
Neural Stem Cells in the Treatment of Alzheimer’s Disease: Current Status, Challenges, and Future Prospects
Abstract
Alzheimer’s disease (AD), a progressive dementia, is one of the world’s most dangerous and debilitating diseases. Clinical trial results of amyloid-β (Aβ) and tau regulators based on the pretext of straightforward amyloid and tau immunotherapy were disappointing. There are currently no effective strategies for slowing the progression of AD. Further understanding of the mechanisms underlying AD and the development of novel therapeutic options are critical. Neurogenesis is impaired in AD, which contributes to memory deficits. Transplanted neural stem cells (NSCs) can regenerate degraded cholinergic neurons, and new neurons derived from NSCs can form synaptic connections with neighboring neurons. In theory, employing NSCs to replace and restore damaged cholinergic neurons and brain connections may offer new treatment options for AD. However there remain barriers to surmount before NSC-based therapy can be used clinically. The objective of this article is to describe recent advances in the treatment of AD models and clinical trials involving NSCs. In addition, we discuss the challenges and prospects associated with cell transplant therapy for AD.
BACKGROUND
Alzheimer’s disease (AD), characterized by progressive dementia, is one of the most life-threatening and burdensome illnesses [1]. Based on the age of onset, AD can be divided into two distinct groups: cases occurring after age 65 are classified as late-onset AD, whereas cases occurring before age 65 are classified as early-onset AD and account for less than 5% of all cases. The majority of cases are characterized by sporadic onset, while 1%–2% of cases are inherited in an autosomal dominant manner [2]. Although countless research has been conducted on AD pathogenesis, there remains a lack of effective disease-modifying therapies [3, 4]. Recently approved pharmacological treatments include the N-methyl-D-aspartate receptor antagonist memantine and cholinesterase inhibitors, which merely relieve associated symptoms [2, 5–7]. Current promising pharmaceutical treatments include anti-inflammatory, anti-amyloid, and immunomodulator therapies [6]. In addition, monoclonal antibodies capable of serving as disease-modifying drugs are being developed and tested, such as bapinezumab, solanezumab, crenezumab, and aducanumab [2]. Among these drugs, only aducanumab, which targets conformation-specific amyloid-β (Aβ) aggregates, has been approved by United States Food and Drug Administration (FDA) [8]. With recent data indicating that the global dementia population will triple by 2050 [6], further understanding of the mechanisms underlying AD and the development of novel therapeutic options are critical.
Tremendous investments have been made to inhibit processes underlying AD, although the results are mixed [1]. There are still no effective ways to curb AD progression [9]. Recently accumulated evidence suggests that stem cell transplantation may aid in maintaining brain homeostasis and enhancing cognition, learning, and memory [10–16]. Transplanted neural stem cells (NSCs) have been demonstrated to regenerate damaged cholinergic neurons, and NSC-derived neurons can form synaptic connections with nearby neurons, suggesting a potential strategy to slow the progression of AD [10]. Simultaneously, neurotrophic support, immunomodulation, and anti-inflammatory effects play a critical role in hindering AD progression [10]. Here, we evaluate recent research on NSC transplantation in AD mouse models and address the obstacles and potential benefits of cell transplant therapy for AD.
PATHOGENESIS AND PATHOPHYSIOLOGY OF AD
The pathophysiology of AD is still a mystery. The amyloid cascade hypothesis has been challenged because it provides no explanation for the silent period of AD [17]. Although the pathological criteria for AD diagnosis remain the formation of extracellular Aβ plaques and intracellular tau accumulation [2], clinical trial results for regulators of Aβ and tau under the guise of straightforward amyloid and tau immunotherapy were disappointing [18–22]. Accordingly, some scholars believe that the focus on Aβ peptide may be inappropriate [1]. Indeed, recent evidence indicates that the real cause of AD may occur upstream of Aβ and tau proteinopathies [17].
Like Aβ and tau, neuroinflammation plays a crucial role in AD pathogenesis [23, 24]. In an intriguing study [25], an analysis of gene expression profiles in the brains of healthy individuals and patients with AD of a variety of ages was carried out. This research found that the rise in expression of immune- or inflammation-related genes with age was considerably greater in patients with AD compared with healthy individuals. The brain becomes more prone to inflammatory processes as it ages, which may explain why ageing is the major driver of AD [17, 26].
Some scientists hold the view that ageing and age-related neurodegeneration, including AD, are characterized by the accumulation of damaged mitochondria [27, 28]. Indeed, mitochondrial dysfunction is central to the pathophysiology of AD. Specifically, mitochondrial dysfunction, such as dysfunction of mitochondrial axonal transport and mutations in mitochondrial DNA, plays imperative roles in the pathogenesis of AD that may result in oxidative stress, ATP depletion, and synaptic dysfunction [29, 30]. Mitophagy enhancement is a possible therapeutic option for AD-related tau hyperphosphorylation because it can increase microglial phagocytosis and decrease neuroinflammation [28].
Because amyloid accumulation may result from vascular defects, the vascular hypothesis has been proposed, which posits that breakdown of the blood-brain barrier (BBB) results in neurotoxic serum protein accumulation and synaptic dysfunction, ultimately leading to defects in Aβ and tau clearance [17, 31]. Numerous risk loci for AD have been identified by genome-wide association studies (GWAS) [32]. However, even after many large-scale GWAS, the apolipoprotein E (APOE) ɛ4 allele remains the largest genetic risk factor for AD [33–35]. Corresponding conclusions have also been confirmed by many studies [36–38]. Over the past 5 years, knowledge about the pathophysiology of APOE has grown beyond Aβ pathways to include tau neurofibrillary degeneration, disruption of the BBB, and microglia and astrocyte responses [33].
In addition, abnormalities in neuronal cholesterol homeostasis and fatty acid metabolism seem to contribute to the induction of AD. Oxidized cholesterol is widely acknowledged as one of the main triggers of AD [39, 40]. Cholesterol 24-hydroxylase and cholesterol 27-hydroxylase (encoded by CYP46a1 and CYP27a1, respectively) play a critical role in regulating brain cholesterol homeostasis [40]. In a study by Djelti et al. [39], the cholesterol concentration in hippocampal neurons of normal mice was increased by decreasing Cyp46a1 expression, which resulted in cognitive deficits and hippocampal atrophy.
Collectively, the results described above reveal that many pathophysiological processes, including neuroinflammation, abnormal microglia activation, metabolic failure, oxidative stress, and sustained cholesterol-associated neuronal distress, may be potential targets for intervention [18].
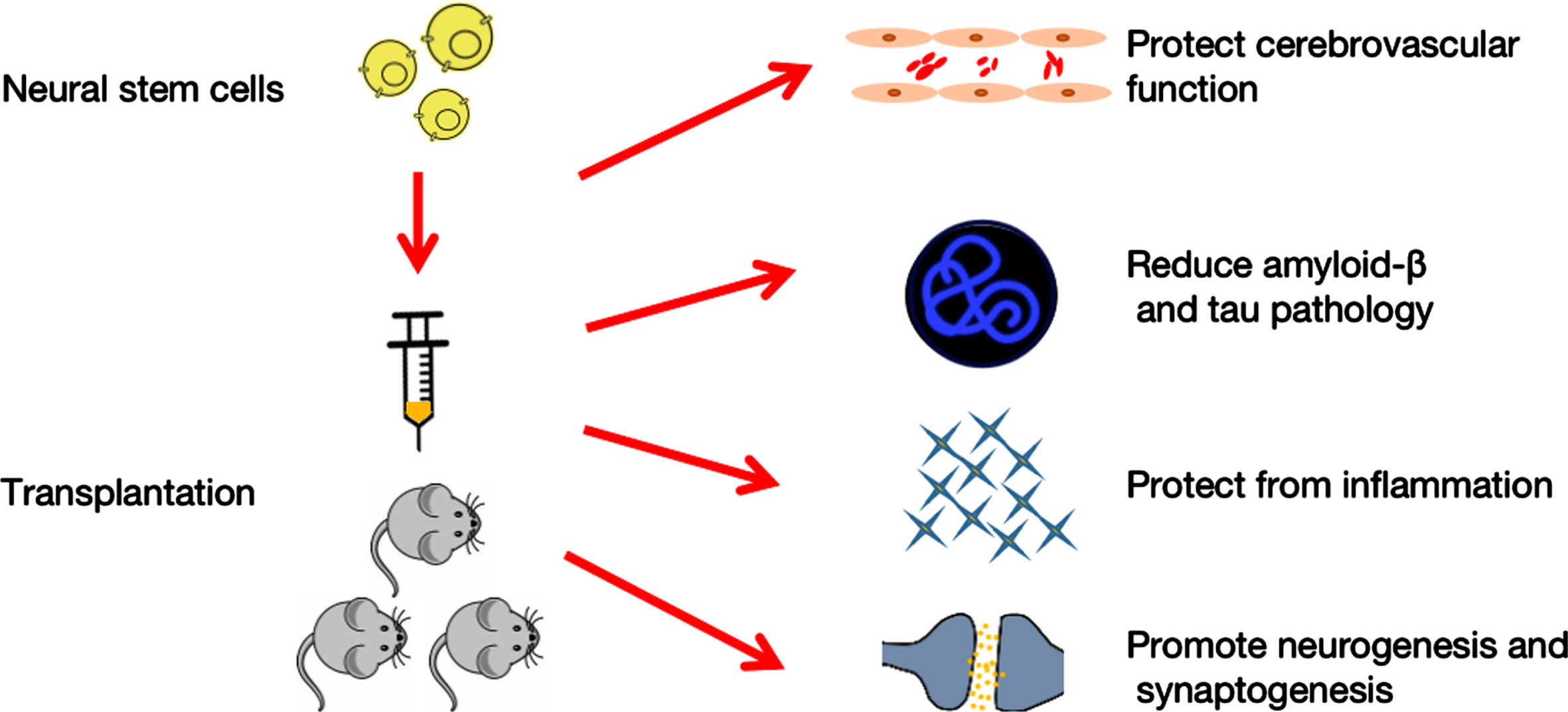
Fig.1
Neural stem cells therapy for Alzheimer’s disease. Transplantation of neural stem cells protects cerebrovascular function, decreases amyloid-β and tau pathology, decreases inflammation, and enhances neurogenesis and synaptogenesis, all of which affect behavioral performance.
Accumulated evidence has confirmed that AD is a complex disease with different interacting phases. Basic scientists divide AD into biochemical, cellular, and clinical phases [17]. Before cognitive symptoms are observed, alterations in cells drive the insidious progression of illness in the cellular phase [6, 17]. Irreversible and significant synaptic and neuronal loss occur during the preclinical phase [41]. Once homeostasis of the cellular phase is broken, the clinical phase is initiated [17]. Preclinical research is continually generating fresh information about parts of the intricate AD jigsaw, and analysis of this data may show patterns of pharmacological interactions rather than single prospective therapeutic targets [3].
NEURAL STEM CELL THERAPY FOR AD
Currently approved pharmacological treatments merely relieve associated symptoms, with a distinct lack of disease-modifying therapies. The disappointing results of Aβ-targeting therapies suggest the inadequacy of single-directed approaches for treatment of AD, while treatments addressing multiple mechanisms should be promoted. Recently, stem cell transplantation has received growing attention for the treatment of neurodegenerative diseases. Such therapies provide the possibility to target multiple mechanisms, potentially including the repair of damaged synapses, modulation inflammation, and neuroprotection through neurotrophic secretion. At present, the most often used stem cell types for the treatment of neurodegenerative illnesses are NSCs, mesenchymal stromal cells (MSCs), and induced pluripotent stem cells (iPSCs) [10]. In particular, NSC transplantation is a potential therapy for AD with tremendous therapeutic potential, such as protection of cerebral capillaries, attenuation of Aβ and tau pathologies, reduction of inflammation, and strengthening of neurogenesis. In addition, NSCs have demonstrated efficacy in treating various preclinical neurodegenerative models and the capacity to develop into central nervous system (CNS)-relevant cell types, despite the ongoing controversy over which kind of stem cell should be used to treat patients suffering from CNS injury [42]. Indeed, NSCs have been transplanted into patients with amyotrophic lateral sclerosis and Parkinson’s disease, and the results were promising [43, 44]. NSCs are ideal candidates for cell transplantation because they have a definite destiny and are relatively similar to functional neuronal types throughout their differentiation.
Cholinergic system impairment is expected to have a significant role in the cognitive and functional difficulties associated with AD [45]. In theory, employing NSCs to replace and restore damaged cholinergic neurons and brain connections may provide a new treatment option for patients with AD.
EFFECTS ON CEREBROVASCULAR FUNCTION
Several investigations have identified relationships between cerebrovascular dysfunction and a variety of AD features [46–48]. However, there is still debate about whether BBB malfunction is merely a consequence of inflammation and Aβ, or whether it is the first pathophysiological step leading to AD [46, 47, 49]. Aβ can be cleared from the CNS into the circulatory system via the BBB and blood-cerebrospinal fluid barrier [46]. Thus, proper function of the BBB and cerebrovasculature is critical for the clearance of Aβ [17, 31, 46]. Moreover, the protection of BBB functioning may promote Aβ clearance to curb AD progression [46]. The underlying vasculoprotective mechanism could be triggered by epidermal growth factor receptor (EGFR) secretion from transplanted genetically modified NSCs [46]. In addition, NSCs have the ability to reduce cerebral amyloid angiopathy and protect cerebral capillaries, especially if they are modified to use neprilysin (NEP) or vascular endothelial growth factor (VEGF) [46].
ATTENUATING AMYLOID-β AND TAU PATHOLOGY
The reduction in Aβ burden following NSC transplantation could be related to lower β-secretase concentrations [50], as well as a much higher presence of NEP [46]. Other pathways could include inducing microglial recruitment and activation, as well as triggering of immune responses to facilitate efficient Aβ phagocytosis and clearance from the CNS [46]. Although research has demonstrated that transplanted human NSCs (hNSCs) in the lateral ventricles of mice can effectively reduce levels of tau phosphorylation [51], the underlying mechanism remains unclear [46]. Neurotrophic and degradation enzymes produced by transplanted NSCs could prevent additional cognitive decline as the disease advances. In fact, clearance of Aβ may only have a minimal impact on global improvements because the environment generally remains conducive for Aβ production and aggregation [52].
ATTENUATING INFLAMMATION
Age-related changes in low-grade chronic inflammation may assist the neurodegenerative process in AD [53]. Transplantation of NSCs for AD appears to have an anti-inflammatory effect in preclinical studies [46]. Transplanted NSCs have been shown to lower pro-inflammatory cytokines including interleukin 1 (IL-1), IL-6, tumor necrosis factor α, and prostaglandin E2, as well as molecular markers associated with a pro-inflammatory environment like Toll-like receptor 4 (TLR-4) [54]. However, decreased inflammation seems to have nothing to do with Aβ levels [46]. Despite reduced inflammation and enhanced cognitive function, Aβ levels remained unchanged following NSC therapy [54]. However, unlike the former study, other research showed that mouse-derived NSCs can switch microglia states from pro-inflammatory to anti-inflammatory, resulting in bidirectional regulation of inflammatory cytokine levels [55].
EFFECTS ON NEUROGENESIS AND SYNAPTOGENESIS
The hippocampus has an imperative role in adult neurogenesis [56] and is one of the most affected areas in AD [57]. In the adult hippocampus, NSCs residing in the subgranular zone (SGZ) of the dentate gyrus (DG) can continually generate new neurons. The surviving cells later integrate into existing neuronal circuits. Abnormal neurogenesis in the adult hippocampus is an early important event in the progression of AD [58]. Dysfunctional neurogenesis may aggravate neuronal vulnerability to AD and lead to cognitive impairment [3]. There are still debates about the neurogenesis in humans. Some believe, for instance, that human hippocampal neurogenesis declines significantly from childhood to adulthood, reaching undetectable levels in adults [59]. Others maintain that human neurogenesis persists throughout the aging process [60]. Even in the face of controversial studies on human neurogenesis, the ability to stimulate NSCs to generate new neurons is a promising prospect for neuroreplacement therapy in AD. Multiple molecular players can attune adult hippocampal neurogenesis (AHN), mainly presenilin (PS1), APOE, amyloid-β protein precursor (AβPP) and its metabolites [58]. A recent study demonstrated that AHN is abundant in neurologically healthy humans, but progressively declines as AD advances [57]. Impaired neurogenesis seems to be the underlying mechanism of memory deficits in AD. The DG of the hippocampus is the primary source of NSCs that create granule neurons throughout life in the human brain [61]. Management and maintenance of the brain stem cell pool may be susceptible to innovative treatment approaches for some neurodegenerative diseases [61]. NSCs are glia-like cells found in the SGZ of the DG that can be quiescent, self-renew, or proliferate [56]. In AD, for instance, the sluggish pace of AHN might be increased by the awakening of quiescent NSCs to reduce cognitive impairments [61].
Recently accumulated evidence indicates a critical role of NSCs in neurogenesis and synaptogenesis [46]. Multiple gene products are synthesized by NSCs, including brain-derived neurotrophic factor (BDNF) and VEGF, which have paracrine effects that may attenuate AD-related neuronal loss and improve cognition [46]. Blurton-Jones et al. used triple-transgenic mice (3xTg-AD) to investigate the effect of NSC transplantation on AD-related cognitive dysfunction [62]. Their findings showed that instead of reducing Aβ and tau pathology, improved cognition resulted from a significant increase in hippocampal synaptic density mediated by BDNF [62]. Thus, both neurotrophin and BDNF released by transplanted NSCs increased synapse density and reversed cognitive impairments [62]. This study, however, did not investigate the effects of transplanted NSCs on AHN, which may benefit from increased axonal outgrowth or synaptic recovery.
NEURAL STEM CELL TRANSPLANTATION IN MOUSE MODELS
Although an increasing number of researchers are investigating the involvement of pathogenic proteins in the development of AD using animal models, the physiological relevance of these models to humans is debatable because they have not yet fully recapitulated human AD [52, 63–65]. Multiple limitations exist in many of these models, such as late onset of pathology, diverse genetic backgrounds, breeding issues, gender disparities in pathology, and substantial variability in Aβ levels [66]. As a result, exact characterizations of the favorable benefits of NSC transplantation are difficult [52]. The development of a transgenic murine model that overcomes the majority of these difficulties is critical. To mimic the microenvironment of patients with AD, 3xTg, Tg2576, APP/PS1, and 5xFAD mice are frequently employed [52] (Table 1).
Table 1
Various mouse models to mimic AD
| Mice model | Carrying transgenes | Express Aβ/age | Express tau/age | Cognitive impairment | Reference |
| 3xTg | APP Swedish, MAPT P301 L, PSEN1 M146 V | Yes/4 months (intracellular) 6 months (extracellular) | Yes/12 months | Yes/4 months | [67] |
| Tg2576 | APP Swedish | Yes/6 months | No | Yes/9 months | [11] |
| APP/ps1 | APP Swedish/PSEN1 (L166P) | Yes/6 weeks (neocortex) 3–4 months (hippocampus) | No | Yes/8 months | [66] |
| 5XFAD | APP Swedish/Florida (I716 V)/London (V717I)/ PSEN1/M146 L /L286 V | Yes/6 weeks (intracellular) 8 weeks (extracellular) | No | Yes/3-6 months | [68] |
In 2003, Oddo et al. developed the 3xTg as a triple-transgenic AD animal model [67]. The model has APP Swedish, MAPT P301 L, and PSEN1 M146 V transgenes associated with familial Alzheimer’s disease (FAD). As the first transgenic AD model to display Aβ aggregation and neurofibrillary tangles derived from hyperphosphorylated tau protein, the 3xTg mouse model represents a breakthrough in the field of AD research. In contrast to the 3xTg model, Tg2576 mice express exclusively human APP Swedish. These mutations result in a substantial rise in the amount of Aβ generated, allowing mice to exhibit progressive pathogenic protein buildup and behavioral impairments despite the absence of neurofibrillary tangles or severe neuronal loss [52]. The APP/PS1 mouse model is one of the most widely used AD rodent models. Several human APP genes, including the Swedish mutation and PSEN1 (L166P) mutation, have been included in this model [52]. Owing to the early start of amyloid lesions in this model, as well as its established genetic background and ease of breeding, APP/PS1 mice are ideal for researching treatment strategies and the pathophysiology of amyloidosis [66]. 5xFAD mice are a subtype of immune-deficient mice. Because of the absence of key constituent cells in their adaptive immune system (specifically, T cells, B cells, and natural killer cells), these animals permit longer durability of transplanted NSCs. Five mutations have been introduced into this model, which is capable of generating Aβ42 and rapidly accumulates huge amounts of Aβ42 in the cerebral cortex [68].
Here, we reviewed recent studies related to transplantation of NSCs in AD mouse models and summarized the details of transplantation, intervention targets, and effects (Table 2).
Table 2
Neural stem cells transplantation in mice models
| Mice model | NSC type | Sex/Age | Transplantation Surgery | Transplant region | Test method/time | Target | Effect | Pathological change (Aβ or Tau) | reference |
| 3xTg mice | hCNS-SCs | Female/19 months | 1.0×105 cells /2μL/injection, 1μL/min | Hippocampus bilaterally | MWM and NOR/ 1 month later | Promote synaptic growth, enhancing endogenous synaptogenesis | AmelioratesContext and SpatialLearning and MemoryImpairments | No | [42] |
| GFP-NSCs mice | NM/18 months | 1.0×105 cells | Hippocampus bilaterally | MWM and NOR/ 1 month later | Increase BDNF, enhance synaptic density | Rescues the spatiallearningand memory deficits | No | [62] | |
| GFP-NSCs mice | Male/ 12months | 1.0×106 cells/μL, 2μL/10 min | Hippocampus bilaterally | MWM/ 2 months later | Neuronal regeneration | Improve spatial learning and memory | NM | [69] | |
| GFP-NSCs mice | NM | 1.0×105 cells, 0.5μL/min | Hippocampus subiculum | Histological examination/3 months later | Secrete neprilysin | Enhance synaptic density/ degrade Aβ | Yes | [73] | |
| Tg2576 mice | hNSCs | Both/6-9 months | 2.5×104 cells/hemisphere, 1μL | Hippocampus bilaterally | MWM/5 weeks later | Increase α7 nAChR-expressing astrocytes | Endogenous neurogenesis | No | [70] |
| Mice embryos | Both/13 months | 1.0×105 cells/2μL, 0.4μl/min | Hippocampus dentate gyrus bilaterally | MWM, Histological examination/2 months later | Upregulating clearance of Aβ, anti-inflammatory cytokines, endogenous neurogenesis, synapse formation | Improve cognition | Yes | [55] | |
| APP/ps1 tg mice | hNSCs | Male/12 weeks | 1.8×105 cells/6μL | Hippocampus fmbria fornix bilaterally | MWM and NOR/4 weeks and 16 weeks respectively | Induced microglial activation and amyloid phagocytosis | Improve cognition | Yes | [74] |
| Mice embryos | Both/12 months | 1.0×105 cells/3μL, 1μL/min | Hippocampus bilaterally | MWM/10 weeks later | Suppress glial and TLR4-mediated inflammatory pathway | Improve cognition, attenuation of inflammatory | No | [54] | |
| GFP-NSCs | Male/ 9 months | 1.0×105 cells/2μL/5min | Hippocampus bilaterally | MWM/4 weeks later | Improve ChAT activity and ACh concentration | Improve cognition | No | [71] | |
| Mice embryos | Both/ 12months | 1.0×106 cells/5μL, 1μL/min | Hippocampus bilaterally | MWM/8 weeks later | Enhance long-term potentiation, neuron expressing protein, synaptogenesis, BDNF | Improve cognition | No | [76] | |
| 5XFAD mice | hCNS-SCs | Both/2 months | 1.0×105 cells total/hemisphere/2μL/2 min | Hippocampus bilaterally | MWM and NOR/ 5 months later | Neuronal regeneration and differentiation | No evidence of improved cognition, no changes in BDNF, no increase in synaptic density | No | [72] |
| hiNPCs | Male/ 4 months | 1.0×105 cells/2μL/hemisphere, 0.4μL/min | Hippocampus dentate gyrus bilaterally | Y-Maze and Barnes Maze/5-6 months later | Enhance synaptic density, increase BDNF, neuronal regeneration | Reinforce synaptic network and rescue cognitive deficits | Yes | [75] |
Aβ, amyloid β; GFP, green fluorescent protein; MWM, Morris water maze; NOR, novel object recognition; ChAT, cholinergic acetyl transferase; hiNPCs, human induced neural progenitor/stem cells; BDNF, Brain-derived neurotrophic factor; NM, not mentioned.
The most popular test used to evaluate the efficiency of transplantation was the Morris water maze. The task is conducted in a 1-m diameter circular pool filled with opaque water [42]. Mice are trained to swim to an invisible platform submerged 1.5 cm beneath the water’s surface.
The most common region for transplantation is the hippocampus. One to six months after transplantation, researchers attempted to evaluate the efficacy of transplantations. Although interventions targeting Aβ and tau seem promising for the treatment of AD, the results of studies shown in Table 2 are mixed. Indeed, there are a number of studies that show cognitive or behavioral improvements without changes in Aβ or tau [42, 54, 62, 69–72]. There could be several reasons for this phenomenon. Neurotrophic and degradation enzymes, such as NEP, matrix metalloproteinases, and insulin-degrading enzyme produced by transplanted NSCs, may aid in the clearance of Aβ or tau [55, 73–75], thereby halting or ameliorating disease progression. However, because the fundamental environment continues to be favorable for Aβ generation and aggregation, the ultimate improvements in cognition are modest [52, 72]. Moreover, it is widely recognized that AD is a multifactorial disease requiring a comprehensive treatment strategy, indicating that treatment for one of the causes is frequently inadequate.
Additionally, researchers have debated the optimal phase of disease for stem cell transplantation. Kim et al. [55] established that administering stem cell therapy at the optimal time is critical for achieving maximum therapeutic effects. In their study, NSCs were transplanted into 12-month-old and 15-month-old Tg2576 mice, and the outcomes were noteworthy. The group injected at 12 months demonstrated significant improvements in both cognitive impairments and neuropathological characteristics. In contrast, the group injected at 15 months demonstrated no significant improvement in cognitive dysfunction or Aβ neuropathology. They concluded that it was too late to intervene in disease progression once the symptoms of memory loss became apparent. Indeed, because AD takes so long to develop, it is crucial to prioritize treatment interventions based on the disease progression in each patient. It remains a major challenge in the treatment of AD to provide the correct medication to the correct patient at the correct time. Future research should focus on when and how to intervene in the AD process, as well as identification of early markers indicative of the onset of AD pathogenesis.
Numerous studies have established the critical role of NSCs in cognitive improvements associated with AD. However, prior studies [42, 54, 55, 62, 69–71, 74, 76] focused exclusively on short-term effects. Marsh et al. examined long-term transplantation of human NSCs using 5xFAD mice and conducted behavioral testing and histological examination 5 months after transplantation [72]. The final analysis revealed no proof of improved cognition, change in BDNF levels, or enhancement of synaptic density. Their research emphasized the critical nature of rigorously evaluating the efficacy and safety of NSC transplantation in appropriate long-term models. Contrary to expectations, a similar experiment conducted 3 years later yielded the opposite findings [75]. The same 5xFAD mouse model was employed by Zhang et al. to investigate the therapeutic potential of human induced neural progenitor/stem cells (hiNPCs) for AD [75]. Unexpectedly, they found that the grafted hiNPCs successfully differentiated into neurons with long-term survival and generated graft-host synaptic connections that ultimately strengthened and restored the host hippocampal neural networks. As a result, AD mice implanted with hiNPCs had increased synaptic plasticity and cognitive abilities. These radically divergent outcomes may be explained by differences in the source of stem cells used in these two studies. Indeed, it is reasonable that reprogrammed somatic cells, such as hiNPCs, have a stronger capacity for brain differentiation [52].
CHALLENGES OF NSC-BASED THERAPY IN AD
Although stem cell treatments have shown efficacy in animal models, various human studies in AD patients have had unfavorable outcomes [77]. To date, there have been no clinical trials involving NSCs the treatment of AD. It remains crucial to accelerate stem cell-based translational research for AD [78]. When it comes to cell transplantation, NSCs should be the best choice because they have a clear fate and are relatively similar to functional neuronal types throughout their differentiation. Because NSCs are capable of differentiating into astrocytes, it is vital to explore how to guarantee preferential development of NSCs into neurons. Moreover, there are restricted cell sources for NSCs, which exhibit variations in differentiation plasticity [45]. Identifying the optimal technique for stem cell transplantation is likewise a formidable obstacle. The proper transplantation technique can increase the number of transplanted cells that survive and function. Accordingly, future clinical trials must investigate transplantation methods, such as intravenous injection, intracerebral transplantation, and the use of biological materials. In addition, investigations attempting to elucidate the function of NSCs in AD must address concerns such as adequate differentiation, isolation, cell dose, time frame, immune rejection, environment, and location of transplantation, all of which need further study [45, 79, 80] (Fig. 2).
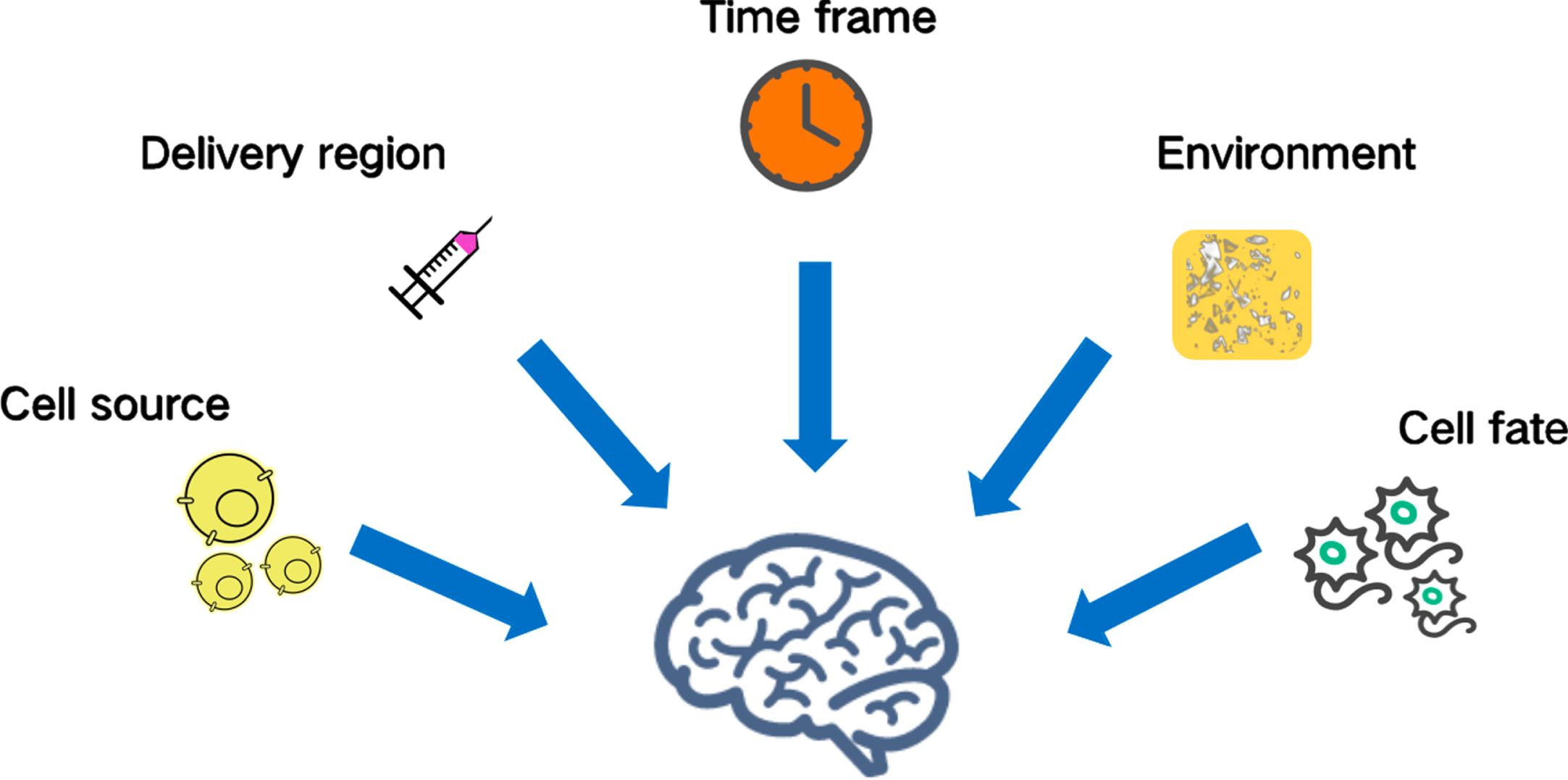
Fig. 2
Challenges of neural stem cell-based therapy in Alzheimer’s disease. The source, adequate differentiation, isolation, cell dose, delivery region, time frame, immune rejection, and location of transplantation are all challenges requiring further study.
SOURCE OF NSCs
Preclinical research on human and mouse NSCs has been conducted to determine the effectiveness of NSC treatment for neurodegenerative diseases. No conclusion on the optimal type of NSCs can be made at present because no clinical trials have evaluated NSCs for the treatment of AD. However, we can provide a list of probable sources that might be used in AD. hNSCs may be produced from embryonic stem cell (ESC) lines or human fetal cadaver brain tissues containing neurogenic areas (like the SVZ) [81]. The safety of ESC-derived NSCs for transplantation has been established [81, 82], as they do not develop tumors when implanted into normal nude animals. However, when ESCs are implanted as donor cells, immunological incompatibility issues must be addressed. Although iPSC-based treatments will have fewer immunological problems, the fact that FAD patients may lack healthy somatic cells that can be used for reprogramming is a concern [83]. iPSCs are a type of pluripotent stem cells, meaning they can differentiate into all cell types. However, incomplete differentiation may cause the failure of transplantation. In addition, the potential risk for tumorigenesis remains a major challenge for iPSC-based clinical trials. Accordingly, high levels of quality control are needed before transplantation to avoid possible uncontrolled differentiation or proliferation within patient tissue. In summary, many characteristics, including safety, immunological response in the host environment, markers to characterize hNPCs, accessibility, and operability must be dealt with to effectively translate NSC-based treatments to clinical investigations.
REGIONS OF TRANSPLANTATION
Because AD affects numerous brain areas, including the parietal lobe, temporal lobe, sections of the cingulate gyrus, and frontal cortex, it is difficult to establish the best target for transplantation [83]. As summarized in Table 2, the most common region for transplantation is the hippocampus. Adult neurogenesis occurs in the DG of the hippocampus, which is critical to learning and memory [57, 84]. Although the basal forebrain and hippocampus are thought to be the best areas for cell transplantation, these regions are also prone to the most microenvironmental modifications [45]. Because of the altered microenvironment, transplanted NSCs may be unable to flourish in these locations. Although increasing the number of transplanted cells or repeating injections may solve the propagation problem, scientists continue to express reservations about the safety and invasiveness of repeated parenchymal injections performed using a stereotactic technique [85]. Consequently, numerous initiatives have been taken to develop promising techniques for minimizing treatment-related injuries [85]. Kim et al. conducted a phase I clinical trial in nine patients with AD to determine the safety and dose-limiting toxicity of MSCs generated from human umbilical cord blood [85]. To minimize transplantation-related harm, researchers implanted an Ommaya reservoir into the lateral ventricle to distribute the MSCs. The results indicated that this strategy was practicable, safe, and well tolerated.
TIMING OF TRANSPLANTATION
Because AD is an age-related disease, the timing of NSC transplantation is also critical. Prior animal studies have established that optimal timing of transplantation is critical for achieving the maximum therapeutic effect [55, 72]. When cognitive impairment and Aβ neuropathology exist, stem cell transplantation may be ineffective [55]. However, once the chronic course of AD is stopped or disrupted, the cognitive function of patients will improve or no longer deteriorate. Patients enrolled in future clinical trials may meet criteria for having a significant amyloid burden or neuronal degeneration, as determined by brain magnetic resonance imaging or positron emission tomography [85]. Because AHN is essential to the progression of AD, it may be possible to use it as an early indicator of AD to determine the optimal intervention window.
ENVIRONMENT
The environment into which NSCs are be transplanted is quite significant. Quiescent NSCs have a low metabolic rate and are sensitive to their surrounding signaling environment [86]. A variety of stressors, including Aβ or inflammatory molecules, can force neurons in the SVZ to leave or remain quiescent, leading to neuronal death rather than division [87]. Inflammation is thought to drive AD pathogenesis because it increases the Aβ burden [88]. The neuroinflammation in AD is considered to be mediated by the immune system [89], in which abnormal microglial activation is thought to play a crucial role [90]. Thus, controlling the level of inflammation or providing a harmonious environment for transplanted NSCs in AD brains might be beneficial to their neural differentiation.
FATE OF TRANSPLANTED NSCs
Even if we overcome all the previously mentioned obstacles, an uncontrollable cell fate would jeopardize the entire effort. To investigate the differentiation and migration of stem cells, immunofluorescence and confocal microscopy are two main technologies. Recent research indicates that modulating the Notch signaling system makes it feasible to precisely control the process and proportion of NSCs that develop into neural functional cells [91]. In addition to their therapeutic effectiveness, monitoring the interactions of implanted NSCs and their reactivity to the tissue environment all crucial considerations.
In addition to differentiating into CNS cells, stem cells can migrate beyond the transplantation region and form synapses with local CNS cells. Relevant synaptic markers, such as synaptophysin, synapsin, and growth-associated protein-43 (GAP-43) are frequently tested to evaluate synaptic integrity. Indeed, many of the experiments in Table 2 observed that transplantation of NSCs could improve cognition. However, the mechanism for this modulation is currently unclear. Moreover, whether transplanted NSCs could join the correct circuits and perform correctly requires more research. Given the incomplete understanding of AD pathophysiology, it remains debatable whether to direct NSC differentiation in only a single direction. Although recent research has established the significance of nanotechnology in monitoring and guided differentiation of NSC transplantation [5, 92–94], the safety of this technology in the human body requires additional investigation. In addition, transplanted cells may exist in the patient for more than 10 years, whereas the majority of current stem cell transplantation studies in mice are observed for only 3–6 months. Potential risks, such as incomplete differentiation of stem cells, might require longer observation periods in future research.
CONCLUSIONS AND FUTURE PROSPECT
Growing numbers of studies employing animal models are examining the role of pathogenic proteins in the development of AD. At present, none of the available animal models has been able to properly imitate human AD. As a result, it is difficult to exactly quantify the positive effects of NSCs in patients with AD. Although stem cell therapies have been demonstrated as effective in animal models, several human research studies on AD patients have had negative results. Thus, additional research and development of better animal or cell models should be encouraged.
It has been shown repeatedly that transplanting of NSCs for AD has some benefits, although the drawbacks and risks cannot be ignored. Ethical issues, immune rejection, genetic mutations, and the potential risk of tumorigenesis are all issues that need to be urgently addressed. Accordingly, no clinical studies employing NSCs in the treatment of AD patients have been conducted and more work must be done before NSC-based therapy can be used to benefit patients. Important factors include the cell source, cell dose, mode of dissemination, and timing of transplantation.
Prior animal research has shown that the appropriate timing of cell transplantation is crucial for attaining the maximal therapeutic impact. Current treatment studies are almost certainly predicated on early detection and intervention to avert future severe synapse and neuronal loss. Because AD is a complicated condition, it needs multiple interventions. Thus, combining NSC transplantation with already authorized medicines may pave a promising road for future treatment of AD.
ACKNOWLEDGMENTS
We thank Liwen Bianji (Edanz) (http://www.liwenbianji.cn/) for basic language editing of a draft of this manuscript.
This work was supported by the National Key Research and Development Program of China (2018YFA0108602, 2021YFE0114300), the National Natural Science Foundation of China (82170799, 82171475) and the CAMS Initiative for Innovative Medicine (2021-1-I2M-019).
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/22-0721r2).
REFERENCES
[1] | Hodson R ((2018) ) Alzheimer’s disease. Nature 559: , 1. |
[2] | Long JM , Holtzman DM ((2019) ) Alzheimer disease: An update on pathobiology and treatment strategies. Cell 179: , 312–339. |
[3] | Mangialasche F , Solomon A , Winblad B , Mecocci P , Kivipelto M ((2010) ) Alzheimer’s disease: Clinical trials and drug development. Lancet Neurol 9: , 702–716. |
[4] | Papaspyropoulos A , Tsolaki M , Foroglou N , Pantazaki AA ((2020) ) Modeling and targeting Alzheimer’s disease with organoids. Front Pharmacol 11: , 396. |
[5] | Srivastava S , Ahmad R , Khare SK ((2021) ) Alzheimer’s disease and its treatment by different approaches: A review. Eur J Med Chem 216: , 113320. |
[6] | Scheltens P , De Strooper B , Kivipelto M , Holstege H , Chételat G , Teunissen CE , Cummings J , van der Flier WM ((2021) ) Alzheimer’s disease. Lancet 397: , 1577–1590. |
[7] | Gao LB , Yu XF , Chen Q , Zhou D ((2016) ) Alzheimer’s disease therapeutics: Current and future therapies. Minerva Med 107: , 108–113. |
[8] | Rabinovici GD ((2021) ) Controversy and progress in Alzheimer’s disease - FDA approval of aducanumab. N Engl J Med 385: , 771–774. |
[9] | Reddy AP , Ravichandran J , Carkaci-Salli N ((2020) ) Neural regeneration therapies for Alzheimer’s and Parkinson’s disease-related disorders. Biochim Biophys Acta Mol Basis Dis 1866: , 165506. |
[10] | Shen Z , Li X , Bao X , Wang R ((2017) ) Microglia-targeted stem cell therapies for Alzheimer disease: A preclinical data review. J Neurosci Res 95: , 2420–2429. |
[11] | Liu XY , Yang LP , Zhao L ((2020) ) Stem cell therapy for Alzheimer’s disease. World J Stem Cells 12: , 787–802. |
[12] | Kang JM , Yeon BK , Cho SJ , Suh YH ((2016) ) Stem cell therapy for Alzheimer’s disease: A review of recent clinical trials. J Alzheimers Dis 54: , 879–889. |
[13] | Duncan T , Valenzuela M ((2017) ) Alzheimer’s disease, dementia, and stem cell therapy. Stem Cell Res Ther 8: , 111. |
[14] | Kim J , Lee Y , Lee S , Kim K , Song M , Lee J ((2020) ) Mesenchymal stem cell therapy and Alzheimer’s disease: Current status and future perspectives. J Alzheimers Dis 77: , 1–14. |
[15] | Andrzejewska A , Dabrowska S , Lukomska B , Janowski M ((2021) ) Mesenchymal stem cells for neurological disorders. Adv Sci (Weinh) 8: , 2002944. |
[16] | Zhang FQ , Jiang JL , Zhang JT , Niu H , Fu XQ , Zeng LL ((2020) ) Current status and future prospects of stem cell therapy in Alzheimer’s disease. Neural Regen Res 15: , 242–250. |
[17] | De Strooper B , Karran E ((2016) ) The cellular phase of Alzheimer’s disease. Cell 164: , 603–615. |
[18] | Sung PS , Lin PY , Liu CH , Su HC , Tsai KJ ((2020) ) Neuroinflammation and neurogenesis in Alzheimer’s disease and potential therapeutic approaches. Int J Mol Sci 21: , 701–. |
[19] | Wang J , Tan L , Yu JT ((2016) ) Prevention trials in Alzheimer’s disease: Current status and future perspectives. J Alzheimers Dis 50: , 927–945. |
[20] | Schilling S , Rahfeld JU , Lues I , Lemere CA ((2018) ) Passive Abeta immunotherapy: Current achievements and future perspectives. Molecules 23: , 1068. |
[21] | Hoskin JL , Sabbagh MN , Al-Hasan Y , Decourt B ((2019) ) Tau immunotherapies for Alzheimer’s disease. Expert Opin Investig Drugs 28: , 545–554. |
[22] | Panza F , Lozupone M , Seripa D , Imbimbo BP ((2019) ) Amyloid-β immunotherapy for Alzheimer disease: Is it now a long shot? Ann Neurol 85: , 303–315. |
[23] | Andreasson KI , Bachstetter AD , Colonna M , Ginhoux F , Holmes C , Lamb B , Landreth G , Lee DC , Low D , Lynch MA , Monsonego A , O’Banion MK , Pekny M , Puschmann T , Russek-Blum N , Sandusky LA , Selenica ML , Takata K , Teeling J , Town T , Van Eldik LJ ((2016) ) Targeting innate immunity for neurodegenerative disorders of the central nervous system. J Neurochem 138: , 653–693. |
[24] | Bronzuoli MR , Iacomino A , Steardo L , Scuderi C ((2016) ) Targeting neuroinflammation in Alzheimer’s disease. J Inflamm Res 9: , 199–208. |
[25] | Cribbs DH , Berchtold NC , Perreau V , Coleman PD , Rogers J , Tenner AJ , Cotman CW ((2012) ) Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: A microarray study. J Neuroinflammation 9: , 179. |
[26] | Ju Y , Tam KY ((2022) ) Pathological mechanisms and therapeutic strategies for Alzheimer’s disease. Neural Regen Res 17: , 543–549. |
[27] | Macdonald R , Barnes K , Hastings C , Mortiboys H ((2018) ) Mitochondrial abnormalities in Parkinson’s disease and Alzheimer’s disease: Can mitochondria be targeted therapeutically? Biochem Soc Trans 46: , 891–909. |
[28] | Fang EF , Hou Y , Palikaras K , Adriaanse BA , Kerr JS , Yang B , Lautrup S , Hasan-Olive MM , Caponio D , Dan X , Rocktaschel P , Croteau DL , Akbari M , Greig NH , Fladby T , Nilsen H , Cader MZ , Mattson MP , Tavernarakis N , Bohr VA ((2019) ) Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat Neurosci 22: , 401–412. |
[29] | Vasic V , Barth K , Schmidt MHH ((2019) ) Neurodegeneration and neuro-regeneration-Alzheimer’s disease and stem cell therapy. Int J Mol Sci 20: , 4272. |
[30] | Chen Z , Zhong C ((2014) ) Oxidative stress in Alzheimer’s disease. Neurosci Bull 30: , 271–281. |
[31] | Zlokovic BV ((2011) ) Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci 12: , 723–738. |
[32] | Wingo AP , Liu Y , Gerasimov ES , Gockley J , Logsdon BA , Duong DM , Dammer EB , Robins C , Beach TG , Reiman EM , Epstein MP , De Jager PL , Lah JJ , Bennett DA , Seyfried NT , Levey AI , Wingo TS ((2021) ) Integrating human brain proteomes with genome-wide association data implicates new proteins in Alzheimer’s disease pathogenesis. Nat Genet 53: , 143–146. |
[33] | Serrano-Pozo A , Das S , Hyman BT ((2021) ) APOE and Alzheimer’s disease: Advances in genetics, pathophysiology, and therapeutic approaches. Lancet Neurol 20: , 68–80. |
[34] | Shi Y , Holtzman DM ((2018) ) Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat Rev Immunol 18: , 759–772. |
[35] | Ishii M , Iadecola C ((2020) ) Risk factor for Alzheimer’s disease breaks the blood-brain barrier. Nature 581: , 31–32. |
[36] | Yan S , Zheng C , Paranjpe MD , Li J , Benzinger TLS , Lu J , Zhou Y ((2020) ) Association of sex and APOE ɛ4 with brain tau deposition and atrophy in older adults with Alzheimer’s disease. Theranostics 10: , 10563–10572. |
[37] | Rasmussen KL , Nordestgaard BG , Frikke-Schmidt R , Nielsen SF ((2018) ) An updated Alzheimer hypothesis: Complement C3 and risk of Alzheimer’s disease-A cohort study of 95,442 individuals. Alzheimers Dement 14: , 1589–1601. |
[38] | Lim YY , Kalinowski P , Pietrzak RH , Laws SM , Burnham SC , Ames D , Villemagne VL , Fowler CJ , Rainey-Smith SR , Martins RN , Rowe CC , Masters CL , Maruff PT ((2018) ) Association of β-amyloid and apolipoprotein E ɛ4 with memory decline in preclinical Alzheimer disease. JAMA Neurol 75: , 488–494. |
[39] | Djelti F , Braudeau J , Hudry E , Dhenain M , Varin J , Bièche I , Marquer C , Chali F , Ayciriex S , Auzeil N , Alves S , Langui D , Potier MC , Laprevote O , Vidaud M , Duyckaerts C , Miles R , Aubourg P , Cartier N ((2015) ) CYP46A1 inhibition, brain cholesterol accumulation and neurodegeneration pave the way for Alzheimer’s disease. Brain 138: , 2383–2398. |
[40] | Testa G , Staurenghi E , Zerbinati C , Gargiulo S , Iuliano L , Giaccone G , Fantò F , Poli G , Leonarduzzi G , Gamba P ((2016) ) Changes in brain oxysterols at different stages of Alzheimer’s disease: Their involvement in neuroinflammation. Redox Biol 10: , 24–33. |
[41] | Gómez-Isla T , Price JL , McKeel DW , Jr. , Morris JC , Growdon JH , Hyman BT ((1996) ) Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J Neurosci 16: , 4491–4500. |
[42] | Ager RR , Davis JL , Agazaryan A , Benavente F , Poon WW , LaFerla FM , Blurton-Jones M ((2015) ) Human neural stem cells improve cognition and promote synaptic growth in two complementary transgenic models of Alzheimer’s disease and neuronal loss. Hippocampus 25: , 813–826. |
[43] | Madrazo I , Kopyov O , Avila-Rodriguez MA , Ostrosky F , Carrasco H , Kopyov A , Avendano-Estrada A , Jimenez F , Magallon E , Zamorano C , Gonzalez G , Valenzuela T , Carrillo R , Palma F , Rivera R , Franco-Bourland RE , Guizar-Sahagun G ((2019) ) Transplantation of human neural progenitor cells (NPC) into putamina of parkinsonian patients: A case series study, safety and efficacy four years after surgery. Cell Transplant 28: , 269–285. |
[44] | Mazzini L , Gelati M , Profico DC , Sgaravizzi G , Projetti Pensi M , Muzi G , Ricciolini C , Rota Nodari L , Carletti S , Giorgi C , Spera C , Domenico F , Bersano E , Petruzzelli F , Cisari C , Maglione A , Sarnelli MF , Stecco A , Querin G , Masiero S , Cantello R , Ferrari D , Zalfa C , Binda E , Visioli A , Trombetta D , Novelli A , Torres B , Bernardini L , Carriero A , Prandi P , Servo S , Cerino A , Cima V , Gaiani A , Nasuelli N , Massara M , Glass J , Soraru G , Boulis NM , Vescovi AL ((2015) ) Human neural stem cell transplantation in ALS: Initial results from a phase I trial. J Transl Med 13: , 17. |
[45] | Li XY , Bao XJ , Wang RZ ((2015) ) Potential of neural stem cell-based therapies for Alzheimer’s disease. J Neurosci Res 93: , 1313–1324. |
[46] | Boese AC , Hamblin MH , Lee JP ((2020) ) Neural stem cell therapy for neurovascular injury in Alzheimer’s disease. Exp Neurol 324: , 113112. |
[47] | Nation DA , Sweeney MD , Montagne A , Sagare AP , D’Orazio LM , Pachicano M , Sepehrband F , Nelson AR , Buennagel DP , Harrington MG , Benzinger TLS , Fagan AM , Ringman JM , Schneider LS , Morris JC , Chui HC , Law M , Toga AW , Zlokovic BV ((2019) ) Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med 25: , 270–276. |
[48] | Sweeney MD , Montagne A , Sagare AP , Nation DA , Schneider LS , Chui HC , Harrington MG , Pa J , Law M , Wang DJJ , Jacobs RE , Doubal FN , Ramirez J , Black SE , Nedergaard M , Benveniste H , Dichgans M , Iadecola C , Love S , Bath PM , Markus HS , Salman RA , Allan SM , Quinn TJ , Kalaria RN , Werring DJ , Carare RO , Touyz RM , Williams SCR , Moskowitz MA , Katusic ZS , Lutz SE , Lazarov O , Minshall RD , Rehman J , Davis TP , Wellington CL , González HM , Yuan C , Lockhart SN , Hughes TM , Chen CLH , Sachdev P , O’Brien JT , Skoog I , Pantoni L , Gustafson DR , Biessels GJ , Wallin A , Smith EE , Mok V , Wong A , Passmore P , Barkof F , Muller M , Breteler MMB , Román GC , Hamel E , Seshadri S , Gottesman RF , van Buchem MA , Arvanitakis Z , Schneider JA , Drewes LR , Hachinski V , Finch CE , Toga AW , Wardlaw JM , Zlokovic BV ((2019) ) Vascular dysfunction-The disregarded partner of Alzheimer’s disease. Alzheimers Dement 15: , 158–167. |
[49] | Montagne A , Barnes SR , Sweeney MD , Halliday MR , Sagare AP , Zhao Z , Toga AW , Jacobs RE , Liu CY , Amezcua L , Harrington MG , Chui HC , Law M , Zlokovic BV ((2015) ) Blood-brain barrier breakdown in the aging human hippocampus. Neuron 85: , 296–302. |
[50] | Nalivaeva NN , Beckett C , Belyaev ND , Turner AJ ((2012) ) Are amyloid-degrading enzymes viable therapeutic targets in Alzheimer’s disease? J Neurochem 120 Suppl 1: , 167–185. |
[51] | Lee IS , Jung K , Kim IS , Lee H , Kim M , Yun S , Hwang K , Shin JE , Park KI ((2015) ) Human neural stem cells alleviate Alzheimer-like pathology in a mouse model. Mol Neurodegener 10: , 38. |
[52] | Hayashi Y , Lin HT , Lee CC , Tsai KJ ((2020) ) Effects of neural stem cell transplantation in Alzheimer’s disease models. J Biomed Sci 27: , 29. |
[53] | Onyango IG , Jauregui GV , Čarná M , Bennett JP Jr. , Stokin GB ((2021) ) Neuroinflammation in Alzheimer’s disease. Biomedicines 9: , 524. |
[54] | Zhang Q , Wu HH , Wang Y , Gu GJ , Zhang W , Xia R ((2016) ) Neural stem cell transplantation decreases neuroinflammation in a transgenic mouse model of Alzheimer’s disease. J Neurochem 136: , 815–825. |
[55] | Kim JA , Ha S , Shin KY , Kim S , Lee KJ , Chong YH , Chang KA , Suh YH ((2015) ) Neural stem cell transplantation at critical period improves learning and memory through restoring synaptic impairment in Alzheimer’s disease mouse model. Cell Death Dis 6: , e1789. |
[56] | Disouky A , Lazarov O ((2021) ) Adult hippocampal neurogenesis in Alzheimer’s disease. Prog Mol Biol Transl Sci 177: , 137–156. |
[57] | Moreno-Jiménez EP , Flor-García M , Terreros-Roncal J , Rábano A , Cafini F , Pallas-Bazarra N , Ávila J , Llorens-Martín M ((2019) ) Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat Med 25: , 554–560. |
[58] | Mu Y , Gage FH ((2011) ) Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Mol Neurodegener 6: , 85. |
[59] | Sorrells SF , Paredes MF , Cebrian-Silla A , Sandoval K , Qi D , Kelley KW , James D , Mayer S , Chang J , Auguste KI , Chang EF , Gutierrez AJ , Kriegstein AR , Mathern GW , Oldham MC , Huang EJ , Garcia-Verdugo JM , Yang Z , Alvarez-Buylla A ((2018) ) Human hippocampal neurogenesis drops sharply in children to undetectable levels in adults. Nature 555: , 377–381. |
[60] | Boldrini M , Fulmore CA , Tartt AN , Simeon LR , Pavlova I , Poposka V , Rosoklija GB , Stankov A , Arango V , Dwork AJ , Hen R , Mann JJ ((2018) ) Human hippocampal neurogenesis persists throughout aging. Cell Stem Cell 22: , 589–599.e585. |
[61] | Gillotin S , Sahni V , Lepko T , Hanspal MA , Swartz JE , Alexopoulou Z , Marshall FH ((2021) ) Targeting impaired adult hippocampal neurogenesis in ageing by leveraging intrinsic mechanisms regulating neural stem cell activity. Ageing Res Rev 71: , 101447. |
[62] | Blurton-Jones M , Kitazawa M , Martinez-Coria H , Castello NA , Muller FJ , Loring JF , Yamasaki TR , Poon WW , Green KN , LaFerla FM ((2009) ) Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc Natl Acad Sci U S A 106: , 13594–13599. |
[63] | Puzzo D , Gulisano W , Palmeri A , Arancio O ((2015) ) Rodent models for Alzheimer’s disease drug discovery. Expert Opin Drug Discov 10: , 703–711. |
[64] | Cenini G , Hebisch M , Iefremova V , Flitsch LJ , Breitkreuz Y , Tanzi RE , Kim DY , Peitz M , Brüstle O ((2021) ) Dissecting Alzheimer’s disease pathogenesis in human 2D and 3D models. Mol Cell Neurosci 110: , 103568. |
[65] | Penney J , Ralvenius WT , Tsai LH ((2020) ) Modeling Alzheimer’s disease with iPSC-derived brain cells. Mol Psychiatry 25: , 148–167. |
[66] | Radde R , Bolmont T , Kaeser SA , Coomaraswamy J , Lindau D , Stoltze L , Calhoun ME , Jaggi F , Wolburg H , Gengler S , Haass C , Ghetti B , Czech C , Holscher C , Mathews PM , Jucker M ((2006) ) Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep 7: , 940–946. |
[67] | Oddo S , Caccamo A , Shepherd JD , Murphy MP , Golde TE , Kayed R , Metherate R , Mattson MP , Akbari Y , LaFerla FM ((2003) ) Triple-transgenic model of Alzheimer’s disease with plaques and tangles. Neuron 39: , 409–421. |
[68] | Oakley H , Cole SL , Logan S , Maus E , Shao P , Craft J , Guillozet-Bongaarts A , Ohno M , Disterhoft J , Van Eldik L , Berry R , Vassar R ((2006) ) Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J Neurosci 26: , 10129–10140. |
[69] | Chen SQ , Cai Q , Shen YY , Wang PY , Li MH , Teng GY ((2014) ) Neural stem cell transplantation improves spatial learning and memory via neuronal regeneration in amyloid-beta precursor protein/presenilin 1/tau triple transgenic mice. Am J Alzheimers Dis Other Demen 29: , 142–149. |
[70] | Lilja AM , Malmsten L , Rojdner J , Voytenko L , Verkhratsky A , Ogren SO , Nordberg A , Marutle A ((2015) ) Neural stem cell transplant-induced effect on neurogenesis and cognition in Alzheimer Tg2576 mice is inhibited by concomitant treatment with amyloid-lowering or cholinergic alpha7 nicotinic receptor drugs. Neural Plast 2015: , 370432. |
[71] | Gu G , Zhang W , Li M , Ni J , Wang P ((2015) ) Transplantation of NSC-derived cholinergic neuron-like cells improves cognitive function in APP/PS1 transgenic mice. Neuroscience 291: , 81–92. |
[72] | Marsh SE , Yeung ST , Torres M , Lau L , Davis JL , Monuki ES , Poon WW , Blurton-Jones M ((2017) ) HuCNS-SC human NSCs fail to differentiate, form ectopic clusters, and provide no cognitive benefits in a transgenic model of Alzheimer’s disease. Stem Cell Reports 8: , 235–248. |
[73] | Blurton-Jones M , Spencer B , Michael S , Castello NA , Agazaryan AA , Davis JL , Müller FJ , Loring JF , Masliah E , LaFerla FM ((2014) ) Neural stem cells genetically-modified to express neprilysin reduce pathology in Alzheimer transgenic models. Stem Cell Res Ther 5: , 46. |
[74] | McGinley LM , Kashlan ON , Bruno ES , Chen KS , Hayes JM , Kashlan SR , Raykin J , Johe K , Murphy GG , Feldman EL ((2018) ) Human neural stem cell transplantation improves cognition in a murine model of Alzheimer’s disease. Sci Rep 8: , 14776. |
[75] | Zhang T , Ke W , Zhou X , Qian Y , Feng S , Wang R , Cui G , Tao R , Guo W , Duan Y , Zhang X , Cao X , Shu Y , Yue C , Jing N ((2019) ) Human neural stem cells reinforce hippocampal synaptic network and rescue cognitive deficits in a mouse model of Alzheimer’s disease. Stem Cell Reports 13: , 1022–1037. |
[76] | Zhang W , Wang PJ , Sha HY , Ni J , Li MH , Gu GJ ((2014) ) Neural stem cell transplants improve cognitive function without altering amyloid pathology in an APP/PS1 double transgenic model of Alzheimer’s disease. Mol Neurobiol 50: , 423–437. |
[77] | Abdi S , Javanmehr N , Ghasemi-Kasman M , Bali HY , Pirzadeh M ((2022) ) Stem cell-based therapeutic and diagnostic approaches in Alzheimer’s disease. Curr Neuropharmacol 20: , 1093–1115. |
[78] | Hunsberger JG , Rao M , Kurtzberg J , Bulte JWM , Atala A , LaFerla FM , Greely HT , Sawa A , Gandy S , Schneider LS , Doraiswamy PM ((2016) ) Accelerating stem cell trials for Alzheimer’s disease. Lancet Neurol 15: , 219–230. |
[79] | Tong LM , Fong H , Huang Y ((2015) ) Stem cell therapy for Alzheimer’s disease and related disorders: Current status and future perspectives.e. Exp Mol Med 47: , e151. |
[80] | Pacheco-Herrero M , Soto-Rojas LO , Reyes-Sabater H , Garcés-Ramirez L , de la Cruz López F , Villanueva-Fierro I , Luna-Muñoz J ((2021) ) Current status and challenges of stem cell treatment for Alzheimer’s disease. J Alzheimers Dis 84: , 917–935. |
[81] | Huang L , Zhang L ((2019) ) Neural stem cell therapies and hypoxic-ischemic brain injury. Prog Neurobiol 173: , 1–17. |
[82] | Daadi MM , Maag AL , Steinberg GK ((2008) ) Adherent self-renewable human embryonic stem cell-derived neural stem cell line: Functional engraftment in experimental stroke model. PLoS One 3: , e1644. |
[83] | Fan X , Sun D , Tang X , Cai Y , Yin ZQ , Xu H ((2014) ) Stem-cell challenges in the treatment of Alzheimer’s disease: A long way from bench to bedside. Med Res Rev 34: , 957–978. |
[84] | Babcock KR , Page JS , Fallon JR , Webb AE ((2021) ) Adult hippocampal neurogenesis in aging and Alzheimer’s disease. Stem Cell Reports 16: , 681–693. |
[85] | Kim HJ , Cho KR , Jang H , Lee NK , Jung YH , Kim JP , Lee JI , Chang JW , Park S , Kim ST , Moon SW , Seo SW , Choi SJ , Na DL ((2021) ) Intracerebroventricular injection of human umbilical cord blood mesenchymal stem cells in patients with Alzheimer’s disease dementia: A phase I clinical trial. Alzheimers Res Ther 13: , 154. |
[86] | Urban N , Blomfield IM , Guillemot F ((2019) ) Quiescence of adult mammalian neural stem cells: A highly regulated rest. Neuron 104: , 834–848. |
[87] | Merlo S , Basile L , Giuffrida ML , Sortino MA , Guccione S , Copani A ((2015) ) Identification of 5-methoxyflavone as a novel DNA polymerase-beta inhibitor and neuroprotective agent against beta-amyloid toxicity. J Nat Prod 78: , 2704–2711. |
[88] | Heppner FL , Ransohoff RM , Becher B ((2015) ) Immune attack: The role of inflammation in Alzheimer disease. Nat Rev Neurosci 16: , 358–372. |
[89] | Irwin MR , Vitiello MV ((2019) ) Implications of sleep disturbance and inflammation for Alzheimer’s disease dementia. Lancet Neurol 18: , 296–306. |
[90] | Baik SH , Kang S , Lee W , Choi H , Chung S , Kim JI , Mook-Jung I ((2019) ) A breakdown in metabolic reprogramming causes microglia dysfunction in Alzheimer’s disease. Cell Metab 30: , 493–507.e496. |
[91] | Harada Y , Yamada M , Imayoshi I , Kageyama R , Suzuki Y , Kuniya T , Furutachi S , Kawaguchi D , Gotoh Y ((2021) ) Cell cycle arrest determines adult neural stem cell ontogeny by an embryonic Notch-nonoscillatory Hey1 module. Nat Commun 12: , 6562–. |
[92] | Zhang B , Yan W , Zhu Y , Yang W , Le W , Chen B , Zhu R , Cheng L ((2018) ) Nanomaterials in neural-stem-cell-mediated regenerative medicine: Imaging and treatment of neurological diseases. Adv Mater 30: , e1705694. |
[93] | Liu L , Wu J , Wang S , Kun L , Gao J , Chen B , Ye Y , Wang F , Tong F , Jiang J , Ou J , Wilson DA , Tu Y , Peng F ((2021) ) Control the neural stem cell fate with biohybrid piezoelectrical magnetite micromotors. Nano Lett 21: , 3518–3526. |
[94] | Formicola B , Cox A , Dal Magro R , Masserini M , Re F ((2019) ) Nanomedicine for the treatment of Alzheimer’s disease. J Biomed Nanotechnol 15: , 1997–2024. |


