
A Scoping Review of Alzheimers Disease Hypotheses: An Array of Uni- and Multi-Factorial Theories
Abstract
Background:
There is a common agreement that Alzheimers disease (AD) is inherently complex; otherwise, a general disagreement remains on its etiological underpinning, with numerous alternative hypotheses having been proposed.
Objective:
To perform a scoping review of original manuscripts describing hypotheses and theories of AD published in the past decades.
Results:
We reviewed 131 original manuscripts that fulfilled our inclusion criteria out of more than 13,807 references extracted from open databases. Each entry was characterized as having a single or multifactorial focus and assigned to one of 15 theoretical groupings. Impact was tracked using open citation tools.
Results:
Three stages can be discerned in terms of hypotheses generation, with three quarter of studies proposing a hypothesis characterized as being single-focus. The most important theoretical groupings were the Amyloid group, followed by Metabolism and Mitochondrial dysfunction, then Infections and Cerebrovascular. Lately, evidence towards Genetics and especially Gut/Brain interactions came to the fore.
Conclusions:
When viewed together, these multi-faceted reports reinforce the notion that AD affects multiple sub-cellular, cellular, anatomical, and physiological systems at the same time but at varying degree between individuals. The challenge of providing a comprehensive view of all systems and their interactions remains, alongside ways to manage this inherent complexity.
INTRODUCTION
After more than a century of research, Alzheimers disease (AD) remains perplexing, a thorn in the side of modern medical science. Its etiological underpinning remains under debate, with numerous alternative hypotheses having been proposed. None were confirmed to a level sufficient for its operationalization into a therapeutic approach unequivocally proven effective and efficient in a clinical setting. Surveying this landscape of ideas—some vibrantly pursued, others nearly forgotten—should provide insights as to future general directions for research.
One aspect commonly agreed upon is that AD is inherently complex. Its most common form is primarily associated with aging, and hence overlaps exist between the wide variability in anatomical and physiological changes that accompany the latter with those of the former, early in the disease process at the very least. Beyond aging but related to it, multiple risk factors have been identified from epidemiological studies, ranging from education in early life; hearing loss, traumatic brain injury, hypertension, alcohol consumption, and obesity at mid-life; to smoking, depression, social isolation, physical inactivity, air pollution, and diabetes, in later years [1]. Yet, the compounding factor of time renders difficult the determination of whether some of these factors are early disease markers, etiological agents, catalysts, or a combination of these roles. The sheer breadth of these identified risk constructs, with impacts at scales ranging from proteins to behaviors, further points to the complexity of uncovering disease pathways at the most fundamental levels—and therefore amenable to therapeutic approaches, pharmacological or otherwise—that compound into the observed pathological cognitive and behavioral presentations.
A common agreement on what constitutes AD seems therefore a first and necessary step to address this complexity and work together towards a solution. In fact, the discordance between the clinical presentation of AD and some of its most accepted biomarkers (e.g., amyloid-β (Aβ), present in AD but also prevalent at all ages without necessarily affecting cognition [2], and histopathological findings (e.g., similar lesions being associated with dementia or not [3]), means that the very definition of the disease remains a question as much of viewpoints as one of evidence. Thus, one could argue that there are three effective, overlapping yet still irreconciled definitions of AD: one arising from a clinical/cognitive viewpoint; a second, informed by \itin vivo biomarkers; and a third, defined by histopathological evidence.
An important aspect of this debate was introduced by Alois Alzheimer himself, when he first described the disease in his seminal paper [4], relating his observations on one patient, who had first come to the clinic at 51 years of age with clear clinical dementia. Based on the age of this patient and her rapid decline, it has been argued that this was a form of autosomal dominant AD. For the first time, Aβ plaques were observed and reported. Yet, this seminal observation has set the parameters of a major argument that still rages more than a century later: are Aβ plaques fundamental or accessory to the pseudo-sporadic (pseudo-, as some would argue that AD is determined by gene combinations and therefore not sporadic per se) form of the disease, form that is by far the most prevalent? Is AD an amyloidosis, first and foremost, in all cases—not only early in life, but in older patients as well? This amyloid hypothesis [5], which has been at the center of research for the past few decades, is the fields major attempt at a “great unifying theory. It also oriented minds into a paradigm often seen in medical research, the search for a single etiological cause, regardless of the complexities inherent to human biology. Consequently, this singular view of the disease set in motion a quest, so far with limited positive results, for a similarly singular cure. Yet, the amyloid hypothesis is now seriously under siege. While it never made unanimity, as theories are initially wont to be, significant drawbacks in the past few years have led many to look anew for alternatives. First is the high prevalence of amyloid deposition in otherwise cognitively healthy individuals [6] and the lack of association between amyloid deposition and transverse and longitudinal cognitive status in otherwise cognitively healthy individuals [7]. Next we can find the highly combinatorial nature of pathologies present (especially cerebrovascular lesions) when one looks at large post-mortem samples [8], meaning that amyloid is likely not the only etiology driving decline. Finally, the accumulated failures of tens of phase III trials focused on Aβ removal to reach meaningful clinical changes [9], even when compared to those few that succeeded with modest (but, to be fair, statistically significant) success [10]. While many see these last trials as vindication of the hypothesis, others notice how the equivocal results cast further doubts on its centrality, given significant levels of side effects [9] and lack of response in many sub-groups [11]. Given their efficacy at removing amyloid but restrained clinical outcomes, combinatorial approaches will likely be necessary [12], circling back to the same issue: what exactly constitutes AD?
Thus, to solve this riddle, a theoretical framework must be sufficiently complex and detailed to be able to explain these multiple events. Many have attempted to do so in the past. It is their work which is reviewed here (using the “Scoping Review frame), in an attempt first to enumerate, then to assess the relative importance of the diversity of hypotheses and theories that have been proposed in the past decades. We aimed to survey this wide array of ideas, listing all hypotheses, and reporting on their meta-characteristics. The sheer size of the literature precludes us from offering but the most cursory overview of each theoretical grouping, as well as delving into the evidence propose to rank, qualitatively or quantitatively, any one theory. Indeed, assessing the quality of evidence of even a single theory adequately deserves an entire manuscript. This being so, we draw some general conclusions to orient the field.
METHODS
Literature search strategy and sources
This review followed the PRISMA guidelines [13] and was conducted using the Web of Science, Embase, Medline, and PsycInfo databases for articles published from their inception through to 14 September 2023. Keywords referred to the variables of interest (AD theory). The search strategy is presented in Supplementary Table 1. Due to the sheer volume of work that has been published on AD, we limited the number of databases and search terms/synonyms. This qualifies our work as a scoping rather than systematic review.
Table 1
Theoretical groupings (Modified from Ambrose 2015)
| Grouping | Description | Studies |
| Amyloid | Abnormal production, accumulation, or removal of Aβ n-meric species and/or plaques | [5, 14–26] |
| Cerebrovascular and BBB | Vascular abnormalities affecting blood flow and perfusion, cerebrovascular lesions (infarcts, microbleeds, white matter hyperintensities), and BBB permeability | [27–38] |
| Genetics | Inherited genetic mutation(s), including APOE | [39–41] |
| Gut/brain axis | Dysbiosis in the gut microbiome | [42–45] |
| Infection | Protein misfolding in cells following infection by transmissible agents such as prion, subviral entities, HSV type 1 | [46–56] |
| Inflammation/Immunological response | Autoimmune response and inflammation | [57–63] |
| Metabolism | Cortical glucose utilization and transport across the BBB | [64–72] |
| Metals | Neurotoxicity (e.g., Al, Hg) or deficiency in trace elements (e.g., Fe, Cu, Se) | [73–80] |
| Mitochondrial dysfunction | Dysfunction in brain mitochondria leading to energy deficits and oxidative stress | [81–88] |
| Neurotransmitter | Deficits in cholinergic, noradrenergic, and other neurotransmitters | [89–93] |
| Oxidative stress | Accumulation of oxidative damage in the brain from free radicals | [94–101] |
| Proteinopathy | Tau, TDP43, and/or other proteins dysregulation | [102–105] |
| White matter disease | Degeneration and disruption of white matter connections | [106, 107] |
| Others | Calcium channel blockers; nerve growth factors; autophagy, neuronal cell cycle abnormalities | [108–141] |
BBB, blood-brain barrier.
Study selection process
Inclusion criteria were established as follows: (a) must be a peer-reviewed, scientific article; (b) must be written in either English or French; (c) must describe a hypothesis or theory related to AD; and (d) must not report solely on a pre-clinical model of AD.
Article selection was performed using the Covidence systematic review software (Veritas Health Innovation). After the initial search (L.S.R., S.D.), duplicates were removed. Next, a first sort was performed based on article titles and abstracts, followed by a second sort based on the full-text articles. All articles were evaluated against the inclusion and exclusion criteria, and some articles were excluded accordingly. The screening was made by independent reviewers (L.S.R.; F.B.; L.A.W.; B.C.; M.A.; V.L.; S.D.; S.M.P.). Articles that were not included by reviewers were reevaluated by a third and included or excluded by common agreement.
Study selection
On 14 September 2023, 13,807 studies were uploaded to Covidence from Embase (n = 4,787), Medline (n = 1,159), Web of Science (n = 4,240), and PsycInfo (n = 1,321) databases. Of these studies, 4,784 duplicates were removed. Three reviewers (L.S.R.; F.B.; S.D.) screened the remaining 9,022 titles and abstracts according to the inclusion and exclusion criteria. The intervention of a third independent reviewer was not required. A total of 8,398 references were excluded. Full-text screening of the 609 articles led to the exclusion of 466 articles after review by seven reviewers (L.S.R., L.A.W., B.C., M.A., S.D., S.M.P., V.L.). The excluded articles were either not describing a model (172), duplicates (86), described a non-etiological model (71), were proceedings (38) or non-peer reviewed articles (30), were in another language than English or French (23), were not applicable to AD (24), were inaccessible (12), were only concerned with the familial form of AD or Down syndrome (3), were not relevant for clinical (i.e. in humans) applications (6), or retracted (1) (see Fig. 1).
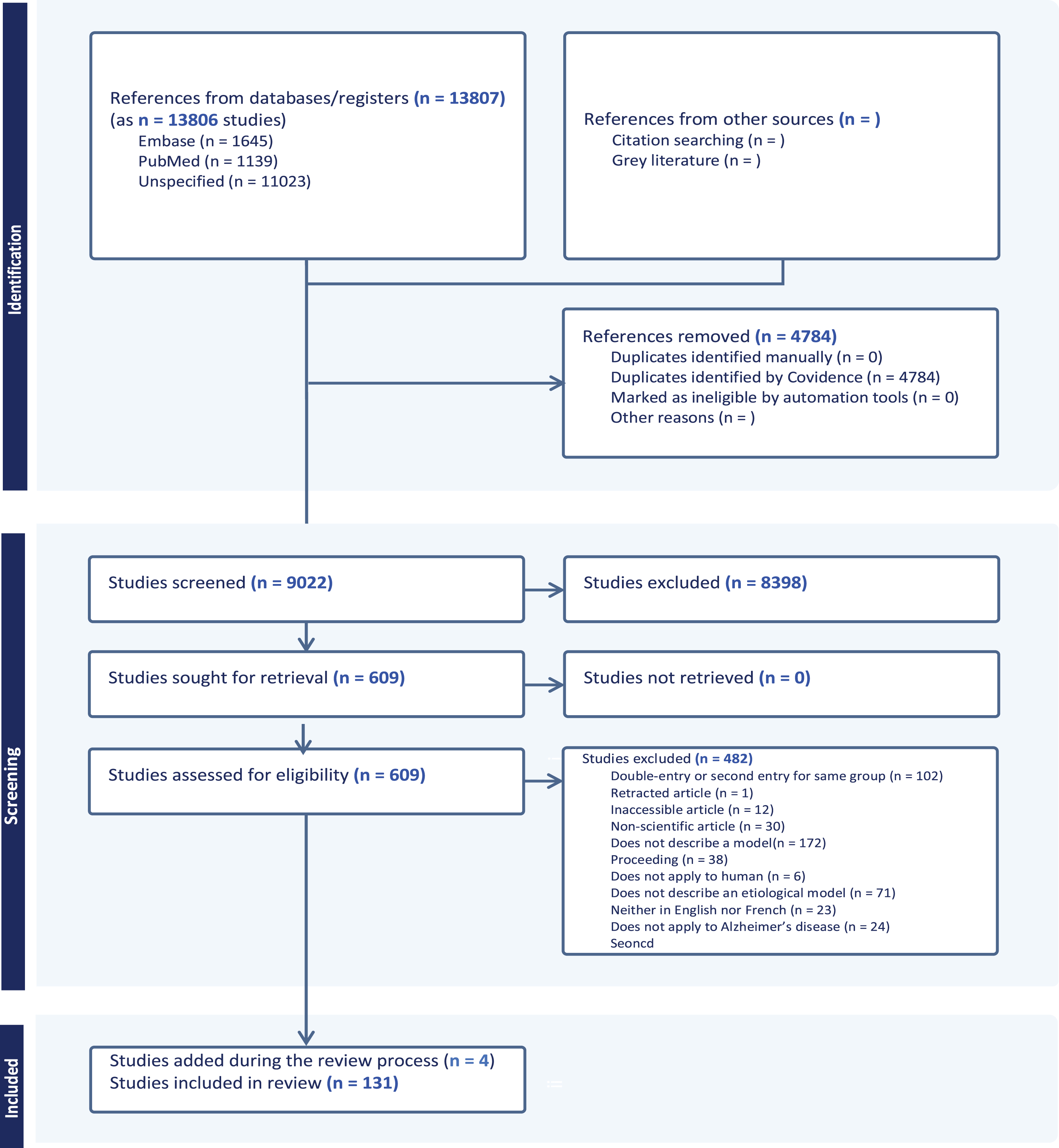
Fig. 1
PRISMA flowchart. Following the search strategy, 13,807 abstracts were extracted from the Embase, Web of Science, Medline, and Psycinfo databases. After removing duplicates and irrelevant studies, applying exclusion criteria and streamlining to keep only seminal and original articles, a total of 131 studies were included in this scoping review.
A final streamlining review of the 143 remaining articles was completed (S.D.) to remove papers from an author or group of authors that published the same hypothesis/theory more than once. We included only the earliest paper for each group in our review, as it was felt to represent the original inception of the idea. It is acknowledged that the latest manuscript, on the other hand, might have proposed the most up to date version of the hypothesis/theory under consideration. Four additional papers were added through the review process.
Following these steps yielded a final tally of 130 papers that were included in this review (Supplementary Table 2).
It must be stated unequivocally that this process, while rigorous, is more likely than not to have missed important papers, published decades ago or contemporary to us. Overall, the selection of manuscripts should be seen as representative of their categories, rather than seminal in the absolute. The emphasis is on identifying ideas within large categories, and not to initiate a subordinate debate on improper attribution of accolades. Thus, we apologize in advance to the authors of any other work that is not listed here and yet deserves to be shown.
Table 2
First author provenance
| Country | N papers |
| United States | 60 |
| Canada | 8 |
| UK | 9 |
| France | 7 |
| Italy | 6 |
| Australia | 5 |
| Germany | 4 |
| Spain | 4 |
| Israel | 3 |
| Brazil | 2 |
| China | 2 |
| India | 2 |
| Poland | 2 |
| Singapore | 2 |
| The Netherlands | 2 |
| Turkey | 2 |
| Chile | 1 |
| Czech Republic | 1 |
| Hong-Kong | 1 |
| Iran | 1 |
| Japan | 1 |
| Malaysia | 1 |
| Portugal | 1 |
| Russia | 1 |
| Saudi Arabia | 1 |
| Slovenia | 1 |
| Switzerland | 1 |
| Total | 131 |
Data extraction and classification
Characteristics of the included studies were extracted and are reported in Supplementary Table 2: lead author; country of lead author; whether there were other authors; title; year of publication; study design; and abstract.
Citations count for each paper as of 22 December 2023 were obtained via the Google Scholar search engine. While it is known that this count may include self-citations, it was the only count that could be reliably obtained for each paper across publishers and other citation engines.
Studies were then classified (S.D.) according to two epistemological dimensions. First was whether the hypothesis/theory being proposed was unifactorial (e.g., Aβ over-production initiates a cascade of downstream effects leading to neuronal death and dementia) or multifactorial (e.g., the co-existence and interactions between amyloid, tau, and inflammation lead to neuronal death and dementia). Secondly, studies were grouped according to the primary factor that was pushed forward in the article, according to the modified theoretical grouping of Ambrose 2015 (see Table 1).
RESULTS
Studies overview
Out of 131 papers, a majority (76/131) was written by multiple, as opposed to a single, authors. First authors were mainly from the United States (60), the UK (9), Canada (8), or continental Europe (37) (see Table 2).
Study categories and impact
Nearly two third of studies proposed a hypothesis characterized as being single-focus (100/131), with the most important theoretical grouping being, unsurprisingly, the Amyloid group (14/131), followed by Metabolism (10/131) (18/131 when joined to Mitochondrial dysfunction articles), Infection (11/131), and Cerebrovascular and BBB (10/131).
Impact, as measured by total citations count, followed a similar pattern. Out of a total of 20,209 citations for the 131 papers, the most cited single-focused grouping was Amyloid (52%), followed by Mitochondrial dysfunction (6%, reaching 10.0% when adding Metabolism articles), then Cerebrovascular and BBB (8.6%) and Oxidative Stress (4%).
Evolution
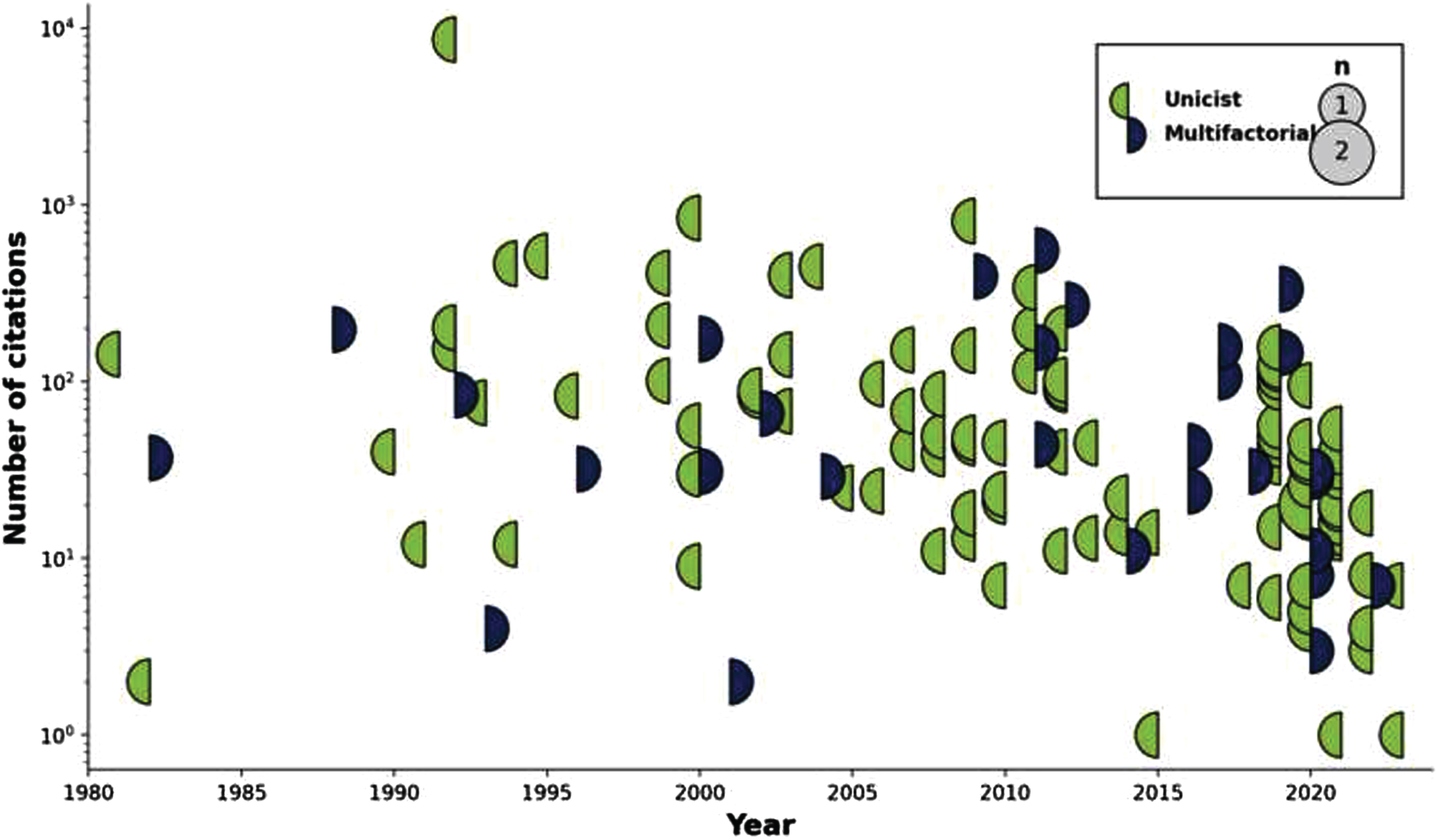
The number of articles proposing, repositioning, or actualizing individual hypotheses and theories across time is shown in Fig. 2, from the very first (King et al.s aluminum toxicity hypothesis in 1981) [79] to the latest entries in 2023. Likewise, a similar timeline but this time showing the impact (i.e., citation count) for each one of these articles is shown in Fig. 3.
Fig. 2
Display of articles by topic through time. Circles are sized according to the number of publications that year, with a color code indicating whether the published articles were unifactorial (left circle hemisphere) or multifactorial (right circle hemisphere).
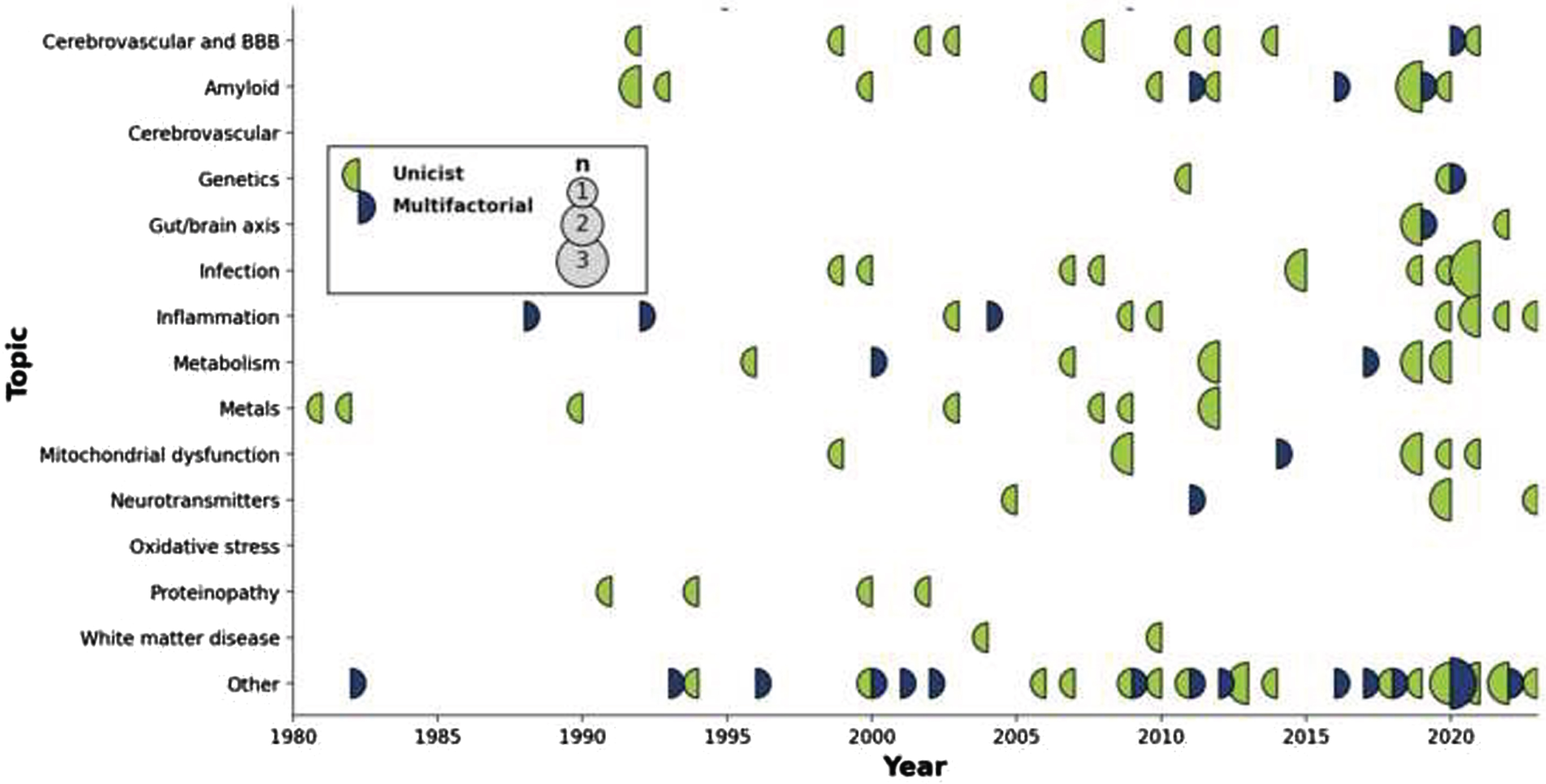
Fig. 3
Display of articles citation count by topic through time. Circles are sized according to the number of citations that the articles achieved that year, with a color code indicating whether the published articles were unifactorial (left circle hemisphere) or multifactorial (right circle hemisphere).
DISCUSSION
Summary of findings
We have conducted a scoping review of AD-related hypothesis or theories in the literature dating from the early 1980 s to the current era. We selected 131 representative papers for review, written mostly by authors based in North America and Europe, and spanning a wide range of conceptual groupings.
Uni- or multi-factorial?
The idea that failure of a single pathway is responsible for the ensuing cascade of anatomical and physiological aberrations has been espoused not only for those hypothesizing about the role of Aβ, but all others as well, be they molecular, proteinic, cellular, or tissular entities, with nearly two-thirds of all theories being unifactorial (cf.). Given the massive increase in neuroscientific knowledge of the last few decades, clearly adding to our previous understanding of the intricate complexity of the brain, and the lack of any clinically meaningful results in therapeutic pursuits when targeting individual pathways, it remains puzzling why there remains such an emphasis on unifactorial hypotheses. This includes the Amyloid hypothesis subgroup, the most studied of them all, for which unifactoriality was de rigueur until 2011; since then, more unifactorial point of view articles have been published than those espousing a multifactorial framework in which Aβ is a strong contributor. In essence, the field seems to have doubled down on the amyloid cascade hypothesis, albeit in a slightly modified form, regardless of disproving evidence as to its centrality. The existence of possible non-scientific reasons as to why this situation has been self-perpetuating is a possibility; a single theory does offer a direct promise of a high reward if proven correct.
Novelty and endurance of hypotheses
Three stages can be discerned in terms of hypotheses generation. The first phase (∼1980–1995) included the establishment of the main thrusts that have endured to this day. Its most prevalent entry was the Amyloid hypothesis, with the field-defining work of Hardy et al. (cited 8,633 times) [5], before the separation into clear autosomal dominant and pseudo-sporadic cases (often conflated or confounded with Early and Late Onset AD designations). In the same period were also introduced initial reports for the Glial [18], Infection [50], Inflammation [97], Metabolism [69], Oxidative stress [97], and Proteinopathies [102] hypotheses; the concept of interdependence across all scales, central to multifactorality [122]; as well as the by now marginal notion of neurotoxicity of trace elements [79]. The importance of BBB integrity was also recognized [28]. In the second phase (1995–2005), the importance of the Cerebrovasculature [35, 37, 38], Mitochondrial dysfunction [84], and Neurotransmitters [91] took center stage. Lately (2005–2020), evidence towards Genetics (outside of the autosomal dominant form) [41], and especially Gut/Brain interactions [42] came to the fore. The latter category has seen a rapid increase in impact when compared to others, with the paper from Angelucci et al. receiving a rate of 66.8 citations/year since its publication in 2019 (second only to the seminal Hardy et al. paper, with 269.8 citations/year since publication).
Biases
There are obvious biases in the impact statistics that must temper any interpretation. First, citation counts from Google Scholar often include self-citations; real numbers will therefore be smaller than recorded. Secondly, articles may be cited for positive (e.g., new evidence corroborating the hypothesis), negative (e.g., new evidence against the hypothesis), or even neutral (e.g., overview of the field) reasons, including positioning of ones hypothesis against the leading one (which, in the case of AD, would favor the citation rates attached to the Amyloid group). Raw citation count therefore may not reflect actual acceptance of any idea. Finally, the field is heavily biased towards ground-breaking, first-in-field articles; while seminal, they may not represent the best or most up to date version of a particular hypothesis. In particular, the article of Hardy et al. [5], already mentioned, established the amyloid cascade hypothesis, and represents 43.2% of all citations from the 131 papers in this scoping review. The power law of distributions is evidently quite at work here as elsewhere; the 10-most cited papers (Amyloid: 2; Cerebrovascular: 2; general theories around aging: 2; and Mitochondrial dysfunction, Oxidative stress, Neurotransmitters, and Glial disease: 1 each) are responsible for 67.0% of all citations.
Finally, we refrained from providing an assessment of the relative merit of each theory, as any such assessment would have been biased. First, since the quality and quantity of evidence for each theory is demonstratively related to its preponderance in the literature (e.g., evidence on amyloid far outweighs that on the impact of gut microbiota). Next, since no theory, including the amyloid hypothesis, has overwhelmingly been demonstrated to explain all aspects of pseudo-sporadic AD, then it implies that either insufficient evidence has been gathered to arrive at a conclusion, or a sufficiently complete theory has yet to be formulated to fit the available evidence. Hence, we felt that any assessment would have been fraught without a clear epistemological direction.
Future considerations
Which hypotheses or theory have notable evidence?
We posit that to answer this question successfully a theory would need to fulfill three criteria: 1) unequivocal clinical benefit from a therapeutic intervention, with 2) unambiguous disease-modifying results confirming a 3) global theoretical framework of the etiology.
As of now, only three classes of interventions have achieved results. First are anti-cholinesterase inhibitors, approved in the late 1990 s [142] following clinical trials that demonstrated a statistically significant yet weak improvement in cognition for a limited time period. Acting on the principle that they enhance the availability of important neurotransmitters, they appear to provide but a temporary respite to clinical disease progression, while not influencing biomarkers of neurodegeneration. They remain seen as symptoms-, rather than disease-, modifiers [143]. Hence, while our first criterion seems met, the latter two are not.
A second class of intervention has gained traction in the past few years. Three Aβ antibodies have been approved by the Food and Drugs Agency in the United States following successful clinical trials, for which the primary outcome was biomarker defined, rather than clinical [144]. While the aptitude of both compounds at Aβ plaque removal is not in question (criterion 2), they had no effect on anatomy or physiology (e.g., neurodegeneration, neuroinflammation, neurometabolism) as well as limited clinically-observable effects (criterion 1), the latter remaining in doubt [9, 145], especially in the face of severe side-effects that point to an incomplete understanding of the role of Aβ [146]. Such antibodies are not the only compounds that have attempted to reduce production, block aggregation, or remove Aβ plaques; as a general rule, these few successes are not strong indicators of the centrality of the amyloid hypothesis in any general theory of AD (criterion 3).
Finally, non-pharmaceutical combinatory approaches targeting lifestyle interventions, nutrition, physical exercise, and aggressive management of co-morbidities (e.g., hypertension, diabetes) have had also significant successes, with effect sizes that are actually equal to pharmacological approaches such as Aβ antibodies [147] (criterion 1). The difficulty here is to understand which one, or many, etiological pathways are affected and with which efficiency (criterion 2). It does however provide strong evidence that multi-scale targeting stands a better chance than other, single-pathway focused interventions (criterion 3).
Theory versus practice: pharmaceutical trials
Hypothesis-driven research, relatively free from the pressure of providing immediate returns to its largely state funders in terms of success or failure, has therefore kept on exploring avenues, new and old, with what can be seem at times steadfastness. Witness to this effect the 33% increase in Aβ-centric theoretical works that have been proposed in the 2010–2020 interval.
On the contrary, the field of pharmaceutical trials seems to have already integrated the notion of multi-factoriality, exploring a host of different avenues, often in combination. This can be seen for example in the 54% decrease (41 versus 19) in the number of Aβ-centric clinical trials between 2010 and 2022 [148, 149].
Single versus multiple factors
On the face of the evidence presented it is therefore hard to refute completely any one of the hypotheses that have been proposed. While many concepts can be proven as extremely unlikely and rejected on this basis, most articles present compelling evidence to this or that effect that is either borne out in a clinical population, either via epidemiological studies or randomized clinical trials, or from translational work in animal models. Viewed differently, they reinforce the notion in fact that AD affects multiple sub-cellular, cellular, anatomical, and physiological systems at the same time but at varying degree between individuals. The simple fact of the existence of the incredible variety of etiological factors itself should makes one realize that we are dealing with a complex multifactorial problem; this is further evidenced by the correlation of dementia with all other health problems [150]. The biggest hurdle to overcome seems rather to provide a comprehensive overview of all these systems at the same time, and their interactions.
Further, any theory being proposed would need to explain a significant body of epidemiological evidence pointing to, for example, sex imbalance in the prevalence of AD [151], as well as in trajectories and therapeutic response in other ethno-centric and socio-economic factors [11].
The emphasis on unifactorial viewpoints is possibly an artefact of research, including the well-known bias towards positive research reporting [152]. The necessity to provide results that have reached statistical significance will naturally be easier to achieve in limited experimental settings, given statistical power considerations, and thus favor the study of individual effects over the logistically much more difficult task of deciphering interactions in a system; complexity increasing factorially with each new factor and parameter being considered.
CONCLUSION
Hence, while new frontiers keep being opened, it could be argued that most of the anatomical and physiological systems that impact or are impacted by AD, at various time and spatial scales, have already been well surveyed. A corollary of this observation is endurance. Indeed, some hypotheses have been proposed and therefore tested for over 30 years. The fact that we are still discussing whether AD is primarily an amyloidopathy, what is the importance of metabolism, or what role plays the cerebrovasculature, attests to both the complexity of the matter and the need for a holistic framework that could reframe the debate in simpler lines. Geometry provides an analogy: problems that are arduous in cartesian space become simpler when expressed in polar coordinates. Such a transformation might be necessary for the field to reach clarity.
AUTHOR CONTRIBUTIONS
Simon Duchesne (Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Resources; Software; Supervision; Validation; Visualization; Writing – original draft; Writing – review & editing); Louis-Simon Rousseau (Conceptualization; Data curation; Formal analysis; Methodology; Validation; Writing – original draft); Florence Belzile (Data curation; Writing – review & editing); Laurie-Ann Welch (Data curation; Writing – review & editing); Beatrice Cournoyer (Data curation; Writing – review & editing); Marianne Arseneau (Data curation; Writing – review & editing); Véronick Lapierre (Data curation; Writing – review & editing); Sara-Maude Poulin (Data curation; Writing – review & editing); Olivier Potvin (Data curation; Formal analysis; Visualization; Writing – original draft; Writing – review & editing); Carol Hudon (Conceptualization; Funding acquisition; Investigation; Methodology; Project administration; Resources; Supervision; Validation; Writing – original draft; Writing – review & editing).
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
S.D. would like to acknowledge funding by the Canadian Institute for Health Research (Operating grant #PJT-159778). L.S.R. would like to acknowledge funding from the Fonds de recherche du Québec – Santé (scholarship #271206).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
DATA AVAILABILITY
The final list of articles selected for this scoping review is available upon request in tabular format.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-230772.
REFERENCES
[1] | Livingston G , Huntley J , Sommerlad A , Ames D , Ballard C , Banerjee S , Brayne C , Burns A , Cohen-Mansfield J , Cooper C , Costafreda SG , Dias A , Fox N , Gitlin LN , Howard R , Kales HC , Kivimaki M , Larson EB , Ogunniyi A , Orgeta V , Ritchie K , Rockwood K , Sampson EL , Samus Q , Schneider LS , Selbaek G , Teri L , Mukadam N ((2020) ) Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 396: , 413–446. |
[2] | Braak H , Thal DR , Ghebremedhin E , Del Tredici K ((2011) ) Stages of the pathologic process in Alzheimer disease: Age categories from 1 to 100 years. J Neuropathol Exp Neurol 70: , 960–969. |
[3] | Boyle PA , Yu L , Wilson RS , Schneider JA , Bennett DA ((2013) ) Relation of neuropathology with cognitive decline among older persons without dementia. Front Aging Neurosci 5: , 50. |
[4] | Alzheimer A ((1907) ) Über eine eigenartige Erkrankung derHirnrinde. Allg Zeitschr Psychiatr 64: , 146–148. |
[5] | Hardy JA , Higgins GA ((1992) ) Alzheimers disease: The amyloid cascade hypothesis. Science 256: , 184–185. |
[6] | Jansen WJ , Ossenkoppele R , Knol DL , Tijms BM , Scheltens P , Verhey FR , Visser PJ , Aalten P , Aarsland D , et al.Amyloid Biomarker Study Group ((2015) ) Prevalence of cerebral amyloid pathology in personswithout dementia: A meta-analysis. JAMA 313: , 1924–1938. |
[7] | Parent C , Rousseau LS , Predovan D , Duchesne S , Hudon C ((2023) ) Longitudinal association between ss-amyloid accumulation and cognitive decline in cognitively healthy older adults: A systematic review. Aging Brain 3: , 100074. |
[8] | Boyle PA , Yu L , Wilson RS , Leurgans SE , Schneider JA , Bennett DA ((2018) ) Person-specific contribution of neuropathologies to cognitive loss in old age. Ann Neurol 83: , 74–83. |
[9] | Knopman DS , Jones DT , Greicius MD ((2021) ) Failure to demonstrate efficacy of aducanumab: An analysis of the EMERGE and ENGAGE trials as reported by Biogen, December 2019. Alzheimers Dement 17: , 696–701. |
[10] | van Dyck CH , Swanson CJ , Aisen P , Bateman RJ , Chen C , Gee M , Kanekiyo M , Li D , Reyderman L , Cohen S , Froelich L , Katayama S , Sabbagh M , Vellas B , Watson D , Dhadda S , Irizarry M , Kramer LD , Iwatsubo T ((2023) ) Lecanemab in early Alzheimers disease. N Engl J Med 388: , 9–21. |
[11] | Manly JJ , Deters KD ((2023) ) Donanemab for Alzheimer disease-whobenefits and who is harmed? JAMA 330: , 510–511. |
[12] | Barbera M , Perera D , Matton A , Mangialasche F , Rosenberg A , Middleton L , Ngandu T , Solomon A , Kivipelto M ((2023) ) Multimodal precision prevention - a new direction in Alzheimers disease. J Prev Alzheimers Dis 10: , 718–728. |
[13] | Page MJ , McKenzie JE , Bossuyt PM , Boutron I , Hoffmann TC , Mulrow CD , Shamseer L , Tetzlaff JM , Akl EA , Brennan SE , Chou R , Glanville J , Grimshaw JM , Hrobjartsson A , Lalu MM , Li T , Loder EW , Mayo-Wilson E , McDonald S , McGuinness LA , Stewart LA , Thomas J , Tricco AC , Welch VA , Whiting P , Moher D ((2021) ) The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. Rev Esp Cardiol (Engl Ed) 74: , 790–799. |
[14] | Anastasio TJ ((2011) ) Data-driven modeling of Alzheimer disease pathogenesis. J Theor Biol 290: , 60–72. |
[15] | Caselli RJ , Knopman DS , Bu G ((2020) ) An agnostic reevaluation of the amyloid cascade hypothesis of Alzheimers disease pathogenesis: The role of APP homeostasis. Alzheimers Dement 16: , 1582–1590. |
[16] | Castellani RJ , Plascencia-Villa G , Perry G ((2019) ) The amyloid cascade and Alzheimers disease therapeutics: Theory versus observation. Lab Invest 99: , 958–970. |
[17] | Ethell DW ((2010) ) An amyloid-notch hypothesis for Alzheimers disease. Neuroscientist 16: , 614–617. |
[18] | Frederickson RC ((1992) ) Astroglia in Alzheimers disease. Neurobiol Aging 13: , 239–253. |
[19] | Gallardo G , Holtzman DM ((2019) ) Amyloid-beta and tau at the crossroads of Alzheimers disease. Adv Exp Med Biol 1184: , 187–203. |
[20] | Han PC , Shi J ((2016) ) A theoretical analysis of the synergy of amyloid and tau in Alzheimers disease. J Alzheimers Dis 52: , 1461–1470. |
[21] | Paroni G , Bisceglia P , Seripa D ((2019) ) Understanding the amyloid hypothesis in Alzheimers disease. J Alzheimers Dis 68: , 493–510. |
[22] | Petrella JR , Hao W , Rao A , Doraiswamy PM ((2019) ) Computational causal modeling of the dynamic biomarker cascade in Alzheimers disease. Comput Math Methods Med 2019: , 6216530. |
[23] | Roberts GW , Nash M , Ince PG , Royston MC , Gentleman SM ((1993) ) On the origin of Alzheimers disease: A hypothesis. Neuroreport 4: , 7–9. |
[24] | Sambamurti K , Suram A , Venugopal C , Prakasam A , Zhou Y , Lahiri DK , Greig NH ((2006) ) A partial failure of membrane protein turnover may cause Alzheimers disease: A new hypothesis. Curr Alzheimer Res 3: , 81–90. |
[25] | Selkoe DJ ((2000) ) Toward a comprehensive theory for Alzheimers disease. Hypothesis: Alzheimers disease is caused by the cerebral accumulation and cytotoxicity of amyloid β-protein. Ann N Y Acad Sci 924: , 17–25. |
[26] | Tam JHK , Pasternak SH ((2012) ) Amyloid and Alzheimers disease: Inside and out. Can J Neurol Sci 39: , 286–298. |
[27] | Ambrose CT ((2014) ) A therapeutic approach for senile dementias: Neuroangiogenesis. J Alzheimers Dis 43: , 1–17. |
[28] | Mann DM ((1985) ) The neuropathology of Alzheimers disease: A review with pathogenetic, aetiological and therapeutic considerations. Mech Ageing Dev 31: , 213–255. |
[29] | Cordonnier C , Van Der Flier WM ((2011) ) Brain microbleeds and Alzheimers disease: Innocent observation or key player? Brain 134: , 335–344. |
[30] | Henry-Feugeas MC ((2008) ) Alzheimers disease in late-life dementia: A minor toxic consequence of devastating cerebrovascular dysfunction. Med Hypotheses 70: , 866–875. |
[31] | Kapadia A , Mirrahimi A , Dmytriw AA ((2020) ) Intersection between sleep and neurovascular coupling as the driving pathophysiology of Alzheimers disease. Med Hypotheses 144: , 110283. |
[32] | Orehek AJ ((2012) ) The micron stroke hypothesis of Alzheimers disease and dementia. Med Hypotheses 78: , 562–570. |
[33] | Pluta R , Ułamek M ((2008) ) Brain ischemia and ischemic blood-brainbarier as etiological factors in sporadic Alzheimers disease. Neuropsychiatric Disease and Treatment 4: , 855–864. |
[34] | Vagnucci AH , Jr. , Li WW ((2003) ) Alzheimers disease and angiogenesis. Lancet 361: , 605–608. |
[35] | Zlokovic BV ((2002) ) Vascular disorder in Alzheimers disease: Role in pathogenesis of dementia and therapeutic targets. Adv Drug Deliv Rev 54: , 1553–1559. |
[36] | Vidal C , Zhang L ((2021) ) An analysis of the neurological and molecular alterations underlying the pathogenesis of Alzheimers disease. Cells 10: , 546. |
[37] | Kalaria RN ((1992) ) The blood-brain barrier and cerebral microcirculation in Alzheimer disease. Cerebrovasc Brain Metab Rev 4: , 226–260. |
[38] | de la Torre JC ((1999) ) Critical threshold cerebral hypoperfusion causes Alzheimers disease? Acta Neuropathol 98: , 1–8. |
[39] | Bruni AC , Bernardi L , Gabelli C ((2020) ) From beta amyloid to altered proteostasis in Alzheimers disease. Ageing Res Rev 64: , 101126. |
[40] | Kaeser GE , Chun J ((2020) ) Mosaic somatic gene recombination as a potentially unifying hypothesis for Alzheimers disease. Front Genet 11: , 390. |
[41] | Yurov YB , Vorsanova SG , Iourov IY ((2011) ) The DNA replication stress hypothesis of Alzheimers disease. ScientificWorldJournal 11: , 2602–2612. |
[42] | Angelucci F , Cechova K , Amlerova J , Hort J ((2019) ) Antibiotics, gut microbiota, and Alzheimers disease. J Neuroinflammation 16: , 108. |
[43] | Bostanciklioglu M ((2019) ) The role of gut microbiota in pathogenesis of Alzheimers disease. J Appl Microbiol 127: , 954–967. |
[44] | Cerovic M , Forloni G , Balducci C ((2019) ) Neuroinflammation and the gut microbiota: Possible alternative therapeutic targets to counteract Alzheimers disease? Front Aging Neurosci 11: , 284. |
[45] | Roe K ((2022) ) An alternative explanation for Alzheimers disease and Parkinsons disease initiation from specific antibiotics, gut microbiota dysbiosis and neurotoxins. Neurochem Res 47: , 517–530. |
[46] | Bastian FO ((2015) ) Is Alzheimers disease infectious? Relative to the CJD bacterial infection model of neurodegeneration. Aims Neurosci 2: , 240–258. |
[47] | Baudron CR , Varon C , Megraud F , SalleS N ((2015) ) Alzheimers disease: The infectious hypothesis. Geriatr Psychol Neuropsychiatr Vieil 13: , 418–424. |
[48] | Block J ((2019) ) Alzheimers disease might depend on enabling pathogens which do not necessarily cross the blood-brain barrier. Med Hypotheses 125: , 129–136. |
[49] | Dezfulian M , Shokrgozar MA , Sardari S , Parivar K , Javadi G ((2008) ) Can phages cause Alzheimers disease? Med Hypotheses 71: , 651–656. |
[50] | Fulop T , Munawara U , Larbi A , Desroches M , Rodrigues S , Catanzaro M , Guidolin A , Khalil A , Bernier F , Barron AE , Hirokawa K , Beauregard PB , Dumoulin D , Bellenger JP , Witkowski JM , Frost E ((2020) ) Targeting infectious agents as a therapeutic strategy in Alzheimers disease. CNS Drugs 34: , 673–695. |
[51] | McDonald RJ ((2002) ) Multiple combinations of co-factors produce variants of age-related cognitive decline: A theory. Can J Exp Psychol 56: , 221–239. |
[52] | Reinscheid F ((2021) ) A new proposal for the causative agent of the sporadic form of Alzheimers disease. Med Hypotheses 146: , 110453. |
[53] | Li F , Hearn M , Bennett LE ((2021) ) The role of microbial infection in the pathogenesis of Alzheimers disease and the opportunity for protection by anti-microbial peptides. Crit Rev Microbiol 47: , 240–253. |
[54] | Sait A , Angeli C , Doig AJ , Day PJR , Viral involvement in Alzheimer’s disease. ACS Chem Neurosci 12: , 1049–1060. |
[55] | Yong SJ , Yong MH , Teoh SL , Soga T , Parhar I , Chew J , Lim WL ((2021) ) The hippocampal vulnerability to herpes simplex virus type I infection: Relevance to Alzheimers disease and memory impairment. Front Cell Neurosci 15: , 695738. |
[56] | Dobson CB , Itzhaki RF ((1999) ) Herpes simplex virus type 1 and Alzheimers disease. Neurobiol Aging 20: , 457–465. |
[57] | Barger SW ((2004) ) An unconventional hypothesis of oxidation in Alzheimers disease: Intersections with excitotoxicity. Front Biosci 9: , 3286–3295. |
[58] | Bermejo-Pareja F , Del Ser T , Valenti M , de la Fuente M , Bartolome F , Carro E ((2020) ) Salivary lactoferrin as biomarker for Alzheimers disease: Brain-immunity interactions. Alzheimers Dement 16: , 1196–1204. |
[59] | Maccioni RB , Farias GA , Rojo LE , Sekler MA , Kujis RO , Maccioni RB , Perry G (2009) What HaveWe Learned from the Tau Hypothesis? Springer, New York. |
[60] | Seaton A , Tran L , Chen R , Maynard RL , Whalley LJ ((2020) ) Pollution, particles, and dementia: A hypothetical causative pathway. Int J Environ Res Public Health 17: , 862. |
[61] | Bowirrat A ((2022) ) Immunosenescence and aging: Neuroinflammation is a prominent feature of Alzheimers disease and is a likely contributor to neurodegenerative disease pathogenesis. J Pers Med 12: , 1817. |
[62] | de Oliveira J , Kucharska E , Garcez ML , Rodrigues MS , Quevedo J , Moreno-Gonzalez I , Budni J ((2021) ) Inflammatory cascade in Alzheimers disease pathogenesis: A review of experimental findings. Cells 10: , 2581. |
[63] | Patil GV , Joshi RS , Kazi RS , Kulsange SE , Kulkarni MJ ((2020) ) A possible role of glycation in the regulation of amyloid beta precursor protein processing leading to amyloid beta accumulation. Med Hypotheses 142: , 109799. |
[64] | Accardi G , Caruso C , Colonna-Romano G , Camarda C , Monastero R , Candore G ((2012) ) Can Alzheimer disease be a form of type 3 diabetes? Rejuvenation Res 15: , 217–221. |
[65] | Demetrius LA , Simon DK ((2012) ) An inverse-Warburg effect and the origin of Alzheimers disease. Biogerontology 13: , 583–594. |
[66] | Folch J , Olloquequi J , Ettcheto M , Busquets O , Sanchez-Lopez E , Cano A , Espinosa-Jimenez T , Garcia ML , Beas-Zarate C , Casadesus G , Bullo M , Auladell C , Camins A ((2019) ) The involvement of peripheral and brain insulin resistance in late onset Alzheimers dementia. Front Aging Neurosci 11: , 236. |
[67] | Lahiri DK , Maloney B , Basha MR , Ge YW , Zawia NH ((2007) ) How and when environmental agents and dietary factors affect the course of Alzheimers disease: The “LEARn model (latent early-life associated regulation) may explain the triggering of AD. Curr Alzheimer Res 4: , 219–228. |
[68] | Lynch C , Mobley W ((2000) ) Comprehensive theory of Alzheimers disease. The effects of cholesterol on membrane receptor trafficking. Ann N Y Acad Sci 924: , 104–111. |
[69] | Meier-Ruge W , Bertoni-Freddari C ((1996) ) The significance of glucose turnover in the brain in the pathogenetic mechanisms of Alzheimers disease. Rev Neurosci 7: , 1–19. |
[70] | Mullins RJ , Diehl TC , Chia CW , Kapogiannis D ((2017) ) Insulin resistance as a link between amyloid-beta and tau pathologies in Alzheimers disease. Front Aging Neurosci 9: , 118. |
[71] | Rorbach-Dolata A , Piwowar A ((2019) ) Neurometabolic evidence supporting the hypothesis of increased incidence of type 3 diabetes mellitus in the 21st Century. Biomed Res Int 2019: , 1435276. |
[72] | Suresh J , Khor IW , Kaur P , Heng HL , Torta F , Dawe GS , Tai ES , Tolwinski NS ((2021) ) Shared signaling pathways in Alzheimers andmetabolic disease may point to new treatment approaches. FEBSJ 288: , 3855–3873. |
[73] | Bjorksten JA ((1982) ) Aluminium as a cause of senile dementia. Compr Ther 8: , 73–76. |
[74] | Brewer GJ ((2012) ) Metals in the causation and treatment of Wilsons disease and Alzheimers disease, and copper lowering therapy in medicine. Inorganica Chim Acta 393: , 135–141. |
[75] | Bush AI ((2003) ) Copper, zinc, and the metallobiology of Alzheimer disease. Alzheimer Dis Assoc Disord 17: , 147–150. |
[76] | Constantinidis J ((1990) ) Alzheimers disease: The zinc theory. Encephale 16: , 231–239. |
[77] | Craddock TJA , Tuszynski JA , Chopra D , Casey N , Goldstein LE , Hameroff SR , Tanzi RE ((2012) ) The zinc dyshomeostasis hypothesis of Alzheimers disease. PLoS One 7: , e33552. |
[78] | Dwyer BE , Zacharski LR , Balestra DJ , Lerner AJ , Perry G , Zhu X , Smith MA ((2009) ) Getting the iron out: Phlebotomy for Alzheimers disease? Med Hypotheses 72: , 504–509. |
[79] | King SW , Savory J , Wills MR ((1981) ) The clinical biochemistry of aluminum. Crit Rev Clin Lab Sci 14: , 1–20. |
[80] | Klevay LM ((2008) ) Alzheimers disease as copper deficiency. Med Hypotheses 70: , 802–807. |
[81] | Albensi BC ((2019) ) Dysfunction of mitochondria: Implications for Alzheimers disease. Int Rev Neurobiol 145: , 13–27. |
[82] | Area-Gomez E , Guardia-Laguarta C , Schon EA , Przedborski S ((2019) ) Mitochondria, OxPhos, and neurodegeneration: Cells are not just running out of gas. J Clin Invest 129: , 34–45. |
[83] | Bonda DJ , Wang X , Gustaw-Rothenberg KA , Perry G , Smith MA , Zhu X ((2009) ) Mitochondrial drugs for Alzheimer disease. Pharmaceuticals (Basel) 2: , 287–298. |
[84] | Bonilla E , Tanji K , Hirano M , Vu TH , DiMauro S , Schon EA ((1999) ) Mitochondrial involvement in Alzheimers disease. Biochim Biophys Acta 1410: , 171–182. |
[85] | Chen M , Nguyen HT ((2014) ) Our “energy-Ca2+signaling deficits hypothesis and its explanatory potential for key features of Alzheimers disease. Front Aging Neurosci 6: , 329. |
[86] | Ebanks B , Ingram TL , Chakrabarti L ((2020) ) ATP synthase and Alzheimers disease: Putting a spin on the mitochondrial hypothesis. Aging 12: , 16647–16662. |
[87] | Swerdlow RH , Khan SM ((2009) ) The Alzheimers disease mitochondrial cascade hypothesis: An update. Exp Neurol 218: , 308–315. |
[88] | Jadiya P , Garbincius JF , Elrod JW ((2021) ) Reappraisal of metabolic dysfunction in neurodegeneration: Focus on mitochondrial function and calcium signaling. Acta Neuropathol Commun 9: , 124. |
[89] | Bi D , Wen L , Wu Z , Shen Y ((2020) ) GABAergic dysfunction in excitatory and inhibitory (E/I) imbalance drives the pathogenesis of Alzheimers disease. Alzheimers Dement 16: , 1312–1329. |
[90] | Craig LA , Hong NS , McDonald RJ ((2011) ) Revisiting the cholinergic in the development of Alzheimers disease. Neurosci Biobehav Rev 35: , 1397–1409. |
[91] | Schmitt HP ((2005) ) Neuro-modulation, aminergic neuro-disinhibition and neuro-degeneration. Draft of a comprehensive theory for Alzheimer disease. Med Hypotheses 65: , 1106–1119. |
[92] | Sordillo LA , Sordillo PP , Alfano RR , Alfano RR , Demos SG , Seddon AB (2020) Abnormal tryptophan metabolism in Alzheimer’s disease (ALZ): Label-free spectroscopy suggests an alternative theory of ALZ causation. Proceedings of the SPIE, Volume 11234, id. 112341P 5 pp. |
[93] | Zorec R , Vardjan N ((2023) ) Adrenergic regulation of astroglial aerobic glycolysis and lipid metabolism: Towards a noradrenergic hypothesis of neurodegeneration. Neurobiol Dis 182: , 106132. |
[94] | Aliev G , Palacios HH , Lipsitt AE , Fischbach K , Lamb BT , Obrenovich ME , Morales L , Gasimov E , Bragin V ((2009) ) Nitric oxide as an initiator of brain lesions during the development of Alzheimer disease. Neurotox Res 16: , 293–305. |
[95] | Benzi G , Moretti A ((1995) ) Are reactive oxygen species involved in Alzheimers disease? Neurobiol Aging 16: , 661–674. |
[96] | Harman D ((1992) ) Free radical theory of aging: A hypothesis on pathogenesis of senile dementia of the Alzheimers type. Age 16: , 23–30. |
[97] | Henderson AS ((1988) ) The risk factors for Alzheimers disease: A review and a hypothesis. Acta Psychiatr Scand 78: , 257–275. |
[98] | Rodrigues R , Bonda DJ , Perry G , Castellani RJ , Casadesus G , Lee HG , Lee HP , Wang X , Zhu X , Petersen RB , Smith MA (2010) Oxidative stress and neurodegeneration: An inevitable consequence of aging? Implications for therapy. In Brain Protection in Schizophrenia, Mood and Cognitive Disorders, Ritsner M, ed. Springer, Dordrecht, pp. 305-323. |
[99] | Veurink G , Fuller SJ , Atwood CS , Martins RN ((2003) ) Genetics, lifestyle and the roles of amyloid β and oxidative stress in Alzheimers disease. Ann Hum Biol 30: , 639–667. |
[100] | Lloret A , Esteve D , Lloret MA , Monllor P , Lopez B , Leon JL , Cervera-Ferri A ((2021) ) Is Oxidative stress the link between cerebral small vessel disease, sleep disruption, and oligodendrocyte dysfunction in the onset of Alzheimers disease? Front Physiol 12: , 708061. |
[101] | Roy RG , Mandal PK , Maroon JC ((2023) ) Oxidative stress occurs prior to amyloid Abeta plaque formation and tau phosphorylation in Alzheimers disease: Role of glutathione and metal ions. ACS Chem Neurosci 14: , 2944–2954. |
[102] | Di Patre PL ((1991) ) Cytoskeletal alterations might account for the phylogenetic vulnerability of the human brain to Alzheimers disease. Med Hypotheses 34: , 165–170. |
[103] | Liautard JP ((1994) ) A hypothesis on the etiology of Alzheimers disease - Description of a model involving a misfolded chaperone. Med Hypotheses 43: , 372–380. |
[104] | Masliah E ((2000) ) The role of synaptic proteins in Alzheimers disease. Ann N Y Acad Sci 924: , 68–75. |
[105] | Torreilles F , Touchon J ((2002) ) Pathogenic theories and intrathecal analysis of the sporadic form of Alzheimers disease. Prog Neurobiol 66: , 191–203. |
[106] | Erol A , et al ((2010) ) Are paradoxical cell cycle activities in neurons and glia related to the metabolic theory of Alzheimers disease? J Alzheimers Dis 19: , 129–135. |
[107] | Streit WJ ((2004) ) Microglia and Alzheimers disease pathogenesis. J Neurosci Res 77: , 1–8. |
[108] | Arendt T , Bruckner MK ((2007) ) Linking cell-cycle dysfunction in Alzheimers disease to a failure of synaptic plasticity. Biochim Biophys Acta 1772: , 413–421. |
[109] | Armstrong RA ((2011) ) The pathogenesis of Alzheimers disease: Areevaluation of the “amyloid cascade hypothesis. Int JAlzheimers Dis 2011: , 630865. |
[110] | Axelsen PH , Komatsu H , Murray IVJ ((2011) ) Oxidative stress and cell membranes in the pathogenesis of Alzheimers disease. Physiology 26: , 54–69. |
[111] | Ball MJ ((1982) ) Alzheimers disease. A challenging enigma. Arch Pathol Lab Med 106: , 157–162. |
[112] | Bugiani O ((2020) ) The puzzle of preserved cognition in the oldest old. Neurol Sci 41: , 441–447. |
[113] | Castello MA , Soriano S ((2013) ) Rational heterodoxy: Cholesterol reformation of the amyloid doctrine. Ageing Res Rev 12: , 282–288. |
[114] | Clarke RP ((2000) ) Does longer-term memory storage never become overloaded, and would such overload cause Alzheimers disease and other dementia? Med Hypotheses 55: , 419–428. |
[115] | Denis PA ((2013) ) Alzheimers disease: A gas model. The NADPH oxidase-Nitric Oxide system as an antibubble biomachinery. Med Hypotheses 81: , 976–987. |
[116] | Fotuhi M , Hachinski V , Whitehouse PJ ((2009) ) Changing perspectives regarding late-life dementia. Nat Rev Neurol 5: , 649–658. |
[117] | Fox M ((2018) ) ‘Evolutionary medicine perspectives on Alzheimers disease: Review and new directions. Ageing Res Rev 47: , 140–148. |
[118] | Khachaturian ZS ((1994) ) Calcium hypothesis of Alzheimers disease and brain aging. Ann N Y Acad Sci 747: , 1–11. |
[119] | Lopes JP , Oliveira CR , Agostinho P ((2009) ) Cell cycle re-entry in Alzheimers disease: A major neuropathological characteristic? Curr Alzheimer Res 6: , 205–212. |
[120] | Maurizi CP ((2010) ) Choroid plexus portals and a deficiency of melatonin can explain the neuropathology of Alzheimers disease. Med Hypotheses 74: , 1059–1066. |
[121] | Mesulam MM ((2000) ) A plasticity-based theory of the pathogenesis of Alzheimers disease. Ann N Y Acad Sci 924: , 42–52. |
[122] | Miller DW ((1993) ) Reflections on the psychobiological nature of reality, with a theory about Alzheimers disease. Advances 9: , 69–76. |
[123] | Moulton PV , Yang W ((2012) ) Air pollution, oxidative stress, and Alzheimers disease. J Environ Public Health 2012: , 9. |
[124] | Nehls M ((2016) ) Unified theory of Alzheimers disease (UTAD): Implications for prevention and curative therapy. J Mol Psychiatry 4: , 3. |
[125] | Offringa-Hup A ((2020) ) Alzheimers disease: The derailed repair hypothesis. Med Hypotheses 136: , 109516. |
[126] | Roccisano D , Henneberg M , Saniotis A ((2014) ) A possible cause of Alzheimers dementia - Industrial soy foods. Med Hypotheses 82: , 250–254. |
[127] | Schiel KA ((2018) ) A new etiologic model for Alzheimer.s disease. Med Hypotheses 111: , 27–35. |
[128] | Tse KH , Herrup K ((2017) ) Re-imagining Alzheimers disease –the diminishing importance of amyloid and a glimpse of what lies ahead. J Neurochem 143: , 432–444. |
[129] | Uleman JF , Melis RJF , Quax R , van der Zee EA , Thijssen D , Dresler M , van de Rest O , van der Velpen IF , Adams HHH , Schmand B , de Kok I , de Bresser J , Richard E , Verbeek M , Hoekstra AG , Rouwette E , Rikkert M ((2021) ) Mapping the multicausality of Alzheimers disease through group model building. Geroscience 43: , 829–843. |
[130] | Webber KM , Casadesus G , Zhu X , Obrenovich ME , Atwood CS , Perry G , Bowen RL , Smith MA ((2006) ) The cell cycle and hormonal fluxes in Alzheimer disease: A novel therapeutic target. Curr Pharm Design 12: , 691–697. |
[131] | Ying W ((1996) ) Deleterious network hypothesis of Alzheimers disease. Med Hypotheses 46: , 421–428. |
[132] | Zuodong S ((2019) ) The theory of dove-like particles. IBRO Reports 6: , S363. |
[133] | Hansen SB ((2023) ) Cholesterols function and origin in the Alzheimers disease brain. J Alzheimers Dis 94: , 471–472. |
[134] | Kawabata S ((2022) ) Excessive/aberrant and maladaptive synaptic plasticity: A hypothesis for the pathogenesis of Alzheimers disease. Front Aging Neurosci 14: , 913693. |
[135] | Lehmann DJ , Elshorbagy A , Hurley MJ ((2023) ) Many paths to Alzheimers disease: A unifying hypothesis integrating biological, chemical, and physical risk factors. J Alzheimers Dis 95: , 1371–1382. |
[136] | Turknett J , Wood TR ((2022) ) Demand coupling drives neurodegeneration: A model of age-related cognitive decline and dementia. Cells 11: , 2789. |
[137] | Ferrer I ((2022) ) Alzheimers disease is an inherent, natural part of human brain aging: An integrated perspective. Free Neuropathol 3: , 3–17. |
[138] | Festa BP , Barbosa AD , Rob M , Rubinsztein DC ((2021) ) The pleiotropic roles of autophagy in Alzheimers disease: From pathophysiology to therapy. Curr Opin Pharmacol 60: , 149–157. |
[139] | Kurakin A , Bredesen DE ((2020) ) Alzheimers disease as a systems network disorder: Chronic stress/dyshomeostasis, innate immunity, and genetics. Aging (Albany NY) 12: , 17815–17844. |
[140] | Polis B , Karasik D , Samson AO ((2021) ) Alzheimers disease as a chronic maladaptive polyamine stress response. Aging (Albany NY) 13: , 10770–10795. |
[141] | Tang BL ((2020) ) Neuropathological mechanisms associated with pesticides in Alzheimers disease. Toxics 8: , 21. |
[142] | McGleenon BM , Dynan KB , Passmore AP ((1999) ) Acetylcholinesterase inhibitors in Alzheimers disease. Br J Clin Pharmacol 48: , 471–480. |
[143] | Marucci G , Buccioni M , Ben DD , Lambertucci C , Volpini R , Amenta F ((2021) ) Efficacy of acetylcholinesterase inhibitors in Alzheimers disease. Neuropharmacology 190: , 108352. |
[144] | Sevigny J , Chiao P , Bussiere T , Weinreb PH , Williams L , Maier M , Dunstan R , Salloway S , Chen T , Ling Y , OGorman J , Qian F , Arastu M , Li M , Chollate S , Brennan MS , Quintero-Monzon O , Scannevin RH , Arnold HM , Engber T , Rhodes K , Ferrero J , Hang Y , Mikulskis A , Grimm J , Hock C , Nitsch RM , Sandrock A ((2016) ) The antibody aducanumab reduces Abeta plaques in Alzheimers disease. Nature 537: , 50–56. |
[145] | Sabbagh MN , Cummings J ((2021) ) Open Peer Commentary to “Failure to demonstrate efficacy of aducanumab: An analysis of the EMERGE and ENGAGE Trials as reported by Biogen December 2019”. Alzheimers Dement 17: , 702–703. |
[146] | Atwood CS , Perry G ((2023) ) Russian roulette with Alzheimers disease patients: Do the cognitive benefits of lecanemab outweigh the risk of edema and stroke? J Alzheimers Dis 92: , 799–801. |
[147] | Ngandu T , Lehtisalo J , Solomon A , Levalahti E , Ahtiluoto S , Antikainen R , Backman L , Hanninen T , Jula A , Laatikainen T , Lindstrom J , Mangialasche F , Paajanen T , Pajala S , Peltonen M , Rauramaa R , Stigsdotter-Neely A , Strandberg T , Tuomilehto J , Soininen H , Kivipelto M ((2015) ) A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): A randomised controlled trial. Lancet 385: , 2255–2263. |
[148] | Mangialasche F , Solomon A , Winblad B , Mecocci P , Kivipelto M ((2010) ) Alzheimers disease: Clinical trials and drug development. Lancet Neurol 9: , 702–716. |
[149] | Cummings J , Lee G , Nahed P , Kambar M , Zhong K , Fonseca J , Taghva K ((2022) ) Alzheimers disease drug development pipeline: 2022. Alzheimers Dement (N Y) 8: , e12295. |
[150] | Song X , Mitnitski A , Rockwood K ((2011) ) Nontraditional risk factors combine to predict Alzheimer disease and dementia. Neurology 77: , 227–234. |
[151] | Mazure CM , Swendsen J ((2016) ) Sex differences in Alzheimers disease and other dementias. Lancet Neurol 15: , 451–452. |
[152] | Dalton JE , Bolen SD , Mascha EJ ((2016) ) Publication bias: The elephant in the review. Anesth Analg 123: , 812–813. |

