
Polyunsaturated Fatty Acids Mend Macrophage Transcriptome, Glycome, and Phenotype in the Patients with Neurodegenerative Diseases, Including Alzheimer’s Disease
Abstract
Background:
Macrophages of healthy subjects have a pro-resolution phenotype, upload amyloid-β (Aβ) into endosomes, and degrade Aβ, whereas macrophages of patients with Alzheimer’s disease (AD) generally have a pro-inflammatory phenotype and lack energy for brain clearance of Aβ.
Objective:
To clarify the pathogenesis of sporadic AD and therapeutic effects of polyunsaturated fatty acids (PUFA) with vitamins B and D and antioxidants on monocyte/macrophage (MM) migration in the AD brain, MM transcripts in energy and Aβ degradation, MM glycome, and macrophage clearance of Aβ.
Methods:
We followed for 31.3 months (mean) ten PUFA-supplemented neurodegenerative patients: 3 with subjective cognitive impairment (SCI), 2 with mild cognitive impairment (MCI), 3 MCI/vascular cognitive impairment, 2 with dementia with Lewy bodies, and 7 non-supplemented caregivers. We examined: monocyte migration in the brain and a blood-brain barrier model by immunochemistry and electron microscopy; macrophage transcriptome by RNAseq; macrophage glycome by N-glycan profiling and LTQ-Orbitrap mass spectrometry; and macrophage phenotype and phagocytosis by immunofluorescence.
Results:
MM invade Aβ plaques, upload but do not degrade Aβ, and release Aβ into vessels, which develop cerebrovascular amyloid angiopathy (CAA); PUFA upregulate energy and Aβ degradation enzyme transcripts in macrophages; PUFA enhance sialylated N-glycans in macrophages; PUFA reduce oxidative stress and increase pro-resolution MM phenotype, mitochondrial membrane potential, and Aβ phagocytosis (p < 0.001).
Conclusion:
Macrophages of SCI, MCI, and AD patients have interrelated defects in the transcriptome, glycome, Aβ phagocytosis, and Aβ degradation. PUFA mend macrophage transcriptome, enrich glycome, enhance Aβ clearance, and benefit the cognition of early-stage AD patients.
INTRODUCTION
Amyloid-β1 - 42 (Aβ) and P-tau accumulation in the brain is a proximal cause of Alzheimer’s disease (AD) neuropathology. The original amyloid hypothesis explained the neuropathology by Aβ deposition in plaques as the primary cause through disruption of neuronal connections [1]. Subsequent work showed that soluble low molecular weight Aβ oligomers are the toxic species damaging synapses and inducing tau hyperphosphorylation, and Aβ oligomers and aggregated P-tau are synergistically toxic at synapses [2].
The accumulation of Aβ and P-tau in the brain induces central nervous system (CNS) and systemic immune and vascular pathologies.
Brain clearance depends upon the innate immune system’s macrophages and microglia [3] and the glymphatic system [4]. Microglial chemokines attract peripheral monocytes into the CNS [5]. Macrophages target both soluble and aggregated Aβ and release soluble fragments in contrast to microglia, which are limited in the uptake and degradation of Aβ in endosomes [6]. Healthy macrophages upload and degrade Aβ [7], but AD patients’ macrophages fail Aβ clearance due to defective glycoproteins [7], low energy [8], inflammatory M1 phenotype [8], defective unfolded protein response (UPR) [9], and other defects. In the AD brain, macrophages participate in the formation of microaneurysms in the most severe cases of cerebrovascular amyloid angiopathy (CAA) [10].
In AD patients, both brain cells and macrophages are defective in energy. The AD brain has a reduction in glucose metabolism of the temporoparietal cortex detected by fluorodeoxyglucose positron emission tomography (FDG PET) associated with reduced expression of nuclear genes encoding the subunits of the mitochondrial electron transport chain [11].
In the process of Aβ phagocytosis, reactive oxygen species (ROS) cause oxidative and nitrosative damage to nuclear and mitochondrial DNA in energy enzymes in glycolysis, tricarboxylic acid cycle (TCA), and OX-PHOS genes. In addition, carbonylation of proteins and peroxidation of lipids increase toxic molecule 4-hydroxy-2-nonenal, endoplasmic reticulum (ER) stress, and intracellular calcium rise [12].
In this study, we highlight the key role of blood-borne macrophages in brain clearance and examine the pathways of monocyte migration from vessels to plaques and back to vessels. We analyze the patchy repair of macrophage transcriptome and glycome by polyunsaturated fatty acid (PUFA) supplementation in a cohort of 10 neurodegenerative patients [5 mild cognitive impairment (MCI), 3 subjective cognitive impairment (SCI), 2 dementia with Lewy bodies (DLB)] supplemented by PUFA for 22–56 months. Our objective has been to identify the defects in macrophage transcriptome, glycome and functions, and the effects of PUFA together with anti-oxidants and vitamins D and B on these defects.
MATERIALS AND METHODS
Study design and diagnostic studies
We performed a prospective study since 2012 of 10 independently-living patients (6 females and 4 males) with the complaints on admission of memory loss and declining cognitive state despite the cholinesterase inhibitor (CI) therapy in five patients (Table 1). All of these patients and/or their caregivers requested and received PUFA supplementation by the SmartfishR (SMF) drink and their caregivers (spouse or child) were enrolled as non-supplemented controls. Mean age: Patients 73.9 years; controls 61.2 years and M/F ratios 0.66 and 0.4.
Table 1
Demographic information and dates of samples collected from patients and control caregivers and time elapsed since the onset of SMF supplementation
| Pt # | Sex | Age at onset (y) | Diagnosis | SMF supplementation start date | Date of sample collection | Time elapsed between supplementation and sampling (mo) | Control caregiver for each patient | Age at onset, sex | MMSE |
| 2 | M | 78 | MCI/VCI | 9/2013 | 8/2015 | 23 | wife | 70,F | 30 |
| 8 | F | 64 | SCI | 6/2014 | 8/2015 | 14 | – | ||
| 9 | M | 79 | DLB | 6/2014 | 5/2015 | 11 | wife | 71, F | 30 |
| 10 | F | 61 | SCI | 6/2014 | 5/2015 | 11 | – | ||
| 18 | F | 71 | SCI | 6/2016 | 1/2017 | 6 | – | ||
| 22A | F | 82 | MCI/AD | – | – | 5 | daughter | 51, F | 30 |
| 14 | F | 87 | MCI/VCI | 9/2014 | 8/2015 | 11 | son | 63, M | 30 |
| 28 | F | 81 | MCI/AD | 10/2017 | 6/2021 | 50 | son | 53, M | 30 |
| 33 | M | 65 | MCI/VCI | 1/2019 | 6/2021 | 30 | wife | 56, F | 30 |
| 37 | M | 71 | DLB | 5/2019 | 3/2021 | 22 | wife | 65, F | 30 |
| M/F=0.6 | Mean=73.9 | M/F=0.4 | Mean=61.2 |
The patients received PUFA supplementation by nightly intake of the SMF drink five days per week (omitting the weekend to prevent saturation of macrophage receptors; Note: In our preceding study, we found that daily supplementation with SMF resulted after several months in a decrease in macrophage phagocytosis of Aβ. We instruct the patients “Supplement five days a week and omit the weekend”). The SMF drink contains in a 200 ml carton marine docosahexaenoic acid (DHA) 1.1 gm and eicosapentaenoic acid (EPA) 0.9 gm per drink from the fish oil (Vesteraalens Co. Norway), the botanical antioxidants pomegranate and chokeberry, 10μg vitamin D3, and 150 mg resveratrol. Lipidomic analysis of the SMF drink showed, in addition to DHA and EPA, other lipids, including omega-6 fatty acids and phospholipids, cholesterol esters, and ceramides, lysophosphatidylcholine, and lysophosphatidylethanolamine. In addition, the patients were supplemented daily by a B complex vitamin tablet.
Following each patient’s entry, we made the diagnosis using Petersen criteria [13], the Mini-Mental State Examination (MMSE), and clinical and radiological criteria, as: a) SCI with the MMSE test score > 28 (patients 8,10,18), b) MCI with the MMSE score 20–28 (patients 22 and 28), c) MCI/VCI with a history of stroke(s) (patients 2,14, 37); e) DLB (patients 9 and 33). We established a specific diagnosis according to the FDG PET scan (not provided to SCI patients per insurance) as AD or MCI (hypometabolism in temporo-parietal areas), or DLB (diffuse hypometabolism) (Table 2).
Table 2
Diagnosis, radiology, history, PUFA supplementation, and cognitive results. Patients are listed by accession number and show clinical diagnosis (Dx), radiology (PET and MRI), history and symptoms, cholinesterase inhibitor (CI) (Aricept) and NMDA blocker (Namenda) therapy, MMSE score at the first and the last visit, onset of dementia, and duration of PUFA supplementation by the SmartfishR drink (SMF) (months)
| Subjective Cognitive Impairment | |||||||||
| Months on AD Therapy | MMSE Score | ||||||||
| Pt # | Dx | Radiology | History | CI | NMDA | First Visit | Last Visit | Onset of Dementia (mo p onset) | PUFA supplement (mo) |
| 8 | SCI | N/D | Memory loss | 0 | 0 | 30 | 30 | neg | 24 |
| 10 | SCI | N/D | Memory loss, familial alcoholism | 0 | 0 | 29 | 30 | neg | 24 |
| 18 | SCI | N/D | Concussion, low exec. function | 2 | 2 | 30 | 30 | neg | 56 |
| Mild Cognitive Impairment (MCI) | |||||||||
| 22 | MCI | N/D | Memory loss | 24 | 24 | 22 | 24 | neg | 25 |
| 28 | MCI | No stenosis (MRA) | Memory loss | 24 | 0 | 16 | 25 | neg | >19 |
| MCI/Vascular Cognitive Impairment (VCI) | |||||||||
| 2 | VCI | Regional hypo metabolism (PET) | Repeated minor strokes | 0 | 24 | 24 | 27 | Neg | 44 |
| 14 | VCI | Post cingulate hypometabolism (PET) | Repeated minor strokes | 24 | 0 | 25 | 27 | 60 | 49 |
| 37 | VCI | Assymetric temporal hypometabolism | Depression | 0 | 0 | 30 | 30 | Neg | 36 |
| Dementia with Lewy Bodies (DLB)/ VCI | |||||||||
| 9 | DLB | Cortical hypometabolism (PET) | Paranoia | 0 | 0 | 26 | 25 | 24 | 22 |
| 33 | DLB | Parieto-temporal, cingulate and visual cortex hypometabolism (PET) | Major depression | 12 | 12 | 26 | 30 | Neg | 36 |
We collected 20 cc of anticoagulated blood and 10 ml of coagulated blood, isolated peripheral blood mononuclear cells (PBMC) and prepared macrophage cultures, as described [8]. We examined phagocytosis of FITC-Aβ, oxidative stress, mitochondrial membrane potential (MMP), and sequenced macrophage transcriptome. The GlycoAnalytics core at UCSD tested the PBMC glycome by monosaccharide analysis and N-glycan profiling. The UCLA Institutional Review Board (IRB) approved the conduct of this study. The study subjects signed Informed Consent in accordance with the Code of Ethics of the World Medical Association.
Isolation of PBMC and preparation of macrophage cultures
We isolated PBMC from heparin-anticoagulated venous blood by centrifugation (3,000 RPM, 25 min) on a Ficoll-Hypaque gradient. We placed 500,000 PBMC into the wells of an 8-well T.C. plate in Iscove’s modified Dulbecco’s medium (IMDM) with 12% autologous serum and cultivated the cells at 37°C in a CO2 incubator for one to two weeks when typical macrophages became differentiated with expression of CD54, CD80, CD163, CD206, and iNOS markers.
Fluorescent fibrillary Aβ
FAM-Aβ (Anaspec) was dissolved in 1x PBS to achieve a final concentration of 1 mg/ml. Insoluble material was removed by centrifugation at 10,000 rpm for 10 min, and fibrils were prepared from the supernatant with shaking at 250 RPM at 37°C. Fibril formation was confirmed by negative-staining electron microscopy.
YFP fibrillary P-tau
We thank Dr. Paul Seidler, UCLA-DOE Institute, for providing purified fibrils of phosphorylated 4R1 N tau (P-tau) fused at the C-terminus to an EYFP reporter from HEK293 tau biosensor cells. HEK293 cells stably expressing 4R1 N tau P301 S tau-EYFP were seeded with recombinant fibrils of 4R2 N tau. Seeded cells were expanded from a 96-well plate sequentially to T25, T75, and finally T225 flasks maintained in DMEM (Life Technologies) supplemented with 10% FBS to yield a 1.7-gram cell pellet from which insoluble tau fibrils were extracted. Cells were resuspended in 5 ml of the Sucrose buffer (0.8 M NaCl, 10% sucrose, 10 mM Tris–HCl, and pH 7.4) supplemented with 1 mM EGTA were lysed in a polytron and clarified by centrifugation at 13,000 rpm for 20 min. The supernatant was incubated with 1% sarkosyl for 1 h at 37°C with gentle shaking, and sarkosyl insoluble fibrils of tau were isolated by ultracentrifugation at 95,000 rpm for 1 h. The pellet was resuspended in 1 ml of the sucrose buffer supplemented with 5 mM EDTA and 1 mM EGTA and spun at 13,000 RPM for 20 min. The supernatant was spun at 95,000 RPM for 1 h, and the pellet resuspended in 0.5 ml of 20 mM Tris–HCl pH 7.4 and 100 mM NaCl. Fibrils were confirmed by negative stain electron microscopy and protein concentration was quantified using the bicinchoninic acid (BCA) assay.
In vitro macrophage assays
Live cell staining: Macrophages were treated as indicated in the IMDM medium, washed and stained. Cells were stained for 30 min with Lysotracker for lysosomes, Cell ROX Orange for ROS, Mito Tracker Red Cell ROX for mitochondrial membrane potential, and Nucl Blue Live for nuclei (all from Fisher Scientific) and examined by immunofluorescence.
Phagocytosis assay: We incubated macrophages with HiLyte fluor 488 amyloid-β1 - 42 (FAM-Aβ) (Anaspec, Fremont, CA) (18 h, 2μg/ml in DMSO) and Omega FF-100-1 (Vesteraalens, Oslo, Norway) containing 50μM DHA and 36μM EPA in the IMDM, as indicate. After incubation, we washed, fixed, stained with phalloidin-tetramethylrhodamine (Sigma-Aldrich, St. Louis, MO), and examined by fluorescence microscopy for FAM-AB phagocytosis in Olympus BX60 microscope and Image-Pro Software.
Omega-3 fatty acids: In vitro, we treated macrophages with a final dilution of omega fatty acids in the medium.
Epoxyeicosatrienoic acid (EET) methyl esters: The EET methyl esters were synthesized from arachidonic acid methyl ester according to the method previously reported [14] and stored at –20°C as a 1 mM DMSO solution [15]. We treated macrophages with 50μM concentrations of EET methyl esters in IMDM.
Dual cyclooxygenase-2/soluble epoxide hydrolase inhibitor: The COX-2/sEH dual inhibitor (4-(5-ph-enyl-3-3-[3-(4-trifluoromethyl-phenyl)-ureido]-pro-pyl-pyrazol-1-yl)-benzenesulfonamide (PTUPB) was synthesized according to the methods reported previously [15]. We treated macrophages with 50μM concentrations of PTUPB.
RNA sequencing
We extracted RNA from 40,000 macrophages using Quick-RNA Miniprep kit (Zymo Research, http://www.zymoresearch.com). Total RNA quality was assessed on a TapeStation 2200 (Agilent Technologies) using an RNA HS ScreenTape Assay; the concentration was measured with Qubit RNA HS Assay (ThermoFisher Scientific). Libraries were prepared with the KAPA mRNA HyperPrep kit (Roche Sequencing) according to the manufacturer’s recommendations on a custom Janus G3 Liquid Handler (Perkin-Elmer). Briefly, 200 ng of total RNA were used as starting input for the library preparation; poly-A RNA was captured and fragmented to 200–300 nt. Reverse transcription, second-strand synthesis, and A-tailing were performed according to the protocol. KAPA Dual Index Adapters (cat #KK8722) were used for the ligation and the reaction was purified twice using KAPA Pure beads. Twelve cycles of PCR amplification and subsequent purification were performed according to the protocol. Final libraries were quantified using the Qubit dsDNA BR Assay and the average size calculated with a D1000 ScreenTape Assay on a 2200 TapeStation (Agilent Technologies). Libraries were diluted and sequenced on a NovaSeq 6000 (Illumina, Inc.) SP lane 2x50 bp. The RNA-sequencing data were first processed to remove adapter sequences and low-quality reads followed by the alignment to the human genome GCHr38 using STAR (v2.6) [16]. Count data from HTSeq were calculated and normalized using DESeq2’s median of ratios method [17]. We compared the normalized counts of treated and untreated macrophage samples for each of the samples used. An unsupervised analysis was performed to assess statistical significance of omega fatty acid supplementation on macrophage transcriptome using the “pheatmap” package in R. This package was used to generate microarray heatmaps of normalized RNASeq data. A supervised analysis of individual genes was performed using R packages “ggplot2”, and “ggprism”. An unpaired two-tailed t-test was used to calculate the p-value applied to bar charts.
Isolation of N-glycans
PBMC were homogenized with ultra-pure water in presence of a protease inhibitor and total protein content was assayed using BCA-assay kit. The total protein amount was used as normalizing factor for subsequent analysis. N-linked glycans were isolated under reduced denaturing condition using PNgaseF enzyme. Further purification of N-glycans were done using C18 and PGC cartridge and used for following analysis [18].
Monosaccharide analysis
Purified N-glycans were hydrolyzed using 2 N TFA at 100°C for 4 h followed by removal of acid. Analysis of monosaccharides was done on high-performance anion-exchange chromatography with pulsed amperometric detection (HPAEC-PAD) (Dionex ICS-3000) using CarboPac PA-1 column (Thermo Fisher). An isocratic solvent mixture of 19 mM NaOH containing 1.33 mM NaOAc was used to separate the monosaccharides followed by detection using a PAD detector [19]. We quantified the constituent monosaccharides fucose, N-acetyl glucosamine, galactose and mannose by comparing with known concentration of each standard (Sigma).
Sialic acid composition analysis
Sialic acids were measured in purified N-glycans using ultra performance liquid chromatography (UPLC) (Acquity, Waters) attached with an online fluorescent detector. Briefly, samples were hydrolyzed using 2M HOAc at 80°C for 3 h, followed by removal of the acid in speed vac. The samples were then reacted with 1, 2-diamino-4, 5-methylenedioxybenzene (DMB) and injected on a BEH-C18 column (Waters). The elution solvent was a gradient mixture consisting of 7% aqueous methanol and ACN both containing 0.1% TFA. The excitation and emission wavelength for fluorescence detector was at 373 nm and 448 nm respectively. Sialic acids were quantified by comparing with authentic standard of N-acetyl neuraminic acid (Sigma).
N-glycan profiling
Known amount of N-glycans were tagged with fluorescent tag 2-aminobenzamide (2-AB) and/or procainamide (Sigma) and profiled using BEH-Amide column using Waters Acquity UPLC system equipped with online fluorescence detector. The excitation and emission wavelengths were set at 320 nm and 420 nm for 2AB and 310 nm and 370 nm for procainamide, respectively. We used a gradient mixture of 100 mM ammonium formate buffer at pH 4.5 and acetonitrile as running buffer for UPLC. The peaks eluted were compared with highly sialylated N-glycans isolated from bovine fetuin. We confirmed the structures by off-line mass spectral identification of the procainamide tagged N-glycans using LTQ-Orbitrap mass spectrometry (Thermo Scientific) in negative ionization mode. Briefly, selected N-glycan peaks were collected from UPLC, dried down by speed-vac, reconstituted in 50% aqueous methanol containing 10 mM ammonium formate (pH 4.5) and injected directly into ion-source from external syringe pump at a flow rate of 5μL/min. We confirmed by Mass Spectrometry the structural assignment given to each peak from UPLC-FL.
Neuropathology
We investigated monocyte migration in the frontal and temporal lobe tissues of a patient with AD (from the UCLA ADRC Brain Bank). After antigen retrieval, the issues were stained using DAKO EnVision Doublestain System or by indirect immunofluorescence, as described previously [20].
Immunofluorescence microscopy
Tissues and macrophage cultures were stained with primary mouse anti-CD68 and Rb anti-Aβ; nuclei with DAPI; secondary donkey anti-mouse Alexa fluor 488 and donkey anti-Rb Alexa fluor 568 (In Vitrogen). The preparations were examined in Olympus BX60 microscope or Leica TCS SP laser scanning confocal microscope (Heidelberg, Germany).
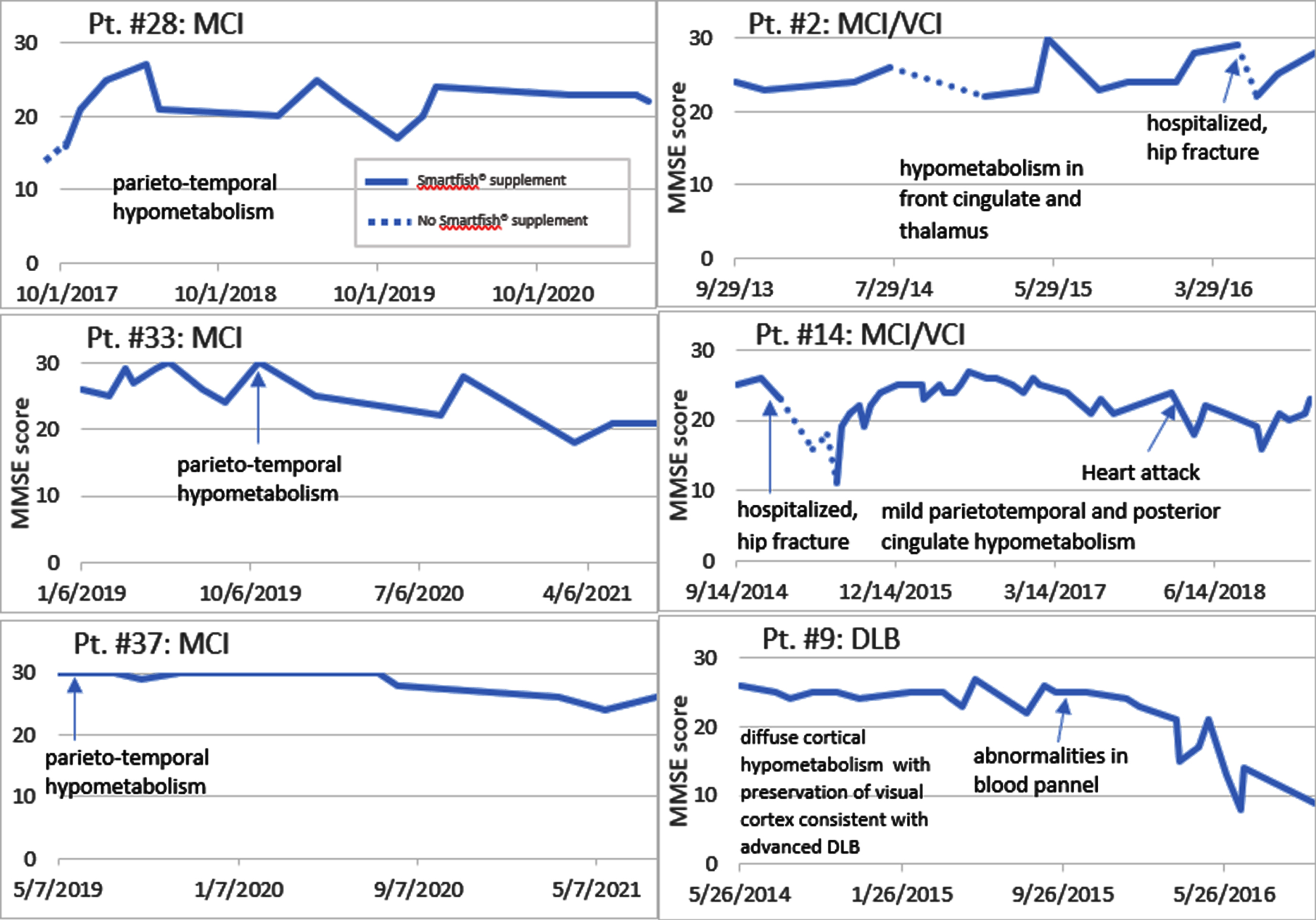
Fig. 1
Disease course on and off PUFA supplementation. Note a cognitive decline (off PUFA) and cognitive recovery (on PUFA) (Patients #28, 2, 14). Note a cognitive stabilization of MCI patients for 3 years but a decline of the DLB patient after one year.
RESULTS
Enrollment of neurodegenerative patients and control caregivers
We enrolled 10 neurodegenerative patients (6 females and 4 males, mean age on entry 73.9 years) into the study of PUFA supplementation by the nutritional supplement SMF [8] (Table 1). The entry criteria involved a loss of memory, confusion, depression, cognitive decline in the MMSE score, and failing CI therapy (CI therapy is not authorized in SCI patients per health insurance). Note: Because each patient and/or caregiver requested PUFA supplementation due to failing CI therapy, no patient received a placebo. Five patients treated by CI therapy continued CI therapy in our study. In the follow-up, the diagnoses were established as SCI (3 patients), MCI (2 patients), MCI with vascular cognitive impairment (VCI) (3 patients), and DLB (2 patients). The diagnoses were confirmed by imaging (PET scan) in 5 patients (Table 2). These patients supplemented by SMF drink and non-supplemented care givers served as controls at visits with cognitive evaluation by MMSE test at 1- to 3-monthly intervals. The control caregivers had normal results of MMSE testing. The results of MMSE testing (Fig. 1) show that a lack or an interruption of supplementation therapy (patients #28, 2, and 14) were associated with a cognitive decline.
Anatomical pathways of monocyte migration from vessels to plaques, back to vessels, and release of Aβ into vessels with CAA
To identify the pathways of monocytes targeting Aβ in the AD brain parenchyma, we performed a series of tissue staining of the brain microvasculature and Aβ plaques in the brain sections of an AD/CAA patient who was supplemented with the SMF drink for several months before death. Confocal microscopy revealed invasion of Aβ plaques by macrophages (CD68 + green) and Aβ (red) (Fig. 2A). The staining showed Aβ phagocytosis randomly defective and plaques irregularly cleared by macrophages (brown) (Fig. 2B). Confocal microscopy showed CD68 + positive (green) macrophages putatively exiting a vessel (free of Aβ (green)) (Fig. 2Ca) but engorged with Aβ (CD68 green/Aβ red/overlap yellow) putatively on return to the vessel. This chronology is supported by a release of Aβ (red) underneath the abutting macrophage (yellow) (Fig. 2Cb). Similar results were obtained in the brain tissues of 4 other patients (one Binswanger encephalopathy, two AD Braak stage VI, and one DLB), as published [20]. According to these confocal microscopic results, we propose a pathway of monocyte emigration from vessels into Aβ plaques, phagocytosis of Aβ, return to vessels, and release of Aβ inducing CAA.
Fig. 2
Anatomical pathways of monocyte/macrophage (MM) migration in the frontal lobe of the PUFA-supplemented MCI patient with cerebrovascular amyloid angiopathy (A to C). MM migration in a blood-brain barrier (BBB) model (D). A) CD-68-positive macrophages (green) invade Aβ (red) plaques (immunofluorescence microscopy of the CAA brain; Olympus BX60 microscope, 20x). B) macrophages invade and asynchronously clear Aβ plaques - Aβ content in macrophages ranges from 720 to 3,140 px; residual Aβ plaque areas range from 262 to 11,584 px (Immunohistochemistry of the frontal lobe of an AD patient using anti-CD68 (brown) and anti-Aβ (red)). C) a) Empty (no Aβ, CD68 (green)) macrophages emigrate from a vessel and b) release Aβ (red) into a congophilic vessel (yellow) (Olympus BX60 microscope, 100x). D) BBB model: MM without Aβ migrate freely through tall brain microvascular endothelial cells (BMVEC) cells, whereas macrophages loaded with Aβ adhere to BMVEC: a) the BBB model (BMVEC on top and astrocytes on bottom of a terephthalate membrane separating upper chamber (blood) from the lower chamber (brain)); b) Transmission electron microscopy: MM stuck on the endothelial cells when loaded with Aβ (left) and passing through freely without Aβ (right); c) Immunohistochemical staining of a CD-68-positive monocyte/macrophages (purple) stuck on the outside of the vascular wall. E) Scheme of macrophage migration pathways for Aβ clearance in the AD brain. Illustration created with Biorender.com.
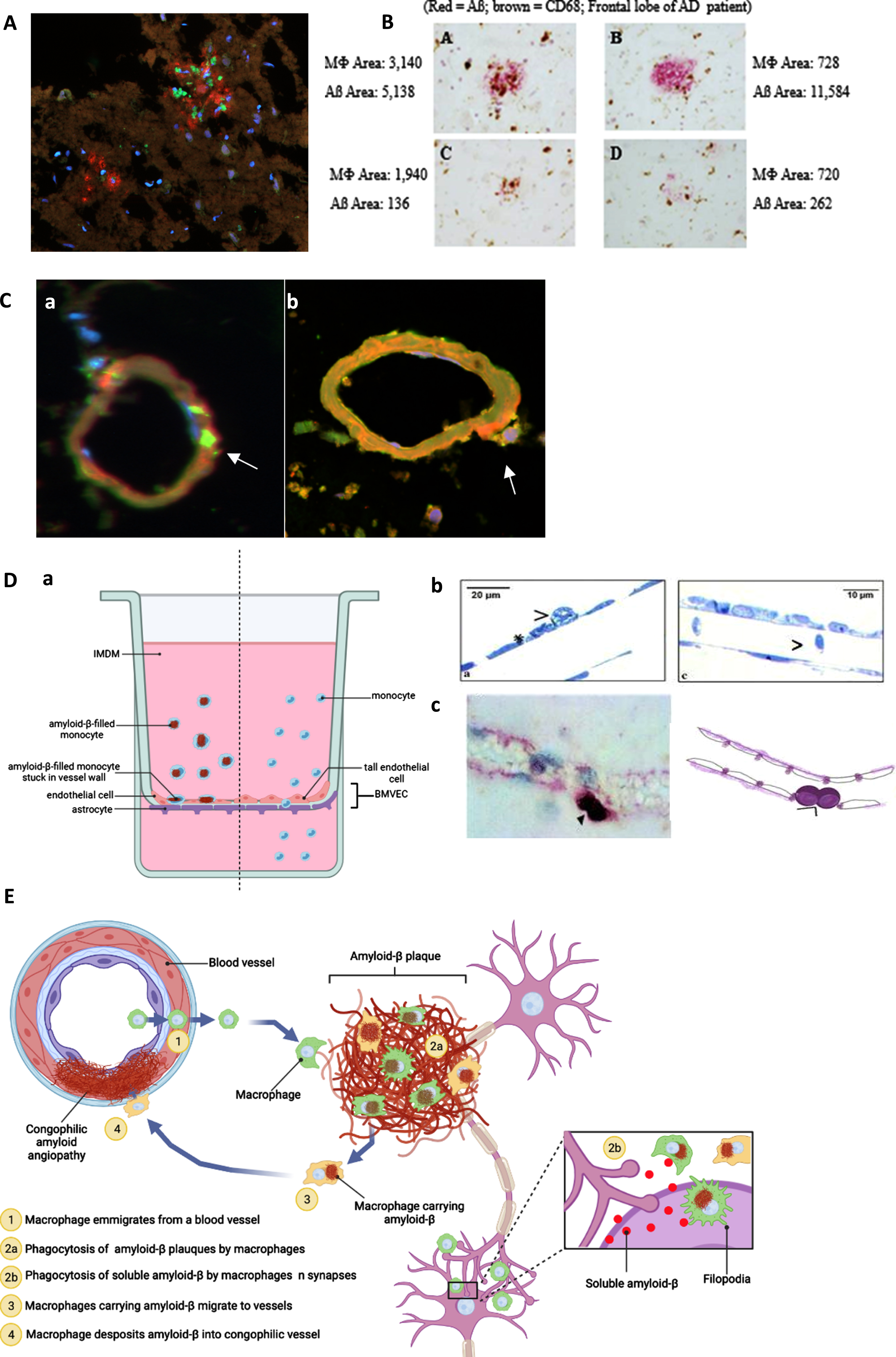
A further study measured trans-endothelial migration of monocyte/macrophage (MM) across brain microvascular endothelial cells (BMVEC) on the top and astrocytes on the bottom of a terephthalate membrane mimicking the human blood-brain barrier (BBB) (Fig. 2Da). Transmission electron microscopy showed that MM migrated seamlessly across BMVEC when free of Aβ, but could not pass and became adherent to BMVEC when loaded with Aβ (Fig. 2Db). This finding was corroborated by an immunohistochemical staining for CD-68 positive macrophages stuck at the vascular border in the brain of a patient with AD (Fig. 2Dc). Our scheme of these pathways in the AD brain is depicted (Fig. 2E).
In vivo PUFA supplementation upregulates key energy and Aβ degradation transcripts in macrophages of AD patients in comparison to non-supplemented controls
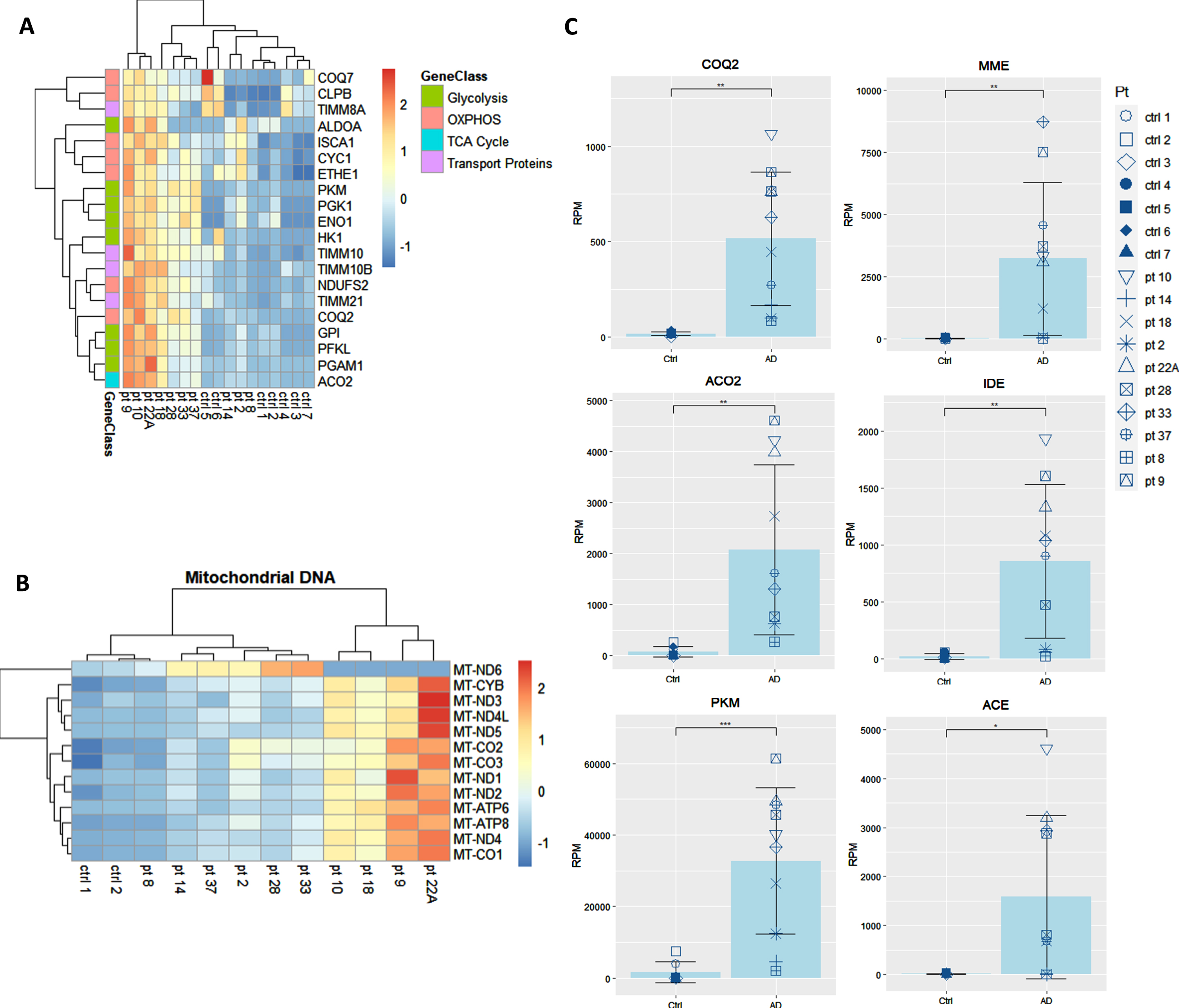
We showed that the transcripts for UPR to ER stress, which are integral to the function of macrophages, are downregulated in AD patients and are upregulated by omega fatty acids supplementation in comparison to baseline [8] and to non-supplemented AD patients [9]. Here, we prospectively followed 10 neurodegenerative patients all supplemented by the omega-3 drink SMF. Following in vivo omega-3 supplementation of these neurodegenerative patients, the expression of key energy genes in the pathways for glycolysis, TCA cycle, OXPHOS, and mitochondrial transport proteins were all significantly upregulated in comparison to their non-supplemented healthy caregivers (spouses and children) (Fig. 3A, B). After the analysis of all of the genes from these energy pathways, the three most significantly upregulated genes were coenzyme Q2 (COQ2), aconitase 2 (ACO2), and pyruvate kinase (PKM) (Fig. 3C). Additionally, PUFA-supplemented patients upregulated the Aβ-degradation enzymes membrane metalloendopeptidase (MME), insulin degrading enzyme (IDE), and angiotensin converting enzyme (ACE) (Fig. 3C).
Fig. 3
Transcriptome of macrophages of PUFA-supplemented AD patients versus non-supplemented controls (caregivers). A) Microarray heatmaps of transcripts in glycolysis, OXPHOS, TCA, and transport; B) mitochondrial DNA-encoded genes in omega-3 supplemented patients versus non-supplemented caregivers. Color scale indicates the normalized level of expression of the gene for each patient and control. C) Individual analysis of the expression of energy and degradation gene transcripts in Reads per Million (RPM) in healthy non-supplemented caregivers versus PUFA-supplemented AD patients (p < 0.01).
PUFA treatment of macrophages reduces oxidative stress, modulates pro-phagocytic macrophage phenotype, and increases MMP
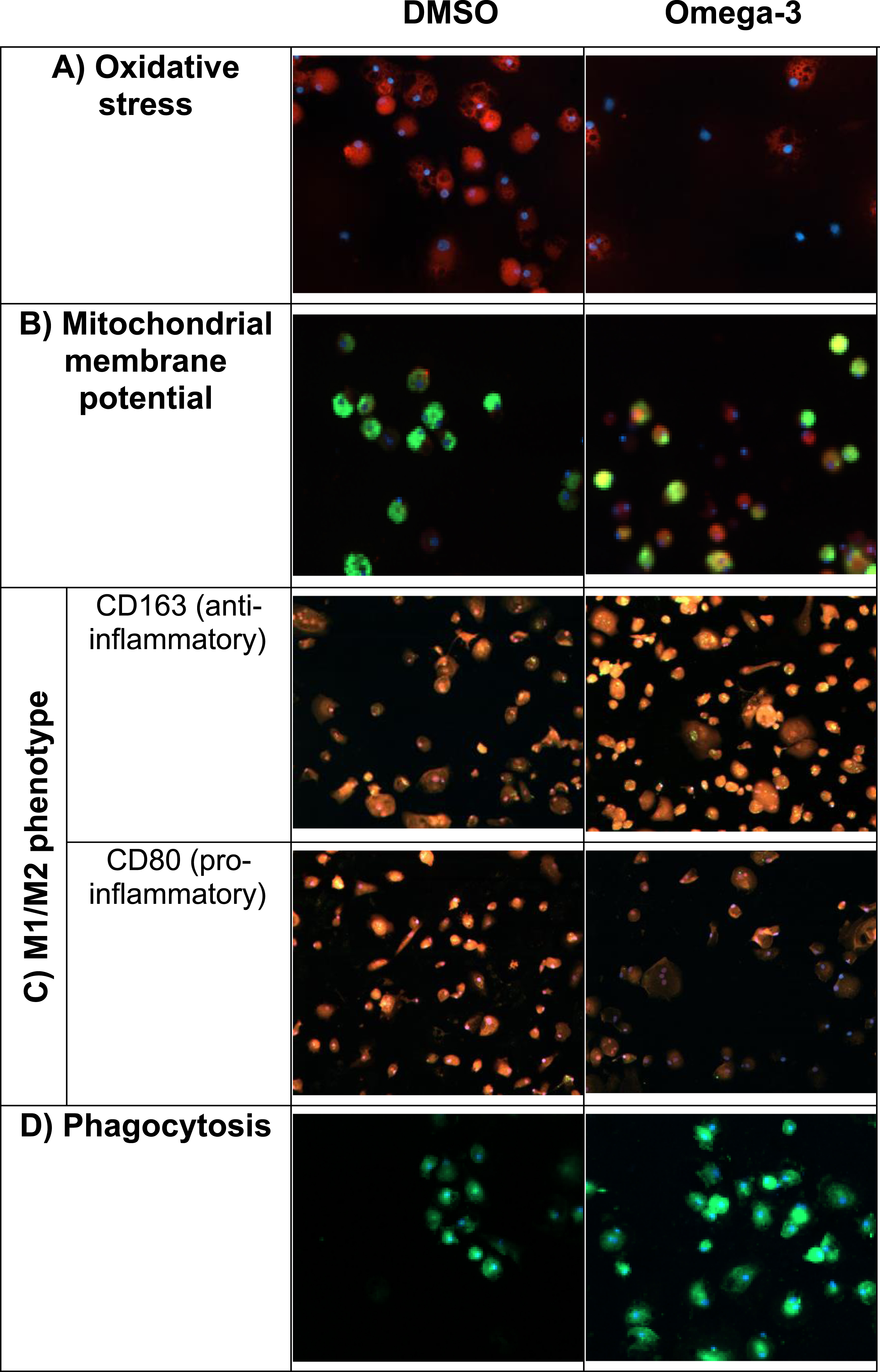
We tested in vitro PUFA effects on essential biophysical functions of the macrophages of MCI patients, including oxidative stress by ROS, MMP, and M1M2 phenotype. ROS, which are produced during oxidative phosphorylation, inhibit macrophage function and are elevated in patients with AD. PUFA stimulation reduced oxidative stress induced by tunicamycin (Fig. 4A). PUFA increased MMP (red) indicating an increased energy of mitochondria (Fig. 4B). Most AD patients’ macrophages have either the pro-inflammatory M1 phenotype or the anti-inflammatory M2 phenotype but not the beneficial pro-resolution phenotype [8]. PUFA treatment decreased the expression of the pro-inflammatory CD80 marker and increased the expression of the anti-inflammatory CD163 marker in macrophages (Fig. 4C). Twenty-four-hour-PUFA treatment of macrophages increased phagocytosis of FAM-Aβ (Fig. 4D).
Fig. 4
In vitro PUFA effects on biophysical and immunological functions of AD patients’ macrophages (macrophages were obtained before in vivo supplementation): A) Oxidative stress. Omega-3 treatment decreased oxidative stress induced by Aβ. Cells stained for cytoplasmic superoxide (red: reactive oxygen species, blue: nuclei); B) Mitochondrial membrane potential (MMP). Omega-3 stimulation increased MMP in macrophages. Cells stained with Mito Tracker Red CMXRos (red: MMP, green: FAM-Aβ, blue: nuclei); C) M1/M2 phenotype. Omega-3 stimulation changed inflammatory M1 phenotype to a pro-resolution M1/M2 phenotype. Note decreased inflammatory CD80 (red) and increased pro-resolution CD163 (red) markers following omega-3 stimulation; D) Aβ phagocytosis. Omega-3 stimulation increased phagocytosis of Aβ (green: FAM-Aβ, blue: nuclei).
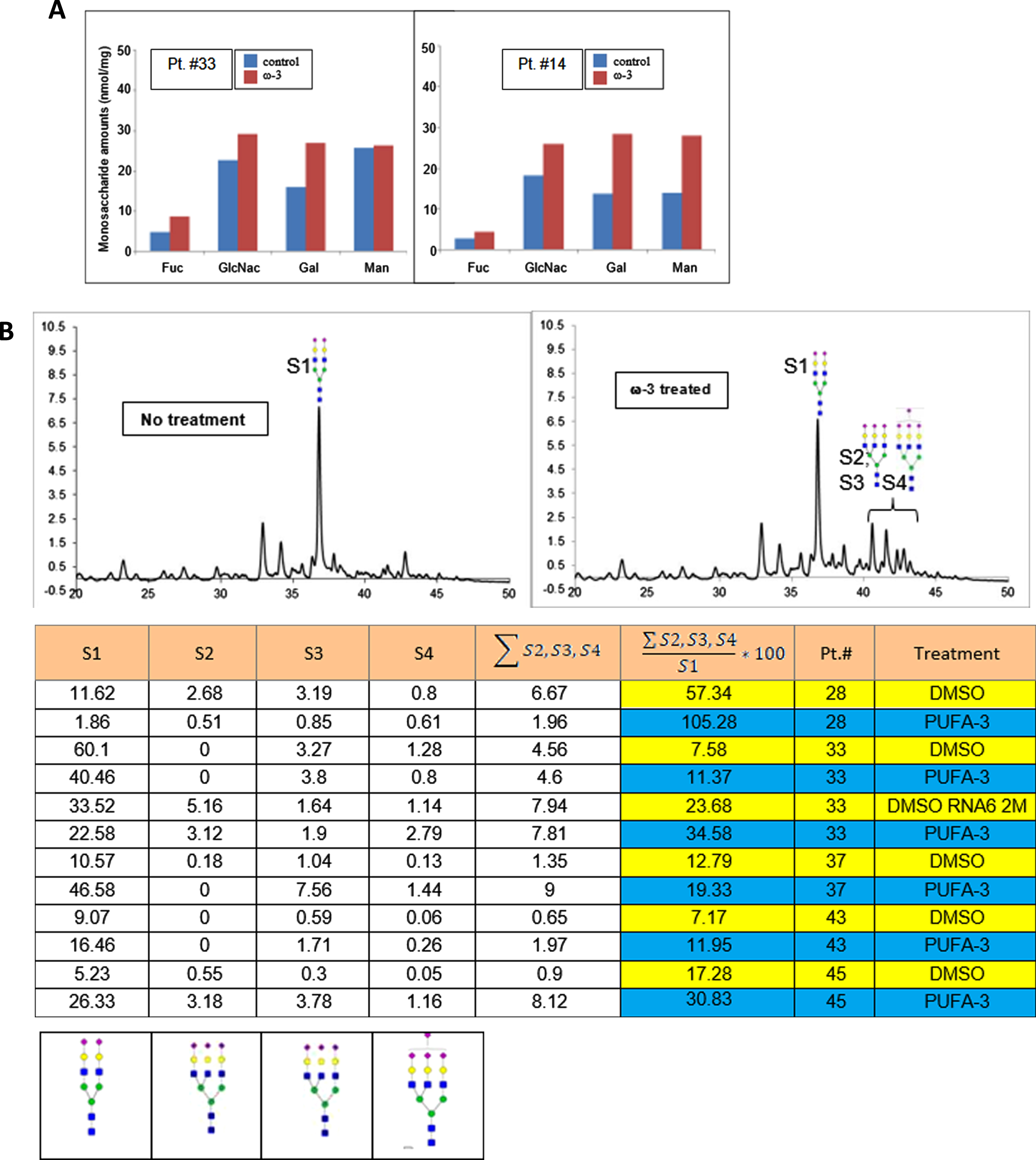
Omega-3 treatment of PBMC increases highly sialylated N-glycans
Using UPLC-FL, we examined the effects of PUFA treatment on N-linked glycans in PBMC of AD patients. In comparison to DMSO treatment, PUFA treatment of PBMC increased the glycan monosaccharides (Fig. 5A). The putative structures of N-glycans found in the samples were: structure-1 (S1) (bi-antennary complex glycan containing two sialic acids at the non-reducing end of the glycans); structure-2 (S2), and structures 3 and 4 (S3 and S4) with tri-antennary tri-sialylated glycans with isomeric forms (S3 and S4) eluting at different retention times on the UPLC. Both of these structures have three sialic acids with different linkages to galactose residues; structure-4 (S4) including tri-antennary tetra-sialylated glycans with four sialic acid residues at the non-reducing end (Fig. 5B). We assigned the structures based on the separation of 2-AB-labeled glycans in the UPLC system using BEH-amide column and comparison to fetuin 2-AB labeled glycans. The structures were confirmed by off-line mass spectral identification of the glycans using LTQ-Orbitrap mass spectrometry in a negative ionization mode.
Fig. 5
In vitro PUFA effects on macrophage glycome: A) increased glycosylation by monosaccharides, and B) highly sialylated N-linked glycoproteins (A) ω-3 treatment of macrophages (DHA 10μg/ml) increased relative area of fucose, GlcNH2, Gal, and Mann in N-glycans of the patients #33 and #14 (p < 0.01); B) ω-3 treatment increased the area of the peaks corresponding to the glycan structures 1 through 4 in the UPLC-FL chromatograms (i and ii): The chromatograms of 2-AB tagged N-linked glycans are compared between i) no treatment (DMSO) and ii) ω-3-treated samples. Columns 1, 2, 3, and 4 represent the area of peaks corresponding to the N-glycans with structure 1, 2, 3, and 4, respectively. Column-5 presents the sum total of areas from tri- and tetra-sialylated glycans. Column-6 presents the ratio (x100) of tri- and tetra- sialylated glycans over bi-antennary glycans containing two sialic acids (Struc-1). iii) The putative structures of N-glycans in the samples: structure-1 (di-antennary complex glycan containing two sialic acids at the non-reducing end of the glycans); structure-2 and structure-3 (tri-antennary tri-sialylated glycans are isomeric forms eluting at different retention time on the UPLC. Both of the structures have three sialic acids with different linkages to subterminal galactose residues; structure-4 (tri-antennary tetra-sialylated glycans with four sialic acid residues at the non-reducing end) (p < 0.01).
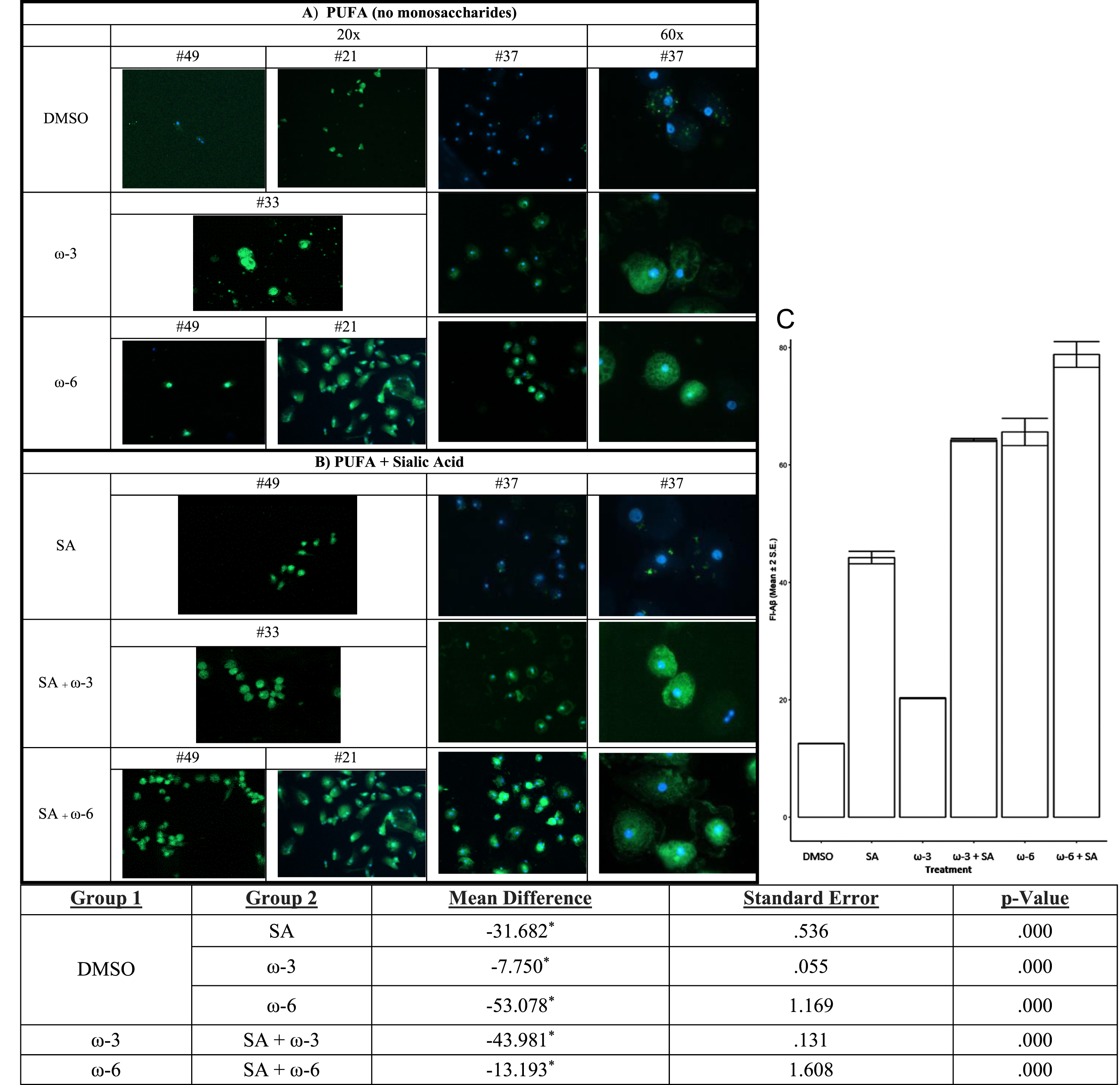
PUFA enhance Aβ phagocytosis alone and with additive effect of sialic acid in the medium
Because PUFA treatment of PBMC increased sialic acid in complex N-glycans, we reasoned that the addition of sialic acid to the medium of macrophages would increase the macrophage phagocytosis of soluble Aβ. Therefore, we tested the effects on Aβ phagocytosis of the addition of ω-3 or ω-6 alone versus ω-3 or ω-6 with sialic acid (Fig. 6). ω-6 alone was more effective than ω-3 alone. Sialic acid in the medium of macrophages amplified the positive effects of both ω-3 and ω-6 on Aβ phagocytosis (Fig. 6C).
Fig. 6
In vitro treatment of MCI patients’ macrophages by PUFA+/- sialic acid increase phagocytosis of soluble FITC-Aβ: Phagocytosis of Aβ is increased by: A) ω-3 (DHA and EPA) or ω-6 (EETs) and B) ω-3 (DHA and EPA) or ω-6 (EET) with sialic acid in the medium. C) The areas of fluorescence were scanned by Image-Pro and the data were analyzed for multiple comparisons of omega fatty acid versus DMSO (Mean and 2 SEmean of the areas of fluorescence (N) scanned by Image-Pro).
DISCUSSION
In this study, we analyzed the defects in macrophage transcriptome, glycome, and Aβ brain clearance by phagocytosis and degradation in neurodegenerative patients with SCI, MCI, AD, and DLB.
Immune brain clearance of Aβ and P-tau
This clearance depends on macrophages together with the G-lymphatic system. AD macrophages are defective in Aβ phagocytosis and subcellular transport of Aβ, leaving undegraded Aβ in plaques and CAA vessels. in vitro PUFA treatment of macrophages upregulated energetic and circadian rhythm transcripts, improved macrophage and T cell immunity. PUFA in vivo supplementation slowed cognitive decline beyond cholinesterase inhibitors [21]. In this study, we clarify a) the pathways of monocyte migration in the brain, b) the defects in macrophage transcriptome, c) the defects in macrophage glycome, and d) PUFA mending macrophage transcriptome and glycome.
Monocyte/macrophage pathways in the brain and Aβ clearance
Confocal microscopic results in the AD brain show macrophages with Aβ in plaques and other macrophages either releasing Aβ (red) to vessels or exiting a vessel without Aβ (green) (Fig. 2). These results and those in the BBB model suggest the following scheme of brain clearance by macrophages: Aβ-free MM emigrate from vessels into Aβ plaques, randomly upload Aβ (red/green overlap = yellow), migrate back to vessels but cannot emigrate across BBB when engorged with Aβ, undergo apoptosis and release Aβ into the vessels, which develop CAA (Fig. 2). Soluble Aβ may still clear into cervical lymphatics.
Macrophage transcriptome and glycome, and their repair by PUFA
The transcriptome of AD patients has multiple defects responsible for faulty function: downregulation of the transcripts for energy, circadian rhythm and protein glycosylation. PUFA supplementation mended, individually and incompletely, certain defects, of which the energetic deficits are the most prominent. PUFA supplementation upregulated glycolytic, TCA cycle, and OXPHOS energy genes in macrophage transcriptome of MCI patients coded by nuclear and mitochondrial DNA (Fig. 3), and repaired energetic deficits. PUFA upregulated the nuclear-encoded glycolysis genes pyruvate kinase (PKM) and phosphoglycerate kinase (PGK1), the TCA cycle genes aconitase (ACO2), and mitochondrial genes the subunit of NADH-Ubiquinone reductase (NDUFS2), the component of electron transport chain cytochrome C1 (CYC) and mitochondrial transport proteins in comparison to their non-supplemented caregivers (Fig. 3). PUFA upregulated mitochondrial DNA-encoded genes in OXPHOS, including MT-ND1, MT-ND5, MT-CYB, MT-CO1, MT-CO2, MT-CO3, and MT-ATP6 in macrophages but not in PBMC. PUFA enriched the glycome of macrophages and, when coupled with sialic acid in the medium, increased phagocytosis of Aβ (Figs. 5 and 6). PUFA upregulated macrophage transcripts of the Aβ degradation enzymes MME, ACE, and IDE (Fig. 3C). The Aβ plaques in the brain of an AD patient supplemented for several months before death with the omega-3 SMF drink were vigorously invaded by macrophages (Fig. 2A immunofluorescence, Fig. 2B immunochemistry), but their clearance was heterogeneous leaving Aβ vestiges in some plaques while clearing other plaques (Fig. 2B).
Inflammation in circulating immune cells
In the course of AD, the phenotype of peripheral MM becomes more inflammatory M1 with reduced Aβ phagocytosis [8]. PUFA increase energy, decrease oxidative stress, increase mitochondrial membrane potential, modulate the inflammatory M1 macrophage phenotype to the pro-resolution M1M2 type, and stimulate phagocytosis of Aβ (Fig. 4).
Therapeutic clearance of Aβ in the mouse model brain
Aβ is cleared in the mouse brain by several pathways involving endothelial cells, macrophages, microglia, glymphatic system, and energy, including: a) Low-density lipoprotein receptor-related protein 1(LRP1)-dependent endocytic pathway in endothelial cells and smooth muscle cells, which involves receptor-mediated transport by LRP1-dependent endocytic pathway [22]. LRP1 levels are significantly decreased in aging and AD and are upregulated by rifampicin and caffeine [23]. b) Trojan macrophage mechanism by peripheral ACE-positive MM [24], c) glymphatic pathway clearing the wastes in paravenous lymphatics into dural lymphatics [25]; cGAS-STING pathway supported by NAD+ supplementation [26]; e) triggering receptor expressed on myeloid cells-2 (TREM2). A missense mutation of this receptor increased risk of AD and was associated with lesser mental performance [27]. The Trojan macrophage transport is supported by the neuropathological results in our study.
Immunization of transgenic mouse model against 42-amino acid Aβ peptide and development of Aβ monoclonal antibodies (mAb) for human therapy
The initial approach to AD therapy was developed in a transgenic mouse model with APP 717Val - phe mutation by immunization with Aβ [28] and was followed by Aβ monoclonal antibodies, which bind the Fc receptor for IgG (FcγR) on hematopoietic cells. Therapeutic trials with the mAb to soluble Aβ solanezumab, the small molecular inhibitor of β-secretase (BACE-1) verubecestat, and the mAb antibody ponezumab [29] were a disappointment for the Aβ hypothesis; ponezumab failed to have treatment effects despite improved cerebrovascular reactivity of the CAA vessels. In 2021, the mAb aducanumab, a fully human IgG1mAb reacting with the conformational epitope involving amino acids 3-6 in the toxic Aβ oligomers and insoluble fibers, cleared brain amyloid and was approved for AD therapy. Aducanumab did not clear CAA in the Tg2576 transgenic mouse model. In the model aducanumab recruited microglia to plaques by binding to Aβ and engagement of Fcγ receptors. In a controlled clinical trial of aducanumab versus a placebo, aducanumab crossed the BBB and cleared soluble and aggregated Aβ in plaques in a dose-responsive fashion according to florbetapir PET imaging. Aducanumab slowed disease progression, however, in association with amyloid-related imaging abnormalities and CAA [30]. CAA is a significant pathology in AD as well as in aging [31]. The results (Fig. 3) show increased transcripts of Aβ degradation enzymes MME, IDE, and ACE, suggesting that PUFA may increase Aβ degradation in the AD brain.
PUFA supplementation of AD patients
In a controlled trial of omega fatty acids (1.7 gm of DHA and 0.6 gm of EPA), cognitive decline was reduced in a subgroup with very early AD patients (MMSE > 27 points). In a large controlled study with mild to moderate AD patients, algal DHA (2 g/day) had no effect on the rate of cognitive and functional decline in the total population but was associated with a lower mental decline in APOE ɛ4-negative group [32]. The effects of PUFA on brain clearance thus depend on the initial condition, APOE genotype, the therapeutic supplement, including quality and quantity of PUFA, antioxidants, vitamins D and B complex and other small molecules, and administration schedule [33].
Signaling effects of PUFA metabolites
Specialized pro-resolving mediators (https://en.wikipedia.org/wiki/Specialized_pro-resolving_mediators) from DHA, EPA, or omega-3 docosapentaenoic acid, such as neuroprotectin D1 (NPD1) and resolvins, exert signaling effects in CNS [34]. In the brain, the cytochrome P450 (CYP) enzymes produce endogenous anti-inflammatory epoxides from a) arachidonic acid called EETs, b) EPA acid called epoxyeicosatetraenoic acids (EEQs), and c) DHA called epoxydocosapentaenoic acids (EDPs). Importantly, soluble epoxide hydrolase metabolizes EETs, EEQs, and EDPs to the corresponding diols, which show reduced or no biological activity [35]. Therefore, we used soluble epoxide hydrolase inhibitors (sEHIs), such as TPPU, or dual-acting sEHIs, such as PTUPB [36] to demonstrate the bioavailability of anti-inflammatory epoxides [37].
Conclusions
Our results concerning the pathologies of macrophage transcriptome, glycome, and functions are limited to a small cohort of neurodegenerative patients, including patients in all stages of sporadic AD, therefore, require further studies. The immunochemical results in the AD brain and the biochemical results in macrophages support the following scheme: MM invade Aβ plaques but, unlike healthy macrophage, are defective in Aβ phagocytosis and Aβ degradation, become stuck and apoptotic around vessels and release Aβ into vessel walls creating CAA vessels with a blocked transport.
PUFA are imported into macrophage mitochondria, upregulate the transcripts of enzymes for energy and Aβ degradation, and increase energy, N-linked glycosylation by sialic acid, and Aβ phagocytosis. Supplementation by omega fatty acids had clinical success in very early MCI patients [38] but were weakened by a negative study with algal omega fatty acids [39]. Our clinical results with supplementation by marine omega fatty acids, antioxidants, resveratrol, and vitamins D and B, and our biochemical results in vitro and in vivo of increased energy and glycosylation by PUFA signaling demonstrate that the composition of the supplement matters in each trial. PUFA supplementation together with a diet low on saturated fat and glucose, and, when available for human therapy, sEHIs could repair in a patchy fashion the critical defects of macrophage transcriptome and glycome.
ACKNOWLEDGMENTS
Smartfish Company supported the studies at UCLA with SmartfishR drink by a donation to UCLA research 2010-2019.
Partial support was provided by NIEHS/Superfund Research Program P42 ES004699 and NIEHS RIVER Award R35ES030443.
We thank J. Sayre, UCLA School of Public Health, for statistical analysis of data in Fig. 4, Paul Seidler for fibrillary P-tau/analytical tools; L. Bentolila UCLA CNSI for the Confocal laser scanning microscopy performed at the Advanced Light Microscopy/Spectroscopy Laboratory and the Leica Microsystems Center of Excellence at the California NanoSystems Institute at UCLA. We prepared the illustrations using the program BioRender. We thank Dr. Paul Seidler, UCLA-DOE Institute, for providing purified fibrils of phosphorylated 4R1 N tau (P-tau).
Authors’ disclosures avaialblbe online (https://www.j-alz.com/manuscript-disclosures/22-0764r1).
REFERENCES
[1] | Hardy J , Selkoe DJ ((2002) ) The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 297: , 353–356. |
[2] | Boyle EA , Li YI , Pritchard JK ((2017) ) An expanded view of complex traits: From polygenic to omnigenic. Cell 169: , 1177–1186. |
[3] | Fiala M , Liu PT , Espinosa-Jeffrey A , Rosenthal MJ , Bernard G , Ringman JM , Sayre J , Zhang L , Zaghi J , Dejbakhsh S , Chiang B , Hui J , Mahanian M , Baghaee A , Hong P , Cashman J ((2007) ) Innate immunity and transcription of MGAT-III and Toll-like receptors in Alzheimer’s disease patients are improved by bisdemethoxycurcumin. Proc Natl Acad Sci U S A 104: , 12849–12854. |
[4] | Louveau A , Plog BA , Antila S , Alitalo K , Nedergaard M , Kipnis J ((2017) ) Understanding the functions and relationships of the glymphatic system and meningeal lymphatics. J Clin Invest 127: , 3210–3219. |
[5] | Persidsky Y , Ghorpade A , Rasmussen J , Limoges J , Liu XJ , Stins M , Fiala M , Way D , Kim KS , Witte MH , Weinand M , Carhart L , Gendelman HE ((1999) ) Microglial and astrocyte chemokines regulate monocyte migration through the blood-brain barrier in human immunodeficiency virus-1 encephalitis. Am J Pathol 155: , 1599–1611. |
[6] | Majumdar A , Chung H , Dolios G , Wang R , Asamoah N , Lobel P , Maxfield FR ((2008) ) Degradation of fibrillar forms of Alzheimer’s amyloid beta-peptide by macrophages. Neurobiol Aging 29: , 707–715. |
[7] | Avagyan H , Goldenson B , Tse E , Masoumi A , Porter V , Wiedau-Pazos M , Sayre J , Ong R , Mahanian M , Koo P , Bae S , Micic M , Liu PT , Rosenthal MJ , Fiala M ((2009) ) Immune blood biomarkers of Alzheimer disease patients. J Neuroimmunol 210: , 67–72. |
[8] | Famenini S , Rigali EA , Olivera-Perez HM , Dang J , Chang MT , Halder R , Rao RV , Pellegrini M , Porter V , Bredesen D , Fiala M ((2017) ) Increased intermediate M1-M2 macrophage polarization and improved cognition in mild cognitive impairment patients on omega-3 supplementation. FASEB J 31: , 148–160. |
[9] | Olivera-Perez HM , Lam L , Dang J , Jiang W , Rodriguez F , Rigali E , Weitzman S , Porter V , Rubbi L , Morselli M , Pellegrini M , Fiala M ((2017) ) Omega-3 fatty acids increase the unfolded protein response and improve amyloid-beta phagocytosis by macrophages of patients with mild cognitive impairment. FASEB J 31: , 4359–4369. |
[10] | Vinters HV , Zarow C , Borys E , Whitman JD , Tung S , Ellis WG , Zheng L , Chui HC ((2018) ) Review: Vascular dementia: Clinicopathologic and genetic considerations. Neuropathol Appl Neurobiol 44: , 247–266. |
[11] | Butterfield DA , Halliwell B ((2019) ) Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat Rev Neurosci 20: , 148–160. |
[12] | Butterfield DA , Boyd-Kimball D ((2018) ) Oxidative stress, amyloid-beta peptide, and altered key molecular pathways in the pathogenesis and progression of Alzheimer’s disease. J Alzheimers Dis 62: , 1345–1367. |
[13] | Petersen RC , Smith GE , Waring SC , Ivnik RJ , Tangalos EG , Kokmen E ((1999) ) Mild cognitive impairment: Clinical characterization and outcome. Arch Neurol 56: , 303–308. |
[14] | Falck JR , Yadagiri P , Capdevila J ((1990) ) Synthesis of epoxyeicosatrienoic acids and heteroatom analogs. Methods Enzymol 187: , 357–364. |
[15] | Hwang SH , Wagner KM , Morisseau C , Liu JY , Dong H , Wecksler AT , Hammock BD ((2011) ) Synthesis and structure-activity relationship studies of urea-containing pyrazoles as dual inhibitors of cyclooxygenase-2 and soluble epoxide hydrolase. J Med Chem 54: , 3037–3050. |
[16] | Dobin A , Davis CA , Schlesinger F , Drenkow J , Zaleski C , Jha S , Batut P , Chaisson M , Gingeras TR ((2013) ) STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29: , 15–21. |
[17] | Anders S , Huber W ((2010) ) Differential expression analysis for sequence count data. Genome Biol 11: , R106. |
[18] | Maley F , Trimble RB , Tarentino AL , Plummer TH Jr., ((1989) ) Characterization of glycoproteins and their associated oligosaccharides through the use of endoglycosidases. Anal Biochem 180: , 195–204. |
[19] | Hardy MR , Townsend RR , Lee YC ((1988) ) Monosaccharide analysis of glycoconjugates by anion exchange chromatography with pulsed amperometric detection. Anal Biochem 170: , 54–62. |
[20] | Fiala M , Liu QN , Sayre J , Pop V , Brahmandam V , Graves MC , Vinters HV ((2002) ) Cyclooxygenase-2-positive macrophages infiltrate the Alzheimer’s disease brain and damage the blood-brain barrier. Eur J Clin Invest 32: , 360–371. |
[21] | Fiala M , Lau YCC , Aghajani A , Bhargava S , Aminpour E , Kaczor-Urbanowicz KE , Mirzoyan H , Nichols I , Ko MW , Morselli M , Santana J , Dang J , Sayre J , Paul K , Pellegrini M ((2020) ) Omega-3 fatty acids increase amyloid-beta immunity, energy, and circadian rhythm for cognitive protection of Alzheimer’s disease patients beyond cholinesterase inhibitors. J Alzheimers Dis 75: , 993–1002. |
[22] | Kanekiyo T , Liu CC , Shinohara M , Li J , Bu G ((2012) ) LRP1 in brain vascular smooth muscle cells mediates local clearance of Alzheimer’s amyloid-beta. J Neurosci 32: , 16458–16465. |
[23] | Qosa H , Abuznait AH , Hill RA , Kaddoumi A ((2012) ) Enhanced brain amyloid-beta clearance by rifampicin and caffeine as a possible protective mechanism against Alzheimer’s disease. J Alzheimers Dis 31: , 151–165. |
[24] | Koronyo-Hamaoui M , Sheyn J , Hayden EY , Li S , Fuchs DT , Regis GC , Lopes DHJ , Black KL , Bernstein KE , Teplow DB , Fuchs S , Koronyo Y , Rentsendorj A ((2020) ) Peripherally derived angiotensin converting enzyme-enhanced macrophages alleviate Alzheimer-related disease. Brain 143: , 336–358. |
[25] | Louveau A , Smirnov I , Keyes TJ , Eccles JD , Rouhani SJ , Peske JD , Derecki NC , Castle D , Mandell JW , Lee KS , Harris TH , Kipnis J ((2015) ) Structural and functional features of central nervous system lymphatic vessels. Nature 523: , 337–341. |
[26] | Hou Y , Wei Y , Lautrup S , Yang B , Wang Y , Cordonnier S , Mattson MP , Croteau DL , Bohr VA ((2021) ) NAD(+) supplementation reduces neuroinflammation and cell senescence in a transgenic mouse model of Alzheimer’s disease via cGAS-STING. Proc Natl Acad Sci U S A 118: , e2011226118. |
[27] | Jonsson T , Stefansson H , Steinberg S , Jonsdottir I , Jonsson PV , Snaedal J , Bjornsson S , Huttenlocher J , Levey AI , Lah JJ , Rujescu D , Hampel H , Giegling I , Andreassen OA , Engedal K , Ulstein I , Djurovic S , Ibrahim-Verbaas C , Hofman A , Ikram MA , van Duijn CM , Thorsteinsdottir U , Kong A , Stefansson K ((2013) ) Variant of TREM2associated with the risk of Alzheimer’s disease. N Engl J Med 368: , 107–116. |
[28] | Lemere CA , Maier M , Jiang L , Peng Y , Seabrook TJ ((2006) ) Amyloid-beta immunotherapy for the prevention and treatment of Alzheimer disease: Lessons from mice, monkeys, and humans. Rejuvenation Res 9: , 77–84. |
[29] | Leurent C , Goodman JA , Zhang Y , He P , Polimeni JR , Gurol ME , Lindsay M , Frattura L , Sohur US , Viswanathan A , Bednar MM , Smith EE , Ponezumab Trial Study Group, Greenberg SM ((2019) ) Immunotherapy with ponezumab for probable cerebral amyloid angiopathy. Ann Clin Transl Neurol 6: , 795–806. |
[30] | Sevigny J , Chiao P , Bussière T , Weinreb PH , Williams L , Maier M , Dunstan R , Salloway S , Chen T , Ling Y , O’Gorman J , Qian F , Arastu M , Li M , Chollate S , Brennan MS , Quintero-Monzon O , Scannevin RH , Arnold HM , Engber T , Rhodes K , Ferrero J , Hang Y , Mikulskis A , Grimm J , Hock C , Nitsch RM , Sandrock A ((2016) ) The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 537: , 50–56. |
[31] | Alakbarzade V , French JM , Howlett DR , Attems J , Francis PT , Stratton S , Clark CN , Pereira AC , Hainsworth AH ((2021) ) Cerebral amyloid angiopathy distribution in older people: A cautionary note. Alzheimers Dement (N Y) 7: , e12145. |
[32] | Freund-Levi Y , Hjorth E , Lindberg C , Cederholm T , Faxen-Irving G , Vedin I , Palmblad J , Wahlund LO , Schultzberg M , Basun H , Eriksdotter Jonhagen M ((2009) ) Effects of omega-3 fatty acids on inflammatory markers in cerebrospinal fluid and plasma in Alzheimer’s disease: The OmegAD study. Dement Geriatr Cogn Disord 27: , 481–490. |
[33] | Fiala M , Restrepo L , Pellegrini M ((2018) ) Immunotherapy of mild cognitive impairment by omega-3 supplementation: Why are amyloid-beta antibodies and omega-3 not working in clinical trials? J Alzheimers Dis 62: , 1013–1022. |
[34] | Bazan NG , Musto AE , Knott EJ ((2011) ) Endogenous signaling by omega-3 docosahexaenoic acid-derived mediators sustains homeostatic synaptic and circuitry integrity. Mol Neurobiol 44: , 216–222. |
[35] | Spector AA , Fang X , Snyder GD , Weintraub NL ((2004) ) Epoxyeicosatrienoic acids (EETs): Metabolism and biochemical function. Prog Lipid Res 43: , 55–90. |
[36] | Zarriello S , Tuazon JP , Corey S , Schimmel S , Rajani M , Gorsky A , Incontri D , Hammock BD , Borlongan CV ((2019) ) Humble beginnings with big goals: Small molecule soluble epoxide hydrolase inhibitors for treating CNS disorders. Prog Neurobiol 172: , 23–39. |
[37] | Morisseau C , Inceoglu B , Schmelzer K , Tsai HJ , Jinks SL , Hegedus CM , Hammock BD ((2010) ) Naturally occurring monoepoxides of eicosapentaenoic acid and docosahexaenoic acid are bioactive antihyperalgesic lipids. J Lipid Res 51: , 3481–3490. |
[38] | Freund-Levi Y , Eriksdotter-Jonhagen M , Cederholm T , Basun H , Faxen-Irving G , Garlind A , Vedin I , Vessby B , Wahlund LO , Palmblad J ((2006) ) Omega-3 fatty acid treatment in 174 patients with mild to moderate Alzheimer disease: OmegAD study: A randomized double-blind trial. Arch Neurol 63: , 1402–1408. |
[39] | Quinn JF , Raman R , Thomas RG , Yurko-Mauro K , Nelson EB , Van Dyck C , Galvin JE , Emond J , Jack CR Jr., Weiner M , Shinto L , Aisen PS ((2010) ) Docosahexaenoic acid supplementation and cognitive decline in Alzheimer disease: A randomized trial. JAMA 304: , 1903–1911. |


