
Extremely Early-Onset Frontotemporal Dementia: A Case Report and Literature Review
Abstract
Background:
In most cases, the onset of frontotemporal dementia (FTD) occurs between the ages of 45 and 65 years. However, some patients experience an extremely early disease onset.
Objective:
To investigate the clinical, genetic, and pathological features of extremely early-onset FTD.
Methods:
We conducted a comprehensive clinical, genetic, and neuropathological analysis of a 25-year-old patient experiencing the onset of behavioral variant frontotemporal dementia (bvFTD). In addition, we conducted a literature review and summarized the clinical, genetic, and pathological features of patients with FTD with onset age≤25 years.
Results:
The patient was diagnosed with bvFTD; however, there was no family history of FTD, no positive genetic test results and no deposition of TDP43, tau, ubiquitin, and synuclein in the brain. Literature screening identified 18 patients with onset age ≤25 years with FTD. The youngest patient was 14 years of age. Most patients (8/14) had a positive family history. The most common clinical phenotype was the behavioral variant (12/14). Genetic results were reported for 11 patients; the most common pathogenic gene was MAPT (10/12), with four cases of G389 R, two cases of P301 S, one case of G335 S, one case of G335A, one case of G335 V, and one case of L315 R. Pathological results were reported for 13 patients; the most common pathological subtype was tau (8/13).
Conclusion:
FTD can start at an extremely early age. The most common phenotype of extremely early onset FTD was the behavioral variant, the most common pathogenic gene was MAPT, and the most common neuropathological type was tau.
INTRODUCTION
Frontotemporal dementia (FTD) is a neurodegenerative disease with behavioral and language variants [1]. One clinical feature of FTD is an early onset; most patients experience disease onset between the ages of 45 and 65 years [2, 3]. However, some cases of extremely early onset have been reported in which the initiation of neurodegeneration occurred in patients in their 20 s to 30 s. The youngest onset age reported thus far is 14 years [4], although this is relatively rare in clinical practice.
The most challenging subtype in young patients is the behavioral variant (bvFTD). Patients with bvFTD present with early personality changes and inappropriate or disruptive behaviors that influence their normal work and daily life, thus causing heavier management burdens for both caregivers and society [1, 5]. In addition, bvFTD can be easily misdiagnosed in young patients thus delaying timely genetic tests and effective treatment; the rate of misdiagnosis is 70% [6]. The potential hazards and diagnostic challenges encountered during the stages of initial presentation highlight the importance of recognizing young patients with FTD as early as possible.
In this study, we conducted a comprehensive clinical, neuroimaging, neuropsychological, genetic, and neuropathological analysis of a 25-year-old patient experiencing the onset of sporadic bvFTD with four years of follow-up. Furthermore, we conducted a literature review of patients with FTD with an onset age of 25 years or younger to summarize their clinical subtype, along with relevant genetic and pathological features.
METHODS
Clinical and neuropsychological workup
The patient was evaluated at Xuanwu hospital in Beijing, China. We collated a range of clinical data relating to initial symptoms, disease progression, family history, and other medical history. A comprehensive neurological physical examination was conducted by a professional neurologist. Longitudinal follow-up was conducted every two years to track disease progression.
A neuropsychologist administered a standardized battery of tests every two years. The neuropsychological test battery consisted of assessments that measured cognitive function in the domains of memory, executive function, language, and behavior. Global cognitive screening measures included the Mini-Mental State Examination (MMSE) [7], the Montreal Cognitive Assessment (MoCA) [8], and the Frontotemporal Lobar Degeneration (FTLD)-Clinical Dementia Rating Scale [9]. Executive function was measured using the Trail Making Test [10]. Visuo-spatial function was assessed using the Rey–Osterrieth Complex Figure Test [11]. Word list memory was evaluated using Rey’s Auditory-Verbal Learning Test [12]. Language was measured using the Boston Naming Test [13]. The severity of behavioral abnormality was assessed using the Frontal Behavior Inventory (FBI) [14] and the Neuropsychiatric Inventory (NPI) [15]. The ability to perform daily living activities was assessed using the Activities of Daily Living Scale (ADL) [16].
This study was conducted according to the Declaration of Helsinki. The clinical protocols were approved by the ethics committee and local institutional review board of Xuanwu Hospital, Capital Medical University, China. The study was conducted in accordance with relevant guidelines and regulations for the use of human subjects in research. Written informed consent was obtained from the participant and his guardians before the start of the study.
Neuroimaging analysis
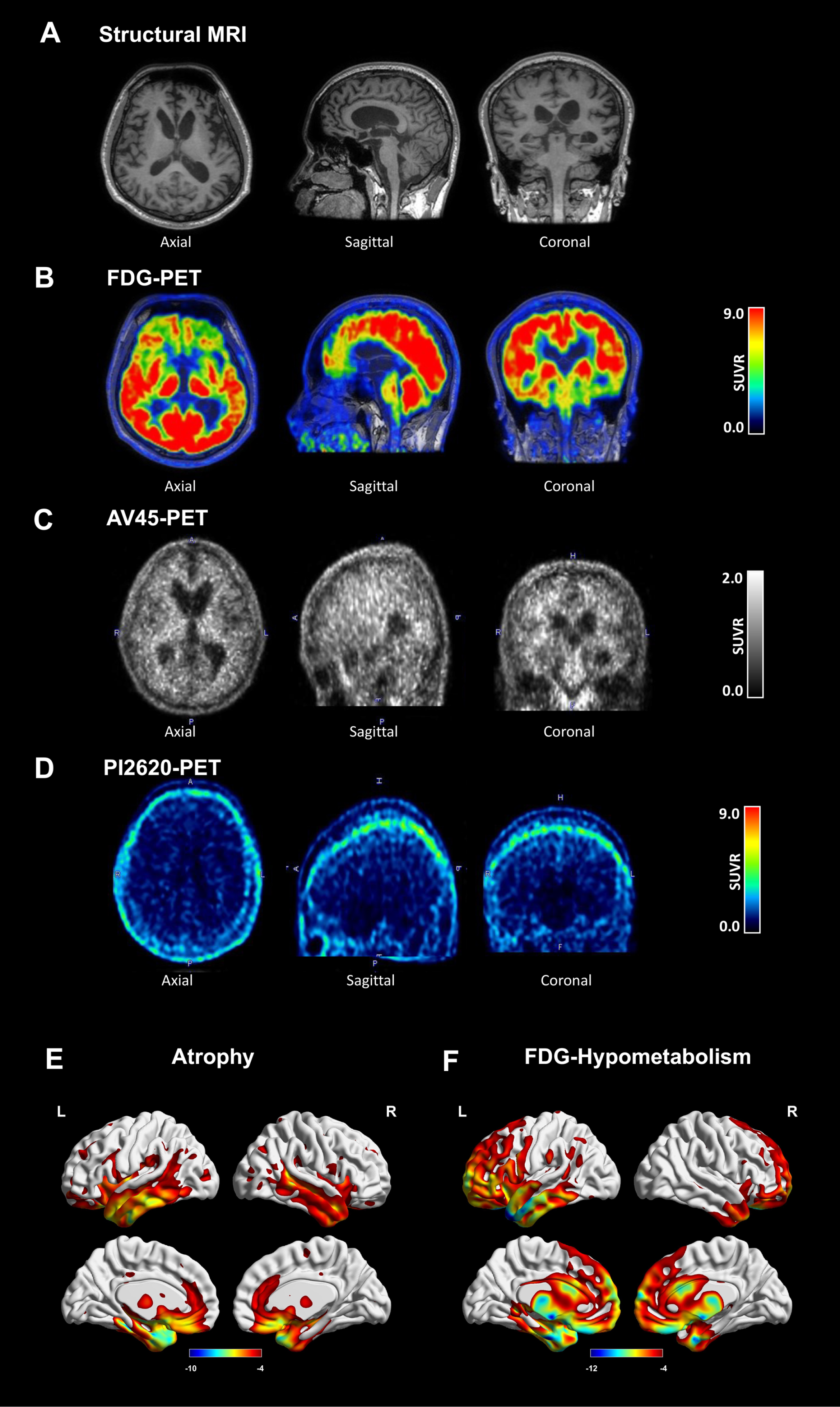
Magnetic resonance imaging (MRI) and positron emission tomography (PET) images were acquired on a hybrid 3.0 T TOF PET/MRI scanner (SIGNA PET/MR, GE Healthcare, WI, USA) at Xuanwu Hospital. MRI and PET data were acquired simultaneously using a vendor-supplied 19-channel head and neck union coil. Images of amyloid PET (AV45), Tau PET (PI2620), and 18F fluoro-deoxy-glucose (18F-FDG)-PET were acquired. PET data were reconstructed using an ordered subset expectation maximization algorithm with weighted attenuation. Images were smoothened using an 8-mm Gaussian kernel with scatter correction and evaluated before conducting an analysis of patient motion and adequacy of statistical counts. Standardized uptake value ratios were calculated using the cerebellar gray matter reference region to normalize mean activity from acquired intervals.
We conducted a two-sample t-test to compare structural MRI and FDG-PET images between the patient and 22 healthy men matched for age and educational level (mean age = 26 years, range = 24-30 years; mean education = 15 years, range = 12-19 years); these tests applied a False Discovery Rate (FDR) corrected p < 0.05 and cluster size > 200. Details relating to the imaging processing steps, analyses and software are given in the Supplementary Material. Patients were diagnosed as AV45-positive or PI2620-positive based on both visual interpretations of elevated binding in the neocortex and semi-quantitative assessment.
Genetic analysis
We extracted genomic DNA from fresh peripheral blood leukocytes taken from the patient and his parents and used an Agilent SureSelect Human All Exon V6 Kit (Agilent Technologies, Santa Clara, CA, USA) to generate a sequencing library for whole-exome sequencing. The prepared libraries were sequenced using the HiSeq-2000 platform (Illumina, San Diego, CA, USA). The sequenced reads were then aligned to the human genome (GRCh37/hg19). Reads were then aligned to the targeted regions and collated for single nucleotide polymorphism calling and subsequent analysis using Burrows-Wheeler Aligner software. Our final analysis included 42 genes (GRN, C9orf72, MAPT, CHMP2B, VCP, TARDBP, SQSTM1, FUS, UBQLN2, OPTN, TREM2, CHCHD10, TBK1, CYLD, TIA1, CCNF, hnRNPA1, hnRNPA2B1, TMEM106B, RAB38, CSF1 R, MATR3, TUBA4A, CFAP410, KIF5A, DCTN1, C21ORF2, ITM2B, PSEN1, PSEN2, APP, AARS2, PLP1, PSAP, PRKAR1B, MOBP, BTNL2, HLA-DRA, CTSC, APOE, TOMM40, ARHGAP35, SERPINA1, GFRA2, UNC13A, SORT1, CIAO1, PRNP, SIGMAR1, GBA, NOTCH3, TRPM7, ABCC1, ABCA7, APBB2, ATP13A2, SPG21, DMT1, VPS2B, ALS17, EIF4G1, SCN8A, COQ2, TSC1, TSC2, HCFC1, ITPR3, PLA2G6, and SLC9A6) that were associated with FTD and other neurodegenerative diseases. Repeat primed polymerase chain reaction was performed to obtain a qualitative estimation of the presence of C9orf72-expanded repeats.
Neuropathological analysis
A biopsy test was conducted in the third year from disease onset. The tissues were extracted from the deep white matter and the borderline between the gray matter and the white matter of the right frontal lobe using robotic stereotactic assistance (ROSA) brain biopsy. Brian tissue samples were fixed with 10% formalin and embedded in paraffin. For immunostaining, deparaffinized sections were incubated with 1% H2O2 in methanol for 10 min to eliminate endogenous peroxidase activity in the tissue. Sections were then pretreated by autoclaving for 10 min in 10 mM sodium citrate buffer (pH 6.0) at 120°C. After washing three times with 0.01 M phosphate-buffered saline (pH 7.4), the sections were processed with the polymer horseradish peroxidase detection system (Polink-1 HRP Broad Spectrum DAB Detection Kit, Golden Bridge International, Mukilteo, WA, USA). The antibodies employed for immunohistochemistry were glial fibrillary acidic protein (GFAP, OriGene USA; monoclonal, clone UMAB129, 1 : 200), neuronal nuclear protein (NeuN, Chemicon, USA; monoclonal, 1 : 4000), Olig-2 (Millipore, USA; Polyclonal, AB9610, 1 : 250), ubiquitin (Abcam, UK; monoclonal, ab140601, 1 : 250), AT8 (hyperphosphorylated tau, Ser202 and Thr205; Thermo, USA; monoclonal, Clone: AT8, 1 : 4000), TDP-43 (Proteintech, USA; monoclonal, Clone 6H6E12, 1 : 10000) and synuclein (Clone 3D5,1 : 10000, a gift from Professor S. Yu) [17].
Literature review
To preliminary describe the clinical, genetic, and pathological features of young patients with FTD with an age of onset similar to the present case, two of the investigators (MC and LL) performed a literature review using PubMed and Embase databases, from inception to September 2021. We included all patients who had been diagnosed with FTD with an onset of≤25-years-of-age. The first author’s name and year of publication, as well as each patient’s age of onset, sex, clinical, neuroimaging, genetics, and pathological results, were extracted from the literature. Two neurologists (MC and LL) independently made a clinical classification of the phenotype involved in each case according to the clinical and neuroimaging features and by referring to the diagnostic criteria for bvFTD [1], non-fluent variant primary progressive aphasia (nfvPPA) [18] and semantic variant PPA (svPPA) [18]. It should be noted that behavioral, language, and motor syndromes overlapped in some patients. The definition of “family history” used in the literature review was 1) a definite report of a family history and 2) some of the relatives presented with dementia, motor dysfunction or other neurodegenerative disease.
RESULTS
Clinical course
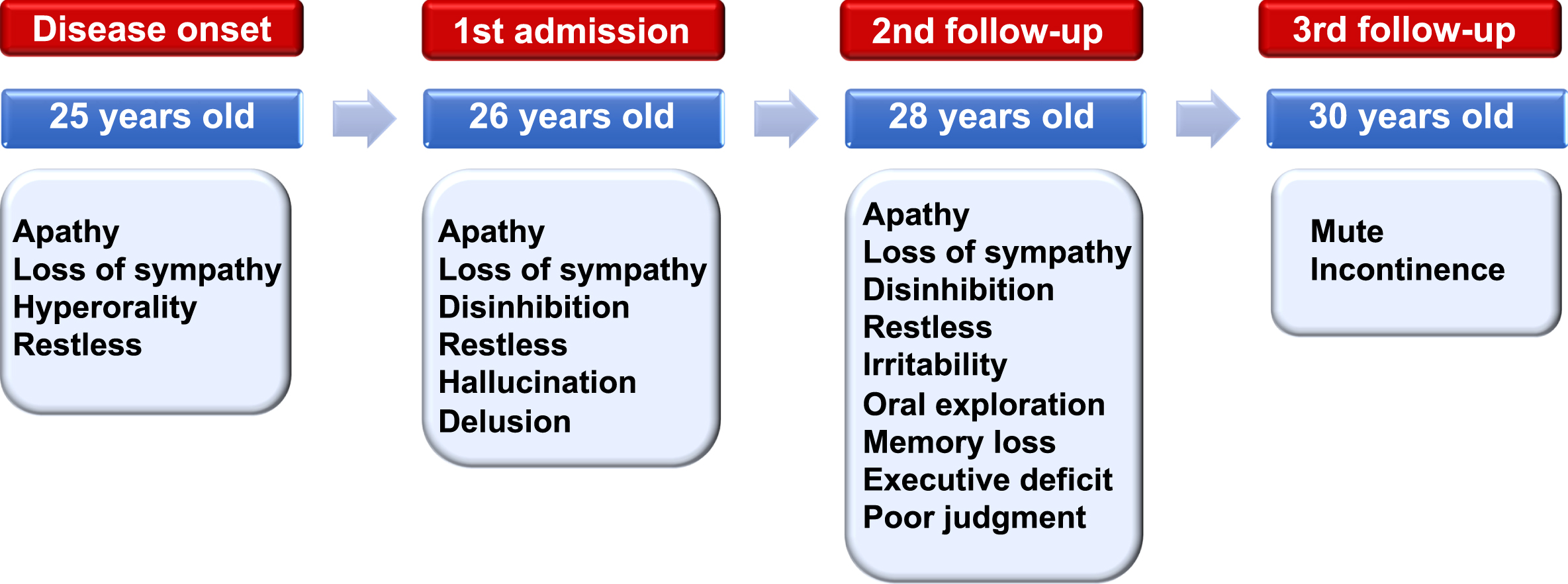
The patient was a 30-year-old male with 14 years of education, who used to be a worker in a company selling mineral water. At the age of 25 years, the patient’s family and friends observed marked personality changes; previously, the patient was outgoing but became introverted over time. He stopped working for the mineral water company, became socially withdrawn and lost sympathy with a reduced response to the feelings of his parents and friends; notably, he was indifferent to his brother’s wedding. He exhibited apathy and self-neglect and did not care about personal hygiene. He was obsessed with computer games and had hyperorality presenting with the increased consumption of excessive snacks and alcohol. Sometimes he was restless and quarreled with his family. Depression was initially suspected but the patient was unresponsive to anti-depressive drugs.
Within one year, his condition worsened. He was still restless, apathetic with a loss of sympathy. Behavioral disinhibition was observed; this manifested as impulsive and socially inappropriate behavior (stealing the possessions of others and scratching cars belonging to others). Hallucination and delusion were identified as the patient reported seeing birds in his room; he also thought he had been discarded by his family. However, the hallucination and delusion were moderate and only occurred on two occasions. He was diagnosed with schizophrenia in another hospital nine months after onset but did not respond to antipsychotic drugs.
Approximately one year after onset, he was admitted to our hospital. Physical examination revealed an abnormal cognitive status with memory and executive dysfunction; this is described in the neuropsychological section in Table 1. The patient had normal cranial signs, and normal limb motor, reflex, sensory and cerebellar function; there were no signs of meningeal irritation. He denied a history of drug use. There was no family history of dementia, motor dysfunction or any other neurodegenerative disease. His parents are still alive and cognitively intact; no other relatives died from neurodegenerative disease. MRI revealed frontal and temporal lobe atrophy and FDG-PET revealed severe hypometabolism in the frontal and temporal lobes. Thus, we considered probable behavior variant frontotemporal dementia as the first diagnosis according to the 2011 diagnostic criteria [1]. To exclude the early onset of Alzheimer’s disease, we conducted amyloid and tau PET and found no deposition of pathological protein. The neuroimaging results of Aβ and tau PET are shown in the neuroimaging section of Fig. 2. Metabolic disease including methylmalonic aciduria was also taken into consideration; however, no abnormality was found in the hematuria organic acid screening and genetic examination.
Table 1
Spatial coordinates and peak values of brain areas showing significant differences in gray matter volume and metabolism of this patient
| Cluster | Brain region | Peak | MNI | Cluster |
| intensity | coordinate | size | ||
| Atrophy on structural MRI (FDR corrected p < 0.05) | ||||
| 1 | Left: middle temporal, superior temporal pole, middle temporal pole, inferior temporal, superior orbital frontal, inferior orbital frontal, medial orbital frontal, rectus, anterior cingulate, putamen, caudate, thalamus, fusiform, para-hippocampus, hippocampus Right: superior orbital frontal, medial orbital frontal, inferior orbital frontal, rectus, anterior cingulate, putamen, caudate | -10.86 | -30 -3 6 | 28707 |
| 2 | Right middle temporal pole | -7.34 | 31.5 19.5 -33 | 2585 |
| 3 | Right hippocampus | -6.65 | 33 -13.5 -12 | 435 |
| 4 | Right superior temporal cortex | -5.05 | -46.5 -45 9 | 249 |
| 5 | Right middle temporal cortex | -4.57 | 48 -22.5 -15 | 243 |
| Hypometabolism on FDG-PET (FDR corrected p < 0.05) | ||||
| 1 | Left: inferior temporal, middle temporal, middle temporal pole, superior temporal pole, superior medial frontal, superior frontal, superior orbital frontal, inferior orbital frontal, middle orbital frontal, medial orbital frontal, middle frontal, rectus, anterior cingulate, insula, caudate, hippocampus, para-hippocampus, thalamus, putamen, fusiform, lingual cortex Right: middle temporal pole, superior medial frontal, superior orbital frontal, medial orbital frontal, middle orbital frontal, rectus, cingulate, caudate, putamen, para-hippocampus, fusiform | -12.60 | -22 -12 -36 | 25293 |
| 2 | Left precentral cortex | -6.00 | -54 12 34 | 332 |
Fig. 2
Neuroimages of the patient. Axial, sagittal, and coronal images of (A) structural MRI, (B) FDG-PET, (C) AV45-PET, and (D) PI2620 PET. (E) Atrophy and (F) Hypometabolism patterns in the patient in comparison with healthy controls. Detailed radiological Montreal Neurosciences Institution (MNI) coordinates of the clusters are shown in Table 1.
Two years after onset, the patient experienced severe memory and cognitive decline. He was still restless, unstable, and easily lost his temper. Apathy and loss of sympathy were severe in that he did not care about his parents and declined to meet any of his relatives and friends. Some social disinhibition behaviors were observed including talking and physical contact with strangers. Oral exploration was observed in that he always took something inedible, such as papers, to his mouth. Cognitive function declined severely, including some of the long-term memory, executive function, and judgement. Compared with the first follow-up, no more changes were found in the two-year follow-up physical examination.
At the third telephone follow-up, he became mute, bed-ridden, incontinent of urine and feces, and lost the ability to take care of himself. The telephone follow-up was conducted without physical examination. The clinical course is summarized in Fig. 1. At the time of writing the patient is still alive.
Fig. 1
Clinical course of the patient. Symptoms began at 25 years of age and the patient was first admitted to our hospital at 26 years of age. A second and third follow-up were conducted at 28 and 30 years of age, respectively. The main clinical presentations were listed at each time point.
Neuroimaging results
Structural MRI, FDG-PET, amyloid PET (AV45), and tau PET (PI2620) were conducted one year after disease onset. Marked frontotemporal atrophy and hypometabolism were observed on raw MRI images (Fig. 2A) and FDG-PET images (Fig. 2B). Negative results were found on the AV45-PET (Fig. 2C) and PI2620-PET (Fig. 2D). The patient showed predominant volume loss and hypometabolism in the frontal, temporal, anterior cingulate, and subcortical brain regions when compared with healthy controls (Fig. 2E). The detailed spatial coordinates and peak values of brain areas showing significant differences in gray matter volume and metabolism in the patient are shown in Table 2. Atrophy and hypometabolism overlapped several brain regions including the left (inferior temporal, middle temporal, middle temporal pole, superior temporal pole, superior orbital frontal, inferior orbital frontal, medial orbital frontal, rectus, anterior cingulate, fusiform, hippocampus, para-hippocampus, thalamus, caudate, and putamen) and right brain regions (superior orbital frontal, medial orbital frontal, rectus, anterior cingulate, middle temporal pole, and caudate). Due to agitation and uncooperative behavior, we were unable to perform follow-up PET/MRI.
Table 2
Neuropsychological tests of the patient with extremely early-onset FTD
| Scales | Baseline | Follow-up 1 | Follow-up 2 |
| General cognitive test | |||
| MMSE | 16 | NA | NA |
| MoCA | 11 | NA | NA |
| Executive function | |||
| TMT-A | 60 s/24 | NA | NA |
| TMT-B | NA | NA | NA |
| Memory | |||
| RAVLT-learning | 11 | NA | NA |
| RAVLT-recall | 0 | NA | NA |
| Visuospatial skill | |||
| ROCFT | 15 | NA | NA |
| Language test | |||
| BNT | 13 | NA | NA |
| Behavior test | |||
| NPI-patient | 69 | 78 | NA |
| FBI (total/negative/positive) | 35/19/16 | 38/18/20 | NA |
| Disease severity | |||
| FTLD-CDR | 12 | NA | NA |
| Activities of daily living | |||
| ADL | 25 | 40 | 80 |
MMSE, Mini-Mental State Examination; MoCA, Montreal Cognitive Assessment; TMT-A, Trail Making Test-A; TMT-B, Trail Making Test-B; RAVLT, Rey’s Auditory-Verbal Learning Test; ROCFT, Rey–Osterrieth Complex Figure Test; BNT, Boston Naming test; FBI, Frontal Behavior Inventory; NPI, neuropsychiatric Inventory; ADL, Activity of Daily Living; FTLD-CDR, Frontotemporal Lobar Degeneration Clinical Dementia Rating Scale.
Neuropsychological tests
Neuropsychological test results are shown in Table 2. At the first admission, the patient could cooperate with some neuropsychological tests. In terms of general mental status, the MMSE score was 16/30 (23/24 for individuals with 7 or more years of education) and the MoCA score was 11/30 (normal range > 24). In terms of executive function, the Trail Making Test-A complete time was 60 s with no error lines (normal range < 78 s); the Trail Making Test-B was not completed. In terms of memory, Rey’s Auditory-Verbal Learning Test total score was 11 (abnormal range≤20); no words were repeated in the recall and recognition phase. With regards to visual and spatial function, the Rey–Osterrieth Complex Figure Test score was 7/36 (normal range > 29). In terms of language skills, the Boston Naming Test score was 13/30 (normal range > 22); sentence-making (0/1) and the reading test were 0/1 and 1/1 in the MMSE, respectively. Sentence repetition (0/1) and word fluency were 0/1 and 1/1 in the MoCA, respectively. In terms of disease severity, the FTLD-Clinical Dementia Rating Scale sum of box score was 12/32 (abnormal > 0). At the second follow-up, the patient’s cognitive function declined; he was restless, irritable and did not cooperate well during the neuropsychological tests. At the third follow-up, the patient’s condition had become more severe; he was bedridden, mute and was losing the ability to communicate with the physicians.
Table 3
Cases of young onset frontotemporal dementia (25-years-of-age or younger) in the literature
| First Author year | Case | Sex | Age at | Age at | Family | Original | Re-classified | Genetics | Pathology |
| onset (y) | death (y) | history | diagnosis | Clinical | |||||
| phenotype | |||||||||
| Ando 2020 [4] | 1 | M | 14 | 34 | No | FTD | bvFTD | MAPT G335A | Tau [Tau (+), GFAP (+), NFT (+), Aβ (-), α-synuclein (-) and TDP-43(-)] |
| Chaunu 2013 [19] | 2 | F | 17 | 24 | Yes | FTDP-17 | bvFTD | MAPT G389R | Tau [Tau (+), TDP-43(-), neurofilaments (-), FUS (-), α-synuclein (-), and Aβ (-)] |
| Pickering-Brown 2000 [20] | 3 | F | 17 | 37 | No | Pick’s disease | bvFTD | MAPT G389R | Tau [Tau (+), NFT (+)] |
| Taniguchi 2004 [21] | 4 | F | 17 | 31 | NA | Nontau-FTD | NA* | NA | Non-Tau |
| Davidson 2007 [22] | 5 | F | 17 | NA | NA | FTD | NA* | NA | Dementia lacking distinctive histology |
| Bermingham 2008 [23] | 6 | F | 20 | 24 | Yes | FTDP-17 | bvFTD | MAPT G389R | Tau [Tau (+), α-synuclein (-), Aβ (-) and ubiquitin (-)] |
| Snowden 2004 [24] | 7 | F | 21 | 30 | No | Sporadic FTD | bvFTD | Negative | Microvacuolar-type degeneration [Tau (-), Aβ (-), ubiquitin (-), neurofilament (-), and α-synuclein (-)] |
| Velakoulis 2009 [25] | 8 | F | 21 | 25 | No | FTDP17 | bvFTD | NA | Tau [Tau (+), ubiquitin (-), TDP 43(-)] |
| Spina 2006 [26] | 9 | M | 22 | 36 | Yes | FTDP17 | bvFTD | MAPT G335S | Tau [Tau (+), Alz50 (+), Aβ (-), α-synuclein (-)] |
| Tacik 2017 [27] | 10 | M | 24 | 31 | No | FTDP17 | nfvPPA | MAPT G389R | Tau [Tau (+), TDP-43(-), IBA-1(-), Aβ (-), PBL (+)] |
| Baba 2007 [28] | 11 | F | 25 | 39 | Yes | FTD | bvFTD | MAPT P301S | NA |
| Mackenzie 2004 [29] | 12 | F | 25 | 29 | Yes | FTD and PLS | bvFTD | NA | Neurofilament-immunoreactive neuronal inclusions [ubiquitin (+), neurofilament proteins (+), tau (-), a-synuclein (-)] |
| Neumann 2005 [30] | 13 | NA | 25 | NA | Yes | FTDP-17 | bvFTD | MAPT G335V | NA |
| van Herpen, 2003 [31] | 14 | NA | 25 | 33 | Yes | FTDP-17 | svPPA | MAPT L315R | Tau [Tau (+), ubiquitin (-), Aβ (-), α-synuclein (-)] |
| Sperfeld, 1999 [32] | 15 | F | 25 | NA | Yes | FTDP-17 | bvFTD | MAPT P301S | NA |
| Baborie 2012 [33]/Stone 2003 [34] | 16 | M | 22 | 29 | No | FTLD | bvFTD | Negative | Ubiquitin [Ubiquitin (+), Tau (-), Aβ (-), SMI31(-), SMI32 (-), NF160 (-), NR4 (-), α-synuclein (-), TDP43 (-), FUS (-)] |
*Two papers did not provide detailed information on the clinical symptoms of the patients and could not be classified to a clinical subtype. M, Male; F, female; NA, not available; FTD, frontotemporal dementia; FTDP-17, frontotemporal dementia with Parkinsonism-17; PLS, primary lateral sclerosis; FTLD, frontotemporal lobe degeneration; bvFTD, behavior variant frontotemporal dementia; nfvPPA, non-fluent variant primary progressive aphasia; svPPA, semantic variant primary progressive aphasia; MAPT microtubule-associated protein tau.
The NPI, FBI, and ADL were conducted. At the first admission, the patient had an NPI score of 69; in addition, the patient showed severe behavioral symptoms, including agitation, depression, apathy, disinhibition, irritability, aberrant motor behavior, and appetite changes. His FBI score was 35 and negative symptoms were scored at 19, including apathy, aspontaneity, indifference, inflexibility, disorganization, inattention, personal neglect, and loss of insight. Positive symptoms were scored at 16, including irritability, poor judgment, inappropriate behavior, impulsivity, restlessness, aggression, and utilization behavior. The ADL score was 25 at admission. At the second follow-up, the patient’s NPI score was 78, the FBI total score was 38, the FBI disinhibition subscale score was 20, the FBI apathy subscale score was 18, and the ADL score was 40. In the third follow-up, the patient became bedridden and mute, thus we only conducted the ADL test; the score was 80.
Genetic tests
Genetic tests of the patient and his parents for hexanucleotide mutation in the chromosome 9 open reading frame 72 gene and whole-exome sequencing for mutations in known genes associated with FTD and other neurodegenerative diseases were negative. No abnormality was found in the genetic results related to metabolic disease.
Neuropathological tests
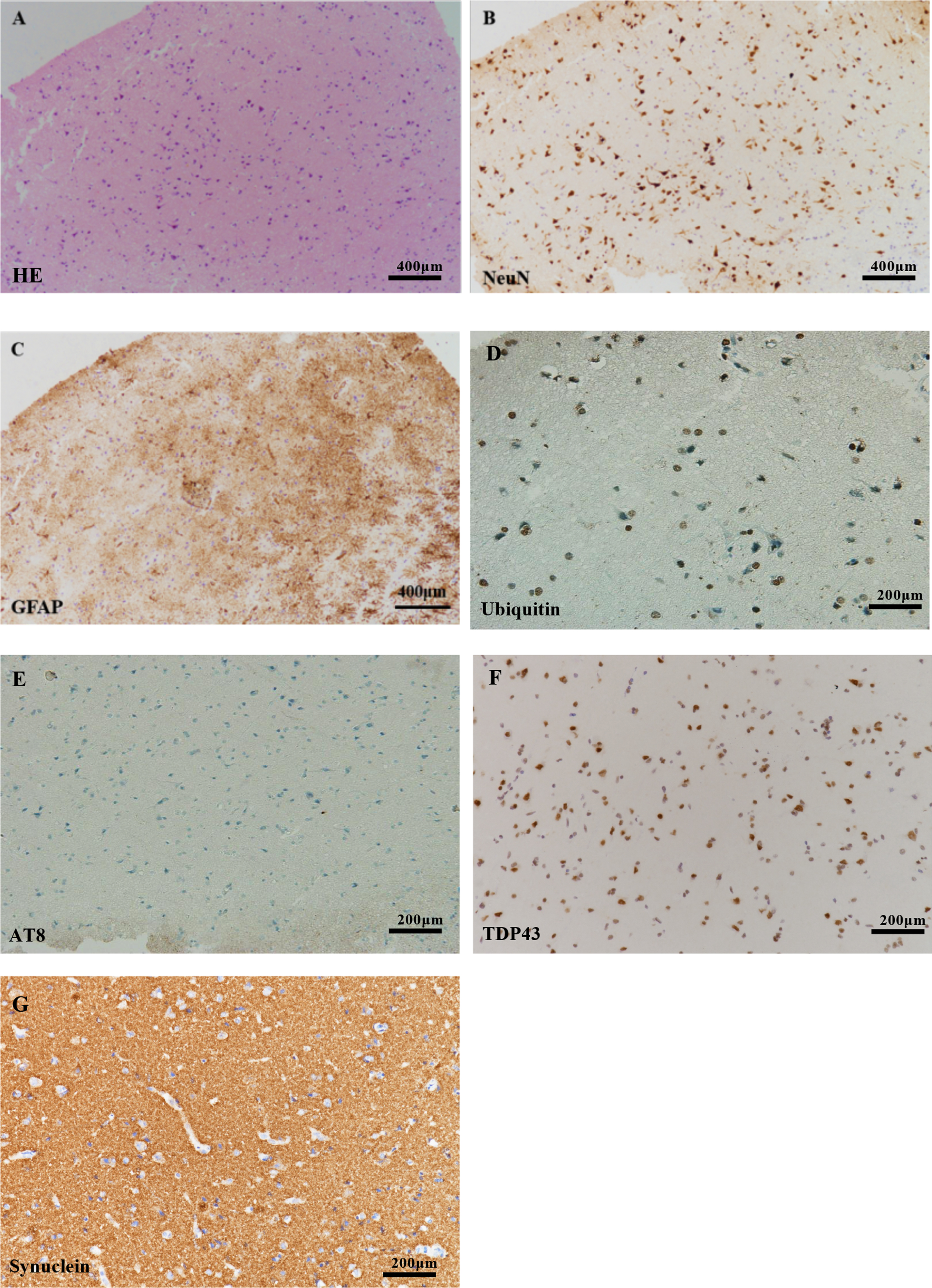
There was mild neuronal loss and shrinkage accompanied by gliosis in the deep cortical layers of the frontal lobe (Fig. 3). The subcortical white matter looked a little sparse, but no inclusion bodies were detected by immunohistochemical staining with ubiquitin, phosphorylated tau protein (AT8), DNA-binding protein of 43 kDa (TDP-43), and synuclein.
Fig. 3
Histopathological findings. Mild neuronal loss was evident in (A) H&E staining and (B) NeuN immunostaining. C) The proliferation of astrocytes was evident in GFAP immunostaining (Scale bar = 400μm). No inclusion bodies were detected by immunohistochemical staining with (D) ubiquitin, (E) AT8, (F) TDP43, and (G) synuclein (Scale bar = 200μm).
Literature review
Through the literature search, we identified 16 patients with FTD with an age of onset of 25 years or younger. The detailed clinical, genetic, and pathological data of these patients are shown in Table 1. The youngest patient was 14-years-of-age. The mean age at onset was 21.06±3.70 years (range: 14-25 years). The age of death was reported for 13 patients; the mean age of death was 30.92±4.84 years (range: 24-39 years). Most of the patients with FTD (8/14) had a positive family history. The most common reclassified clinical phenotype was the behavioral variant (12/14); two papers did not report detailed information relating to clinical symptoms. Genetic results were reported for 12 patients; the most common pathogenic gene was MAPT (10/12), with four cases of G389 R, two cases of P301 S, one case of G335 S, one case of G335A, one case of G335 V and one case of L315 R. Pathological results were reported in 13 patients; the most common pathological subtype was tau (8/13). Detailed data extracted from the literature is shown in Supplementary Table 1.
DISCUSSION
In this study, we comprehensively investigated the clinical, neuroimaging, genetic, and pathological features of a 25-year-old patient who met the diagnosis of probable bvFTD. We also delineated some unique features of extremely young patients with FTD. The most common clinical subtype was the behavioral variant, most of the reported mutations were on the MAPT gene and the most common pathological subtype was tau pathology. This young-onset patient adds to our knowledge of extremely early-onset FTD and serves as a reminder that FTD should be taken into consideration when young patients present with behavioral deficits.
Our patient met the diagnosis for probable bvFTD with a typical clinical pattern that manifested as disinhibition, apathy, loss of sympathy, and oral exploration. The early-onset patient described herein presented with behavioral and cognitive deficits, which could be differentiated from Alzheimer’s disease, some psychiatric disease (including schizophrenia or depression) and metabolic disease, such as methylmalonic acidemia. The patient was negative for amyloid and tau PET, this helping us to differentiate from AD. Psychiatric disease, including depression or schizophrenia, was considered at first, although obvious brain atrophy and hypometabolism were detected by MRI and FDG-PET. Furthermore, the patient did not respond to anti-depression and anti-psychotic drugs. In addition, metabolic diseases, including methylmalonic aciduria were taken into consideration; however, no abnormality was detected by hematuria organic acid screening and genetic analysis.
Marked fronto-temporal impairment was observed on MRI and FDG-PET, thus supporting the diagnosis of bvFTD that was consistent with the typical patterns of bvFTD atrophy and hypometabolism [35]. However, we did not find any pathogenic mutations in the patient or his parents, and no family history was reported; therefore, we concluded that the patient was experiencing sporadic bvFTD. No positive inclusion bodies were detected by immunohistochemical staining with ubiquitin, phosphorylated tau protein (AT8), DNA-binding protein of 43 kDa (TDP-43), and synuclein when we conducted a frontal biopsy. However, because of laboratory limitations, we were unable to stain for FTLD-FET, thus limiting our diagnosis to probable bvFTD. The neuropathological results should be interpreted with caution because the pathological brain tissue was acquired through a stereotactic biopsy that was limited to the bilateral frontal tissue. Further post-mortem autopsy needs to be conducted when possible.
Patients with FTD and an age of onset that is 25 years or younger appear to have unique and characteristic features. Most patients had a positive family history (55.6%); this was higher than the 30-50% with typical FTD [36]. This pedigree investigation represents a significant step forward in terms of identifying the features of young patients with FTD. If a patient has pathogenic mutations, then blood samples from the proband’s biological parents are important to identify whether a patient carries a de novo mutation or whether autosomal dominant inheritance is involved.
Most patients exhibited a behavioral clinical phenotype (77.8%); this is a higher frequency than for typical FTD (in which bvFTD was reported to account for half of such cases) [37]. The clinical manifestations of these patients are the same as those of typical bvFTD, including apathy and behavior disinhibition; most patients had personality changes as their initial symptoms [19, 20, 23-26, 28, 30, 32-34]. These might be considered as psychiatric disorders, including depression, schizophrenia, or solvent abuse at the first evaluation and had undergone extensive tests to reach an accurate diagnosis of FTD [4, 19, 23–25, 30, 33]. The behavioral deficit of these extremely young patients causes heavy social and family burdens and is a huge challenge for patient care and management [25]. This issue therefore deserves more attention.
All the reported pathogenic mutations were located on the MAPT gene [4, 19, 20, 23, 26–28, 30–32]; this differs from the genetic distribution of typical FTD in that C9 has been reported as the most common type in Western countries [38]; MAPT abnormalities are more common in China [39]. In addition, we also found some specific mutation sites that might contribute to extremely early onset; the most common mutations were MAPT G389 R [19, 20, 23, 27] and G335A [4]/S [26]/V [30] which are located in exons 13 and 12. We found some discrepancies in the frequency of MAPT mutations between extremely early onset and typical FTD. In cases with typical FTD, the most common MAPT mutations were P301 L (rs63751273; 234 individuals and 59 families), IVS10 + 16C⟶T (rs63751011; 149 individuals and 48 families), R406 W (rs63750424; 67 individuals and 9 families), and N279K (rs63750756; 44 individuals, 17 families) [2]. However, the mechanisms underlying the more significant contributions of G389 R or G335A/S/V to an early onset need further investigation. Most patients had tau pathology; this differed from the main pathology of typical FTD, in which the most common subtype was reported to be TDP43 [40]. The pathological distribution discrepancy of high tau proportion might be related to a genetic-pathology MAPT-Tau corresponding relationship [41].
This study had some limitations that need to be considered. First, this is a case report and literature review; the papers which report positive results from patients with pathogenic mutations or specific pathological results tend to be more easily accepted than those without pathogenic mutations or non-specific pathological results. This might create some bias when summarizing the frequencies in the literature. Second, due to the rarity of this condition, the sample of patients with FTD with a disease onset of 25 years or younger was small. Therefore, the results reported in this study should be considered as exploratory. Third, the pathological tests were not comprehensive as they lacked FTLD-FET due to laboratory limitations, thus limiting the diagnosis to probable bvFTD. Finally, our understanding of the genetics and pathologies of FTD has progressed over the last 20 years, thus, the results reported herein should be interpreted with caution and validated in a larger cohort.
Conclusions
Our analysis showed that FTD can occur at an extremely early age; the youngest patient ever reported was 14 years of age. The most common mutations reported in cases of extremely early onset were involved in the MAPT gene. The most common mutation site was G389 R, and the most common pathological subtype was tau pathology.
ACKNOWLEDGMENTS
The authors are grateful to all subjects for their participation in the study.
This work was supported by grants from the National Natural Science Foundation of China [no.81971011].
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/22-0679r1).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-220679.
REFERENCES
[1] | Rascovsky K , Hodges J , Knopman D , Mendez M , Kramer J , Neuhaus J , van Swieten J , Seelaar H , Dopper E , Onyike C , Hillis A , Josephs K , Boeve B , Kertesz A , Seeley W , Rankin K , Johnson J , Gorno-Tempini M , Rosen H , Prioleau-Latham C , Lee A , Kipps C , Lillo P , Piguet O , Rohrer J , Rossor M , Warren J , Fox N , Galasko D , Salmon D , Black S , Mesulam M , Weintraub S , Dickerson B , Diehl-Schmid J , Pasquier F , Deramecourt V , Lebert F , Pijnenburg Y , Chow T , Manes F , Grafman J , Cappa S , Freedman M , Grossman M , Miller B ((2011) ) Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 134: , 2456–2477. |
[2] | Moore K , Nicholas J , Grossman M , McMillan C , Irwin D , Massimo L , VanDeerlin V , Warren J , Fox N , Rossor M , Mead S , Bocchetta M , Boeve B , Knopman D , Graff-Radford N , Forsberg L , Rademakers R , Wszolek Z , vanSwieten J , Jiskoot L , Meeter L , Dopper E , Papma J , Snowden J , Saxon J , Jones M , Pickering-Brown S , Le Ber I , Camuzat A , Brice A , Caroppo P , Ghidoni R , Pievani M , Benussi L , Binetti G , Dickerson B , Lucente D , Krivensky S , Graff C , Öijerstedt L , Fallström M , Thonberg H , Ghoshal N , Morris J , Borroni B , Benussi A , Padovani A , Galimberti D , Scarpini E , Fumagalli G , Mackenzie I , Hsiung G , Sengdy P , Boxer A , Rosen H , Taylor J , Synofzik M , Wilke C , Sulzer P , Hodges J , Halliday G , Kwok J , Sanchez-Valle R , Lladó A , Borrego-Ecija S , Santana I , Almeida M , Tábuas-Pereira M , Moreno F , Barandiaran M , Indakoetxea B , Levin J , Danek A , Rowe J , Cope T , Otto M , Anderl-Straub S , de Mendonça A , Maruta C , Masellis M , Black S , Couratier P , Lautrette G , Huey E , Sorbi S , Nacmias B , Laforce R , Tremblay M , Vandenberghe R , Damme P , Rogalski E , Weintraub S , Gerhard A , Onyike C , Ducharme S , Papageorgiou S , Ng A , Brodtmann A , Finger E , Guerreiro R , Bras J , Rohrer J ((2020) ) Age at symptom onsetand death and disease duration in genetic frontotemporal dementia:An international retrospective cohort study. Lancet Neurol 19: , 145–156. |
[3] | Khan I , De Jesus O ((2022) ) Frontotemporal lobe dementia. In, StatPearls, StatPearls Publishing LLC, Treasure Island (FL). StatPearls. |
[4] | Ando K , Ferlini L , Suain V , Yilmaz Z , Mansour S , Le Ber I , Bouchard C , Leroy K , Durr A , Clot F , Sarazin M , Bier JC , Brion JP ((2020) ) de novo MAPT mutation G335A causes severe brain atrophy, 3R and 4R PHF-tau pathology and early onset frontotemporal dementia. Acta Neuropathol Commun 8: , 94. |
[5] | SilvermanHE, AkeJM, ManoochehriM, ApplebyBS, BrushaberD, DevickKL, DickersonBC, FieldsJA, ForsbergLK, GhoshalN, Graff-RadfordNR, GrossmanM, HeuerHW, KornakJ, LapidMI, LitvanI, MackenzieIR, MendezMF, OnyikeCU, PascualB, TartagliaMC, BoeveBF, BoxerAL, RosenHJ, CosentinoS, HueyED, BarkerMS, GoldmanJS; ALLFTD consortium ((2022) ) The contribution of behavioral features to caregiver burden in FTLD spectrum disorders. Alzheimers Dement 18: , 1635–1649. |
[6] | Chemali Z , Withall A , Daffner KR ((2010) ) The plight of caring for young patients with frontotemporal dementia. Am J Alzheimers Dis Other Demen 25: , 109–115. |
[7] | Folstein MF , Folstein SE , McHugh PR ((1975) ) “Mini-mental state”. Apractical method for grading the cognitive state of patients for theclinician. , J Psychiatr Res 12: , 189–198. |
[8] | Nasreddine ZS , Phillips NA , Bédirian V , Charbonneau S , Whitehead V , Collin I , Cummings JL , Chertkow H ((2005) ) The Montreal CognitiveAssessment, MoCA: A brief screening tool for mild cognitiveimpairment. J Am Geriatr Soc 53: , 695–699. |
[9] | Knopman DS , Kramer JH , Boeve BF , Caselli RJ , Graff-Radford NR , Mendez MF , Miller BL , Mercaldo N ((2008) ) Development of methodology for conducting clinical trials in frontotemporal lobar degeneration. Brain 131: , 2957–2968. |
[10] | Reitan RM ((1958) ) Validity of the Trail Making Test as an indicator of organic brain damage. Percept Mot Skills 8: , 271–276. |
[11] | Osterrieth PA ((1944) ) Le test test de copie d’une figure complexe (A test of copying a complex figure)., Arch Psychol 30: , 206–356. |
[12] | Boone KB , Salazar X , Lu P , Warner-Chacon K , Razani J ((2002) ) The Rey 15-item recognition trial: A technique to enhance sensitivity of the Rey 15-item memorization test. J Clin Exp Neuropsychol 24: , 561–573. |
[13] | Cheung RW , Cheung MC , Chan AS ((2004) ) Confrontation naming in Chinese patients with left, right or bilateral brain damage. J Int Neuropsychol Soc 10: , 46–53. |
[14] | Kertesz A , Davidson W , Fox H ((1997) ) Frontal behavioral inventory: Diagnostic criteria for frontal lobe dementia. Can J Neurol Sci 24: , 29–36. |
[15] | Kaufer DI , Cummings JL , Ketchel P , Smith V , MacMillan A , Shelley T , Lopez OL , DeKosky ST ((2000) ) Validation of the NPI-Q, a brief clinical form of the Neuropsychiatric Inventory. J Neuropsychiatry Clin Neurosci 12: , 233–239. |
[16] | Katz S ((1983) ) Assessing self-maintenance: Activities of daily living, mobility, and instrumental activities of daily living. J Am Geriatr Soc 31: , 721–727. |
[17] | Yu S , Li X , Liu G , Han J , Zhang C , Li Y , Xu S , Liu C , Gao Y , Yang H , Uéda K , Chan P ((2007) ) Extensive nuclear localization ofalpha-synuclein in normal rat brain neurons revealed by a novelmonoclonal antibody. Neuroscience 145: , 539–555. |
[18] | Gorno-Tempini ML , Hillis AE , Weintraub S , Kertesz A , Mendez M , Cappa SF , Ogar JM , Rohrer JD , Black S , Boeve BF , Manes F , Dronkers NF , Vandenberghe R , Rascovsky K , Patterson K , Miller BL , Knopman DS , Hodges JR , Mesulam MM , Grossman M ((2011) ) Classification of primary progressive aphasia and its variants. Neurology 76: , 1006–1014. |
[19] | Chaunu MP , Deramecourt V , Buée-Scherrer V , Le Ber I , Brice A , Ehrle N , El Hachimi K , Pluot M , Maurage CA , Bakchine S , Buée L ((2013) ) Juvenile frontotemporal dementia with parkinsonism associatedwith tau mutation G389R. J Alzheimers Dis 37: , 769–776. |
[20] | Pickering-Brown S , Baker M , Yen SH , Liu WK , Hasegawa M , Cairns N , Lantos PL , Rossor M , Iwatsubo T , Davies Y , Allsop D , Furlong R , Owen F , Hardy J , Mann D , Hutton M ((2000) ) Pick’s disease is associated with mutations in the tau gene. Ann Neurol 48: , 859–867. |
[21] | Taniguchi S , McDonagh AM , Pickering-Brown SM , Umeda Y , Iwatsubo T , Hasegawa M , Mann DM ((2004) ) The neuropathology of frontotemporal lobar degeneration with respect to the cytological and biochemical characteristics of tau protein. Neuropathol Appl Neurobiol 30: , 1–18. |
[22] | Davidson Y , Kelley T , Mackenzie IR , Pickering-Brown S , Du Plessis D , Neary D , Snowden JS , Mann DM ((2007) ) Ubiquitinated pathological lesions in frontotemporal lobar degeneration contain the TAR DNA-binding protein, TDP-43. Acta Neuropathol 113: , 521–533. |
[23] | Bermingham N , Cowie TF , Paine M , Storey E , McLean C ((2008) ) Frontotemporal dementia and Parkinsonism linked to chromosome 17 in a young Australian patient with the G389R Tau mutation. Neuropathol Appl Neurobiol 34: , 366–370. |
[24] | Snowden JS , Neary D , Mann DM ((2004) ) Autopsy proven sporadic frontotemporal dementia due to microvacuolar-type histology, with onset at 21 years of age. J Neurol Neurosurg Psychiatry 75: , 1337–1339. |
[25] | Velakoulis D , Walterfang M , Mocellin R , Pantelis C , McLean C ((2009) ) Frontotemporal dementia presenting as schizophrenia-like psychosis in young people: Clinicopathological series and review of cases. Br J Psychiatry 194: , 298–305. |
[26] | Spina S , Murrell JR , Yoshida H , Ghetti B , Bermingham N , Sweeney B , Dlouhy SR , Crowther RA , Goedert M , Keohane C ((2007) ) The novel Tau mutation G335S: Clinical, neuropathological and molecular characterization.Acta Neuropathol 113: , 461–470. |
[27] | Tacik P , DeTure MA , Carlomagno Y , Lin WL , Murray ME , Baker MC , Josephs KA , Boeve BF , Wszolek ZK , Graff-Radford NR , Parisi JE , Petrucelli L , Rademakers R , Isaacson RS , Heilman KM , Petersen RC , Dickson DW , Kouri N ((2017) ) FTDP-17 with Pick body-like inclusions associated with a novel tau mutation, p.E372G. Brain Pathol 27: , 612–626. |
[28] | Baba Y , Baker MC , Le Ber I , Brice A , Maeck L , Kohlhase J , Yasuda M , Stoppe G , Bugiani O , Sperfeld AD , Tsuboi Y , Uitti RJ , Farrer MJ , Ghetti B , Hutton ML , Wszolek ZK ((2007) ) Clinical and genetic features of families with frontotemporal dementia and parkinsonism linked to chromosome 17 with a P301S tau mutation. J Neural Transm (Vienna) 114: , 947–950. |
[29] | Mackenzie IR , Feldman H ((2004) ) Neurofilament inclusion body disease with early onset frontotemporal dementia and primary lateral sclerosis. Clin Neuropathol 23: , 183–193. |
[30] | Neumann M , Diekmann S , Bertsch U , Vanmassenhove B , Bogerts B , Kretzschmar HA ((2005) ) Novel G335V mutation in the tau gene associated with early onset familial frontotemporal dementia. Neurogenetics 6: , 91–95. |
[31] | van Herpen E , Rosso SM , Serverijnen LA , Yoshida H , Breedveld G , van de Graaf R , Kamphorst W , Ravid R , Willemsen R , Dooijes D , Majoor-Krakauer D , Kros JM , Crowther RA , Goedert M , Heutink P , van Swieten JC ((2003) ) Variable phenotypic expression and extensive tau pathology in two families with the novel tau mutation L315R. Ann Neurol 54: , 573–581. |
[32] | Sperfeld AD , Collatz MB , Baier H , Palmbach M , Storch A , Schwarz J , Tatsch K , Reske S , Joosse M , Heutink P , Ludolph AC ((1999) ) FTDP-17: An early-onset phenotype with parkinsonism and epileptic seizures caused by a novel mutation. Ann Neurol 46: , 708–715. |
[33] | Baborie A , Jaros E , Griffiths TD , Momeni P , Perry R , Mann DM ((2012) ) Frontotemporal lobar degeneration in a very young patient is associated with fused in sma (FUS) pathological changes. Neuropathol Appl Neurobiol 38: , 101–104 arco. |
[34] | Stone J , Griffiths TD , Rastogi S , Perry RH , Cleland PG ((2003) ) Non-Picks frontotemporal dementia imitating schizophrenia in a 22-year-old man. J Neurol 250: , 369–370. |
[35] | Chu M , Liu L , Wang J , Liu L , Kong Y , Jing D , Xie K , Cui Y , Cui B , Zhang J , Ye H , Li J , Wang L , Rosa-Neto P , Gauthier S , Wu L ((2021) ) Investigating the roles of anterior cingulate in behavioral variant frontotemporal dementia: A PET/MRI study. J Alzheimers Dis 84: , 1771–1779. |
[36] | Onyike CU , Diehl-Schmid J ((2013) ) The epidemiology of frontotemporal dementia. Int Rev Psychiatry 25: , 130–137. |
[37] | Johnson JK , Diehl J , Mendez MF , Neuhaus J , Shapira JS , Forman M , Chute DJ , Roberson ED , Pace-Savitsky C , Neumann M , Chow TW , Rosen HJ , Forstl H , Kurz A , Miller BL ((2005) ) Frontotemporal lobar degeneration: Demographic characteristics of 353 patients. Arch Neurol 62: , 925–930. |
[38] | Sirkis DW , Geier EG , Bonham LW , Karch CM , Yokoyama JS ((2019) ) Recent advances in the genetics of frontotemporal dementia. Curr Genet Med Rep 7: , 41–52. |
[39] | Liu L , Cui B , Chu M , Cui Y , Jing D , Li D , Xie K , Kong Y , Xia T , Wang C , Wu L ((2021) ) The frequency of genetic mutations associated with behavioral variant frontotemporal dementia in Chinese Han patients. Front Aging Neurosci 13: , 699836. |
[40] | Seeley WW ((2019) ) Behavioral variant frontotemporal dementia. Continuum (Minneap Minn) 25: , 76–100. |
[41] | Leveille E , Ross OA , Gan-Or Z ((2021) ) Tau and MAPT genetics in tauopathies and synucleinopathies. Parkinsonism Relat Disord 90: , 142–154. |


