
Associations Between Sub-Threshold Amyloid-β Deposition, Cortical Volume, and Cognitive Function Modulated by APOE ɛ4 Carrier Status in Cognitively Normal Older Adults
Abstract
Background:
There has been renewed interest in the deteriorating effects of sub-threshold amyloid-β (Aβ) accumulation in Alzheimer’s disease (AD). Despite evidence suggesting a synergistic interaction between the APOE ɛ4 allele and Aβ deposition in neurodegeneration, few studies have investigated the modulatory role of this allele in sub-threshold Aβ deposition during the preclinical phase.
Objective:
We aimed to explore the differential effect of the APOE ɛ4 carrier status on the association between sub-threshold Aβ deposition, cortical volume, and cognitive performance in cognitively normal older adults (CN).
Methods:
A total of 112 CN with sub-threshold Aβ deposition was included in the study. Participants underwent structural magnetic resonance imaging, [18F] flutemetamol PET-CT, and a neuropsychological battery. Potential interactions between APOE ɛ4 carrier status, Aβ accumulation, and cognitive function for cortical volume were assessed with whole-brain voxel-wise analysis.
Results:
We found that greater cortical volume was observed with higher regional Aβ deposition in the APOE ɛ4 carriers, which could be attributed to an interaction between the APOE ɛ4 carrier status and regional Aβ deposition in the posterior cingulate cortex/precuneus. Finally, the APOE ɛ4 carrier status-neuropsychological test score interaction demonstrated a significant effect on the gray matter volume of the left middle occipital gyrus.
Conclusion:
There might be a compensatory response to initiating Aβ in APOE ɛ4 carriers during the earliest AD stage. Despite its exploratory nature, this study offers some insight into recent interests concerning probabilistic AD modeling, focusing on the modulating role of the APOE ɛ4 carrier status during the preclinical period.
INTRODUCTION
Alzheimer’s disease (AD) is distinguished from other neurodegenerative diseases by amyloid-β (Aβ) deposition [1]. This Aβ accumulation has been shown to nonlinearly increase decades before the first clinical symptoms in AD [2]. In this regard, there is a period during which only Aβ exists without overt clinical symptoms, and this period has been defined as the preclinical AD phase [3]. In preclinical AD patients, the rate of conversion to mild cognitive impairment (MCI) and dementia is five times higher than that of the control group without Aβ [4]. In addition, because this period is attracting attention as a suitable time to perform a primary intervention, several attempts to identify Aβ pathology have been carried out through in vivo brain imaging, such as Aβ-positron emission tomography (PET) [5].
However, questions surrounding the clinical implication of Aβ accumulation below the threshold for a Aβ-PET positive scan have recently resurfaced [6]. Previous postmortem study has demonstrated that 65% of individuals with a negative [18F] flutemetamol (FMM) PET scan display an early autopsy stage of Aβ accumulation, and 15% exhibit an advanced stage [7]. Additionally, Aβ has shown a regional spreading pattern in the accumulating trajectory even during the preclinical phase [8], and specific brain regions relevant to the default-mode neural network have been demonstrated to be prone to early Aβ deposits [9]. Furthermore, even in cognitively unimpaired older adults, sub-threshold Aβ deposits were related to cognitive and functional decline [10] and further accumulation of Aβ and tau pathology [11, 12].
Brain cortical atrophy, an early neurodegenerative biomarker during preclinical phase [13], predicts further cognitive decline [13] and is correlated with soluble as well as fibrillar Aβ accumulation [14, 15]. Moreover, a previous study has found a differential atrophy pattern between positive and negative Aβ scans in the CN group [16].
The APOE genotype has been demonstrated to perform a modulatory role in the penetrance of the Aβ-dependent pathway and the effect of environmental factors and other low-risk genes in sporadic AD [17]. Among APOE genotypes, the APOE ɛ4 allele has been shown to increase Aβ production [18], decrease Aβ clearance [19], and contribute to the increased risk of AD in a dose-dependent manner [20]. These APOE ɛ4 fragments have also been reported to interact synergistically with AD pathology, deteriorating the degree of neurodegeneration [21, 22]. In this regard, previous AD studies have reported that APOE ɛ4 carrier status affects medial temporal lobe atrophy as well as that of the frontal and parietal lobes in a dose-dependent manner [23–25]. However, other research findings pertinent to APOE ɛ4 have been inconsistent, demonstrating no significant difference in cortical volume compared with other APOE genotypes in AD patients [26, 27]. In addition, APOE ɛ4 carriers in the CN group have displayed accelerating hippocampus atrophy [28] but modest cortical brain region atrophy [23]. However, in another study within CN subjects, the APOE ɛ4 allele has been associated with faster cortical thinning in AD-related cortical brain regions [29].
Despite the synergistic interaction between APOE ɛ4 allele and Aβ deposition for the neurodegeneration [22], most aforementioned studies did not evaluate the presence of Aβ deposits. Moreover, although sub-threshold Aβ accumulation has clinical implications during the earliest phase of AD, few studies have explored interactions with APOE ɛ4 allele for cortical atrophy in patients with sub-threshold-level Aβ deposition. In addition, given that the predefined signature regions of AD tend not to exhibit atrophy during the preclinical phase [10], there might be undetermined effects in other brain regions, as many previous studies have only evaluated cortical atrophy in predefined brain regions. Furthermore, in the preclinical phase, although sub-threshold Aβ shows a regional spreading pattern [8] that has been a more accurate predictor of cognitive decline than global Aβ [30], previous studies have mainly focused on the global accumulation.
In this regard, the present study aimed to evaluate the differential impact of the APOE ɛ4 carrier status on the association between sub-threshold Aβ deposition, cortical volume, and cognitive performance in preclinical phase AD patients. Additionally, the present study assessed the effect of the APOE ɛ4 carrier status on cortical volume by whole-brain analysis. Furthermore, the study examined the impact of subtle changes in global and regional Aβ deposition and evaluated both memory performance and executive functions affected during the earliest trajectory of AD.
MATERIALS AND METHODS
Participants
Subjects were recruited from volunteers registered in the Catholic Aging Brain Imaging database, which contains brain scans of patients who visited the outpatient clinic at the Catholic Brain Health Center, Yeouido St. Mary’s Hospital, The Catholic University of Korea, from 2017 to 2021. In all 2,219 subjects, cognitive function was assessed using the Korean version of the Consortium to Establish a Registry for AD (CERAD-K) [31]. The measurements included assessments regarding the Korean version of the verbal fluency (VF) test, the 15-item Boston Naming Test, Mini-Mental State Examination (MMSE-K) [32], word list memory (WLM), word list recall (WLR), word list recognition (WLRc), constructional praxis, and constructional recall. In addition, total memory (TM) scores were obtained by summing the respective z-scores from the WLM, WLR, and WLRc tests. The total CERAD-K scores were calculated by summing all subcategory z-scores, excluding the MMSE-K and constructional recall scores. Additionally, the Stroop Word-Color Interference test, Trail Making Test B, and VF test were used to assess executive function [33, 34]. Higher Trail Making Test B scores mean lower executive function. To calculate the executive function composites, we summed respective z-scores of the Stroop Word-Color Interference test, VF test, and inverse value of Trail Making Test B. Details regarding the use of specific tests and the reviewing process are described in the Supplementary Material. The inclusion criteria were as follows: 1) unimpaired memory function, quantified by scoring above age-, sex-, and education-adjusted cut-offs on the WLM, WLR, and WLRc domains; 2) MMSE-K score between 24 and 30; 3) Clinical Dementia Rating score of 0; 4) Memory Box score of 0; 5) normal cognitive function based on the absence of significant impairment in cognitive function or activities of daily living; and 6) no family history of AD. We excluded participants with a history of alcoholism, drug abuse, head trauma, or psychiatric disorders and those taking any psychotropic medications (e.g., cholinesterase inhibitors, antidepressants, benzodiazepines, and antipsychotics), those with uncontrolled multiple cardiovascular risk factors (e.g., uncontrolled arterial hypertension, diabetes mellitus, dyslipidemia, cardiac disease including coronary heart disease, arrhythmia, etc.), and those with evidence of subcortical ischemic changes corresponding to a score ≥2 on the Fazeka’s scale [35]. T2-weighted fluid-attenuated inversion recovery data were acquired to objectively exclude vascular lesions or other diseases. Participants underwent FMM PET-CT within 3 months of the magnetic resonance imaging (MRI) scan and those with positive FMM PET scan were excluded. The procedures for APOE genotyping are described in the Supplementary Material. Considering the protective effect of APOE ɛ2 allele [36], we excluded participants with the APOE ɛ2 allele (Fig. 1). If a participant had at least one APOE ɛ4 allele, they were categorized as an APOE ɛ4 carrier; if they had no APOE ɛ4 allele, they were categorized as an APOE ɛ4 non-carrier. The study was conducted under the ethical and safety guidelines set forth by the Institutional Review Board of The Catholic University of Korea, which approved all research activities (SC18TESI0143). Written informed consent was obtained from all participants.
Fig. 1
Flow chart describing the selection process of the participants in our cohort study. *Exclusion Criteria: a history of alcoholism, drug abuse, head trauma, or psychiatric disorders and those taking any psychotropic medications; uncontrolled multiple cardiovascular risk factors; evidence of subcortical ischemic changes corresponding to a score ≥2 on the Fazeka’s scale. FMM, [18F] flutemetamol.
![Flow chart describing the selection process of the participants in our cohort study. *Exclusion Criteria: a history of alcoholism, drug abuse, head trauma, or psychiatric disorders and those taking any psychotropic medications; uncontrolled multiple cardiovascular risk factors; evidence of subcortical ischemic changes corresponding to a score ≥2 on the Fazeka’s scale. FMM, [18F] flutemetamol.](https://content.iospress.com:443/media/jad/2022/89-3/jad-89-3-jad220427/jad-89-jad220427-g001.jpg)
Structural MRI data acquisition
Imaging data were collected by the Yeouido Saint Mary’s Hospital Department of Radiology at The Catholic University of Korea using a 3T Siemens Skyra MRI machine and a 32-channel Siemens head coil (Siemens Medical Solutions, Erlangen, Germany). The parameters used for the T1-weighted volumetric magnetization-prepared rapid gradient echo scan sequences were TE = 2.6 ms, TR = 1,940 ms, inversion time = 979 ms, FOV = 230 mm, matrix = 256×256, and voxel size = 1.0×1.0×1.0 mm3.
Structural MRI preprocessing
Image preprocessing was conducted using SPM12 (Wellcome Trust Centre for Neuroimaging, London, UK;http://www.fil.ion.ucl.ac.uk/spm) using the CAT12 toolbox (http://www.neuro.uni-jena.de/cat/). We utilized an optimized voxel-based morphometry (VBM) process [37] that included 1) segmentation and extraction of the brain in native space, 2) normalization of the images to a Montreal Neurological Institute (MNI) standard space, 3) segmentation and extraction of the normalized brain (extraction was repeated to ensure that no non-brain tissue remained); 4) modulation of the normalized images to correct for tissue volume differences due to the normalization procedure, and 5) sample homogeneity evaluation to identify any outliers in the study population. The gray matter probability values were smoothed using an 8-mm full-width half-maximum isotropic Gaussian kernel. The smoothed gray matter images were used for statistical analysis using SPM12.
[18F]-flutemetamol PET image acquisition and processing
FMM was manufactured, and FMM-PET data were collected and analyzed as described previously [5]. Static PET scans were acquired from 90 to 110 min after 185 MBq of FMM injection. MRI for each participant was used to co-register and define the ROIs and correct partial volume effects that arose from expansion of the cerebrospinal spaces accompanying cerebral atrophy using a geometric transfer matrix.
SUVR calculation
The semi-quantification of FMM uptake on PET/CT scan was performed by obtaining the standardized uptake value ratios (SUVRs). The volumes of interest (VOIs) were restricted to gray matter, covering the frontal, superior parietal, lateral temporal, anterior, and posterior cingulate cortex/precuneus regions. These VOIs were also considered in a previous study [5]. The reference region for SUVR calculations was pons. The mean uptake counts of each VOIs and reference region were measured on the preprocessed image. A regional SUVR was calculated as the ratio of each cortical regional mean count to the pons mean count (SUVRPONS). The global cortical average (composite SUVR) was calculated by averaging regional cortical SUVRs weighted for size. We used a cut-off of 0.62 for “positive” versus ‘sub-threshold’ neocortical SUVR, consistent with the cut-off values used in a previous FMM PET study [5]. PET scans classified with sub-threshold Aβ accumulation also exhibited normal visual reading.
Statistical analysis
Statistical analyses were performed using R software (version 4.0.5), jamovi (version 1.6.23) (https://www.jamovi.org), and SPM 12. Assumptions of normality were tested for continuous variables using the Kolmogorov–Smirnov test in R software; all data demonstrated a normal distribution. The two-sample t-test and chi-square (χ2) tests were used to probe for differences in demographic variables, clinical data, regional and global Aβ deposition, and cognitive function between APOE ɛ4 carriers and non-carriers in each CN sub-Aβ group. All statistical analyses were conducted considering a two-tailed p-value < 0.05 to define statistical significance.
Structural image analysis was performed using SPM12 (Wellcome Trust Centre for Neuroimaging, London, UK; http://www.fil.ion.ucl.ac.uk/spm). To compare the difference in the cortical volume depending on the APOE ɛ4 carrier status, ANCOVA on a voxel-by-voxel basis was carried out between the APOE ɛ4 carriers and non-carriers on the individual smoothed gray matter images. Age, sex, years of education, and total intracranial volume were included as covariates in the statistical tests. Additionally, a general linear model (GLM, a flexible factorial model in SPM12) based on whole-brain analysis was performed to evaluate the impact of Aβ deposits-APOE ɛ4 carrier status interaction on cortical volume using the smoothed gray matter images in the CN sub-Aβ group. APOE ɛ4 carrier status, regional, and global FMM SUVRPONS were the independent variables. We controlled for the effects of age, sex, years of education, and total intracranial volume using GLM analysis implemented in SPM 12. The threshold was set at p < 0.05 [false discovery rate (FDR)] to control for multiple comparisons.
Furthermore, a GLM based on whole-brain analysis was performed on the smoothed gray matter images to evaluate the impact of APOE ɛ4 carrier status-by-neuropsychological test scores interaction on the cortical volume. APOE ɛ4 carrier status, CERAD-K subdomain, TM, executive function, and total CERAD-K scores were the independent variables. We performed correction for multiple comparisons using the family-wise-error correction based on Gaussian Random Field Theory (GRFT) [38] at cluster level (p < 0.05) combined with a primary uncorrected voxel-level threshold of p < 0.001 in DPABI_V5.1_201201 (http://rfmri.org/dpabi, GNU GENERAL PUBLIC LICENSE, Beijing, China). GRFT finds right threshold for a smooth statistical map which gives the required family-wise-error correction.
RESULTS
Baseline demographic and clinical data
Table 1 shows the baseline demographic data for APOE ɛ4 non-carriers and carriers in the CN sub-Aβ group. There were no significant differences in age, sex, number of years of education, and total intracranial volume between APOE ɛ4 carriers and non-carriers. Regarding global and regional FMM SUVRPONS, no significant differences were found between APOE ɛ4 carriers and non-carriers. Among the neuropsychological test scores, the CERAD-K VF, BNT, and CR subdomains were significantly higher in the APOE ɛ4 non-carriers (p < 0.05). However, the scores in the remaining domains, total memory, executive function, and total CERAD-K scores showed no significant difference between APOE ɛ4 non-carriers and carriers. Additionally, whole-brain analysis of the CN sub-Aβ group did not reveal any significant differences in gray matter volume between APOE ɛ4 carriers and non-carriers.
Table 1
Demographic and clinical characteristics of the study participants
| APOE ɛ4 carrier status | Non-carrier (N = 80) | Carrier (N = 32) | p |
| Age (y) | 66.9±6.1 | 67.3±7.5 | 0.945 |
| Gender | 0.974 | ||
| Male | 23 (28.8%) | 10 (31.2%) | |
| Female | 57 (71.2%) | 22 (68.8%) | |
| Years of education | 12.8±3.6 | 12.5±4.2 | 0.724 |
| TIV (mm3) | 1503.1±141.2 | 1511.5±138.5 | 0.780 |
| Global SUVRPONS | 0.56±0.03 | 0.55±0.04 | 0.293 |
| Regional SUVRPONS | |||
| ACC | 0.57±0.04 | 0.58±0.04 | 0.218 |
| FL | 0.44±0.04 | 0.43±0.04 | 0.309 |
| PL | 0.37±0.04 | 0.38±0.06 | 0.434 |
| PCC/Precuneus | 0.49±0.04 | 0.48±0.04 | 0.474 |
| TL | 0.51±0.03 | 0.51±0.04 | 0.958 |
| CERAD-K | |||
| VF | 17.0±4.2 | 15.5±3.0 | 0.037 |
| BNT | 13.2±1.4 | 12.2±1.7 | 0.001 |
| MMSE | 28.3±1.4 | 28.2±1.2 | 0.966 |
| WLM | 20.2±3.2 | 20.4±3.0 | 0.781 |
| CP | 10.8±0.6 | 10.7±0.9 | 0.665 |
| WLR | 7.0±1.5 | 7.0±1.4 | 0.920 |
| WLRc | 9.5±0.7 | 9.5±0.8 | 0.805 |
| CR | 8.4±2.6 | 7.3±2.8 | 0.038 |
| TMT B | 118.2±61.6 | 142.7±73.3 | 0.074 |
| Stroop word-color | 41.7±10.8 | 39.9±8.3 | 0.386 |
| Total memory | 0.11±2.42 | –0.28±2.45 | 0.438 |
| Executive function | 0.27±2.57 | –0.68±1.91 | 0.060 |
| Total CERAD-K | 0.33±3.90 | –0.83±3.64 | 0.150 |
Data are presented as the mean±SD unless indicated otherwise. SUVRPONS, standardized uptake value ratio of [18F] flutemetamol, using the pons as a reference region; ACC, anterior cingulate cortex; FL, frontal lobes; PL, parietal lobes; PCC/Precuneus, posterior cingulate cortex and precuneus; TL, lateral temporal lobes; CERAD-K, Korean version of Consortium to Establish a Registry for Alzheimer’s Disease; VF, verbal fluency; BNT, Boston Naming Test; MMSE, the Korean version of the Mini-Mental Status Examination; WLM, Word List Memory; CP, Constructional Praxis; WLR, Word List Recall; WLRc, Word List Recognition; CR, constructional recall; TIV, total intracranial volume; Total memory, composite score summing respective z-scores of the WLM, WLR, and WLRc tests; Executive function, composite score summing respective z-scores of the Stroop Word-Color Interference test, VF test, and inverse value of Trail Making Test B; Total CERAD-K, composite score summing respective z-scores of the CERAD-K VF, BNT, WLM, CP, WLR, and WLRc domains; TMT B, Trail Making Test B.
Association between quantitative value of Aβ deposition and cortical volume according to the APOE ɛ4 carrier status
Figure 2A shows brain regions with a significant interaction between regional Aβ deposition in the PCC/precuneus and APOE ɛ4 carrier status in terms of cortical volume after adjusting for age, sex, years of education, and total intracranial volume (FDR-adjusted p < 0.001). The results can be attributed to APOE ɛ4 carriers showing greater cortical volume with higher regional Aβ deposition. The results can be attributed to APOE ɛ4 carriers showing greater cortical volume with higher regional Aβ deposition. Scatter plots visualize these results in Fig. 2B. Table 2 displays the anatomical locations and cluster size of the brain regions with significant Aβ deposition-APOE ɛ4 carrier status interactions regarding gray matter volume. Considering global and regional Aβ deposits in ROIs other than the PCC/precuneus, there were no significant Aβ deposition-APOE ɛ4 carrier status interactions on cortical volume.
Fig. 2
A) Brain regions showing a significant interaction between Aβ deposition in the posterior cingulate cortex/precuneus and APOE ɛ4 carrier status on gray matter volume in cognitively normal older adults with sub-threshold Aβ deposition. B) Scatter plots visualizing relationships between sub-threshold Aβ deposition in the posterior cingulate cortex/precuneus and gray matter volume in regions of interest according to APOE ɛ4 carrier status. General linear model analysis adjusting for age, sex, years of education, and total intracranial volume, FDR-adjusted p < 0.001, cluster p < 0.05. The cluster size of the regions of interest was greater than 100 voxels. SUVRPONS, standardized uptake value ratio of [18F] flutemetamol.
![A) Brain regions showing a significant interaction between Aβ deposition in the posterior cingulate cortex/precuneus and APOE ɛ4 carrier status on gray matter volume in cognitively normal older adults with sub-threshold Aβ deposition. B) Scatter plots visualizing relationships between sub-threshold Aβ deposition in the posterior cingulate cortex/precuneus and gray matter volume in regions of interest according to APOE ɛ4 carrier status. General linear model analysis adjusting for age, sex, years of education, and total intracranial volume, FDR-adjusted p < 0.001, cluster p < 0.05. The cluster size of the regions of interest was greater than 100 voxels. SUVRPONS, standardized uptake value ratio of [18F] flutemetamol.](https://content.iospress.com:443/media/jad/2022/89-3/jad-89-3-jad220427/jad-89-jad220427-g002.jpg)
Table 2
Anatomical locations of the regions showing a significant sub-threshold Aβ deposition-APOE ɛ4 carrier status interaction on gray matter volume in cognitively normal older adults with sub-threshold Aβ deposition
| Region | L/R | Cluster (Voxel count) | Peak F value | Peak MNI coordinates (x, y, z) | ||
| PCC/Precuneus SUVRPONS-by-APOE ɛ4 carrier | ||||||
| status interaction on gray matter volume | ||||||
| Hippocampus | R | 606 | 34.0823 | 28 | –14 | –14 |
| Middle temporal gyrus | R | 295 | 28.1768 | 40 | –34 | –8 |
| Amygdala | L | 292 | 30.6923 | –18 | 2 | –16 |
| Hippocampus | L | 278 | 45.5994 | –16 | –34 | 0 |
| Cerebellum 8 | L | 256 | 35.2305 | –22 | –54 | –48 |
| Cerebellum 8 | R | 177 | 32.7515 | 18 | –58 | –48 |
| Temporal pole: middle temporal gyrus | L | 176 | 28.3857 | –30 | 4 | –46 |
| Putamen | R | 120 | 27.6089 | 16 | 16 | –8 |
| Postcentral gyrus | L | 112 | 26.6399 | –38 | –16 | 46 |
| Rectus gyrus | L | 108 | 27.9905 | 2 | 34 | –14 |
General linear model analysis adjusted for age, sex, years of education, and total intracranial volume, FDR-adjusted p < 0.001, cluster p < 0.05. SUVRPONS, standardized uptake value ratios of [18F] flutemetamol; L, left; R, right.
APOE ɛ4 carrier status-by-neuropsychological performance scores interaction on cortical volume
After adjusting for age, sex, years of education, and total intracranial volume, the APOE ɛ4 carrier status-total CERAD-K score interaction demonstrated a significant effect on gray-matter volume of the left middle occipital gyrus in the CN sub-Aβ group (GRFT correction at a p < 0.05, voxel p < 0.001). As expected from the visualizing scatter plot shown in Fig. 3, cortical volume in the left middle occipital gyrus was larger in APOE ɛ4 carriers that showed higher total CERAD-K scores. Table 3 displays the anatomical locations and cluster sizes of the brain regions showing a significant APOE ɛ4 carrier status-by-global cognitive function interaction. There was no significant interaction effect with other CERAD-K subdomains, TM, and executive function scores.
Fig. 3
Impact of the interaction between global cognitive function and APOE ɛ4 carrier status on cortical volume in cognitively normal older adults with sub-threshold Aβ deposition. General linear model analysis adjusting for age, sex, years of education, and total intracranial volume. Thresholds were set using GRFT correction at a p < 0.05, voxel p < 0.001. The statistical threshold of cluster size > 44. Total CERAD-K, composite score summing respective z-scores of the CERAD-K VF, BNT, WLM, CP, WLR, and WLRc domains.
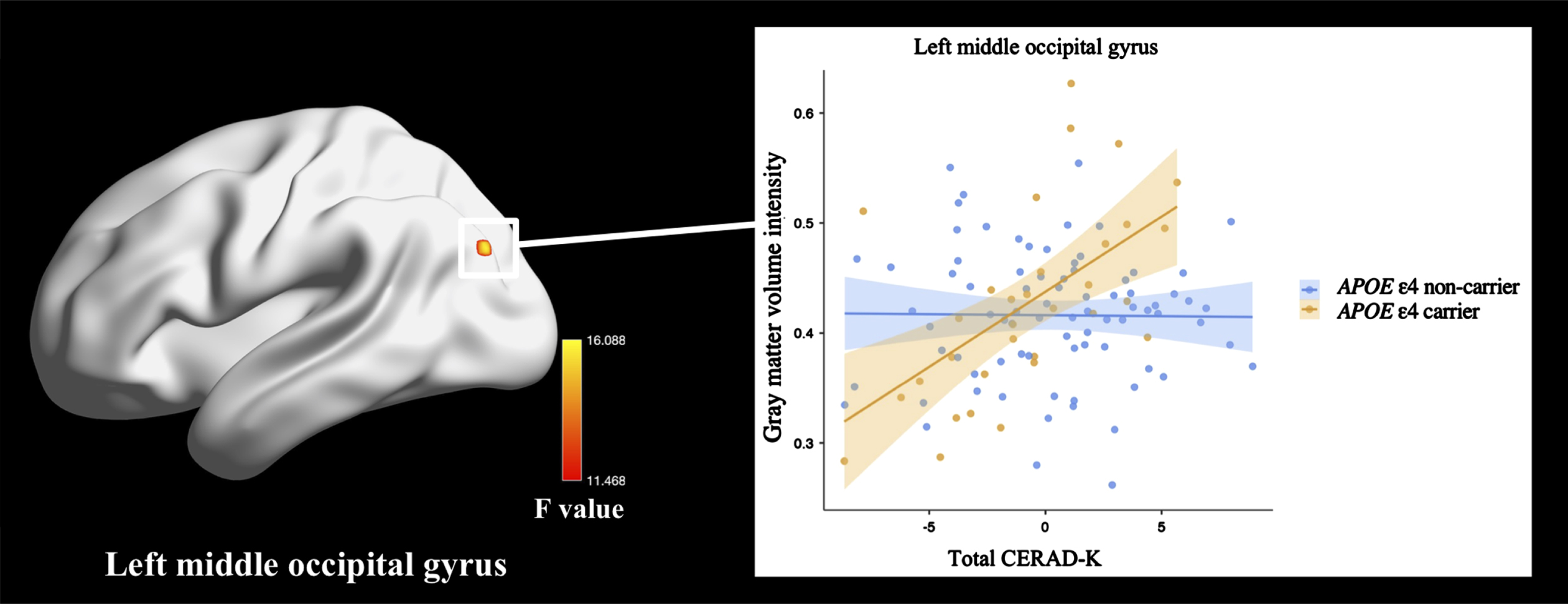
Table 3
Anatomical locations of the regions showing a significant interaction between global cognitive function and APOE ɛ4 carrier status on gray matter volume in cognitively normal older adults with sub-threshold Aβ deposition
| Region | L/R | Cluster (Voxel count) | Peak F value | Peak MNI coordinates (x, y, z) | ||
| Middle occipital gyrus | L | 63 | 16.0884 | –40 | –78 | 30 |
General linear model analysis adjusted for age, sex, years of education, and total intracranial volume. Thresholds were set using GRFT correction at a p < 0.05, voxel p < 0.001. The statistical threshold of the cluster size was 44. L, left; R, right.
DISCUSSION
The current study aimed to evaluate differential associations between regional and global Aβ accumulation, cortical volume, and cognitive performance scores depending on the APOE ɛ4 carrier status in the CN sub-Aβ group.
In the current study, there were no significant differences in regional and global Aβ deposition between APOE ɛ4 carriers and non-carriers in the CN sub-Aβ group. While some studies with AD patients have displayed increased Aβ deposition in APOE ɛ4 carriers compared to non-carriers [26, 39, 40], other studies have reported no difference between APOE ɛ4 carriers and non-carriers [41, 42]. Moreover, there are limited studies examining the significant difference in Aβ deposition according to the APOE ɛ4 allele in the CN sub-Aβ group. In particular, considering that Aβ accumulation increases most steeply during the middle-aged [43, 44], the characteristics of the subjects whose Aβ deposition level remained below the threshold until their mid-60 s also might have influenced the non-significant difference in Aβ level according to APOE ɛ4 carrier status.
With regard to cortical volume, we found no significant difference in gray matter volume between APOE ɛ4 carriers and non-carriers in the CN sub-Aβ group. Although the impact of the APOE genotype on cortical structure is understudied in the preclinical phase, the current results differ from previous research documenting thicker cortices and faster cortical thinning in APOE ɛ4 carriers than non-carriers [29]. Additionally, another prior study suggested APOE ɛ4 fragments to be associated with a neuron-specific proteolytic mechanism for cortical atrophy [45]. However, these findings are somewhat limited by the lack of information on Aβ deposits in the CN group. Additionally, the impact of APOE ɛ4 carrier status on the cortical volume might be more limited because the current study was conducted in subjects at a stage before Aβ deposition progressed significantly. Given the limitation of the previous paper, the current study explored the differential effect of sub-threshold Aβ deposition on cortical volume according to APOE ɛ4 carrier status. The results of this study showed that APOE ɛ4 carriers in the CN sub-Aβ group had a greater cortical volume with higher regional Aβ deposition of the PCC/precuneus. Although these results differ from those of earlier studies [46], the results presented herein support evidence from clinical observations showing a positive association between Aβ accumulation and cortical volume in CN participants [47]. Another prior study suggests a different pattern in the association between Aβ deposition and cortical thickness depending on the level of Aβ deposition in CN subjects, only showing a positive association between the sub-threshold Aβ deposition value and cortical thickness [48]. Moreover, the previous findings showed thicker cortices in temporoparietal regions with an increase in early AD pathology index, which incorporates Aβ and tau levels in the trajectory of AD [25]. In this research, APOE ɛ4 carriers displayed faster hippocampus atrophy with an increase in AD pathology index during the whole AD spectrum [25]. Considering their research methods, these studies measured AD pathology through cerebrospinal fluid, which cannot be used to assess regional AD pathology. Additionally, they also did not examine the interaction effect between AD pathology and APOE genotype on cortical volume. Moreover, some of the findings were observed during the AD spectrum period rather than during the preclinical phase. Therefore, these limitations suggest that prior study findings need to be interpreted cautiously.
Among the ROIs showing a larger cortical volume with higher regional Aβ deposition in the PCC/precuneus in APOE ɛ4 carriers within the present study, the cerebellum has been demonstrated to be structurally and functionally connected to the cerebral cortex [49, 50]. In particular, the posterior cerebellum, including the cerebellum 8, has been reported to be related to the known ROIs of the default mode network, which is vulnerable to Aβ and tau deposition [51, 52]. Additionally, AD and MCI patients have exhibited greater atrophy in the posterior cerebellum than healthy controls [53–55]. Considering the previous findings, although it is assumed that the APOE ɛ4 allele can modulate the association between the portions of the posterior cerebellum and the cerebral cortex vulnerable to AD pathology, research on the effect of the APOE ɛ4 genotype on cerebellar volume has not yet been conducted in CN individuals.
Concerning larger cortical volume and higher AD pathology during the preclinical phase, post-mortem studies considering preclinical AD patients have found nuclear and cellular hypertrophy [56, 57], followed by cellular atrophy [58]. Additionally, transgenic mice studies of AD have found neural hypertrophy and increased synapses [59]. Moreover, another animal research model of AD showed amyloid precursor protein to increase cortical neuron size [60]. These findings have been explained by the reactive neuronal hypertrophy or inflammatory response to initial Aβ accumulation [58]. With regard to the reactive inflammatory response, the APOE ɛ4 allele is associated with enhanced microglial activation and greater astrogliosis in the frontal and temporal lobe compared with other APOE genotypes [61, 62]. Additionally, transgenic mouse models of AD pathology have shown the APOE ɛ4 genotype to increase the inflammatory response of microglia and astrocytes [63, 64], modulating cytokine release depending on the glial cell type and disease stage [65, 66]. Furthermore, the APOE ɛ4 allele was found to increase inflammatory gene transcription, such as by triggering the receptor expressed on myeloid cells 2 (TREM2) [64, 67]. Considering all of this evidence, the principal theoretical implication of the present findings is that the inflammatory response to the earliest stage of Aβ accumulation could affect neuronal and glial hypertrophy in CN participants, with this effect being enhanced by the APOE ɛ4 carrier status.
Another important finding of the present investigation was that the PCC/precuneus was the ROI where Aβ deposition exhibited a distinctive effect on cortical volume according to the APOE ɛ4 carrier status. In addition, the PCC/precuneus has been reported to be vulnerable to initial Aβ accumulation in the course of AD [68]. Furthermore, prior research in AD patients has demonstrated that Aβ does not significantly deposit in the medial temporal and temporal poles, although atrophy progressed in these regions [69]. On the contrary, in the PCC/precuneus, atrophy did not progress clearly, but Aβ deposition was significant [69]. This discrepancy could be partially explained by the fact that Aβ affects remote brain regions through transsynaptic spread via afferent connections [70]. In the current study, no differences in Aβ deposition were observed in the PCC/precuneus compared with other regions. This might be because the current study participants were in the preclinical phase. However, the interaction of Aβ accumulation with APOE ɛ4 carrier status in terms of cortical volume was not observed in areas other than the PCC/precuneus. Additionally, despite subthreshold Aβ deposition in the present study, Aβ deposition in the PCC/precuneus region of APOE ɛ4 carriers differentially affected the cortical volume of the temporal lobe, including the hippocampus, middle temporal gyrus, and temporal pole. Another possible explanation is tauopathy, which affects atrophy rate, interacting with Aβ deposition during the preclinical phase [71]. However, it has been demonstrated that the proportion of the CN sub-Aβ group with positive tau deposition is only 12% [72], and the effect of tauopathy on cortical atrophy starts in the early stages of MCI [46]. In this respect, it could be estimated that the effect of tau on atrophy in the preclinical phase of AD in those without Aβ deposition is limited. Moreover, the ROIs showing the sub-threshold Aβ-APOE ɛ4 carrier status interaction on the cortical volume are part of the cortico-hippocampal systems [73]. This system consists of the posterior medial (PM) and anterior temporal (AT) systems, which are involved in processing contexts and item information, respectively [73]. Additionally, the hippocampus integrates these two sub-systems for supporting memory-guided behavior [73]. While the PCC/precuneus constitutes the PM system, the ROIs of the current study correspond to the AT system and hippocampus. Taken together, the earliest Aβ deposition in the hub region of the PM system could differentially affect the cortical volume in brain regions of the AT system according to the APOE ɛ4 carrier status. However, we did not detect any interaction between cortical volume and APOE ɛ4 carrier status on the cognitive performance scores in these ROIs. Instead, we explored the interaction between cognitive function and APOE ɛ4 carrier status on cortical volume by whole-brain analysis and found greater left middle occipital gyrus volume to be associated with higher global cognition scores in APOE ɛ4 carriers compared with non-carriers. The left middle occipital gyrus volume has predicted AD occurrence con-jointing with AD-associated single nucleotide polymorphism data [74] and has exhibited a significant decrease in MCI patients compared to the CN group [75]. However, this region has displayed greater volume in the APOE ɛ4 carriers of healthy middle-aged individuals [76]. Although there is a paucity of studies examining the APOE ɛ4 allele-cognitive function interaction on brain structure, including the left middle occipital gyrus in the earliest phase of AD, given that greater cortical volume was found in APOE ɛ4 carriers with initial Aβ deposits, this finding might reflect the compensatory response to the earliest Aβ in APOE ɛ4 carriers within a normal deposition level. However, previous research has indicated that the relationship between structural biomarkers and memory performance in the preclinical phase starts to appear from the age of 70 years [77]. Moreover, the current study has evaluated the association with subtle cognitive differences within the normal range. In this regard, these factors could have contributed to the relative lack of statistical robustness for the interactions described in the present paper.
Finally, several limitations to this study need to be acknowledged. First, we did not evaluate causative factors, including tau, TAR DNA-binding protein-43, neuronal injury marker, etc., known to affect cortical atrophy in the trajectory of AD [78–80]. Second, since the atrophy rate depending on AD pathology and APOE genotype could have contributed to a higher cortical volume in a cross-sectional study [71], further longitudinal studies need to be carried out to clarify the aforementioned associations in the CN sub-Aβ group. In addition, the possible effect of the duration of Aβ accumulation on cortical atrophy also supports the need for further longitudinal studies [81]. Thirdly, since the current study has focused on the CN sub-Aβ group, there is a bias toward including APOE ɛ4 non-carriers, considering the increased chance of Aβ pathology affected by this AD high-risk allele. Therefore, the APOE ɛ4 carriers whose Aβ deposition level remained below the threshold until their mid-60 s could have a higher possibility of having other characteristics, including protective genes [82] and higher cognitive reserve [83], etc. Finally, the current study’s proportion of APOE ɛ4 carriers was 28.6%, which is relatively high compared to the global proportion of 23.9% [84]. Therefore, this selection bias could have contributed to the significant interaction with APOE ɛ4 carrier status observed in the present study.
This study set out to explore the differential effects of the APOE ɛ4 carrier status on the associations between Aβ deposition, cortical volume, and cognitive performance during the preclinical phase of AD within those with sub-threshold Aβ deposition level. We found the possibility of a compensatory response to the initiating Aβ in APOE ɛ4 carriers at the earliest stage of AD, which is overlooked due to the absence of clinical symptoms. Despite its exploratory nature, this study offers some insight into recent interests concerning probabilistic AD modeling, focusing on the modulating role of APOE ɛ4 carrier status by showing the earliest impact of this allele during the preclinical period. Finally, the current findings contribute to a deeper understanding of the earliest stage of Aβ accumulation within the progression of AD with a compliment to the aforementioned limitations.
ACKNOWLEDGMENTS
This work was supported by National Research Foundation of Korea grants funded by the Korean government (Ministry of Science and ICT) (No. 2019R1A2C2009100 and 2019R1C1C1007608) and Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education (No. 2022R1I1A1A01053710). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/22-0427r1).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-220427.
REFERENCES
[1] | Jack CR Jr , Bennett DA , Blennow K , Carrillo MC , Dunn B , Haeberlein SB , Holtzman DM , Jagust W , Jessen F , Karlawish J ((2018) ) NIA-AA research framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement 14: , 535–562. |
[2] | Jack CR Jr , Knopman DS , Jagust WJ , Petersen RC , Weiner MW , Aisen PS , Shaw LM , Vemuri P , Wiste HJ , Weigand SD , Lesnick TG , Pankratz VS , Donohue MC , Trojanowski JQ ((2013) ) Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol 12: , 207–216. |
[3] | Sperling RA , Aisen PS , Beckett LA , Bennett DA , Craft S , Fagan AM , Iwatsubo T , Jack CR Jr , Kaye J , Montine TJ ((2011) ) Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7: , 280–292. |
[4] | Vos SJ , Xiong C , Visser PJ , Jasielec MS , Hassenstab J , Grant EA , Cairns NJ , Morris JC , Holtzman DM , Fagan AM ((2013) ) Preclinical Alzheimer’s disease and its outcome: A longitudinal cohort study. Lancet Neurol 12: , 957–965. |
[5] | Thurfjell L , Lilja J , Lundqvist R , Buckley C , Smith A , Vandenberghe R , Sherwin P ((2014) ) Automated quantification of 18F-flutemetamol PET activity for categorizing scans as negative or positive for brain amyloid: Concordance with visual image reads. J Nucl Med 55: , 1623–1628. |
[6] | Bischof GN , Jacobs HIL ((2019) ) Subthreshold amyloid and its biological and clinical meaning: Long way ahead. Neurology 93: , 72–79. |
[7] | Salloway S , Gamez JE , Singh U , Sadowsky CH , Villena T , Sabbagh MN , Beach TG , Duara R , Fleisher AS , Frey KA ((2017) ) Performance of [18F] flutemetamol amyloid imaging against the neuritic plaque component of CERAD and the current (2012) NIA-AA recommendations for the neuropathologic diagnosis of Alzheimer’s disease. Alzheimers Dement (Amst) 9: , 25–34. |
[8] | Cho H , Choi JY , Hwang MS , Kim YJ , Lee HM , Lee HS , Lee JH , Ryu YH , Lee MS , Lyoo CH ((2016) ) In vivo cortical spreading pattern of tau and amyloid in the Alzheimer disease spectrum. Ann Neurol 80: , 247–258. |
[9] | Palmqvist S , Schöll M , Strandberg O , Mattsson N , Stomrud E , Zetterberg H , Blennow K , Landau S , Jagust W , Hansson O ((2017) ) Earliest accumulation of β-amyloid occurs within the default-mode network and concurrently affects brain connectivity. Nat Commun 8: , 1–13. |
[10] | Insel PS , Ossenkoppele R , Gessert D , Jagust W , Landau S , Hansson O , Weiner MW , Mattsson N ((2017) ) Time to amyloid positivity and preclinical changes in brain metabolism, atrophy, and cognition: Evidence for emerging amyloid pathology in Alzheimer’s disease. Front Neurosci 11: , 281. |
[11] | Farrell ME , Jiang S , Schultz AP , Properzi MJ , Price JC , Becker JA , Jacobs HIL , Hanseeuw BJ , Rentz DM , Villemagne VL , Papp KV , Mormino EC , Betensky RA , Johnson KA , Sperling RA , Buckley RF ((2021) ) Defining the lowest threshold for amyloid-PET to predict future cognitive decline and amyloid accumulation. Neurology 96: , e619–e631. |
[12] | Leal SL , Lockhart SN , Maass A , Bell RK , Jagust WJ ((2018) ) Subthreshold amyloid predicts tau deposition in aging. J Neurosci 38: , 4482–4489. |
[13] | Pacheco J , Goh JO , Kraut MA , Ferrucci L , Resnick SM ((2015) ) Greater cortical thinning in normal older adults predicts later cognitive impairment. Neurobiol Aging 36: , 903–908. |
[14] | Llado-Saz S , Atienza M , Cantero JL ((2015) ) Increased levels of plasma amyloid-beta are related to cortical thinning and cognitive decline in cognitively normal elderly subjects. Neurobiol Aging 36: , 2791–2797. |
[15] | Becker JA , Hedden T , Carmasin J , Maye J , Rentz DM , Putcha D , Fischl B , Greve DN , Marshall GA , Salloway S , Marks D , Buckner RL , Sperling RA , Johnson KA ((2011) ) Amyloid-β associated cortical thinning in clinically normal elderly. Ann Neurol 69: , 1032–1042. |
[16] | Nosheny RL , Insel PS , Mattsson N , Tosun D , Buckley S , Truran D , Schuff N , Aisen PS , Weiner MW ((2019) ) Associations among amyloid status, age, and longitudinal regional brain atrophy in cognitively unimpaired older adults. Neurobiol Aging 82: , 110–119. |
[17] | Frisoni GB , Altomare D , Thal DR , Ribaldi F , van der Kant R , Ossenkoppele R , Blennow K , Cummings J , van Duijn C , Nilsson PM , Dietrich PY , Scheltens P , Dubois B ((2022) ) The probabilistic model of Alzheimer disease: The amyloid hypothesis revised. Nat Rev Neurosci 23: , 53–66. |
[18] | DeMattos RB , Cirrito JR , Parsadanian M , May PC , O’Dell MA , Taylor JW , Harmony JA , Aronow BJ , Bales KR , Paul SM , Holtzman DM ((2004) ) ApoE and clusterin cooperatively suppress Abeta levels and deposition: Evidence that ApoE regulates extracellular Abeta metabolism}. Neuron 41: , 193–202. |
[19] | Castellano JM , Kim J , Stewart FR , Jiang H , DeMattos RB , Patterson BW , Fagan AM , Morris JC , Mawuenyega KG , Cruchaga C , Goate AM , Bales KR , Paul SM , Bateman RJ , Holtzman DM ((2011) ) Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci Transl Med 3: , 89ra57. |
[20] | Corder EH , Saunders AM , Strittmatter WJ , Schmechel DE , Gaskell PC , Small GW , Roses AD , Haines JL , Pericak-Vance MA ((1993) ) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261: , 921–923. |
[21] | Andrews-Zwilling Y , Bien-Ly N , Xu Q , Li G , Bernardo A , Yoon SY , Zwilling D , Yan TX , Chen L , Huang Y ((2010) ) Apolipoprotein E4 causes age- and Tau-dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J Neurosci 30: , 13707–13717. |
[22] | Bien-Ly N , Andrews-Zwilling Y , Xu Q , Bernardo A , Wang C , Huang Y ((2011) ) C-terminal-truncated apolipoprotein (apo) E4 inefficientlyclears amyloid-beta (Abeta) and acts in concert with Abeta to elicitneuronal and behavioral deficits in mice. Proc Natl Acad Sci US A 108: , 4236–4241. |
[23] | Liu Y , Paajanen T , Westman E , Wahlund LO , Simmons A , Tunnard C , Sobow T , Proitsi P , Powell J , Mecocci P , Tsolaki M , Vellas B , Muehlboeck S , Evans A , Spenger C , Lovestone S , Soininen H ((2010) ) Effect of APOE ɛ4 allele on cortical thicknesses andvolumes: The AddNeuroMed study. J Alzheimers Dis 21: , 947–966. |
[24] | Agosta F , Vossel KA , Miller BL , Migliaccio R , Bonasera SJ , Filippi M , Boxer AL , Karydas A , Possin KL , Gorno-Tempini ML ((2009) ) Apolipoprotein E epsilon4 is associated with disease-specific effects on brain atrophy in Alzheimer’s disease and frontotemporal dementia. Proc Natl Acad Sci U S A 106: , 2018–2022. |
[25] | Gispert JD , Rami L , Sánchez-Benavides G , Falcon C , Tucholka A , Rojas S , Molinuevo JL ((2015) ) Nonlinear cerebral atrophy patternsacross the Alzheimer’s disease continuum: Impact of APOE4 genotype. Neurobiol Aging 36: , 2687–2701. |
[26] | Drzezga A , Grimmer T , Henriksen G , Mühlau M , Perneczky R , Miederer I , Praus C , Sorg C , Wohlschläger A , Riemenschneider M , Wester HJ , Foerstl H , Schwaiger M , Kurz A ((2009) ) Effect of APOE genotype on amyloid plaque load and gray matter volume in Alzheimer disease. Neurology 72: , 1487–1494. |
[27] | Leung KK , Bartlett JW , Barnes J , Manning EN , Ourselin S , Fox NC ((2013) ) Cerebral atrophy in mild cognitive impairment and Alzheimer disease: Rates and acceleration. Neurology 80: , 648–654. |
[28] | Cohen RM , Small C , Lalonde F , Friz J , Sunderland T ((2001) ) Effect of apolipoprotein E genotype on hippocampal volume loss in aging healthy women. Neurology 57: , 2223–2228. |
[29] | Espeseth T , Westlye LT , Fjell AM , Walhovd KB , Rootwelt H , Reinvang I ((2008) ) Accelerated age-related cortical thinning in healthy carriers of apolipoprotein E ɛ4. Neurobiol Aging 29: , 329–340. |
[30] | Farrell ME , Chen X , Rundle MM , Chan MY , Wig GS , Park DC ((2018) ) Regional amyloid accumulation and cognitive decline in initially amyloid-negative adults. Neurology 91: , e1809–e1821. |
[31] | Lee JH , Lee KU , Lee DY , Kim KW , Jhoo JH , Kim JH , Lee KH , Kim SY , Han SH , Woo JI ((2002) ) Development of the Korean Version of the Consortium to Establish a Registry for Alzheimer’s Disease Assessment Packet (CERAD-K) clinical and neuropsychological assessment batteries. J Gerontol B Psychol Sci Soc Sci 57: , P47–P53. |
[32] | Park J-H ((1989) ) Standardization of Korean version of the Mini-Mental State Examination (MMSE-K) for use in the elderly. Part II. Diagnostic validity. J Korean Neuropsychiatr Assoc 28: , 508–513. |
[33] | Stroop JR ((1992) ) Studies of interference in serial verbal reactions. J Exp Psychol Gen 121: , 15. |
[34] | Tombaugh TN ((2004) ) Trail Making Test A and B: Normative data stratified by age and education. Arch Clin Neuropsych 19: , 203–214. |
[35] | Fazekas F , Chawluk JB , Alavi A , Hurtig HI , Zimmerman RA ((1987) ) MR signal abnormalities at 1.5 T in Alzheimer’s dementia and normal aging. Am J Neuroradiol 8: , 421–426. |
[36] | Li Z , Shue F , Zhao N , Shinohara M , Bu G ((2020) ) APOE2: Protective mechanism and therapeutic implications for Alzheimer’s disease. Mol Neurodegener 15: , 1–19. |
[37] | Good CD , Johnsrude IS , Ashburner J , Henson RN , Friston KJ , Frackowiak RS ((2001) ) A voxel-based morphometric study of ageing in 465 normal adult human brains. Neuroimage 14: , 21–36. |
[38] | Nichols TE ((2012) ) Multiple testing corrections, nonparametric methods, and random field theory. Neuroimage 62: , 811–815. |
[39] | Tiraboschi P , Hansen L , Masliah E , Alford M , Thal L , Corey-Bloom J ((2004) ) Impact of APOE genotype on neuropathologic and neurochemical markers of Alzheimer disease. Neurology 62: , 1977–1983. |
[40] | Berg L , McKeel DW , Miller JP , Storandt M , Rubin EH , Morris JC , Baty J , Coats M , Norton J , Goate AM ((1998) ) Clinicopathologic studies in cognitively healthy aging and Alzheimer disease: Relation of histologic markers to dementia severity, age, sex, and apolipoprotein E genotype. Arch Neurol 55: , 326–335. |
[41] | Rowe CC , Ellis KA , Rimajova M , Bourgeat P , Pike KE , Jones G , Fripp J , Tochon-Danguy H , Morandeau L , O’Keefe G ((2010) ) Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging 31: , 1275–1283. |
[42] | Landen M , Thorsell A , Wallin A , Blennow K ((1996) ) The apolipoprotein E allele epsilon 4 does not correlate with the number of senile plaques or neurofibrillary tangles in patients with Alzheimer’s disease. J Neurol Neurosurg Psychiatry 61: , 352–356. |
[43] | Bischof GN , Rodrigue KM , Kennedy KM , Devous MD , Park DC ((2016) ) Amyloid deposition in younger adults is linked to episodic memory performance. Neurology 87: , 2562–2566. |
[44] | Rodrigue K , Kennedy K , Devous M , Rieck J , Hebrank A , Diaz-Arrastia R , Mathews D , Park D ((2012) ) β-Amyloid burden in healthy aging: Regional distribution and cognitive consequences. Neurology 78: , 387–395. |
[45] | Mahley RW , Weisgraber KH , Huang Y ((2006) ) Apolipoprotein E4: A causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc Natl Acad Sci U S A 103: , 5644–5651. |
[46] | Fletcher E , Filshtein TJ , Harvey D , Renaud A , Mungas D , DeCarli C ((2018) ) Staging of amyloid β, t-tau, regional atrophy rates, and cognitive change in a nondemented cohort: Results of serial mediation analyses. Alzheimers Dement (Amst) 10: , 382–393. |
[47] | Chételat G , Villemagne VL , Pike KE , Baron JC , Bourgeat P , Jones G , Faux NG , Ellis KA , Salvado O , Szoeke C , Martins RN , Ames D , Masters CL , Rowe CC ((2010) ) Larger temporal volume in elderly withhigh versus low beta-amyloid deposition. Brain 133: , 3349–3358. |
[48] | Fortea J , Sala-Llonch R , Bartrés-Faz D , Lladó A , Solé-Padullés C , Bosch B , Antonell A , Olives J , Sanchez-Valle R , Molinuevo JL , Rami L ((2011) ) Cognitively preservedsubjects with transitional cerebrospinal fluid β-amyloid1-42 values have thicker cortex in Alzheimer’s disease vulnerableareas. Biol Psychiat 70: , 183–190. |
[49] | Buckner RL ((2013) ) The cerebellum and cognitive function: 25 years of insight from anatomy and neuroimaging. Neuron 80: , 807–815. |
[50] | Buckner RL , Krienen FM , Castellanos A , Diaz JC , Yeo BT ((2011) ) The organization of the human cerebellum estimated by intrinsic functional connectivity. J Neurophysiol 106: , 2322–2345. |
[51] | Gellersen HM , Guell X , Sami S ((2021) ) Differential vulnerability of the cerebellum in healthy ageing and Alzheimer’s disease. Neuroimage Clin 30: , 102605. |
[52] | Insel PS , Mormino EC , Aisen PS , Thompson WK , Donohue MC ((2020) ) Neuroanatomical spread of amyloid β and tau in Alzheimer’s disease: Implications for primary prevention. Brain Commun 2: , fcaa007. |
[53] | Lim TS , Iaria G , Moon SY ((2010) ) Topographical disorientation in mild cognitive impairment: A voxel-based morphometry study. J Clin Neurol 6: , 204–211. |
[54] | Zhang H , Sachdev PS , Wen W , Kochan NA , Crawford JD , Brodaty H , Slavin MJ , Reppermund S , Draper B , Zhu W , Kang K , Trollor JN ((2012) ) Gray matter atrophy patterns of mild cognitive impairment subtypes. J Neurol Sci 315: , 26–32. |
[55] | Risacher SL , Shen L , West JD , Kim S , McDonald BC , Beckett LA , Harvey DJ , Jack CR Jr. , Weiner MW , Saykin AJ ((2010) ) Longitudinal MRI atrophy biomarkers: Relationship to conversion in the ADNI cohort. Neurobiol Aging 31: , 1401–1418. |
[56] | Iacono D , O’Brien R , Resnick SM , Zonderman AB , Pletnikova O , Rudow G , An Y , West MJ , Crain B , Troncoso JC ((2008) ) Neuronal hypertrophy in asymptomatic Alzheimer disease. J Neuropathol Exp Neurol 67: , 578–589. |
[57] | Riudavets MA , Iacono D , Resnick SM , O’Brien R , Zonderman AB , Martin LJ , Rudow G , Pletnikova O , Troncoso JC ((2007) ) Resistance to Alzheimer’s pathology is associated with nuclear hypertrophy in neurons. Neurobiol Aging 28: , 1484–1492. |
[58] | Fortea J , Sala-Llonch R , Bartrés-Faz D , Bosch B , Lladó A , Bargalló N , Molinuevo JL , Sánchez-Valle R ((2010) ) Increasedcortical thickness and caudate volume precede atrophy in PSEN1mutation carriers. J Alzheimers Dis 22: , 909–922. |
[59] | West MJ , Bach G , Søderman A , Jensen JL ((2009) ) Synaptic contact number and size in stratum radiatum CA1 of APP/PS1DeltaE9 transgenic mice. Neurobiol Aging 30: , 1756–1776. |
[60] | Oh ES , Savonenko AV , King JF , Fangmark Tucker SM , Rudow GL , Xu G , Borchelt DR , Troncoso JC ((2009) ) Amyloid precursor protein increases cortical neuron size in transgenic mice. Neurobiol Aging 30: , 1238–1244. |
[61] | Egensperger R , Kösel S , von Eitzen U , Graeber MB ((1998) ) Microglial activation in Alzheimer disease: Association with APOE genotype. Brain Pathol 8: , 439–447. |
[62] | Overmyer M , Helisalmi S , Soininen H , Laakso M , Riekkinen P Sr. , Alafuzoff I ((1999) ) Astrogliosis and the ApoE genotype. an immunohistochemical study of postmortem human brain tissue. Dement Geriatr Cogn Disord 10: , 252–257. |
[63] | Rodriguez GA , Tai LM , LaDu MJ , Rebeck GW ((2014) ) Human APOE4increases microglia reactivity at Aβ plaques in a mousemodel of Aβ deposition. J Neuroinflamm 11: , 111;. |
[64] | Shi Y , Yamada K , Liddelow SA , Smith ST , Zhao L , Luo W , Tsai RM , Spina S , Grinberg LT , Rojas JC , Gallardo G , Wang K , Roh J , Robinson G , Finn MB , Jiang H , Sullivan PM , Baufeld C , Wood MW , Sutphen C , McCue L , Xiong C , Del-Aguila JL , Morris JC , Cruchaga C , Fagan AM , Miller BL , Boxer AL , Seeley WW , Butovsky O , Barres BA , Paul SM , Holtzman DM ((2017) ) ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549: , 523–527. |
[65] | Guo L , LaDu MJ , Van Eldik LJ ((2004) ) A dual role for apolipoprotein e in neuroinflammation: Anti- and pro-inflammatory activity. J Mol Neurosci 23: , 205–212. |
[66] | Dorey E , Bamji-Mirza M , Najem D , Li Y , Liu H , Callaghan D , Walker D , Lue LF , Stanimirovic D , Zhang W ((2017) ) Apolipoprotein E isoforms differentially regulate Alzheimer’s disease and amyloid-β-induced inflammatory response} and}. J Alzheimers Dis 57: , 1265–1279. |
[67] | Krasemann S , Madore C , Cialic R , Baufeld C , Calcagno N , El Fatimy R , Beckers L , O’Loughlin E , Xu Y , Fanek Z , Greco DJ , Smith ST , Tweet G , Humulock Z , Zrzavy T , Conde-Sanroman P , Gacias M , Weng Z , Chen H , Tjon E , Mazaheri F , Hartmann K , Madi A , Ulrich JD , Glatzel M , Worthmann A , Heeren J , Budnik B , Lemere C , Ikezu T , Heppner FL , Litvak V , Holtzman DM , Lassmann H , Weiner HL , Ochando J , Haass C , Butovsky O ((2017) ) The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47: , 566–581.e569. |
[68] | Sojkova J , Driscoll I , Iacono D , Zhou Y , Codispoti KE , Kraut MA , Ferrucci L , Pletnikova O , Mathis CA , Klunk WE , O’Brien RJ , Wong DF , Troncoso JC , Resnick SM ((2011) ) In vivo fibrillar beta-amyloid detected using [11C]PiB positron emission tomography and neuropathologic assessment in older adults. Arch Neurol 68: , 232–240. |
[69] | La Joie R , Perrotin A , Barré L , Hommet C , Mézenge F , Ibazizene M , Camus V , Abbas A , Landeau B , Guilloteau D , de LaSayette V , Eustache F , Desgranges B , Chételat G ((2012) ) Region-specific hierarchy between atrophy, hypometabolism, andβ-amyloid (Aβ) load in Alzheimer’s disease dementia. J Neurosci 32: , 16265–16273. |
[70] | Harris JA , Devidze N , Verret L , Ho K , Halabisky B , Thwin MT , Kim D , Hamto P , Lo I , Yu GQ , Palop JJ , Masliah E , Mucke L ((2010) ) Transsynaptic progression of amyloid-β-induced neuronal dysfunction within the entorhinal-hippocampal network. Neuron 68: , 428–441. |
[71] | Pegueroles J , Vilaplana E , Montal V , Sampedro F , Alcolea D , Carmona-Iragui M , Clarimon J , Blesa R , Lleó A , Fortea J ((2017) ) Longitudinal brain structural changes in preclinical Alzheimer’s disease. Alzheimers Dement 13: , 499–509. |
[72] | Jack CR Jr. , , Wiste HJ , Weigand SD , Therneau TM , Knopman DS , Lowe V , Vemuri P , Mielke MM , Roberts RO , Machulda MM , Senjem ML , Gunter JL , Rocca WA , Petersen RC ((2017) ) Age-specific and sex-specific prevalence of cerebral β-amyloidosis, tauopathy, and neurodegeneration in cognitively unimpaired individuals aged 50-95 years: A cross-sectional study. Lancet Neurol 16: , 435–444. |
[73] | Ranganath C , Ritchey M ((2012) ) Two cortical systems for memory-guided behaviour. Nat Rev Neurosci 13: , 713–726. |
[74] | Ning K , Chen B , Sun F , Hobel Z , Zhao L , Matloff W , Toga AW , Alzheimer’s Disease Neuroimaging Initiative ((2018) ) Classifying Alzheimer’s disease with brain imaging and genetic data using a neural network framework. Neurobiol Aging 68: , 151–158. |
[75] | Singh V , Chertkow H , Lerch JP , Evans AC , Dorr AE , Kabani NJ ((2006) ) Spatial patterns of cortical thinning in mild cognitive impairment and Alzheimer’s disease. Brain 129: , 2885–2893. |
[76] | Cacciaglia R , Molinuevo JL , Falcón C , Brugulat-Serrat A , Sánchez-Benavides G , Gramunt N , Esteller M , Morán S , Minguillón C , Fauria K ((2018) ) Effects of APOE-ɛ4allele load on brain morphology in a cohort of middle-aged healthyindividuals with enriched genetic risk for Alzheimer’s disease. Alzheimers Dement 14: , 902–912. |
[77] | Knopman DS , Lundt ES , Therneau TM , Vemuri P , Lowe VJ , Kantarci K , Gunter JL , Senjem ML , Mielke MM , Machulda MM , Boeve BF , Jones DT , Graff-Radford J , Albertson SM , Schwarz CG , Petersen RC , Jack CR ((2019) ) Entorhinal cortex tau, amyloid-β, cortical thickness and memory performance in non-demented subjects. Brain 142: , 1148–1160. |
[78] | Harrison TM , Du R , Klencklen G , Baker SL , Jagust WJ ((2021) ) Distinct effects of beta-amyloid and tau on cortical thickness in cognitively healthy older adults. Alzheimers Dement 17: , 1085–1096. |
[79] | Wisse LEM , de Flores R , Xie L , Das SR , McMillan CT , Trojanowski JQ , Grossman M , Lee EB , Irwin D , Yushkevich PA , Wolk DA ((2021) ) Pathological drivers of neurodegeneration in suspected non-Alzheimer’s disease pathophysiology. Alzheimers Res Ther 13: , 100. |
[80] | Kang MS , Aliaga AA , Shin M , Mathotaarachchi S , Benedet AL , Pascoal TA , Therriault J , Chamoun M , Savard M , Devenyi GA , Mathieu A , Chakravarty MM , Sandelius Å , Blennow K , Zetterberg H , Soucy JP , Cuello AC , Massarweh G , Gauthier S , Rosa-Neto P ((2021) ) Amyloid-beta modulates the association between neurofilament light chain and brain atrophy in Alzheimer’s disease. Mol Psychiatr 26: , 5989–6001. |
[81] | Insel PS , Mattsson N , Donohue MC , Mackin RS , Aisen PS , Jack CR Jr. , , Shaw LM , Trojanowski JQ , Weiner MW ((2015) ) The transitional association between β-amyloid pathology and regional brain atrophy. Alzheimers Dement 11: , 1171–1179. |
[82] | Seto M , Weiner RL , Dumitrescu L , Hohman TJ ((2021) ) Protective genes and pathways in Alzheimer’s disease: Moving towards precision interventions. Mol Neurodegener 16: , 1–16. |
[83] | Stern Y ((2009) ) Cognitive reserve. Neuropsychologia 47: , 2015–2028. |
[84] | Wang Y-Y , Ge Y-J , Tan C-C , Cao X-P , Tan L , Xu W ((2021) ) The proportion of APOE4 carriers among non-demented individuals: A pooled analysis of 389,000 community-dwellers. J Alzheimers Dis 81: , 1331–1339. |


