
Extracellular Tau Oligomers Damage the Axon Initial Segment
Abstract
Background:
In Alzheimer’s disease (AD) brain, neuronal polarity and synaptic connectivity are compromised. A key structure for regulating polarity and functions of neurons is the axon initial segment (AIS), which segregates somatodendritic from axonal proteins and initiates action potentials. Toxic tau species, including extracellular oligomers (xcTauOs), spread tau pathology from neuron to neuron by a prion-like process, but few other cell biological effects of xcTauOs have been described.
Objective:
Test the hypothesis that AIS structure is sensitive to xcTauOs.
Methods:
Cultured wild type (WT) and tau knockout (KO) mouse cortical neurons were exposed to xcTauOs, and quantitative western blotting and immunofluorescence microscopy with anti-TRIM46 monitored effects on the AIS. The same methods were used to compare TRIM46 and two other resident AIS proteins in human hippocampal tissue obtained from AD and age-matched non-AD donors.
Results:
Without affecting total TRIM46 levels, xcTauOs reduce the concentration of TRIM46 within the AIS and cause AIS shortening in cultured WT, but not TKO neurons. Lentiviral-driven tau expression in tau KO neurons rescues AIS length sensitivity to xcTauOs. In human AD hippocampus, the overall protein levels of multiple resident AIS proteins are unchanged compared to non-AD brain, but TRIM46 concentration within the AIS and AIS length are reduced in neurons containing neurofibrillary tangles.
Conclusion:
xcTauOs cause partial AIS damage in cultured neurons by a mechanism dependent on intracellular tau, thereby raising the possibility that the observed AIS reduction in AD neurons in vivo is caused by xcTauOs working in concert with endogenous neuronal tau.
INTRODUCTION
A hallmark of Alzheimer’s disease (AD) is the accumulation of neurofibrillary tangles (NFTs) in neurons, which correspond to tightly packed filament bundles of tau, a microtubule-associated protein normally enriched in axons [1–3]. Prior to NFT formation, misfolded forms of tau, including monomers, oligomers, and short filaments, can escape from diseased neurons, and then enter nearby neurons provoking toxic misfolding of their endogenous tau [4–8]. This prion-like mechanism of tau misfolding may explain how secreted tau propagates intracellular tau pathology from neuron to neuron [9–14].
While the mechanisms of inter-neuronal tau exchange have been extensively studied, knowledge of how neuronal structure and behavior are affected by this process is limited. What is known, however, points to profound effects on neuronal polarity and neurotransmission. For example, we previously showed that one conspicuous effect of extracellular tau oligomers (xcTauOs) on cultured mouse cortical neurons is the accumulation of endogenous tau in the somatodendritic compartment [15]. Prior studies have demonstrated that excess somatodendritic tau can cause hyperexcitability of NMDA receptors, synapse loss, and neuron death [16–20], and that xcTauOs can alter action potential dynamics, disrupt synaptic transmission, and impair neuronal plasticity [21].
The loss of endogenous tau polarity caused by xcTauOs [15] prompted us to test the hypothesis that the axon initial segment (AIS) is a target of xcTauOs. The AIS serves two major functions. It is the site of action potential initiation, and it serves as a diffusion barrier that helps to segregate somatodendritic from axonal molecules and organelles [22–26]. The AIS is located within the first 20–60μm of the proximal end of the axon, and its length and distance from the soma are instrumental for initiating and shaping action potentials [27–29]. The AIS is organized by the sub-membranous cytoskeletal proteins, ankyrin-G [22, 30–36] and βIV-spectrin [37–40]; the transmembrane cell adhesion protein, neurofascin-186 (NF-186) [26, 41–43]; and the microtubule-associated protein, TRIM46 [41, 44–46]; among other proteins. Notably, depletion of AIS structural proteins results in AD-like tau accumulation in the somatodendritic compartment [44, 47].
Dysregulation or partial loss of the AIS, as evidenced by even small morphometric changes in AIS structure, has been implicated in various adverse effects both in vitro in cultured neurons and in vivo in brain [24, 29, 47–53]. However, evidence regarding AIS integrity in human AD brain is contradictory. Sohn and colleagues reported that AIS structure is reduced in AD neurons [52], whereas Antón-Fernández and colleagues provided evidence that hyperphosphorylated tau in AD neurons correlates with a longer AIS [48]. One reason this discrepancy has not been resolved is that direct effects of tau aggregates, like xcTauOs and filaments, on AIS protein content and morphology had never been investigated to the best of our knowledge. Accordingly, we decided to address the following questions: 1) Is AIS morphology affected by xcTauOs? 2) Are AIS protein levels downregulated in AD hippocampus? 3) Is AIS length correlated with the presence of NFTs in the same neuron?
To address these questions, we quantified AIS protein content and morphology in primary mouse cortical neuron cultures and postmortem human hippocampal tissue using quantitative western blotting and immunofluorescence microscopy. For cultured neurons, we found that xcTauOs cause a reduction of the TRIM46 concentration within the AIS and AIS shortening from its distal end, but no changes in overall TRIM46 protein levels. Similarly, in human AD hippocampus, we found that the overall levels of multiple resident AIS proteins are unchanged compared to non-AD brain, but TRIM46 concentration within the AIS and AIS length are reduced in NFT-containing neurons. Collectively, these findings prompt the hypothesis that xcTauOs work in concert with endogenous intraneuronal tau to degrade the AIS in vivo AD brain.
MATERIALS AND METHODS
Mice
Wild type (WT; C57/BL6J, https://www.jax.org/strain/000664) and tau knockout (KO, https://www.jax.org/strain/007251) mice were used for this study. Animals were maintained, bred, and euthanized in compliance with all policies of the Animal Care and Use Committee of the University of Virginia.
Cultured neurons
Primary mouse cortical neuron cultures were prepared from WT and tau KO E16.5 embryonic brains using a standard protocol [54]. Briefly, brain cortices were dissected from embryos and minced with a scalpel in Hank’s balanced salt solution (HBSS, Gibco 14175-095) containing 0.25% trypsin (Gibco 15090-046) and deoxyribonuclease I (Worthington LK003170). The minced tissue suspension was incubated at 37°C for 30 min, the dissociated tissue was then pelleted in a clinical centrifuge, re-suspended in HBSS, and triturated using a plastic P1000 pipette tip or a glass Pasteur pipette. After centrifugation at 900 RPM for 10 min, the cells were suspended in Neurobasal medium (Gibco 21103-049) supplemented with 5% fetal bovine serum (Optima FBS, R&D Systems S12450), B-27 (Gibco 17504-044) or B-27+ (Gibco A35828-01), 2 mM L-glutamine (Gibco 25030-081), 50μg/ml gentamicin (Gibco 15710-064), and 15 mM D-(+)-glucose monohydrate (Sigma-Aldrich 16301). The cells were then plated at a density of 1.125 million cells/cm2 on #1 thickness, 12 mm round glass coverslips (Chem Glass CLS-1760-012) in 24-well dishes (Corning 353504), or without coverslips in 12-well (Costar, Cat# 3513) or 6-well (CELLSTAR 657-160) plates following coating of the cell attachment surfaces with poly-D-lysine hydrobromide (Sigma-Aldrich P0899). Cultures were placed in a humidified incubator maintained at 37°C and 5% CO2, the following day the medium was replaced with a fresh medium that lacked serum, and finally the cells were grown for a total of 7–22 days with 50% medium changes at least once per week. Primary cortical neurons were treated at 6–21 days in vitro for 24 hours with xcTauOs diluted to 125 nM total tau or with vehicle.
Tau lentivirus
Lentiviral particles encoding WT human 2N4R tau were prepared using the TRIPZ Trans-Lentiviral Open Reading Frame kit [55] in conjunction with the pBOB-NepX or pLKO.1 expression plasmids, and the packaging vectors, pSPAX2 and pMD2.G (Addgene plasmids 12260 and 12259, respectively; deposited by Dr. Didier Trono, École Polytechnique Fédérale de Lausanne). These collective plasmids were transfected using Lipofectamine 3000 (Thermo Fisher Scientific L3000-015) into HEK293T cells grown in Dulbecco’s Modified Eagle Medium (Gibco 11965-092) supplemented with Cosmic Calf Serum (HyClone SH30087.03) to 10% and gentamycin to 50μg/ml. Lentivirus-conditioned medium was collected 48 h and 72 h after the start of transfection. The lentiviral particles were pelleted out of medium using a Beckman Coulter Optima LE-80K Ultracentrifuge and an SW28 rotor spun at 23,000 RPM for 2 h at 4°C. The final pellet was resuspended in 400μl Neurobasal medium, dispensed into 40μl volumes, and stored at –80°C.
Primary cortical neuron cultures derived from tau KO mice were transduced with the tau-encoding lentivirus. Experimental manipulations of the cultures were begun 48 h after initial exposure to lentivirus.
Purification and oligomerization of recombinant human tau
BL21 E. coli cells were transformed with expression vectors for 1N4R or 2N4R WT human tau, each with a C-terminal his tag (kindly provided by Nicholas Kanaan of Michigan State University). Tau was purified out of soluble extracts of transformed BL21 cells using TALON Metal Affinity Resin (TaKaRa 635502) and imidazole (Sigma-Aldrich I-0125) according to the vendors’ instructions. The purified 1N4R and 2N4R solutions were concentrated in Amicon Ultra-4 Centrifugal Filters 10K (Sigma-Aldrich UFC801024) spun in a clinical centrifuge at 3,000 RPM at 4°C for 15-min intervals until the concentrate volume was ∼500μl [56]. Dithiothreitol (DTT, GE Healthcare 17-1318-02) was added to 1 mM, and the tau was then dispensed into 40–50μl aliquots that were stored at –80°C. The protein concentration was measured using reducing agent-compatible BCA Protein Assay Kit (Abcam 207003).
The 1N4R and 2N4R oligomers were made as previously described [56] and summarized here. First, the buffer in which the purified tau was dissolved was changed to 0.1 M phosphate at pH 7.4 using Amicon Ultra-4 Centrifugal Filters 10K (Sigma-Aldrich UFC801024) according to the manufacturer’s instructions. Next, the tau was diluted to 8μM into a solution of 5 mM DTT, 100 mM NaCl, 10 mM HEPES (Gibco 15630-080) at pH 7.6, 0.1 mM EDTA and 300μM arachidonic acid (Cayman Chemical Company 90010.1), and was incubated in the dark at room temperature for 6–18 h. Vehicle controls were prepared identically, except that 0.1 M phosphate buffer was used in place of tau.
Protein electrophoresis and western blotting
Primary and secondary antibodies are listed in Supplementary Table 1, and all blots were scanned using a LI-COR Odyssey infrared imager.
Tau oligomer
Tau oligomer aliquots were mixed 3:1 with 4X NuPAGE LDS Sample Buffer (Thermo Fisher Scientific NP0007) and stored at –20°C. Thawed samples were electrophoresed in NuPAGE 4–12% Bis-Tris Gels (Thermo Fisher Scientific NP0323) using 1X MES running buffer (Thermo Fisher Scientific NP0002). After electrophoresis, gels were transferred for 1.5 h at 100 V to 0.2μm pore size nitrocellulose membranes (Bio-Rad 1620112) at 4°C. The membranes were then blocked with Intercept Blocking Buffer TBS (TBS blocking buffer, LI-COR 927-60001) at room temperature for 1 h and incubated at 4°C overnight with the pan-tau mouse monoclonal antibody, Tau-5. Next, blots were washed twice with TBS/Tween 20 (TBST, VWR 0777) for 10 min each and incubated at room temperature for 1 h with secondary antibody: IRDye 800 CW goat anti-mouse IgG. Following two more 10-min washes in TBST and one rinse with TBS, blots were scanned.
Cultured neurons
Cultured neurons were collected in NuPAGE LDS Sample Buffer and 25% glycerol (Sigma-Aldrich G7893), and samples were stored at –20°C until use. Electrophoresis and western blotting were performed as described above for tau oligomers, except for the primary and secondary antibodies that were used (see Supplementary Table 1).
Human hippocampus
Human hippocampus samples were classified as non-AD or AD based on criteria of the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD), Braak staging or both. Unfractionated tissue lysates were prepared as described [30]. Tissue was homogenized manually using a plastic pestle (VWR 47747-358) in 10 ml/g tissue mass of a solution of 8M urea (Sigma-Aldrich U5378), 5 mM N-ethylmaleimide (Sigma-Aldrich E3876), 5% SDS, and 1:100 HALT protease inhibitor cocktail (Thermo Fisher Scientific 78430). The homogenate was mixed 1:1 with 5X PAGE buffer (5% SDS, 25% sucrose, 50 mM Tris at pH 8.5, 1 mM EDTA, 30 mg/ml DTT and bromophenol blue as a tracking dye). Ankyrin-G blots were made as described earlier [30] using 3.5–17.5% gradient gels in 1X Tris buffer, pH 7.4. Gels were transferred overnight at 4°C at 300 mA to 0.45μm pore size nitrocellulose membranes in 0.5X Tris buffer with 0.01% SDS. Membranes were blocked in TBS containing 5% BSA for 1 h followed by incubation overnight with primary antibody (rabbit anti-ankyrin-G [57] plus mouse monoclonal anti-α-tubulin), and then for 1 h with secondary antibodies (IRDye 800 goat anti-rabbit IgG and IRDye 680 goat anti-mouse IgG diluted into TBST supplemented with 5% bovine serum albumin - BSA, Sigma-Aldrich A3803). Blots were washed 3 times in TBST for 15 min each after each antibody step, and finally once each in TBS and H2O after the secondary antibodies. The Tau (pS202/T205) , neurofascin-186, TRIM46, and mouse monoclonal anti-βIII-tubulin (clone Tuj1) blots were processed as described above for tau oligomers and cultured neurons. Secondary antibodies for these blots were IRDye 680RD goat anti-rabbit IgG and IRDye 800 CW goat anti-mouse IgG.
Immunofluorescence microscopy
Primary and secondary antibodies are listed in Supplementary Table 1. Primary mouse neuron cultures grown on 12 mm glass coverslips in 24-well plates were fixed in PBS containing 4% paraformaldehyde for 15 min. The cultures were then washed sequentially with the following ice-cold solutions: twice in PBS for 2 min each, once in PBS containing 0.1 M glycine for 5 min, and once in PBS for 2 min. Next, the fixed cells were permeabilized and blocked for 1 h in PBS containing 1% BSA, 0.05% saponin (Sigma-Aldrich 84510), and 0.1% normal goat serum (Southern Biotech 0060-01). Then, they were incubated with various combinations of primary antibodies (chicken polyclonal anti-TRIM46, chicken polyclonal anti-MAP2, rabbit polyclonal anti-TRIM46, and mouse monoclonal anti-tau (clone 5) and mouse monoclonal anti-GFAP) followed by secondary antibodies (Alexa Fluor 568 goat anti-chicken IgY, Alexa Fluor 647 goat anti-rabbit IgG, Alexa Fluor 488 goat anti-mouse IgG) for 1 h. Following each antibody step the coverslips were washed twice in PBS containing 0.05% saponin for 2 min each. Immediately prior to sealing the coverslips to glass slides with Fluoromount G (Southern Biotech 0100-01), the cells were incubated with 0.2μg/ml Hoechst 33342 trihydrochloride trihydrate (Thermo Fisher Scientific H3570) in PBS for 2 min, then with H2O to remove excess dye and salts.
Antibody-labeled neuron cultures
Antibody-labeled neuron cultures were observed on an inverted Nikon Eclipse Ti microscope equipped with a Yokogawa CSU-X1 spinning disk confocal head, a Hamamatsu Flash 4.0 scientific C-MOS camera, a Nikon 40X oil 1.4 NA Plan Apo objective, and 405 nm, 488 nm, 561 nm, and 640 nm lasers. From each coverslip, 20 unique fields of cells with at least 20 cells per treatment group were imaged. All analyses were performed by 4 examiners (MNB, YL, NNF, and NK) blinded to the treatments using the FIJI version of ImageJ. More specifically, we analyzed: 1) the shortest distance between the nucleus and the AIS (nucleus-AIS gap) measured using the “straight line tool”; 2) mean TRIM46 immunofluorescence intensity within the AIS measured using an “AIS Analysis” plugin (https://github.com/ksiller/aisanalyzer) created by KHS; and 3) AIS length (region-of-interest; ROI outlining the AIS thresholded at 50% fluorescence intensity of a skeletonized line measuring the length of the AIS) measured using the “AIS Analysis” plugin. The plugin processes pairs of fluorescent images and corresponding FIJI/ImageJ ROI files. The output includes automated nucleus and AIS ROIs that aid in the quantification of AIS morphometric parameters (Supplementary Figure 1).
Human brain hippocampus sections
Human brain hippocampus sections were first examined by JWM to assess quality of fixation. Brains that did not show autolysis by loss of nuclear integrity were embedded in paraffin and cut into 4μm thick sections from coronal blocks. The sections were deparaffinized using xylenes (Fischer Chemical X5-4), and antigen retrieval was accomplished using antigen unmasking solution, citrate-based (Vector Laboratories H-3300-250), and heat, according to the vendor’s instructions. After cooling, the sections were washed twice in PBS for 5 min each, and then were permeabilized and blocked as described above for cultured mouse neurons. The primary antibodies included chicken polyclonal anti-MAP2, rabbit polyclonal anti-TRIM46, and Tau-5, and the secondary antibodies were as described above for cultured mouse neurons. The sections were washed twice in PBS for 5 min each after the primary and secondary antibody steps. Immediately prior to sealing the slides to glass coverslips with fluoromount G, lipofuscin autofluorescence was eliminated using the autofluorescence eliminator reagent (Sigma-Aldrich 2160) following the vendor’sprotocol.
The brain tissue sections were imaged with a Lecia Thunder Microscope equipped with a 40X dry objective, and 488 nm, 568 nm, and 640 nm lasers. All images were acquired using the same camera settings and laser intensity for consistent image analysis. From the CA1 region of each brain, 55 single plane widefield images were captured and stitched together and accounted for an average of 72 neurons per brain. All images were analyzed using FIJI software. All analyses were done with the investigator blinded to the age, sex, and neuropathological diagnosis of the case. First, image files were loaded into FIJI. Second, image files were evaluated according to inclusion criteria: neuronal somas (MAP2 channel = 568 nm) and connecting AIS (TRIM46 channel = 647 nm) in the field of view. If criteria were met, the image was saved as a TIFF file for further analyses. Third, using the TRIM46 channel, background fluorescence intensity levels were measured as the average median gray value of three ROIs in areas void of AIS channel signal using “Measure” option under the drop-down menu labeled “Analyze”. Fourth, on the same image the freehand line tool was used to generate a manual tracing of the full AIS. The plot profile of the tracing was displayed using “Plot profile” option under the drop-down menu labeled “Analyze”. The plot values were displayed, and the distance (microns) and gray values were copied into a spreadsheet. Fifth, the Tau-5 antibody, which recognizes all tau isoforms independently of aggregation state or any post-translational modifications they may harbor, was used to determine whether each neuronal soma associated with an AIS contained “no tau accumulation” (absence of Tau-5 signal), “tau accumulation without NFTs” (diffuse Tau-5 signal) or “NFTs present” (Tau-5 positive, perinuclear flame-shaped structure that does not occupy the entirety of the cell; Supplementary Figure 2). The data from all the neurons in an individual brain section were aggregated into a single spreadsheet that was exported into R for statistical analysis (as described below: statistics).
Analysis Scripts
The “AIS Analysis” (https://github.com/ksiller/aisanalyzer) and statistical analysis (https://github.com/mariekekjones/Alzheimer_AIS_data_analysis) codes are available online at the websites indicated here.
Statistics
Statistical analyses were performed in GraphPad Prism or in R. For cell culture western blots, data were analyzed across vehicle and xcTauOs groups using two-tailed independent Student’s t-tests, pooled variance. For cell culture immunocytochemistry, data were analyzed at the neuron level using linear mixed effects models that consider the measurement of several neurons within each coverslip. AIS morphometric parameters were explained by treatment condition (vehicle and xcTauOs) and the interaction term. Human brain tissue sample selection was based on tissue availability, and experiments were completed with 5–9 biologically distinct samples per group. For human brain western blots, the data were analyzed across non-AD and AD groups using two-tailed independent Student’s t-tests, unpooled variance. For human brain immunohistochemistry, data were analyzed at the neuron level using linear mixed effects models that consider the measurement of several neurons within each brain. AIS morphometric parameters were explained by disease state (nonAD and AD), tau pathology (“no tau accumulation”, “tau accumulation without NFTs” or “NFTs present”), and the interaction term. Because neurons from both non-AD and AD brains had tau accumulation and tangles, it was critical to analyze our data at the neuron level.
RESULTS
xcTauOs cause partial AIS loss in cultured neurons
Considering the evidence that xcTauOs cause accumulation of endogenous tau in the somatodendritic compartment [15, 20], a likely sign of AIS functional impairment [24, 47, 49, 51, 52], we tested the hypothesis that AIS structure is compromised by xcTauOs. To do so, we exposed primary mouse cortical neuron cultures to xcTauOs made from purified recombinant human 1N4R or 2N4R tau (Supplementary Figure 3) and used immunofluorescence microscopy and western blotting as quantitative morphometric and biochemical readouts, respectively, to monitor effects on the AIS. For the experiments described here, we used xcTauOs made from 1N4R and 2N4R tau interchangeably since both affected the AIS indistinguishably without any obvious effects on overall neuronal morphology, the density of axons and dendrites, or cell viability (see, for example, Figs. 1A and 2A).
Fig. 1
xcTauOs reduce clustering of TRIM46 within the AIS and AIS length. A) Localization of TRIM46 (white arrows indicate proximal AIS ends), tau (Tau-5), somatodendritic compartment (MAP2), and nuclei (Hoechst) in primary WT mouse cortical neuron cultures. B) Quantitative western blotting of unfractionated WT cultures probed with anti-TRIM46, and as a loading control, the neuron-specific anti-βIII-tubulin. p-value is based on two-tailed unpaired t-tests, pooled variance. Bar graph with mean and error bars represent±standard error of the mean. Plots of (C) Nucleus-AIS gap; (D) mean TRIM46 immunofluorescence intensity within the AIS, a surrogate measure of TRIM46 concentration; and (E) AIS length. Graph data were measured from 3–5 biological replicates (independent neuronal culture preparations). Immunofluorescence was performed with 1 technical replicate (independent coverslip) each, which accounted for a total of ∼200 neurons per treatment condition (vehicle or xcTauOs). p-values from mixed model linear regression are indicated. Large dot indicates the predicted mean and error bars represent the 95% confidence interval.
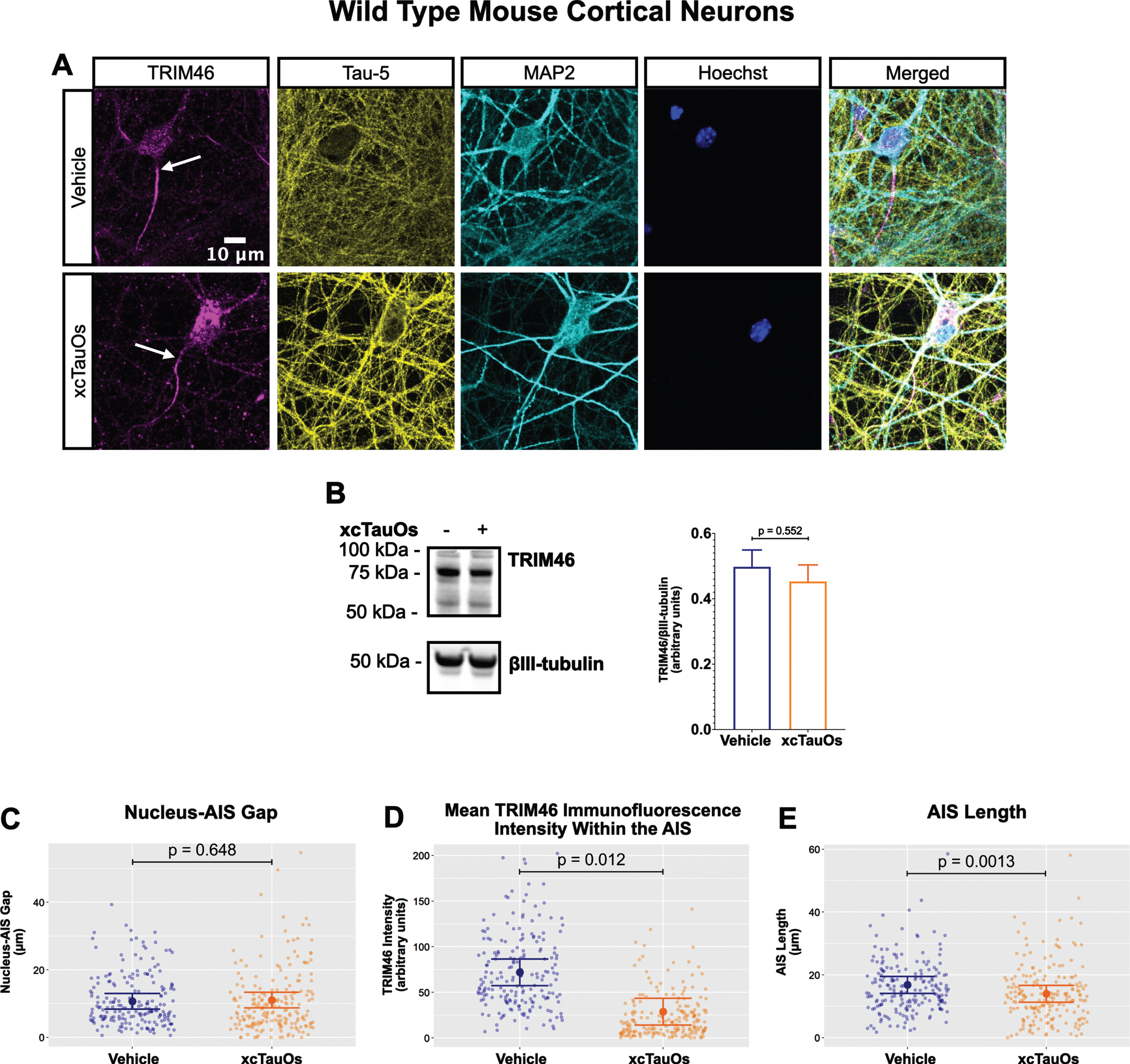
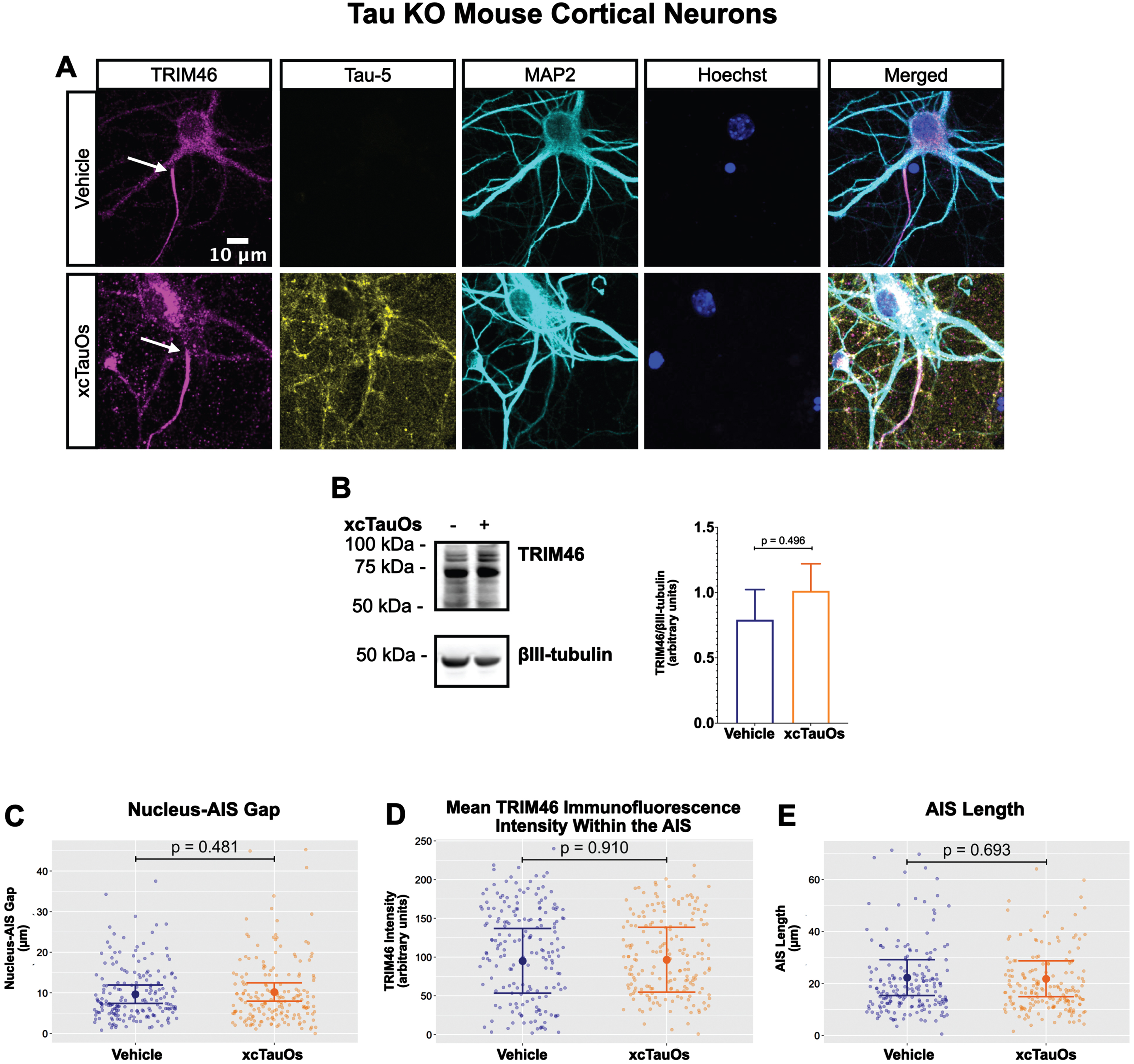
Fig. 2
xcTauOs do not affect the AIS in tau KO neurons. A) Localization of TRIM46 (white arrows indicate proximal AIS ends), tau (Tau-5), somatodendritic compartment (MAP2), and nuclei (Hoechst) in primary tau KO mouse cortical neuron cultures. B) Quantitative western blotting of unfractionated tau KO cultures probed with anti-TRIM46, and as a loading control, the neuron-specific anti-βIII-tubulin. p-values are based on two-tailed unpaired t-tests, pooled variance. Bar graph with mean and error bars represent±standard error of the mean. Plots of (C) Nucleus-AIS gap; (D) mean TRIM46 immunofluorescence intensity within the AIS, a surrogate measure of TRIM46 concentration; and (E) AIS length. Graph data were measured from 5-6 biological replicates (independent neuronal culture preparations). Immunofluorescence was performed with 1 technical replicate (independent coverslip) each, which accounted for a total of ∼184 neurons per treatment condition (vehicle or xcTauOs). p-values from mixed model linear regression are indicated. Large dot indicates the predicted mean and error bars represent the 95% confidence interval.
Robust immunofluorescence labeling of the AIS in primary neurons was observed with anti-TRIM46 (Fig. 1A). Exposing the cells to xcTauOs at a total tau concentration of 125 nM for 24 h resulted in the modest accumulation of tau and of punctate TRIM46 in the somatodendritic compartment of neurons. xcTauOs did not alter the intraneuronal TRIM46 protein levels (Fig. 1B), nor the shortest distance between the nucleus and proximal end of the AIS (Nucleus-AIS gap; Fig. 1C). By contrast, xcTauOs induced an ∼60% decrease in the average TRIM46 staining intensity within the AIS (Fig. 1D). Similarly, xcTauOs induced an ∼19% decrease in AIS length, from an average of 17.1μm to an average of 13.8μm (Fig. 1E). It is possible that this loss of TRIM46 from the AIS reflects the corresponding appearance of somatodendritic TRIM46 puncta in xcTauO-treated neurons. It is important to note that only ∼1/3 of the total extracellular tau was in the form of oligomers (see Supplementary Figure 3B), predominantly putative dimers and trimers [58], so the true concentration of tau oligomers was in the range of 10–20 nM. Thus, we conclude that while low nanomolar concentrations of xcTauOs do not reduce the overall neuronal protein levels of TRIM46, they cause an ∼19% decrease in AIS length and an ∼60% reduction of TRIM46 concentration within what remains of the AIS. These data extend previous findings that AIS intensity and length can be modulated by external stimuli [47, 59, 60] and provide the first evidence of AIS sensitivity to xcTauOs.
Intriguingly, xcTauOs also induced perinuclear puncta of TRIM46 in the astrocytes that were present in primary mouse brain cell cultures, even though TRIM46 was undetectable in astrocytes that had not been exposed to xcTauOs (Supplementary Figure 4). We do not know if the TRIM46 in xcTauO-treated astrocytes had been synthesized by those cells or originated in neurons before being taken up byastrocytes.
Tau is necessary for xcTauO-induced AIS damage
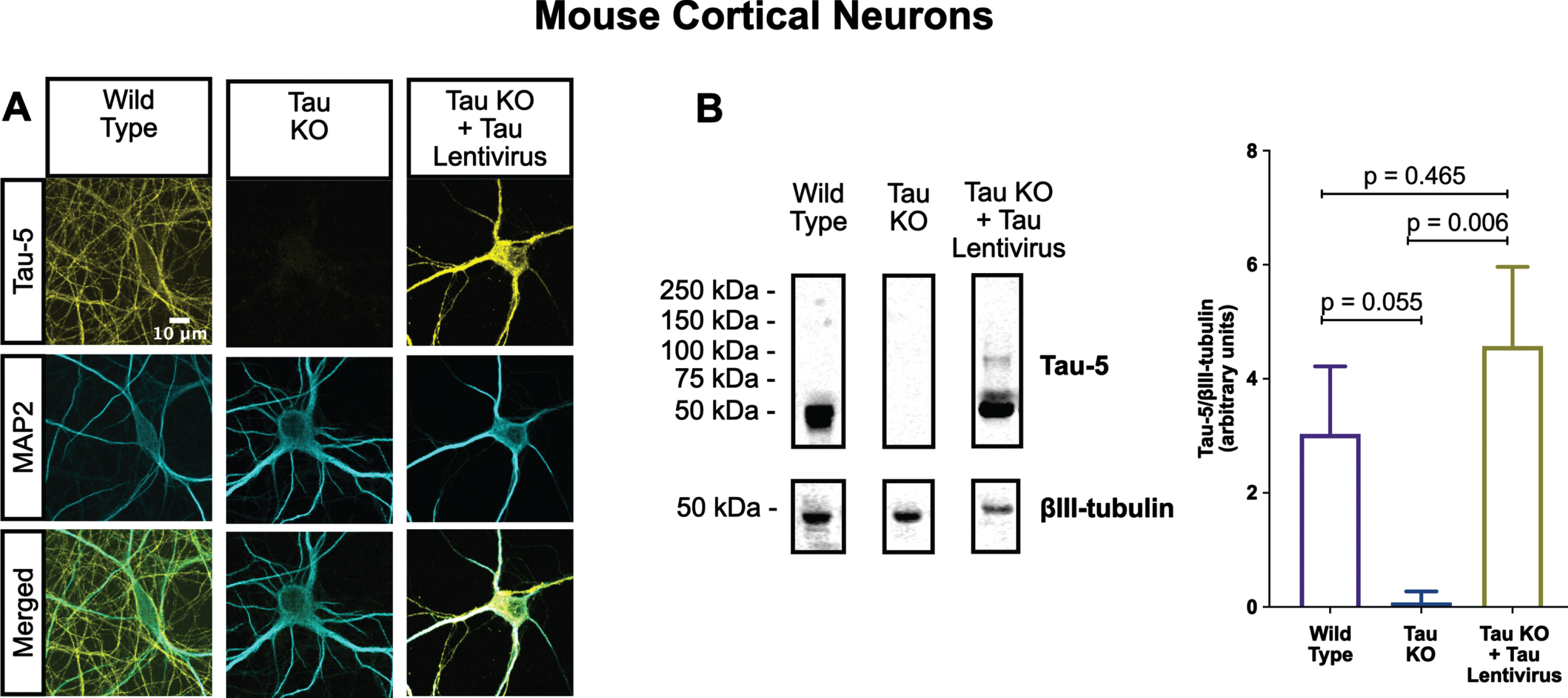
One of the most conspicuous targets of extracellular tau aggregates, like short filaments and xcTauOs, is intracellular tau, which responds by aggregating in the cytoplasm. We therefore hypothesized that intracellular tau is required for the partial AIS breakdown caused by xcTauOs. To test our hypothesis, we repeated the experiments shown in Fig. 1 using primary cortical neurons derived from tau KO mice [61]. As shown in Fig. 2, treatment of tau KO neurons with xcTauOs at a total tau concentration of 125 nM for 24 h did not significantly alter the cellular level of TRIM46, the distance between the nucleus and the proximal boundary of the AIS, the length of the AIS, or the concentration of TRIM46 within the AIS. When WT human 2N4R tau was expressed in tau KO neurons by lentiviral transduction, however, sensitivity to xcTauOs was restored for AIS length, but not for the TRIM46 concentration within the remainder of the AIS (Fig. 3). In such cells, the AIS shortened by an average of ∼24%, from 20μm to 15.3μm (Fig. 3E). Although the average expression level of lentiviral-encoded human tau in tau KO mouse neurons was ∼1.3-fold higher than endogenous tau in WT mouse neurons, that difference was not statistically significant (Fig. 4). Taken together, these results are consistent with the hypothesis that xcTauOs cause partial disassembly of the AIS by a mechanism dependent on intracellular tau.
Fig. 3
Expression of human tau in tau KO neurons restores AIS length sensitivity to xcTauOs. A) Localization of TRIM46 (white arrows indicate proximal AIS ends), tau (Tau-5), somatodendritic compartment (MAP2), and nuclei (Hoechst) in primary tau KO mouse cortical neuron cultures expressing lentiviral encoded human 2N4R tau. B) Quantitative western blotting of unfractionated tau KO cultures with lentiviral tau expression probed with anti-TRIM46, and as a loading control, the neuron-specific anti-βIII-tubulin. p-values are based on two-tailed unpaired t-tests, pooled variance. Bar graph with mean and error bars represent±standard error of the mean. Plots of (C) Nucleus-AIS gap; (D) mean TRIM46 immunofluorescence intensity within the AIS, a surrogate measure of TRIM46 concentration; and (E) AIS length. Graph data were measured from 3–5 biological replicates (independent neuronal culture preparations). Immunofluorescence was performed with 1 technical replicate (independent coverslip) each, which accounted for a total of ∼243 neurons per treatment condition (vehicle or xcTauOs). p-values from mixed model linear regression are indicated. Large dot indicates the predicted mean and error bars represent the 95% confidence interval.
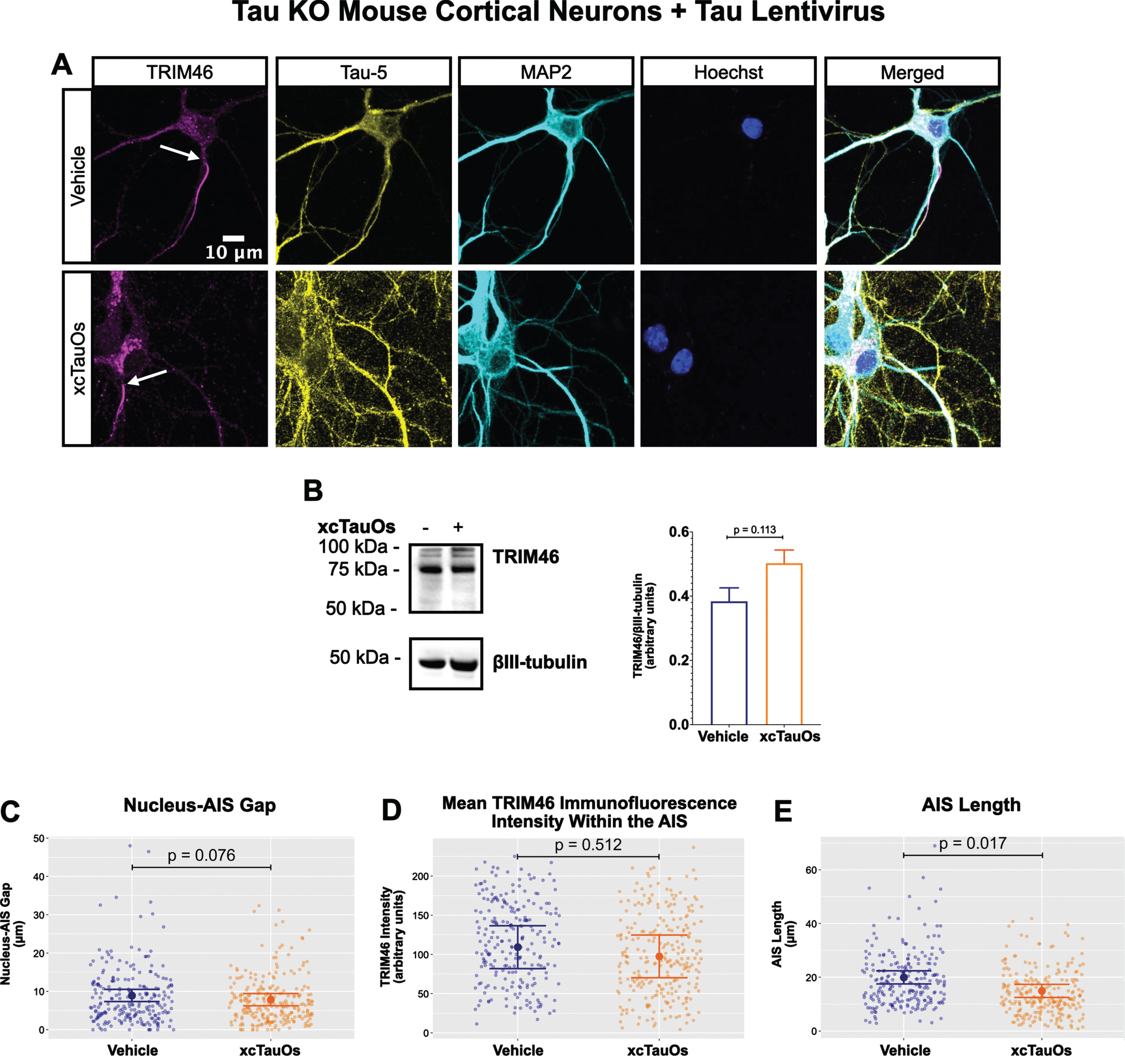
Fig. 4
Lentiviral-encoded tau is expressed within normal levels in tau KO neurons. Shown here are detection of lentiviral-encoded human 2N4R tau in cultured tau KO neurons by immunofluorescence microscopy (A) and quantitative western blotting (B). Note that the average human tau expression level was ∼30% higher than the endogenous mouse tau expression level in cultured WT neurons, but the difference was not statistically different. Data were obtained from the following number of biological replicates: 3 for Wild Type, 6 for Tau KO and 3 for Tau KO + Tau Lentivirus. p-values are based on two-tailed unpaired t-tests, pooled variance. Error bars on the graph represent±standard error of the mean.
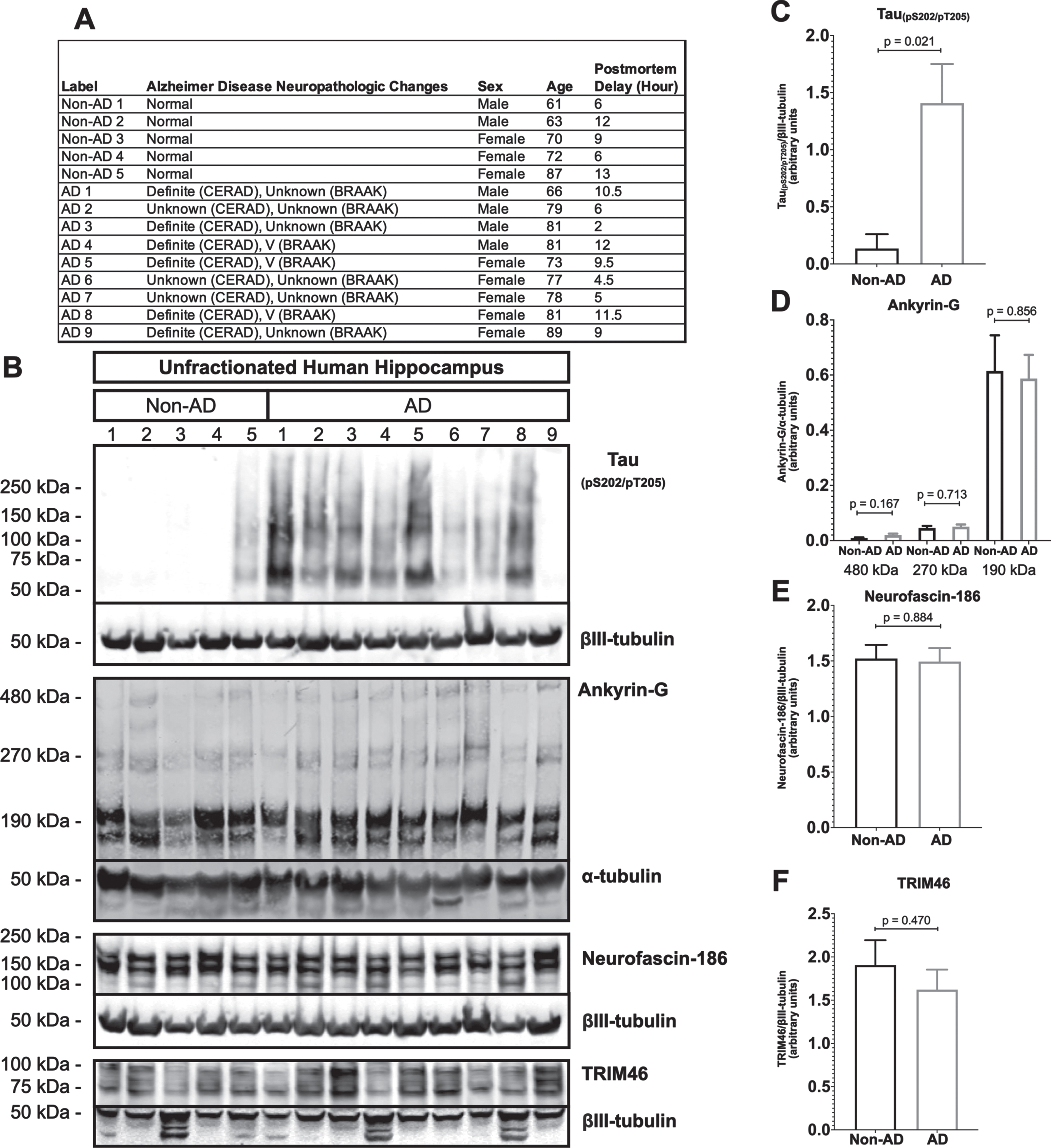
AIS protein levels are unchanged in human AD hippocampus
A prior study claimed that the levels of two major structural proteins of the AIS, ankyrin-G and βIV-spectrin, are reduced in human AD frontal cortex [52]. To determine if AIS protein loss also occurs elsewhere in human AD brain, we performed western blotting of unfractionated human hippocampal tissue derived from postmortem AD and age matched non-AD donors (Fig. 5). As expected, most AD samples (8 of 9), but not non-AD samples (1 of 5), were positive for the paired helical filament tau marker, tau(pS202/pT205) (Fig. 5B, C). In contrast, none of the three AIS proteins that we examined, ankyrin-G, neurofascin-186, or TRIM46, were reduced in AD versus non-AD brain. In the case of ankyrin-G, there are three major splice isoforms, 190 kDa, 270 kDa, and 480 kDa, and only the latter two are highly enriched in the AIS. As shown in Fig. 5B and 5D, there were no differences detected in levels of any ankyrin-G isoform between AD and non-AD hippocampus.
Fig. 5
Resident AIS proteins levels are not reduced in AD hippocampus. A) Clinical characterization of human hippocampus samples used for quantitative western blotting. B) Western blotting of unfractionated human hippocampus probed with antibodies to tau (pS202/pT205) and the resident AIS proteins, ankyrin-G, neurofascin-186, and TRIM46. Loading controls, βIII-tubulin or α-tubulin, are shown for each of the aforementioned proteins. C-F) Quantitation of western blots. Graphs were generated from 5–9 biological replicates (independent tissue samples) per blot. p-values are based on two-tailed unpaired t-tests, unpooled variance. Bar graph with mean and error bars represent±standard error of the mean.
AIS damage in human AD neurons with NFTs
The western blotting results for ankyrin-G, neurofascin-186, and TRIM46 (Fig. 5) in human hippocampus resemble the cultured neuron results for TRIM46 (Figs. 1 and 3) in the sense that total protein levels were not reduced in AD versus non-AD by xcTauOs in vitro. Because xcTauO treatment of cultured neurons caused AIS shortening and reduced the concentration of TRIM46 by ∼60% within the remainder of the AIS (Fig. 1), we searched for AIS structural changes in human AD brain. To that end, we performed immunohistochemistry of paraffin-embedded human hippocampal tissue using anti-TRIM46 as an AIS marker and Tau-5 (Fig. 6). Eight tissue sections from both AD and age-matched non-AD controls were examined, and individual neurons were classified as “no tau accumulation”, “tau accumulation without NFTs” (see asterisk), or “NFTs present” (see arrowhead). Matching the analysis of cultured neurons shown in Figs. 1–3, we measured the nucleus-AIS gap length, mean TRIM46 intensity within the AIS, and AIS length for both AD and non-AD samples. Note that these human brain sections were 4μm thick, but the AIS typically ranges in length from 20–60μm, so most length measurements did not account for the entire span of an AIS.
Fig. 6
In AD brain, NFTs are associated with reduced clustering of TRIM46 within the AIS and AIS shortening. A) Clinical characterization of human hippocampus samples used for quantitative immunofluorescence microscopy. B) Localization of TRIM46 (white arrows indicate proximal AIS ends) and tau (Tau-5, tau accumulation without NFTs = asterisk, NFTs present = white arrowhead) in non-AD and AD hippocampus. Plots of (C) Nucleus-AIS gap, (D) mean TRIM46 immunofluorescence intensity within the AIS, a surrogate measure of TRIM46 clustering, and (E) AIS length. Graphs were generated from 8 biological replicates (independent tissue samples), which accounted for a total of ∼580 neurons per group (non-AD or AD). p-values from mixed model linear regression are indicated. Large dot indicates the predicted mean and error bars represent the 95% confidence interval.
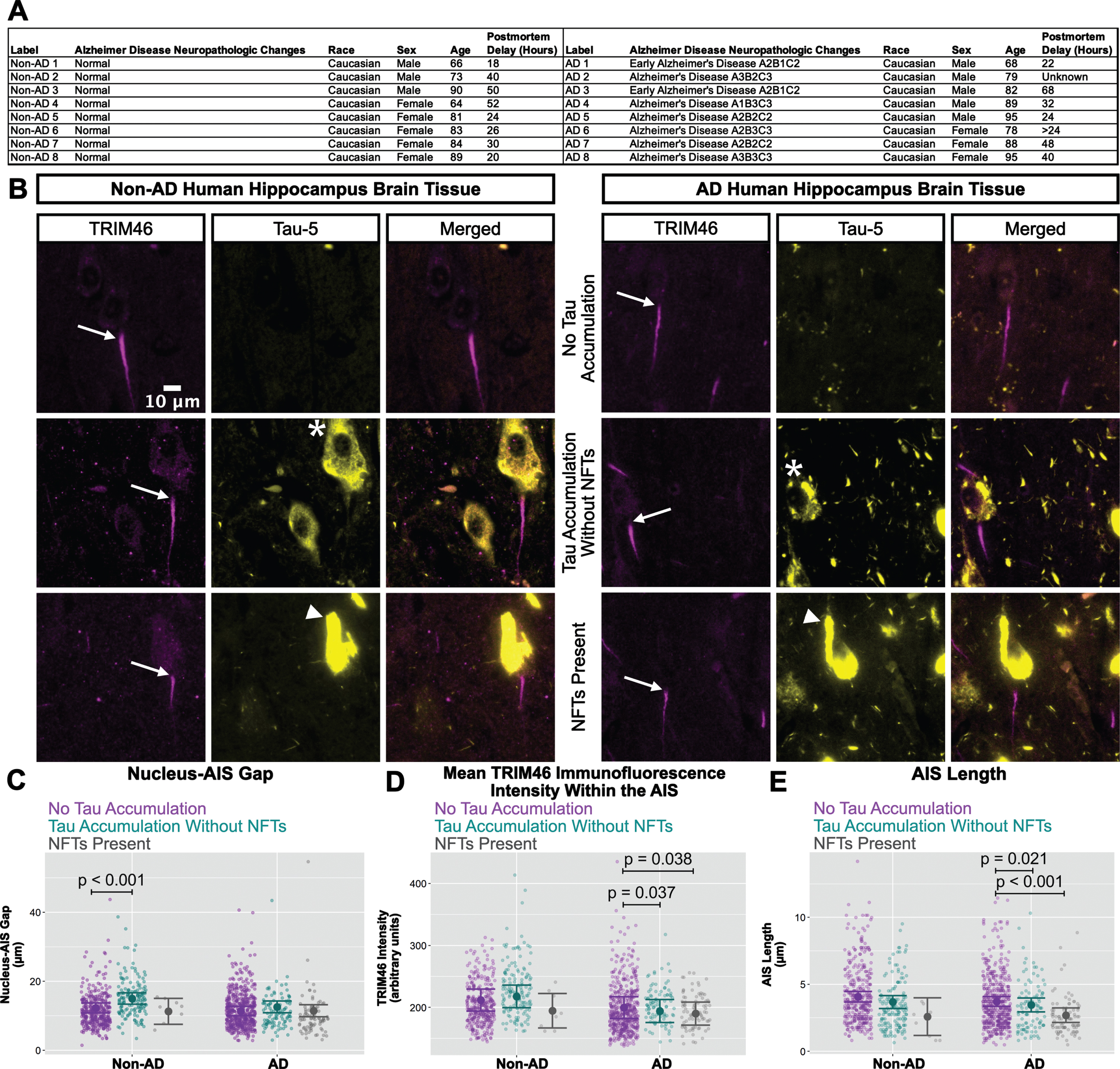
Interestingly, as shown in Fig. 6C, the nucleus-AIS gap in non-AD hippocampus was slightly, but statistically significantly shorter in neurons with “no tau accumulation” compared to neurons with “tau accumulation without NFTs". AIS length and mean TRIM46 intensity within the AIS were not affected by tau aggregation status in non-AD hippocampus, with the caveat that the number of neurons with NFTs that we observed in the non-AD group is small. In the AD samples, however, both mean TRIM46 concentration within the AIS (Fig. 6D) and AIS length (Fig. 6E) were statistically significantly greater in neurons with “no tau accumulation” compared to neurons with “tau accumulation without NFTs” or with “NFTs present".
Finally, we also analyzed the raw data for nucleus-AIS gap length, mean TRIM46 intensity within the AIS, and AIS length in the context of disease stage without regard for tau aggregation status. As shown in Supplementary Figure 5, by doing so we could not discriminate early Braak, late Braak and age-matched non-AD from each other by this set of criteria. In other words, decreases in AIS length and TRIM46 concentration within the AIS are correlated with tau aggregation state on a neuron-by-neuron basis, but not with disease stage.
DISCUSSION
Neurons are arguably one of the most structurally polarized animal cell types, and the AIS, which is found only in neurons, is essential for organizing neuronal cytoarchitecture. For example, the AIS encompasses a diffusion barrier that helps to segregate axonal from somatodendritic molecules, and it houses tightly packed voltage-gated ion channels that generate action potentials. Given the important functionality of the AIS, it follows naturally that damage of the AIS could have profound negative effects on the structural polarity of neurons, and by extension, on neuronal function.
Several prior studies have revealed AIS damage in neurodegenerative disorders, including AD [24, 29, 47–50, 52, 62–71], and there is evidence for possible involvement of amyloid-β oligomers [47] and plaques [29] in triggering that damage. Here we identify xcTauOs, which are hallmarks of AD and other tauopathies, as a potent inducer of AIS damage. Using TRIM46 as an AIS marker, we show that exposure of primary mouse cortical neurons to xcTauOs causes AIS shortening and reduces the concentration of TRIM46 within the remainder of the AIS by more than half (Fig. 1A, D, E). The total neuronal protein levels of TRIM46 were not altered by xcTauOs, however, indicating that TRIM46 is redistributed from the AIS to presumably the cytoplasm in response to xcTauO exposure (Fig. 1B). Effects of xcTauOs on the AIS were not observed in cultured tau KO neurons (Fig. 2) unless human tau was expressed in the cells by lentiviral transduction (Fig. 3). xcTauOs thus work in concert with intracellular tau to damage the AIS in cultured neurons.
The in vivo relevance of these cultured neuron findings was established by showing that reduced AIS length and clustering of TRIM46 within the AIS is associated with the presence of NFTs in hippocampal neurons (Fig. 6), but that the overall hippocampal levels of TRIM46 and two other AIS proteins, ankyrin-G, and neurofascin-186, are not significantly different in AD versus age-matched tissue without AD (Fig. 5). Altogether, these new results establish the AIS as a previously unknown target of xcTauOs, identify intracellular tau as an essential co-factor for the AIS damage induced by xcTauOs, and identify xcTauOs as likely causes of AIS damage in vivo.
Expression of human 2N4R tau in tau KO neurons caused AIS length to be even more sensitive to xcTauOs than in WT neurons that expressed only endogenous mouse tau (Figs. 1 and 3). Average AIS shortening after a 24-h exposure to xcTauOs was statistically significant: 19% in WT neurons and 24% in human tau-expressing tau KO neurons. In contrast, while the intensity of the TRIM46 immunofluorescence signal, a quantitative indicator of TRIM46 concentration, dropped ∼60% after xcTauO exposure for WT neurons, it dropped only 10% for tau KO neurons that expressed human tau and that figure did not reach statistical significance.
One possible explanation for the differential AIS sensitivities to xcTauOs in WT versus tau KO neurons expressing human tau concerns intracellular tau isoforms. The WT neurons we used expressed approximately equal levels of 0N3R and 0N4R mouse tau (unpublished observations), whereas the human tau expressed in tau KO neurons was 2N4R, and like all human tau isoforms contains an 11 amino acid sequence, residues 19–29, that is not present in mouse tau. Perhaps species-specific or isoform-specific structural features of intracellular tau influence how the AIS responds to xcTauOs, and to the isoform composition of the xcTauOs as well. Regardless, the AIS shortening we observed in xcTauO-treated tau KO neurons that expressed human tau was slightly greater than that of WT neurons, while the TRIM46 concentration in the remainder of the WT AIS trended lower. While it is clear that AIS sensitivity to xcTauOs requires intracellular tau, the mechanistic basis for this requirement remains to be determined.
We chose to focus on TRIM46 as an indicator of AIS structure for two main reasons. The first was simply practical. Although TRIM46 co-localized in our experiments with the well-known AIS structural proteins, ankyrin-G, βIV-spectrin and neurofascin-186 (Supplementary Figure 6), the TRIM46 immunofluorescence signal was observed to be brighter and more confined to the AIS than the signals for the other two proteins. Secondly, TRIM46, which bundles microtubules in the AIS [44, 45], has not been extensively studied so we were inspired to investigate it in the context of AD and other tauopathies. That said, it would be interesting to extend the characterization of AIS protein responses to xcTauOs to additional resident AIS proteins.
A prior report of AIS protein content in human brain by Sohn and colleagues included evidence of a dramatic reduction of ankyrin-G and βIV-spectrin in AD compared to non-AD brain [52], which seems to contradict what we report here for ankyrin-G (no change; see Fig. 5). Several factors might account for this discrepancy. Whereas Sohn and colleagues examined RIPA buffer-soluble extracts of frontal cortex clarified by ultracentrifugation for western blotting and quantified only the 270 kDa isoform of ankyrin-G, for western blotting we used uncentrifuged, SDS-soluble extracts of hippocampus and quantification of the 190 kDa, 270 kDa, and 480 kDa isoforms of ankyrin-G, the latter two of which are highly enriched in the AIS. It is possible that differences in tissue source (frontal cortex versus hippocampus), sample extraction methods and ankyrin-G isoform quantification, alone or in combination, explain why an AD-associated drop in ankyrin-G levels were found by Sohn and colleagues, but not in our study.
Similarly, our data for AIS shortening in human AD hippocampus (Fig. 6) are inconsistent with a report by Antón-Fernández and colleagues that the AIS is longer in AD compared to non-AD brain [48]. A few factors might account for this discrepancy. Whereas Antón-Fernández and colleagues examined βIV-spectrin in 50μm thick frontal cortex, we used TRIM46 as an AIS marker in 4μm thick sections of hippocampus. It is thus possible that differences in AIS marker (βIV-spectrin versus TRIM46), tissue thickness (50μm versus 4μm), and brain region (frontal cortex versus hippocampus), alone or in combination, explain why we observed AIS shortening, and Antón-Fernández and colleagues observed AIS lengthening in AD neurons in vivo.
Notably, our human brain tissue observations were performed only on Caucasian (Fig. 6) and indeterminate race (Fig. 5) donors. As demonstrated in previous studies, the inclusion of non-Caucasian donors [72–76], particularly non-Hispanic Black groups [76], may increase experimental validity as this group has a higher incidence of AD [77]. Future studies should therefore strive to include individuals from ethnically and racially diverse populations. This increase in sample size may help untangle the correlation of NFTs with AIS changes statistically.
ACKNOWLEDGMENTS
This paper represents partial fulfillment of the Ph.D. requirements for MNB. The authors would like to thank MNB’s dissertation committee (Drs. Mark Beenhakker, George Bloom, Thurl Harris, James Mandell,Edward Perez-Reyes, Bettina Winckler, and Scott Zeitlin); Drs. Silvia Blemker, Mete Civelek, Barry Condron, Jasmine Crenshaw, Carl Creutz, Heather Ferris, Gordon Laurie, Yi Hao, Marlit Hayslett, Keisha John, Kimberly Kelly, Joshua Kulas, John Lazo, Sonali Majumdar, Elizabeth Sharlow, Anthony Spano, Ann Sutherland, and Cedric Williams; past and current members of the Bloom lab (Drs. Guillermo Eastman, Shahzad Khan, Erin Kodis, Andrés Norambuena, Evelyn Pardo, Binita Rajbanshi, Lauren Rudenko, Antonia Silva, Eric Swanson and Edward John, Taylor Kim, Lisa Post, Nutan Shivange, Victoria Sun, and Horst Wallrabe); past and current members of the University of Virginia’s Advanced Microscopy facility (Stacey Criswall and Natalia Dworak); past and current members of the University of Virginia’s Writing Center (Grace Tavakkol, CJ Oswald, and Sydney Anderson), Academic English Now (Dr. Marek Kiczkowiak), and departmental administrators (Phillis Hynes, Sherrie Jones, Antoinette Reid, Deborah Steele, and Carrie Walker) for their support and encouragement.
FUNDING
This work was supported by NIH Grant AG051085 (GSB); the Owens Family Foundation (GSB); the Cure Alzheimer’s Fund (GSB); NIH Training Grant T32GM008715 (MNB); and NSF Training Grant EXPAND Program 2021791 (MNB).
CONFLICT OF INTEREST
George Bloom is an Editorial Board Member of this journal but was not involved in the peer-review process nor had access to any information regarding its peer-review. All other authors have no conflicts of interest to report.
DATA AVAILABILITY
All original data supporting the findings of this study are available on request from the corresponding author. The study described in this report does not include large datasets that warrant filing on publicly available websites.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-221284.
REFERENCES
[1] | Andorfer C , Kress Y , Espinoza M , De Silva R , Tucker KL , Barde YA , Duff K , Davies P ((2003) ) Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem 86: , 582–590. |
[2] | Bennett DA , Schneider JA , Wilson RS , Bienias JL , Arnold SE ((2004) ) Neurofibrillary tangles mediate the association of amyloid load with clinical Alzheimer disease and level of cognitive function. Arch Neurol 61: , 378–384. |
[3] | Lacosta AM , Insua D , Badi H , Pesini P , Sarasa M ((2017) ) Neurofibrillary tangles of Aβx-40 in Alzheimer’s disease brains. J Alzheimers Dis 58: , 661–667. |
[4] | Iqbal K , Gong C-X , Liu F ((2013) ) Hyperphosphorylation-induced tau oligomers. Front Neurol 4: , 112. |
[5] | Holmes BB , Furman JL , Mahan TE , Yamasaki TR , Mirbaha H , Eades WC , Belaygorod L , Cairns NJ , Holtzman DM , Diamond MI ((2014) ) Proteopathic tau seeding predicts tauopathy in vivo. Proc Natl Acad Sci U S A 111: , E4376–E4385. |
[6] | Frost B , Diamond M ((2010) ) Prion-like mechanisms in neurodegenerative diseases. Nat Rev Neurosci 11: , 155–159. |
[7] | Holmes BB , Diamond MI ((2014) ) Prion-like properties of tau protein: The imortance of extracellular tau as a therapeutic target. J Biol Chem 289: , 19855–19861. |
[8] | Mirbaha H , Chen D , Mullapudi V , Terpack SJ , White CL , Joachimiak LA , Diamond MI ((2022) ) Seed-competent tau monomer initiates pathology in a tauopathy mouse model. J Biol Chem 298: , https://doi.org.10.1016/j.jbc.2022.102163. |
[9] | Karch CM , Jeng AT , Goate AM ((2013) ) Calcium phosphatase calcineurin influences tau metabolism. Neurobiol Aging 34: , 374–386. |
[10] | Bright J , Hussain S , Dang V , Wright S , Cooper B , Byun T , Ramos C , Singh A , Parry G , Stagliano N , Griswold-Prenner I ((2015) ) Human secreted tau increases amyloid-beta production. Neurobiol Aging 36: , 693–709. |
[11] | Kanmert D , Cantlon A , Muratore CR , Jin M , O’Malley TT , Lee G , Young-Pearse TL , Selkoe DJ , Walsh DM ((2015) ) C-terminally truncated forms of tau, but not full-length tau or its C-terminal fragments, are released from neurons independently of cell death. J Neurosci 35: , 10851–10865. |
[12] | Chai X , Dage JL , Citron M ((2012) ) Constitutive secretion of tau protein by an unconventional mechanism. Neurobiol Dis 48: , 356–366. |
[13] | Yamada K , Cirrito JR , Stewart FR , Jiang H , Finn MB , Holmes BB , Binder LI , Mandelkow EM , Diamond MI , Lee VMY , Holtzman DM ((2011) ) In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice. J Neurosci 31: , 13110–13117. |
[14] | Magnoni S , Esparza TJ , Conte V , Carbonara M , Carrabba G , Holtzman DM , Zipfel GJ , Stocchetti N , Brody DL ((2012) ) Tau elevations in the brain extracellular space correlate with reduced amyloid-β levels and predict adverse clinical outcomes after severe traumatic brain injury. Brain 135: , 1268–1280. |
[15] | Swanson E , Breckenridge L , McMahon L , Som S , McConnell I , Bloom GS ((2017) ) Extracellular tau oligomers induce invasion of endogenous tau into the somatodendritic compartment and axonal transport dysfunction. J Alzheimers Dis 58: , 803–820. |
[16] | Li C , Götz J ((2017) ) Somatodendritic accumulation of tau in Alzheimer’s disease is promoted by Fyn-mediated local protein translation. EMBO J 36: , 3120–3138. |
[17] | Kobayashi S , Tanaka T , Soeda Y , Almeida OFX , Takashima A ((2017) ) Local somatodendritic translation and hyperphosphorylation of tau protein triggered by AMPA and NMDA receptor stimulation. EBioMedicine 20: , 120–126. |
[18] | Ittner A , Ittner LM ((2018) ) Dendritic tau in Alzheimer’s disease. Neuron 99: , 13–27. |
[19] | Ittner LM , Ke YD , Delerue F , Bi M , Gladbach A , van Eersel J , Wölfing H , Chieng BC , Christie MJ , Napier IA , Eckert A , Staufenbiel M , Hardeman E , Götz J ((2010) ) Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 142: , 387–397. |
[20] | Usenovic M , Niroomand S , Drolet RE , Yao L , Gaspar RC , Hatcher NG , Schachter J , Renger JJ , Parmentier-Batteur S ((2015) ) Internalized tau oligomers cause neurodegeneration by inducing accumulation of pathogenic tau in human neurons derived from induced pluripotent stem cells. J Neurosci 35: , 14234–14250. |
[21] | Hill E , Karikari TK , Moffat KG , Richardson MJE , Wall MJ ((2019) ) Introduction of tau oligomers into cortical neurons alters action potential dynamics and disrupts synaptic transmission and plasticity. eNeuro 6: , https://doi.org/10.1523/ENEURO.0166-19.2019. |
[22] | Sobotzik JM , Sie JM , Politi C , Del Turco D , Bennett V , Deller T , Schultz C ((2009) ) AnkyrinG is required to maintain axo-dendritic polarity in vivo . Proc Natl Acad Sci U S A 106: , 17564–17569. |
[23] | Winckler B , Forscher P , Mellman I ((1999) ) A diffusion barrier maintains distribution of membrane proteins in polarized neurons. Nature 397: , 698–701. |
[24] | Li X , Kumar Y , Zempel H , Mandelkow E-M , Biernat J , Mandelkow E ((2011) ) Novel diffusion barrier for axonal retention of tau in neurons and its failure in neurodegeneration. EMBO J 30: , 4825–4837. |
[25] | Foust A , Popovic M , Zecevic D , McCormick DA ((2010) ) Action potentials initiate in the initial segment and propagate through axon collaterals reliably in cerebellar purkinje neurons. J Neurosci 30: , 6891–6902. |
[26] | Zonta B , Desmazieres A , Rinaldi A , Tait S , Sherman DL , Nolan MF , Brophy PJ ((2011) ) A critical role for neurofascin in regulating action potential initiation through maintenance of the axon initial segment. Neuron 69: , 945–956. |
[27] | Gulledge AT , Bravo JJ ((2016) ) Neuron morphology influences axon initial segment plasticity. eNeuro 3: , 255–265. |
[28] | Höfflin F , Jack A , Riedel C , Mack-Bucher J , Roos J , Corcelli C , Schultz C , Wahle P , Engelhardt M ((2017) ) Heterogeneity of the axon initial segment in interneurons and pyramidal cells of rodent visual cortex. Front Cell Neurosc https://doi.org/10.3389/fncel.2017.00332. |
[29] | Marin MA , Ziburkus J , Jankowsky J , Rasband MN ((2016) ) Amyloid-β plaques disrupt axon initial segments. Exp Neurol 281: , 93–98. |
[30] | Nelson AD , Caballero-Florán RN , Rodríguez Díaz JC , Hull JM , Yuan Y , Li J , Chen K , Walder KK , Lopez-Santiago LF , Bennett V , McInnis MG , Isom LL , Wang C , Zhang M , Jones KS , Jenkins PM ((2020) ) Ankyrin-G regulates forebrain connectivity and network synchronization via interaction with GABARAP. Mol Psychiatry 25: , 2800–2817. |
[31] | Jenkins PM , Kim N , Jones SL , Tseng WC , Svitkina TM , Yin HH , Bennett V ((2015) ) Giant ankyrin-G: A critical innovation in vertebrate evolution of fast and integrated neuronal signaling. Proc Natl Acad Sci U S A 112: , 957–964. |
[32] | Zhou D , Lambert S , Malen PL , Carpenter S , Boland LM , Bennett V ((1998) ) AnkyrinG is required for clustering of voltage-gated Na channels at axon initial segments and for normal action potential firing. J Cell Biol 143: , 1295–1304. |
[33] | Kordeli E , Lambert S , Bennett V ((1995) ) AnkyrinG: A new ankyrin gene with neural-specific isoforms localized at the axonal initial segment and node of Ranvier. J Biol Chem 270: , 2352–2359. |
[34] | Hedstrom KL , Ogawa Y , Rasband MN ((2008) ) AnkyrinG is required for maintenance of the axon initial segment and neuronal polarity. J Cell Biol 183: , 635–640. |
[35] | Salzer JL ((2019) ) An unfolding role for ankyrin-G at the axon initial segment. Proc Natl Acad Sci U S A 116: , 19228–19230. |
[36] | Farías GG , Fréal A , Tortosa E , Stucchi R , Pan X , Portegies S , Will L , Altelaar M , Hoogenraad CC ((2019) ) Feedback-driven mechanisms between microtubules and the endoplasmic reticulum instruct neuronal polarity. Neuron 102: , 184–201. |
[37] | Berghs S , Aggujaro D , Dirkx R , Maksimova E , Stabach P , Hermel JM , Zhang JP , Philbrick W , Slepnev V , Ort T , Solimena M , Solimena M ((2000) ) βIV spectrin, a new spectrin localized at axon initial segments and nodes of ranvier in the central and peripheral nervous system. J Cell Biol 151: , 985–1002. |
[38] | Yang Y , Ogawa Y , Hedstrom KL , Rasband MN ((2007) ) βIV spectrin is recruited to axon initial segments and nodes of Ranvier by ankyrinG. J Cell Biol 176: , 509–519. |
[39] | Komada M , Soriano P ((2002) ) βIV-spectrin regulates sodium channel clustering through ankyrin-G at axon initial segments and nodes of Ranvier. J Cell Biol 156: , 337–348. |
[40] | Yang Y , Lacas-Gervais S , Morest DK , Solimena M , Rasband MN ((2004) ) BetaIV spectrins are essential for membrane stability and the molecular organization of nodes of Ranvier. J Neurosci 24: , 7230–7240. |
[41] | Fréal A , Rai D , Tas RP , Pan X , Katrukha EA , van de Willige D , Stucchi R , Aher A , Yang C , Altelaar AFM , Vocking K , Post JA , Harterink M , Kapitein LC , Akhmanova A , Hoogenraad CC ((2019) ) Feedback-driven assembly of the axon initial segment. Neuron 104: , 305–321. |
[42] | Alpizar SA , Baker AL , Gulledge AT , Hoppa MB ((2019) ) Loss of neurofascin-186 disrupts alignment of ankyrinG relative to its binding partners in the axon initial segment. Front Cell Neurosci 13: , https://doi.org/10.3389/fncel.2019.00001. |
[43] | Hedstrom KL , Xu X , Ogawa Y , Frischknecht R , Seidenbecher CI , Shrager P , Rasband MN ((2007) ) Neurofascin assembles a specialized extracellular matrix at the axon initial segment. J Cell Biol 178: , 875–886. |
[44] | Van Beuningen SFB , Will L , Harterink M , Chazeau A , Van Battum EY , Frias CP , Franker MAM , Katrukha EA , Stucchi R , Vocking K , Antunes AT , Slenders L , Doulkeridou S , Sillevis Smitt P , Altelaar AFM , Post JA , Akhmanova A , Pasterkamp RJ , Kapitein LC , de Graaff E , Hoogenraad CC ((2015) ) TRIM46 controls neuronal polarity and axon specification by driving the formation of parallel microtubule arrays. Neuron 88: , 1208–1226. |
[45] | Harterink M , Vocking K , Pan X , Soriano Jerez EM , Slenders L , Fréal A , Tas RP , van de Wetering WJ , Timmer K , Motshagen J , van Beuningen SFB , Kapitein LC , Geerts WJC , Post JA , Hoogenraad CC ((2019) ) TRIM46 organizes microtubule fasciculation in the axon initial segment. J Neurosci 39: , 4864–4873. |
[46] | Ichinose S , Ogawa T , Jiang X , Hirokawa N ((2019) ) The spatiotemporal construction of the axon initial segment via KIF3/KAP3/TRIM46 transport under MARK2 signaling. Cell Rep 28: , 2413–2426. |
[47] | Zempel H , Dennissen F , Kumar Y , Luedtke J , Biernat J , Mandelkow E-M , Mandelkow E ((2017) ) Axodendritic sorting and pathological missorting of Tau is isoform specific and determined by axon initial segment architecture. J Biol Chem 292: , 12192–12207. |
[48] | Antón-Fernández A , León-Espinosa G , DeFelipe J , Muñoz A ((2022) ) Pyramidal cell axon initial segment in Alzheimer’s disease. Sci Rep 12: , https://doi.org/10.1038/s41598-022-12700-9. |
[49] | Sun X , Wu Y , Gu M , Liu Z , Ma Y , Li J , Zhang Y ((2014) ) Selective filtering defect at the axon initial segment in Alzheimer’s disease mouse models. Proc Natl Acad Sci U S A 111: , 14271–14276. |
[50] | Sun X , Wu Y , Gu M , Zhang Y ((2014) ) miR-342-5p decreases ankyrin G levels in Alzheimer’s disease transgenic mouse models. Cell Rep 6: , 264–270. |
[51] | Tsushima H , Emanuele M , Polenghi A , Esposito A , Vassalli M , Barberis A , Difato F , Chieregatti E ((2015) ) HDAC6 and RhoA are novel players in Abeta-driven disruption of neuronal polarity. Nat Commun https://doi.org/10.1038/ncomms8781. |
[52] | Sohn PD , Tracy TE , Son H-I , Zhou Y , Leite REP , Miller BL , Seeley WW , Grinberg LT , Gan L ((2016) ) Acetylated tau destabilizes the cytoskeleton in the axon initial segment and is mislocalized to the somatodendritic compartment. Mol Neurodegener 11: , https://doi.org/10.1186/s13024-016-0109-0. |
[53] | León-Espinosa G , DeFelipe J , Muñoz A ((2012) ) Effects of amyloid-β plaque proximity on the axon initial segment of pyramidal cells. J Alzheimers Dis 29: , 841–852. |
[54] | Kaech S , Banker G ((2006) ) Culturing hippocampal neurons. Nat Protoc 1: , 2406–2415. |
[55] | Seward ME , Swanson E , Norambuena A , Reimann A , Cochran JN , Li R , Roberson ED , Bloom GS ((2013) ) Amyloid-β signals through tau to drive ectopic neuronal cell cycle re-entry in Alzheimer’s disease. J Cell Sci 126: , 1278–1286. |
[56] | Combs B , Tiernan CT , Hamel C , Kanaan NM ((2017) ) Production of recombinant tau oligomers in vitro. Methods Cell Biol 141: , 45–64. |
[57] | Jenkins PM , Vasavda C , Hostettler J , Davis JQ , Abdi K , Bennett V ((2013) ) E-cadherin polarity is determined by a multifunction motif mediating lateral membrane retention through ankyrin-G and apical-lateral transcytosis through clathrin. J Biol Chem 288: , 14018–14031. |
[58] | Patterson KR , Remmers C , Fu Y , Brooker S , Kanaan NM , Vana L , Ward S , Reyes JF , Philibert K , Glucksman MJ , Binder LI ((2011) ) Characterization of prefibrillar tau oligomers in vitro and in Alzheimer disease. J Biol Chem 286: , 23063–23076. |
[59] | Kuba H , Oichi Y , Ohmori H ((2010) ) Presynaptic activity regulates Na(+) channel distribution at the axon initial segment. Nature 465: , 1075–1078. |
[60] | Grubb MS , Burrone J ((2010) ) Activity-dependent relocation of the axon initial segment fine-tunes neuronal excitability. Nature 465: , 1070–1074. |
[61] | Dawson HN , Ferreira A , Eyster MV , Ghoshal N , Binder LI , Vitek MP ((2001) ) Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J Cell Sci 114: , 1179–1187. |
[62] | Sohn PD , Huang CT-L , Yan R , Fan L , Tracy TE , Camargo CM , Montgomery KM , Arhar T , Mok S-A , Freilich R , Baik J , He M , Gong S , Roberson ED , Karch CM , Gestwicki JE , Xu K , Kosik KS , Gan L ((2019) ) Pathogenic tau impairs axon initial segment plasticity and excitability homeostasis. Neuron 104: , 458–470. |
[63] | Hatch RJ , Wei Y , Xia D , Götz J ((2017) ) Hyperphosphorylated tau causes reduced hippocampal CA1 excitability by relocating the axon initial segment. Acta Neuropathol 133: , 717–730. |
[64] | Bonnevie VS , Dimintiyanova KP , Hedegaard A , Lehnhoff J , Grøndahl L , Moldovan M , Meehan CF ((2020) ) Shorter axon initial segments do not cause repetitive firing impairments in the adult presymptomatic G127X SOD-1 Amyotrophic Lateral Sclerosis mouse. Sci Rep 10: , https://doi.org/10.1038/s41598-019-57314-w. |
[65] | León-Espinosa G , Defelipe J , Muoz A ((2012) ) Effects of amyloid-β plaque proximity on the axon initial segment of pyramidal cells. J Alzheimers Dis 29: , 841–852. |
[66] | Sasaki S , Maruyama S ((1992) ) Increase in diameter of the axonal initial segment is an early change in amyotrophic lateral sclerosis. J Neurol Sci 110: , 114–120. |
[67] | Lüningschrör P , Werner G , Stroobants S , Kakuta S , Dombert B , Sinske D , Wanner R , Lüllmann-Rauch R , Wefers B , Wurst W , D’Hooge R , Uchiyama Y , Sendtner M , Haass C , Saftig P , Knöll B , Capell A , Damme M ((2020) ) The FTLD risk factor TMEM106B regulates the transport of lysosomes at the axon initial segment of motoneurons. Cell Rep 30: , 3506–3519. |
[68] | Meza RC , López-Jury L , Canavier CC , Henny P ((2018) ) Role of the axon initial segment in the control of spontaneous frequency of nigral dopaminergic neurons in vivo. J Neurosci 38: , 733–744. |
[69] | Sasaki S , Warita H , Abe K , Iwata M ((2005) ) Impairment of axonal transport in the hillock and the initial segment of anterior horn neurons in transgenic mice with a G93A mutant SOD1 gene. Acta Neuropathol 110: , 48–56. |
[70] | Jørgensen HS , Jensen DB , Dimintiyanova KP , Bonnevie VS , Hedegaard A , Lehnhoff J , Moldovan M , Grondahl L , Meehan CF ((2021) ) Increased axon initial segment length results in increased Na+ currents in spinal motoneurones at symptom onset in the G127X SOD1 mouse model of amyotrophic lateral sclerosis. Neuroscience 468: , 247–264. |
[71] | Martínez-Silva M de L , Imhoff-Manuel RD , Sharma A , Heckman CJ , Shneider NA , Roselli F , Zytnicki D , Manuel M ((2018) ) Hypoexcitability precedes denervation in the large fast-contracting motor units in two unrelated mouse models of ALS. Elife 7: , https://doi.org/10.7554/eLife.30955.001. |
[72] | Elliott CL , Ryan L , Silverberg N ((2021) ) Building inclusive and open Alzheimer disease and Alzheimer disease– related dementias research programs. JAMA Neurol 78: , 1177–1178. |
[73] | Raman R , Quiroz YT , Langford O , Choi J , Ritchie M , Baumgartner M , Rentz D , Aggarwal NT , Aisen P , Sperling R , Grill JD ((2021) ) Disparities by race and ethnicity among adults recruited for a preclinical Alzheimer disease trial. JAMA Network Open https://doi.org/10.1001/jamanetworkopen.2021.14364. |
[74] | Dilworth-Anderson P ((2011) ) Introduction to the science of recruitment and retention among ethnically diverse populations, Gerontologist 51: , S1–S4. |
[75] | Dilworth-Anderson P , Cohen MD ((2010) ) Beyond diversity to inclusion: Recruitment and retention of diverse groups in Alzheimer research, Alzheimer Dis Assoc Disord 24: , S14–S18. |
[76] | Deters KD , Napolioni V , Sperling RA , Greicius MD , Mayeux R , Hohman T , Mormino EC ((2021) ) Amyloid PET imaging in self-identified non-Hispanic Black participants of the Anti-Amyloid in Asymptomatic Alzheimer’s Disease (A4) Study. Neurology 96: , e1491–e1500. |
[77] | Plassman BL , Langa KM , Fisher GG , Heeringa SG , Weir DR , Ofstedal MB , Burke JR , Hurd MD , Potter GG , Rodgers WL , Steffens DC , Willis RJ , Wallace RB ((2007) ) Prevalence of dementia in the United States: The Aging, Demographics, and Memory Study. Neuroepidemiology 29: , 125–132. |


