
Advanced Alzheimer’s Disease Patients Show Safe, Significant, and Persistent Benefit in 6-Month Bryostatin Trial
Abstract
Background:
In pre-clinical studies, Bryostatin, MW (molecular weight) 904, has demonstrated synaptogenic, anti-apoptotic, anti-amyloid, and anti-tau tangle efficacies.
Objective:
To identify AD patients who show significant cognitive benefit versus placebo when treated in a trial with chronic Bryostatin dosing.
Methods:
In this 6-month 122 AD patient Bryostatin trial, there were two cohorts: the Moderate Cohort (MMSE, Mini-Mental Status Exam: 15-18) and the Moderately Severe Cohort (MMSE 10-14) as pre-specified secondary endpoints. Patient randomization was stratified by baseline SIB to insure balance in baseline cognitive ability between treatment arms.
Results:
With no safety events noted by the data safety and monitoring board, the Moderately Severe (MMSE 10-14) Bryostatin-treated patients were significantly improved above the placebo patients for Weeks #13 through Week #42. After two cycles of 7 x i.v. Bryostatin doses over a 26-week period, the 10-14 Cohort Severe Impairment Battery (SIB), measured every 2 weeks, showed significant benefit using a Mixed Model Repeated Measures model (MMRM, 2-tailed, p < 0.05) for Weeks #13 through #42, even 16 weeks after dosing completion by Week #26. Placebo 10-14 patients showed no benefit, declining to negative 12.8 points by Week #42. Trend analyses confirmed the MMRM data for this Cohort, with a significant downward slope (equivalent to Cognitive Decline) for the placebo group, p < 0.001, 2-tailed, but no significant decline for the Bryostatin-treated group (p = 0.409, NS), treatment versus placebo p < 0.007. The Moderate Cohort patients showed no significant benefit.
Conclusions:
The Bryostatin-treated MMSE 10-14 patients showed no significant cognitive decline throughout the 10-month trial, versus placebo patients’ decline of -12.8 SIB points.
INTRODUCTION
Alzheimer’s disease (AD) has been defined [1, 2] by the National Institute of Neurological Disorders and Stroke (NINDS of the National Institutes of Health) as requiring three “Gold Standard” criteria: 1) Dementia in life (<26/30 Mini-Mental Status Exam (MMSE) score), 2) Amyloid plaques at autopsy, and 3) neurofibrillary tangles at autopsy.
The broad range of AD risk factors such as genetics, age, diabetes, and hypertension, suggests that multiple pathologic pathways lead to AD neurodegeneration, as defined by these accepted NINDS criteria. There are also many diverse types of AD co-morbidity at autopsy, such as AD comorbid with multi-infarct dementia, Parkinson’s disease, frontal lobe disease, and/or Lewy body disease. This diversity of co-morbidity also suggests that a complex sequence of etiologic pathophysiologic steps can lead to diverse combinations of degenerative disorders. AD, nevertheless, is the most common cause of dementia for increasingly aged populations worldwide—notwithstanding the complexity of etiology and co-morbidity. Given these complexities, it is not surprising that definitive diagnosis and treatment of AD have been notoriously difficult to conclusively achieve.
What are not included in the criteria that define AD are some of the important pathologic consequences of the degeneration, such as the loss of the synapses and neurons in the brain. These consequences also cross diagnostic boundaries for neurodegeneration. Yet the loss of synapses is the single clearest pathologic correlation of cognitive decline [3]. The drug tested in the study presented here, Bryostatin, has among its primary, proposed mechanisms of action synaptogenesis, prevention and/or restoration of lost synapses, anti-apoptosis, prevention of neuronal death. While AD biomarkers have improved diagnostic accuracy, few have provided an unequivocal AD diagnosis, as ultimately validated with autopsy. One study that was validated with seventy autopsies for an AD biomarker [4] indicated that clinical diagnosis alone is highly inaccurate in the first several years of AD progression but becomes much more accurate after more than 4 years of AD progression. Clinical diagnosis alone of “probable AD,” as recommended by the NINCDS committee [1], requires both demonstrated dementia (e.g., MMSE < 26/30) as well as an observed or recorded period of cognitive deterioration.
The difficulties of conducting definitive therapeutic trials for AD are thought to arise, therefore, from 1) inaccurate diagnosis, 2) lack of safety and unwanted side-effects, and 3) lack of clinically significant efficacy at preventing cognitive decline over sufficient time intervals. For these reasons, a drug that unequivocally treats the underlying AD pathophysiology and progression has never been approved by the FDA for patients with clearly identified and advanced AD. Recently, some progress has been made with antibodies to treat mild cognitive impairment (MCI) patients with fewer patients for whom there was suggestion of early AD, although none of the latter diagnoses were validated with autopsies. These patients were shown to have positive PET imaging scans for brain amyloid. However, the majority of patients who were treated with these candidate antibody treatments (aducanumab, lecanemab, and donanemab) were MCI patients and showed efficacy for slowing the rate of cognitive decline by 22–30% but did not prevent the continued cognitive decline throughout the trial. In contrast to these patients, the majority of whom were not demented, patients with advanced AD (for example, with MMSE < 15) have, for the most part, not been the subject of successful therapeutic trials for many years. In fact, the conventional wisdom has attributed the failure of so many therapeutic trials, including with antibodies, to the lack of drug benefits for AD patients who have more serious AD pathology, including amyloid plaques, neurofibrillary tangles, and significant loss of brain synapses. By treating much less advanced AD pathology in MCI and early AD patients, it was thought, AD drugs could have greater benefit.
Because the Synaptogenix drug, Bryostatin, had shown clear pre-clinical (animal models) efficacy for generating newly mature synapses and efficacy for preventing neuronal death (anti-apoptosis), we hypothesized that Bryostatin might have efficacy for reversing the loss of synapses even in advanced AD patients [5, 6]. Given these potential synaptogenic and anti-apoptotic efficacies as well as the dearth of therapeutic candidates for advanced AD patients, the focus of the present study, as well as two previous pilot studies [7–9], was the development of an effective therapeutic to address this huge unmet medical need, consisting potentially of millions of advanced AD patients in the U.S. alone. On this basis, the major inclusion criteria for this recently completed trial, Study #204, consisted of the following:
1) MMSE scores in two Cohorts, 10-14/30 and 15-18/30, indicating moderately severe or moderate dementia;
2) A magnetic resonance imaging test showing no localized lesions such as tumor, stroke, or hydrocephalus;
3) No baseline medication with the glutamatergic blocker, Namenda (or any other glutamatergic blockers), within the past 90 days;
4) Absence of serious chronic heart, lung, and/or renal disorders and creatinine clearance values of > 45 gm/l;
5) No other proscribed medications such as major sedatives, acetaminophen, gabapentin, and/or valproic acid.
Patients in the trial reported here were at least 50 years old, lived as outpatients in the community and were considered, by the principal investigators at each site, to have probable AD according to the criteria of the Diagnostic and Statistical Manual of Mental Disorders, fifth edition (DSM-V) [2] and of the NINDS Committee [1].
Patients in two pilot Phase II trials, with MMSE scores from 10-15 (Moderately Severe Cohort), when treated with Bryostatin, showed significant improvement over baseline of the Severe Impairment Battery (SIB). A similar Moderately Severe Cohort (MMSE 10-14) was then included in the present study together with an additional Cohort with MMSE scores from 15-18 (Moderate Cohort). Responses to the SIB of both the Moderate Cohort and the Moderately Severe Cohort were then monitored throughout the 26-week period of dose administration and for up to 16 weeks after all dosing had been completed. The SIB has been considered by many to be the most accurate psychometric for advanced AD patients in past trials [10]. Standard of care administration of the glutamate blocker, memantine, was prohibited in the present 6-month trial because in the first 3-month trial, drug benefit was only observable in its absence [7, 8]. This finding was consistent with the interpretation that synaptogenesis, one of Bryostatin’s principle putative mechanisms of action, previously demonstrated in pre-clinical trials [5, 6, 11–13], was thought to depend on fully functional glutamatergic synapses (blocked by memantine).
As described below, the Moderate Cohort showed no significant benefit in this 6-month dosing trial, while, in contrast, the Moderately Severe Cohort patients, treated with Bryostatin, were protected from any significant cognitive decline throughout the dosing regimen even 4 months after all dosing had been completed (see below for further discussion). The Moderately Severe Cohort placebo patients, on the other hand, declined, on average, by 12.8 SIB score points over the 10 months of periodic measurement. The evidence below, therefore, suggests the possibility that with Bryostatin treatment, there was no cognitive deterioration for patients with advanced AD, even 4 months of completion of the drug regimen.
METHODS
The protocol followed here was the same as previously used in the two pilot Bryostatin trials [6] but was extended from 3 months to 6 months. The two previous trials included one cycle of Bryostatin (45 min. i.v. infusion) administered with 7 doses over an 11-week dosing regimen [7]. The present study included two cycles of the same 7 dosing-regimen, the cycles separated by a 4-week interval. Measurement of patient performance on the SIB psychometric was assessed at two-week intervals throughout these two dosing cycles and, in addition, at other weeks over a four- month period without any further dosing, ultimately reaching a 42-week SIB assessment. The psychometric measure MMSE was used for the initial enrollment of patients into two cohorts: Moderately Severe Cohort (MMSE 10-14) and Moderate Cohort (MMSE 15-18). The SIB was used to monitor cognitive performance throughout the trial. While there was not precise correspondence of the MMSE to the SIB for each patient at the beginning of the trial, as would be expected since SIB is more appropriate for advanced AD patients, there was a general correspondence of the two scales. Other psychometric measures and medical monitoring was as previously described for the two pilot phase II trials.
Statistical methods
All patients were enrolled, evaluated, and treated as previously described [7, 8]. Conditions of enrollment, including the absence of standard of care medication with memantine, were also previously described [7, 8]. Because a primary mechanism of action of Bryostatin for treating AD patients has been suggested to be synaptogenesis [5], based on extensive pre-clinical data, the need for functional glutamatergic synapses would be expected. This is based on the previously implicated involvement of such synapses in synaptogenesis and on the demonstrated regulation of such synapses by PKC epsilon–mediated regulation of these synapses [7].
A total of 221 patients were enrolled in the study, of whom 122 were randomly assigned (1 : 1): 61 to placebo and 61 to Bryostatin. The majority in both the placebo and Bryostatin groups completed the study (40 [65.6%] and 33 [54.1%] subjects, respectively). The most common reason for dropout from the study was withdrawal by subject (10 [16.4%] and 14 [23.0%] for the placebo and Bryostatin subjects, respectively). Comparable baseline SIB scores were regularly checked by an independent observer.
Once all eligibility criteria for the study were met, a patient was randomized via Interactive Response Technology. Patient randomization was stratified by baseline SIB to insure balance in baseline cognitive ability between treatment arms. The principal measure of cognitive function, the SIB has been widely accepted for its appropriate use in patients with advanced AD. Treated patients received i.v.-Bryostatin on a bi-weekly basis after two weekly “loading” doses on trial initiation.
Eligible subjects received 7 doses of Bryostatin (i.v., 20 μg) or matching placebo during the first 12 weeks. A second course of treatment consisting of 7 doses began 30 days after the final dose of the first treatment period. Cognitive tests were assessed at two-week intervals during the study and 30 days after the final dose of the study drug. The primary endpoint was the total SIB score assessment obtained at Week 28, following completion of two courses of treatment. Eligible subjects were stratified based on baseline SIB total scores and were randomized 1 : 1 to one of two treatment arms: 20 μg Bryostatin or placebo for twelve weeks, the first treatment period. The first two doses of study drug were a loading dose 20% higher (i.e., 24 μg) than the assigned dose and were administered one week apart. Thereafter, the assigned dose of 20 μg was commenced with the third dose and, thereafter, administered every other week. The second course of treatment was identical to the first, beginning 30 days after completion of the first 7 dose cycle. Other secondary tests, ongoing medical monitoring, etc., were also previously described.
Data was analyzed by the modified intention-to-treat paradigm, meaning that all patients who received at least one dose of study drug and who had at least one post-baseline SIB assessment were analyzed according to the treatment arm to which they were assigned. The primary efficacy analysis was based on the linear mixed model with repeated measures (MMRM) model. This model was implemented using the SAS Proc Mixed command with the repeated option as specified in the Statistical Analysis Plan (SAP). Correlation of repeated SIB measurements within person were accounted for by using the unstructured covariance matrix. The method of Kenward and Roger [14] was used in the inference of the fixed effects to adjust for small sample bias. Time of follow-up visit was treated as a linear and continuous predictor with the actual time of SIB assessment used (rounded to the nearest week) rather than the scheduled time of the SIB assessment. In addition, a time by treatment interaction and baseline SIB terms were included in the model. Treatment differences in the predicted SIB obtained from the MMRM analysis were assessed at Weeks 5, 9, 13, 15, 20, 24, 28, 30, and 42, with the treatment difference at 28 weeks considered as the primary endpoint. Initially, the MMRM models were applied to all patients, and then broken down by MMSE-2 strata. Exploratory trend analyses were also considered using the Stata 17.0 mixed command which compared the differences in slopes between the two treatment arms over all 42 weeks of the trial. In these, both the MMRM and trend analyses, missing data was assumed to be missing at random, and hence no missing SIB values were imputed.
Demographic background
The majority of subjects in the Safety Analysis Set were White (105 [89.7%] subjects) not Hispanic or Latino (89 [76.1%] subjects). The mean (SD) age was 73.9 (7.67) years, and the mean (SD) duration of AD diagnosis was 3.7 (2.00) years prior to screening. The mean (SD) MMSE-2 score was 14.6 (2.52) and the mean (SD) Rosen-modified Hachinski score was 0.7 (0.74) overall. Demographics and baseline characteristics were generally well balanced between groups. Demographics and baseline characteristics in the Full Analysis Set were similar to those in the Safety Analysis Set. The limited number of total patients, 122, did not allow greater population diversity, particularly during the COVID epidemic. This will be more carefully planned in subsequent trials.
RESULTS
Safety
The overall treatment emergent adverse event (TEAE) profile was similar between the placebo and Bryostatin groups. A similar number of subjects had treatment-related TEAEs in the placebo and Bryostatin groups (13 events in 8 [13.8%] subjects and 15 events in 10 [16.9%] subjects, respectively). Most TEAEs were not considered treatment related. There were no obvious treatment-related TEAEs and no fatal TEAEs. With higher doses of Bryostatin as were used in cancer trials, myalgia was occasionally observed. No myalgia or any other related side-effects were observed in the current trial. The lack of side-effects is, of course, important for the eventual clinical use of the drug.
There were 4 events of COVID-19 in 4 (6.8%) subjects in the Bryostatin group and 1 event of SARS-CoV-2 test positive in 1 (1.7%) subject in the placebo group. The events of COVID19 were considered mild, unlikely related to study drug, and had an outcome of recovered/resolved. Study drug dose was not changed for these events. The event of SARS-CoV-2 test positive was considered mild, unlikely related to study drug, and had an outcome of recovering/resolving. Study drug was interrupted for this event.
Of the two cohorts, only the Moderately Severe Cohort patients showed significant benefit for the Bryostatin-treatment versus the placebo patients. All patients were randomized with respect to treatment groups, with safety checked by an independent data safety and monitoring board. In the absence of significant benefit for the Moderate Cohort, statistics for the combination of both cohorts were not significant (see below for further discussion). For the Moderately Severe Cohort, however, statistics were significant for all SIB measurements taken from Week 13 throughout the trial and including Week 42.
As Fig. 1 demonstrates, the benefit of the Bryostatin-treatment versus placebo is apparent from the two lines determined by all the treatment points and all the placebo points. All these points were pre-specified in Study #204 Clinical Protocol as reported in clinicaltrials.gov. A subset of these same points was pre-specified in the SAP, or, alternatively, were determined, still closely adhering to the Study #204 Clinical Protocol, as exploratory or post-hoc end-points (see Fig. 1). Weeks 9, 15, 20, and 24 were pre-specified in the SAP as secondary endpoints. Weeks 13, 28, 30, and 42 were pre-specified in the Study #204 Clinical Protocol, but not in the SAP, and were, therefore, exploratory or post-hoc endpoints.
Fig. 1
Expected mean (±se) differences in SIB from baseline for patients in the Moderately Severe 10-14 Cohort as obtained by the MMRM model for all time point assessments; placebo (blue) and Bryostatin (red).
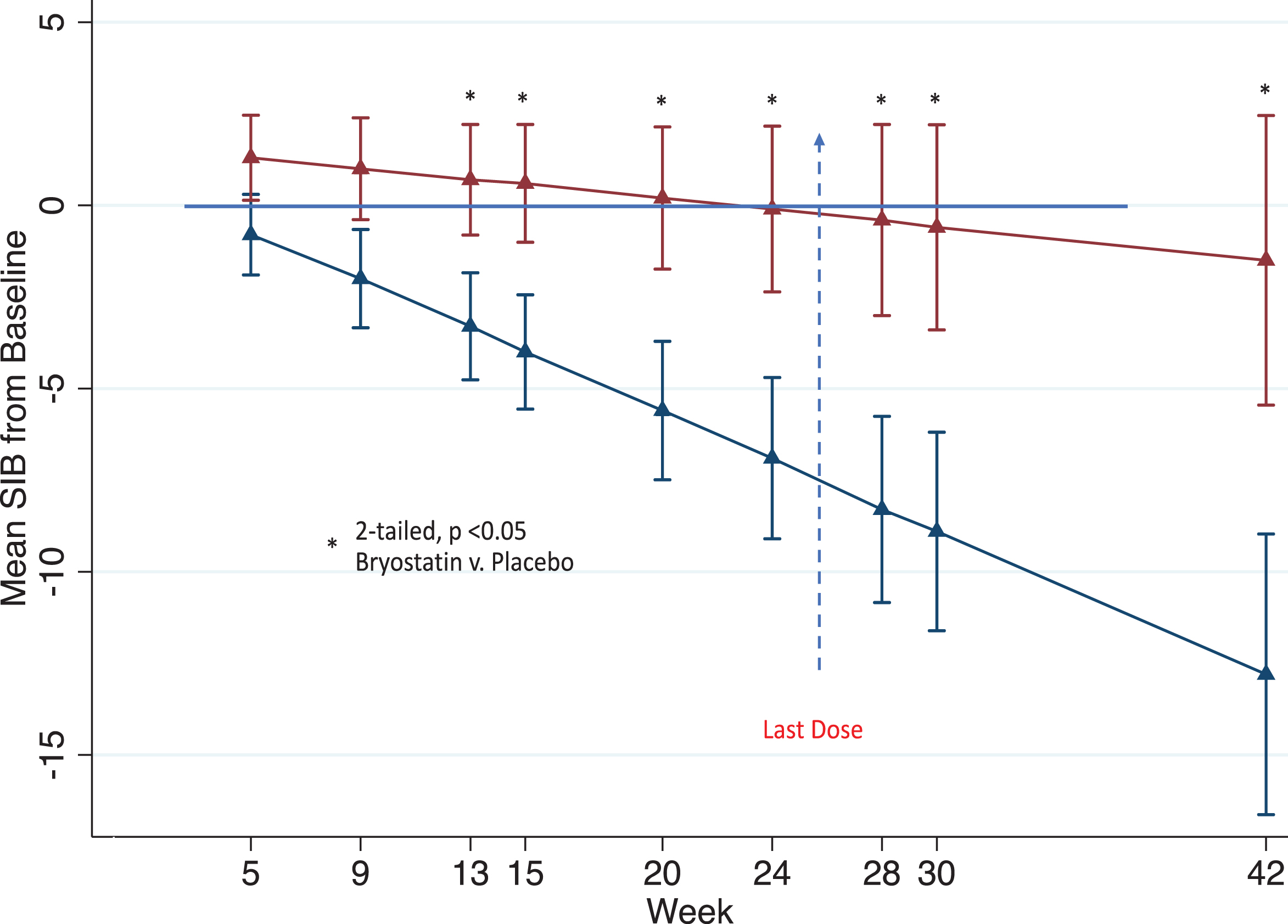
As shown in Fig. 1, there is a significant benefit of Bryostatin versus placebo that persisted at all weeks after the last dose was administered (vertical blue line). At each measurement point from Week 13 on, the treatment group was significantly greater than the placebo group as determined by a two-sided alpha < 0.05 level. In addition, the separation of the Bryostatin cognitive measures from placebo at early endpoints increased at end points in the last one-third of the trial.
Table 1 gives the estimated SIB means from baseline obtained from the MMRM model for each treatment arm as well as the difference in these means. Values are shown for Weeks 9, 15, 20, and 24 as pre-specified in the SAP as secondary endpoints as well as Weeks 13, 28, 30, and 42 as exploratory endpoints.
Table 1
Estimated least square (LS) mean SIBs from baseline and 95% confidence intervals as well as treatment differences in mean SIBs from baseline for the Moderately Severe MMSE 10-14 Cohort as determined by the MMRM model. Estimated means for Week 9 through Week 30 were determined by including follow-up times in the MMRM model up to scheduled visit at Week 30 as specified in the SAP. The mean estimates at Week 42 were obtained by including follow-up times to the time point in the MMRM model. The number of patients at each time point are those that had an SIB at the scheduled follow-up time points
| Baseline MMSE-2 score: 10-14 | |||
| Week | Statistics | Placebo | Bryostatin |
| Week 9 * | LS Mean (SE) | –2.6 (1.32) | 0.8 (1.37) |
| No. of Patients | N = 24 | N = 22 | |
| Difference (SE) | 3.4 (1.91) | ||
| 95% CI | (–0.5, 7.2) | ||
| p | 0.084 | ||
| Week 13** | LS Mean (SE) | –3.7 (1.42) | 0.5 (1.47) |
| No. of Patients | N = 23 | N = 21 | |
| Difference (SE) | 4.2 (2.05) | ||
| 95% CI | (0.1, 8.4) | ||
| p | 0.045 | ||
| Week 15* | LS Mean (SE) | –4.2 (1.51) | 0.4 (1.56) |
| No. of Patients | N = 22 | N = 21 | |
| Difference (SE) | 4.7 (2.18) | ||
| 95% CI | (0.3, 9.1) | ||
| p | 0.038 | ||
| Week 20* | LS Mean (SE) | –5.6 (1.81) | 0.1 (1.87) |
| No. of Patients | N = 21 | N = 18 | |
| Difference (SE) | 5.7 (2.61) | ||
| 95% CI | (0.5, 11.0) | ||
| p | 0.033 | ||
| Week 24* | LS Mean (SE) | –6.7 (2.11) | –0.1 (2.18) |
| No. of Patients | N = 20 | N = 18 | |
| Difference (SE) | 6.6 (3.04) | ||
| 95% CI | (0.5, 12.7) | ||
| p | 0.036 | ||
| Week 28** | LS Mean (SE) | –7.8 (2.44) | –0.4 (2.70) |
| No. of Patients | N = 19 | N = 18 | |
| Difference (SE) | 7.5 (3.51) | ||
| 95% CI | (0.4, 14.6) | ||
| p | 0.040 | ||
| Week 30** | LS Mean (SE) | –8.3 (2.62) | –0.4 (2.70) |
| No. of Patients | N = 18 | N = 16 | |
| Difference (SE) | 7.9 (3.76) | ||
| 95% CI | (0.3, 15.5) | ||
| p | 0.042 | ||
| Week 42** | LS Mean (SE) | –12.8 (3.83) | –1.5 (3.95) |
| No. of Patients | N = 16 | N = 11 | |
| Difference (SE) | 11.3 (5.51) | ||
| 95% CI | (0.17, 22.5) | ||
| p | 0.047 | ||
*Pre-specified in the SAP as secondary endpoints for the 10-14 Cohort. **Pre-specified in the Study Protocol, but not the SAP for the 10-14 Cohort.
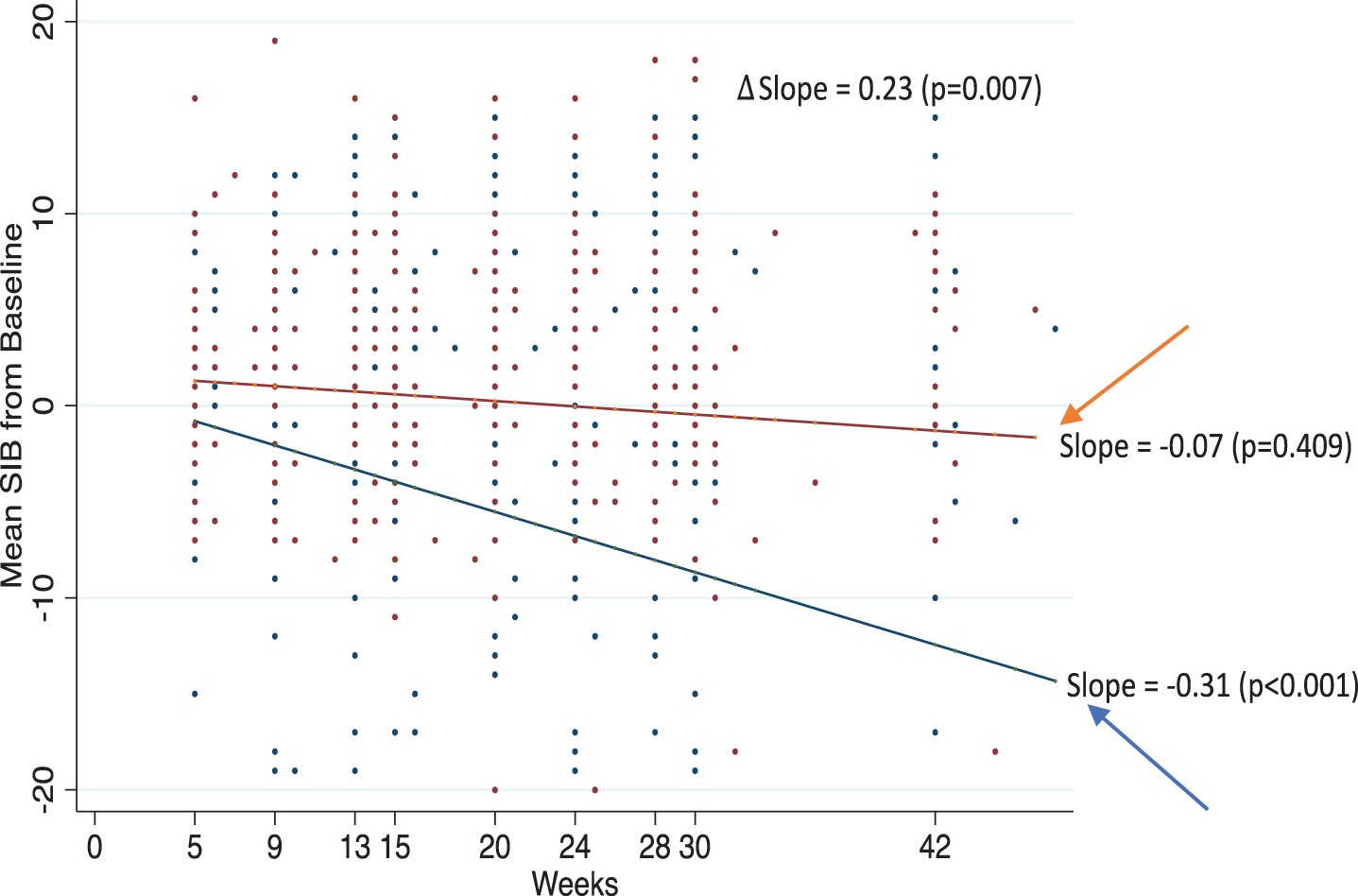
The post-hoc trend analysis as shown in Fig. 2 confirmed the MMRM data for the Moderately Severe Cohort (i.e., MMSE 10-14). This post-hoc trend analysis demonstrated a significant downward slope, equivalent to cognitive decline, for the placebo arm (p < 0.001), but no significant decline for the Bryostatin-treated group (p = 0.40). In this Post-hoc analysis of all points pre-specified in the Clinical Protocol, the difference in slopes between the Bryostatin-treatment group and the placebo was highly significant (p < 0.007). Placebo, but not Bryostatin Moderately Severe, patients showed significant SIB decline below baseline up to negative 12.8 points by Week 42.
Fig. 2
Trend analyses. Estimate slopes over time in mean SIB from baseline for Bryostatin (red) and placebo (blue) with actual data points given for patients in the Moderately Severe 10-14 Cohort. Twenty-five SIB values< -20 are not shown in the plot but were included in the trend analysis.
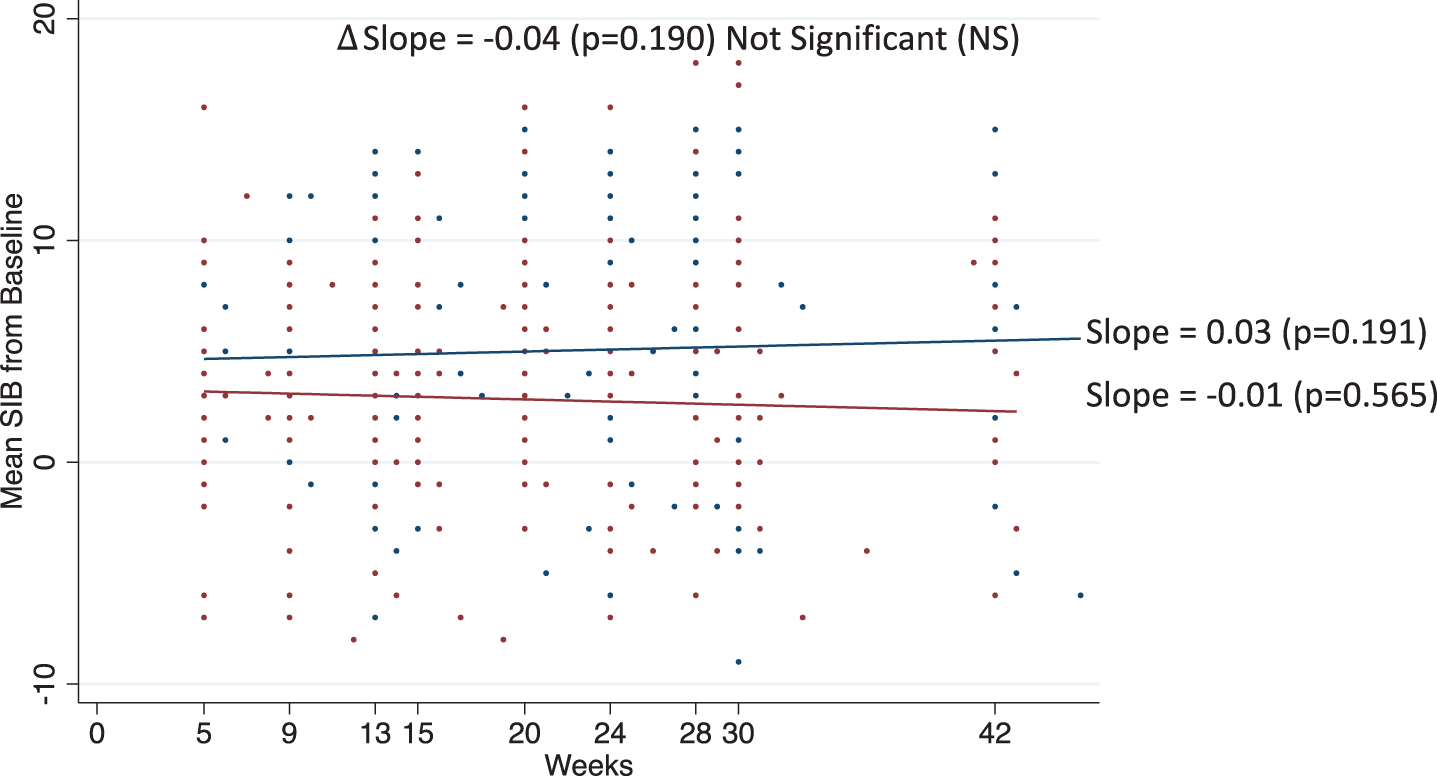
Results of the trend analyses for the Moderate 15-18 Cohort are given in Fig. 3. For this cohort, the trend in SIB scores over time was not statistically different from zero for either the Bryostatin-treated patients (p = 0.565) or the placebo (p = 0.191). In addition, there was no statistical difference in the slopes between treatment arms (p = 0.190).
Fig. 3
Trend analyses. Estimate slopes over time in mean SIB from baseline for Bryostatin (red) and placebo (blue) with actual data points given for patients in the Moderate10-14 Cohort.
Consistency with previous trials
The previous two Bryostatin trials, Study #202 and Study #203, also included Moderately Severe Patient Cohorts, but were not exact replications of Study #204. However, all three studies showed benefit for Severe AD Patients (10-14) at Week 13 which was included in all three trials. Pooling of Study #202 and 203 for patients with MMSE 10-14 (to offset the baseline imbalance of Study #203, and the resulting placebo benefit at early weeks in #203) showed that this improvement was significant with respect to placebo at p < 0.001, 2-tailed [8].
DISCUSSION
These data above demonstrated that Bryostatin-treated patients showed statistically significant improvement of cognitive performance (SIB) over placebo patients in the Moderately Severe Cohort for Weeks 13 through 42, with the last dose administered at Week 26. As mentioned above, while all the SIB endpoint data had been pre-specified in the Study Protocol, some of these had not been identified in the SAP and were thus considered to be post-hoc. Nevertheless, the integrated plots of all the SIB measurements (Figs. 1 and 2) illustrate a clear, consistent, and persistent benefit for the Moderately Severe MMSE 10-14 Cohort. Benefit after the final dose in the two pilot phase II studies were shown to persist at least 30 days beyond drug dosing. Here, the persistence of benefit at least 16 weeks beyond drug dosing suggests the possibility of a long-lasting change in the brain networks of the Moderately Severe Cohort patients. The putative mechanisms of action, synaptogenesis and anti-apoptosis, observed and implicated in extensive pre-clinical testing, are consistent with such persistent benefit of the Bryostatintreatment.
We can only speculate as to why there was no significant benefit in the Moderate MMSE 15-18 Cohort. Data analyses revealed that the placebo patients in the Moderate Cohort did not show progressive cognitive deterioration throughout the trial, but actually a 5-point SIB improvement (see Fig. 3). Those results suggest that some of the Moderate Cohort patients did not perform as would be expected and required for patients diagnosed to have “probable AD”—as defined by the NINDS criteria [12]. Increased duration beyond 10 months as conducted in the present trial might ultimately reveal some decline in placebo patients and possibly Bryostatin benefit in the ModerateCohort.
On the other hand, the Moderately Severe MMSE 10-14 Cohort placebo patients did show the expected progressive cognitive deterioration, reaching a deficit of negative 12.8 SIB points by Week 42. This deteriorating performance of the Moderately Severe Cohort placebo patients was entirely consistent with the NINDS criteria for the diagnosis of “probable AD”. In stark contrast to the 10-14 placebo patients, those 10-14 patients treated with Bryostatin showed no significant cognitive deterioration throughout the entire 10 months of the trialduration.
With improved diagnostic biomarkers, strict adherence to the NINDS clinical diagnostic criteria, and larger patient numbers, it may very well be possible to identify AD patients more accurately in the more moderate cohorts. It is worth noting that diagnosing AD by clinical criteria alone has been very difficult to achieve accurately during the first several years of progression, while clinical diagnosis of more advanced AD can be much more accurate [3]. In any case, the significant benefit demonstrated for the Moderately Severe Cohort does support the need for follow-up testing, with increased numbers of patients, possibly resulting in a confirmed therapeutic for these patients with advanced AD, for whom no disease-modifying drug is currently available. The absence of any significant cognitive decline for the Moderately Severe Cohort over the entire 10-month trial versus the clearly significant decline of the placebo patients suggests that Bryostatin may benefit those patients who have not been part of the recent trials with aducanumab, lecanemab, and donanemab. While these drugs reduced the rate of cognitive decline for MCI patients and putative early AD patients, Bryostatin shows the potential for complimentary benefits for Moderately Severe AD patients at a much later stage in disease progression. No such clear and sustained benefit for advanced AD patients has been previously observed in the many trials that have heretofore been conducted. Pre-clinical studies have demonstrated that Bryostatin causes significantly increased numbers of mature, mushroom-spine synapses [5] in AD transgenic mice and in mice exposed to global hypoxia and/or cerebral infarction. The prolonged absence of any significant cognitive decline in Bryostatin-treated patients versus placebo patients—even 16 weeks after the final dose of Bryostatin—suggests a long-lasting positive change in the treated patients’ brains. This appears to be consistent with the synaptogenic and anti-apoptotic efficacies, as well as Bryostatin’s anti-amyloid and anti-tau efficacies, of Bryostatin in the pre-clinical studies. With additional cycles of Bryostatin treatment, even further improvement in the advanced AD patients might be possible. We might also speculate that with more accurate AD diagnosis for earlier AD patients [15], who remain difficult to identify by clinical criteria alone, and with increased numbers of patient participants, Bryostatin may also demonstrate synaptogenic and anti-apoptotic efficacies earlier in AD disease progression aswell.
ACKNOWLEDGMENTS
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
FUNDING
Research reported in this publication was supported by the National Institute on Aging of the National Institutes of Health under Award Number R44AG066366 (MKS).
The study was sponsored by Synaptogenix, Inc.
CONFLICT OF INTEREST
Drs. Alkon, Sun, Tuchman, and Thompson are all paid consultants of the sponsoring company, Synaptogenix, Inc.
Dr. Alkon is also affiliated with SynapsDx, Inc.
Dr. Alkon is an Editorial Board Member of this journal but was not involved in the peer-review process nor had access to any information regarding its peer-review.’
DATA AVAILABILITY
The data supporting the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
[1] | McKhann GM , Knopman DS , Chertkow H , Hyman BT , Jack CR Jr , Kawas CH , Klunk WE , Koroshetz WJ , Manly JJ , Mayeux R , Mohs RC , Morris JC , Rossor MN , Scheltens P , Carrillo MC , Thies B , Weintraub S , Phelps CH ((2011) ) The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7: , 263–269. |
[2] | American Psychiatric Association ((2013) ) Diagnostic and Statistical Manual of Mental Disorders, 5th Edition: DSM-5. American Psychiatric Association, Washington DC. |
[3] | Terry RD , Masliah E , Salmon DP , Butters N , DeTeresa R , Hill R , Hansen LA , Katzman R ((1991) ) Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann Neurol 30: , 572–580. |
[4] | Khan TH , Alkon DL ((2006) ) An internally controlled peripheral biomarker for Alzheimer’s disease: Erk1 and Erk2 responses to the inflammatory signal bradykinin. Proc Natl Acad Sci U S A 103: , 13203–13207. |
[5] | Hongpaisan J , Sun MK , Alkon DL ((2011) ) PKC epsilon activation prevents synaptic loss, A Beta elevation, and cognitive deficit in Alzheimer’s disease transgenic mice. J Neurosci 31: , 630–643. |
[6] | Sen A , Alkon DL , Nelson TJ ((2012) ) ApoE3 but not ApoE4 protects against synaptic loss through increased expression of PKC epsilon. J Biol Chem 12: , 15947–15958. |
[7] | Farlow MR , Thompson RE , Wei LJ , Tuchman AJ , Grenier E , Crockford D , Wilke S , Benison J , Alkon DL ((2019) ) A randomized, double-blind, placebo-controlled, phase II study assessing safety, tolerability, and efficacy of Bryostatin in the treatment of moderately severe to severe Alzheimer’s disease. J Alzheimers Dis 67: , 555–570. |
[8] | Thompson RE , Tuchman AJ , Alkon DL ((2022) ) Bryostatin placebo-controlled trials indicate cognitive restoration above baseline for advanced Alzheimer’s disease in the absence of memantine. J Alzheimers Dis 86: , 1221–1229. |
[9] | Nelson TJ , Sun MK , Lim C , Sen A , Khan T , Chirila FV , Alkon DL ((2017) ) Bryostatin effects on cognitive function and PK in Alzheimer’s phase II and expanded access trials. J Alzheimers Dis 58: , 521–535. |
[10] | Reisberg B , Doody R , Stöffler A , Schmitt F , Ferris S , Möbius HJ ; Memantine Study Group ((2003) ) Memantine in moderate-to-severe Alzheimer’s disease. N Engl J Med 348: , 1333–1341. |
[11] | Nelson TJ , Cui C , Luo Y , Alkon DL ((2009) ) Reduction of beta-amyloid levels by novel PKC (epsilon) activators. J Biol Chem 284: , 34514–34521. |
[12] | Quattrone A , Pascale A , Nogues X , Zhao W , Gusev P , Pacini A , Alkon DL ((2001) ) Posttranscriptional regulation of gene expression in learning by the neuronal ELAV-like mRNA stabilizing proteins. Proc Natl Acad Sci U S A 98: , 11668–11673. |
[13] | Lim CS , Alkon DL ((2012) ) Protein kinase C stimulates HuD-mediated mRNA stability and protein expression of neurotrophic factors and enhances dendritic maturation of hippocampal neurons in culture. Hippocampus 22: , 2303–2319. |
[14] | Kenward MG , Roger JH ((1997) ) Small sample inference for fixed effects from restricted maximum likelihood. Biometrics 53: , 983–997. |
[15] | Chirila FV , Xu G , Fontaine D , Kern G , Khan TK , Brandt J , Konishi Y , Nebe-von-Caron G , White CL 3rd , Alkon DL ((2022) ) Morphometric imaging biomarker identifies Alzheimer’s disease even among mixed dementia patients. Sci Rep 12: , 17675. |


