggest therapeutic possibilities using spermidine. AD murine models have demonstrated that spermidine can reduce neuroinflammation and levels of soluble Aβ [130]. Spermidine is essential for translation since spermidine is conjugated to a lysine residue on elongation factor ElF5A (eukaryotic translation initiation factor 5A) and converted to hypusine to activate EIF5A. Spermidine preserves mitrochondrial functioning and supports proper autophagy to clear damaged cells from tissues [131]. In addition, supplemental spermidine reduces the demands on SAM since the putrescine to spermidine step is not needed.
Suppression of Alu expression may be a difficult task since RNA pol III is needed for expression of many other RNAs. Dampening RNA pol III activity, such as using weak inhibitors, may have some benefit. Increasing nucleolin expression and/or increasing expression of structural RNAs involved in stabilizing the nucleolar shell are other possibilities.
Since EBV is suspected of involvement in autoimmune diseases, EBV entry could be targeted with a neutralizing antibody to prevent EBV’s spread to neighboring cells [132]. To stop activity from reactivation of viral genes, specific viral and endogenous genes could be targeted, such as EBNA1 and other EBV-related genes, the host’s c-Myc activity, and polyamine genes. The cGAS/STING pathway that monitors for DNA and RNA abnormalities could also be targeted since the cGAS/STING pathway can induce ODC activity.
EXPERIMENTAL TESTING OF THE HYPOTHESIS
As with any hypothesis, this hypothesis should be amenable to testing. Testing will involve monitoring nucleolar dynamics, polyamine metabolism, Alu expression and expression of Alzheimer’s related biomarkers (e.g., tau and Aβ). In vitro assays, such as dynamic light scattering analysis of polyamines and other cations (e.g., metals) can test the concept of these cations causing aggregation of various forms of tau (e.g., acetylated, phosphorylated). Also, competition assays for nucleolin binding between Alu RNA transcripts and nucleolar structural RNA transcripts could be performed under proper RNase-free conditions. The hypothesis implies that murine models for human AD have shortcomings since the murine genome lacks the abundance of Alu elements present in the human genome and there are human versus mouse differences in chromosome structure and content that further complicate analysis. As far as structural differences, potential differences in the stability of X chromosome inactivation in the human submetacentric X chromosome versus the murine telocentric X chromosome have been discussed above. Regarding chromosome content, difficulties in modeling Down’s syndrome (chromosome 21 trisomy in humans) in mice exemplify similar difficulties for AD models. Chromosome 21 (with its NOR, A βPP gene, and other chromosome 21 genes) is intimately involved with nucleolar dynamics and possible epigenetic disruption whereas the equivalent genes in the mouse map to three different murine chromosomes (10, 16, and 17) with potentially less vulnerability to nucleolar dynamics [133].
Fortunately, there have been recent advances in human organoid techniques which will allow for potentially greater options and fidelity in modeling human AD events [134]. It can also sidestep issues with blood-brain barrier permeability, allowing for administration and monitoring of compounds that may otherwise be excluded. This can allow, for example, testing with the large number of drug candidates originally developed as cancer therapeutics to target polyamine metabolism, but using them as molecular probes: diminazene aceturate could be used to inhibit SAT1 [135], diflouromethylornithine to inhibit ODC [136], and valbenazine or verubecestat to inhibit AMD1 [137]. Activity of p38 MAP kinase and tau phosphorylation could also be monitored in an organoid scenario using a variety of inhibitors and activators [138]. In these various experiments, we would want to monitor changes in SAM, acetyl CoA, and acetylcholine levels.
An organoid system could also be used for testing the effects of the great number of neuropeptides that are believed to exist to determine their potential neuroprotective effects [139]. We can take advantage of autoimmune disease tautology in a brain organoid simulation of AD by considering photosensitivity in SLE. UV light can cause DNA damage and trigger SLE-related skin rashes. This suggests that DNA repair genes, such as in chromosomes 17 and 19, could be opened to assist in nucleolar DNA repair. This could also open the high concentrations of Alu elements and disrupt epigenetic control on those chromosomes and disrupt the nucleolus by Alu RNA competition for nucleolin with release of sequestered SAT1 RNA from nucleolin as proposed by the hypothesis in AD. AD brain cells in vivo would not have this exposure to UV light, but this could be a useful trigger in brain organoid cells to study such events. Changes in DNase sensitivity at specific sites (e.g., TAU and MPO alleles on chromosome 17) could be monitored. Also, florescent labeled anti-nucleolin antibodies could show changes in nucleolin localization. Other events that could be explored in brain organoids are activation of cGAS and the cGAS/STING pathway that can induce increased ODC activity. Formation of autoantigens may be another observable feature in brain organoids. Many autoantigens observed in SLE are nucleolar components and the systemic nature of SLE allows exposure of the autoantigens to the immune system. Similar antigens may arise in AD brain cells, but the blood-brain barrier prevents sufficient exposure to the immune system to provoke an autoimmune response. The possibility that such antigens are occurring in an AD scenario can be explored with brain organoids. Further testing could involve transfection of inducible polyamine enzyme genes (e.g., AMD1), RNA pol III, and/or Alu elements to test overexpression of those genes in brain organoids.
CONCLUSIONS
Changes in polyamine levels are highly suspected of an adverse role in autoimmune diseases even though polyamines are essential in cells. Herein, four routes of altered polyamine levels have been described that may occur in AD and some other autoimmune diseases. First, polyamine synthesis and recycling can be increased by stress, including intracellular viral or bacterial infection or latent viral gene activation (e.g., EBV stimulation of c-MYC activity that induces increased SAT1, SMS, and ODC activity). Second, polyamines stored in neighboring cells, such as glial cells, can be released to counter oxidative stress. Third, microbiota in the gut or tissue infections can be a source of additional polyamines. Fourth, disruption of the nucleoli, such as by RNA pol III generated Alu transcripts, could release the high concentrations of nucleolar polyamines.
Discussing possible involvement of polyamines in AD can also provide insights that carry over to other neurodegenerative diseases such as Huntington’s disease (HD), PD, and ALS. Keep in mind though that, despite possible common mechanistic aspects, each of these diseases differ, for example, in the cell type and tissue setting (e.g., SLE is systemic, whereas the main damage in AD is primarily behind the blood-brain barrier) and they differ in autoantigens targeted (e.g., citrullinated proteins being major targets in MS and RA while tau and Aβ are major targets in AD, and chromatin and/or nucleolar components are targets in SLE). Considering AD, HD, PD, and ALS, they may have similar polyamine and nucleolar complications, but we might think of AD as primarily affecting memory whereas HD, PD, and ALS appear to affect motor skills directed from other sites in the brain.
There are alternate routes to polyamine dysregulation to consider. For example, as an initiating event, chromatin disruption could open extensive RNA pol III Alu expression that fragments the nucleoli, releasing nucleolar polyamines, and the Alu transcripts overwhelm the nucleolin supply with release of nucleolin sequestered SAT1 transcripts. Expression of SAT1 could initiate polyamine recycling and putrescine induction of AMD1, thereby reducing SAM and acetyl-CoA, and only later inducing increased polyamine synthesis. In effect, activation of ODC, SRM, and SMS are not necessarily the initial events, in contrast to the pattern described for EBV initiation. We should also consider different results if only SMS (polyamine synthesis) or only SAT1 (polyamine recycling), as opposed to both, escape sequestration in the Xi. However overall, the different routes would have many similarities.
In conclusion, the “polyamine” hypothesis brings new concepts for AD: Alu disruption of nucleoli; release of nucleolar polyamines; and the reduction of SAM and acetyl-CoA that can occur from excessive polyamine synthesis and recycling. Abnormal fluctuations in polyamines and acetylated polyamines can be quite disruptive. And the hypothesis points to the need for more consideration of systems biology approaches and autoimmune disease tautology in autoimmune and neurodegenerative disease research. New approaches, such as using brain organoids for experimentation, and computational drug development, can provide promising means of testing and implementing the hypothesis.
AUTHOR CONTRIBUTIONS
Wesley H Brooks (Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Visualization; Writing – original draft; Writing – review & editing).
ACKNOWLEDGMENTS
This work was created solely by the author and developed in memory of Thomas Garrett Brooks, Sr. and Florence Anne “Polly” Boone Brooks.
FUNDING
The author has no funding to report.
CONFLICT OF INTEREST
The author has no conflict of interest to report.
REFERENCES
[1] | Morgan SL , Naderi P , Koler K , Pita-Juarez Y , Prokopenko D , Vlachos IS , Tanzi RE , Bertram L , Hide WA ((2022) ) Most pathways can be related to the pathogenesis of Alzheimer’s disease. Front Aging Neurosci 14: , 846902. |
[2] | Chen Z-R , Huang J-B , Yang S-L , Hong F-F ((2022) ) Role of cholinergic signaling in Alzheimer’s disease. Molecules 27: , 1816. |
[3] | Anaya JM ((2017) ) The autoimmune tautology. A summary of evidence. Joint Bone Spine 84: , 251–253. |
[4] | Samuels H , Malov M , Saha Detroja T , Ben Zaken K , Bloch N , Gal-Tanamy M , Avni O , Polis B , Samson AO ((2022) ) Autoimmune disease classification based on PubMed text mining. J Clin Med 11: , 4345. |
[5] | Li X , Sundquist J , Zöller B , Sundquist K ((2018) ) Dementia and Alzheimer’s disease risks in patients with autoimmune disorders. Geriatr Gerontol Int 18: , 1350–1355. |
[6] | Chou RC , Kane M , Ghimire S , Gautam S , Gui J ((2016) ) Treatment for rheumatoid arthritis and risk of Alzheimer’s disease: A nested case-control analysis. CNS Drugs 30: , 1111–1120. |
[7] | Boukhvalova MS , Kastrukoff L , Blanco JCG ((2023) ) Alzheimer’s disease and multiple sclerosis: A possible connection through the viral demyelinating neurodegenerative trigger (vDENT). Front Aging Neurosci 15: , 1204852. |
[8] | Zhao Z , Rocha NP , Salem H , Diniz BS , Teixeira AL ((2018) ) The association between systemic lupus erythematosus and dementia A meta-analysis. Dement Neuropsychol 12: , 143–151. |
[9] | Lin Y-R , Chou L-C , Chen H-C , Liou T-H , Huang S-W , Lin H-W ((2016) ) Increased risk of dementia in patients with systemic lupus erythematosus: A nationwide population-based cohort study. Arthritis Care Res (Hoboken) 68: , 1774–1779. |
[10] | Liliang P-C , Liang C-L , Lu K , Yang S-N , Hsieh M-T , Tai Y-C , Wang K-W ((2018) ) Population-based study suggests an increased risk of Alzheimer’s disease in Sjögren’s syndrome. Clin Rheum 37: , 935–941. |
[11] | Pasoto SG , Martins VAO , Bonfa E ((2019) ) Sjögren’s syndrome and systemic lupus erythematosus: Links and risks. Open Access Rheum Res Rev 11: , 33–45. |
[12] | Trzeciak P , Herbet M , Dudka J ((2021) ) Common factors of Alzheimer’s disease and rheumatoid arthritis— pathomechanism and treatment. Molecules 26: , 6038. |
[13] | Masi G , Annunziata P ((2016) ) Sjögren’s syndrome and multiple sclerosis: Two sides of the same coin? . Autoimm Rev 15: , 457–461. |
[14] | Tseng C-C , Yen J-H , Tsai W-C , Ou T-T , Wu C-C , Sung W-Y , Hsieh M-C , Chang S-J ((2015) ) Increased incidence of Sjogren’s syndrome in systemic sclerosis: Anationwide population study. Autoimmunity 48: , 438–44. |
[15] | Hsu H-C , Hou T-Y , Lin T-M , Chang Y-S , Chen W-S , Kuo P-I , Lin Y-C , Chang C-C , Chen J-H ((2020) ) Higher risk of Parkinson disease in patients with primary Sjögren’s syndrome. Clin Rheumatol 39: , 2999–3007. |
[16] | Calingasan NY , Uchida K , Gibson GE ((1999) )Proteinbound acrolein: A novel marker of oxidative stress in Alzheimer’s disease. J Neurochem 72: , 751–756. |
[17] | Higashi K , Yoshida M , Igarashi A , Ito K , Wada Y , Murakami S , Kobayashi D , Nakano M , Sohda M , Nakajima T , Narita I , Toida T , Kashiwagi K , Igarashi K ((2010) ) Intense correlation between protein-conjugated acrolein and primary Sjögren’s syndrome. Clin Chim Acta 411: , 359–363. |
[18] | Li JZ , Steinman CR ((1989) ) Plasma DNA in systemic lupus erythematosus. Characterization of cloned base sequences. Arthritis Rheum 32: , 726–733. |
[19] | Larsen PA , Hunnicutt KE , Larsen RJ , Yoder AD , Saunders AM ((2018) ) Warning SINEs: Alu elements, evolution of the human brain, and the spectrum of neurological disease. Chromosome Res 26: , 93–111 . |
[20] | Rubin LH , Sundermann EE , Moore DJ ((2019) ) The current understanding of overlap between characteristics of HIV-associated neurocognitive disorders and Alzheimer’sdisease. J Neurovirol 25: , 661–672. |
[21] | Padron GT , Hernandez-Trujillo VP ((2023) ) Autoimmunity in primaryimmunodeficiencies (PID). Clin Rev Allerg Immunol 65: , 1–18. |
[22] | Brooks WH , Renaudineau Y ((2015) ) Epigenetics and autoimmune diseases: The Xchromosome-nucleolus nexus. Front Genet 16: , 22–41. |
[23] | Igarashi K , Kashiwagi K ((2019) ) The functional role of polyamines in eukaryotic cells. Int J Biochem Cell Biol 107: , 104–115. |
[24] | Goyns MH ((1981) ) Relationship between polyamine accumulation and RNA biosynthesis and content during the cell cycle. Experientia 37: , 34–35. |
[25] | Igarashi K , Kashiwagi K ((2010) ) Modulation of cellular function by polyamines. Int J Biochem Cell Biol 42: , 39–51. |
[26] | Xaplanteri MA , Petropoulos AD , Dinos GP , Kalpaxis DL ((2005) ) Localization of spermine binding sites in 23S rRNA by photoaffinity labeling: Parsing the spermine contribution to ribosomal 50S subunit functions. Nucleic Acids Res 33: , 2792–2805. |
[27] | Jovine L , Djordjevic S , Rhodes D ((2000) ) The crystal structure of yeast phenylalanine tRNA at 2.0 A resolution: Cleavage by Mg(2+) in 15-year old crystals. J Mol Biol 301: , 401–414. |
[28] | Lee Y-H , Ren D , Jeon B , Liu H-W ((2023) ) S-adenosylmethionine: More than just a methyl donor. Nat Prod Rep 40: , 1521–1549. |
[29] | Wang Y , Yang H , Geerts C , Furtos A , Waters P , Cyr D , Wang S , Mitchell GA ((2023) ) The multiple facets of acetyl-CoA metabolism: Energetics, biosynthesis, regulation, acylation and inborn errors. Mol Genet Metab 138: , 106966. |
[30] | Gfeller E , Stern DN , Russell DH , Levy CC , Taylor RL ((1972) ) Ultrastructural changes of rat liver nucleoli in response to polyamines. Z Zellforsch Mikrosk Anat 129: , 447–454. |
[31] | Whelly SM ((1991) ) Role of polyamines in the regulation of RNA synthesis in uterine nucleoli. J Steroid Biochem Mol Biol 39: , 161–167. |
[32] | D’Agostino L , Di Pietro M , DiLuccia A ((2006) ) Nuclear aggregates of polyamines. IUBMB Life 58: , 75–82. |
[33] | Brooks WH ((2013) ) Increased polyamines alter chromatin and stabilize autoantigens in autoimmune diseases. Front Immunol 4: , 91. |
[34] | Smirnova OA , Isaguliants MG , Hyvonen MT , Keinanen TA , Tunitskaya VL , Vepsalainen J , Alhonen L , Kochetkov SN , Ivanov AV ((2012) ) Chemically induced oxidative stress increases polyamine levels by activating the transcription of ornithine decarboxylase and spermidine/spermine-N1-acetyltransferase in human hepatoma HUH7 cells. Biochimie 94: , 1876–1883. |
[35] | Morrison JD , Smith DD , Kish SJ ((1996) ) Brain S-adenosylmethionine levels are severely decreased in Alzheimer’s disease. J Neurochem 67: , 1328–1331. |
[36] | Lin DW , Chung BP , Kaiser P ((2014) ) S-adenosylmethionine limitation induces p38 mitogen-activated protein kinase and triggers cell cycle arrest in G1. J Cell Sci 127: , 50–59. |
[37] | Wallin C , Hiruma Y , Wärmländer SKTS , Huvent I , Jarvet J , Abrahams JP , Gräslund , Lippens G , Luo J ((2018) ) The neuronal tau protein blocks fibrillation of theamyloid-β (Aβ) peptide at the oligomeric stage. J Am Chem Soc 140: , 8138–8146. |
[38] | Min S-W , Cho S-H , Zhou Y , Schroeder S , Haroutunian V , Seeley WW , Huang EJ , Shen Y , Masliah E , Mukherjee C , Meyers D , Cole PA , Ott M , Gan L ((2010) ) Acetylation of Tau inhibits its degradation and contributes to tauopathy. Neuron 67: , 953–966. |
[39] | Cohen TJ , Guo JL , Hurtado DE , Kwong LK , Mills IP , Trojanowski JQ , Lee VM ((2011) ) The acetylation of tau inhibits its function and promotes pathological tauaggregation. Nat Commun 2: , 252. |
[40] | Irwin DJ , Cohen TJ , Grossman M , Arnold SE , Xie SX , Lee VM , Trojanowski JQ ((2012) ) Acetylated tau, a novel pathological signature in Alzheimer’s disease and other tauopathies. Brain 135: , 807–818. |
[41] | Sandusky-Beltran LA , Kovalenko A , Placides DS , Ratnasamy K , Ma C , Hunt JB , Ma C , Hunt JB , Liang H , Calahatian JIT , Michalski C , Fahnestock M , Blair LJ , Darling AL , Baker JD , Fontaine SN , Dickey CA , Gamsby JJ , Nash KR , Abner E , Selenica MB , Lee DC ((2021) ) Aberrant AZIN2 and polyamine metabolism precipitates tau neuropathology. J Clin Invest 131: , e126299. |
[42] | Perez-Leal O , Barrero CA , Clarkson AB , Casero RA , Merali S ((2012) ) Polyamine-regulated translation of spermidine/spermine-N1 –acetyltransferase. Mol Cell Biol 32: , 1453–1467. |
[43] | Chaudhry U , Malik DA , Saleem N , Malik MT ((2018) ) Nucleolin: Role in bacterial and viral infections"EC Microbiol 14: , 631–640 . |
[44] | Jia W , Yao Z , Zhao J , Guan Q , Gao L ((2017) ) New perspectives of physiological and pathological functions of nucleolin (NCL). Life Sci 186: , 1–10. |
[45] | Stewart TM , Dunston TT , Woster PM , Casero RA ((2018) ) Polyamine catabolism and oxidative damage. J Biol Chem 293: , 18736–18745. |
[46] | Hansmannel F , Sillaire A , Kamboh MI , Lendon C , Pasquier F , Hannequin D , Laumet G , Mounier A , Ayral AM , DeKosky ST , Hauw JJ , Berr C , Mann D , Amouyel P , Campion D , Lambert JC ((2010) ) Is the urea cycle involved in Alzheimer’s disease? . J Alzheimers Dis 21: , 1013–1021. |
[47] | Savarin P , Barbet A , Delga S , Hoshi V , Hamon L , Lefevre J , Nakib S , De Bandt J-P , Moinard C , Curmi PA , Pastré D ((2010) ) A central role for polyamines in microtubule assembly in cells. Biochem J 430: , 151–159. |
[48] | Luo J , Yu CH , Yu H , Borstnar R , Kamerlin SCL , Gräslund A , Abrahams JP , Wärmländer SKTS ((2013) ) Cellular polyamines promote amyloid-beta (Aβ) peptide fibrillation and modulate the aggregation pathways. ACS Chem Neurosci 4: , 454–462. |
[49] | Morrison LD , Kish SJ ((1995) ) Brain polyamine levels are altered in Alzheimer’s disease. Neurosci Lett 197: , 5–8. |
[50] | Nilsson JA , Keller UB , Baudino TA , Yang C , Norton S , Old JA , Nilsson LM , Neale G , Kramer DL , Porter CW , Cleveland JL ((2005) ) Targeting ornithine decarboxylase in Myc-induced lymphoma genesis prevents tumor formation. Cancer Cell 7: , 433–444. |
[51] | Makitie LT , Kanerva K , Polvikoski T , Paetau A , Andersson LC ((2010) ) Brain neurons express ornithine decarboxylase-activating antizyme inhibitor 2 with accumulation in Alzheimer’s disease. Brain Pathol 20: , 571–580. |
[52] | Mahajan UV , Varma VR , Griswold ME , Blackshear CT , An Y , Oommen AM , Varma S , Troncoso JC , Pletnikova O , O’Brien R , Hohman TJ , Legido-Quigley C , Thambisetty M ((2020) ) Dysregulation of multiple metabolic networks related to brain transmethylation and polyamine pathways in Alzheimer disease: A targeted metabolomic and transcriptomic study. PLoS Med 17: , e1003012. |
[53] | Polis B , Karasik D , Samson AO ((2021) ) Alzheimer’s disease as a chronic maladaptive polyamine stress response. Aging 13: , 10770–10795. |
[54] | Yatin SM , Yatin M , Varadarajan S , Ain KB , Butterfield DA ((2001) ) Role of spermine in amyloid beta-peptide-associated free radical-induced neurotoxicity. J Neurosci Res 63: , 395–401. |
[55] | Weiss T , Bernard R , Laube G , Rieck J , Eaton MJ , Skatchkov SN , Veh RW ((2023) ) Asverified with the aid of biotinylated spermine, the brain cannot take up polyamines from the bloodstream leaving it solely dependent on local biosynthesis. Biomolecules 13: , 1114. |
[56] | Rozov A , Zakharova Y , Vazetdinova A , Valiullina-Rakhmatullina F ((2018) ) The role of polyamine-dependent facilitation of calcium permeable AMPARs in short-term synaptic enhancement. Front Cell Neurosci 12: , 345. |
[57] | Ahmad Y , Boisvert FM , Gregor P , Cobley A , Lamond AI ((2009) ) NOPdb: Nucleolar Proteome Database— 2008 update. Nuc Acids Res 37: , D181–D184. |
[58] | Dubois ML , Boisvert FM (2016) The nucleolus: Structure and function. In The Functional Nucleus; Bazett-JonesDP, Gellaire G, eds. Springer International Publication, Cham, Switzerland, pp. 29-49. |
[59] | Cerqueira AV , Lemos B ((2019) ) Ribosomal DNA and the nucleolus as keystones of nuclear architecture, organization, and function. Trends Genet 35: , 710–723. |
[60] | Iarovaia OV , Minina EP , Sheval EV , Onichtchouk D , Dokudovskaya S , Razin SV , Vassetzky YS ((2019) ) Nucleolus: A central hub for nuclear functions. Trends Cell Biol 29: , 647–659. |
[61] | Boulon S , Westman BJ , Hutten S , Boisvert FM , Lamond AI ((2010) ) The nucleolus under stress. Mol Cell 40: , 216–227. |
[62] | Nemeth A , Langst G ((2011) ) Genome organization in and around the nucleolus. Trends Genet 27: , 149–156. |
[63] | Lyon MF ((2000) ) LINE-1 elements and X chromosome inactivation: A function for “junk” DNA? . Proc Natl Acad Sci U S A 97: , 6248–6249 . |
[64] | Ross MT , Grafham DV , Coffey AJ , Scherer S , McLay K , Muzny D , Platzer M , Howell GR , Burrows C , Bird CP , et al ((2005) ) The DNA sequence of the human X chromosome. Nature 434: , 325–337. |
[65] | Bost C , Arleevskaya MI , Brooks WH , Plaza S , Guerya JC , Renaudineau Y ((2022) ) Long non-coding RNA Xist contribution in systemic lupus erythematosus and rheumatoid arthritis. Clin Immunol 236: , 108937. |
[66] | Iulita MF , Chavez DG , Christensen MK , Tamayo NV , Plana-Ripoll O , Rasmussen SA , Figuls MR , Alcolea D , Videla L , Barroeta I , Benejam B , Altuna M , Padilla C , Pegueroles J , Fernandez S , Belbin O , Carmona-Iragui M , Blesa R , Lleó A , Bejanin A , Fortea J ((2022) ) Association of Alzheimer Disease with life expectancy in people with Down syndrome. JAMA Network Open 5: , e2212910. |
[67] | Reynolds WF , Maki RA (2021) The role of myeloperoxidase in neurodegenerative disease. In Mammalian Heme Peroxidases, 1st Edition, Hawkins C, Nauseef WM, eds. CRC Press, Boca Raton, pp. 249-274. |
[68] | Liu CC , Zhao N , Fu Y , Wang N , Linares C , Tsai CW , Bu G ((2017) ) ApoE4 accelerates early seeding of amyloid pathology. Neuron 96: , 1024–1032. |
[69] | Polesskaya O , Kananykhina E , Roy-Engel AM , Nazarenko O , Kulemzina I , Baranova A , Vassetsky Y , Myakishev-Rempel M ((2018) ) The role of Alu-derived RNAs in Alzheimer’s and other neurodegenerative conditions. Med Hypotheses 115: , 29–34. |
[70] | Deininger P ((2011) ) Alu elements: Know the SINEs. Genome Biol 12: , 236–247. |
[71] | Brouha B , Schustak J , Badge RM , Lutz-Prigge S , Farley AH , Moran JV , Kazazian HH Jr ((2003) ) Hot L1s account for the bulk of retrotransposition in the human population. Proc Natl Acad Sci U S A 100: , 5280–5285. |
[72] | Dewannieux M , Esnault C , Heidmann T ((2003) ) LINE-mediated retrotransposition of marked Alu sequences. Nat Genet 35: , 41–48. |
[73] | Akopian D , Shen K , Zhang X , Shan S ((2013) ) Signal recognition particle: An essential protein targeting machine. Annu Rev Biochem 82: , 693–721. |
[74] | Walters RD , Kugel JF , Goodrich JA ((2009) ) InvAluable junk: The cellular impact and function of Alu and B2 RNAs. IUBMB Life 61: , 831–837. |
[75] | Gu S , Yuan B , Campbell IM , Beck CR , Carvalho CMB , Nagamani SCS , Erez A , Patel A , Bacino CA , Shaw CA , Stankiewicz P , Cheung SW , Bi W , Lupski JR , ((2015) ) Alu-mediated diverse and complex pathogenic copy-number variants within human chromosome 17 at p13.3. Hum Mol Genetics 24: , 4061–4077. |
[76] | Grimwood J , Gordon L , Olsen A , Terry A , Schmutz J , Lamerdin J , Hellsten U , Goodstein D , Couronne O , Tran-Gyamfi M , Aerts A , Altherr M , Ashworth L , Bajorek E , Black S , Branscomb E , Caenepeel S , Carrano A , Caoile C , Chan YM , Christensen M , Cleland CA , Copeland A , Dalin E , Dehal P , Denys M , Detter JC , Escobar J , Flowers D , Fotopulos D , Garcia C , Georgescu AM , Glavina T , Gomez M , Gonzales E , Groza M , Hammon N , Hawkins T , Haydu L , Ho I , Huang W , Israni S , Jett J , Kadner K , Kimball H , Kobayashi A , Larionov V , Leem SH , Lopez F , Lou Y , Lowry S , Malfatti S , Martinez D , McCready P , Medina C , Morgan J , Nelson K , Nolan M , Ovcharenko I , Pitluck S , Pollard M , Popkie AP , Predki P , Quan G , Ramirez L , Rash S , Retterer J , Rodriguez A , Rogers S , Salamov A , Salazar A , She X , Smith D , Slezak T , Solovyev V , Thayer N , Tice H , Tsai M , Ustaszewska A , Vo N , Wagner M , Wheeler J , Wu K , Xie G , Yang J , Dubchak I , Furey TS , DeJong P , Dickson M , Gordon D , Eichler EE , Pennacchio LA , Richardson P , Stubbs L , Rokhsar DS , Myers RM , Rubin EM , Lucas SM ((2004) ) The DNA sequence and biology of human chromosome 19. Nature 428: , 529–535. |
[77] | Chen C , Gentles AJ , Jurka J , Karlin S ((2002) ) Genes, pseudogenes, and Alu sequence organization across human chromosomes 21 and 22. Proc Natl Acad Sci U S A 99: , 2930–2935. |
[78] | Chaudhuri AR , Nussenzweig A ((2017) ) The multifaceted roles of PARP1 in DNA repair and chromatin remodeling. Nat Rev Mol Cell Biol 18: , 610–621. |
[79] | Matveeva EA , Mathbout LF , Fondufe-Mittendorf YN ((2019) ) PARP1 is a versatile factor in the regulation of mRNA stability and decay. Sci Rep 9: , 3722. |
[80] | Zeng J , Libien J , Shaik F , Wolk J , Hernández AI ((2016) ) Nucleolar PARP1expression is decreased in Alzheimer’s disease: Consequences for epigenetic regulation of rDNA and cognition. Neural Plasticity 2016: , 8987928. |
[81] | Caudron-Herger M , Pankert T , Seiler J , Németh A , Voit R , Grummt I , Rippe K ((2015) ) Alu element-containing RNAs maintain nucleolar structure and function. EMBO J 34: , 2758–2774. |
[82] | Miller-Fleming L , Olin-Sandoval V , Campbell K , Raiser M ((2015) ) Remaining mysteries of molecular biology: The role of polyamines in the cell. J Mol Biol 427: , 3389–3406. |
[83] | Krasnoslobodtsev AV , Peng J , Asiago JM , Hindupur J , Rochet J-C , Lyubchenko YL ((2012) ) Effect of spermidine on misfolding and interactions of alpha-synuclein. PLoS One 7: , e38099. |
[84] | Saiki S , Sasazawa Y , Fujimaki M , Kamagata K , Kaga N , Taka H , Li Y , Souma S , Hatano T , Imamichi Y , Furuya N , Mori A , Oji Y , Ueno SI , Nojiri S , Miura Y , Ueno T , Funayama M , Aoki S , Hattori N ((2019) ) A metabolic profile of polyamines in Parkinson disease: A promising biomarker. Ann Neurol 86: , 251–263. |
[85] | Bowie D ((2018) ) Polyamine-mediated channel block of ionotropic glutamate receptors and its regulation by auxiliary proteins. J Biol Chem 293: , 18789–18802. |
[86] | Brooks WH ((2012) ) Autoimmune diseases and polyamines. Clin Rev Allerg Immunol 42: , 58–70. |
[87] | Liu F , Zhang Z , Zhang L , Meng RN , Gao J , Jin M , Li M , Wang XP ((2022) ) Effect of metal ions on Alzheimer’s disease. Brain Behav 12: , e2527. |
[88] | Megur A , Baltriukienė D , Bukelskienė V , Burokas A ((2020) ) The microbiota-gut-brain axis and Alzheimer’s disease: Neuroinflammation Is to blame? . Nutrients 13: , 37. |
[89] | Eimer WA , Vijaya Kumar DK , Navalpur Shanmugam NK , Rodriguez AS , Mitchell T , Washicosky KJ , György B , Breakefield XO , Tanzi RE , Moir RD ((2018) ) Alzheimer’s disease-associated β-amyloid Is rapidly seeded by herpesviridae to protect against brain Infection. Neuron 99: , 56–63.e3. |
[90] | Johnson NH , Vaccari JP , Bramlett HM , Keane RW , Dietrich WD ((2023) ) Inflammasome activation in traumatic brain injury and Alzheimer’s disease. Transl Res 254: , 1–12. |
[91] | Das N , Raymick J , Sarkar S ((2021) ) Role of metals in Alzheimer’s disease. Metab Brain Dis 36: , 1627–1639. |
[92] | Watad A , Sharif K , Schoenfeld Y ((2017) ) The ASIA syndrome: Basic concepts. Mediterr J Rheumatol 28: , 64–69. |
[93] | Baroli G , Sanchez JR , Agostinelli E , Mariottini P , Cervelli M ((2020) ) Polyamines: The possible missing link between mental disorders and epilepsy (Review). Intl J Mol Med 45: , 3–9. |
[94] | Firpo MR , Mounce BC ((2020) ) . Diverse functions of polyamines in virus infection. Biomolecules 10: , 628. |
[95] | Takei M , Kitamura N , Nagasawa Y , Tsuzuki H , Iwata M , Nagatsuka Y , Nakamura H , Imai K , Fujiwara S ((2022) ) Are viral infections key inducers of autoimmune diseases? Focus on Epstein–Barr virus. Viruses 14: , 1900. |
[96] | Houen G , Trier NH , ((2021) ) Epstein-Barr virus and systemic autoimmune diseases. Front Immunol 11: , 587380. |
[97] | Bajaj BG , Murakami M , Cai Q , Verma SC , Lan K , Robertson ES ((2008) ) Epstein-Barr Virus nuclear antigen 3C interacts with and enhances the stability of the c-Myconcoprotein. J Virology 82: , 4082–4090. |
[98] | Schlosser I , Hölzel M , Mürnseer M , Burtscher H , Weidle UH , Eick D ((2003) ) A role for c-Myc in the regulation of ribosomal RNA processing. Nucleic Acids Res 31: , 6148–6156. |
[99] | Bello-Fernandez C , Packham GG , Cleveland JL ((1993) ) The ornithine decarboxylase gene is a transcriptional target of c-Myc. Proc Natl Acad Sci U S A 90: , 7804–7808. |
[100] | Dang CV ((1999) ) c-Myc targets genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol 19: , 1–11. |
[101] | Watt KE , Macintosh J , Bernard G , Trainor PA ((2023) ) RNA polymerase I and III indevelopment and disease. Sem Cell Dev Biol 136: , 49–63. |
[102] | Yeganeh M , Hernandez N ((2020) ) RNA polymerase III transcription as a disease factor. Genes Dev 34: , 865–882. |
[103] | Sutter SO , Lkharrazi A , Schraner EM , Michaelsen K , Meier AF , Marx J , Vogt B , Büning H , Fraefel C ((2022) ) Adeno-associated virus type 2 (AAV2) uncoating is a stepwise process and is linked to structural reorganization of the nucleolus. PLoS Pathogens 18: , e1010187. |
[104] | Volkman HE , Cambier S , Gray EE , Stetson DB ((2019) ) Tight nuclear tethering of cGAS is essential for preventing autoreactivity. eLife 8: , e47491. |
[105] | Han W , Du C , Zhu Y , Ran L , Wang Y , Xiong J , Wu Y , Lan Q , Wang Y , Wang L , Wang J , Yang K , Zhao J ((2022) ) Targeting myocardial mitochondria-STING-polyamine axis prevents cardiac hypertrophy in chronic kidney disease. JACC Basic Transl Sci 7: , 820–840. |
[106] | Carrel L , Park C , Tyekucheva S , Dunn J , Chiaromonte F , Makova KD ((2006) ) Genomic environment predicts expression patterns on the human inactive X chromosome. PLoS Genet 2: , e151. |
[107] | Satoh M , Ceribelli A , Hasegawa T , Tanaka S ((2022) ) Clinical significance of antinucleolar antibodies: Biomarkers for autoimmune diseases, malignancies, and others. Clin Rev Allergy Immunol 63: , 210–239. |
[108] | Sibley JT , Lee JS , Decoteau WE ((1984) ) Left-handed “Z” DNA antibodies in rheumatoid arthritis and systemic lupus erythematosus. J Rheumatol 11: , 633–637. |
[109] | Thomas TJ , Thomas T ((1994) ) Polyamine-induced Z-DNA conformation in plasmids containing (dA-dC)n.(dG-dT)n inserts and increased binding of lupus autoantibodies to the Z-DNA form of plasmids. Biochem J 298: , 485–491. |
[110] | Brooks WH ((2017) ) Viral impact in autoimmune diseases: Expanding the “Xchromosome-nucleolus nexus” hypothesis. Front Immunol 8: , 1657–71. |
[111] | Brooks WH ((2018) ) Review of the “X chromosome-nucleolus nexus” hypothesis of autoimmune diseases with an update explaining disruption of the nucleolus. Immunologic Res 66: , 790–799. |
[112] | Brooks WH ((2020) ) An epigenetics-based hypothesis of auto antigen development in systemic lupus erythematosus. Epigenomes 4: , 6. |
[113] | Hernández-Ortega K , Garcia-Esparcia P , Gil L , Lucas JJ , Ferrer I ((2016) ) Alteredmachinery of protein synthesis in Alzheimer’s: From the nucleolus to the ribosome.. Brain Pathol 26: , 593–605. |
[114] | Nyhus C , Pihl M , Hyttel P , Hall VJ ((2019) ) Evidence for nucleolar dysfunction in Alzheimer’s disease. Rev Neurosci 30: , 685–700. |
[115] | Inoue K , Tsutsui H , Akatsu H , Hashizume Y , Matsukawa N , Yamamoto T , Toyo’oka T ((2013) ) Metabolic profiling of Alzheimer’s disease brains. Sci Rep 3: , 2364. |
[116] | Joaquim HPG , Costa AC , Forlenza OV , Gattaz WF , Talib LL ((2019) ) Decreased plasmatic spermidine and increased spermine in mild cognitive impairment and Alzheimer’s disease patients. Arch Clin Psychiatry 46: , 120–4. |
[117] | Ivanov SM , Atanasova M , Dimitrov I , Doytchinova IA ((2020) ) Cellular polyamines condense hyperphosphorylated Tau, triggering Alzheimer’s disease. Sci Rep 10: , 10098. |
[118] | Ghosh I , Sankhe R , Mudgal J , Arora D , Nampoothiri M ((2020) ) Spermidine, an autophagy inducer, as a therapeutic strategy in neurological disorders. Neuropeptides 83: , 102083. |
[119] | Frühauf-Perez PK , Temp FR , Pillat MM , Signor C , Wendel AL , Ulrich H , Mello CF , Rubin MA ((2018) ) Spermine protects from LPS-induced memory deficit via BDNF and TrkB activation. Neurobiol Learn Mem 149: , 135–143. |
[120] | Ni YQ , Liu YS ((2021) ) New insights into the roles and mechanisms of spermidine inaging and age-related diseases. Aging Dis 12: , 1948–1063. |
[121] | Tiwari D , Singh VK , Baral B , Pathak DK , Jayabalan J , Kumar R , Tapryal S , Jha HC ((2021) ) Indication of neurodegenerative cascade initiation by amyloid-like aggregate-forming EBV proteins and peptide in Alzheimer’s disease. ACS Chem Neurosci 12: , 3957–3967. |
[122] | Harley JB , Chen X , Pujato M , Miller D , Maddox A , Forney C , Magnusen AF , Lynch A , Chetal K , Yukawa M , Barski A , Salomonis N , Kaufman KM , Kottyan LC , Weirauch MT ((2018) ) Transcription factors operate across disease loci, with EBNA2 implicated in autoimmunity. Nat Genet 50: , 699–707. |
[123] | Janjetovic S , Hinke J , Balachandran S , Akyüz N , Behrmann P , Bokemeyer C , Dierlamm J , Penas EMM ((2022) ) Non-random pattern of integration for Epstein-Barr virus with preference for gene-poor genomic chromosomal regions into the genome of Burkitt lymphoma cell lines. Viruses 14: , 86. |
[124] | Kanda T (2018) EBV-encoded latent genes. In Human Herpesviruses. Advances inExperimental Medicine and Biology, vol 1045. Kawaguchi Y, Mori Y, Kimura H, eds. Springer, Singapore, pp. 377-394. |
[125] | Zhu X , Rottkamp CA , Boux H , Takeda A , Perry G , Smith MA ((2000) ) Activation of p38 kinase links tau phosphorylation, oxidative stress, and cell cycle-related events in Alzheimer disease. J Neuropathol Exp Neurol 59: , 880–888. |
[126] | Salvi M , Toninello A ((2004) ) Effects of polyamines on mitochondrial Ca2 + transport. Biochim Biophys Acta 1661: , 113–124. |
[127] | Lee JK , Kim N-J ((2017) ) Recent advances in the inhibition of p38 MAPK as a potential strategy for the treatment of Alzheimer’s disease. Molecules 22: , 1287. |
[128] | Pekar T , Wendzel A , Jarisch R , ((2024) ) The positive effect of spermidine in older adults suffering from dementia after 1 year. Wien Klin Wochenschr 136: , 64–66. |
[129] | Gutierrez GEJ , Jiménez FVB , Muñoz LG , Guerrero YST , Melo NMM , Luna JMC , Garcia NL , Martínez JC , Magaña JJ ((2023) ) The molecular role of polyamines in age-related diseases: An update. Int J Mol Sci 24: , 16469. |
[130] | Freitag K , Sterczyk N , Wendlinger S , Obermayer B , Schulz J , Farztdinov V , Mülleder M , Ralser M , Houtman J , Fleck L , Braeuning C , Sansevrino R , Hoffmann C , Milovanovic D , Sigrist SJ , Conrad T , Beule D , Heppner FL , Jendrach M ((2022) ) Spermidine reduces neuroinfammation and soluble amyloid beta in an Alzheimer’s disease mouse model. J Neuroinflamm 19: , 172. |
[131] | Hofer SJ , Liang YT , Zimmermann A , Schroeder S , Dengiel J , Kroemer G , Eisenberg T , Sigrist SJ , Madeo F ((2021) ) Spermidine-induced hypusination preserves mitochondrial and cognitive function during aging. Autophagy 17: , 2037–2039. |
[132] | Hong J , Zhong L , Zheng Q , Wu Q , Zha Z , Wei D , Chen H , Zhang W , Zhang S , Huang Y , Chen K , Chen J , Li S , Zeng M-S , Zeng Y-X , Xia N , Zhang X , Xu M , Chen Y ((2022) ) A neutralizing antibody targeting gH provides potent protection against EBV challengein in vivo. J Virol 96: , e00075–22. |
[133] | Gupta M , Dhanasekaran AR , Gardiner KJ ((2016) ) Mouse models of Down syndrome: Gene content and consequences. Mamm Genome 27: , 538–555. |
[134] | Qian X , Song H , Ming G-L ((2019) ) Brain organoids: Advances, applications and challenges. , dev. Development 146: , 166074. |
[135] | Neidhart M , Karouzakis E , Jüngel A , Gay RE , Gay S ((2014) ) Inhibition of spermidine/spermine N1-acetyltransferase activity: A new therapeutic concept in rheumatoid arthritis. Arthritis Rheumatol 66: , 1723–1733. |
[136] | Alexiou GA , Lianos GD , Ragos V , Galani V , Kyritsis A ((2017) ) Difluoromethylornithine in cancer: New advances. Future Oncol 13: , 809–819. |
[137] | Zhang Y , Zheng Q , Zhou Y , Liu S ((2020) ) Repurposing clinical drugs as AdoMetDC inhibitors using the SCAR strategy. Front Pharmacol 11: , 248. |
[138] | Denny WA ((2022) ) Inhibitors and activators of the p38 mitogen-activated MAP kinase (MAPK) family as drugs to treat cancer and inflammation. Curr Cancer Drug Targets 22: , 209–220. |
[139] | Chen X-Y , Du Y-F , Chen L ((2019) ) Neuropeptides exert neuroprotective effects in Alzheimer’s disease. Front Mol Neurosci 11: , 493. |

