
Long-Term Depression-Inducing Low Frequency Stimulation Enhances p-Tau181 and p-Tau217 in an Age-Dependent Manner in Live Rats
Abstract
Background:
Cognitive decline in Alzheimer’s disease (AD) correlates with the extent of tau pathology, in particular tau hyperphosphorylation, which is strongly age-associated. Although elevation of cerebrospinal fluid or blood levels of phosphorylated tau (p-Tau) at residues Thr181 (p-Tau181), Thr217 (p-Tau217), and Thr231 (p-Tau231) are proposed to be particularly sensitive markers of preclinical AD, the generation of p-Tau during brain activity is poorly understood.
Objective:
To study whether the expression levels of p-Tau181, p-Tau217, and p-Tau231 can be enhanced by physiological synaptic long-term depression (LTD) which has been linked to the enhancement of p-Tau in hippocampus.
Methods:
In vivo electrophysiology was performed in urethane anesthetized young adult and aged male rats. Low frequency electrical stimulation (LFS) was used to induce LTD at CA3 to CA1 synapses. The expression level of p-Tau and total tau was measured in dorsal hippocampus using immunofluorescent staining and/or western blotting.
Results:
We found that LFS enhanced p-Tau181 and p-Tau217 in an age-dependent manner in the hippocampus of live rats. In contrast, phosphorylation at residues Thr231, Ser202/Thr205, and Ser396 appeared less sensitive to LFS. Pharmacological antagonism of either N-methyl-D-aspartate or metabotropic glutamate 5 receptors inhibited the elevation of both p-Tau181 and p-Tau217. Targeting the integrated stress response, which increases with aging, using a small molecule inhibitor ISRIB, prevented the enhancement of p-Tau by LFS in aged rats.
Conclusion:
Together, our data provide a novel in vivo means to uncover brain plasticity-related cellular and molecular processes of tau phosphorylation at key sites in health and aging.
INTRODUCTION
Clinical evidence indicates that age-associated progressive cognitive decline in Alzheimer’s disease (AD) patients correlates with the extent of tau pathology, in particular the degree and nature of tau phosphorylation [1–4]. Among the latter, elevation of phosphorylated tau (p-Tau) on residues of Thr181 (p-Tau181), Thr217 (p-Tau217), and Thr231 (p-Tau231) in cerebrospinal fluid (CSF) or blood were recently proposed to be particularly sensitive markers of early AD, long before the diagnosis of clinical dementia [5–11]. P-Tau in the brain and its subsequent release into CSF and blood is a dynamic process that changes during disease evolution [5, 12]. Although reports from various memory clinics indicate that p-Tau181, p-Tau217, and p-Tau231 distinguish AD from controls with high accuracy for very early AD diagnosis [13, 14], the generation of these p-Tau species in patients’ brains, in particular learning and memory processes, is still unclear. Also, the phosphorylation patterns of physiological and pathological tau are surprisingly similar and heterogenous, making it difficult to identify specific modifications as therapeutic targets [3, 15–17]. Whether the phosphorylation of tau at those residues is physiological or pathological should be carefully distinguished before embarking on the costly development of treatments targeting individual forms of abnormal tau.
Hyperphosphorylation of tau, together with other post-translational modifications, causes it to form species more likely to aggregate, which likely leads to the deposition of fibrillar tau in neurofibrillary tangles, the main intraneuronal lesions in AD [18, 19]. The lesions primarily accumulate in the transentorhinal and entorhinal regions (Braak stages I-II), and subsequently progress to the limbic allocortex and adjoining neocortex (Braak stages III-IV) and later the neocortex including the secondary and primary fields (Braak stages V-VI) [20, 21]. The hippocampus is one of the areas that p-Tau first appears during Braak stage II of AD [21] and this region of brain is particularly vulnerable to age-related changes [22–26]. Activity-dependent persistent increases and decreases in synaptic strength, long-term potentiation (LTP), and long-term depression (LTD), respectively, are the main forms of synaptic plasticity in the hippocampus and are used to study the neurophysiological substrates of normal learning and memory function [27, 28]. Interestingly, recent studies indicate that LTD induction enhances tau phosphorylation at Ser396 (p-Tau396) [29, 30] and, when repeated chronically, Ser202/Thr205 (p-Tau202/205) [31] in hippocampus in vitro. It is still unknown whether the expression levels of p-Tau181, p-Tau217, and p-Tau231 can also be enhanced by physiological LTD induction. Whether or not their enhancement is more sensitive compared with other reported residues and the age dependence of tau phosphorylation remains elusive.
Both N-methyl-D-aspartate receptors (NMDARs) and metabotropic glutamate receptors (mGluRs) are required for the induction of most forms of LTD by low frequency conditioning stimulation (LFS) [32]. Recent evidence implicates a particular role for extrasynaptic NMDAR [33, 34] and metabotropic glutamate receptor subtype 5 (mGluR5) [35–38], both also involved in tau pathology [39–41], in LTD induction.
The integrated stress response (ISR) is aberrantly elevated in aged mouse brain and a small molecule ISR inhibitor, ISRIB (trans-isomer), restores age-related changes in hippocampal neuron function [42]. Increasing the ISR can facilitate mGluR-LTD [43, 44] while suppressing it has the opposite effect [43–46]. We therefore hypothesized that the ISR may mediate any increased tau phosphorylation triggered by LFS in aged animals.
Using LFS to induce LTD at CA3 to CA1 synapses in live rats [37, 47, 48], the current study investigated 1) the age dependence ((2-3-months and 17-18-months) of tau phosphorylation at specific sites (p-Tau181, p-Tau217, p-Tau231, p-Tau202/205, and p-Tau396), 2) the role of NMDARs and mGluR5, and 3) the involvement of the ISR.
MATERIALS AND METHODS
Animals
All experiments were performed following the guidelines of the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines 2.0 [49] and were approved by the Animal Care and Use Committee of Zhengzhou University, China. All efforts were made to minimize the number of animals used and their suffering.
Young adult (2-3-month-old) and aged (17-18-month-old) male Sprague Dawley rats were provided by the Laboratory Animal Center of Zhengzhou University. We conducted the experiments in accordance with local and international guidelines on the ethical use of laboratory animals and made efforts to minimize the number of rats used in this study. Totally, 20 young adult and 40 aged rats were used in this study. The number of animals allocated to each experimental group was 4–8 based on previous publications [37, 50]. All efforts were made to minimize the number of animals used as well as their suffering.
The animals were housed under a 12 h light-dark cycle at room temperature (19–22°C) with continuous access to food and water ad libitum. Prior to the acute experiments, animals were anaesthetized with urethane (1.5-1.6 g/kg, i.p.). Lignocaine (10 mg, 1% adrenaline, s.c.) was injected over the area of the skull where electrodes and screws were to be implanted. The body temperature of the rats was maintained at 37-38°C with a feedback-controlled heating blanket during the whole period of surgery and recording.
Electrophysiology
Electrodes were made and implanted as described previously [37]. Briefly, monopolar recording electrodes were constructed from Teflon-coated tungsten wires (75μm inner core diameter, 112μm external diameter) and twisted bipolar stimulating electrodes were constructed from Teflon-coated tungsten wires (50μm inner core diameter, 75μm external diameter) separately. Field excitatory postsynaptic potentials (EPSPs) were recorded from the stratum radiatum in the CA1 area of left or right hippocampus in response to stimulation of the Schaffer collateral-commissural pathway. Electrode implantation sites were identified using stereotaxic coordinates relative to bregma, with the recording site located 3.4 mm posterior to bregma and 2.5 mm lateral to midline, and stimulating site 4.2 mm posterior to bregma and 3.8 mm lateral to midline. The final placement of electrodes was optimized by using electrophysiological criteria and confirmed via postmortem analysis.
Test EPSPs were evoked by a single square wave pulse (0.2 ms duration) at a frequency of 0.033 Hz and an intensity that triggered a 50% maximum EPSP response. LTD was induced using 1 Hz low frequency stimulation (LFS) consisting of 900 pulses (0.2 ms duration). During the LFS the intensity was raised to trigger EPSPs of 95% maximum amplitude. LTP was induced using 200 Hz high frequency stimulation (HFS) consisting of one set of 10 trains of 20 pulses (inter-train interval of 2 s). The stimulation intensity was raised to trigger EPSPs of 75% maximum during the HFS. None of the conditioning stimulation protocols elicited any detectible abnormal changes in background EEG, which was recorded from the hippocampus throughout the experiments.
Immunofluorescent staining
The rats were sacrificed 30 min post-LFS in the LFS-treated group or and equivalent time in the baseline recording control group. After electrophysiological recording under urethane anesthesia, rats were transcardially perfused with pre-warmed normal saline followed by cold 4% paraformaldehyde in PBS at pH 7.4. Brains were carefully removed and post-fixed in 4% paraformaldehyde for 6 h. Brains were cryoprotected in 20% sucrose followed by 30% sucrose. Tissues were rapidly frozen and cut coronally (50μm). Sections near the stimulating and recording electrodes were reserved and stored in cryoprotective solution (150 mM ethylene, 100 mM glycerol, 250 mM PBS) at –20°C. For total tau and phosphorylated-tau staining, sections were washed three times with PBS for 5 min and then permeabilized with 0.1% Triton X-100 diluted in PBS for 30 min at room temperature. Then sections were blocked in a PBS solution containing 10% normal goat serum, 3% BSA, and 0.1% Triton X-100 for 1 h at room temperature. Sections were incubated respectively with the primary antibodies (Supplementary Table 1) (Tau5, Cell Signaling Technology, 46687 S, 1:200; Tau46, Cell Signaling Technology, 4019 S, 1:200; Phospho-Tau (Thr181), Cell Signaling Technology, 12885 S, 1:200; Phospho-Tau (Thr217), ThermoFisher, 44–744, 1:200; Phospho-Tau (Thr231), Abcam, ab151559, 1:200; Phospho-Tau (Ser202, Thr205), ThermoFisher, MN1020, 1:200; Phospho-Tau (Ser396), Affinty, AF3148, 1:200) in a humidified chamber overnight at 4°C. After 24 h, sections were washed three times with PBS for 5 min, followed by incubation with species-specific secondary antibodies conjugated to 488 nm or 568 nm fluorophores (Alexa Fluor® 488, Abcam, 150113, 1:500; Alexa Fluor® 568, Abcam, 175471, 1;500) for 1 h at room temperature. Sections were subsequently washed and stained with 4′,6-diamidino-2-phenylindole (DAPI) solution (Solarbio, C0065) for 10 min at room temperature. After washing with PBS, sections were mounted on the glass slide with antifade mountant (Southern Biotech, 0100-35) and then processed for imaging using an Olympus fluorescent microscope (BX53F, Olympus, Japan). ImageJ version 1.52a software (National Institute of Mental Health, Bethesda, MD, USA) was used to analyze the intensity of total tau and p-Tau of immunofluorescent staining.
Western blot
The rats were sacrificed 30 min post-LFS in the LFS-treated group or the same timeline under urethane anaesthesia in the naïve control group. The whole brain was taken out and the hippocampus from both sides was separated. Approximately 2-mm-thick hippocampal tissue surrounding the sites of electrodes was kept and frozen immediately in liquid nitrogen and stored at –80°C. The tissues were homogenized in lysis buffer (10 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 0.5% Triton X-100, 0.1 mM PMSF) containing 1% protease inhibitor cocktail (CWBiO, CW2200S) and 1% phosphatase inhibitor cocktail (CWBiO, CW2383S). The protein concentrations were determined by the BCA Protein Assay Kit (Glpbio, GK10009), and samples were separated by 10% Tris-glycine SDS-PAGE. The proteins were transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, IPVH00010). Then the membranes were blocked with 5% non-fat milk for 1 h at room temperature. After blocking, the membranes were incubated respectively with the primary antibodies (Supplementary Table 1) overnight at 4°C. After primary antibody incubation, the membranes were washed three times in TBST and then incubated with HRP-conjugated goat anti-rabbit IgG (ZSGB-BIO, ZB-2301, 1:25000) for 2 h at room temperature. Finally, the target protein bands were visualized with chemiluminescence reagents (Shanghai Willget Biotech, F03) and then detected with FluorChem E System (ProteinSimple, USA). Quantification of the protein expression was calculated with ImageJ (version 1.52a).
Pharmacological agents
(R,S)-3-(2-carboxypiperazin-4-yl)propyl-1-phosphonic acid ((±)-CPP, Alomone Labs, C-175) and 3-((2-methyl-1,3-thiazol-4-yl)ethynyl)pyridine hydrochloride (MTEP hydrochloride, Abcam, ab120035) were prepared in distilled water and diluted with saline to the required concentration. Trans-N,N′-(Cyclohexane-1,4-diyl)bis(2-(4-chlorophenoxy) acetamide (ISRIB, Sigma, SML0843) was dissolved in dimethyl sulfoxide (DMSO) with gentle warming in a 40°C water bath and vortexed until the solution became clear. Then the solution was diluted in polyethylene glycol 400 (PEG400) with gentle warming in a 40°C water bath and vortexed. The solution was prepared freshly and diluted in warm saline (37°C) before injection. 1:1 DMSO and PEG400 in saline was used as vehicle control. The choice of dose and timing of ISRIB administration was based on previous reports [42] and our study of the pharmacokinetics of ISRIB in live rats [44]. Four pair-housed aged (17-18-month-old) rats received a single daily injection of ISRIB (2.5 mg/kg, i.p.) on 3 consecutive days. Then the rats stayed pair-housed for 18 days after the third injection of ISRIB.
Data analysis
Values are expressed as the mean±s.e.m. For the electrophysiology experiments, the last 10 min prior to LFS was used to calculate the “Pre” -induction EPSP amplitude. Unless otherwise stated the magnitude of LTD was measured over the last 10 min at the end of recording after (“Post”) LFS. To compare between two group, repeated measures two-way ANOVA with Bonferroni post hoc test was used. To compare between groups of three or more, one-way ANOVA with Bonferroni multiple comparisons was used. A two-tailed paired Student’s t-test (paired t) was used to compare between “Pre” and “Post” within groups. For the immunofluorescence staining, internal comparison was used. Thus, immunofluorescent staining signal was quantified in the entire and selected subregions of the ipsilaterally stimulated hippocampi and the contralateral one from the same animals. The average of the contralateral control was standardized to 1. A two-tailed paired Student’s t-test was used to compare between ipsilateral and contralateral. Data of western blotting between conditions were compared using one-way ANOVA with Bonferroni multiple comparisons. A value of p < 0.05 was considered statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). All data were evaluated and graphed using Prism 9.0 (GraphPad Inc, San Diego, CA, USA).
RESULTS
Induction of LTD by LFS enhances p-Tau181 and p-Tau217 in an age-dependent manner in live rats
Having developed stimulation protocols to reliably induce LTD at CA3 to CA1 synapses in live rats [37, 38, 47, 48], we confirmed [37] that this protocol (LFS-900, 900 pulses at 1 Hz) triggered a robust persistent form of LTD that, like certain forms of long-term memory formation, is protein synthesis-dependent (Supplementary Figure 1). We used the same protocol (LFS-900, 900 pulses at 1 Hz) in this study (Fig. 1A). LFS depressed field EPSPs to 63.8±7.8% of baseline in 2-3-month-old rats, and 62.4±3.4% in 17-18-month-old rats. The magnitude of LFS-induced synaptic depression is thus comparable at both ages (Fig. 1B, C).
Fig. 1
LFS promotes p-Tau181 in an age-dependent manner in live rats. A) Schematic of the field EPSP recording configuration in CA1 stratum radiatum (REC) overlaid with a schematic of a bipolar stimulation electrode (STIM) for Schaffer collateral axon fibers (black) in the ipsilateral hemisphere. Additional excitatory projections from CA3 include local recurrent connections (blue) of CA3 pyramidal cells onto other CA3 pyramidal cells, and the back projection (pink) of CA3 pyramidal neurons to the dentate gyrus (DG). B) Application of LFS (horizontal bar, LFS-900; 900 pulses at 1Hz) induced robust LTD at CA3-CA1 synapses in anaesthetized rats at two different ages (2-3-months and 17-18-months). Calibration bars: vertical, 2 mV; horizontal, 10 ms. C) Summarized EPSP amplitude 30 min post LFS. The EPSP decreased to 63.8±7.8% in 2-3-month-old rats (n = 5, p = 0.0147 compared with Pre), and 62.4±3.4% in 17-18-month-old rats (n = 7, p < 0.0001 compared with Pre) respectively; paired t test. The amplitude of LTD is comparable in both groups (two-way ANOVA, age, F (1, 10) = 0.09427, p = 0.7651). D) The upper panel shows p-Tau181 (red) immunofluorescent staining in dorsal hippocampus (Scale bar: 200μm), CA1, CA3, and hilus of DG (scale bars: 50μm) from 2-3-month-old rats. The corresponding statistical results compared with contralateral side are displayed in the lower panel. The expression level of p-Tau181 was not affected by LFS in dorsal hippocampus (p = 0.4611), CA1 (p = 0.3792), CA3 (p = 0.6842), and DG (p = 0.1953); paired t test. E) Immunofluorescent staining of p-Tau181 (red) in ipsilateral dorsal hippocampus, CA1, CA3, and DG from 17-18-month-old rats. LTD induction by LFS significantly enhanced the level of p-Tau181 in dorsal hippocampus (p < 0.0001), CA1 (p = 0.0220), CA3 (p = 0.0059), and DG (p = 0.0202); paired t test. Values are mean±s.e.m.
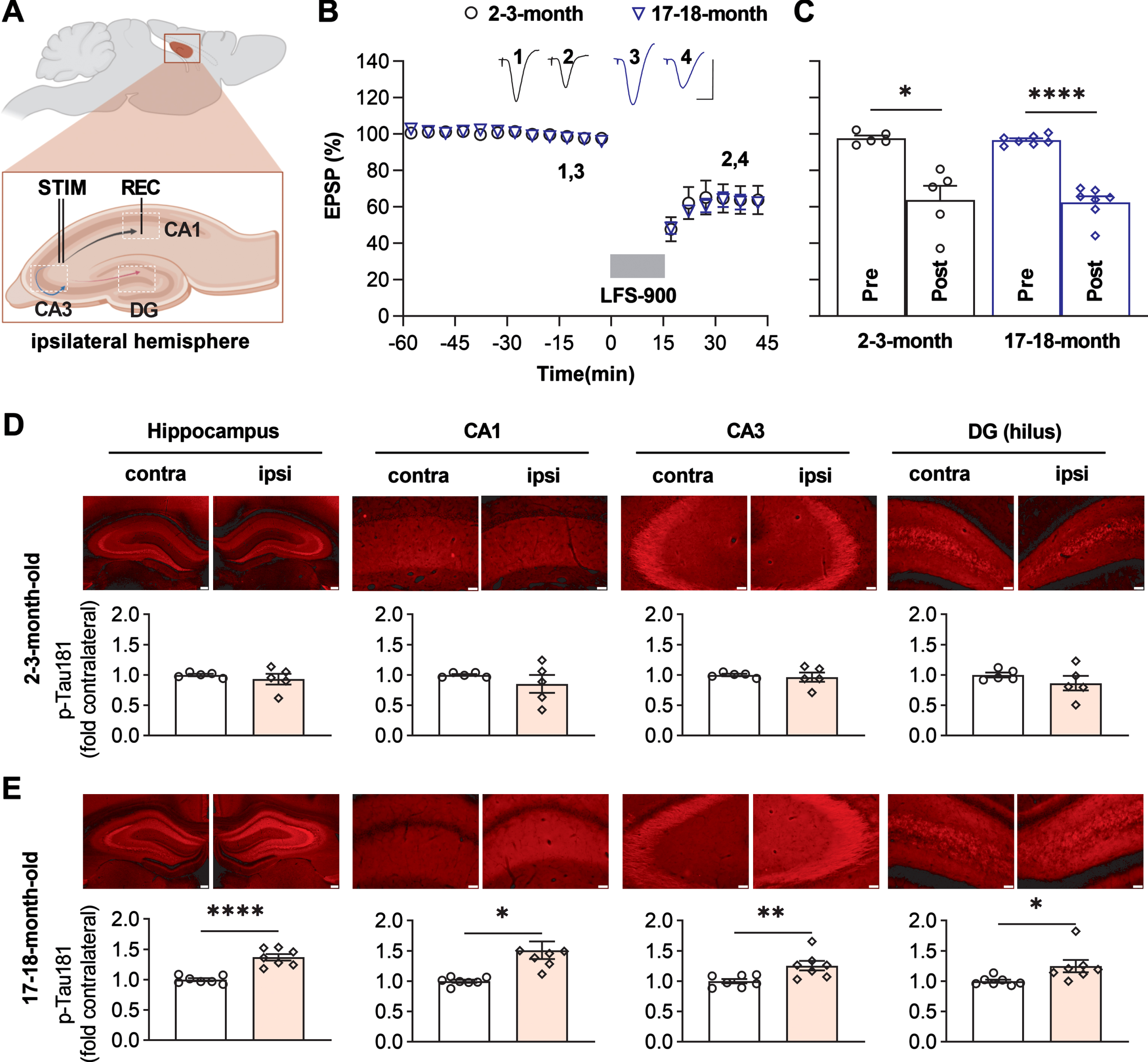
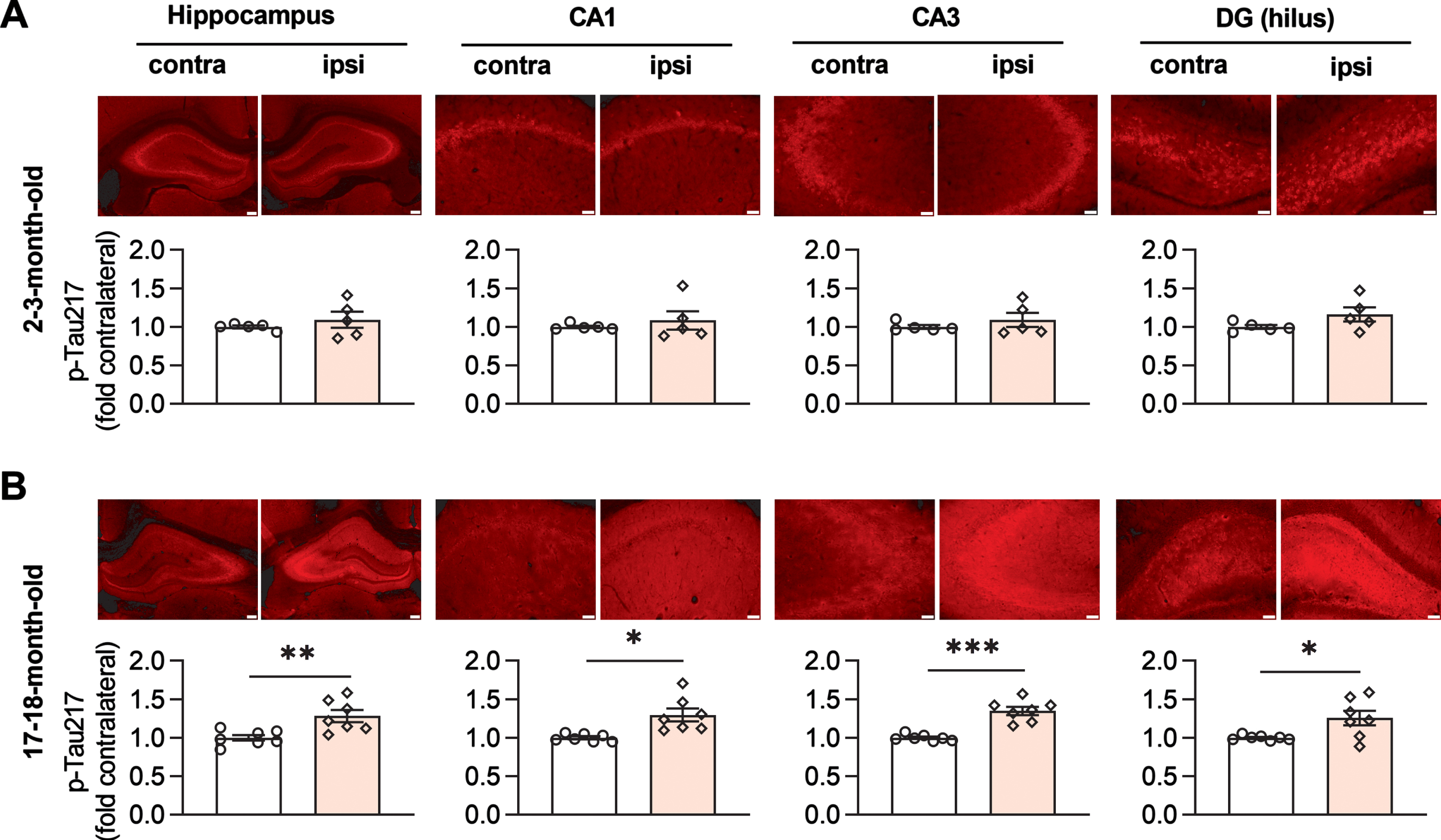
Assessed by immunofluorescent staining, the application of LFS did not lead to a change in total tau expression level in either age group (Supplementary Figure 2). Whereas no difference of p-Tau181 level was apparent at 2-3-months of age (Fig. 1D), in 17-18-month-old animals tau phosphorylation at Thr181 was obviously enhanced in all three hippocampal fields ipsilateral to the side of LFS, relative the contralateral hippocampus (Fig. 1E, Supplementary Figure 3). We then assayed the expression level of p-Tau217 in adjacent brain slices from the same animals. Similar to p-Tau181, no difference was observed in 2-3-month-old rats (Fig. 2A), while an enhancement of p-Tau217 was seen in CA1, CA3, and the hilus of dentate gyrus (DG) in 17-18-month-old rats (Fig. 2B, Supplementary Figure 4). In contrast, the expression levels of p-Tau231, p-Tau202/205, and p-Tau396 did not appear to change in adjacent brain slices from young (Supplementary Figure 5) or aged rats overall, with the exception of a significant enhancement of p-Tau202/205 in the CA1 area of older rats (Supplementary Figure 6).
Fig. 2
LFS promotes p-Tau217 in an age-dependent manner in live rats. A) The upper panel shows immunofluorescent staining of p-Tau217 (red) in dorsal hippocampus (Scale bar: 200μm), CA1, CA3, and hilus of DG (scale bars: 50μm) from 2-3-month-old rats. The corresponding statistical results compared with contralateral side are displayed in the lower panel. LFS did not affect the expression level of p-Tau217 in dorsal hippocampus (p = 0.3820), CA1 (p = 0.4488), CA3 (p = 0.3409), and DG (p = 0.1567); paired t test. B) Immunofluorescent staining of p-Tau217 (red) in dorsal hippocampus, CA1, CA3, and DG from 17-18-month-old rats. LFS ipsilaterally enhanced the level of p-Tau217 in dorsal hippocampus (p < 0.0019), CA1 (p = 0.0153), CA3 (p = 0.0003), and DG (p = 0.0251); paired t test. Values are mean±s.e.m.
To confirm the changes in p-Tau by ipsilateral LTD-inducing LFS detected using immunofluorescence, we measured the expression level of the same phospho-tau species using western blotting after the same conditioning LFS was applied in another cohort of 17-18-month-old rats (Fig. 3A, B). β-actin was used to ensure equal protein loading on gels. The total tau expression level, normalized to β-actin, in either the ipsilateral or contralateral hippocampus did not differ from age-matched naïve control rats (Supplementary Figure 7). Consistent with the immunofluorescent staining, LTD-inducing LFS significantly enhanced p-Tau181 (Fig. 3C; Supplementary Figure 8) and p-Tau217 (Fig. 3D; Supplementary Figure 8) in the ipsilateral hippocampus, while it had no overall significant effect compared with naïve controls on the expression level of p-Tau at the other residues investigated (Supplementary Figure 9). Western blotting indicated that the expression levels of p-Tau181 and p-Tau217, slightly, although not significantly, also increased in the contralateral hippocampus when compared with naïve controls. This indicates that application of LFS activates the commissural pathway sufficiently to weakly enhance the expression levels of both p-Tau181 and p-Tau217 in parts of the contralateral hippocampus.
Fig. 3
LFS enhances p-Tau181 and p-Tau217 in aged rats. A) Application of LFS-900 induced robust LTD at CA3-CA1 synapses in anaesthetized 17-18-month-old rats. Calibration bars: vertical, 2 mV; horizontal, 10 ms. B) Summarized EPSP amplitude 30 min post LFS. The EPSP decreased to 54.8±10.2% (n = 5, p = 0.0110 compared with Pre, paired t). C) Left panels show representative blotting band of p-Tau181 and total tau (Tau5) in the hippocampus of age-matched naïve control group and the experimental group either contralateral or ipsilateral to LFS. Statistical results of p-Tau181 over total tau was quantified and normalized to naïve control (n = 5 per group, ipsilateral versus naïve, p = 0.0292; contralateral versus naïve, p = 0.5075; contralateral versus ipsilateral, p = 0.4022; one-way ANOVA-Bonferroni). D) Left panels show representative blotting band of p-Tau217 and total tau (tau5) in age-matched naïve control group, contralateral hippocampus, and ipsilaterally stimulated hippocampus. Statistical results of p-Tau217 over total tau was quantified and normalized to naïve control (n = 5 per group, ipsilateral versus naïve, p = 0.0049; contralateral versus naïve, p = 0.0979; contralateral versus ipsilateral, p = 0.3905; one-way ANOVA-Bonferroni). Values are mean±s.e.m.
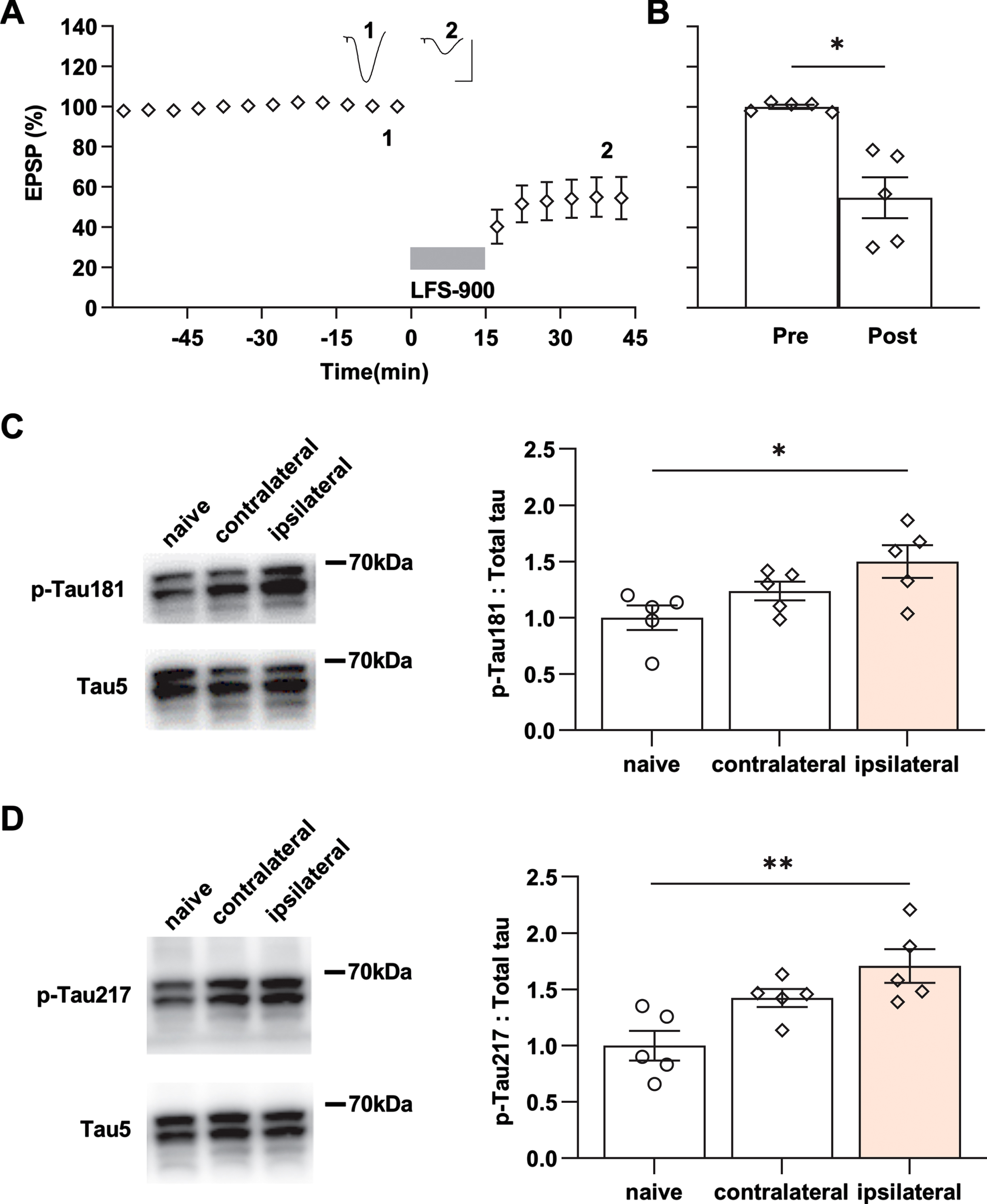
Enhanced neuronal activity accelerates tauopathy in vivo in tau mouse models [51] and mechanical injury of neurons induces tau mislocalization to dendritic spines [52]. In order to exclude any influence of electrode implantation and baseline recording of evoked field EPSPs on the local expression level of p-Tau, the same experimental processes were performed but no LFS was applied in another cohort of 17-18-month-old rats (Supplementary Figure 10A, B). The immunofluorescent staining results confirmed that no change of p-Tau181 and p-Tau217 levels were detectible in hippocampus from these rats (Supplementary Figure 10C, D).
Antagonists of NMDAR or mGluR5 inhibit the elevation of both p-Tau181 and p-Tau217
Previously, we found that the induction of LTD by LFS was not blocked by standard systemic doses of either NMDAR or mGluR5 antagonists when applied alone [37, 47], while combined systemic injection of both antagonists completely abrogated the induction of synaptic LTD in young adult rats [47]. Therefore, we wondered if synaptic LTD could be blocked by standard systemic doses of either NMDAR or mGluR5 antagonists when applied alone in relatively aged (17-18-month-old) rats. Similar to our previous finding in young rats [37, 47], a standard systemic dose (10 mg/kg, i.p.) of the competitive NMDAR antagonist CPP which completely blocked synaptic LTP in CA3-CA1 synapses (Supplementary Figure 11), did not affect the induction of LTD in aged rats. Thus 1 h post the injection of CPP, the application of LFS suppressed the field EPSPs to 70.2±2.5% in 17-18-month-old rats (Fig. 4A, B). Next, we tested the standard dose of MTEP (3 mg/kg, i.p.) which is known to non-competitively block mGluR5, with a rat hippocampal receptor occupancy of >90% [53]. The application of LFS induced a depression of field EPSPs to 79.6±3.7% in these MTEP-treated animals (Fig. 4A, B).
Fig. 4
Antagonists of either NMDAR or mGluR5 prevent the elevation of both p-Tau181 and p-Tau217 induced by LFS in aged rats. A) Systemic injection of the competitive NMDAR antagonist CPP (10 mg/kg, i.p.) alone 1 h prior to the application of LFS did not affect LTD induction in 17-18-month-old rats. Similarly, intraperitoneal injection of the mGluR5 negative allosteric modulator MTEP (3 mg/kg) alone did not affect LTD induction by LFS 1 h later in 17-18-month-old rats. Calibration bars: vertical, 2 mV; horizontal, 10 ms. B) Summary of the mean EPSP amplitude pre and post -LFS. The EPSP decreased to 70.2±2.5% (n = 6, p < 0.0001 compared with pre, paired t test) and 79.6±3.7% (n = 5, p = 0.0068 compared with pre, paired t test) 30 min post-LFS in CPP or MTEP treated rats respectively (two-way RM ANOVA, Treatment F1,9 = 6.999, p = 0.0267). C) The upper panel shows the fluorescent images of p-Tau181 labeling (red) and the lower panel displays the corresponding statistical results in CPP-treated 17-18-month-old rats (Scale bar = 200μm in dorsal hippocampus; scale bars = 50μm in CA1, CA3, and DG regions). LTD induction by LFS did not affect the level of p-Tau181 in dorsal hippocampus (p = 0.1218), CA3 (p = 0.6147), and DG (p = 0.0578) except for CA1 region (p = 0.0353); paired t test. D) The upper panel shows the fluorescent images of p-Tau217 labeling (red) and the corresponding statistical results are displayed in the lower panel in CPP-treated 17-18-month-old rats. No significant difference was found in p-Tau217 levels compared with that in contralateral dorsal hippocampus (p = 0.8075), CA1 (p = 0.0920), CA3 (p = 0.5811), and DG (p = 0.7041); paired t test. E) In MTEP-treated aged rats, LFS failed to promote the levels of p-Tau181 (red as shown in the upper panel). Summarized mean fluorescence intensities (fold to contralateral) in dorsal hippocampus (p = 0.9154), CA1 (p = 0.3161), CA3 (p = 0.1839), and DG (p = 0.8417); paired t test. F) MTEP treatment also prevented the enhancement of p-Tau217 by LFS in aged rats. The upper panel shows the fluorescent images of p-Tau217 (red). The corresponding statistical results are displayed in the lower panel and no significant difference was found in dorsal hippocampus (p = 0.7702), CA1 (p = 0.1598), CA3 (p = 0.2262), and DG (p = 0.5311); paired t test. Values are mean±s.e.m.
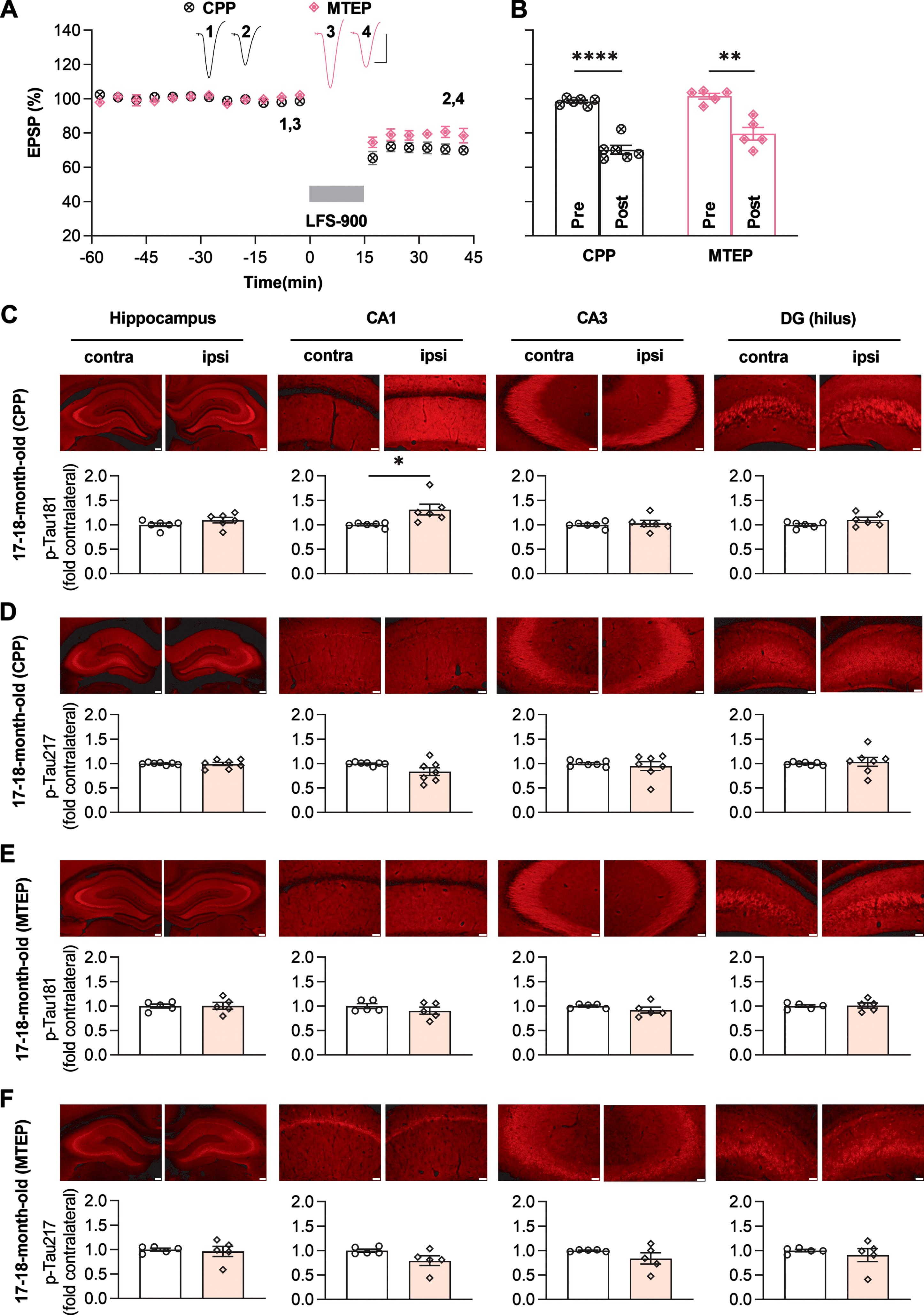
Given that neither block of NMDAR with CPP nor block of mGluR5 with MTEP prevented the induction of LTD in aged rats, we hypothesized that neither of these treatments on their own would affect the enhancement of p-Tau181 and p-Tau217 by LFS. To test this, we assessed total tau and p-Tau on both residues in the hippocampus of the rats treated with CPP or MTEP. Total tau immunofluorescence with Tau46 confirmed that neither agent affected total tau expression level after induction of LTD by LFS in aged rats (Supplementary Figure 12). Surprisingly, even though neither of these treatments on their own affected the induction of LTD, elevation of p-Tau181 and p-Tau217 by LFS were abolished/inhibited in both CPP and MTEP-treated aged rats. After systemic treatment of CPP, the enhancement of p-Tau181 by LFS was strongly inhibited overall although some enhancement was seen in the CA1 area (Fig. 4C), while the elevation of p-Tau217 by LFS was completely abolished (Fig. 4D). Somewhat similarly, the elevation of both p-Tau181 and p-Tau217 triggered by LFS was completely prevented by systemic administration of MTEP (Fig. 4E, F). CPP is a competitive NMDAR antagonist, so its ability to block NMDARs depends on the magnitude of stimulus-evoked transmitter release during the LFS period. This may help explain why the enhancement of p-Tau181 was resistant to CPP in the CA1 area.
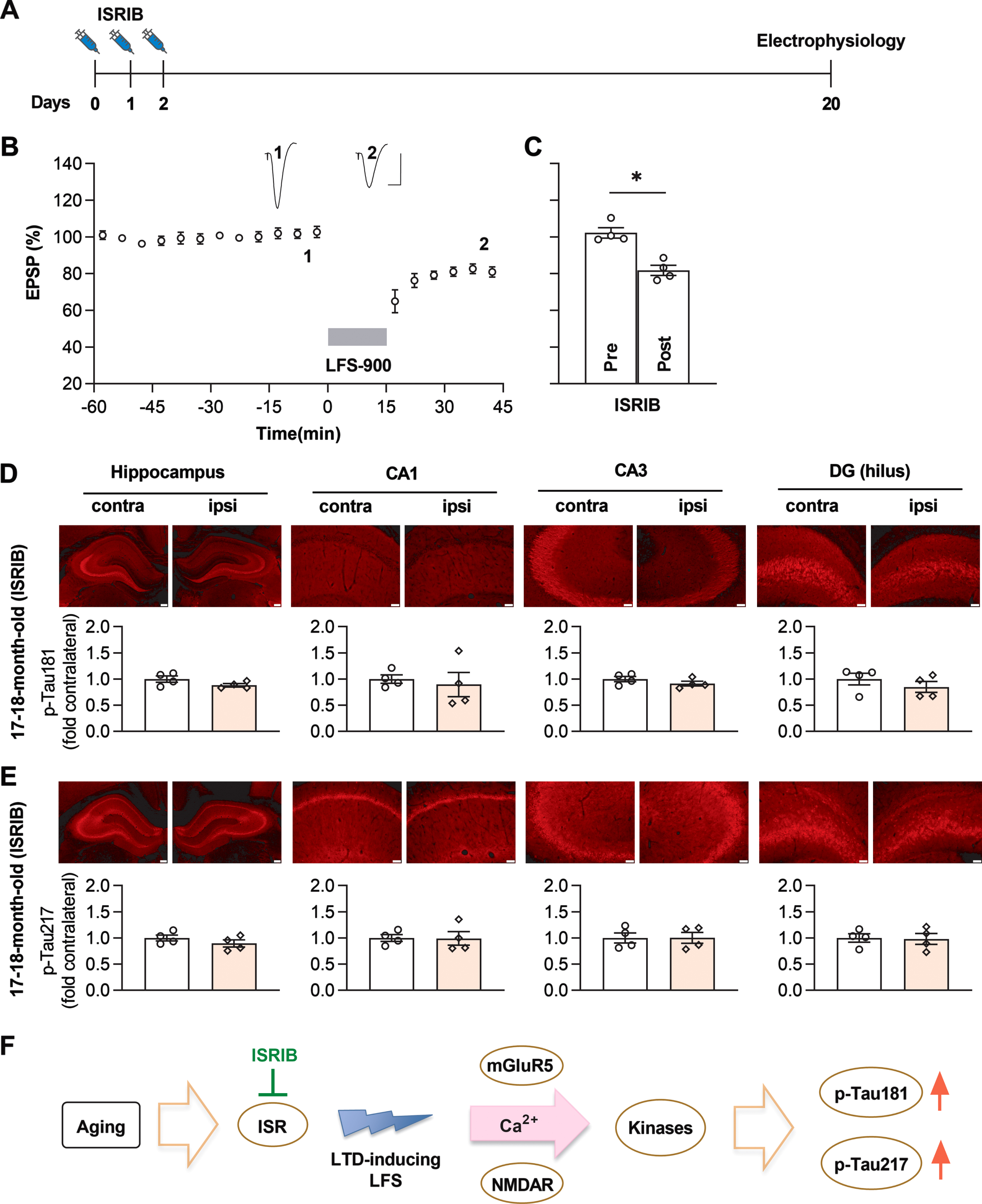
Targeting the ISR with a small molecule inhibitor ISRIB blocks the enhancement of p-Tau181 and p-Tau217 by LFS in aged rats
Given that induction of LTD by LFS enhances p-Tau181 and p-Tau217 in an age-dependent manner and the ISR is aberrantly elevated in aged brain [42], we hypothesized that suppressing the ISR with ISRIB [42] might prevent the elevation of p-Tau181 and p-Tau217 by LFS. To test this hypothesis, we treated relatively aged (17-18-month-old) rats with the same ISRIB treatment paradigm of daily injections (2.5 mg/kg, i.p.) on 3 consecutive days as reported [42], and in vivo electrophysiological experiments were carried out on these animals 18 days after the last injection of ISRIB (Fig. 5A). LFS induced stable and robust LTD in all these rats (Fig. 5B, C). Assessed by immunofluorescence, induction of LTD by LFS did not lead to a change in total tau expression level in ISRIB-treated rats (Supplementary Figure 13). Intriguingly, neither p-Tau181 (Fig. 5D) nor p-Tau217 (Fig. 5E) were increased by the same LFS conditioning protocol which triggered enhancement of both p-Tau181 and p-Tau217 in untreated age-matched rats (Figs. 1E and Fig. 2B).
Fig. 5
ISRIB blocks LFS-induced enhancement of p-Tau181 and p-Tau217 in aged rats. A) Experimental paradigm for ISRIB injections and electrophysiology experiments. ISRIB (2.5 mg/kg, i.p.) was systemically injected for 3 days and in vivo electrophysiology experiments were performed 18 days after the last injection. B) LFS induced robust LTD in ISRIB-treated aged rats. Calibration bars: vertical, 2 mV; horizontal, 10 ms. C) The EPSP decreased to 81.8±2.7% at 30 min post LFS (n = 4, p = 0.0211 compared with pre, paired t test). D) Treatment of ISRIB completely prevented the increase of p-Tau181 induced by LFS in aged rats. The upper panel shows the fluorescent images of p-Tau181 labeling (red). As summarized in the lower panel, no significant difference was found in the dorsal hippocampus (p = 0.0579), CA1 (p = 0.7006), CA3 (p = 0.0715), and DG (p = 0.2696); paired t test. E) Treatment with ISRIB also successfully blocked the enhancement of p-Tau217 induced by LFS in aged rats. The upper panel shows the fluorescent images of p-Tau217 labeling (red). Summarized statistical results in the lower panel show no significant difference in all regions, including dorsal hippocampus (p = 0.1185), CA1 (p = 0.9049), CA3 (p = 0.9172), and DG (p = 0.6636); paired t test. Scale bar = 200μm in hippocampus, scale bar = 50μm in CA1, CA3 and DG regions. F) Schematic diagram showing proposed mechanism of LTD-inducing LFS-triggered tau phosphorylation in aged rats. The ISR is enhanced in aged brain, resulting in increased susceptibility to cellular processes underlying the induction of mGluR and NMDAR-dependent LTD either directly or indirectly. Facilitation of certain LTD-associated kinases, such as GSK3α/β or Cdk5, may phosphorylate tau at residues Thr181 and Thr217 with high sensitivity. Values are mean±s.e.m.
DISCUSSION
This study reveals that aging enables the elevation of p-Tau181 and p-Tau217 triggered by LTD-inducing conditioning stimulation in live rats. The enhancement of tau phosphorylation at these residues by LFS appeared more sensitive compared with residues Thr231, Ser202/Thr205, and Ser396. Pharmacological antagonism of either NMDAR or mGluR5, or a small molecule ISR inhibitor ISRIB inhibited/prevented the elevation of both p-Tau181 and p-Tau217 by LFS in aged rats.
Growing evidence from many different memory center cohorts indicates that p-Tau181 and p-Tau217 distinguish AD from controls with high accuracy very early in the disease process [9, 54]. These findings encourage the prospect that these markers are good enough to become early AD diagnostic tools. Our data provide a novel in vivo means to uncover brain plasticity-related cellular and molecular processes of tau phosphorylation at these key sites in health and aging. Because LTD induction by LFS at CA3-CA1 synapses in the hippocampus usually necessitates transient elevated synaptic glutamate release and subsequent activation of NMDARs and/or mGluRs [32], we wondered if the enhanced tau phosphorylation triggered by LFS entails these receptors also. Pretreatment with either the NMDAR or mGluR5 antagonist alone completely abrogated the LFS-induced tau phosphorylation. If the activation of either glutamate receptor subtype alone was sufficient to trigger the increased tau phosphorylation by LFS, co-application of both antagonists together would have been required to prevent the increase. Therefore, given our finding that either antagonist alone was effective, it seems likely that both receptors need to be co-activated for LFS to trigger enhanced p-Tau181 and p-Tau217. Although we only examined one time point, it is clear that LFS triggers an NMDAR and mGluR5-dependent increase in tau phosphorylation that persists for at least 30 min in all hippocampal subfields examined. Whereas the induction of in vivo LTD is only blocked by combined pretreatment with standard doses of CPP and MTEP [47], injecting either agent alone prevented LFS-triggered tau phosphorylation. In view of these findings, it seems unlikely that enhanced tau phosphorylation at either residues Thr181 or Thr217 is essential for LTD at CA3-CA1 synapses in vivo. In apparent contrast, a tau-dependent form of NMDAR-dependent LTD has been reported to require tau phosphorylation at Thr396 in vitro [29, 30]. However, our findings do not exclude the possibility that background, rather than LFS-triggered, tau phosphorylation at these or other sites may be sufficient to enable LTD induction in vivo. Also, it remains to be determined if the LFS protocol used in the present study triggers LTD at CA3 recurrent collaterals [55] or back projections as reported previously. Recent evidence implicates a particular role for extrasynaptic NMDAR in LTD induction [33, 34]. In the hippocampal CA1 area, mGluR5 is found predominantly located perisynaptically and extrasynaptically on postsynaptic spines of pyramidal cells [56, 57]. Co-activation of mGluR5 with NMDARs located in this vicinity is known to strongly enhance the function of the NMDARs, in particular GluN2B-containing NMDARs [58, 59]. Numerous studies have shown that synaptic NMDARs are mainly involved in normal cognitive function, while extrasynaptic NMDARs are important mediators of neurotoxicity [60]. Glutamate-induced excitotoxicity increases tau phosphorylation [61] and more recent evidence indicates the involvement of extrasynaptic NMDARs activation in tau pathology [39, 40]. Tau is phosphorylated at many different sites by different protein kinases. Both glycogen synthase kinase 3β (GSK3β) and cyclin-dependent kinase-5 (Cdk5) can phosphorylate tau at Thr181 and Thr217 [62]. The activity of GSK3β is significantly enhanced during hippocampal LTD [63] but very recent evidence indicates the involvement of glycogen synthase kinase 3α (GSK3α) in both NMDAR-dependent [64] and mGluR5-dependent [65] LTD. Cdk5 activation appears to be required for NMDAR-dependent LTD at CA3-CA1 synapses also [66].
Age-related changes in brain make it especially susceptible to most neurodegenerative diseases including AD [67]. Age-related reduction in glutamate uptake is associated with extrasynaptic NMDAR and mGluR activation at hippocampal CA1 synapses [68]. Very recent evidence indicates that imbalanced synaptic weights underlie the aberrant elevated firing characteristics of both CA3 and CA1 pyramidal neurons in aged, learning-impaired rats [25]. Intriguingly, Krukowski et al. discovered that ISRIB reverses the aberrantly elevated ISR in aged mice brain and restores age-related changes in hippocampal neuron function [42]. In the present study, the same ISRIB treatment paradigm successfully prevented the elevation of both p-Tau181 and p-Tau217 by LFS in aged rats (Fig. 5F).
As noted in the Introduction, increasing the ISR can facilitate mGluR-LTD [43, 44] while suppressing it has the opposite effect [43–46]. Enhanced mGluR-LTD has also been found in models of different neurological diseases including AD [37, 44, 69] and aberrant elevated ISR has been found in sporadic AD patients’ brains [70–76]. Moreover, ISRIB has been found to have beneficial effects in neurodegeneration models [77] including AD models [44, 76]. Although the safety of ISRIB in humans has yet to be evaluated in clinical trials, given that the action of ISRIB is state-dependent and it lacks the pancreatic toxicity of the protein kinase R-like endoplasmic reticulum kinase inhibitors [78], targeting the ISR with ISRIB-like agents may provide a feasible therapeutic strategy for AD [79].
Previous reports indicate that p-Tau396 can be enhanced by LFS-900 (1 Hz) in acute hippocampal slices [29, 30]. In contrast, no change in p-Tau202/205 was triggered by LFS except under the extreme condition of 2 h LTD induction over a period of 7 days in slice cultures [31]. The experimental conditions used to detect changes in p-Tau396 in hippocampal slices differ very significantly from our live brain studies, including relatively young age and non-physiological temperature. It is well recognized that p-Tau is developmentally regulated [80–84] and can be hypothermia-induced [85–88]. Although not statistically significant, we found slight elevation of p-Tau396 triggered by LFS-900 in vivo in this study. Nevertheless, future studies should elucidate if, like the present in vivo studies, changes in p-Tau181 and p-Tau217 are more sensitive to LTD-inducing LFS in vitro. Phosphorylated tau species have faster tau turnover rates and shorter half-lives in cultured human and rodent neurons compared with that in human and rodent brains [89]. LTD of synaptic transmission can be induced in human hippocampal slices from apparently non-pathological tissue resected from patients undergoing surgery for epilepsy and brain tumors [90]. LTD-like plasticity can also be induced in intact human brain cortex by non-invasive brain stimulation techniques, such as transcranial magnetic stimulation [91] or transcranial direct current stimulation but how this LTD-like plasticity relates to synaptic LTD remains to be determined [92, 93]. Thus, our findings in live animals provide a valuable experimental model to directly study the generation of p-Tau during learning and memory-related synaptic plasticity.
We want to also note some limitations of this study. AD is a complex disease and the neuropathological hallmarks of AD include extracellular amyloid-β (Aβ) plaques and cerebral amyloid angiopathy, intracellular hyperphosphorylated tau-containing neurofibrillary tangles, and glial responses [94]. Accumulating evidence suggests that AD most likely involves the interplay between Aβ and tau [95]. Taylor et al. recently discovered that tau hyperphosphorylation caused by either Aβ or LTD-inducing LFS share similar mechanisms in organotypic hippocampal slices [31]. The current study presents the age dependence of the preferential tau phosphorylation triggered by LTD-inducing LFS in the absence of synaptotoxic Aβ conditions in vivo. Previously, we and others reported that Aβ oligomers facilitate a novel form of LTD in hippocampus [35, 37, 38, 44]. Future studies should examine the mechanisms of tau phosphorylation triggered by LTD-inducing LFS in the presence of synaptotoxic Aβ in vivo.
Further highlighting that AD is a chronic disease, it is feasible to carry out more detailed time course analysis of phosphorylated tau in our live animal models. To conclude, our data in this study show that, similar to clinical biomarker findings, tau phosphorylation is preferentially triggered at certain sites including Thr181 and Thr217 by LTD-inducing conditioning stimulation in aged brain in live rats. Thus, our experimental models provide a valuable means to 1) directly study the generation of p-Tau during learning and memory-related synaptic plasticity, and 2) aid in the selection of p-Tau directed therapies in early AD.
ACKNOWLEDGMENTS
This study has been funded by National Natural Science Foundation of China (U2004134) and Zhengzhou University (140/32310295) to NWH, and by Science Foundation Ireland (19/FFP/6437 and 14/IA/2571) to MJR. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We thank Professor Seán Kennelly (Tallaght University Hospital, Trinity College Dublin) and Professor Tim Lynch (Mater Misericordiae University Hospital, University College Dublin) for advice.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/22-0351r1).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-220351.
REFERENCES
[1] | Wegmann S , Bennett RE , Delorme L , Robbins AB , Hu M , McKenzie D , Kirk MJ , Schiantarelli J , Tunio N , Amaral AC , Fan Z , Nicholls S , Hudry E , Hyman BT ((2019) ) Experimental evidence for the age dependence of tau protein spread in the brain, Sci Adv 5: , eaaw6404. |
[2] | Wesseling H , Mair W , Kumar M , Schlaffner CN , Tang S , Beerepoot P , Fatou B , Guise AJ , Cheng L , Takeda S , Muntel J , Rotunno MS , Dujardin S , Davies P , Kosik KS , Miller BL , Berretta S , Hedreen JC , Grinberg LT , Seeley WW , Hyman BT , Steen H , Steen JA ((2020) ) Tau PTM profiles identify patient heterogeneity and stages of Alzheimer’s disease. Cell 183: , 1699–1713. |
[3] | Chang CW , Shao E , Mucke L ((2021) ) Tau: Enabler of diverse brain disorders and target of rapidly evolving therapeutic strategies., Science 371: , eabb8255. |
[4] | Nies SH , Takahashi H , Herber CS , Huttner A , Chase A , Strittmatter SM ((2021) ) Spreading of Alzheimer tau seeds is enhanced by aging and template matching with limited impact of amyloid-beta, J Biol Chem 297: , 101159. |
[5] | Barthelemy NR , Horie K , Sato C , Bateman RJ ((2020) ) Blood plasma phosphorylated-tau isoforms track CNS change in Alzheimer’s disease., J Exp Med 217: , e20200861. |
[6] | Barthelemy NR , Li Y , Joseph-Mathurin N , Gordon BA , Hassenstab J , Benzinger TLS , Buckles V , Fagan AM , Perrin RJ , Goate AM , Morris JC , Karch CM , Xiong C , Allegri R , Mendez PC , Berman SB , Ikeuchi T , Mori H , Shimada H , Shoji M , Suzuki K , Noble J , Farlow M , Chhatwal J , Graff-Radford NR , Salloway S , Schofield PR , Masters CL , Martins RN , O’Connor A , Fox NC , Levin J , Jucker M , Gabelle A , Lehmann S , Sato C , Bateman RJ , McDade E , Dominantly Inherited Alzheimer Network ((2020) ) A soluble phosphorylated tau signature links tau, amyloid and the evolution of stages of dominantly inherited Alzheimer’s disease. Nat Med 26: , 398–407. |
[7] | Karikari TK , Pascoal TA , Ashton NJ , Janelidze S , Benedet AL , Rodriguez JL , Chamoun M , Savard M , Kang MS , Therriault J , Scholl M , Massarweh G , Soucy JP , Hoglund K , Brinkmalm G , Mattsson N , Palmqvist S , Gauthier S , Stomrud E , Zetterberg H , Hansson O , Rosa-Neto P , Blennow K ((2020) ) Blood phosphorylated tau 181 as a biomarker for Alzheimer’s disease: A diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol 19: , 422–433. |
[8] | O’Connor A , Karikari TK , Poole T , Ashton NJ , Lantero Rodriguez J , Khatun A , Swift I , Heslegrave AJ , Abel E , Chung E , Weston PSJ , Pavisic IM , Ryan NS , Barker S , Rossor MN , Polke JM , Frost C , Mead S , Blennow K , Zetterberg H , Fox NC ((2020) ) Plasma phospho-tau181 in presymptomatic and symptomatic familial Alzheimer’s disease: A longitudinal cohort study. Mol Psychiatry 26: , 5967–5976. |
[9] | Hansson O ((2021) ) Biomarkers for neurodegenerative diseases. Nat Med 27: , 954–963. |
[10] | Palmqvist S , Tideman P , Cullen N , Zetterberg H , Blennow K , Alzheimer’s Disease Neuroimaging Initiative, Dage JL , Stomrud E , Janelidze S , Mattsson-Carlgren N , Hansson O ((2021) ) Prediction of future Alzheimer’s disease dementia using plasma phospho-tau combined with other accessible measures. Nat Med 27: , 1034–1042. |
[11] | Wegmann S , Biernat J , Mandelkow E ((2021) ) A current view on Tau protein phosphorylation in Alzheimer’s disease. Curr Opin Neurobiol 69: , 131–138. |
[12] | Zetterberg H , Blennow K ((2021) ) Moving fluid biomarkers for Alzheimer’s disease from research tools to routine clinical diagnostics. Mol Neurodegener 16: , 10. |
[13] | Leuzy A , Cullen NC , Mattsson-Carlgren N , Hansson O ((2021) ) Current advances in plasma and cerebrospinal fluid biomarkers in Alzheimer’s disease. Curr Opin Neurol 34: , 266–274. |
[14] | Karikari TK , Ashton NJ , Brinkmalm G , Brum WS , Benedet AL , Montoliu-Gaya L , Lantero-Rodriguez J , Pascoal TA , Suarez-Calvet M , Rosa-Neto P , Blennow K , Zetterberg H ((2022) ) Blood phospho-tau in Alzheimer disease: Analysis, interpretation, and clinical utility. Nat Rev Neurol 18: , 400–418. |
[15] | Imbimbo BP , Ippati S , Watling M , Balducci C ((2021) ) A critical appraisal of tau-targeting therapies for primary and secondary tauopathies. Alzheimers Dement 18: , 1008–1037. |
[16] | Xia Y , Prokop S , Giasson BI ((2021) ) “Don’t Phos Over Tau": Recent developments in clinical biomarkers and therapies targeting tau phosphorylation in Alzheimer’s disease and other tauopathies. Mol Neurodegener 16: , 37. |
[17] | Zhang Y , Wu KM , Yang L , Dong Q , Yu JT ((2022) ) Tauopathies: New perspectives and challenges. Mol Neurodegener 17: , 28. |
[18] | Ballatore C , Lee VM , Trojanowski JQ ((2007) ) Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci 8: , 663–672. |
[19] | Aragao Gomes L , Uytterhoeven V , Lopez-Sanmartin D , Tome SO , Tousseyn T , Vandenberghe R , Vandenbulcke M , von Arnim CAF , Verstreken P , Thal DR ((2021) ) Maturation of neuronal AD-tau pathology involves site-specific phosphorylation of cytoplasmic and synaptic tau preceding conformational change and fibril formation. Acta Neuropathol 141: , 173–192. |
[20] | Braak H , Braak E ((1991) ) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82: , 239–259. |
[21] | Braak H , Alafuzoff I , Arzberger T , Kretzschmar H , Del Tredici K ((2006) ) Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112: , 389–404. |
[22] | Driscoll I , Hamilton DA , Petropoulos H , Yeo RA , Brooks WM , Baumgartner RN , Sutherland RJ ((2003) ) The aging hippocampus: Cognitive, biochemical and structural findings. Cereb Cortex 13: , 1344–1351. |
[23] | Ianov L , De Both M , Chawla MK , Rani A , Kennedy AJ , Piras I , Day JJ , Siniard A , Kumar A , Sweatt JD , Barnes CA , Huentelman MJ , Foster TC ((2017) ) Hippocampal transcriptomic profiles: Subfield vulnerability to age and cognitive impairment. Front Aging Neurosci 9: , 383. |
[24] | McKiernan EC , Marrone DF ((2017) ) CA1 pyramidal cells have diverse biophysical properties, affected by development, experience, and aging, PeerJ 5: , e3836. |
[25] | Buss EW , Corbett NJ , Roberts JG , Ybarra N , Musial TF , Simkin D , Molina-Campos E , Oh KJ , Nielsen LL , Ayala GD , Mullen SA , Farooqi AK , D’Souza GX , Hill CL , Bean LA , Rogalsky AE , Russo ML , Curlik DM , Antion MD , Weiss C , Chetkovich DM , Oh MM , Disterhoft JF , Nicholson DA ((2021) ) Cognitive aging is associated with redistribution of synaptic weights in the hippocampus, Proc Natl Acad Sci U S A 118: , e1921481118. |
[26] | Veldsman M , Nobis L , Alfaro-Almagro F , Manohar S , Husain M ((2021) ) The human hippocampus and its subfield volumes across age, sex and APOE ɛ4 status, Brain Commun 3: , fcaa219. |
[27] | Connor SA , Wang YT ((2016) ) A place at the table: LTD as a mediator of memory genesis. Neuroscientist 22: , 359–371. |
[28] | Magee JC , Grienberger C ((2020) ) Synaptic plasticity forms and functions. Annu Rev Neurosci 43: , 95–117. |
[29] | Kimura T , Whitcomb DJ , Jo J , Regan P , Piers T , Heo S , Brown C , Hashikawa T , Murayama M , Seok H , Sotiropoulos I , Kim E , Collingridge GL , Takashima A , Cho K ((2014) ) Microtubule-associated protein tau is essential for long-term depression in the hippocampus. Philos Trans R Soc Lond B Biol Sci 369: , 20130144. |
[30] | Regan P , Piers T , Yi JH , Kim DH , Huh S , Park SJ , Ryu JH , Whitcomb DJ , Cho K ((2015) ) Tau phosphorylation at serine 396 residue is required for hippocampal LTD. J Neurosci 35: , 4804–4812. |
[31] | Taylor HBC , Emptage NJ , Jeans AF ((2021) ) Long-term depression links amyloid-beta to the pathological hyperphosphorylation of tau. Cell Rep 36: , 109638. |
[32] | Collingridge GL , Peineau S , Howland JG , Wang YT ((2010) ) Long-term depression in the CNS. Nat Rev Neurosci 11: , 459–473. |
[33] | Papouin T , Ladepeche L , Ruel J , Sacchi S , Labasque M , Hanini M , Groc L , Pollegioni L , Mothet JP , Oliet SH ((2012) ) Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell 150: , 633–646. |
[34] | Liu DD , Yang Q , Li ST ((2013) ) Activation of extrasynaptic NMDA receptors induces LTD in rat hippocampal CA1 neurons. Brain Res Bull 93: , 10–16. |
[35] | Li S , Hong S , Shepardson NE , Walsh DM , Shankar GM , Selkoe D ((2009) ) Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 62: , 788–801. |
[36] | Luscher C , Huber KM ((2010) ) Group 1 mGluR-dependent synaptic long-term depression: Mechanisms and implications for circuitry and disease. Neuron 65: , 445–459. |
[37] | Hu NW , Nicoll AJ , Zhang D , Mably AJ , O’Malley T , Purro SA , Terry C , Collinge J , Walsh DM , Rowan MJ ((2014) ) mGlu5 receptors and cellular prion protein mediate amyloid-beta-facilitated synaptic long-term depression in vivo. Nat Commun 5: , 3374. |
[38] | O’Riordan KJ , Hu NW , Rowan MJ ((2018) ) Aβ facilitates LTD at Schaffer collateral synapses preferentially in the left hippocampus. Cell Rep 22: , 2053–2065. |
[39] | Tackenberg C , Grinschgl S , Trutzel A , Santuccione AC , Frey MC , Konietzko U , Grimm J , Brandt R , Nitsch RM ((2013) ) NMDA receptor subunit composition determines beta-amyloid-induced neurodegeneration and synaptic loss, Cell Death Dis 4: , e608. |
[40] | Sun XY , Tuo QZ , Liuyang ZY , Xie AJ , Feng XL , Yan X , Qiu M , Li S , Wang XL , Cao FY , Wang XC , Wang JZ , Liu R ((2016) ) Extrasynaptic NMDA receptor-induced tau overexpression mediates neuronal death through suppressing survival signaling ERK phosphorylation, Cell Death Dis 7: , e2449. |
[41] | Benarroch EE ((2018) ) Glutamatergic synaptic plasticity and dysfunction in Alzheimer disease: Emerging mechanisms. Neurology 91: , 125–132. |
[42] | Krukowski K , Nolan A , Frias ES , Boone M , Ureta G , Grue K , Paladini MS , Elizarraras E , Delgado L , Bernales S , Walter P , Rosi S ((2020) ) Small molecule cognitive enhancer reverses age-related memory decline in mice, Elife 9: , e62048. |
[43] | Di Prisco GV , Huang W , Buffington SA , Hsu CC , Bonnen PE , Placzek AN , Sidrauski C , Krnjevic K , Kaufman RJ , Walter P , Costa-Mattioli M ((2014) ) Translational control of mGluR-dependent long-term depression and object-place learning by eIF2alpha. Nat Neurosci 17: , 1073–1082. |
[44] | Hu Z , Yu P , Zhang Y , Yang Y , Zhu M , Qin S , Xu JT , Duan D , Wu Y , Wang D , Rowan MJ , Hu NW ((2022) ) Inhibition of the ISR abrogates mGluR5-dependent long-term depression and spatial memory deficits in a rat model of Alzheimer’s disease. Transl Psychiatry 12: , 96. |
[45] | Trinh MA , Ma T , Kaphzan H , Bhattacharya A , Antion MD , Cavener DR , Hoeffer CA , Klann E ((2014) ) The eIF2alpha kinase PERK limits the expression of hippocampal metabotropic glutamate receptor-dependent long-term depression. Learn Mem 21: , 298–304. |
[46] | Huang W , Placzek AN , Viana Di Prisco G , Khatiwada S , Sidrauski C , Krnjevic K , Walter P , Dani JA , Costa-Mattioli M ((2016) ) Translational control by eIF2alpha phosphorylation regulates vulnerability to the synaptic and behavioral effects of cocaine, Elife 5: , e12052. |
[47] | O’Riordan KJ , Hu NW , Rowan MJ ((2018) ) Physiological activation of mGlu5 receptors supports the ion channel function of NMDA receptors in hippocampal LTD induction in vivo. Sci Rep 8: , 4391. |
[48] | Ondrejcak T , Hu NW , Qi Y , Klyubin I , Corbett GT , Fraser G , Perkinton MS , Walsh DM , Billinton A , Rowan MJ ((2019) ) Soluble tau aggregates inhibit synaptic long-term depression and amyloid beta-facilitated LTD in vivo. Neurobiol Dis 127: , 582–590. |
[49] | Percie du Sert N , Hurst V , Ahluwalia A , Alam S , Avey MT , Baker M , Browne WJ , Clark A , Cuthill IC , Dirnagl U , Emerson M , Garner P , Holgate ST , Howells DW , Karp NA , Lazic SE , Lidster K , MacCallum CJ , Macleod M , Pearl EJ , Petersen OH , Rawle F , Reynolds P , Rooney K , Sena ES , Silberberg SD , Steckler T , Wurbel H ((2020) ) The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research, PLoS Biol 18: , e3000410. |
[50] | Kandimalla R , Manczak M , Yin X , Wang R , Reddy PH ((2018) ) Hippocampal phosphorylated tau induced cognitive decline, dendritic spine loss and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum Mol Genet 27: , 30–40. |
[51] | Wu JW , Hussaini SA , Bastille IM , Rodriguez GA , Mrejeru A , Rilett K , Sanders DW , Cook C , Fu H , Boonen RA , Herman M , Nahmani E , Emrani S , Figueroa YH , Diamond MI , Clelland CL , Wray S , Duff KE ((2016) ) Neuronal activity enhances tau propagation and tau pathology in vivo. Nat Neurosci 19: , 1085–1092. |
[52] | Braun NJ , Yao KR , Alford PW , Liao D ((2020) ) Mechanical injuries of neurons induce tau mislocalization to dendritic spines and tau-dependent synaptic dysfunction. Proc Natl Acad Sci U S A 117: , 29069–29079. |
[53] | Busse CS , Brodkin J , Tattersall D , Anderson JJ , Warren N , Tehrani L , Bristow LJ , Varney MA , Cosford ND ((2004) ) The behavioral profile of the potent and selective mGlu5 receptor antagonist 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]pyridine (MTEP) in rodent models of anxiety. Neuropsychopharmacology 29: , 1971–1979. |
[54] | Teunissen CE , Verberk IMW , Thijssen EH , Vermunt L , Hansson O , Zetterberg H , van der Flier WM , Mielke MM , Del Campo M ((2022) ) Blood-based biomarkers for Alzheimer’s disease: Towards clinical implementation. Lancet Neurol 21: , 66–77. |
[55] | Debanne D , Gahwiler BH , Thompson SM ((1998) ) Long-term synaptic plasticity between pairs of individual CA3 pyramidal cells in rat hippocampal slice cultures. J Physiol 507: (Pt 1), 237–247. |
[56] | Lujan R , Nusser Z , Roberts JD , Shigemoto R , Somogyi P ((1996) ) Perisynaptic location of metabotropic glutamate receptors mGluR1 and mGluR5 on dendrites and dendritic spines in the rat hippocampus. Eur J Neurosci 8: , 1488–1500. |
[57] | Lujan R , Roberts JD , Shigemoto R , Ohishi H , Somogyi P ((1997) ) Differential plasma membrane distribution of metabotropic glutamate receptors mGluR1 alpha, mGluR2 and mGluR5, relative to neurotransmitter release sites. J Chem Neuroanat 13: , 219–241. |
[58] | Kotecha SA , Jackson MF , Al-Mahrouki A , Roder JC , Orser BA , MacDonald JF ((2003) ) Co-stimulation of mGluR5 and N-methyl-D-aspartate receptors is required for potentiation of excitatory synaptic transmission in hippocampal neurons. J Biol Chem 278: , 27742–27749. |
[59] | Sarantis K , Tsiamaki E , Kouvaros S , Papatheodoropoulos C , Angelatou F ((2015) ) Adenosine A(2)A receptors permit mGluR5-evoked tyrosine phosphorylation of NR2B (Tyr1472) in rat hippocampus: A possible key mechanism in NMDA receptor modulation. J Neurochem 135: , 714–726. |
[60] | Vieira M , Yong XLH , Roche KW , Anggono V ((2020) ) Regulation of NMDA glutamate receptor functions by the GluN2 subunits. J Neurochem 154: , 121–143. |
[61] | Sindou P , Lesort M , Couratier P , Yardin C , Esclaire F , Hugon J ((1994) ) Glutamate increases tau phosphorylation in primary neuronal cultures from fetal rat cerebral cortex. Brain Res 646: , 124–128. |
[62] | Liu F , Iqbal K , Grundke-Iqbal I , Gong CX ((2002) ) Involvement of aberrant glycosylation in phosphorylation of tau by cdk5 and GSK-3beta. FEBS Lett 530: , 209–214. |
[63] | Peineau S , Taghibiglou C , Bradley C , Wong TP , Liu L , Lu J , Lo E , Wu D , Saule E , Bouschet T , Matthews P , Isaac JT , Bortolotto ZA , Wang YT , Collingridge GL ((2007) ) LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron 53: , 703–717. |
[64] | Draffin JE , Sanchez-Castillo C , Fernandez-Rodrigo A , Sanchez-Saez X , Avila J , Wagner FF , Esteban JA ((2021) ) GSK3alpha, not GSK3beta, drives hippocampal NMDAR-dependent LTD via tau-mediated spine anchoring. , e, EMBO J 40: , 105513. |
[65] | McCamphill PK , Stoppel LJ , Senter RK , Lewis MC , Heynen AJ , Stoppel DC , Sridhar V , Collins KA , Shi X , Pan JQ , Madison J , Cottrell JR , Huber KM , Scolnick EM , Holson EB , Wagner FF , Bear MF ((2020) ) Selective inhibition of glycogen synthase kinase 3alpha corrects pathophysiology in a mouse model of fragile X syndrome, Sci Transl Med 12: , eaam8572. |
[66] | Mishiba T , Tanaka M , Mita N , He X , Sasamoto K , Itohara S , Ohshima T ((2014) ) Cdk5/p35 functions as a crucial regulator of spatial learning and memory. Mol Brain 7: , 82. |
[67] | Hou Y , Dan X , Babbar M , Wei Y , Hasselbalch SG , Croteau DL , Bohr VA ((2019) ) Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol 15: , 565–581. |
[68] | Potier B , Billard JM , Riviere S , Sinet PM , Denis I , Champeil-Potokar G , Grintal B , Jouvenceau A , Kollen M , Dutar P ((2010) ) Reduction in glutamate uptake is associated with extrasynaptic NMDA and metabotropic glutamate receptor activation at the hippocampal CA1 synapse of aged rats. Aging Cell 9: , 722–735. |
[69] | Nosyreva ED , Huber KM ((2006) ) Metabotropic receptor-dependent long-term depression persists in the absence of protein synthesis in the mouse model of fragile X syndrome. J Neurophysiol 95: , 3291–3295. |
[70] | Chang RC , Wong AK , Ng HK , Hugon J ((2002) ) Phosphorylation of eukaryotic initiation factor-2alpha (eIF2alpha) is associated with neuronal degeneration in Alzheimer’s disease. Neuroreport 13: , 2429–2432. |
[71] | Hoozemans JJ , Veerhuis R , Van Haastert ES , Rozemuller JM , Baas F , Eikelenboom P , Scheper W ((2005) ) The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol 110: , 165–172. |
[72] | Kim HS , Choi Y , Shin KY , Joo Y , Lee YK , Jung SY , Suh YH , Kim JH ((2007) ) Swedish amyloid precursor protein mutation increases phosphorylation of eIF2alpha in vitro and in vivo. J Neurosci Res 85: , 1528–1537. |
[73] | O’Connor T , Sadleir KR , Maus E , Velliquette RA , Zhao J , Cole SL , Eimer WA , Hitt B , Bembinster LA , Lammich S , Lichtenthaler SF , Hebert SS , De Strooper B , Haass C , Bennett DA , Vassar R ((2008) ) Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron 60: , 988–1009. |
[74] | Mouton-Liger F , Paquet C , Dumurgier J , Bouras C , Pradier L , Gray F , Hugon J ((2012) ) Oxidative stress increases BACE1 protein levels through activation of the PKR-eIF2alpha pathway. Biochim Biophys Acta 1822: , 885–896. |
[75] | Natunen T , Parrado AR , Helisalmi S , Pursiheimo JP , Sarajarvi T , Makinen P , Kurkinen KM , Mullin K , Alafuzoff I , Haapasalo A , Bertram L , Soininen H , Tanzi RE , Hiltunen M ((2013) ) Elucidation of the BACE1 regulating factor GGA3 in Alzheimer’s disease. J Alzheimers Dis 37: , 217–232. |
[76] | Oliveira MM , Lourenco MV , Longo F , Kasica NP , Yang W , Ureta G , Ferreira DDP , Mendonca PHJ , Bernales S , Ma T , De Felice FG , Klann E , Ferreira ST ((2021) ) Correction of eIF2-dependent defects in brain protein synthesis, synaptic plasticity, and memory in mouse models of Alzheimer’s disease, Sci Signal 14: , eabc5429. |
[77] | Halliday M , Radford H , Sekine Y , Moreno J , Verity N , le Quesne J , Ortori CA , Barrett DA , Fromont C , Fischer PM , Harding HP , Ron D , Mallucci GR ((2015) ) Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity, Cell Death Dis 6: , e1672. |
[78] | Rabouw HH , Langereis MA , Anand AA , Visser LJ , de Groot RJ , Walter P , van Kuppeveld FJM ((2019) ) Small molecule ISRIB suppresses the integrated stress response within a defined window of activation. Proc Natl Acad Sci U S A 116: , 2097–2102. |
[79] | Costa-Mattioli M , Walter P ((2020) ) The integrated stress response: From mechanism to disease, Science 368: , eaat5314. |
[80] | Bramblett GT , Goedert M , Jakes R , Merrick SE , Trojanowski JQ , Lee VM ((1993) ) Abnormal tau phosphorylation at Ser396 in Alzheimer’s disease recapitulates development and contributes to reduced microtubule binding. Neuron 10: , 1089–1099. |
[81] | Brion JP , Smith C , Couck AM , Gallo JM , Anderton BH ((1993) ) Developmental changes in tau phosphorylation: Fetal tau is transiently phosphorylated in a manner similar to paired helical filament-tau characteristic of Alzheimer’s disease. J Neurochem 61: , 2071–2080. |
[82] | Goedert M , Jakes R , Crowther RA , Six J , Lubke U , Vandermeeren M , Cras P , Trojanowski JQ , Lee VM ((1993) ) The abnormal phosphorylation of tau protein at Ser-202 in Alzheimer disease recapitulates phosphorylation during development. Proc Natl Acad Sci U S A 90: , 5066–5070. |
[83] | Yu Y , Run X , Liang Z , Li Y , Liu F , Liu Y , Iqbal K , Grundke-Iqbal I , Gong CX ((2009) ) Developmental regulation of tau phosphorylation, tau kinases, and tau phosphatases. J Neurochem 108: , 1480–1494. |
[84] | Hefti MM , Kim S , Bell AJ , Betters RK , Fiock KL , Iida MA , Smalley ME , Farrell K , Fowkes ME , Crary JF ((2019) ) Tau phosphorylation and aggregation in the developing human brain. J Neuropathol Exp Neurol 78: , 930–938. |
[85] | Avila J , Diaz-Nido J ((2004) ) Tangling with hypothermia. Nat Med 10: , 460–461. |
[86] | Planel E , Miyasaka T , Launey T , Chui DH , Tanemura K , Sato S , Murayama O , Ishiguro K , Tatebayashi Y , Takashima A ((2004) ) Alterations in glucose metabolism induce hypothermia leading to tau hyperphosphorylation through differential inhibition of kinase and phosphatase activities: Implications for Alzheimer’s disease. J Neurosci 24: , 2401–2411. |
[87] | Bretteville A , Marcouiller F , Julien C , El Khoury NB , Petry FR , Poitras I , Mouginot D , Levesque G , Hebert SS , Planel E ((2012) ) Hypothermia-induced hyperphosphorylation: A new model to study tau kinase inhibitors. Sci Rep 2: , 480. |
[88] | Gratuze M , El Khoury NB , Turgeon A , Julien C , Marcouiller F , Morin F , Whittington RA , Marette A , Calon F , Planel E ((2017) ) Tau hyperphosphorylation in the brain of ob/ob mice is due to hypothermia: Importance of thermoregulation in linking diabetes and Alzheimer’s disease. Neurobiol Dis 98: , 1–8. |
[89] | Sato C , Barthelemy NR , Mawuenyega KG , Patterson BW , Gordon BA , Jockel-Balsarotti J , Sullivan M , Crisp MJ , Kasten T , Kirmess KM , Kanaan NM , Yarasheski KE , Baker-Nigh A , Benzinger TLS , Miller TM , Karch CM , Bateman RJ ((2018) ) Tau kinetics in neurons and the human central nervous system. Neuron 97: , 1284–1298. |
[90] | Beck H , Goussakov IV , Lie A , Helmstaedter C , Elger CE ((2000) ) Synaptic plasticity in the human dentate gyrus. J Neurosci 20: , 7080–7086. |
[91] | Baur D , Galevska D , Hussain S , Cohen LG , Ziemann U , Zrenner C ((2020) ) Induction of LTD-like corticospinal plasticity by low-frequency rTMS depends on pre-stimulus phase of sensorimotor mu-rhythm. Brain Stimul 13: , 1580–1587. |
[92] | Lefaucheur JP , Antal A , Ayache SS , Benninger DH , Brunelin J , Cogiamanian F , Cotelli M , De Ridder D , Ferrucci R , Langguth B , Marangolo P , Mylius V , Nitsche MA , Padberg F , Palm U , Poulet E , Priori A , Rossi S , Schecklmann M , Vanneste S , Ziemann U , Garcia-Larrea L , Paulus W ((2017) ) Evidence-based guidelines on the therapeutic use of transcranial direct current stimulation (tDCS). Clin Neurophysiol 128: , 56–92. |
[93] | Mansvelder HD , Verhoog MB , Goriounova NA ((2019) ) Synaptic plasticity in human cortical circuits: Cellular mechanisms of learning and memory in the human brain? , Curr Opin Neurobiol 54: , 186–193. |
[94] | Serrano-Pozo A , Frosch MP , Masliah E , Hyman BT ((2011) ) Neuropathological alterations in Alzheimer disease, Cold Spring Harb Perspect Med 1: , a006189. |
[95] | Busche MA , Hyman BT ((2020) ) Synergy between amyloid-beta and tau in Alzheimer’s disease. Nat Neurosci 23: , 1183–1193. |


