
Pharmaceutical Potential of Casein-Derived Tripeptide Met-Lys-Pro: Improvement in Cognitive Impairments and Suppression of Inflammation in APP/PS1 Mice
Abstract
Background:
Tripeptide Met-Lys-Pro (MKP), a component of casein hydrolysates, has effective angiotensin-converting enzyme (ACE) inhibitory activity. Brain angiotensin II enzyme activates the NADPH oxidase complex via angiotensin II receptor type 1 (AT1) and enhances oxidative stress injury. ACE inhibitors improved cognitive function in Alzheimer’s disease (AD) mouse models and previous clinical trials. Thus, although undetermined, MKP may be effective against pathological amyloid-β (Aβ) accumulation-induced cognitive impairment.
Objective:
The current study aimed to investigate the potential of MKP as a pharmaceutical against AD by examining MKP’s effect on cognitive function and molecular changes in the brain using double transgenic (APP/PS1) mice.
Methods:
Experimental procedures were conducted in APP/PS1 mice (n = 38) with a C57BL/6 background. A novel object recognition test was used to evaluate recognition memory. ELISA was used to measure insoluble Aβ40, Aβ42, and TNF-α levels in brain tissue. Immunohistochemical analysis allowed the assessment of glial cell activation in MKP-treated APP/PS1 mice.
Results:
The novel object recognition test revealed that MKP-treated APP/PS1 mice showed significant improvement in recognition memory. ELISA of brain tissue showed that MKP significantly reduced insoluble Aβ40, Aβ42, and TNF-α levels. Immunohistochemical analysis indicated the suppression of the marker for microglia and reactive astrocytes in MKP-treated APP/PS1 mice.
Conclusion:
Based on these results, we consider that MKP could ameliorate pathological Aβ accumulation-induced cognitive impairment in APP/PS1 mice. Furthermore, our findings suggest that MKP potentially contributes to preventing cognitive decline in AD.
INTRODUCTION
According to a 2021 report from Alzheimer’s Disease International, 55 million people worldwide have dementia. The number of affected patients will continue to increase and is estimated to reach about 78 million in 2030 [1, 2]. The high prevalence of dementia leads to physical, psychological, social, and economic burden, not only on those with dementia, but also on their families and society [2]. Against this background, it would be important to take efforts to prevent dementia in an aging society.
Alzheimer’s disease (AD) is the most common type of dementia, accounting for about 60–70% of patients with dementia [2]. Pathophysiological changes in AD, including the formation of amyloid-β (Aβ) plaques and neurofibrillary tangles, are thought to begin years before clinical symptoms appear, which bring on a very long “preclinical” phase of AD [3]. Currently, the amyloid hypothesis that Aβ accumulation in the brain is the trigger for AD progression has been proposed based on the studies in familial AD (FAD) cases [4, 5]. In addition, according to the amyloid hypothesis, Aβ deposition is thought to cause pathological changes leading to inflammation, synaptic malfunctioning, and synaptic degeneration [6].
The renin-angiotensin system (RAS) is considered an essential circulating fluid system for controlling blood pressure and maintaining sodium and water homeostasis. Angiotensin II (Ang II), which plays a primary role in RAS, affects cell function via Ang II receptor types 1 (AT1) and 2 (AT2) [7]. All RAS components have been found locally in the brain, indicating the presence of brain local RAS independent from circulating RAS [8, 9]. Furthermore, local Ang II reportedly activates the NADPH oxidase complex via AT1 and enhances oxidative stress injury [10]. Thus, modulation of angiotensin-converting enzyme (ACE) activity, which converts angiotensin I to angiotensin II, would be useful for neuroprotection against Ang II-mediated oxidative stress injury in the brain [11, 12]. Histopathological studies have reported increased ACE expression in the cerebrums of AD patients [13–15] and elevated hippocampal ACE activity in the aged rats subjected to chronic mild stress and the Tg2576 transgenic AD model mice [16, 17]. In contrast, ACE inhibitors and angiotensin II receptor blockers (ARBs) improved cognitive function in mouse models of AD [17–20] and clinical trials [21–24]. Furthermore, AbdAlla et al. reported that an ACE inhibitor captopril normalized hippocampal ACE activity and delayed the sign of neurodegeneration in AD mice [17]. Several observational and clinical studies also support that ACE inhibitors with blood-brain barrier (BBB) permeability can reduce the rate of functional deterioration in AD patients and decelerate AD progression [25, 26].
Tripeptide Met-Lys-Pro (MKP) is a component of casein hydrolysates with effective ACE inhibitory activity (IC50 = 0.43μM). MKP has shown antihypertensive effects in spontaneously hypertensive rats and people with mildly high blood pressure [27, 28]. Furthermore, MKP has been shown to inhibit brain injury in stroke-prone spontaneously hypertensive rats [29] and improve cognitive function, specifically orientation as measured by the respective ADAS-cog subscale, in subjects whose range of cognitive function was healthy to mild cognitive impairment (MCI) [30]. Additionally, MKP has shown beneficial effects on cognitive function, inflammation, and oxidative stress in mice with an intracerebroventricular injection of Aβ42 [31]. However, the effect of MKP on preclinical pathological changes in AD, such as Aβ accumulation, has not yet been examined. Therefore, to elucidate the effects of MKP on cognitive impairment induced by pathological Aβ accumulation and examine its potential as preventive medicine, we conducted experiments using an APP/PS1 transgenic mouse model of AD.
MATERIALS AND METHODS
Animals
We obtained APPswe/PS1dE9 double transgenic (APP/PS1) mice with a C57BL/6 background from Jackson Laboratory (Bar Harbor, ME, USA) and bred the mice at the Shiga University of Medical Science. Mice were housed in standard cages (2 to 5 mice per cage), with the temperature controlled at 23°C and maintained on a 12-h light/dark cycle (lights on 8:00–20:00) with free access to food and water. Heterozygous (n = 38) mice at 6 months of age were per os (p.o.) given a standard chow diet (AIN-93M; Oriental Yeast, Tokyo, Japan) with or without MKP (Peptide Institute, Osaka, Japan; 10 ppm, which is equivalent to 0.83 mg/kg body weight/day) ad libitum for 9 months. Behavioral tests were performed at 14.5 months of age, and the mice were sacrificed at 15 months under deep anesthesia with sodium pentobarbital (50 mg/kg, i.p.). The brains of each mouse were separated into two hemispheres in the sagittal plane. One hemisphere was prepared for immunohistochemistry by immersing in 4% paraformaldehyde in 0.1 M phosphate buffer (PB) for 24 h at 4°C followed by 15% sucrose in 0.1 M PB with 0.1% sodium azide. The other hemisphere was snap-frozen in liquid nitrogen and further processed for biological examination.
All experimental procedures in this study were approved by the Animal Care and Use Committee of Shiga University of Medical Science (Ethical committee approval number: 2018-7-9H3) and the Institutional Animal Care and Use Committee of Morinaga Milk Industry Co., Ltd. (Ethical committee approval number: 18-023).
Novel object recognition test
The novel object recognition (NOR) test was performed as previously described [32]. The test was performed in a 25 cm radius circular chamber with 40 cm high walls. The test was conducted on two consecutive days and consisted of habituation, training, and testing. On day 1, for habituation, mice were placed in the open chamber without any object for 20 min. On day 2, each mouse was given a training trial followed by a testing trial. During the training trial, the mouse was placed in the chamber with two identical objects and allowed to explore the objects for 10 min. To exclude the effects of location preference on object familiarity/novelty, the proportion of exploration times of the two identical objects was measured. After 1 h, the test trial was performed, for which the mice were again placed in the same chamber with one previous object and one novel object for 10 min. The chamber and all objects were cleansed after each testing. Exploration was defined as sniffing (within 2 cm), pawing, or biting the object, while leaning against and standing on the object were not included. Exploration time around each object was counted, and a recognition index was calculated as the ratio of time spent exploring the novel object. Location preference and recognition index were calculated using the formulas described below.
Location preference = Time spent exploring one of the identical objects/Total time spent exploring both identical objects×100%
Recognition index = Time spent exploring novel object/(Time spent exploring novel object + Time spent exploring familiar object)×100%
Morris water maze test
The Morris water maze (MWM) test was conducted as previously described [32]. The test was performed in a 60 cm radius, 36 cm deep pool filled with temperature-controlled water (20±1°C) covered with white non-toxic poster paint to maintain the opacity. The pool was practically separated into four quadrants with a clear platform of 5 cm radius submerged approximately 1 cm beneath the surface in a defined target quadrant. The MWM experiment included three parts: place navigation, a probe test, and cued navigation, and several performance parameters were recorded using CompACT VAS Ver.3.0x software. The place navigation test was performed for six consecutive days. Each mouse performed 5 trials per day with 30 min inter-trial intermissions. During the test period, a mouse was placed into the pool facing the wall and was given 90 s to find and climb onto the hidden platform. Any mouse that could not locate the platform within the allotted time was gently led to it. After reaching the platform, the mouse was given a 20-s rest period on the platform to orientate itself to the visual cues in the room. In the next trial, the mouse was released in a different quadrant facing the wall. After finishing the test, the escape latency and swim speed (cm/s) were analyzed. After the place navigation test, a probe test was performed. In the probe test, the platform was withdrawn, and mice were placed in the pool and allowed to swim freely for 90 s. Then the time spent in the objective quadrant and the number of platform points were counted. In the cued navigation test, the platform was moved to the other quadrant and raised from the surface. The mark was placed on the platform, and the optical features on the walls were removed to confirm mice were using cues to identify the platform. Mice that did not reach the platform in the last three trials were excluded from all analyses.
RNA sequencing (RNA-seq) analysis
Total RNA was extracted from the hippocampus with the RNeasy Plus Universal Mini Kit (Qiagen, Venlo, Netherlands) according to the manufacturer’s instructions. All samples met RIN numbers of 8.3 or higher. Library generation and sequencing were performed at Takara Bio Inc (Kusatsu, Japan). RNA-seq libraries were prepared with the SMART-Seq v4 Ultra Low Input RNA Kit for Sequencing (Takara Bio USA, San Jose, CA, USA), and 150-bp paired-end sequencing was performed on NovaSeq 6000 (Illumina, San Diego, CA, USA). The DRAGEN Bio-IT Platform (version 3.7.5, Illumina) was used to align the reads from each sample to the Mus musculus genome GRCm38 assembly. Transcripts per million (TPM) was used as the normalization method. Differentially expressed genes (DEG) were identified using a combination of p < 0.05 and transcript abundance >2-fold cut-offs. To determine functional annotations of the DEG list and perform statistical assessment of annotations, the Database for Annotation, Visualization, and Integrated Discovery (DAVID) was utilized.
Immunohistochemical analysis
Immunohistochemical analysis was conducted as described previously [33]. Briefly, 20-μm free-floating sagittal sections of the brain were washed with 0.1 M phosphate-buffered saline (PBS) containing 0.3% Triton X-100 (PBS-T, pH 7.4) and subsequently soaked in 0.3% hydrogen peroxide in PBS-T for 20 min at room temperature (RT) to inactivate endogenous peroxidase. The sections were then washed three times for 10 min in PBS-T and then blocked with 2% bovine serum albumin (BSA) in PBS-T for 30 min at RT. Afterward, the blocked sections were incubated with rabbit polyclonal anti-human amyloid β (N) (1:500; Immuno-Biological Laboratories, Fujioka, Japan), rabbit polyclonal anti-GFAP (1:10,000; Dako, Glostrup, Denmark) or rabbit polyclonal anti-Iba1 (1:4,000; FUJIFILM Wako, Osaka, Japan) antibodies in PBS-T containing 0.2% BSA overnight at 4°C. The following day, after washes with PBS-T, the immunolabeled sections were incubated with biotinylated goat polyclonal anti-rabbit IgG antibody (1:1,000; Vector Laboratories, Burlingame, CA, USA) for 60 min at RT. After three PBS-T washes, sections were soaked in avidin-biotin complex solution (Vectastain ABC Elite kit, 1:3,000; Vector Laboratories) for 60 min at RT. Finally, after several washes with PBT-T, 3,3’-diaminobenzidine (Dojindo Laboratories, Kumamoto, Japan) with nickel ammonium sulfate was used to visualize the immunolabeling. Immunohistochemistry images were captured with a camera (DP27, Olympus) and analyzed using ImageJ software to measure the immunoreactive areas.
Enzyme-linked immunosorbent assay (ELISA)
Tissues collected from the cerebral cortex were homogenized in cold tris-buffered saline (TBS) (1 mL/150 mg wet tissue weight) with a protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany). After sonicated, the homogenates were centrifuged at 104,300× g for 60 min at 4°C. The supernatant was collected as a TBS-soluble fraction, while the pellet was resuspended in 70% formic acid (FA) in water, sonicated, and centrifuged at 104,300× g for 60 min at 4°C. After neutralizing with 1 M Tris base, the supernatants were collected as FA fractions containing the insoluble Aβ. These samples were then stored at –80°C until further processing. Protein concentration was measured using a Protein Assay BCA Kit (Nacalaitesque, Kyoto, Japan). Commercially available ELISA kits were used to determine Aβ40 (FUJIFILM Wako), Aβ42 (FUJIFILM Wako), IL-1β (RayBiotech, San Diego, GA, USA), IL-6 (RayBiotech), and TNF-α(RayBiotech).
Statistical analysis
All data are presented as mean±SEM. Student’s unpaired t-test was conducted for single comparisons, and Mann-Whitney U-test was used as a nonparametric method. The escape latency in MWM test was assessed with two-way repeated measures analysis of variance (ANOVA) followed by Tukey’s multiple comparisons test and one-way repeated measures ANOVA followed by Dunnett’s multiple comparison test. p-value of <0.05 was considered statistically significant. Analyses were performed using SPSS 25.0 for Windows (SPSS Inc., Chicago, IL, USA). Statistics for RNA-seq analysis are described in the “RNA sequencing (RNA-seq) analysis”section.
RESULTS
MKP improves recognition memory in APP/PS1 mice
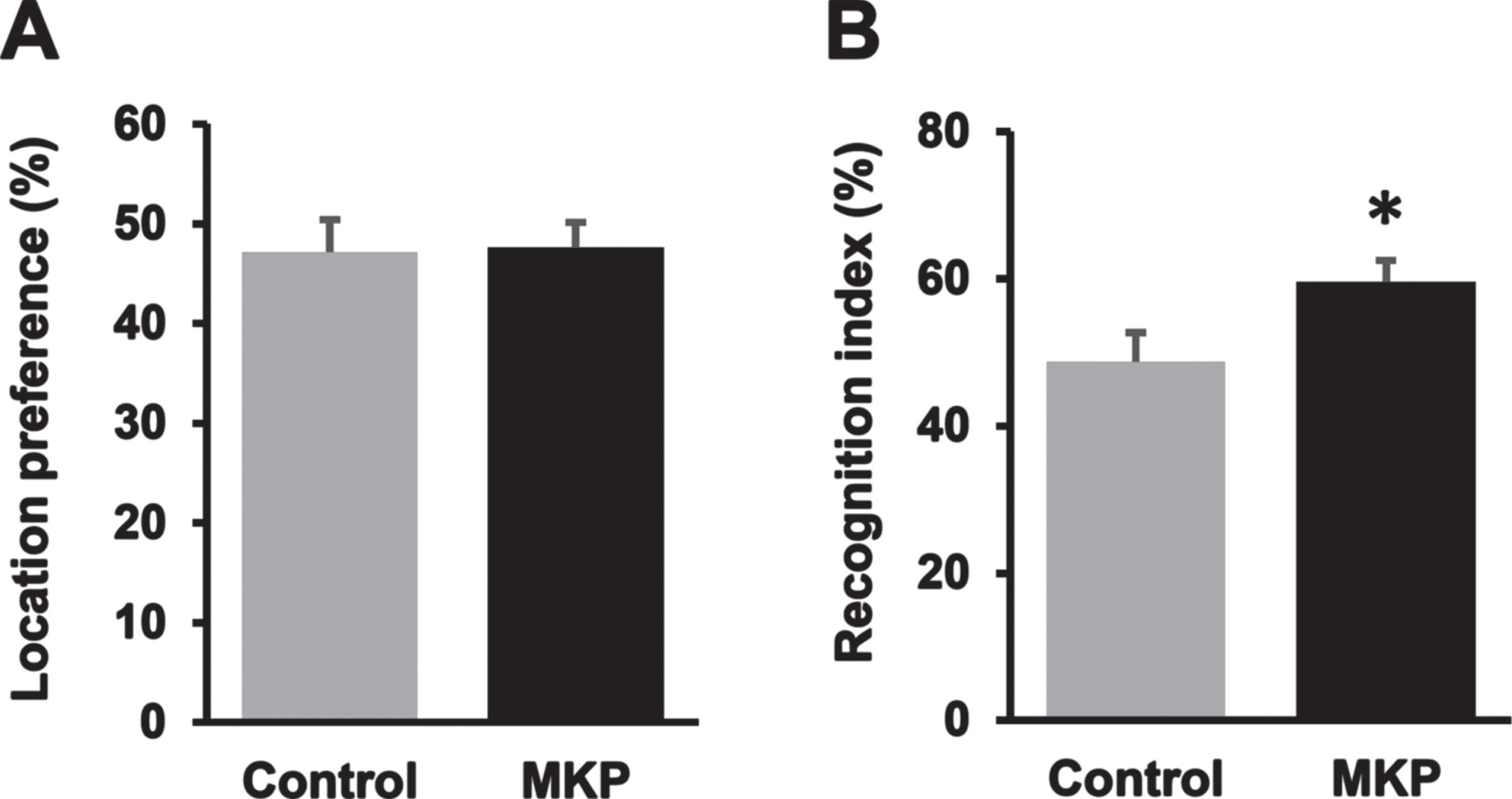
To evaluate the recognition memory of APP/PS1 mice, NOR test was conducted. In the training period, mice of each group spent almost the same time exploring identical objects, suggesting no location bias (U = 187, p = 0.92; Fig. 1A). Subsequently, for the testing period on day 2, one of the identical objects was exchanged for a novel object, and the time spent exploring the novel object was calculated as the recognition index (Fig. 1B). There was a significant difference in the recognition index between MKP-treated APP/PS1 mice and control APP/PS1 mice (57.8±5.9% and 46.1±5.0%, respectively; t = –2.20, df = 36, p < 0.05), suggesting that the MKP group showed a better preference for the novelobject.
Fig. 1
Met-Lys-Pro (MKP) p.o. administration prevents working memory deficit in APP/PS1 mice as measured by the novel object recognition task. A) Both groups spent approximately the same time exploring two identical objects during the training trial, indicating no inherent place preference. B) MKP-treated APP/PS1 mice spent a significantly longer time exploring the novel object compared to control APP/PS1 mice, as indicated by the higher mean recognition index in 10 min measurement. Data are presented as mean±SEM. Data were analyzed with the Student’s t-test or Mann–Whitney U-test. *p < 0.05 compared to Control. (Control, n = 19; MKP, n = 19).
Effect of MKP on spatial learning and memory of APP/PS1 mice
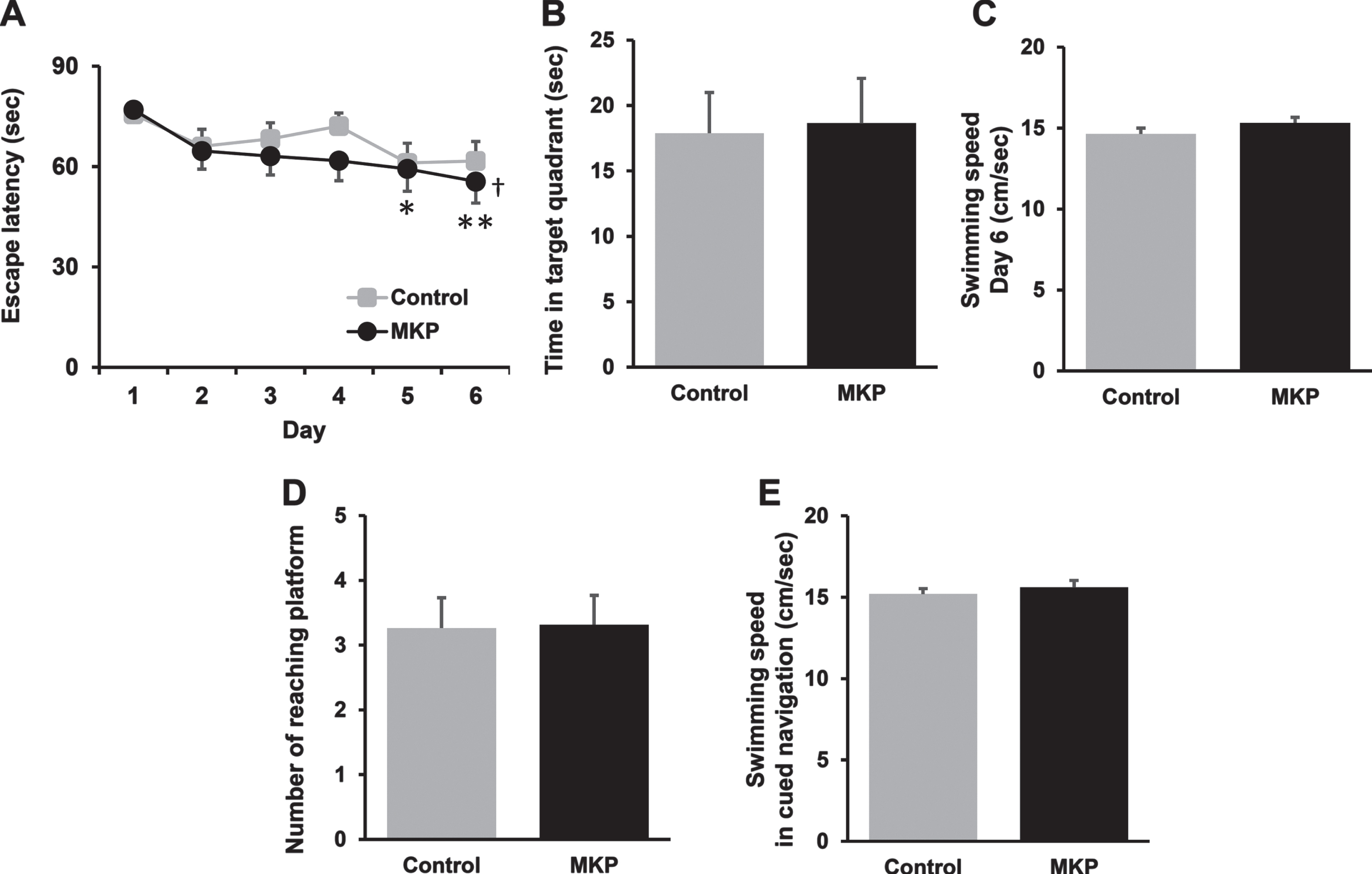
The MWM test was performed to evaluate the effects of MKP on the spatial learning and memory deficiency of APP/PS1 mice. In Fig. 2A, we found a significant main effect of time [F (5, 90) = 8.90, p < 0.001], but no effect of treatment [F (1, 18) = 0.837, p = 0.372] and an interaction between treatment and time [F (5, 90) = 0.559, p = 0.731] using two-way repeated measures ANOVA. In the main effect of time, we detected significant differences in the latency in the MKP group on day 5 (df = 90, p < 0.05) and day 6 (df = 90, p < 0.01) compared to day 1 by post hoc Tukey’s test. In addition, we detected significant differences in the latency in the MKP group between day 1 and 6 [df = 2.37, F = 5.70, p < 0.05] using one-way repeated measures ANOVA followed by post hoc Dunnett’s test, although there were no difference in the control APP/PS1 group at any time point [df = 2.76, F = 2.23, p = 0.102]. A probe trial conducted on day 7 in which the platform was withdrawn from the pool showed no statistically significant difference in time spent in the target quadrant between the groups (U = 182.5, p = 0.95; Fig. 2B). On the cued navigation test (visible platform test), no significant differences were detected between groups (U = 178, p = 0.95; Fig. 2D). There were no group differences in the swimming speed for place navigation training on day 6 (t = –1.40, df = 36, p = 0.17; Fig. 2C) and in the cue navigation tests (t = –0.74, df = 36, p = 0.46; Fig. 2E).
Fig. 2
Met-Lys-Pro (MKP) p.o. administration enhances spatial learning in APP/PS1 mice as measured by the Morris water maze test at 14.5 months of age. A) Each mouse performed 5 trials per day for 5 consecutive days to locate and climb onto the hidden platform in training trials. MKP-treated APP/PS1 mice showed significant reductions in latencies to the platform from day 5 to day 6, compared with day 1. Data are presented as mean±SEM. Significance (Tukey’s multiple comparison test after two-way repeated measures ANOVA): *p < 0.05, **p < 0.01 versus day 1; (Dunnett’s multiple comparison test after one-way repeated measures ANOVA): †p < 0.05 versus day 1. (Control, n = 19; MKP, n = 19). B) Probe test 1 day after the last trial. The time spent in the target quadrant in both groups was approximately the same. C) On day 6 of place navigation training, no significant differences in the swim speed were observed between groups. D) The cued navigation test (visible platform test) was conducted. No significant differences were detected between groups. E) On the cued navigation test, no significant differences in the swim speed were observed between groups. Data are presented as mean±SEM. Data were analyzed with the Student’s t-test or Mann–Whitney U-test. *p < 0.05 compared to Control. (Control, n = 19; MKP, n = 19).
MKP-related changes in Aβ levels in the cerebral cortex
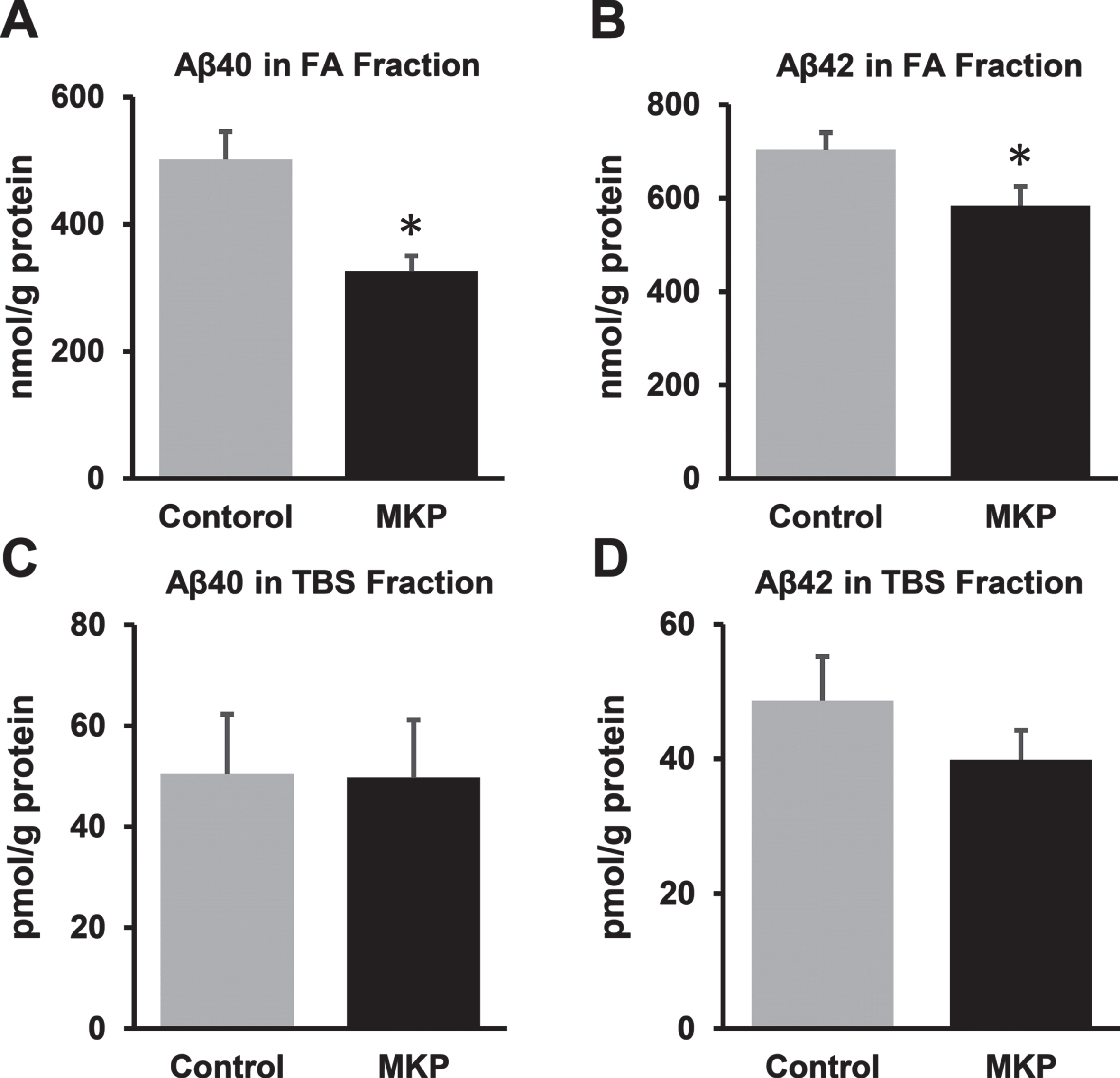
Insoluble Aβ40 levels in the FA fraction were 502.2±43.8 nmol/g protein and 325.9±24.5 nmol/g protein in MKP-treated APP/PS1 mice and control APP/PS1 mice, respectively. Insoluble Aβ42 levels in the FA fraction were 703.9±36.6 nmol/g protein and 584.2±40.8 nmol/g protein, respectively. Both the species in the FA fraction were significantly decreased in MKP-treated APP/PS1 mice compared with control mice (U = 84, p < 0.01; Fig. 3A and t = 2.17, df = 36, p < 0.05; Fig. 3B, respectively). In contrast, there were no significant differences in soluble Aβ40 and Aβ42 levels in the TBS fraction between control APP/PS1 mice and APP/PS1 mice treated with MKP (U = 130, p = 0.22; Fig. 3C and t = 1.12, df = 36, p = 0.27; Fig. 3D), while Aβ42 levels showed a slight reduction in MKP-treated APP/PS1 mice.
Fig. 3
Changes in Aβ levels in the brains of APP/PS1 mice after Met-Lys-Pro (MKP) p.o. administration. One hemisphere of the forebrain was homogenized and sequentially extracted with TBS (TBS fraction) and FA (FA fraction) in APP/PS1 mice. Levels of Aβ40 (A) and Aβ42 (B) in the FA fraction. Levels of Aβ40 (C) and Aβ42 (D) in the TBS fraction. MKP-treated APP/PS1 mice showed significant reductions in Aβ40 and Aβ42 levels in the FA fraction. Data are presented as mean±SEM. Data were analyzed using the Student’s t-test or Mann–Whitney U-test. *p < 0.05 compared to Control. (Control, n = 19; MKP, n = 19).
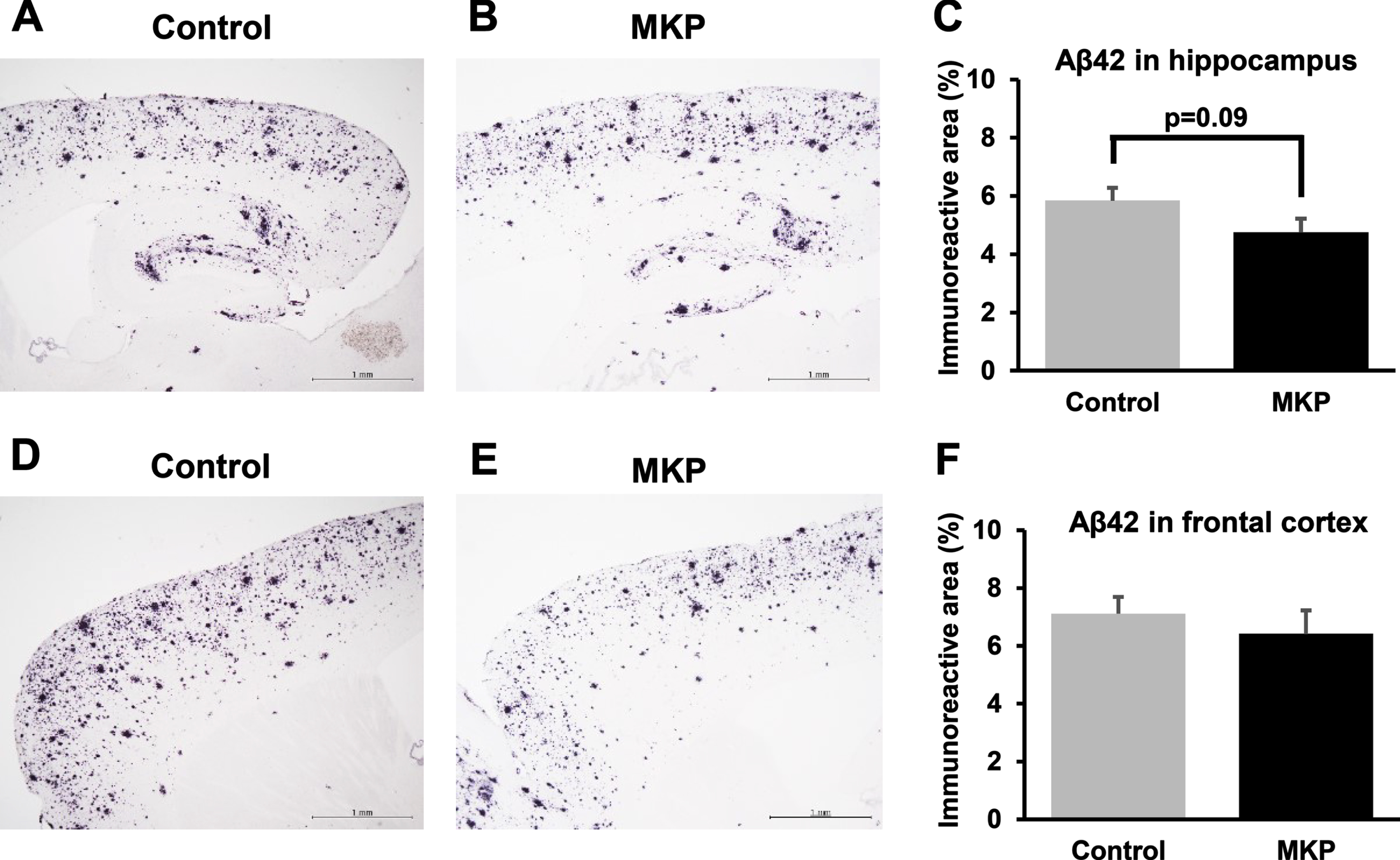
Additionally, we performed immunohistochemistry to evaluate the Aβ burden in the hippocampus and cerebral cortex of the mice (Fig. 4). Immunohistochemistry for Aβ in the hippocampus showed a downward trend in the MKP-treated mice, but it was not statistically significant (t = 1.71, df = 36, p = 0.096; Fig. 4C). Similarly, in the frontal cortex, no significant difference was observed in Aβ-immunoreactivity between control and MKP-treated APP/PS1 mice (t = 0.70, df = 36, p = 0.49; Fig. 4F).
Fig. 4
Aβ42-immunoreactivities in APP/PS1 mice after Met-Lys-Pro (MKP) p.o. administration. Free-floating brain sections in the sagittal plane were immunostained with Abeta (N) antibodies in the hippocampus (A, B, and C) and frontal cortex (D, E, and F). Immunohistochemistry images of control (A) and MKP (B) in the hippocampus. Percentage of immunoreactive areas in each group in the hippocampus (C). Immunohistochemistry images of control (D) and MKP (E) in the frontal cortex. Percentage of immunoreactive areas in each group in the frontal cortex (F). Scale bar: 1 mm. Data represent mean±SEM. Data were analyzed using the Student’s t-test. *p < 0.05 compared to Control. (Control, n = 19; MKP, n = 19).
MKP-related alterations in hippocampal gene expression
To evaluate the gene expression changes in response to oral MKP administration, transcriptional profiling was conducted on hippocampal tissues. In total, 135 genes (45 upregulated, 90 downregulated) were significantly altered in mice that had consumed MKP compared with control mice. GO enrichment analysis of the RNA-seq data using DAVID suggested that the DEGs were involved in immune response-related processes, such as “interleukin-1 production” and “negative regulation of defense responses” (Table 1).
Table 1
List of top 10 GO terms for DEGs
| GO Term (GOTERM_BP_4) | Count | Gene Name | % | p |
| regulation of epithelial cell proliferation | 8 | OVOL2, SNAI2, IQGAP3, EAF2, HPN, SCN5A, MMP12, CDKN2B | 8.99 | 0.00 |
| organ morphogenesis | 13 | OVOL2, RS1, EYA4, SNAI2, FGFR4, HPN, ISL1, SLITRK6, PLAG1, GRHL2, SCUBE2, CRB1, ACP5 | 14.61 | 0.00 |
| extracellular matrix organization | 6 | COL6A4, FGFR4, COL4A6, HPN, RXFP1, MMP12 | 6.74 | 0.00 |
| interleukin-1 production | 4 | NLRP1B, NLRC4, ISL1, ACP5 | 4.49 | 0.00 |
| negative regulation of epithelial cell proliferation | 4 | SNAI2, EAF2, HPN, CDKN2B | 4.49 | 0.02 |
| positive regulation of macromolecule metabolic process | 19 | PLA2G5, OVOL2, LRRC32, EYA4, NLRC4, SNAI2, IQGAP3, FGFR4, EAF2, EBF3, HPN, ISL1, NLRP1B, PLAG1, GRHL2, PIWIL2, ARHGEF5, HJV, RBPJL | 21.35 | 0.02 |
| negative regulation of response to external stimulus | 5 | PLA2G5, MILL2, SNAI2, ISL1, ACP5 | 5.62 | 0.02 |
| negative regulation of defense response | 4 | PLA2G5, MILL2, ISL1, ACP5 | 4.49 | 0.03 |
| sensory organ morphogenesis | 5 | RS1, EYA4, CRB1, HPN, SLITRK6 | 5.62 | 0.03 |
| animal organ development | 22 | OVOL2, RS1, LGR5, BGLAP3, EYA4, SNAI2, IQGAP3, FGFR4, EAF2, HPN, LY9, ISL1, SLITRK6, PLAG1, GRHL2, SCUBE2, CRB1, SCN5A, RXFP1, CDKN2B, ACP5, MYLK3 | 24.72 | 0.03 |
MKP treatment attenuated glial cell activity
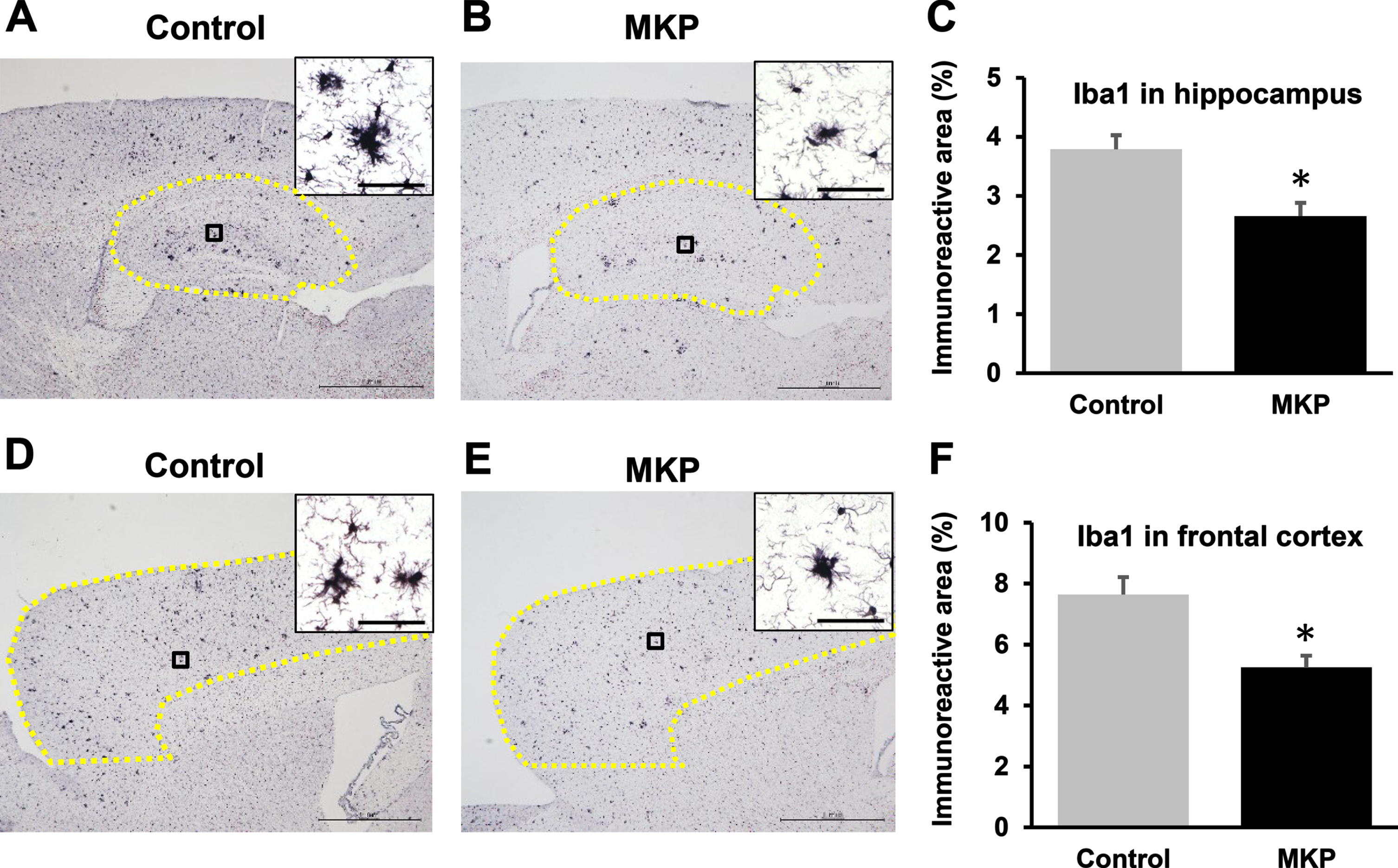
Immunoreactivities for GFAP and Iba1 were used to assess the activities of astrocyte and microglia in the brain, respectively (Figs. 5 and 6). The percentages of GFAP-immunoreactive areas in the hippocampus and the cerebral cortex were significantly reduced in MKP-treated APP/PS1 mice compared with control APP/PS1 mice (t = 3.47, df = 36, p < 0.005; Fig. 5C and t = 2.59, df = 36, p < 0.05; Fig. 5F, respectively). In addition, the levels of Iba1-immunoreactive area in the hippocampus and cerebral cortex in MKP-treated APP/PS1 mice were significantly ameliorated, compared to control APP/PS1 mice (t = 3.44, df = 36, p < 0.005; Fig. 6C and t = 3.48, df = 36, p < 0.005; Fig. 6F, respectively).
Fig. 5
GFAP-immunoreactivity in APP/PS1 mice after Met-Lys-Pro (MKP) p.o. administration. Free-floating brain sections in the sagittal plane immunostained with GFAP antibodies for activated astrocytes in the hippocampus (A, B, and C) and frontal cortex (D, E, and F). Immunohistochemistry images of control (A) and MKP (B) in the hippocampus. Percentage of immunoreactive areas in each group in the hippocampus (C). Immunohistochemistry images of control (D) and MKP (E) in the frontal cortex. Percentage of the immunoreactive area in each group in the frontal cortex (F). Scale bar: 1 mm. To calculate GFAP-immunoreactive area (C and F), the areas outlined in yellow (A, B, D, and E) were normalized. High-magnification images revealed single astroglial cell morphology in the cortex and hippocampus of the APP/PS1 mice (A, B, D, and E). Scale bars: 20μm (insets). Data represent mean±SEM. Data were analyzed using the Student’s t-test. *p < 0.05 compared to Control. (Control, n = 19; MKP, n = 19).
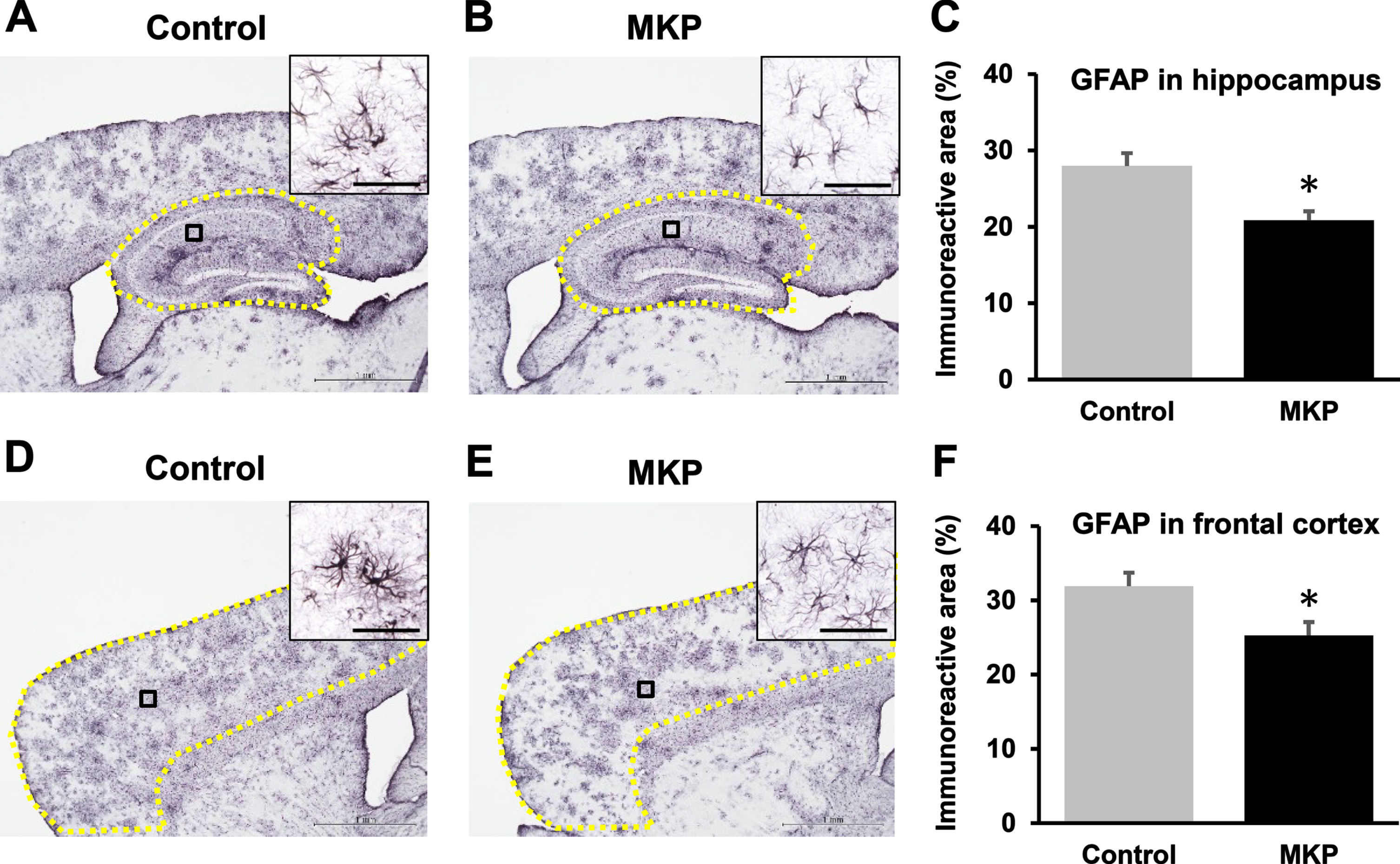
Fig. 6
Iba1-immunoreactivities in APP/PS1 mice after Met-Lys-Pro (MKP) p.o. administration. Free-floating brain sections in the sagittal plane were immunostained with Iba1 antibodies for activated microglia in the hippocampus (A, B, and C) and frontal cortex (D, E, and F). Immunohistochemistry images of control (A) and MKP (B) in the hippocampus. Percentage of the immunoreactive area in each group in the hippocampus (C). Immunohistochemistry images of control (D) and MKP (E) in the frontal cortex. Percentage of the immunoreactive area in each group in the frontal cortex (F). Scale bar: 1 mm. To calculate Iba1-immunoreactive area (C and F), the area outlined in yellow (A, B, D, and E) were normalized. High-magnification images revealed a single microglial cell morphology in the cortex and hippocampus of the APP/PS1 mice (A, B, D, and E). Scale bars: 20μm (insets). Data represent mean±SEM. Data were analyzed using the Student’s t-test. *p < 0.05 compared to Control. (Control, n = 19; MKP, n = 19).
MKP treatment attenuated inflammatory cytokine production in the brain
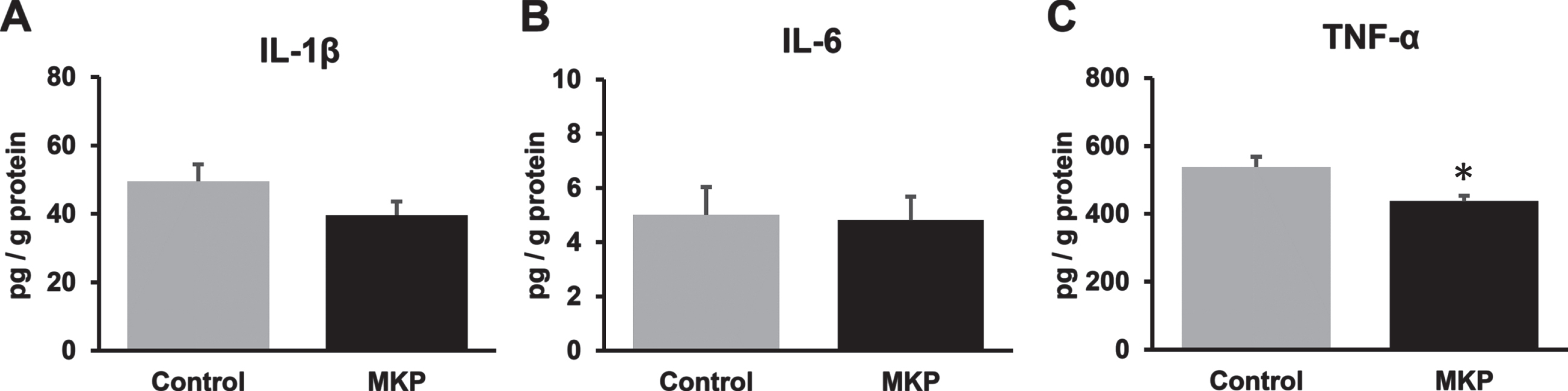
We also investigated neuroinflammation in the brain of APP/PS1 mice after oral MKP administration. The expression of inflammatory mediators, IL-1β, IL-6, and TNF-α, in APP/PS1 mice was determined using ELISA (Fig. 7). The results showed that oral MKP administration did not affect IL-1β and IL-6 levels (t = 1.56, df = 36, p = 0.13; Fig. 7A and U = 159, p = 0.73; Fig. 7B). In contrast, the TNF-α levels in MKP-treated APP/PS1 mice were significantly decreased, compared to control APP/PS1 mice, with the MKP-treated and control groups showing 537.9±29.6 pg/g protein and 437.3±15.9 pg/g protein, respectively (U = 82, p < 0.01; Fig. 7C).
Fig. 7
Changes in the levels of cytokines in the brain of APP/PS1 mice after Met-Lys-Pro (MKP) p.o. administration. One hemisphere of the forebrain was homogenized and sequentially extracted with TBS (TBS fraction) in APP/PS1 mice. Levels of IL-1β (A), IL-6 (B), and TNF-α (C) in the TBS fraction. MKP-treated APP/PS1 mice showed a significant decrease in TNF-α in the TBS fraction. Data represent mean±SEM. Data were analyzed using the Student’s t-test or Mann–Whitney U-test. *p < 0.05 compared to Control. (Control, n = 19; MKP, n = 19).
DISCUSSION
In this study, we investigated the effects of tripeptide MKP on cognitive impairment induced by pathological Aβ accumulation in an AD mouse model. We found that MKP significantly improved the recognition memory in APP/PS1 mice in the NOR test. Oral MKP administration decreased the level of insoluble Aβ40 and Aβ42 in the cerebral cortex, while there were no significant differences in soluble Aβ40 and Aβ42 between groups. Gene expression analysis indicated a change in immune response-associated processes. In addition, immunohistochemical analysis demonstrated that the markers for microglia and reactive astrocytes were decreased in the MKP group. Furthermore, changes in the levels of cytokines also reflected the anti-inflammatory effect of MKP. Based on the present study findings, we considered that MKP could improve the cognitive impairment induced by pathological Aβ accumulation in APP/PS1 mice.
In the present study, MKP was effective for recognition memory in the NOR test and partly for spatial learning and memory in the MWM test (Figs. 1 and 2). It has been shown that the ACE inhibitor perindopril improved recognition memory in Aβ25–35-injected mice [20]. Further, the angiotensin-(1–7)/Mas axis, which includes RAS components that compete with Ang II and mainly exert a tissue-protecting effect, has been reported to be important for maintaining object recognition memory [34]. On the other hand, in the MWM test, two-way repeated measures ANOVA showed a significant main effect of time between day 1 and 5 and between day 1 and 6 in the MKP group, but no significant difference was found in the control group at any time point. In addition, one-way repeated measures ANOVA showed that there was a significant difference in the latency in the MKP group between day 1 and 6, while there was no difference in the control APP/PS1 group. These results suggest that the MKP group learned by trial, but not the control group. However, the effect of MKP on special learning and memory deficiency is likely to be limited, as there were no differences between groups. Although we used this statistical approach to avoid missing potential beneficial effects of MKP treatment on behavior, the risk of overinterpretation of data should be considered. A study by Durani et al. [32] indicated that the reduction in the latency times was less than approximately 55% in the wild type after 6 days of consecutive training. However, the reduction in latency times was less than approximately 25% on day 6 versus day 1 in both control APP/PS1 mice and MKP-treated APP/PS1 mice in our study (Fig. 2A). Although the MWM test is demonstrated to evaluate mouse cognitive function, the radial-arm maze task was more demanding than the water maze in disoriented models [35]. Thus, these results suggested that oral MKP administration improved the recognition memory in the AD model and had little effect on spatial and learning memory. However, to elucidate the effects of MKP on cognitive function, a future study in a comprehensive cognitive decline model should be conducted.
In the present study, both Aβ40 and Aβ42 in the FA fraction were decreased in MKP-treated mice (Fig. 3). In addition, Aβ42 in the TBS fraction tended to decrease in the MKP group (Fig. 3D). These results indicate that MKP significantly reduced hydrophobic Aβ in APP/PS1 mice. In a previous study, ACE was reported to act as an Aβ degrading enzyme in vitro, and ACE inhibition possibly aggravated AD symptoms by increasing Aβ accumulation [36]. However, in vivo studies did not support increased Aβ generation by ACE inhibition [37]. For example, ACE inhibition by a BBB-penetrating ACE inhibitor, such as captopril, ameliorated central AD symptoms and prevented signs of neurodegeneration in AD mice [16, 17]. Several studies suggested that ACE inhibitors prevented AT1 activation by inhibiting Ang II production, leading to decreasing brain reactive oxygen species (ROS) levels [11, 17]. Brain ROS enhances the activation of enzymes, such as β- and γ-secretases, related to Aβ generation [38–40]. Thus, ACE inhibition in the brain may decelerate AD progression via ROS reduction. In our previous study, we reported that radioactivity was detected by autoradiography in the brain of spontaneously hypertensive rats after oral administration of 14C-labeled MKP, which suggested that MKP may reach the brain via BBB [31]. In addition, this previous study also showed that bovine casein-derived peptide with MKP treatment markedly reduced Aβ42-induced gene expression related to oxidative stress. Therefore, in elucidating the mechanism of action of MKP, it will be important to examine the possibility that MKP accesses the brain and reduces Aβ42 via the AT1 cascade and ROS inhibition. Interestingly, Khayer et al. reported that “positive regulation of macromolecule metabolic process,” which was identified in their study, could be ascribed to oxidative stress-related processes that are differentially regulated in AD-like pathology in transgenic AD mice [41]. The mechanism of action of MKP in the brain due to modulation of oxidative stress needs to be carefully examined in future studies. Furthermore, the effects of AT1 receptor blockers (ARBs) and ACE-inhibitors on Aβ-degrading enzymes have been reported [42, 43], and the possible involvement of Aβ-degradation as a mechanism of action of MKP should be considered. A previous study by Drews et al. [42] reported that intranasal losartan in spontaneously hypertensive stroke-prone rats reduced mortality, neuroinflammation, and perivascular Aβ content by enhancing key moderators of Aβ metabolism and clearance, such as insulin-degrading enzyme, neprilysin, and transthyretin. In addition, the oral administration of perindopril, a brain-penetrating ACE inhibitor, to AD-induced rats significantly decreased hippocampal ACE1 expression and increased ACE2, neprilysin (NEP), and insulin-degrading enzyme (IDE) expression [43]. Perindopril treatment induced a significant decrease in Aβ42; improved lipid profile; and ameliorated histopathological damage, cognitive impairment, and depressive behavior. This previous study indicated that improvement of cognitive impairment with an ACE inhibitor may involve the activation of ACE2/NEP/IDE and inhibition of ACE1, followed by the modulation of amyloidogenesis/hyperlipidemia/lipid raft signaling and oxido-nitrosative stress.
In this study, MKP intake may have regulated glial cell activation in APP/PS1 mice. In AD pathology, one of the most meaningful functions of glial cells is thought to be the regulation of Aβ levels in the brain [44]. The changes in gene expression in the hippocampus indicated that MKP intake induced modulation of immune response genes (Table 1). Regarding glial cells, we found that Iba-1-immunoreactive area and GFAP-immunoreactive area were decreased in the MKP group compared to that in the control APP/PS1 group (Figs. 5 and 6), implying that MKP suppressed the proliferation in microglia and reactive astrocytes. According to previous studies, RAS regulation can modulate microglia activation and polarization [45, 46]. Captopril decreased lipopolysaccharide (LPS)-induced microglial activation, both primary glial cells and the BV2 microglial cell line, and nitric oxide and TNF-α production regulation, whereas IL-10 was upregulated. Furthermore, activated astrocytes produced pro-inflammatory cytokines and aggravated the self-destructive environment [47, 48], while another study indicated that brain-penetrating ACE inhibitors improved cognition in APP/PS2-transgenic mice, which was associated with inhibition of astrocyte activation and superoxide production [11]. In addition, it is reported that astrocytes could interact with microglia and affect their activation, including phagocytic capacity and ability to produce inflammatory mediators [49–52]. Therefore, MKP may suppress glial cell activation, which is related to the induction of neurodegenerative processes and Aβ clearance disorder. However, in future studies, it would be necessary to explore the underlying mechanisms, including glial cell activation, the morphological changes occurring in glial cells, and interactions among different cell types. Especially since approximately 40% of DEGs were involved in organ development and morphogenetic processes (Table 1), the examination of morphological changes and related factors would be important.
Increased Aβ concentration is related to an increase in inflammatory cytokines, including TNF-α, in aged transgenic AD model mice [53]. Meanwhile, several studies have indicated that RAS modulation potentially suppresses TNF-α production by microglia [45, 54]. In this study, MKP significantly suppressed the production of TNF-α (Fig. 7C). On the other hand, despite the identification of DEGs associated with interleukin-1 production by RNA-seq (Table 1), IL-1β did not show significant differences in protein-level expression (Fig. 7A). Considering the earlier findings, our data showing that MKP suppressed the production of inflammatory cytokines in MKP-treated APP/PS1 mice suggest that the anti-inflammatory effect of MKP is involved in the improvement of cognitive decline in the AD model.
The present data suggest that MKP improved cognitive impairments, decreased brain Aβ levels, and suppressed inflammatory responses, such as glial cell activity and production of pro-inflammatory cytokines in APP/PS1 mice, indicating its potential as a prophylactic medicine. However, some limitations of this study should be noted. First, although the present study indicated the inhibitory effect of MKP on the expression of markers for microglia and reactive astrocytes, it remains unclear whether MKP mediates glial cell activation and the complicated interactions among glial cells. We need to evaluate more detailed features of microglia and astrocytes when affected by MKP in the AD pathological state. Secondly, the underlying mechanisms of Aβ reduction remain unclear, and whether they are controlled through clearance by glial cells or other systems such as the expression of enzymes related to Aβ production or degradation remains undetermined. For improving the cognitive function, the effect on cholinergic activity is also an important target for elucidating the mechanism of action of MKP. Several studies suggest the augmentation of cholinergic neurotransmission by ACE inhibitors and ARBs [55, 56]. Yamada et al. reported that oral administration of perindopril inhibited ACE activity in the rat brain and increased extracellular levels of ACh in the perirhinal cortex, suggesting enhanced cholinergic neurotransmission [55]. Losartan has been reported to induce changes in the expression of cholinergic markers, indicating that AT1-receptor blockade favors increased cholinergic activity and decreased inflammation in the AD brain [18]. Also, treatment with losartan or enalapril reportedly increases cerebrovascular reactivity to acetylcholine, calcitonin gene-related peptide, and endothelin-1 [56]. Thus, cholinergic activity may be a potential mechanism for cognitive improvement in RAS regulation. Furthermore, the actions of MKP, including its peripheral mechanisms, warrant a closer examination. Zhuang et al. [57] reported that patients in moderate-severe AD stages showed significantly higher ACE in comparison to MCI and controls. Their study also showed that age was associated with ACE activities, which suggests that serum ACE activity correlated with age is a potential risk factor for AD. The possibility that ACE inhibitors, including MKP, may inhibit the progression of AD by suppressing plasma ACE activity via peripheral RAS modulation needs to be looked into. In addition, an association between ACE inhibitor use and elevated plasma Aβ42 levels and Aβ42/Aβ40 ratio has been reported in patients with dementia [58]. This previous study discussed the possibility that decreased clearance of Aβ from the brain due to reduced cerebral blood flow could be ameliorated by maintaining cerebral blood flow with ACE inhibitors. Although additional validation is needed to support this mechanism, the ACE inhibitors used in the study included those non-permeant through the brain. Therefore, we believe that improved Aβ clearance may be a candidate mechanism for decreasing AD risk through peripheral RAS modulation of MKP. These issues might be related to the essential mechanisms by which MKP improves cognitive functions in this AD model; therefore, it is necessary to verify this in a future study.
In conclusion, this study demonstrated that long-term MKP ingestion improved recognition memory in an AD mouse model and that it mediated the expression of markers for microglia and reactive astrocytes and the production of inflammatory mediators, such as TNF-α. Our findings, therefore, suggest that MKP potentially contributes to preventing cognitive decline in AD as a prophylactic medicine.
ACKNOWLEDGMENTS
This study was funded by Morinaga Milk Industry Co., Ltd. The authors would like to thank the staff of the Central Research Laboratory, Shiga University of Medical Science, for their technical assistance.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/22-0192r2).
REFERENCES
[1] | Alzheimer’s Disease International (2021)World Alzheimer Report 2021. Journey through the diagnosis of dementia. https://www.alzint.org/u/World-Alzheimer-Report-2021.pdfhttps://www.alzint.org/u/World-Alzheimer-Report-2021.pdf, Last updated September 21, 2021, Accessed on May 23, 2022. |
[2] | World Health Organization (2021) Dementia. https://www.who.int/news-room/fact. |
[3] | Sperling RA , Aisen PS , Beckett LA , Bennett DA , Craft S , Fagan AM , Iwatsubo T , Jack CR Jr , Kaye J , Montine TJ , Park DC , Reiman EM , Rowe CC , Siemers E , Stern Y , Yaffe K , Carrillo MC , Thies B , Morrison-Bogorad M , Wagster MV , Phelps CH ((2011) ) Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7: , 280–292. |
[4] | Citron M , Oltersdorf T , Haass C , McConlogue L , Hung AY , Seubert P , Vigo-Pelfrey C , Lieberburg I , Selkoe DJ ((1992) ) Mutation of the beta-amyloid precursor protein in familial Alzheimer’s disease increases beta-protein productionNature 360: , 672–674. |
[5] | Hardy J , Selkoe DJ ((2002) ) The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics297, 353-356. Erratum in: Science 297: , 2209. |
[6] | Walsh DM , Selkoe DJ ((2004) ) Deciphering the molecular basis of memory failure in Alzheimer’s disease. Neuron 44: , 181–193. |
[7] | Jones ES , Vinh A , McCarthy CA , Gaspari TA , Widdop RE ((2008) ) AT2 receptors: Functional relevance in cardiovascular disease. Pharmacol Ther 120: , 292–316. |
[8] | McKinley MJ , Albiston AL , Allen AM , Mathai ML , May CN , McAllen RM , Oldfield BJ , Mendelsohn FA , Chai SY ((2003) ) The brain renin-angiotensin system: Location and physiological roles. Int J Biochem Cell Biol 35: , 901–918. |
[9] | Wright JW , Harding JW ((2013) ) The brain renin-angiotensin system: A diversity of functions and implications for CNS diseases. Pflugers Arch 465: , 133–151. |
[10] | Garrido AM , Griendling KK ((2009) ) NADPH oxidases and angiotensin II receptor signaling. Mol Cell Endocrinol 302: , 148–158. |
[11] | Dong YF , Kataoka K , Tokutomi Y , Nako H , Nakamura T , Toyama K , Sueta D , Koibuchi N , Yamamoto E , Ogawa H , Kim-Mitsuyama S ((2011) ) . Perindopril, a centrally active angiotensin-converting enzyme inhibitor, prevents cognitive impairment in mouse models of Alzheimer’s disease. FASEB J 25: , 2911–2920. |
[12] | Zimmerman MC , Lazartigues E , Lang JA , Sinnayah P , Ahmad IM , Spitz DR , Davisson RL ((2002) ) Superoxide mediates the actions of angiotensin II in the central nervous system. Circ Res 91: , 1038–1045. |
[13] | Arregui A , Perry EK , Rossor M , Tomlinson BE ((1982) ) Angiotensin converting enzyme in Alzheimer’s disease increased activity in caudate nucleus and cortical areas. J Neurochem 38,: , 1490–1492. |
[14] | Miners S , Ashby E , Baig S , Harrison R , Tayler H , Speedy E , Prince JA , Love S , Kehoe PG ((2009) ) Angiotensin-converting enzyme levels and activity in Alzheimer’s disease: Differences in brain and CSF ACE and association with ACE1 genotypes. Am J Transl Res 1: , 163–177. |
[15] | Savaskan E , Hock C , Olivieri G , Bruttel S , Rosenberg C , Hulette C , Müller-Spahn F ((2001) ) Cortical alterations of angiotensin converting enzyme, angiotensin II and AT1 receptor in Alzheimer’s dementia. Neurobiol Aging 22: , 541–546. |
[16] | AbdAlla S , El Hakim A , Abdelbaset A , Elfaramawy Y , Quitterer U ((2015) ) Inhibition of ACE retards tau hyperphosphorylation and signs of neuronal degeneration in aged rats subjected to chronic mild stress. Biomed Res Int 2015: , 917156. |
[17] | AbdAlla S , Langer A , Fu X , Quitterer U ((2013) ) ACE inhibition with captopril retards the development of signs of neurodegeneration in an animal model of Alzheimer’s disease. Int J Mol Sci 14: , 16917–16942. |
[18] | Drews HJ , Klein R , Lourhmati A , Buadze M , Schaeffeler E , Lang T , Seferyan T , Hanson LR , Frey WH , de Vries TCGM , Thijssen-van Loosdregt IAEW , Gleiter CH , Schwab M , Danielyan L ((2021) ) Losartan improves memory, neurogenesis and cell motility in transgenic Alzheimer’s mice. Pharmaceuticals (Basel) 14: , 166. |
[19] | Hemming ML , Selkoe DJ , Farris W ((2007) ) Effects of prolonged angiotensin-converting enzyme inhibitor treatment on amyloid beta-protein metabolism in mouse models of Alzheimer disease. Neurobiol Dis 26: , 273–281. |
[20] | Yamada K , Uchida S , Takahashi S , Takayama M , Nagata Y , Suzuki N , Shirakura S , Kanda T ((2010) ) Effect of a centrally active angiotensin-converting enzyme inhibitor, perindopril, on cognitive performance in a mouse model of Alzheimer’s disease. Brain Res 1352: , 176–186. |
[21] | Ohrui T , Tomita N , Sato-Nakagawa T , Matsui T , Maruyama M , Niwa K , Arai H , Sasaki H ((2004) ) Effects of brain-penetrating ACE inhibitors on Alzheimer disease progression. Neurology 63: , 1324–1325. |
[22] | Rozzini L , Vicini Chilovi B , Trabucchi M , Padovani A ((2008) ) Antihypertensive medications influence the rate of conversion from mild cognitive impairment to Alzheimer disease. Arch Neurol 65: , 993–994; author reply, 994. |
[23] | Sink KM , Leng X , Williamson J , Kritchevsky SB , Yaffe K , Kuller L , Yasar S , Atkinson H , Robbins M , Psaty B , Goff DC Jr ((2009) ) Angiotensin-converting enzyme inhibitors and cognitive decline in older adults with hypertension: Results from the Cardiovascular Health Study. Arch Intern Med 169: , 1195–11202. |
[24] | Sohn SI , Kim CJ ((2014) ) Modulation of renin-angiotensin system and arterial stiffness: Evidence from clinical trials. Curr Hypertens Rev 10: , 37–40. |
[25] | de Oliveira FF , Bertolucci PH , Chen ES , Smith MC ((2014) ) Brain-penetrating angiotensin-converting enzyme inhibitors and cognitive change in patients with dementia due to Alzheimer’s disease. J Alzheimers Dis 42: (Suppl 3), S321–S324. |
[26] | O’Caoimh R , Healy L , Gao Y , Svendrovski A , Kerins DM , Eustace J , Kehoe PG , Guyatt G , Molloy DW ((2014) ) Effects of centrally acting angiotensin converting enzyme inhibitors on functional decline in patients with Alzheimer’s disease. J Alzheimers Dis 40: , 595–603. |
[27] | YamadaA, SakuraiT, OchiD, MitsuyamaE, YamauchiK, AbeF ((2015) ) Antihypertensive effect of the bovine casein-derived peptide Met-Lys-Pro. Food Chem 172: , 441–446. |
[28] | Yuda N , Tanaka M , Yamada A , Ochi D , Yamauchi K , Abe F ((2018) ) Antihypertensive effect of the casein-derived peptide Met-Lys-Pro in individuals with high-normal blood pressure or grade 1 hypertension-a randomized, double-blind, placebo-controlled, parallel-group trial. Jpn Pharmacol Ther 46: , 529–537. |
[29] | Tada AM , Hamezah HS , Yanagisawa D , Morikawa S , Tooyama I ((2020) ) Neuroprotective effects of casein-derived peptide Met-Lys-Pro (MKP) in a hypertensive model. Front Neurosci 14: , 845. |
[30] | Yuda N , Tanaka M , Yamauchi K , Abe F , Kakiuchi I , Kiyosawa K , Miyasaka M , Sakane N , Nakamura M ((2020) ) Effect of the casein-derived peptide Met-Lys-Pro on cognitive function in community-dwelling adults without dementia: A randomized, double-blind, placebo-controlled trial. Clin Interv Aging 15: , 743–754. |
[31] | Min LJ , Kobayashi Y , Mogi M , Tsukuda K , Yamada A , Yamauchi K , Abe F , Iwanami J , Xiao JZ , Horiuchi M ((2017) ) Administration of bovine casein-derived peptide prevents cognitive decline in Alzheimer disease model mice. PLoS One 12: , e0171515. |
[32] | Durani LW , Hamezah HS , Ibrahim NF , Yanagisawa D , Nasaruddin ML , Mori M , Azizan KA , Damanhuri HA , Makpol S , Wan Ngah WZ , Tooyama I ((2018) ) Tocotrienol-rich fraction of palm oil improves behavioral impairments and regulates metabolic pathways in AβPP/PS1 mice. J Alzheimers Dis 64: , 249–267. |
[33] | Ibrahim NF , Yanagisawa D , Durani LW , Hamezah HS , Damanhuri HA , Wan Ngah WZ , Tsuji M , Kiuchi Y , Ono K , Tooyama I ((2017) ) Tocotrienol-rich fraction modulates amyloid pathology and improves cognitive function in AβPP/PS1 mice. J Alzheimers Dis 55: , 597–612. |
[34] | Lazaroni TL , Raslan AC , Fontes WR , de Oliveira ML , Bader M , Alenina N , Moraes MF , Dos Santos RA , Pereira GS ((2012) ) Angiotensin-(1-7)/Mas axis integrity is required for the expression of object recognition memory. Neurobiol Learn Mem 97: , 113–123. |
[35] | Gibson BM , Shettleworth SJ , McDonald RJ ((2001) ) Finding a goal on dry land and in the water: Differential effects of disorientation on spatial learning. Behav Brain Res 123: , 103–111. |
[36] | Hemming ML , Selkoe DJ ((2005) ) . Amyloid beta-protein is degraded by cellular angiotensin-converting enzyme (ACE) and elevated by an ACE inhibitor. J Biol Chem 280: , 37644–37650. |
[37] | Eckman EA , Adams SK , Troendle FJ , Stodola BA , Kahn MA , Fauq AH , Xiao HD , Bernstein KE , Eckman CB ((2006) ) Regulation of steady-state beta-amyloid levels in the brain by neprilysin and endothelin-converting enzyme but not angiotensin-converting enzyme. J Biol Chem 281: , 30471–30478. |
[38] | Shen C , Chen Y , Liu H , Zhang K , Zhang T , Lin A , Jing N ((2008) ) Hydrogen peroxide promotes Abeta production through JNK-dependent activation of gamma-secretase. J Biol Chem 283: , 17721–17730. |
[39] | Tan JL , Li QX , Ciccotosto GD , Crouch PJ , Culvenor JG , White AR , Evin G ((2013) ) Mild oxidative stress induces redistribution of BACE1 in non-apoptotic conditions and promotes the amyloidogenic processing of Alzheimer’s disease amyloid precursor protein. PLoS One 8: , e61246. |
[40] | Zhu D , Shi J , Zhang Y , Wang B , Liu W , Chen Z , Tong Q ((2011) ) Central angiotensin II stimulation promotes β amyloid production in Sprague Dawley rats. , . PLoS One 6: , e16037. |
[41] | Khayer N , Marashi SA , Mirzaie M , Goshadrou F ((2017) ) Three-way interaction model to trace the mechanisms involved in Alzheimer’s disease transgenic mice. PLoS One 12: , e0184697. |
[42] | Drews HJ , Yenkoyan K , Lourhmati A , Buadze M , Kabisch D , Verleysdonk S , Petschak S , Beer-Hammer S , Davtyan T , Frey WH 2nd, Gleiter CH , Schwab M , Danielyan L ((2019) ) Intranasal losartan decreases perivascular beta amyloid, inflammation, and the decline of neurogenesis in hypertensive rats.. Neurotherapeutics 16: , 725–740. |
[43] | Messiha BAS , Ali MRA , Khattab MM , Abo-Youssef AM ((2020) ) Perindopril ameliorates experimental Alzheimer’s disease progression: Role of amyloid β degradation, central estrogen receptor and hyperlipidemic-lipid raft signaling. Inflammopharmacology 28: , 1343–1364. |
[44] | Ries M , Sastre M ((2016) ) Mechanisms of Aβ clearance and degradation by glial cells. Front Aging Neurosci 8: , 160. |
[45] | Asraf K , Torika N , Apte RN , Fleisher-Berkovich S ((2018) ) Microglial activation is modulated by captopril: and studies. Front Cell Neurosci 12: , 116. |
[46] | Qie S , Ran Y , Lu X , Su W , Li W , Xi J , Gong W , Liu Z ((2020) ) Candesartan modulates microglia activation and polarization via NF-κB signaling pathway. Int J Immunopathol Pharmacol 34: , 2058738420974900. |
[47] | Choi SS , Lee HJ , Lim I , Satoh J , Kim SU ((2014) ) Human astrocytes: Secretome profiles of cytokines and chemokines. PLoS One 9: , e92325. |
[48] | Deng Y , Xie D , Fang M , Zhu G , Chen C , Zeng H , Lu J , Charanjit K ((2014) ) Astrocyte-derived proinflammatory cytokines induce hypomyelination in the periventricular white matter in the hypoxic neonatal brain. PLoS One 9: , e87420. |
[49] | DeWitt DA , Perry G , Cohen M , Doller C , Silver J ((1998) ) Astrocytes regulate microglial phagocytosis of senile plaque cores of Alzheimer’s disease. Exp Neurol 149: , 329–340. |
[50] | Hailer NP , Wirjatijasa F , Roser N , Hischebeth GT , Korf HW , Dehghani F ((2001) ) Astrocytic factors protect neuronal integrity and reduce microglial activation in an model of N-methyl-D-aspartate-induced excitotoxic injury in organotypic hippocampal slice cultures. Eur J Neurosci 14: , 315–326. |
[51] | Smits HA , van Beelen AJ , de Vos NM , Rijsmus A , van der Bruggen T , Verhoef J , van Muiswinkel FL , Nottet HS ((2001) ) Activation of human macrophages by amyloid-beta is attenuated by astrocytes. J Immunol 166: , 6869–6876. |
[52] | Solà C , Casal C , Tusell JM , Serratosa J ((2002) ) Astrocytesenhance lipopolysaccharide-induced nitric oxide production bymicroglial cells. Eur J Neurosci 16: , 1275–1283. |
[53] | Patel NS , Paris D , Mathura V , Quadros AN , Crawford FC , Mullan MJ ((2005) ) Inflammatory cytokine levels correlate with amyloid load in transgenic mouse models of Alzheimer’s disease. J Neuroinflammation 2: , 9. |
[54] | Torika N , Asraf K , Roasso E , Danon A , Fleisher-Berkovich S ((2016) ) Angiotensin converting enzyme inhibitors ameliorate brain inflammation associated with microglial activation: Possible implications for Alzheimer’s disease. J Neuroimmune Pharmacol 11: , 774–785. |
[55] | Yamada K , Horita T , Takayama M , Takahashi S , Takaba K , Nagata Y , Suzuki N , Kanda T ((2011) ) Effect of a centrally active angiotensin converting enzyme inhibitor, perindopril, on cognitive performance in chronic cerebral hypo-perfusion rats. Brain Res 3: , 110–20. |
[56] | Ongali B , Nicolakakis N , Tong XK , Lecrux C , Imboden H , Hamel E ((2018) ) Transforming growth factor-β1 induces cerebrovascular dysfunction and astrogliosis through angiotensin II type 1 receptor-mediated signaling pathways. Can J Physiol Pharmacol 96: , 527–534. |
[57] | Zhuang S , Wang X , Wang HF , Li J , Wang HY , Zhang HZ , Xing CM ((2016) ) Angiotensin converting enzyme serum activities: Relationship with Alzheimer’s disease. Brain Research 1650: , 196–202. |
[58] | Regenold WT , Blumenthal JB , Loreck DJ , Mordecai KL , Scarinzi G , Doddi SR , Adler L ((2017) ) Elevated plasma Aβ42 in cognitively impaired individuals taking ACE inhibitor antihypertensives. Am J Alzheimers Dis Other Demen 32: , 347–352. |

