
Treatment with 1, 10 Phenanthroline-5-Amine Reduced Amyloid Burden in a Mouse Model of Alzheimer’s Disease
Abstract
Background:
Alzheimer’s disease (AD) is the most prevalent age-related dementia, and, despite numerous attempts to halt or reverse its devastating progression, no effective therapeutics have yet been confirmed clinically. However, one class of agents that has shown promise is certain metal chelators.
Objective:
For the novel assessment of the effect of oral administration of 1,10-phenanthroline-5-amine (PAA) on the severity of amyloid plaque load, we used a transgenic (Tg) mouse model with inserted human autosomally dominant (familial) AD genes: amyloid-β protein precursor (AβPP) and tau.
Methods:
AβPP/Tau transgenic mice that model AD were allotted into one of two groups. The control group received no treatment while the experimental group received PAA in their drinking water starting at 4 months of age. All animals were sacrificed at 1 year of age and their brains were stained with two different markers of amyloid plaques, Amylo-Glo+ and HQ-O.
Results:
The control animals exhibited numerous dense core plaques throughout the neo- and allo- cortical brain regions. The experimental group treated with PAA, however, showed 62% of the amyloid plaque burden seen in the control group.
Conclusions:
Oral daily dosing with PAA will significantly reduce the amyloid plaque burden in transgenic mice that model AD. The underlying mechanism for this protection is not fully known; however, one proposed mechanism involves inhibiting the “metal-seeding” of Aβ.
INTRODUCTION
Being the most common age-related dementia, Alzheimer’s disease (AD) has been researched extensively yet, despite the wealth of information learned, only a few drugs have shown promise, and nothing has translated into a treatment that will stop or even slow the progression of this devastating disease. However, these failures provide opportunities for new drug development discoveries and hope for AD sufferers [1]. The etiology of AD is unknown but presumably involves the interaction of genes and environment [2, 3]. Despite the disease’s pathologies being well characterized, their mechanism of generation is not fully understood. The primary pathologies are extracellular aggregation of Aβ peptides into amyloid plaques and intraneuronal accumulation of hyperphosphorylated microtubule-associated protein tau in neurofibrillary tangles (NFTs).
A number of different targets have been proposed for possible therapeutic intervention against amyloid plaque formation [4]. Such potential interventions include: 1) inhibiting cleavage of Aβ from the amyloid-β protein precursor (AβPP) via secretase inhibitors; 2) preventing oligomerization of Aβ; 3) facilitating clearance of Aβ; 4) reducing cortical inflammatory response; 5) modulating cholesterol homeostasis; and 6) chelating certain transition metals of biological relevance to inhibit aggregation of Aβ. Although some of these approaches may have had promise in transgenic mouse models of AD, none translated to any human AD therapeutics. Some agents, such as γ-secretase inhibitors were, as Notch inhibitors, toxic to the liver and other organs [5]. Inoculation with Aβ oligomers resulted in occurrence of fatal meningoencephalitis in some AD patients [6]. Inhibiting cortical inflammation with anti-inflammatory agents can result in serious opportunistic infections [7, 8]. The use of statin drugs in an attempt to inhibit the formation of amyloid plaques had little efficacy in humans [9, 10]. Finally, despite their clinical application for treating acute metal toxicity, the use of metal chelators for the treatment of AD was not definitive [11]. This underscores the importance of testing different approaches and newer drugs. In this context, we present the novel finding that a non-amphoteric metal chelator 1,10-phenanthroline-5-amine (PAA, CAS: 54258-41-2) can inhibit the aggregation with the following rationale.
Phenanthroline (PA) has a long history of use as a metal chelator [12] and has also been shown to be a metalloprotease inhibitor [13]. Relatively recently a number of different PA derivatives have been developed, including PAA, the focus of this study. Like PA, PAA is capable of chelating a variety of metals including iron [14], copper [15], iridium [16], and europium [17].
The strategy of using chemical chelators to reduce amyloidosis is based on several observations. Specifically, the transition metals, zinc, iron, manganese, and copper facilitate the aggregation of Aβ in vitro [18–26]. They are localized within amyloid plaques in both transgenic mouse and human brain tissue sections [23, 27–32]. Zinc dysregulation plays an especially prominent role in the formation of amyloid plaques [33–35]. The heterocyclic organic compound PAA differs from previously studied chelators which are all amphoteric by virtue of containing both positively charged nitrogen atoms and negatively charged carboxyl or hydroxyl groups. Since PAA contains only positively charged primary and secondary nitrogen atoms, it could have significantly different bioavailability, efficacy, or toxicity than other classes of chelators previously studied.
A variety of metal chelators may be potentially useful for attenuating amyloid lesions in transgenic mice [36–46], but few are tested in human subjects. Nevertheless, several chelators showed promise in reducing amyloidosis in clinical studies, the first being deferoxamine [47]. Although it could dissolve amyloid aggregates, it has not been used extensively for treatment of AD due to low bioavailability, limited stability, and a propensity to cause anemia. However, some promising clinical results have been obtained by intranasal administration of deferoxamine [48]. Another metal chelator reported to reduce amyloid plaques in humans is clioquinol [45, 49] and its analogues VK-28, HLA-20, PBT2 and M-30. One limitation to the clinical use of clioquinol is its tendency to cause subacute myelopathic neuropathy (SMON) in humans [50].
Here our aim is to further test the chelation strategy in AD. For this, we initially selected PAA because it represents a class of metal chelators not previously investigated. Most metal chelators previously studied contain anionic groups, such as carboxylic or hydroxy groups, which impede transport across the blood-brain barrier (BBB). However, PAA only has primary and secondary cationic nitrogen groups. Our hypothesis is two-fold. First, the metal’s chelation by PAA, can significantly reduce overall amyloid plaque burden in transgenic mouse models of AD. Second, PAA results in reducing plaque seeding more than plaque growth. This is the first report to test the effects of a phenanthroline derivative, or any other cationic metal chelator, in tackling the growth and seeding of amyloid plaques. Although phenanthroline and its derivatives have been shown to have an affinity for numerous multivalent metals, those of biological relevance which have been implicated in AD are Zn, Fe, and Cu.
MATERIALS AND METHODS
Animals
The study used double transgenic female APPSWE-Tau P301L mice (Taconic, Rensselaer, NY). These mice were reported to develop brain lesions typical of AD, specifically amyloid plaques and NFTs [51]. The animals were used in accordance with animal use protocol HC-AD-029 and were overseen by the Histo-Chem’s Institutional Animal Care and Use Committee. Initially 12 animals were allotted into two groups of six. Within the first two months of dosing, two animals died in the control group and one animal in the experimental group died. Necropsy indicated that the cause of death was littermate aggression and, after providing individual housing, no more deaths occurred prior to sacrifice at one year of age. Animals were put on a 12 h on/12 h off light cycle, had full time access to rodent chow, and were given a weekly supplement of peanuts to optimize nutrition and vitality. The mice were allotted into an untreated control group (water), or a group orally dosed with a PAA solution from 4 months of age until their sacrifice at 12 months of age.
PAA dosing
1,10-phenanthroline-5-amine (Sigma Aldrich, St Louis, MO) was administered via the animal’s drinking water as a solution of 610 mg of PAA in 1l of water, with 1.7 ml of 25% HCL added to solubilize the compound. Treated animals were given free access to the dosed drinking water. Both groups of animals consumed an average of 3.5 ml of water or dosing solution daily. Thus, the experimental group received an average oral daily dose of 23 mg/kg of PAA. The dosing solution was clear, amber colored, had a pH of 4.8 and was mildly tart to the taste.
Histology
At 1 year of age all animals were euthanized via carbon dioxide at ambient temperature and perfused vascularly with 10% formalin. Their brains were removed and placed in the same fixative solution plus 20% sucrose. The brains were cut on a Leica CM 3050S cryostat into 25 μm thick sections through the extent of the cortex. Tissue sections were mounted onto gelatin coated slides and then a 1 in 3 series was stained with 8-hydroxyquinoline oxalate (HQ-O) and another 1 in 3 series was stained with Amylo-Glo (AG).
Tissue staining
Two different fluorescent tracers were used to localize amyloid plaques, specifically HQ-O and Amylo-Glo. HQ-O staining employed the original method [23]. Briefly, this involved immersing the slides successively in 100% ethanol then 70% ethanol and distilled water for 3 min each. Slides were then transferred to a 0.025% solution of HQ-O (Histo-Chem Inc., Jefferson AR), prepared by dissolving 25 mg of the tracer in 100 ml distilled water and heating the solution to 60°C in an incubator. Slides were stained at this temperature for 2 h and then rinsed for 3 min in two changes of distilled water. Slides were then dehydrated either by air drying on a slide warmer or by immersion in one change of 70% ethanol followed by two changes of 100% ethanol for 3 min per solution. Dehydrated slides were cleared briefly in xylene then cover-slipped with DPX non-polar mounting media (Sigma-Aldrich).
AG+ staining was essentially the same as originally described [52]. Briefly, tissue mounted slides were first immersed in 70% ethanol for 5 min and then in distilled water for 5 min. The slides were then transferred to the AG staining solution for 10 min. The staining solution was made by diluting the AG stock solution (Histo-Chem Inc., Jefferson AR) 1:10 with distilled water. Slides were then rinsed in distilled water, dehydrated, cleared in xylene and cover slipped with DPX mounting media.
Tissue analysis
For each of the two stains, a 1 in 3 series of tissue sections from the anterior prefrontal cortex to the posterior visual and entorhinal cortex was analyzed. All neo-cortical and allo-cortical (hippocampus, piriform cortex, and cingulate cortex) regions were examined except for the most anterior prefrontal cortex and the most caudal visual and entorhinal cortex. All regions were captured at 10X using a Nikon Eclipse 50i microscope equipped with Hg vapor lamp, blue and ultraviolet excitation filters, and a Nikon DSRi2 digital color camera. Once all images were captured, the Nikon NIS Elements program was used to determine both the number of plaques per animal and their total plaque areas in square microns.
Data analysis
The area was analyzed by mixed-level generalized linear model, with treatment and stain as fixed effects and animal as a random intercept effect. The “Area ∼ Treatment + Stain + Treatment×Stain” model was tested to determine whether specific stain choice would produce different results in addition to chelation. A similar model was applied to plaque counts, using a Poisson distribution to account for discontinuous data type (count). Dispersion ratio for this model was 34.15, indicating severe overdispersion. The model was repeated with a negative binomial distribution to correct for overdispersion. Finally, to determine if a change in plaque load was due more to smaller plaques or fewer plaques, the mean area per plaque (area ÷ count) was evaluated. After estimation, models were evaluated by analysis of deviance, commonly called “ANOVA” in deference to historical practices. Mixed-level models cannot generate conventional sums of squares, so χ2 statistics were used for hypothesis testing. Effect sizes of Treatment and Stain were estimated by ω2, which is a less-biased estimator of coefficients of determination than is η2 [53].
RESULTS
Condensed and dense core amyloid plaques were seen in virtually all neo- and allo-cortical regions in both experimental and control transgenic animals used in our experiments (Fig. 1). Notably, transgenic mice that had received daily oral administration of PAA exhibited significantly lower amyloid plaque areas and numbers than did age-matched control animals. This effect was seen in both AG+ and HQ-O-stained tissue sections. Specifically, AG+ stained amyloid plaques from the treated animals exhibited an average of 68% fewer plaques and 69% less plaque areas than the control group. Similarly, the HQ-O-stained tissue sections from the treated animals showed an average of 58% fewer plaques and a 62% reduction in average plaque areas (Fig. 2).
Fig. 1
Photomicrography of amyloid plaques in untreated and treated mice. Photomicrographs illustrate the typical differences in amyloid plaque numbers and areas in control (left column) and PAA treated (right column) AβPP/Tau mice. Amyloid plaques were localized by staining with either AG (top row) or HQ-O (bottom row). Shown are A, B) perirhinal cortex, C) dorsal hippocampus, and D) ventral sensory-motor cortex. Amylo-Glo and HQ-O required ultraviolet or blue light excitation, respectively. Magnification bars = 600 μm.
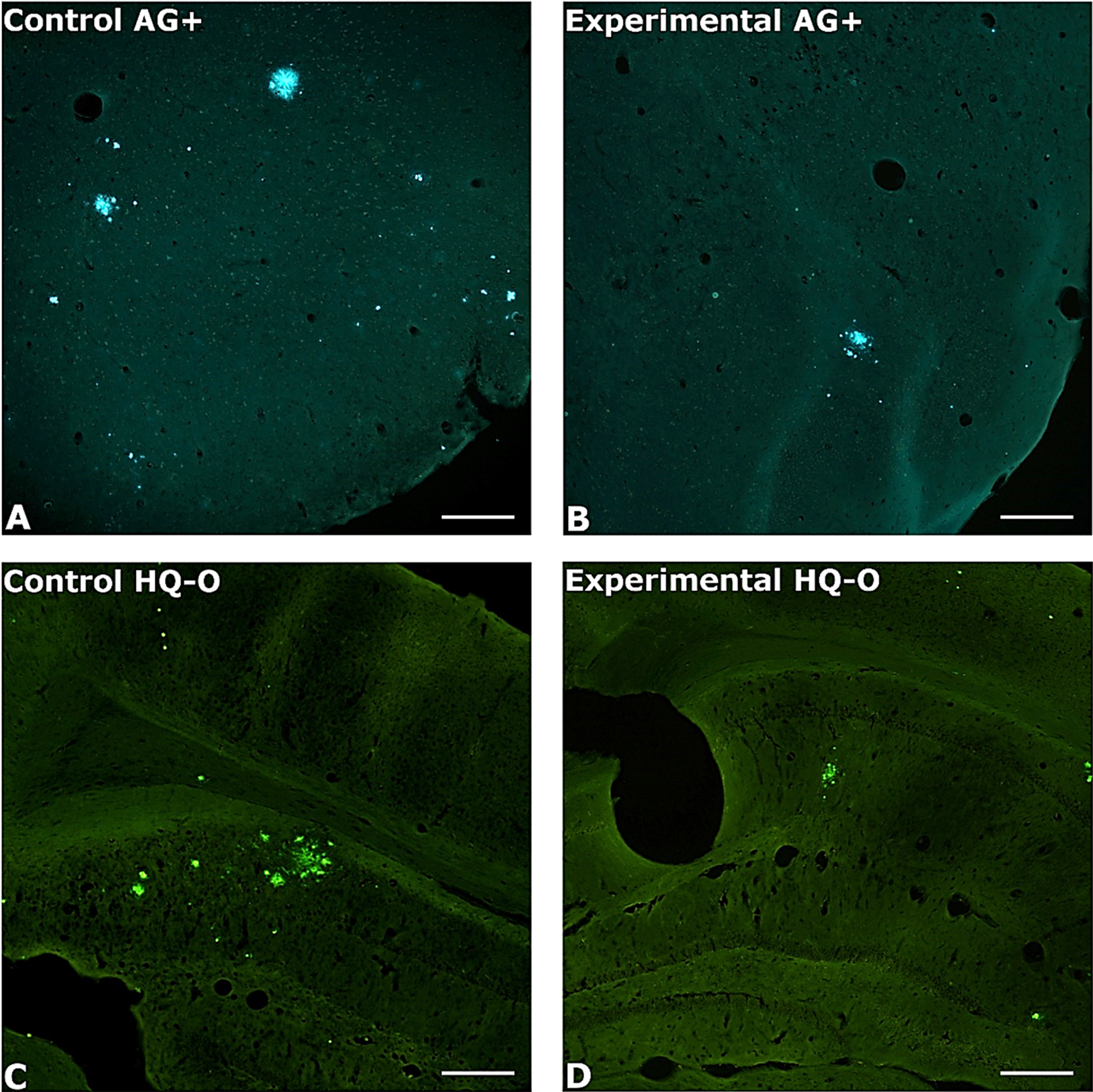
Fig. 2
Total area, total plaques, and area per plaque for untreated versus PAA treated mice. A) Total area, B) plaque counts, C) and area per plaque are shown. Both HQ-O and AG staining results are shown. Error bars represent standard errors of means. Symbols indicate pairwise comparisons (false discovery rate). Bars sharing a symbol did not significantly differ at p≤0.05.
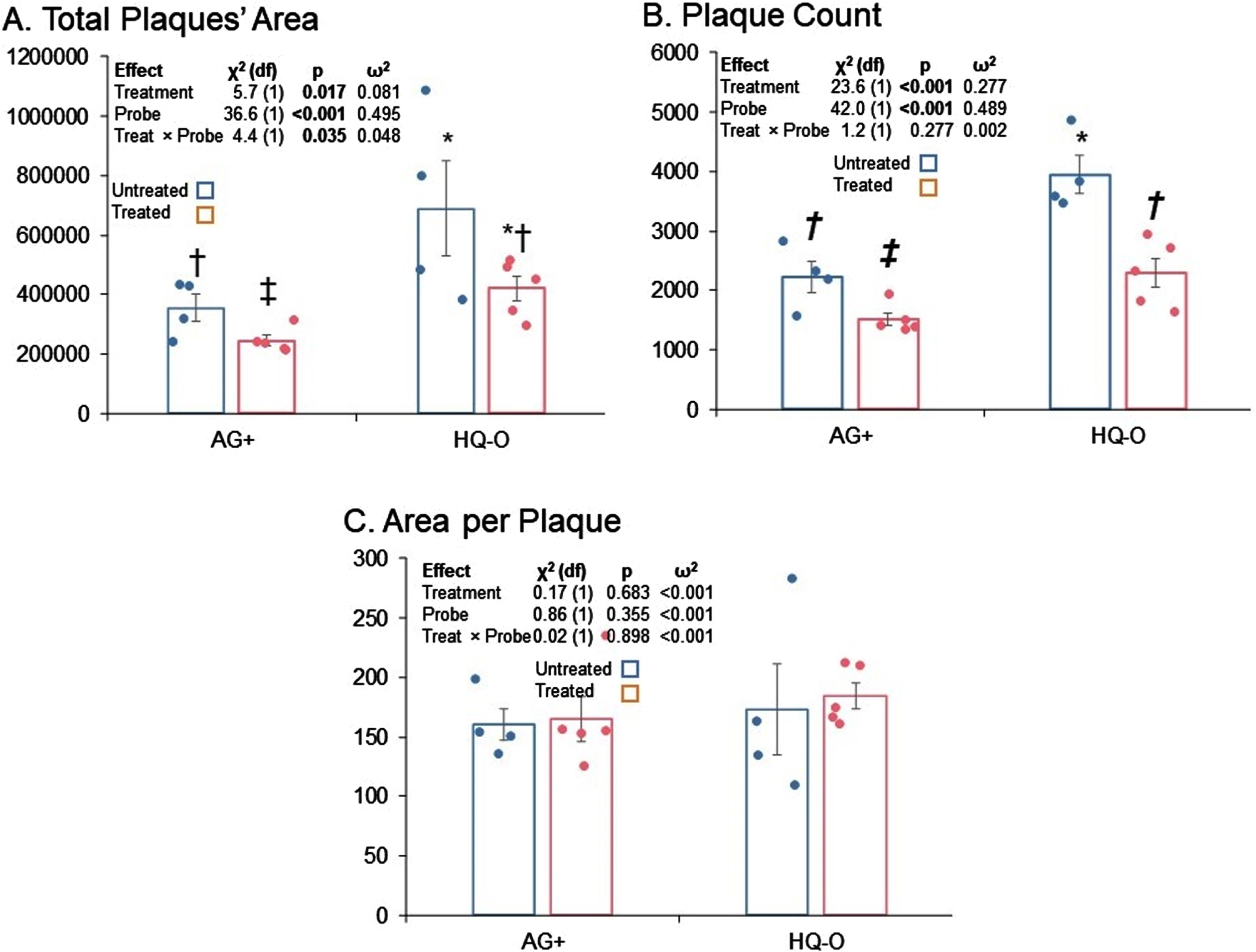
Treatment with PAA resulted in significantly reduced total plaque area; however, comparison of total areas by both treatment and stain type indicated that total plaque area estimated by HQ-O was higher than if estimated by AG+. Furthermore, the estimated difference between treatments was significantly greater for HQ-O than for AG+ (Fig. 2A). On the other hand, while total counts of plaques imaged by HQ-O were higher than if imaged by AG+, the significant reduction in total plaque counts associated with PAA treatment was essentially the same regardless of stain (Fig. 2B).
In order to determine whether apparent reduction in plaque load was due more to reduction in plaque seeding or in plaque growth, we also tested potential differences in mean area per plaque by treatment and stain (Fig. 2C). We found no significant differences. We interpret this to mean that, once a plaque seeded, PAA treatment did not alter its rate of growth. Thus, apparent differences in total plaque area by treatment reflected a difference in plaque seeding as we propose in Fig. 3.
DISCUSSION
Herein, we report the first positive findings with PAA in an AD mouse model. The implications of our results and possible underlying mechanisms need to be addressed. It is plausible that when certain polyvalent cationic metals bind to charged portions of the Aβ molecule they can form a scaffold that facilitates further aggregation. Studies of the coordination chemistry of zinc and Aβ suggest that the transition metal is bound in a tetrahedral position between positively charged histidine and negatively charged aspartate and glutamate amino acids on the non-helical portion of Aβ [54–56]. Zinc has an affinity for AβPP, Aβ, and tau fibrils [9]. Thus, chelators may exert therapeutic benefit by reducing the concentration of “plaque seeding metals” in the brain that facilitate initial aggregation of Aβ (Fig. 3). The precise anatomical locus of aggregation is unknown, although it may form within the synaptic clefts of cortical glutaminergic neurons that release both Aβ and zinc into the synaptic clefts, which is sometimes referred to as the “metal hypothesis” [57, 58].
Fig. 3
Seeding of amyloid plaques by transition metals. A) Schematic diagram depicts how certain transition metals, such as zinc (Zn +2), may act as a bridge or mordant to facilitate the auto aggregation of Aβ. Columns from left to right show the amino acid number, specific amino acid, charge of the amino acid and potential zinc binding sites. Charged amino acids in Aβ are histidine (H), glutamate (E), and aspartate (D). B) Proposed mechanism of metal chelation by phenanthroline amine (PAA), by binding to transition metals and thus inhibiting the aggregation of Aβ is illustrated.
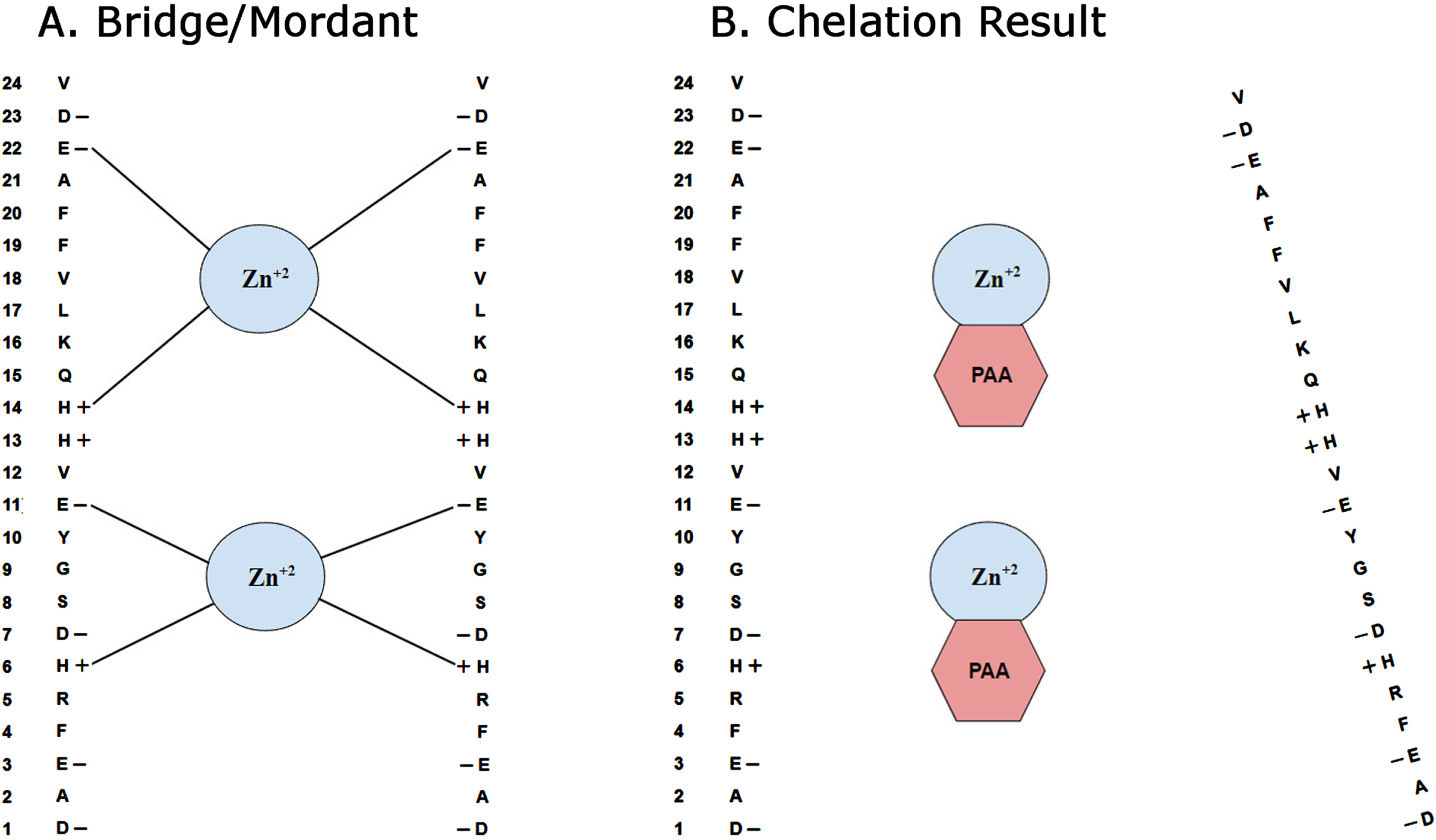
Although the underlying mechanism of action is not known definitively, our results support the hypothesis that the chelation of certain transition metals such as zinc, iron and copper can significantly reduce overall amyloid plaque burden in transgenic mouse models of AD. Particularly, when we look at the mean area per plaque versus total plaque area and numbers of plaques, we see that while both total area and counts were reduced, area per plaque is not. Thus, we conclude that chelation with PAA resulted in reduction of plaque seeding but not plaque growth. If both growth and seeding had been reduced, then area per plaque would have been reduced. If growth but not seeding had been reduced, then counts of plaques would not have been reduced.
Our outcome is a promising start, but several follow-up studies are needed before PAA could be considered as a treatment for humans with AD. In particular, no cognitive assessment was performed. Measuring effects of PAA treatment via Morris water maze performance would be a minimum necessary next step, since lack of cognitive improvement would indicate a treatment was effectless regardless of apparent changes in pathology. Unfortunately, no NFTs were seen in either group at 12 months of age. If, however, a more reliable model of tauopathies were to be found, it would be interesting to see if PAA might also reduce NFTs since zinc facilitates aggregation of neurofilaments as well [35, 59].
The focus of the present study is limited to understanding the pathobiochemical effects of PAA in an AD mice model. The present study resulted in no treatment related mortalities, weight loss or gross behavioral changes, but we did not conduct a comprehensive toxicology/pathology evaluation. However, examination of the liver, lung, spleen and lymph nodes in the rat following a single 10 mg i.p. injection of PAA indicated no pathological changes [60]. We maintain it would be necessary to also study the effects of chronic oral PAA dosing. Even though metal chelators have a long history of therapeutic use for treatment of metal intoxication, Zn, Fe, and Cu are all essential for health of the brain and other organs and therefore their levels should be monitored in any later-phase preclinical, and certainly any human study.
Pharmacokinetics and BBB permeability of PAA would also be important to determine between rodents and humans. It would seem probable that PAA gains access to the brain by virtue of its relative hydrophobicity and lack of anionic charged groups, but this remains to be proved. The present study employed a dosing regimen that started at 4 months of age, which is right around the time the amyloid plaques first become visible microscopically. This is roughly equivalent to beginning treatment in a 23-year-old human. Plaque load was assessed at 12 months, roughly equivalent to 43 years of age in humans [61]. It may be useful to consider animals that naturally develop age-related plaque, tangle, and neurodegeneration, such as domestic cat or dog [62, 63].
In short, we show novel properties of PAA in reducing amyloid burden in a mouse model of AD. The molecular mechanism, particularly the interaction of PAA with other targets, warrants further investigation. Translational control of APPmRNA, including the role of untranslated region (UTR), by iron regulatory protein-1, cytokines, and microRNA, is also emerging [64, 65]. In this context, future work on PAA’s effect, directly or indirectly, on APPmRNA translation, would be interesting.
In conclusion, our study supports the theory that dyshomeostasis of certain transition metals, particularly zinc, in the microenvironment of the brain contributes to amyloidosis [34, 58, 66]. Although the dysregulation may be attributable to impaired metal transporter function, there is presently no known treatment that would modify this. Therefore, reducing regional metal levels via chelation with PAA may be a feasible and viable strategy for suppressing the formation of amyloid plaques in AD patients.
ACKNOWLEDGMENTS
This study received funding from Histo-Chem Inc.
FUNDING
LS received funding from Histo-Chem Inc., DKL is partially supported by NIH grants R01AG051086-06, R56AG072810, R21AG056007 and R21AG076202.
CONFLICT OF INTEREST
Dr. Schmued is an employee of Histo-Chem Inc., which produces and markets both the AG+ and HQ-O stains. Dr. Lahiri is chief scientific advisor for Peptide Therapeutics Provaidya, LLC.
Debomoy K. Lahiri is also an Editorial Board Member of this journal but was not involved in the peer-review process nor had access to any information regarding its peer-review.
DATA AVAILABILITY
The data supporting the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
[1] | Lahiri DK ((2020) ) There is no failure, only discovery-the year ahead for CARving new paths. Curr Alzheimer Res 17: , 1–2. |
[2] | Lahiri DK , Maloney B , Song W , Sokol DK ((2022) ) Crossing the “birth border” for epigenetic effects. Biol Psychiatry 92: , e21–e23. |
[3] | Maloney B , Lahiri DK ((2016) ) Epigenetics of dementia: Understanding the disease as a transformation rather than a state. Lancet Neurol 15: , 760–774. |
[4] | Hardy J ((2009) ) The amyloid hypothesis for Alzheimer’s disease: A critical reappraisal. J Neurochem 110: , 1129–1134. |
[5] | Fleisher AS , Raman R , Siemers ER , Becerra L , Clark CM , Dean RA , Farlow MR , Galvin JE , Peskind ER , Quinn JF , Sherzai A , Sowell BB , Aisen PS , Thal LJ ((2008) ) Phase 2 safety trial targeting amyloid beta production with a gamma-secretase inhibitor in Alzheimer disease. Arch Neurol 65: , 1031–1038. |
[6] | Check E ((2002) ) Nerve inflammation halts trial for Alzheimer’s drug. Nature 415: , 462. |
[7] | Gomez-Gutierrez R , Sanchez-Varo R , Jimenez S , Navarro V , Vizuete M , Davila JC , Vitorica J , Gutierrez A ((2019) ) O4-09-03: Immunosuppression exacerbates Alzheimer’s disease pathology in APP/PS1 transgenic mice (Conference Abstract). Alzheimers Dement 15: , 1256–1257. |
[8] | Aisen PS , Davis KL , Berg JD , Schafer K , Campbell K , Thomas RG , Weiner MF , Farlow MR , Sano M , Grundman M , Thal LJ ((2000) ) A randomized controlled trial of prednisone in Alzheimer’s disease. Alzheimer’s Disease Cooperative Study. Neurology 54: , 588–593. |
[9] | Höglund K , Wiklund O , Vanderstichele H , Eikenberg O , Vanmechelen E , Blennow K ((2004) ) Plasma levels of beta-amyloid(1-40), beta-amyloid(1-42), and total beta-amyloid remain unaffected in adult patients with hypercholesterolemia after treatment with statins. Arch Neurol 61: , 333–337. |
[10] | Padala KP , Padala PR , McNeilly DP , Geske JA , Sullivan DH , Potter JF ((2012) ) The effect of HMG-CoA reductase inhibitors on cognition in patients with Alzheimer’s dementia: A prospective withdrawal and rechallenge pilot study. Am J Geriatr Pharmacother 10: , 296–302. |
[11] | Regland B , Lehmann W , Abedini I , Blennow K , Jonsson M , Karlsson I , Sjögren M , Wallin A , Xilinas M , Gottfries CG ((2001) ) Treatment of Alzheimer’s disease with clioquinol. Dement Geriatr Cogn Disord 12: , 408–414. |
[12] | Brandt WW , Dwyer FP , Gyarfas ED ((1954) ) Chelate complexes of 1,10-phenanthroline and related compounds. Chem Rev 54: , 959–1017. |
[13] | Nam KH ((2021) ) Structural analysis of metal chelation of the metalloproteinase thermolysin by 1,10-phenanthroline. J Inorg Biochem 215: , 111319. |
[14] | Matos CP , Adiguzel Z , Yildizhan Y , Cevatemre B , Onder TB , Cevik O , Nunes P , Ferreira LP , Carvalho MD , Campos DL , Pavan FR , Pessoa JC , Garcia MH , Tomaz AI , Correia I , Acilan C ((2019) ) May iron(III) complexes containing phenanthroline derivatives as ligands be prospective anticancer agents? Eur J Med Chem 176: , 492–512. |
[15] | Marino N , Ikotun OF , Julve M , Lloret F , Cano J , Doyle RP ((2011) ) Pyrophosphate-mediated magnetic interactions in Cu(II) coordination complexes. Inorg Chem 50: , 378–389. |
[16] | Song XD , Kong X , He SF , Chen JX , Sun J , Chen BB , Zhao JW , Mao ZW ((2017) ) Cyclometalated iridium(III)-guanidinium complexes as mitochondria-targeted anticancer agents. Eur J Med Chem 138: , 246–254. |
[17] | Schmued L , Raymick J ((2017) ) Introducing Euro-Glo, a rare earth metal chelate with numerous applications for the fluorescent localization of myelin and amyloid plaques in brain tissue sections. J Neurosci Methods 279: , 79–86. |
[18] | Bush AI , Pettingell WH , Multhaup G , d Paradis M , Vonsattel JP , Gusella JF , Beyreuther K , Masters CL , Tanzi RE ((1994) ) Rapid induction of Alzheimer A beta amyloid formation by zinc. Science 265: , 1464–1467. |
[19] | Faller P ((2009) ) Copper and zinc binding to amyloid-beta: Coordination, dynamics, aggregation, reactivity and metal-ion transfer. Chembiochem 10: , 2837–2845. |
[20] | Mantyh PW , Ghilardi JR , Rogers S , DeMaster E , Allen CJ , Stimson ER , Maggio JE ((1993) ) Aluminum, iron, and zinc ions promote aggregation of physiological concentrations of beta-amyloid peptide. J Neurochem 61: , 1171–1174. |
[21] | McLachlan DR , Smith WL , Kruck TP ((1993) ) Desferrioxamine and Alzheimer’s disease: Video home behavior assessment of clinical course and measures of brain aluminum. Ther Drug Monit 15: , 602–607. |
[22] | Pithadia AS , Lim MH ((2012) ) Metal-associated amyloid-β species in Alzheimer’s disease. Curr Opin Chem Biol 16: , 67–73. |
[23] | Schmued L , Raymick J , Sarkar S ((2019) ) High contrast and resolution labeling of amyloid plaques in tissue sections from APP-PS1 mice and humans with Alzheimer’s disease with the zinc chelator HQ-O: Practical and theoretical considerations. Curr Alzheimer Res 16: , 577–586. |
[24] | Tõugu V , Karafin A , Palumaa P ((2008) ) Binding of zinc(II) and copper(II) to the full-length Alzheimer’s amyloid-beta peptide. J Neurochem 104: , 1249–1259. |
[25] | Tõugu V , Tiiman A , Palumaa P ((2011) ) Interactions of Zn(II) and Cu(II) ions with Alzheimer’s amyloid-beta peptide. Metal ion binding, contribution to fibrillization and toxicity. Metallomics 3: , 250–261. |
[26] | Venkataramani V , Doeppner TR , Willkommen D , Cahill CM , Xin Y , Ye G , Liu Y , Southon A , Aron A , Au-Yeung HY , Huang X , Lahiri DK , Wang F , Bush AI , Wulf GG , Ströbel P , Michalke B , Rogers JT ((2018) ) Manganese causes neurotoxic iron accumulation via translational repression of amyloid precursor protein and H-Ferritin. J Neurochem 147: , 831–848. |
[27] | Lovell MA , Robertson JD , Teesdale WJ , Campbell JL , Markesbery WR ((1998) ) Copper, iron and zinc in Alzheimer’s disease senile plaques. J Neurol Sci 158: , 47–52. |
[28] | James SA , Churches QI , de Jonge MD , Birchall IE , Streltsov V , McColl G , Adlard PA , Hare DJ ((2017) ) Iron, copper, and zinc concentration in Aβ plaques in the APP/PS1 mouse model of Alzheimer’s disease correlates with metal levels in the surrounding neuropil. ACS Chem Neurosci 8: , 629–637. |
[29] | Curtain CC , Ali F , Volitakis I , Cherny RA , Norton RS , Beyreuther K , Barrow CJ , Masters CL , Bush AI , Barnham KJ ((2001) ) Alzheimer’s disease amyloid-beta binds copper and zinc to generate an allosterically ordered membrane-penetrating structure containing superoxide dismutase-like subunits. J Biol Chem 276: , 20466–20473. |
[30] | Danielsson J , Pierattelli R , Banci L , Gräslund A ((2007) ) High-resolution NMR studies of the zinc-binding site of the Alzheimer’s amyloid beta-peptide. FEBS J 274: , 46–59. |
[31] | Hare DJ , Lee JK , Beavis AD , van Gramberg A , George J , Adlard PA , Finkelstein DI , Doble PA ((2012) ) Three-dimensional atlas of iron, copper, and zinc in the mouse cerebrum and brainstem. Anal Chem 84: , 3990–3997. |
[32] | Lee JY , Mook-Jung I , Koh JY ((1999) ) Histochemically reactive zinc in plaques of the Swedish mutant beta-amyloid precursor protein transgenic mice. J Neurosci 19: , Rc10. |
[33] | Cuajungco MP , Fagét KY ((2003) ) Zinc takes the center stage: Its paradoxical role in Alzheimer’s disease. Brain Res Brain Res Rev 41: , 44–56. |
[34] | Tyszka-Czochara M , Grzywacz A , Gdula-Argasińska J , Librowski T , Wiliński B , Opoka W ((2014) ) The role of zinc in the pathogenesis and treatment of central nervous system (CNS) diseases. Implications of zinc homeostasis for proper CNS function. Acta Pol Pharm 71: , 369–377. |
[35] | Huang Y , Wu Z , Cao Y , Lang M , Lu B , Zhou B ((2014) ) Zinc binding directly regulates tau toxicity independent of tau hyperphosphorylation. Cell Rep 8: , 831–842. |
[36] | Cuajungco MP , Frederickson CJ , Bush AI ((2005) ) Amyloid-beta metal interaction and metal chelation. Subcell Biochem 38: , 235–254. |
[37] | Robert A , Liu Y , Nguyen M , Meunier B ((2015) ) Regulation of copper and iron homeostasis by metal chelators: A possible chemotherapy for Alzheimer’s disease. Acc Chem Res 48: , 1332–1339. |
[38] | Cuajungco MP , Fagét KY , Huang X , Tanzi RE , Bush AI ((2000) ) Metal chelation as a potential therapy for Alzheimer’s disease. Ann N Y Acad Sci 920: , 292–304. |
[39] | Sampson EL , Jenagaratnam L , McShane R ((2014) ) Metal protein attenuating compounds for the treatment of Alzheimer’s dementia. Cochrane Database Syst Rev,CD005380. |
[40] | Rogers JT , Lahiri DK ((2004) ) Metal and inflammatory targets for Alzheimer’s disease. Curr Drug Targets 5: , 535–551. |
[41] | Bandyopadhyay S , Huang X , Lahiri DK , Rogers JT ((2010) ) Novel drug targets based on metallobiology of Alzheimer’s disease. Expert Opin Ther Targets 14: , 1177–1197. |
[42] | Sharma A , Pachauri V , Flora SJS ((2018) ) Advances in multi-functional ligands and the need for metal-related pharmacology for the management of Alzheimer disease. Front Pharmacol 9: , 1247. |
[43] | Fasae KD , Abolaji AO , Faloye TR , Odunsi AY , Oyetayo BO , Enya JI , Rotimi JA , Akinyemi RO , Whitworth AJ , Aschner M ((2021) ) Metallobiology and therapeutic chelation of biometals (copper, zinc and iron) in Alzheimer’s disease: Limitations, and current and future perspectives. J Trace Elem Med Biol 67: , 126779. |
[44] | Lakey-Beitia J , Burillo AM , La Penna G , Hegde ML , Rao KS ((2021) ) Polyphenols as potential metal chelation compounds against Alzheimer’s disease. J Alzheimers Dis 82: , S335–s357. |
[45] | Zhang YH , Raymick J , Sarkar S , Lahiri DK , Ray B , Holtzman D , Dumas M , Schmued LC ((2013) ) Efficacy and toxicity of clioquinol treatment and A-beta42 inoculation in the APP/PSI mouse model of Alzheimer’s disease. Curr Alzheimer Res 10: , 494–506. |
[46] | Budimir A ((2011) ) Metal ions, Alzheimer’s disease and chelation therapy. Acta Pharm 61: , 1–14. |
[47] | Crapper McLachlan DR , Dalton AJ , Kruck TP , Bell MY , Smith WL , Kalow W , Andrews DF ((1991) ) Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet 337: , 1304–1308. |
[48] | Guo C , Hao LJ , Yang ZH , Chai R , Zhang S , Gu Y , Gao HL , Zhong ML , Wang T , Li JY , Wang ZY ((2016) ) Deferoxamine-mediated up-regulation of HIF-1α prevents dopaminergic neuronal death via the activation of MAPK family proteins in MPTP-treated mice. Exp Neurol 280: , 13–23. |
[49] | Cherny RA , Atwood CS , Xilinas ME , Gray DN , Jones WD , McLean CA , Barnham KJ , Volitakis I , Fraser FW , Kim Y , Huang X , Goldstein LE , Moir RD , Lim JT , Beyreuther K , Zheng H , Tanzi RE , Masters CL , Bush AI ((2001) ) Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron 30: , 665–676. |
[50] | Tateishi J ((2000) ) Subacute myelo-optico-neuropathy: Clioquinol intoxication in humans and animals.S. Neuropathology 20 Suppl: , 20–24. |
[51] | Ribé EM , Pérez M , Puig B , Gich I , Lim F , Cuadrado M , Sesma T , Catena S , Sánchez B , Nieto M , Gómez-Ramos P , Morán MA , Cabodevilla F , Samaranch L , Ortiz L , Pérez A , Ferrer I , Avila J , Gómez-Isla T ((2005) ) Accelerated amyloid deposition, neurofibrillary degeneration and neuronal loss in double mutant APP/tau transgenic mice. Neurobiol Dis 20: , 814–822. |
[52] | Schmued L ((2022) ) Use of a stable and solvent-soluble variant of Amylo-Glo, Amylo-Glo+, for the high-contrast and resolution fluorescent localization of amyloid plaques in murine and human brain tissue sections. J Neurosci Methods 365: , 109381. |
[53] | Olejnik S , Algina J ((2003) ) Generalized eta and omega squared statistics: Measures of effect size for some common research designs. Psychol Methods 8: , 434–447. |
[54] | Atrián-Blasco E , Gonzalez P , Santoro A , Alies B , Faller P , Hureau C ((2018) ) Cu and Zn coordination to amyloid peptides: From fascinating chemistry to debated pathological relevance. Coord Chem Rev 375: , 38–55. |
[55] | Miller Y , Ma B , Nussinov R ((2010) ) Zinc ions promote Alzheimer Abeta aggregation via population shift of polymorphic states. Proc Natl Acad Sci U S A 107: , 9490–9495. |
[56] | Pan L , Patterson JC ((2013) ) Molecular dynamics study of Zn(aβ) and Zn(aβ)2. PLoS One 8: , e70681. |
[57] | Bush AI , Tanzi RE ((2008) ) Therapeutics for Alzheimer’s disease based on the metal hypothesis. Neurotherapeutics 5: , 421–432. |
[58] | Crouch PJ , White AR , Bush AI ((2007) ) The modulation of metal bio-availability as a therapeutic strategy for the treatment of Alzheimer’s disease. FEBS J 274: , 3775–3783. |
[59] | Mo ZY , Zhu YZ , Zhu HL , Fan JB , Chen J , Liang Y ((2009) ) Low micromolar zinc accelerates the fibrillization of human tau via bridging of Cys-291 and Cys-322. J Biol Chem 284: , 34648–34657. |
[60] | Wesselinova D , Kaloyanov N , Dimitrov G ((2009) ) Cytotoxicity and effects of 1,10-phenanthroline and 5-amino-1,10-phenanthroline derivatives on some immunocompetent cells. Eur J Med Chem 44: , 5099–5102. |
[61] | Hagan C, When are mice considered old? Jackson Laboratory. https://www.jax.org/news-and-insights/jax-blog/2017/november/when-are-mice-considered-old. |
[62] | McKean NE , Handley RR , Snell RG ((2021) ) A review of the current mammalian models of Alzheimer’s disease and challenges that need to be overcome. Int J Mol Sci 22: , 13168. |
[63] | Chambers JK , Tokuda T , Uchida K , Ishii R , Tatebe H , Takahashi E , Tomiyama T , Une Y , Nakayama H ((2015) ) The domestic cat as a natural animal model of Alzheimer’s disease. Acta Neuropathol Commun 3: , 78. |
[64] | Cho HH , Cahill CM , Vanderburg CR , Scherzer CR , Wang B , Huang X , Rogers JT ((2010) ) Selective translational control of the Alzheimer amyloid precursor protein transcript by iron regulatory protein-1. J Biol Chem 285: , 31217–31232. |
[65] | Long JM , Maloney B , Rogers JT , Lahiri DK ((2019) ) Novel upregulation of amyloid-β precursor protein (APP) by microRNA-346 via targeting of APP mRNA 5’-untranslated region: Implications in Alzheimer’s disease. Mol Psychiatry 24: , 345–363. |
[66] | Xu Y , Xiao G , Liu L , Lang M ((2019) ) Zinc transporters in Alzheimer’s disease. Mol Brain 12: , 106. |


