
Genetic Spectrum and Clinical Heterogeneity of Chinese Frontotemporal Dementia Patients: Data from PUMCH Dementia Cohort
Abstract
Background:
There are relatively few data on the genetic spectrum of Chinese frontotemporal dementia (FTD) population.
Objective:
With the dementia cohort of Peking Union Medical College Hospital, we aim to illustrate the genetic spectrum of FTD patients, as well as the phenotypic heterogeneity of FTD-gene variant carriers.
Methods:
204 unrelated, clinically diagnosed FTD patients of Chinese ancestry were enrolled. All the participants received demographic survey, history inquiry, physical examination, cognitive assessment, blood biochemical test, brain CT/MRI, and gene sequencing.
Results:
56.4% (115/204) participants were clinically diagnosed with behavioral variant of FTD, 20.6% (42/204) with nonfluent/agrammatic variant primary progressive aphasia (PPA), 20.1% (41/204) with semantic variant PPA, and 2.9% (6/204) with mixed variant PPA. 11.8% (24/204) subjects harbored the potential causative variants in FTD-related genes, including the MAPT (n = 7), TBK1 (n = 7), GRN (n = 2), TBK1+GRN (n = 1), VCP (n = 1), TARDBP (n = 1), UBQLN2 (n = 1), SQSTM1 (n = 1), DCTN1 (n = 1), HNRNPA1 (n = 1), and C9orf72 GGGGCC repeats (n = 1). The TBK1 T31fs, T457fs, K622fs, c.359-1G>A, the VCP P188T, and the GRN P50fs, P439fs were novel pathogenic/likely pathogenic variants. The TBK1 carriers showed a later disease onset and a higher incidence of parietal atrophy relative to the MAPTcarriers.
Conclusion:
There is genetic and clinical heterogeneity among Chinese FTD population. The TBK1 has a high mutation frequency in Chinese FTD patients.
INTRODUCTION
Frontotemporal dementia (FTD) represents a group of neurodegenerative diseases related to frontotemporal lobar degeneration. It is heterogeneous in phenotype, pathology, and genotype. The common phenotypes are behavioral variant of FTD (bvFTD) and primary progressive aphasia (PPA). The latter mainly comprises nonfluent/agrammatic variant PPA (nvPPA) and semantic variant PPA (svPPA). The common molecular bases include tau, TDP-43, and FUS proteins.
The MAPT, GRN, and C9orf72 are the main causative genes of FTD. However, the genetic variants in the MAPT, GRN, and C9orf72 can only account for about half of the familial FTD patients [1]. The previous studies reveal that some other genes are linked to FTD, including the VCP, CHCHD10, TBK1, CHMP2B, TARDBP, SQSTM1, FUS, UBQLN2, OPTN, TREM2, CYLD, PRKAR1B, TIA1, TUBA4A, CCNF, DCTN1, HNRNPA2B1, and HNRNPA1 [1, 2].
There are relatively few data on the genetic spectrum of Chinese FTD population. In 2021, Dr. Shen reviewed the 97 genetic studies of Chinese FTD population. 32 rare variants in the MAPT, GRN, C9orf72, CHCHD10, VCP, and TBK1 were found [3]. These mutations are definitely not sufficient to cover the genetic spectrum of Chinese FTD patients.
This is a retrospective study from the dementia cohort of Peking Union Medical College Hospital (PUMCH). Herein, we aim to expand the genetic reservoir of Chinese FTD population by interpreting the genetic spectrum in the clinically diagnosed FTD patients. In addition to the common MAPT, GRN, C9orf72, CHCHD10, VCP, and TBK1 genes, we also focus on some rare FTD-related genes, including the CHMP2B, TARDBP, SQSTM1, FUS, UBQLN2, OPTN, TREM2, CYLD, PRKAR1B, TIA1, TUBA4A, CCNF, DCTN1, HNRNPA2B1, and HNRNPA1. Furthermore, we aim to illustrate the phenotypic characteristics of the FTD-gene mutation carriers. These will help us to better understand the phenotypic and genotypic heterogeneity of FTD patients.
METHODS
Participant enrollment
204 unrelated participants of Chinese ancestry were enrolled from the PUMCH dementia cohort between 2007 and 2021. The inclusion criteria were as the following: 1) Intact data on demographic survey, history inquiry, physical examination, cognitive assessment, blood biochemical test, brain CT/MRI, and gene sequencing; 2) Met the clinical diagnostic criteria for probable bvFTD, nvPPA, svPPA, and mixed variant PPA (mvPPA) [4–7]; 3) Informed consent for participation in this research wasobtained.
This study was approved by ethics committee of PUMCH (No. JS-1836).
Gene sequencing and interpretation
The genomic DNA was extracted from the peripheral blood. The DNA libraries were sequenced on the Illumina platform (Illumina Inc., San Diego, CA, USA). The clean reads were aligned to the reference human genome (GRCh37/hg19) with the Burrows-Wheeler Aligner [8]. The annotation was performed with ANNOVAR (2017June8) [9]. The GGGGCC repeat expansion in the C9orf72 was determined by the triplet repeat primed PCR with capillary electrophoresis.
This report focused on the GGGGCC repeat expansion in the C9orf72, as well as the rare variants in the MAPT, GRN, CHMP2B, VCP, TARDBP, SQSTM1, FUS, UBQLN2, OPTN, CHCHD10, TBK1, TREM2, CYLD, PRKAR1B, TIA1, TUBA4A, CCNF, DCTN1, HNRNPA2B1, and HNRNPA1. The rare variants met the following: 1) The minor allele frequency <0.0001 in the 1000genome, ESP6500, and GnomAD databases; 2) Nonsynonymous mutations located in the coding region or±1/2 splice site; 3) Interpreted as pathogenic, likely pathogenic, or uncertain significance according to the standards of American College of Medical Genetics and Genomics (ACMG) [10].
Statistical analysis
The categorical variables were compared with the Chi-square or Fisher’s exact test. The continuous variables were assessed with the Analysis of Variance.
RESULTS
Cohort introduction
204 unrelated participants were enrolled. They were randomly recruited from different pedigrees all over China. 52.0% (106/204) were males and 48.0% (98/204) were females. 73.5% (150/204) subjects were early-onset (age of onset (AOO) <65 years old), while 26.5% (54/204) were late-onset (AOO ≥65 years old). 39.7% (81/204) subjects had a positive family history of dementia. That is, at least one of their first-degree or second-degree family members had dementia.
56.4% (115/204) subjects were clinically diagnosed with bvFTD, 20.6% (42/204) with nvPPA, and 20.1% (41/204) with svPPA. 2.9% (6/204) subjects showed both agrammatism and word comprehension impairment within two years of disease onset. They were diagnosed with mvPPA. Motor neuron disease (MND) coexisted in 2.5% (5/204) subjects, including three bvFTD, one nvPPA and one svPPA patient. None of the subjects developed schizophrenia, corticobasal syndrome or progressive supranuclear palsy.
The demographic data was shown in Table 1. The svPPA patients showed a higher frequency of sporadic cases than bvFTD, nvPPA, and mvPPA patients (80.5% versus 52.2%, 61.9%, 66.7%, p = 0.012). The gender, AOO, disease course, and APOE-ɛ4 status did not differ among the four subgroups.
Table 1
Demographics of the FTD participants
| FTD (n = 204) | bvFTD (n = 115) | nvPPA (n = 42) | svPPA (n = 41) | mvPPA (n = 6) | p* | |
| Male/Female | 106 (52.0)/98 (48.0) | 59 (51.3)/56 (48.7) | 20 (47.6)/22 (52.4) | 24 (58.5)/17 (41.5) | 3 (50.0)/3 (50.0) | 0.793 |
| Age (y) | 62.5±10.6 | 63.2±11.3 | 62.5±10.2 | 60.6±9.4 | 61.3±6.4 | 0.584 |
| AOO (y) | 59.1±10.1 | 59.7±10.7 | 59.2±9.9 | 57.9±9.3 | 57.3±5.9 | 0.778 |
| Disease course (y) | 3.3±2.6 | 3.6±2.7 | 3.3±3.1 | 2.7±1.4 | 4.0±2.0 | 0.490 |
| Early/late-onset n (%) | 150 (73.5)/54 (26.5) | 80 (69.6)/35 (30.4) | 31 (73.8)/11 (26.2) | 34 (82.9)/7 (17.1) | 5 (83.3)/1 (16.7) | 0.400 |
| FHD – /+ n(%) | 123 (60.3)/81 (39.7) | 60 (52.2)/55 (47.8) | 26 (61.9)/16 (38.1) | 33 (80.5)/8 (19.5) | 4 (66.7)/2 (33.3) | 0.012 |
| APOE ɛ4 – /+ n(%) | 156 (76.5)/48 (23.5) | 89 (77.4)/26 (22.6) | 30 (71.4)/12 (28.6) | 31 (75.6)/10 (24.4) | 6 (100.0)/0 (0.0) | 0.557 |
| PCV +/– (%) | 24 (11.8)/180 (88.2) | 7 (6.1)/108 (93.9) | 8 (19.0) /34 (81.0) | 9 (22.0)/32 (78.0) | 0 (0.0)/6 (100.0) | 0.016 |
FTD, frontotemporal dementia; bvFTD, behavioral variant of FTD; nvPPA, nonfluent/agrammatic variant primary progressive aphasia; svPPA, semantic variant primary progressive aphasia; mvPPA, mixed variant primary progressive aphasia; AOO, age of onset; FHD, family history of dementia; APOE, apolipoprotein E; PCV, potential causative variant; *The variables were compared among the bvFTD, nvPPA, svPPA, and mvPPA patients.
Mutation interpretation
20 rare variants in nine FTD-related genes were found (Table 2). According to the ACMG criteria, there were 11 pathogenic or likely pathogenic variants (PLPV) and nine variants of uncertain significance (VUS). Among the 11 PLPV, the MAPT L583V, P618L, the TBK1 T462fs and the TARDBP I383V were reported before [11–13]. Seven PLPV were novel, including the TBK1 T31fs, T457fs, K622fs, c.359-1G>A, the VCP P188T, and the GRN P50fs, P439fs. Among the nine VUS, the MAPT Q561P, SQSTM1 E362K, and UBQLN2 P500S were reported before [11, 14, 15]. Six VUS were novel, including the TBK1 T331N, M719V, R271W, the GRN T220I, the HNRNPA1 N50S and the DCTN1 R292H. No rare variants were found in the CHCHD10, CHMP2B, FUS, OPTN, TREM2, CYLD, PRKAR1B, TIA1, TUBA4A, CCNF, and HNRNPA2B1.
Table 2
Mutation interpretation
| Gene | Variant | 1000genome/ESP6500/GnomAD | SIFT/Polyphen/Mutationtaster | Interpretation | Clinvar | ACMG | Publication (PMID) |
| MAPT | L583V | -/-/- | D/D/A | PHA03247 domain | P | LP | 12509859 |
| P618L | -/-/- | D/D/D | PHA03247 domain | P | P | 9641683 | |
| Q561P | -/-/- | T/D/D | PHA03247 super family domain | – | VUS | 33006106 | |
| TBK1 | T457fs | -/-/- | TBK1_CCD1 domain | – | LP | – | |
| K622fs | -/-/- | TBK1_CCD1 domain | – | LP | – | ||
| c.359-1G>A | -/-/- | dbscsnv: 1.000 | – | P | – | ||
| T462fs | -/-/- | TBK1_CCD1 domain | – | P | 28008748 | ||
| T31fs | -/-/- | STKc_TBK1 domain | – | LP | – | ||
| T331N | -/-/- | D/P/D | Ubl_TBK1 domain | – | VUS | – | |
| M719V | -/-/- | T/B/N | TBK1_CCD1 domain | – | VUS | – | |
| R271W | -/-/- | D/B/D | STKc_TBK1 domain | – | VUS | – | |
| GRN | P439fs | -/-/- | – | LP | – | ||
| P50fs | -/-/- | – | LP | – | |||
| T220I | -/-/- | D/D/D | GRAN domain | – | VUS | – | |
| VCP | P188T | -/-/- | D/D/D | CDC48 super family domain | – | LP | – |
| TARDBP | I383V | -/-/0.00005 | T/B/N | C-terminal site | LP | LP | 18802454 |
| SQSTM1 | E362K | -/-/- | D/B/D | – | VUS | 31859009 | |
| UBQLN2 | P500S | -/-/- | T/B/D | – | VUS | 28716533 | |
| DCTN1 | R292H | -/-/- | D/B/D | Smc and Agg_substance | – | VUS | – |
| HNRNPA1 | N50S | -/-/- | T/B/D | RRM domain | – | VUS | – |
SIFT: D = deleterious, T = tolerated; Polyphen: D = probably damaging, P = possibly damaging, B = benign; Mutationtaster: A = disease_causing_automatic, D = disease_causing, N = polymorphism; Clinvar/ACMG: P = pathogenic, LP = likely pathogenic, VUS = variants of uncertain significance.
Mutation frequency
11.8% (24/204) participants harbored the potential causative variants in FTD-related genes. Of them, 3.5% (7/204) subjects had the MAPT variants, and 3.5% (7/204) had the TBK1 variants. The remaining 10 cases carried the rare variants in the GRN (n = 2, 1.0%), GRN+TBK1 (n = 1, 0.5%), VCP (n = 1, 0.5%), TARDBP (n = 1, 0.5%), UBQLN2 (n = 1, 0.5%), SQSTM1 (n = 1, 0.5%), DCTN1 (n = 1, 0.5%), and HNRNPA1 (n = 1, 0.5%), as well as GGGGCC repeats in the C9orf72 (n = 1, 0.5%) (Fig. 1).
Fig. 1
Genetic spectrum of the FTD participants.
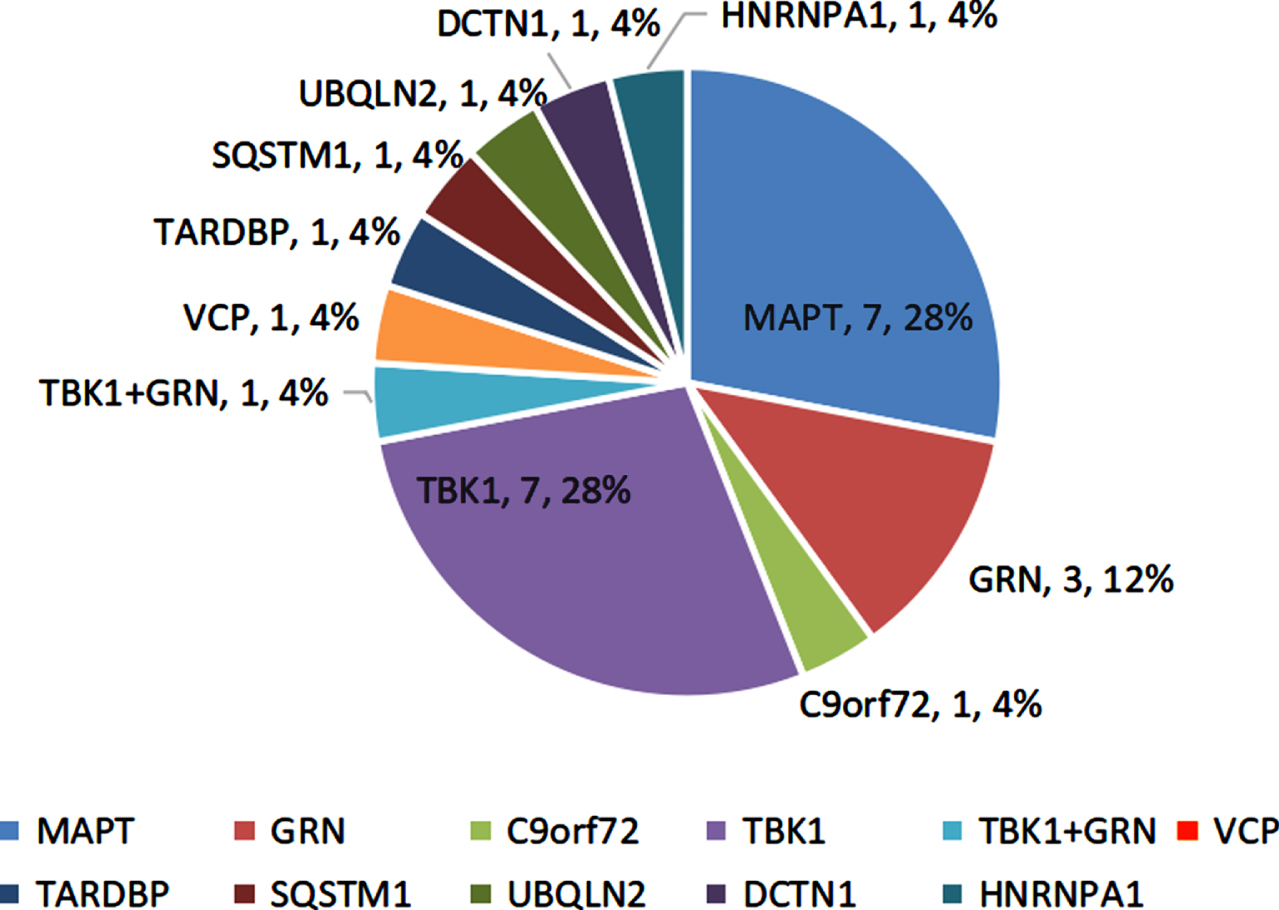
The mutation frequencies of the FTD-genes were higher in the early-onset and familial patients relative to the late-onset and sporadic subjects (14.0%, 16.0% versus 5.6%, 8.9%). The svPPA and nvPPA patients showed higher mutation frequencies than bvFTD patients (22.0%, 19.0% versus 6.1%).
Phenotype of FTD-gene variant carriers
The clinical characteristics of the 24 FTD-gene variant carriers are shown in Supplementary Table 1. Of them, 54.2% (13/24) subjects had a family history of dementia. 12.5% (3/24) had a family history of MND or schizophrenia.
29.2% (7/24) cases had behavioral/psychiatric symptom as their initial symptom. They were clinically diagnosed with bvFTD. 70.8% (17/24) subjects presented with language impairment in the early disease course. They were diagnosed with nvPPA (n = 8) and svPPA (n = 9), respectively.
During the disease progression, 88.9% (8/9) svPPA and 62.5% (5/8) nvPPA patients developed behavioral/psychiatric symptom, such as irritability, anxiety, depression, paranoid, apathy, loss of empathy, disinhibition, and stereotyped and compulsive behavior. Conversely, 28.6% (2/7) bvFTD patients showed expressive aphasia. Moreover, 28.6% (2/7) bvFTD, 37.5% (3/8) nvPPA, and 11.1% (1/9) svPPA subjects had pyramidal symptoms. 42.9% (3/7) bvFTD and 11.1% (1/9) svPPA subjects showed extrapyramidal symptoms. One bvFTD and one svPPA patient progressed into MND. 14.3% (1/7) bvFTD, 37.5% (3/8) nvPPA, and 77.8% (7/9) svPPA subjects developed memory deficit. In addition, urinary or fecal symptom was observed in 33.3% (8/24) subjects. Dietary change and sleep disorder were in 16.7% (4/24) and 25.0% (6/24) cases, respectively.
On structural imaging, the temporal and frontal atrophy were the most common, which occurred in 95.8% (23/24) and 62.5% (15/24) subjects, respectively. 16.7% (4/24) cases had parietal atrophy. 54.2% (13/24) subjects showed left hemisphere-predominant atrophy and 25.0% (6/24) showed bilateral symmetrical atrophy, while 20.8% (5/24) exhibited right hemisphere-predominant atrophy or hypometabolism.
Phenotype of TBK1 variant carriers
Five subjects harbored the PLPV in the TBK1. The TBK1 T457fs carrier started with stereotyped behavior at 50. In the past ten years, at four o’clock every morning, he got up and bicycled to the same restaurant five kilometers away from home and ordered the same food. Gradually, he developed apathy, loss of empathy, and expressive aphasia. CSF testing showed normal Aβ42 (746 pg/ml), p-tau (24pg/ml), and t-tau (150 pg/ml). The TBK1 T31fs carrier was characterized by an early-onset effortful speech with pyramidal sign. Brain MRI showed left temporal predominant atrophy (Fig. 2). The TBK1 K622fs carrier started with impaired naming and comprehension at 69. Brain MRI showed right temporal predominant atrophy (Fig. 2). PIB-PET was negative. The TBK1 c.359-1G>A carrier started with semantic aphasia at 54. His father had cognitive decline after cerebral hemorrhage in his 70 s. The TBK1 T462fs carrier started with semantic aphasia at 53. Two years later, she developed MND.
Fig. 2
Clinical data of the TBK1 variant carriers. A1-2) Sanger sequencing indicated the TBK1 p.K622fs (c.1866_1872del) mutation. Brain MRI showed right temporal predominant atrophy. B1-2) Sanger sequencing indicated the TBK1 p.T31fs (c.92delC). Brain MRI showed left temporal predominant atrophy with periventricular white matter hyperintensities, fazekas grade 2. A3, B3) Sanger sequencing indicated the TBK1 p.T457fs (c.1371_1372del), p.T462fs (c.1385_1388del). C1-3) Sanger sequencing indicated the TBK1 p.T331N (c.992C>A) mutation. Brain MRI showed bilateral frontal and parietal atrophy with mild left insular atrophy. II-d was the index patient. Her mother developed cognitive decline in her 80s and died ten years later. Her father died at 70 without cognitive impairment. Her cognitively healthy brother had a sudden death at 60.
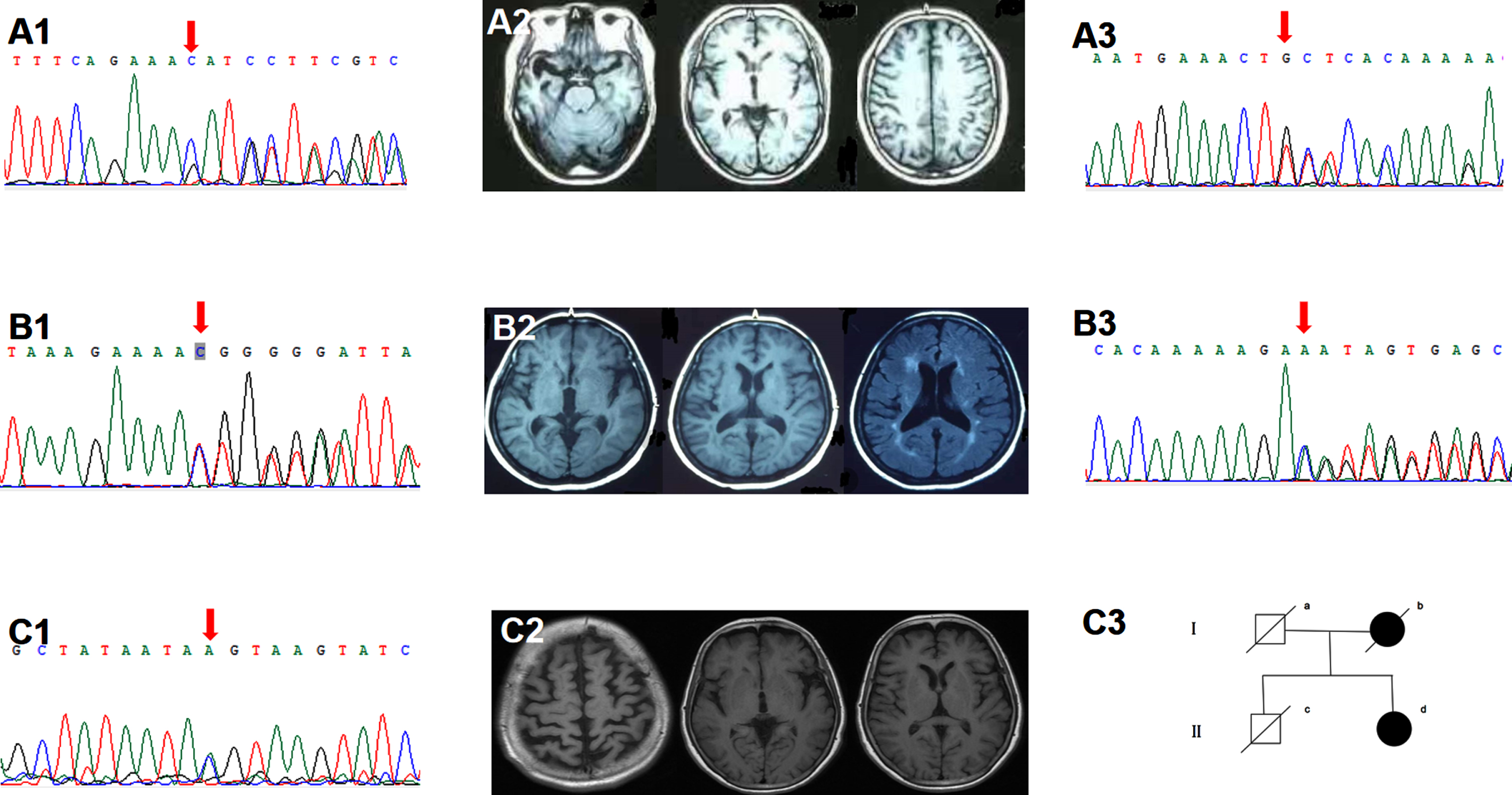
Two subjects carried the TBK1 VUS. The TBK1 T331N carrier started with an effortful speech at 54. Three years later, she developed personality change, dyscalculia, and gait instability. Physical examination suggested pyramidal involvement. Brain MRI showed bilateral frontal, parietal and left insular atrophy (Fig. 2). CSF testing indicated normal Aβ42 (1040 pg/ml) and elevated p-tau (57 pg/ml) and t-tau (243pg/ml). The TBK1 M719V carrier started with word-finding difficulty and agrammatism at 50. Three years later, she developed loss of empathy and stereotyped behavior. Brain FDG-PET showed left cerebral hypometabolism. Her brother suffered from MND.
Phenotype of other FTD-gene variant carriers
Seven subjects harbored the MAPT L583V, P618L, and Q561P. They were reported by our group before [8]. A 57-year-old female with the C9orf72 GGGGCC repeats presented with behavioral/psychiatric disturbance. The UBQLN2 P500S carrier started with apathy, loss of empathy, and dietary change at 57. One year later, she developed MND. Her two sisters suffered from schizophrenia. The TARDBP I383V carrier started with semantic aphasia at 53. He became a heavy smoker and drinker. He picked up the cigarette ends on the ground. His mother and brother presented with language impairment and psychotic disorder in their 40 s. The SQSTM1 E362K carrier started with semantic aphasia at 53. CSF testing indicated normal Aβ42 (666 pg/ml), elevated p-tau (58 pg/ml) and t-tau (430 pg/ml).
Six patients carried the novel variants in other FTD genes. The DCTN1 R292H carrier presented with semantic aphasia at 59. Brain FDG-PET showed left-temporal predominant hypometabolism. CSF testing indicated normal Aβ42 (659 pg/ml), p-tau (33 pg/ml), and t-tau (153 pg/ml). The HNRNPA1 N50S started with loss of empathy, apathy, paranoid, and repetitive behavior at 54. Gradually he developed parkinsonism. FDG-PET showed bilateral frontal hypometabolism. PIB-PET was negative. The VCP P188T carrier started with semantic aphasia at 57. Two years later, he developed expressive disorder. He collected rubbish in the public. The GRN P50fs carrier presented with non-fluent aphasia, while the GRN P439fs carrier exhibited semantic aphasia. The GRN T220I carrier also carried the TBK1 R271W. His mother had cognitive decline in her 70 s. His brother suffered from schizophrenia. He showed loss of empathy, disinhibition, and compulsive behavior at 71.
Comparisons between MAPT and TBK1 variant carriers
Compared with the MAPT patients, the TBK1 variant carriers showed an older AOO (55.0±6.0 versus 46.1±16.2) and a higher proportion of sporadic cases (71.4% (5/7) versus 14.3% (1/7)). On structural MRI, the TBK1 carriers showed less frontal but more parietal involvement relative to the MAPT carriers (42.9% (3/7) versus 100.0% (7/7); 42.9% (3/7) versus 0.0% (0/7)). Accordingly, 42.9% (3/7) of the TBK1 variant carriers had parietal damage associated symptom, such as dyscalculia, visuospatial dysfunction.
DISCUSSION
In this study, the FTD-gene mutations are detected in 11.8% (24/204) FTD patients. These is close to the previous findings. In a Chinese south cohort, 10.9% FTD patients harbored a pathogenic/likely pathogenic variant [16]. Moreover, we find that svPPA and nvPPA patients have higher mutation frequencies than bvFTD patients (22.0%, 19.0% versus 6.1%).
Like the previous genetic studies of Chinese FTD population, the rare variants in the MAPT, GRN, TBK1, VCP, and the GGGGCC repeats in the C9orf72 are found in this study. In addition, the rare variants in the TARDBP, UBQLN2, SQSTM1, DCTN1, and HNRNPA1 are involved in the mutation spectrum.
Seven novel PLPV are found. The TBK1 T31fs, T457fs, K622fs, c.359-1G>A and the GRN P50fs, P439fs are frameshift and splicing mutations. The VCP P188T is a missense mutation in the cdc48_2 domain. They are absent in the 1000genome, ESP6500, and GnomAD databases. We reviewed the mutation distribution of the TBK1, GRN, and VCP in the clinvar database (https://www.ncbi.nlm.nih.gov/clinvar). 60.4% (29/48) TBK1 and 75.8% (75/99) GRN PLPV are frameshift, splicing, or stopgain mutations. 57.7% (15/26) VCP missense PLPV are located in the cdc48_2 domain. According to the ACMG criteria, the seven variants are novel PLPV.
The MAPT, GRN, and C9orf72 account for half of familial FTD cases. Among 58 PPA subjects with autopsies, Mesulam found genetic mutations only in the GRN. However, in this study, the TBK1 shows a high mutation frequency. It is supposed to be one gene frequently harboring FTD-causing variants after the MAPT, GRN, and C9orf72 [17–19]. FTD and MND are the most prevalent phenotypes [19]. Herein, the seven TBK1 variant carriers present with bvFTD or PPA. One case develops MND. Compared with the MAPT carriers, the TBK1 carriers show a later disease onset, as well as a higher incidence of parietal atrophy and parietal damage associatedsymptoms.
The DCTN1 mutations are mainly linked to Perry syndrome. The HNRNPA1 mutations are primarily detected in the subjects with inclusion body myopathy, Paget disease. They also account for a small percentage of FTD or MND cases [20, 21]. In this report, the DCTN1 R292H carrier presents with semantic aphasia and psychiatric symptom. The HNRNPA1 N50S carrier shows behavioral/psychiatric symptom and parkinsonism. Neither of them developed MND.
The TARDBP I383V, UBQLN2 P500S, and SQSTM1 E362K were reported in familial MND patients before [12, 14, 15]. In this study, the UBQLN2 P500S carrier presents with bvFTD and MND. The TARDBP I383V and SQSTM1 E362K are in two svPPA patients. The MAPT L583V, P618L, and Q561P were reported by our group. Dr. Mao described the phenotypic heterogeneity of the MAPT variant carriers [11]. Taken together, our results suggest the phenotypic heterogeneity of these variants.
Of the 24 FTD-gene variant carriers, 54.2% (13/24) subjects have a family history of dementia. In addition, the TBK1 M719V carrier has a family history of MND. The UBQLN2 P500S and the TBK1 R271W/GRN T220I carriers have a family history of schizophrenia. MND and FTD belong to a neurodegenerative disease spectrum. They have some common genetic and pathological bases [22]. FTD and schizophrenia may also have some genetic links in common [23]. The previous studies have confirmed the association between the GRN mutations and schizophrenia [24]. The relevance between the UBQLN2, TBK1, and schizophrenia is unknown.
Compared with nvPPA patients, svPPA cases present with more behavioral/psychiatric and memory symptoms (88.9% (8/9), 77.8% (7/9) versus 62.5% (5/8), 37.5% (3/8)), as well as less pyramidal symptoms (11.1% (1/9) versus 37.5% (3/8)). These are similar to Ulugut’s findings [25] Moreover, we find that bvFTD subjects show more motor symptoms relative to PPA cases (71.4% (5/7) versus 29.4% (5/17)).
Mesulam noted that the most distinctive feature of PPA on structural imaging was left hemisphere-predominant atrophy [26]. However, in this study, the TBK1 K622fs, the GRN P439fs and the SQSTM1 E362K carriers exhibit right hemisphere-predominant atrophy or hypometabolism. They are all right-handed and present with semantic aphasia, behavioral/psychiatric symptoms, and memory deficit. Ulugut found that 11.3% FTD patients showed right temporal-predominant atrophy [27]. These patients might not be purely TDP type C pathology [28].
In conclusion, this study further confirms the genetic and clinical heterogeneity of Chinese FTD population. The TBK1 has a high mutation frequency in Chinese FTD patients. The main limitation of this study is the lack of neuropathological confirmation. Functional analysis of the novel rare variants should be performed next. Besides, the subjects in this paper are all Chinese native speakers. However, the PPA diagnostic criteria we used are from English-speaking population. Unlike English, Chinese is a hieroglyphic language. In writing, Chinese PPA patients might show character structural errors instead of letter omissions. Therefore, the diagnostic criteria for Chinese PPA population should be established in thefuture.
ACKNOWLEDGMENTS
Dr. Jing Gao was supported by grants from National Key Research and Development Program of China (2020YFA0804500, 2016YFC1306300), CAMS Innovation fund for Medical Sciences (2016-I2M-1-004), National Natural Science Foundation of China (81550021, 30470618).
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/22-0594r2).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-220594.
REFERENCES
[1] | Rainero I , Rubino E , Michelerio A , D’Agata F , Gentile S , Pinessi L ((2017) ) Recent advances in the molecular genetics of frontotemporal lobar degeneration. Funct Neurol 32: , 7–16. |
[2] | Guerreiro R , Gibbons E , Tabuas-Pereira M , Kun-Rodrigues C , Santo GC , Bras J ((2020) ) Genetic architecture of common non-Alzheimer’s disease dementias. Neurobiol Dis 142: , 104946. |
[3] | Jiang Y , Jiao B , Xiao X , Shen L ((2021) ) Genetics of frontotemporal dementia in China. Amyotroph Lateral Scler Frontotemporal Degener 22: , 321–335. |
[4] | Neary D , Snowden JS , Gustafson L , Passant U , Stuss D , Black S , Freedman M , Kertesz A , Robert PH , Albert M , Boone K , Miller BL , Cummings J , Benson DF ((1998) ) Frontotemporal lobar degeneration: A consensus on clinical diagnostic criteria. Neurology 51: , 1546–1554. |
[5] | Rascovsky K , Hodges JR , Knopman D , Mendez MF , Kramer JH , Neuhaus J , van Swieten JC , Seelaar H , Dopper EG , Onyike CU , Hillis AE , Josephs KA , Boeve BF , Kertesz A , Seeley WW , Rankin KP , Johnson JK , Gorno-Tempini ML , Rosen H , Prioleau-Latham CE , Lee A , Kipps CM , Lillo P , Piguet O , Rohrer JD , Rossor MN , Warren JD , Fox NC , Galasko D , Salmon DP , Black SE , Mesulam M , Weintraub S , Dickerson BC , Diehl-Schmid J , Pasquier F , Deramecourt V , Lebert F , Pijnenburg Y , Chow TW , Manes F , Grafman J , Cappa SF , Freedman M , Grossman M , Miller BL ((2011) ) Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 134: , 2456–2477. |
[6] | Gorno-Tempini ML , Hillis AE , Weintraub S , Kertesz A , Mendez M , Cappa SF , Ogar JM , Rohrer JD , Black S , Boeve BF , Manes F , Dronkers NF , Vandenberghe R , Rascovsky K , Patterson K , Miller BL , Knopman DS , Hodges JR , Mesulam MM , Grossman M ((2011) ) Classification of primary progressive aphasia and its variants. Neurology 76: , 1006–1014. |
[7] | Mesulam MM , Wieneke C , Thompson C , Rogalski E , Weintraub S ((2012) ) Quantitative classification of primary progressive aphasia at early and mild impairment stages. Brain 135: , 1537–1553. |
[8] | Li H , Durbin R ((2010) ) Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26: , 589–595. |
[9] | Wang K , Li M , Hakonarson H ((2010) ) ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38: , e164. |
[10] | Richards S , Aziz N , Bale S , Bick D , Das S , Gastier-Foster J , Grody WW , Hegde M , Lyon E , Spector E , Voelkerding K , Rehm HL ((2015) ) Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: , 405–424. |
[11] | Mao C , Dong L , Li J , Huang X , Lei D , Wang J , Chu S , Liu C , Peng B , Cui L , Gao J ((2021) ) Phenotype heterogeneity and genotype correlation of MAPT mutations in a Chinese PUMCH Cohort. J Mol Neurosci 71: , 1015–1022. |
[12] | Rutherford NJ , Zhang YJ , Baker M , Gass JM , Finch NA , Xu YF , Stewart H , Kelley BJ , Kuntz K , Crook RJ , Sreedharan J , Vance C , Sorenson E , Lippa C , Bigio EH , Geschwind DH , Knopman DS , Mitsumoto H , Petersen RC , Cashman NR , Hutton M , Shaw CE , Boylan KB , Boeve B , Graff-Radford NR , Wszolek ZK , Caselli RJ , Dickson DW , Mackenzie IR , Petrucelli L , Rademakers R ((2008) ) Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. Plos Genet 4: , e1000193. |
[13] | van der Zee J , Gijselinck I , Van Mossevelde S , Perrone F , Dillen L , Heeman B , Baumer V , Engelborghs S , De Bleecker J , Baets J , Gelpi E , Rojas-Garcia R , Clarimon J , Lleo A , Diehl-Schmid J , Alexopoulos P , Perneczky R , Synofzik M , Just J , Schols L , Graff C , Thonberg H , Borroni B , Padovani A , Jordanova A , Sarafov S , Tournev I , de Mendonca A , Miltenberger-Miltenyi G , Simoes DCF , Ramirez A , Jessen F , Heneka MT , Gomez-Tortosa E , Danek A , Cras P , Vandenberghe R , De Jonghe P , De Deyn PP , Sleegers K , Cruts M , Van Broeckhoven C , Goeman J , Nuytten D , Smets K , Robberecht W , Damme PV , Bleecker J , Santens P , Dermaut B , Versijpt J , Michotte A , Ivanoiu A , Deryck O , Bergmans B , Delbeck J , Bruyland M , Willems C , Salmon E , Pastor P , Ortega-Cubero S , Benussi L , Ghidoni R , Binetti G , Hernandez I , Boada M , Ruiz A , Sorbi S , Nacmias B , Bagnoli S , Sorbi S , Sanchez-Valle R , Llado A , Santana I , Rosario AM , Frisoni GB , Maetzler W , Matej R , Fraidakis MJ , Kovacs GG , Fabrizi GM , Testi S ((2017) ) TBK1 mutation spectrum in an extended European patient cohort with frontotemporal dementia and amyotrophic lateral sclerosis. Hum Mutat 38: , 297–309. |
[14] | Yilmaz R , Muller K , Brenner D , Volk AE , Borck G , Hermann A , Meitinger T , Strom TM , Danzer KM , Ludolph AC , Andersen PM , Weishaupt JH ((2020) ) SQSTM1/p62 variants in 486 patients with familial ALS from Germany and Sweden. Neurobiol Aging 87: , 139. |
[15] | Teyssou E , Chartier L , Amador MD , Lam R , Lautrette G , Nicol M , Machat S , Da BS , Moigneu C , Mairey M , Larmonier T , Saker S , Dussert C , Forlani S , Fontaine B , Seilhean D , Bohl D , Boillee S , Meininger V , Couratier P , Salachas F , Stevanin G , Millecamps S ((2017) ) Novel UBQLN2 mutations linked to amyotrophic lateral sclerosis and atypical hereditary spastic paraplegia phenotype through defective HSP70-mediated proteolysis. Neurobiol Aging 58: , 211–239. |
[16] | Jiao B , Liu H , Guo L , Xiao X , Liao X , Zhou Y , Weng L , Zhou L , Wang X , Jiang Y , Yang Q , Zhu Y , Zhou L , Zhang W , Wang J , Yan X , Li J , Tang B , Shen L ((2021) ) The role of genetics in neurodegenerative dementia: A large cohort study in South China. NPJ Genom Med 6: , 69. |
[17] | Freischmidt A , Muller K , Ludolph AC , Weishaupt JH , Andersen PM ((2017) ) Association of mutations inwith sporadic and familial amyotrophic lateral sclerosis and frontotemporal dementia. JAMA Neurol 74: , 110–113. |
[18] | Guerreiro R , Ross OA , Kun-Rodrigues C , Hernandez DG , Orme T , Eicher JD , Shepherd CE , Parkkinen L , Darwent L , Heckman MG , Scholz SW , Troncoso JC , Pletnikova O , Ansorge O , Clarimon J , Lleo A , Morenas-Rodriguez E , Clark L , Honig LS , Marder K , Lemstra A , Rogaeva E , St GP , Londos E , Zetterberg H , Barber I , Braae A , Brown K , Morgan K , Troakes C , Al-Sarraj S , Lashley T , Holton J , Compta Y , Van Deerlin V , Serrano GE , Beach TG , Lesage S , Galasko D , Masliah E , Santana I , Pastor P , Diez-Fairen M , Aguilar M , Tienari PJ , Myllykangas L , Oinas M , Revesz T , Lees A , Boeve BF , Petersen RC , Ferman TJ , Escott-Price V , Graff-Radford N , Cairns NJ , Morris JC , Pickering-Brown S , Mann D , Halliday GM , Hardy J , Trojanowski JQ , Dickson DW , Singleton A , Stone DJ , Bras J ((2018) ) Investigating the genetic architecture of dementia with Lewy bodies: A two-stage genome-wide association study. Lancet Neurol 17: , 64–74. |
[19] | Helgason E , Phung QT , Dueber EC ((2013) ) Recent insights into the complexity of Tank-binding kinase 1 signaling networks: The emerging role of cellular localization in the activation and substrate specificity of TBK1. FEBS Lett 587: , 1230–1237. |
[20] | Dulski J , Konno T , Wszolek Z ((1993) ) DCTN1-related neurodegeneration. In GeneReviews®, AdamMP, MirzaaGM, PagonRA, WallaceSE, BeanLJH, GrippKW, AmemiyaA, eds. University of Washington, Seattle. |
[21] | Kimonis V ((1993) ) Inclusion body myopathy with paget disease of bone and/or frontotemporal dementia. In GeneReviews®, AdamMP, MirzaaGM, PagonRA, WallaceSE, BeanLJH, GrippKW, AmemiyaA, eds. University of Washington, Seattle. |
[22] | Lillo P , Matamala JM , Valenzuela D , Verdugo R , Castillo JL , Ibanez A , Slachevsky A ((2014) ) Overlapping features of frontotemporal dementia and amyotrophic lateral sclerosis. Rev Med Chil 142: , 867–879. |
[23] | Harciarek M , Malaspina D , Sun T , Goldberg E ((2013) ) Schizophrenia and frontotemporal dementia: Shared causation? Int Rev Psychiatry 25: , 168–177. |
[24] | Galimberti D , Dell’Osso B , Fenoglio C , Villa C , Cortini F , Serpente M , Kittel-Schneider S , Weigl J , Neuner M , Volkert J , Leonhard C , Olmes DG , Kopf J , Cantoni C , Ridolfi E , Palazzo C , Ghezzi L , Bresolin N , Altamura AC , Scarpini E , Reif A ((2012) ) Progranulin gene variability and plasma levels in bipolar disorder and schizophrenia. PLoS One 7: , e32164. |
[25] | Ulugut H , Stek S , Wagemans L , Jutten RJ , Keulen MA , Bouwman FH , Prins ND , Lemstra AW , Krudop W , Teunissen CE , van Berckel B , Ossenkoppele R , Barkhof F , van der Flier WM , Scheltens P , Pijnenburg Y ((2022) ) The natural history of primary progressive aphasia: Beyond aphasia. J Neurol 269: , 1375–1385. |
[26] | Mesulam MM , Weintraub S , Rogalski EJ , Wieneke C , Geula C , Bigio EH ((2014) ) Asymmetry and heterogeneity of Alzheimer’s and frontotemporal pathology in primary progressive aphasia. Brain 137: , 1176–1192. |
[27] | Ulugut EH , Groot C , Heilbron R , Nelissen A , van Rossum J , Jutten R , Koene T , van der Flier WM , Wattjes MP , Scheltens P , Ossenkoppele R , Barkhof F , Pijnenburg Y ((2020) ) A clinical-radiological framework of the right temporal variant of frontotemporal dementia. Brain 143: , 2831–2843. |
[28] | Ulugut H , Dijkstra AA , Scarioni M , Barkhof F , Scheltens P , Rozemuller A , Pijnenburg Y ((2021) ) Right temporal variant frontotemporal dementia is pathologically heterogeneous: A case-series and a systematic review. Acta Neuropathol Commun 9: , 131. |


