
Cerebrospinal Fluid Panel of Synaptic Proteins in Cerebral Amyloid Angiopathy and Alzheimer’s Disease
Abstract
Background:
Alzheimer’s disease (AD) and cerebral amyloid angiopathy (CAA) share pathogenic pathways related to amyloid-β deposition. Whereas AD is known to affect synaptic function, such an association for CAA remains yet unknown.
Objective:
We therefore aimed to investigate synaptic dysfunction in CAA.
Methods:
Multiple reaction monitoring mass spectrometry was used to quantify cerebrospinal fluid (CSF) concentrations of 15 synaptic proteins in CAA and AD patients, and age- and sex-matched cognitively unimpaired controls.
Results:
We included 25 patients with CAA, 49 patients with AD, and 25 controls. Only neuronal pentraxin-2 levels were decreased in the CSF of CAA patients compared with controls (p = 0.04). CSF concentrations of 12 other synaptic proteins were all increased in AD compared with CAA or controls (all p≤0.01) and were unchanged between CAA and controls. Synaptic protein concentrations in the subgroup of CAA patients positive for AD biomarkers (CAA/ATN+; n = 6) were similar to AD patients, while levels in CAA/ATN- (n = 19) were comparable with those in controls. A regression model including all synaptic proteins differentiated CAA from AD at high accuracy levels (area under the curve 0.987).
Conclusion:
In contrast to AD, synaptic CSF biomarkers were found to be largely unchanged in CAA. Moreover, concomitant AD pathology in CAA is associated with abnormal synaptic protein levels. Impaired synaptic function in AD was confirmed in this independent cohort. Our findings support an apparent differential involvement of synaptic dysfunction in CAA and AD and may reflect distinct pathological mechanisms.
INTRODUCTION
Deposition of abnormally folded amyloid-β (Aβ) peptides is a common pathologic mechanism in both Alzheimer’s disease (AD) and cerebral amyloid angiopathy (CAA). In AD, Aβ deposits as plaques in the brain parenchyma, whereas in CAA, Aβ aggregates are found within cortical and leptomeningeal blood vessel walls [1, 2]. Involvement of the same protein fits the high comorbidity of moderate-to-severe CAA observed in almost 50% of AD patients [3]. Symptoms of cognitive impairment and dementia are central to both diseases.
Cognitive dysfunction correlates strongly with synaptotoxicity in AD [2, 4], where synaptic loss is recognized as one of the earliest detectable events in AD pathogenesis. Elevated levels of synaptotagmin-1, growth-associated protein 43, synaptosomal-associated protein 25, and neurogranin have repeatably been demonstrated in the cerebrospinal fluid (CSF) of AD patients [5–7]. Similarly, increased levels of synaptic proteins involved in vesicular transport and synaptic stability have been observed in patients with mild cognitive impairment, particularly those who progressed to AD [8]. A meta-analysis confirmed widespread synaptic loss in AD, with endosomal pathways, vesicular assembly mechanisms, glutamate receptors, and axonal transport being primarily affected [9]. Liquid chromatography-mass spectrometry (LC-MS) methods demonstrated that CSF levels of beta- and gamma-synuclein, neurogranin, phosphatidylethanolamine-binding protein-1 (PEBP-1), 14–3–3 proteins, and neuronal pentraxins levels were altered in AD compared with healthy controls [10], suggesting these proteins may serve as synaptic biomarkers for AD.
In contrast to AD, possible synaptic dysfunction in CAA remains understudied. Vascular amyloid might inflict synaptic degeneration, since a mouse model of non-Aβ Danish CAA demonstrated impaired inhibitory synaptic pathways, and increased tau hyperphosphorylation and misfolding [11, 12]. In contrast, it has previously been demonstrated that CSF levels of the synaptic protein neurogranin are similar in controls and patients with CAA [13]. Studying synaptic dysfunction in CAA may elucidate underlying mechanisms leading to cognitive decline in CAA and reveal yet unknown interactions or differences with AD pathophysiology.
We therefore aimed to investigate synaptic dysfunction in CAA by employing the analysis of a synaptic protein panel in CSF in cohorts including patients with clinical CAA and AD, and controls. Furthermore, we aimed to explore the relation of synaptic protein CSF levels to cerebrovascular imaging markers and cognitive decline in CAA.
MATERIALS AND METHODS
Cohorts
We included CSF samples from 25 patients with probable CAA, 49 patients with AD, and 25 control participants from the Radboud University Medical Center (Radboudumc, Nijmegen, the Netherlands; Table). CSF was collected via lumbar puncture according to a standardized protocol. See the Supplementary Material for details on CSF sample collection and ethical statements for all study participants.
Probable CAA diagnosis was obtained via magnetic resonance imaging (MRI) analysis based on the modified Boston criteria [14]. Cognitive function was assessed using the Montreal Cognitive Assessment (MoCA) in 21 of the CAA patients [15]. AD patients had a positive amyloid/pathological tau/neurodegeneration (ATN) biomarker profile [16, 17], as defined by CSF Aβ42 < 659 pg/ml (A+), phosphorylated tau181 > 64 pg/ml (T+), and total tau > 400 pg/ml (N+) quantified by automated immunoassays using a Lumipulse apparatus (Fujirebio, Ghent, Belgium). Details on the selection of control participants are provided in the Supplementary Material. Age- and sex-matched control participants were cognitively unimpaired. Information on CAA imaging markers was available neither for AD patients, nor for controls.
Magnetic resonance imaging
All CAA patients underwent an MRI scan of the brain. Of those, twenty participants underwent a 3.0 Tesla MRI scan (Siemens Magnetom Prisma, Siemens Healthineers, Erlangen, Germany) using a 32-channel head coil. Participants were examined using a comprehensive protocol, and we analyzed the 3D multi-echo gradient echo T2*-weighted sequence (voxel size 0.8×0.8×0.8 mm), the 3D T2-weighted sequence (voxel size 0.8×0.8×0.8 mm), and 3D fluid-attenuated inversion recovery (FLAIR) sequence (voxel size 0.8×0.8×0.8 mm). Magnitude and phase data from the multi-echo gradient sequence was processed to a susceptibility-weightedimaging (SWI) using the Contrast-weighted, Laplace-unwrapped, bipolar multi-Echo, ASPIRE-combined, homogeneous, improved Resolution SWI (CLEAR-SWI) method [18]. The remaining five participants underwent a 3.0 Tesla MRI using different Tesla systems with varying protocols in either the Radboud University Medical Center (Radboudumc, Nijmegen, the Netherlands) or referral hospitals, which at least included T2*-weighted images or SWI sequence images, FLAIR and T2 sequences.
The imaging markers that were assessed included lobar cerebral microbleeds (CMB), enlarged perivascular spaces (EPVS) in the centrum semi-ovale, cortical superficial siderosis (cSS), and white matter hyperintensities (WMH). CMBs within the parenchyma followed consensus criteria as small, rounded, well-defined hypointense lesions of 2–10 mm in size [19]. Number of lobar CMB was categorized for statistical comparisons (0; 1–5; 6–10; 11–15; 16–50; ≥51). EPVS were characterized as fluid-filled spaces in the centrum semi-ovale that follow the typical course of a vessel as it goes through grey or white matter, with signal intensity similar to CSF on all sequences [20]. EPVS were dichotomously scored as a low (≤20) or high incidence (≥21) [21]. cSS was characterized as linear residues of chronic blood products in the superficial cerebral cortical layers showing a distinctive gyriform pattern of low signal on blood-sensitive images [22]. cSS was scored as either absent, focal (≤3 sulci) or disseminated (≥4 sulci) [23]. Deep and periventricular WMH were ordinally scored using the Fazekas scale, ranging from 0–3 (none, punctuate, early confluent, confluent) [24].
The total burden of small vessel disease (SVD) ordinal score was computed based on the individual scores obtained from all four abovementioned parameters, ranging from 0–6 [21]. One point was granted for 2–5 lobar CMBs, and two points if≥5 CMBs. One point was granted for a high incidence (≥21) of EPVS. One point was granted for focal cSS, and two points if disseminated cSS. One point was granted for either (early) confluent deep WMH (i.e., the region between juxtacortical and ventricular areas; Fazekas score≥2), or irregular periventricular WMH spreading out into deep white matter (Fazekasscore 3).
LC-MS/MS analysis
The panel of synaptic biomarkers for simultaneous quantification included 14–3–3 epsilon, 14–3–3 eta, 14–3–3 zeta/delta, activating protein-2 (AP-2) complex subunit beta, complexin-2, beta-synuclein, gamma-synuclein, neurogranin, neuronal pentraxin-1 (NPTX1), neuronal pentraxin-2 (NPTX2), neuronal pentraxin receptor (NPTXR), rab GDP dissociation inhibitor (GDI) alpha, PEBP-1, syntaxin-1B, and syntaxin-7. The applied methodology has been described in detail elsewhere [10]. See Supplementary Table 1 for all analyzed peptides. In brief, 100μL CSF was mixed with stable isotope labeled peptide standards (internal standard), followed by sample preparation in a consecutive four-step process consisting of reduction, alkylation, tryptic digestion, and purification by solid-phase extraction. For multiple reaction monitoring MS quantitation, a micro-high-performance LC-MS system (6495 Triple Quadrupole LC/MS system, Agilent Technologies, Santa Clara, CA, USA), equipped with a Hypersil Gold reversed phase column (dim. 100×2.1 mm, particle size 1.9μm, Thermo Fisher Scientific, Waltham, MA, USA) was used. Pooled CSF samples were used as quality control and injected at regular intervals to monitor assay performance over time and to assess inter- and intra-assay variation.
Table 1
Cohort demographics
| Controls | AD | CAA | p | |
| Demographics | ||||
| Number of patients | 25 | 49 | 25 | |
| Age (y) | 71.9±7.3 | 69.6±7.2 | 73.2±6.6 | 0.10a |
| Sex, M/F (% male) | 12/13 (48%) | 22/27 (45%) | 13/12 (52%) | 0.84b |
| Aβ/tau levels (pg/ml) | ||||
| Aβ40 | 10,876 [6,419–14,737] | 9,837 [7,574–11,727] | 7,530 [6,125–8,391] | 0.002c, # , & |
| Aβ42 | 641 [530–1,116] | 419 [307–482] | 346 [288–410] | < 0.0001c, $, & |
| t-tau | 256 [177–387] | 856 [726–952] | 391 [272–537] | < 0.0001c, # , $ |
| p-tau181 | 32.8 [25.8–51.6] | 132 [115–148] | 47.2 [34.6–61.8] | < 0.0001c, # , $ |
Age is presented as means±standard deviations. Aβ and tau levels are presented as medians [interquartile range]. Bold p values indicate statistical significance. Aβ, amyloid-β peptide; AD, Alzheimer’s disease; CAA, cerebral amyloid angiopathy; F, female; M, male; p-tau181, phosphorylated tau; t-tau, total tau. aAnalysis of variance with Bonferroni’s post hoc test. bChi-square test. cKruskal-Wallis test with Dunn’s post hoc test. #Statistically significant for AD versus CAA $Statistically significant for controls versus AD &Statistically significant for controls versus CAA
Data processing and statistical analyses
Skyline version 20.1 (MacCoss Lab, University of Washington, USA) was used for chromatographic spectra peak assessment and adjustment. Ratio of the total peak areas for each peptide and corresponding internal standard, multiplied by the amount of standard added per μL CSF, was used as relative peptide concentration for each peptide.
Data was analyzed using GraphPad Prism software version 9.0.0 (GraphPad Software, Inc., San Diego, CA, USA). Shapiro-Wilk tests were used to analyze data normality. Parametric data were analyzed with a Student’s t-test or analysis of variance with Bonferroni’s post hoc test. Non-parametric data were analyzed with a Kruskal-Wallis with Dunn’s post hoc test. Categorical variables were analyzed with a Chi-square test. For proteins with multiple quantified peptides, the peptide with the lowest coefficients of variation (CV) was used for statistical analysis ( Supplementary Table 1). Multiple logistic regression and receiver operating curve analyses were used to analyze diagnostic accuracy in differentiating CAA from AD including all synaptic proteins. Spearman rank correlations were used to evaluate associations between MoCA score and imaging markers with synaptic protein concentrations in CAA patients. p values≤0.05 were consideredsignificant.
To explore the association of synaptic dysfunction with the presence of AD pathology, we performed a subgroup analysis where CAA patients were subdivided into having a positive (CAA/ATN+) or negative (CAA/ATN-) ATN biomarker status.
RESULTS
The intra-assay CV was < 10% and the inter-assay CV < 14% for all peptides ( Supplementary Table 1).
Synaptic protein differences
NPTX2 levels were decreased in CAA compared with controls (p = 0.04), but not in AD (Fig. 1 and Supplementary Table 2). All other synaptic proteins were present at similar concentrations in CAA and controls.
Fig. 1
Cerebrospinal fluid levels of synaptic proteins in controls, AD, and CAA. Concentrations (fmol/ml) were obtained after multiple reaction monitoring analysis of the synaptic proteins. Statistical comparison was performed with analysis of variance with Bonferroni’s post hoc test, or Kruskal-Wallis with Dunn’s post hoc test, as appropriate. p values: *p≤0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001. Median values and interquartile range are indicated. AD, Alzheimer’s disease; AP-2, activating protein 2; CAA, cerebral amyloid angiopathy; GDI, GDP dissociation inhibitor; PEBP-1, phosphatidylethanolamine-binding protein 1.
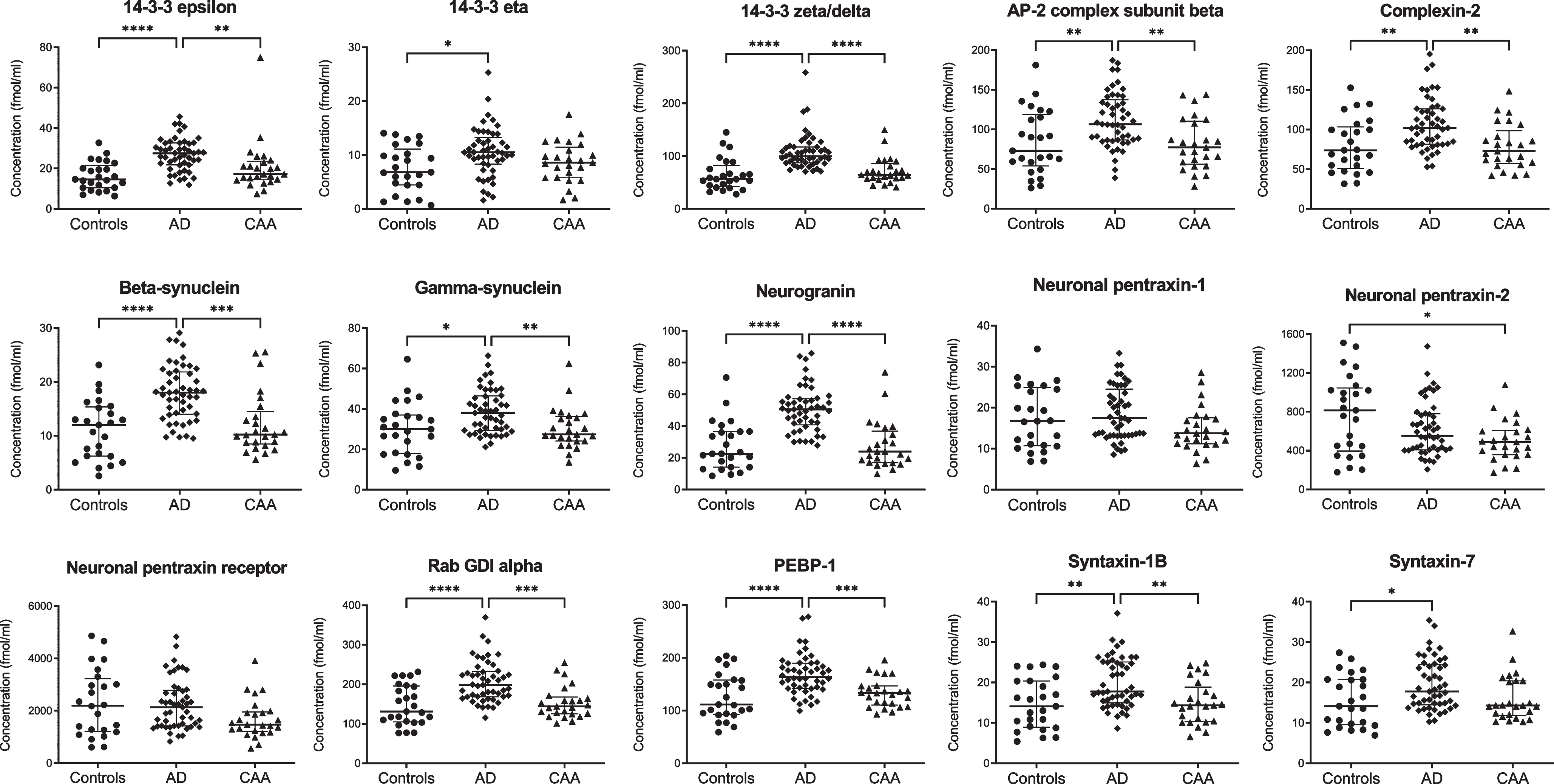
Twelve synaptic proteins displayed higher levels in AD compared with controls (p < 0.01 for syntaxin-7, gamma-synuclein, and 14–3–3 eta; p < 0.001 for syntaxin-1B, complexin-2, AP-2 complex subunit beta; p < 0.0001 for beta-synuclein, rab GDI alpha, PEBP-1, neurogranin, 14–3–3 epsilon and zeta/delta). Compared to CAA, levels in AD were also higher for gamma-synuclein (p < 0.01), syntaxin-1B, complexin-2, AP-2 complex subunit beta (all p < 0.001), beta-synuclein, rab GDI alpha, PEBP-1, neurogranin, 14–3–3 epsilon and zeta/delta (all p < 0.0001). Levels of NPTX1 and NPTXR were similar for all groups. All synaptic proteins combined differentiated CAA from AD with an area under the curve of 0.987 (95% confidence interval: 0.97–1.00, p < 0.0001).
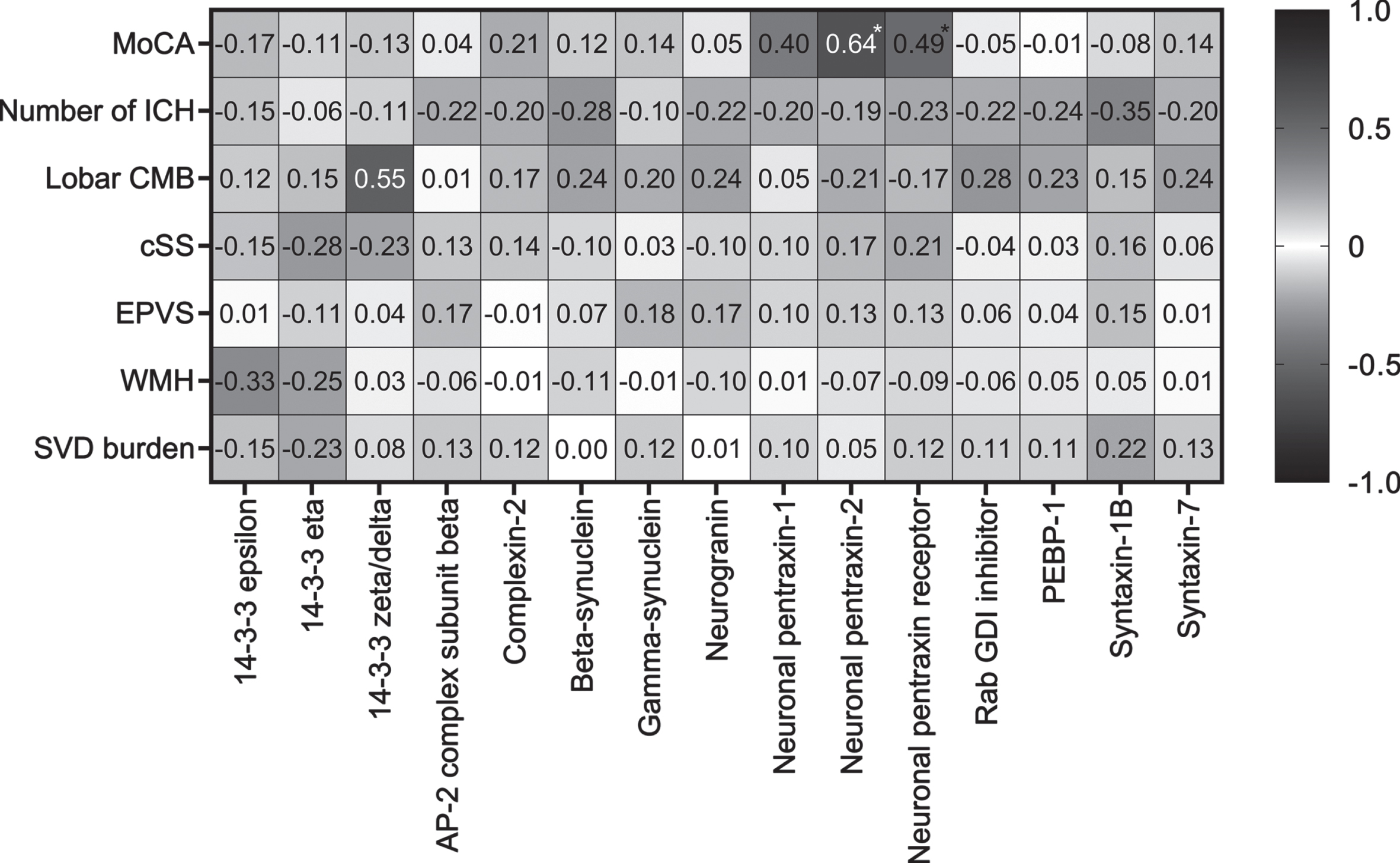
Correlations with MoCA and imaging markers in CAA
Both NPTX2 (rs = 0.64, p = 0.002) and NPTXR (rs = 0.49, p = 0.03) correlated with MoCA score in CAA (Fig. 2). None of the imaging parameters correlated with any of the synaptic proteins.
Fig. 2
Correlations of synaptic proteins with MoCA score and cerebrovascular imaging markers in CAA patients. Spearman rank correlation coefficients are displayed. MoCA was available in a subset (n = 21) of CAA patients. Asterix indicates a significant p value. AP-2, activating protein 2; CMB, cerebral microbleeds; cSS, cortical superficial siderosis; EPVS, enlarged perivascular spaces; GDI, GDP dissociation inhibitor; ICH, intracerebral hemorrhages; MoCA, Montreal Cognitive Assessment; PEBP-1, phosphatidylethanolamine-binding protein 1; SVD, small vessel disease; WMH, white matter hyperintensities.
Exploratory analysis including CAA/ATN+and CAA/ATN-
When we stratified CAA patients according to their ATN biomarker status, six CAA patients were AD biomarker positive (CAA/ATN+) and nineteen were negative (CAA/ATN-). The exploratory analysis showed that compared with CAA/ATN-, levels in AD were increased for NPTX1 (p < 0.05), gamma-synuclein, syntaxin-7 (both p < 0.01), 14–3–3 epsilon, AP-2 complex subunit beta, complexin-2, PEBP-1, syntaxin-1B (all p < 0.001), 14–3–3 zeta/delta, beta-synuclein, neurogranin, and rab GDI alpha (all p < 0.0001; Supplementary Figure 1). Five synaptic proteins displayed increased levels in CAA/ATN+compared with CAA/ATN- (p < 0.05 for 14–3–3 zeta/delta, gamma-synuclein, neurogranin, and rab GDI alpha; p < 0.01 for beta-synuclein).
Levels of 14–3–3 epsilon and zeta/delta, and beta-synuclein were increased in CAA/ATN+compared with controls (all p < 0.05). All synaptic proteins were present at similar concentrations in CAA/ATN- and controls, and in CAA/ATN+ and AD.
MoCA scores and cerebrovascular imaging markers did not differ between CAA/ATN+ and CAA/ATN- (Supplementary Table 3).
DISCUSSION
In this study, we demonstrated a decrease of NPTX2 concentrations in the CSF of CAA patients, but other synaptic protein levels were unchanged in CAA. Moreover, we were able to confirm previous findings of synaptic degeneration in AD in our cohort. The synaptic panel differentiated CAA from AD at high accuracy levels. Finally, the exploratory analysis regarding CAA subgroups showed that CAA/ATN+ patients have a synaptic protein profile resembling that of AD patients, whereas CAA/ATN- were similar to controls.
Several synaptic proteins displayed higher CSF levels in AD as compared with both CAA and controls, coinciding with previous observations. Synapse loss is one of the main neurodegenerative mechanisms in AD, preceding indicators of neuronal death [25] and presenting during the preclinical disease stage [8]. Since synaptotoxicity and cognitive decline correlate well [2, 4], synaptic protein CSF levels may serve as indicators of disease severity in AD. In contrast to this synaptotoxicity in AD driven by parenchymal Aβ, growing evidence suggests that neurodegeneration induced by vascular Aβ in CAA more prominently presents as ischemic brain injury and vascular integrity loss, leading to progressive atrophy and cognitive decline [1, 26]. Thus, unchanged synaptic proteins levels in CAA might reflect mechanistic differences in the pathological pathways of AD and CAA. The observed similar synaptic protein concentrations in CAA/ATN- and controls underline the lack of synaptic loss in CAA in the absence of AD pathology.
Decreased levels of NPTXs secreted into the synaptic cleft, such as NPTX2, could reflect a different mechanisms of synaptic dysfunction in patients with CAA, in the form of inducing short-term or long-term depression by interacting with AMPA receptors to modulate synaptic plasticity [27, 28]. NPTX2 CSF levels correlate the best with cognitive status compared with other synaptic markers [10, 29]. Decreased NPTX2 CSF levels were previously reported in AD, however differentially expressed pentraxins have also been associated with other neurodegenerative diseases, including multiple sclerosis, frontotemporal dementia, and Parkinson’s disease [27, 29]. Moreover, NPTX2 levels have been associated with processes like neuroinflammatory responses [30] and blood-brain barrier dysfunction [31]. Although NPTX2 levels correlated with MoCA scores in CAA, it is questionable whether the lowered NPTX2 levels in CAA display disease-specific synaptotoxicity, or rather reflect universal neurodegeneration. Additionally, synaptic protein levels of CAA/ATN+ resembled that of AD patients, indicating that AD pathology may be driving synaptic dysfunction. As opposed to NPTXs, all other synaptic proteins included in the current study are located at presynaptic and postsynaptic terminals, and not secreted into the synaptic cleft [10]. Unchanged concentrations of these synaptic proteins might consequently point to more conserved synapses in CAA, in contrast to what is commonly seen in AD. Of all currently investigated synaptic proteins, only neurogranin was previously studied in patients with CAA [13], yielding CSF concentrations similar to controls like in our study, further corroborating our hypothesis on more conserved synapses in CAA than AD.
Our results should be considered in light of several limitations. The relatively small sample sizes may lead to a limited study power; however, we could confirm previously reported results of altered levels in AD compared with controls. The APOE ɛ4 allele is a known major risk factor for the development of both vascular and parenchymal Aβ deposits [2], but APOE genotype status was not available to include as confounder. Moreover, AD diagnosis was solely based on ATN-biomarker status, without knowledge of clinical phenotype. Finally, general cognitive assessment scores and MRI data were only available for patients with CAA. A major strength includes using a robust and validated LC-MS quantification method, with high sensitivity and selectivity for the targeted synaptic proteins.
To conclude, our findings show that synaptic functioning may be more conserved in CAA compared with AD. CSF levels of synaptic markers could serve as biomarkers of synaptic pathology in AD. Our findings support a possible differential involvement of synaptic dysfunction in CAA and AD, which is particularly pronounced in the presence of AD pathology. However, since CSF levels are an indirect reflection of pathological processes occurring in the central nervous system, neuropathology studies assessing regional differences will aid in verifying the lack of synaptotoxicity in CAA. Furthermore, longitudinal studies including cognitive assessments are warranted to examine the association of synaptic dysfunction with disease severity.
ACKNOWLEDGMENTS
We thank Hugo van Berckel-Smit for his help with rating the MRIs.
FUNDING
This work was supported by the Eivind and Elsa K: Son Sylvan Foundation, the Märta and Gustaf Ågren Foundation, the Herbert and Karin Jacobsson Foundation, the Gun and Bertil Stohne Foundation, the Foundation for Gamla Tjänarinnor, the Felix Neubergh Foundation, Demensfonden, Rune and Ulla Almlöv Foundation, an anonymous donor and a grant from The Galen and Hilary Weston Foundation (NR170024). MMV is supported by the BIONIC project (no. 733050822, which has been made possible by ZonMW as part of ‘Memorabel’, the research and innovation program for dementia, as part of the Dutch national ‘Deltaplan for Dementia’: zonmw.nl/dementiaresearch) and the CAFÉ project (the National Institutes of Health, USA, grant number 5R01NS104147-02). The BIONIC project is a consortium of Radboudumc, LUMC, ADX Neurosciences, and Rhode Island University. CJMK receives funding for research outside the submitted work of the Netherlands Cardiovascular Research Initiative, which is supported by the Dutch Heart Foundation, CVON2015–01: CONTRAST, and the support of the Brain Foundation Netherlands (HA2015.01.06). CONTRAST is additionally financed by the Ministry of Economic Affairs by means of the PPP Allowance made available by the Top Sector Life Sciences & Health to stimulate public-private partnerships (LSHM17016) and was funded in part through unrestricted funding by Stryker, Medtronic and Cerenovus. The funding sources were not involved in study design, monitoring, data collection, statistical analyses, interpretation of results, or manuscript writing; Radboudumc and Erasmus MC received additional unrestricted funding on behalf of CONTRAST, for the execution of the Dutch ICH Surgery Trial pilot study and for the Dutch ICH Surgery Trial from Penumbra Inc. FHBMS is supported by a senior clinical scientist grant of the Dutch Heart Foundation (grant 2019T060).
HZ is a Wallenberg Scholar supported by grants from the Swedish Research Council (#2018-02532), the European Research Council (#681712), Swedish State Support for Clinical Research (#ALFGBG-720931), the Alzheimer Drug Discovery Foundation (ADDF), USA (#201809-2016862), the AD Strategic Fund and the Alzheimer’s Association (#ADSF-21-831376-C, #ADSF-21-831381-C and #ADSF-21-831377-C), the Olav Thon Foundation, the Erling-Persson Family Foundation, Stiftelsen för Gamla Tjänarinnor, Hjärnfonden, Sweden (#FO2019-0228), the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 860197 (MIRIADE), European Union Joint Program for Neurodegenerative Disorders (JPND2021-00694), and the UK Dementia Research Institute at UCL (UKDRI-1003). KB is supported by the Swedish Research Council (#2017-00915), the Alzheimer Drug Discovery Foundation (ADDF), USA (#RDAPB-201809-2016615), the Swedish Alzheimer Foundation (#AF-742881), Hjärnfonden, Sweden (#FO2017-0243), the Swedish state under the agreement between the Swedish government and the County Councils, the ALF-agreement (#ALFGBG-715986), European Union Joint Program for Neurodegenerative Disorders (JPND2019-466-236), and the Alzheimer’s Association 2021 Zenith Award (ZEN-21-848495). JG is supported by Alzheimerfonden (AF-930934) and the Foundation of Gamla Tjänarinnor. AB is supported by European Union Joint Program for Neurodegenerative Disorders (“PreSSAD” JPND2021-650-272).
CONFLICT OF INTEREST
HZ has served at scientific advisory boards and/or as a consultant for Abbvie, Alector, Annexon, Artery Therapeutics, AZTherapies, CogRx, Denali, Eisai, Nervgen, Pinteon Therapeutics, Red Abbey Labs, Passage Bio, Roche, Samumed, Siemens Healthineers, Triplet Therapeutics, and Wave, has given lectures in symposia sponsored by Cellectricon, Fujirebio, Alzecure, Biogen, and Roche, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program. KB has served as a consultant, at advisory boards, or at data monitoring committees for Abcam, Axon, Biogen, JOMDD/Shimadzu. Julius Clinical, Lilly, MagQu, Novartis, Roche Diagnostics, and Siemens Healthineers, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program. MMV serves as a consultant for Vico. The other authors declare no conflict of interest.
DATA AVAILABILITY
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-220977.
REFERENCES
[1] | Charidimou A , Boulouis G , Gurol ME , Ayata C , Bacskai BJ , Frosch MP , Viswanathan A , Greenberg SM ((2017) ) Emerging concepts in sporadic cerebral amyloid angiopathy. Brain 140: , 1829–1850. |
[2] | Greenberg SM , Bacskai BJ , Hernandez-Guillamon M , Pruzin J , Sperling R , van Veluw SJ ((2020) ) Cerebral amyloid angiopathy and Alzheimerdisease - One peptide, two pathways. Nat Rev Neurol 16: , 30–42. |
[3] | Jäkel L , De Kort AM , Klijn CJM , Schreuder F , Verbeek MM ((2021) ) Prevalence of cerebral amyloid angiopathy: A systematic review andmeta-analysis. Alzheimers Dement 18: , 10–28. |
[4] | Terry RD , Masliah E , Salmon DP , Butters N , DeTeresa R , Hill R , Hansen LA , Katzman R ((1991) ) Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann Neurol 30: , 572–580. |
[5] | Brinkmalm A , Brinkmalm G , Honer WG , Frolich L , Hausner L , Minthon L , Hansson O , Wallin A , Zetterberg H , Blennow K , Ohrfelt A ((2014) ) SNAP-25 is a promising novel cerebrospinal fluid biomarker for synapse degeneration in Alzheimer’s disease. Mol Neurodegener 9: , 53. |
[6] | Ohrfelt A , Brinkmalm A , Dumurgier J , Brinkmalm G , Hansson O , Zetterberg H , Bouaziz-Amar E , Hugon J , Paquet C , Blennow K ((2016) ) The pre-synaptic vesicle protein synaptotagmin is a novel biomarker for Alzheimer’s disease. Alzheimers Res Ther 8: , 41. |
[7] | Kvartsberg H , Duits FH , Ingelsson M , Andreasen N , Öhrfelt A , Andersson K , Brinkmalm G , Lannfelt L , Minthon L , Hansson O ((2015) ) Cerebrospinal fluid levels of the synaptic protein neurogranincorrelates with cognitive decline in prodromal Alzheimer’s disease. Alzheimers Dement 11: , 1180–1190. |
[8] | Duits FH , Brinkmalm G , Teunissen CE , Brinkmalm A , Scheltens P , Vander Flier WM , Zetterberg H , Blennow K ((2018) ) Synaptic proteins inCSF as potential novel biomarkers for prognosis in prodromalAlzheimer’s disease. Alzheimers Res Ther 10: , 5. |
[9] | de Wilde MC , Overk CR , Sijben JW , Masliah E ((2016) ) Meta-analysis of synaptic pathology in Alzheimer’s disease reveals selective molecular vesicular machinery vulnerability. Alzheimers Dement 12: , 633–644. |
[10] | Nilsson J , Gobom J , Sjödin S , Brinkmalm G , Ashton NJ , Svensson J , Johansson P , Portelius E , Zetterberg H , Blennow K ((2021) ) Cerebrospinal fluid biomarker panel for synaptic dysfunction in Alzheimer’s disease. Alzheimers Dement 13: , e12179. |
[11] | Cisternas P , Taylor X , Perkins A , Maldonado O , Allman E , Cordova R , Marambio Y , Munoz B , Pennington T , Xiang S , Zhang J , Vidal R , Atwood B , Lasagna-Reeves CA ((2020) ) Vascular amyloid accumulation alters the gabaergic synapse and induces hyperactivity in a model of cerebral amyloid angiopathy. Aging Cell 19: , e13233. |
[12] | You Y , Perkins A , Cisternas P , Muñoz B , Taylor X , You Y , Garringer HJ , Oblak AL , Atwood BK , Vidal R , Lasagna-Reeves CA ((2019) ) Tau as a mediator of neurotoxicity associated to cerebral amyloid angiopathy. Acta Neuropathol Commun 7: , 26. |
[13] | Banerjee G , Ambler G , Keshavan A , Paterson RW , Foiani MS , Toombs J , Heslegrave A , Dickson JC , Fraioli F , Groves AM , Lunn MP , Fox NC , Zetterberg H , Schott JM , Werring DJ ((2020) ) Cerebrospinal fluid biomarkers in cerebral amyloid angiopathy. J Alzheimers Dis 74: , 1189–1201. |
[14] | Linn J , Halpin A , Demaerel P , Ruhland J , Giese AD , Dichgans M , van Buchem MA , Bruckmann H , Greenberg SM ((2010) ) Prevalence of superficial siderosis in patients with cerebral amyloid angiopathy. Neurology 74: , 1346–1350. |
[15] | Nasreddine ZS , Phillips NA , Bédirian V , Charbonneau S , Whitehead V , Collin I , Cummings JL , Chertkow H ((2005) ) The Montreal CognitiveAssessment, MoCA: A brief screening tool for mild cognitive impairment. J Am Geriatr Soc 53: , 695–699. |
[16] | Jack CR Jr , Bennett DA , Blennow K , Carrillo MC , Dunn B , Haeberlein SB , Holtzman DM , Jagust W , Jessen F , Karlawish J , Liu E , Molinuevo JL , Montine T , Phelps C , Rankin KP , Rowe CC , Scheltens P , Siemers E , Snyder HM , Sperling R ((2018) ) NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement 14: , 535–562. |
[17] | Vos SJ , Visser PJ , Verhey F , Aalten P , Knol D , Ramakers I , Scheltens P , Rikkert MG , Verbeek MM , Teunissen CE ((2014) ) Variability of CSFAlzheimer’s disease biomarkers: Implications for clinical practice. PLoS One 9: , e100784. |
[18] | Eckstein K , Bachrata B , Hangel G , Widhalm G , Enzinger C , Barth M , Trattnig S , Robinson SD ((2021) ) Improved susceptibility weighted imaging at ultra-high field using bipolar multi-echo acquisition and optimized image processing: CLEAR-SWI. Neuroimage 237: , 118175. |
[19] | Gregoire SM , Chaudhary UJ , Brown MM , Yousry TA , Kallis C , Jäger HR , Werring DJ ((2009) ) The Microbleed Anatomical Rating Scale (MARS): Reliability of a tool to map brain microbleeds. Neurology 73: , 1759–1766. |
[20] | Wardlaw JM , Smith EE , Biessels GJ , Cordonnier C , Fazekas F , Frayne R , Lindley RI , O’Brien JT , Barkhof F , Benavente OR , Black SE , Brayne C , Breteler M , Chabriat H , Decarli C , de Leeuw FE , Doubal F , Duering M , Fox NC , Greenberg S , Hachinski V , Kilimann I , Mok V , Oostenbrugge R , Pantoni L , Speck O , Stephan BC , Teipel S , Viswanathan A , Werring D , Chen C , Smith C , van Buchem M , Norrving B , Gorelick PB , Dichgans M ((2013) ) Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol 12: , 822–838. |
[21] | Charidimou A , Martinez-Ramirez S , Reijmer YD , Oliveira-Filho J , Lauer A , Roongpiboonsopit D , Frosch M , Vashkevich A , Ayres A , Rosand J , Gurol ME , Greenberg SM , Viswanathan A ((2016) ) Total magnetic resonance imaging burden of small vessel disease in cerebral amyloid angiopathy: An imaging-pathologic study of concept validation. JAMA Neurol 73: , 994–1001. |
[22] | Charidimou A , Jäger RH , Fox Z , Peeters A , Vandermeeren Y , Laloux P , Baron JC , Werring DJ ((2013) ) Prevalence and mechanisms of cortical superficial siderosis in cerebral amyloid angiopathy. Neurology 81: , 626–632. |
[23] | Charidimou A , Boulouis G , Roongpiboonsopit D , Auriel E , Pasi M , Haley K , van Etten ES , Martinez-Ramirez S , Ayres A , Vashkevich A , Schwab KM , Goldstein JN , Rosand J , Viswanathan A , Greenberg SM , Gurol ME ((2017) ) Cortical superficial siderosis multifocality in cerebral amyloid angiopathy: A prospective study. Neurology 89: , 2128–2135. |
[24] | Fazekas F , Chawluk JB , Alavi A , Hurtig HI , Zimmerman RA ((1987) ) MR signal abnormalities at 1.5 T in Alzheimer’s dementia and normal aging. AJR Am J Roentgenol 149: , 351–356. |
[25] | Lleó A , Núñez-Llaves R , Alcolea D , Chiva C , Balateu-Paños D , Colom-Cadena M , Gomez-Giro G , Muñoz L , Querol-Vilaseca M , Pegueroles J , Rami L , Lladó A , Molinuevo JL , Tainta M , Clarimón J , Spires-Jones T , Blesa R , Fortea J , Martínez-Lage P , Sánchez-Valle R , Sabidó E , Bayés À , Belbin O ((2019) ) Changes in synaptic proteins precede neurodegeneration markers in preclinical Alzheimer’s disease cerebrospinal fluid. Mol Cell Proteomics 18: , 546–560. |
[26] | Smith EE ((2018) ) Cerebral amyloid angiopathy as a cause of neurodegeneration. J Neurochem 144: , 651–658. |
[27] | Camporesi E , Nilsson J , Brinkmalm A , Becker B , Ashton NJ , Blennow K , Zetterberg H ((2020) ) Fluid biomarkers for synaptic dysfunction and loss. Biomark Insights 15: , 1177271920950319. |
[28] | Chapman G , Shanmugalingam U , Smith PD ((2019) ) The role of neuronal pentraxin 2 (NP2) in regulating glutamatergic signaling and neuropathology. Front Cell Neurosci 13: , 575. |
[29] | van der Ende EL , Xiao M , Xu D , Poos JM , Panman JL , Jiskoot LC , Meeter LH , Dopper EG , Papma JM , Heller C , Convery R , Moore K , Bocchetta M , Neason M , Peakman G , Cash DM , Teunissen CE , Graff C , Synofzik M , Moreno F , Finger E , Sánchez-Valle R , Vandenberghe R , Laforce R Jr , Masellis M , Tartaglia MC , Rowe JB , Butler CR , Ducharme S , Gerhard A , Danek A , Levin J , Pijnenburg YA , Otto M , Borroni B , Tagliavini F , de Mendonca A , Santana I , Galimberti D , Seelaar H , Rohrer JD , Worley PF , van Swieten JC ((2020) ) Neuronalpentraxin 2: A synapse-derived CSF biomarker in geneticfrontotemporal dementia. J Neurol Neurosurg Psychiatry 91: , 612–621. |
[30] | Moreno-Rodriguez M , Perez SE , Nadeem M , Malek-Ahmadi M , Mufson EJ ((2020) ) Frontal cortex chitinase and pentraxin neuroinflammatory alterations during the progression of Alzheimer’s disease. J Neuroinflammation 17: , 58. |
[31] | Cummings DM , Benway TA , Ho H , Tedoldi A , Fernandes Freitas MM , Shahab L , Murray CE , Richard-Loendt A , Brandner S , Lashley T , Salih DA , Edwards FA ((2017) ) Neuronal and peripheral pentraxins modify glutamate release and may interact in blood-brain barrier failure. Cereb Cortex 27: , 3437–3448. |


