
COVID-19 and Neurodegenerative Diseases: Prion-Like Spread and Long-Term Consequences
Abstract
COVID-19 emerged as a global pandemic starting from Wuhan in China and spread at a lightning speed to the rest of the world. One of the potential long-term outcomes that we speculate is the development of neurodegenerative diseases as a long-term consequence of SARS-CoV-2 especially in people that have developed severe neurological symptoms. Severe inflammatory reactions and aging are two very strong common links between neurodegenerative diseases and COVID-19. Thus, patients that have very high viral load may be at high risk of developing long-term adverse neurological consequences such as dementia. We hypothesize that people with neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, and aged people are at higher risk of getting the COVID-19 than normal adults. The basis of this hypothesis is the fact that SARS-CoV-2 uses as a receptor angiotensin-converting enzyme 2 to enter the host cell and that this interaction is calcium-dependent. This could then suggest a direct relationship between neurodegenerative diseases, ACE-2 expression, and the susceptibility to COVID-19. The analysis of the available literature showed that COVID-19 virus is neurotropic and was found in the brains of patients infected with this virus. Furthermore, that the risk of having the infection increases with dementia and that infected people with severe symptoms could develop dementia as a long-term consequence. Dementia could be developed following the acceleration of the spread of prion-like proteins. In the present review we discuss current reports concerning the prevalence of COVID-19 in dementia patients, the individuals that are at high risk of suffering from dementia and the potential acceleration of prion-like proteins spread following SARS-CoV-2 infection.
INTRODUCTION
On December 31, 2019, a new infection reported to the World Health Organization was identified as caused by a novel coronavirus that is different from agents that cause severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS) and was later named SARS-CoV-2 and referred to as coronavirus disease 19 (COVID-19). Since its first appearance, the virus has spread at a lightning speed and reached all over the world, taking many lives and badly affecting the world economy. SARS-CoV-2 belongs to the family of coronaviruses that were first identified in 1965 in humans from a child with an upper respiratory infection [2]. Depending on the season, they were later identified as causing 1% to 35% of upper respiratory infections [2]. These community-acquired coronaviruses are known to cause mild respiratory infections. SARS that appeared in 2002– 2003, however, affected more than 8,000 people and caused nearly 10% of mortality [3], and the MERS epidemic that was reported in 2012 affected 2,500 persons with a mortality rate of nearly 35% [4]. They are classified into four genera: alpha, beta, gamma, and delta; SARS-CoV-2 belongs to the beta genus [5]. They are named coronaviruses due to the spike projections that measure 20 nm in length and give them the crown-like appearance seen through the electron microscope. They are enveloped viruses with a diameter of 100– 150 nm and a positive-sense single-stranded RNA genome [2]. The recent genome sequencing of the SARS-CoV-2 showed that the size of its genome is ∼29.9 Kb, sharing ∼78% sequence homology with SARS-CoV [6– 8]. The spike proteins are the one responsible for the virus’s tropism since they are the primary to attach to the angiotensin-converting enzyme receptor type 2 (ACE-2) to infect the cells [5, 9]. The distribution of the ACE-2 receptor in the body includes several organs such as the lungs, oral and nasal mucosa, bone morrow and spleen, skin, heart, arteries, kidneys, adipose tissue, reproductive system, and brain [10]. In the central nervous system, ACE-2 receptor is expressed mainly in thalamic nuclei, cerebellum, and inferior olivary nuclei [11]. It is now becoming widely accepted that the virus could invade the central nervous system (CNS) and cause neurological problems since postmortem studies revealed the presence of both SARS-CoV-2 antigen and RNA in the brain tissue of COVID-19 patients [12, 13]. Besides, the rate of neurological symptoms was reported to be 0.04% in SARS and a 0.2% in MERS [14]. For SARS-COV-2, the presence of neurological manifestations was reported to occur in most hospitalized patients and was associated with increased morbidity and mortality [15, 16]. It is this link between coronaviruses and the CNS that lead to the development of several new theories concerning the potential implication of coronaviruses especially SARS-CoV-2 in the long-term development of neurodegenerative diseases (ND). In fact, neurological manifestations were reported to occur in 36% of COVID-19 patients [17, 18]. Nevertheless, it is unknown if neurological symptoms are a direct result of SARS-CoV-2 infection of brain cells or a consequence of systemic illness [14]. Besides, the detection of coronaviruses in the CNS of patients with Parkinson’s disease (PD), and Alzheimer’s disease (AD) is very well established [19, 20]. A very important and provocative study dating back to 1992 reported the identification of a high level of Beta-murine-CoV antibodies in cerebrospinal fluid (CSF) of PD patients [21]. Furthermore, going back to history, an overlap between the “Spanish flu” (1918- 1920) and the lethargic encephalitis epidemic of 1916 to 1926 was very well documented [22]. Therefore, like previous pandemics, ND could mark the aftermath of this viral infection and it could be the potential cause of a delayed epidemic as was suggested by Serrano-Castro and his team (2020) [23]. Hence, on one hand the infection with the virus could result in acceleration of the neurodegeneration process through the acceleration of the spreading of prion-like proteins [24, 25]. On the other hand, the presence of dementia could make patients more vulnerable to the disease with the presence of a higher fatality rate among them and the great influence on the routine processes of diagnoses, treatments, and daily care. For this reason, in the present review we mainly focus on discussing potential links between SARS-CoV-2 and several ND based on single case reports and the available literature either as an accelerating agent of these diseases through an increase of the prion-like proteins seeding or as a causative agent of dementia development later in life. We further discuss the role of the presence of dementia in the susceptibility to COVID-19.
SARS-COV-2 PENETRATION ROUTES AND INFECTION MECHANISMS
Major penetration routes of COVID-19 into human cells
Three major mechanisms are known as the major routes for the virus’s entry into the CNS. The first one is through the peripheral nervous system. Indeed, COVID-19 may first invade the peripheral nerve terminals and then reach the CNS via a synapse-connected route [26– 28]. This occurs via active transport within the neurons, especially through the motor proteins, kinesin and dynein [29], via microtubules and involves a retrograde axonal transport, reaching the CNS [30]. These neurons could be either motor, sensory, or autonomic neurons, but are most often olfactory neurons [29, 31– 33]. Trans-synaptic transmission is very well documented in some Corona viruses CoVs such as HEV67 [26, 27, 34, 35]. Furthermore, other studies reported that human coronavirus OC43 (HCoV-OC43) might invade the CNS via cranial peripheral nerves [36] as it occurs with other respiratory viruses and influenza virus [31].
The second route could be through the olfactory nerves. The olfactory pathway seems to be an efficient pathway for neuroinvasion of respiratory viruses since it communicates with both the nasal epithelium and the olfactory bulb [29, 37]. Indeed, when given intranasally to transgenic mice, CoVs can reach the brain [38, 39]. Furthermore, following the infection 85.6% and 88.0% of COVID-19 patients reported olfactory and gustatory dysfunction, respectively and 11% of the patients had anosmia before any other clinical symptoms [40]. SARS-CoV-2 entry receptor ACE-2 was found to be ubiquitously expressed in nasal goblet and ciliated cells [41] which further corroborates the hypothesis that SARS-CoV-2 could enter the brain via the olfactory nerves [12].
The third route could be through the hematogenous pathway where the virus can disrupt the epithelium barrier and invade the bloodstream [32]. Indeed, type II alveolar epithelial cells are the one that highly express ACE-2 and as the mainly infected cells they may allow the penetration of the virus into the bloodstream [38]. Once in the blood circulation SARS-CoV-2 could bind to ACE2 receptors of the endothelium and disrupt the blood-brain-barrier (BBB) causing edema, intracranial hypertension, and/or penetration of the virus in the CNS. Indeed, a very recent study in mice showed that the S1 spike protein of SARS-CoV-2 injected intravenously can cross the BBB, enter the parenchymal tissue of the brain, and invade all brain regions examined. This widespread entry could be the basis for brain encephalitis, respiratory difficulties, and anosmia [42– 45]. The viral invasion of the BBB could promote its permeability to the cytokine storm following inflammation or hypoxemia induced by respiratory distress syndrome [33, 45– 47]. However, newly emerging data suggests that other routes could also be involved in the penetration of the virus such as the trigeminal nerve, which innervates nociceptive cells in the nasal cavity, the conjunctiva, and the taste buds. Indeed, SARS-CoV-2 RNA fragments were reported to be found in a patient with conjunctivitis [33, 48]. Also, the nasal epithelium in general, lymphatic tissue, and the CSF may also play roles in SARS-CoV-2 invasion into the CNS [33].
Infection mechanisms
Calcium (Ca2+) has been proposed recently to play a major role in the mediation of the fusion and the penetration of the virus [49]. Indeed, the first point of connection between SARS-COV-2 and the human cell membrane is at the ACE receptors and mediated by the spike protein which is a multidomain homotrimer glycoprotein anchored in the viral envelope membrane [50, 51]. Each monomer of the spike protein has two subunits, S1 and S2. S1 contains the receptor binding domain (RBD) which binds to the ACE2 receptor [52, 53]. S2 subunit includes the segment named fusion peptide (FP) which is essential for the fusion process and leads to the infection by SARS-CoV-2 [50, 54, 55]. Once the RBD of the spike protein interacts with the ACE2 receptor, a furin-like protease recognizes a cleavage site that is composed of 12-nucleotide or four-amino acid insertion, proline– arginine– arginine– alanine (PRRA), at amino acid residues 682– 685 located at the S1/S2 boundary and separates the two components of the spike monomer (152– 154) [6]. Once furin cleavage is achieved, another cleavage site S2’ is exposed at the S2 domain and a second cleavage is mediated by the cell surface enzymes primarily transmembrane serine protease 2 (TMPRSS2) [52, 55]. This cleavage occurs at the N-terminal of the FP and causes a brutal change in the conformation of the S2 domain and the insertion of the virus into the host cell occurs. Hence, it is the interaction between the spike protein and ACE2 receptors what mediates the tissue tropism in the presence of the Ca2+ [55]. Indeed, it has been suggested that SARS-CoV-2 S protein is slightly more positively charged than SARS-CoV [56, 57] and that the binding domain of the ACE-2 receptors has a negative electrostatic potential. This electrostatic interaction may allow a stronger binding affinity between the two proteins and this may be what contributes to the SARS-CoV-2 high virulence and global spread [58– 60].
MECHANISMS INVOLVED IN THE DETRIMENTAL EFFECT OF COVID-19 ON CNS
There are at least four pathological mechanisms that could account for the detrimental effect of COVID-19 on the CNS: 1) direct viral encephalitis, 2) systemic inflammation, 3) peripheral organ dysfunction (liver, kidney, and lung), and 4) cerebrovascular changes (Fig. 2). However, neurological manifestations could be due to a combination of all the above factors. Any complication of this kind could put COVID-19 survivors at a risk of long-term neurological complications either by aggravating a pre-existing neurological disorder or by developing new ones such as that of dementia. This concern is raised since at least one third of patients have evidence of cognitive impairment and motor deficits at the time of discharge which may threaten from the initiation of another potential delayed pandemic such as that of dementia [23, 61, 62].
Fig. 1
Mechanism of penetration of COVID-19 into the nervous system and other organs. The COVID-19 virus is composed of the nucleocapsid, viral envelop, membrane protein, the envelop protein, and the spike protein. The first point of connection between SARS-COV2 and the ACE-2 receptor is mediated by the spike protein. Each monomer of the spike protein is composed of two domains S1 and S2. S1 contains the receptor binding domain (RBD) which binds to the ACE-2 receptor and S2 which contains the fusion peptide (FP). After the interaction of RBD with the ACE-2 receptor, a furin-like protease cleaves the spike protein at the S1/S2 boundary and then separates the two components of the spike protein. Once furin cleavage is achieved, a second cleavage is mediated by the transmembrane serine protease 2. The cleavage occurs at the N-terminal of the FP and causes a brutal change in the conformation of the S2 domain and the insertion into the host cell occurs. This penetration of the virus is enhanced by the calcium binding to the FP. Hence, the encapsulated genetic material of the virus is uncoated, deposited in the cytoplasm and then ready for replication. Finally, viral genes are translated into genomic RNA and viral proteins which unite to form viral particles. The penetration of the virus could be through the olfactory nerve pathway, hematogenous pathway, peripheral nervous system (PNS) pathway, or other pathways like trigeminal nerve, conjunctiva, or taste buds.
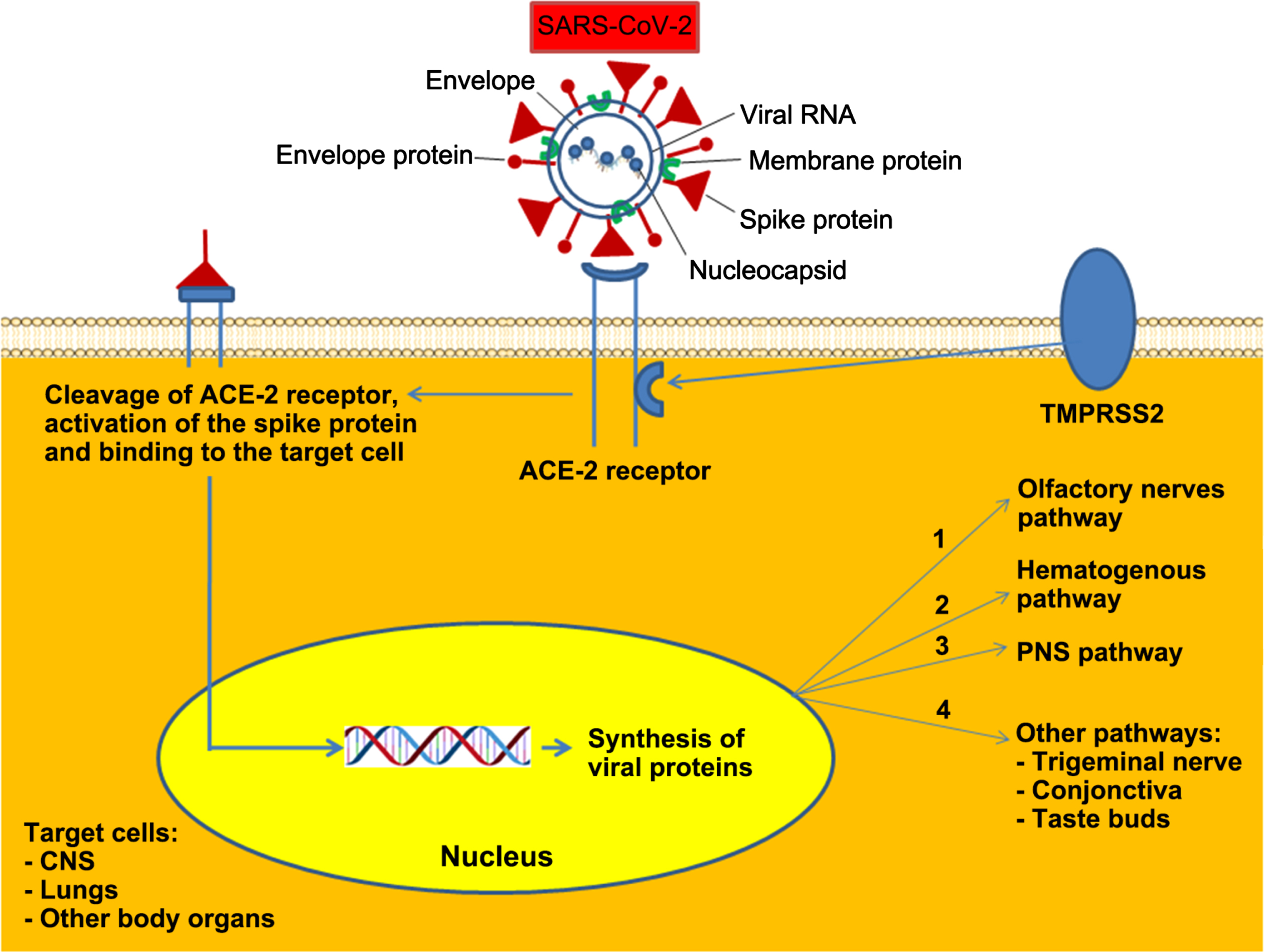
Fig. 2
Possible links between COVID-19 and neurodegenerative diseases. Three major mechanisms are known as the major routes for the virus’s entry into the central nervous system (CNS): it could be either through the peripheral nervous system, the olfactory nerves, or the hematogenous pathway. At least four pathological mechanisms could account for the detrimental effect of COVID-19 on the CNS: (1) direct viral encephalitis, (2) systemic inflammation, (3) peripheral organ dysfunction (liver, kidney, and lung), (4) cerebrovascular changes, or (5) it could be a combination of all the above. These complications could have a long-term neurological effect either by aggravating a pre-existing neurological disorder or by developing new ones such as that of dementia including Alzheimer’s disease or Parkinson’s disease.
SARS-CoV2 infection comprises three main stages; the first step is the vermia which is characterized by high viral replication accompanied by fever, cough, and general discomfort for several days. In the second step, there is a low rate of viral replication, but the symptoms include high fever, hypoxemia, and progression of respiratory symptoms to bilateral pneumonia. In the final stage, viral replication continues to decrease while approximately 20% of the patients develop SARS that is in most cases fatal. Hence, in this stage the pathogenicity is explained more by the cytokine storm which promotes the development of fatal secondary sepsis [23]. Since the human immune system never encountered this type of coronavirus before, the innate immune system is the first line of defense. This response is indeed excessive and dysregulated which may explain the severity of the symptoms associated with it. Hence, the association of the cytokine storm with COVID-19 results in very poor outcomes [23].
COVID-19 and dementia
Although initially COVID-19 was thought to be confined to the respiratory system causing a severe respiratory syndrome, it is now becoming increasingly evident from recent studies that the virus could invade other organs, especially the CNS [12]. For this reason, several researchers are raising the concern about its long-term neurological manifestations since ∼36% of the cases showed neurological complications such as stroke, cognitive dysfunction, depression, psychosis, and delirium [61]. This suggests the predisposition of these patients to NDs [63]. Indeed, it may not be just a simple coincidence that the brain regions, cortex and substantia nigra with the highest risk of SARS-CoV-2 infections through ACE2 are the same associated with the most frequent NDs [20, 64].
Dementia itself has become a worldwide pandemic with high rate of mortality among aged people. Hence, dealing with a pandemic like dementia within the COVID-19 pandemic raises a serious concern. Age is a risk factor for COVID-19, and COVID-19 is a potential agent that may cause an outbreak of dementia in the long-term [20]. Indeed, recent data from Italy reported that out of 627 subjects accepted in acute phase, patients with dementia showed a 40% higher mortality rate than in patients without dementia [20, 65].
In advanced stages dementia could be a major risk factor by itself for causing severity and mortality in COVID-19 patients [20, 66]. In older adults in the UK Biobank, it was reported that pre-existing dementia is a major risk factor for COVID-19 severity with an odds ratio of 3.07 [67].
Role of COVID-19 in the development of dementia
In fact, in a nationwide patient electronic health record database in the United States (US), COVID-19 patients with dementia had significantly worst outcomes compared to patients without dementia and those with vascular dementia are first in the row followed by presenile dementia and AD [68]. Meanwhile, although the long-term neurological consequences of COVID-19 are not yet known; there is a potential risk that COVID-19 survivors may develop in the long run dementia and NDs [20]. Besides, for most patients in severe cases, the high level of cytokines causes the so called cytokine storm and required assisted ventilation. All these factors lead to cognitive decline or the aggravation of a pre-existing one [61]. Considering the overlap between the age of the risk of the SARS-CoV-2 infected subjects and the age when people typically develop neurodegenerative or cerebrovascular disease, the neuroinvasive potential of the virus might lead/contribute to the development of NDs [61]. Indeed, recently three research groups independently discovered that SARS-CoV-2 infects human cells only when they express the lysosomal receptor TMEM106B [69– 71]. This same gene is known as a risk factor for the development of dementia. AD represents one of the most common forms of dementia since it represents 60– 70% of the total dementia cases [12].
Additionally, similar shared features were found between microglia and astrocytes activated in COVID-19 infection and those associated with several ND such as AD, Huntington’s disease, and autism spectrum disorder [72]. Furthermore, synaptic signaling that is associated with upper layer excitatory neurons that is related to higher cognitive functions was found to be preferentially affected by COVID-19. Indeed, three major symptoms were reported to persist in COVID-19 patients after release from the hospital which are “brain fog”, difficulty in concentrating and fatigue [72– 74]. Recently at the Alzheimer’s Association International Conference (AAIC) the presence of brain fog was proposed as a high risk of having AD [75]. Besides, the activation of the complement pathway from the choroid plexus to microglia was reported following COVID-19 infection and was previously linked to deficient synaptic pruning in ND diseases [72]. In a recent postmortem study of COVID-19 patients, it was reported that the virus remained in the postmortem brains and that the small blood vessels in different brain regions were leaking in a manner akin to mini-strokes. Mini-strokes and inflammation were reported to put the patients at risk of developing ND diseases. In a study reported by Jaywant et al. 2021 [76], it was found that attention and executive functions were frequently impaired in COVD-19 patients and that this impairment lasted even after the discharge from the hospital. Indeed, at the time of the discharge, nearly 81% of the survivors of an acute respiratory distress syndrome exhibited cognitive impairment and that after 3 months 40% of them who were critically ill had at least mild cognitive deficits and 26% had moderate cognitive deficits [76]. This suggests that the greater the number of COVID-19 survivors with cognitive function, the greater the likelihood of the development of ND diseases in the next 10 years [77].
Role of dementia as a risk factor for mortality in COVID-19 patients
Age and the diagnosis of dementia in a retrospective study of 627 COVID-19 patients admitted to hospitals with critical case of pneumonia were found to be the greatest risk factor for mortality. Dementia was diagnosed in 13.1% of the cases and within that percentage the mortality rate was 62.2% compared to 26.2% in subjects without dementia [65]. The diagnosis of dementia in the most advanced stage was found to be an important risk factor for mortality and the most frequently accompanied symptoms were delirium and worsening of the functional symptoms, but the classic symptoms of COVID-19 were less frequent [65]. In addition, in a UK biobank study, dementia was found to represent the greatest risk factor for the development of dementia [78]. In another study, dementia patients were found to have twice the risk of developing COVID-19 compared to those without dementia [79, 80].
Besides, pre-existing dementia was reported to represent a high risk of infection and both severity and mortality were associated with subsequent delirium and altered mental status [81, 82]. In fact, delirium is so common in COVID-19 cases and long-term research studies showed that a single episode of delirium can increase the risk of developing dementia years later [83]. Furthermore, it can accelerate the rate of cognitive decline in those who already have the condition [84]. A Brazilian study of 309 patients with an average age of 78 years showed that those who showed delirium at hospital stay, 32% of them developed dementia compared to only 16% of those who did not become delirious. Additionally, in a meta-analysis study, delirium when diagnosed in a hospital stay was found to be associated with 2.3 times greater odds of developing dementia [85, 86]. In fact, several studies now in the US and internationally plan to study the link between COVID-19 survivors who developed delirium during their hospital stay and the likelihood of developing dementia as a long-term consequence [85].
COVID-19 and AD
AD and COVID-19 share several risk factors and comorbidities such as aging, hypertension, diabetes, gender, and APOE4 expression [66]. Furthermore, systemic inflammation is a common feature between AD and COVID-19. Indeed, NLRP3 inflammasome-mediated inflammation was shown to cause the accumulation of fibrillar amyloid-β which may directly exacerbate the neurodegenerative process causing functional impairment or fasten the progression of the disease through the spread of pathology [61, 87, 88]. This hypothesis is further corroborated from findings of animal studies that have shown that NLRP3-driven and interleukin-1β-mediated modulation of phosphokinases and phosphatases largely accounts for the pathological formation of neurofibrillary tangles in mouse models of tauopathies [61]. The NLRP3 inflammasome-mediated inflammation was also reported to be activated in COVID-19 patients [89– 91].
Another plausible hypothesis is that SARS-CoV-2 can cause damage to the CNS either through direct neurotoxicity or the induction of systemic inflammation leading to a cytokine storm. The overactivation of the immune system could lead to demyelination, neurodegeneration, and cellular senescence, hence, leading to neurodegeneration and dementia [66, 92]. Interleukin 6 (IL-6), interleukin 1 (IL-1), cytoskeleton-associated protein 4 (CKAP4), and galectin-9 (GAL-9 or Gal-9) are among the cytokines that have received much attention since they are a common link between COVID-19 and AD manifestations [93, 94].
Because AD is a disease of aging, a direct link was found between AD, ACE-2 expression, and oxidative stress. In fact, aging causes an imbalance in the redox state, causing oxidative stress and hence an upregulation of ACE-2 expression [66, 95]. The increase in ACE-2 expression is also found in patients with low severity AD [18]. Since ACE-2 is the receptor of SARS-CoV-2, this may result in a higher viral load and may explain the high vulnerability of AD patients and the high prevalence of infections in AD patients [18, 23]. Thus, a mutualistic relationship seems to exist between AD and COVID-19. On one hand AD patients are more prone to the infection by the virus and on the other hand COVID-19 survivors are at increased risk of developing AD [66].
Currently, the aftermath of COVID-19 on brain insult and acceleration of aging leading to NDs is still unknown [66]. In fact, in a 3-month follow up neuroradiological study it was shown that in the brain of COVID-19 survivors there was a loss of integrity especially in the hippocampal area. Hippocampal atrophy is indeed related to cognitive decline and is a major characteristic of AD [96, 97].
Potential role of COVID-19 in the acceleration of AD pathology
In a study reported in the Alzheimer’s Association International Conference (AAIC®) 2021, an association was found between COVID-19 infection and persistent cognitive deficits including the acceleration of AD symptoms and pathology. Indeed, in infected patients who present toxic-metabolic encephalopathy (TME) an increase was found in the level of t-tau, NfL, GFAP, pTau-181, UCH-L1, and the ratio of pTau/Aβ42 in COVID-19 patients with TME compared to COVID-19 patients without TME. The increase in the above mentioned AD markers was accompanied by an increase in the markers of inflammation that accompany the BBB disruption due to neuronal/glial injury [98]. Furthermore, several reports showed that there was a white matter ischemic damage after hypoperfusion due to hypercoagulability and disseminated intravascular coagulation in critical cases of COVID-19 [66]. Cerebral hypoperfusion may accelerate the phosphorylation of tau. Likewise, in AD ischemic white matter, damage has been reported at the early stages contributing to the acceleration of the disease and to cognitive decline [99, 100]. Ischemia was also proposed to cause an induction of α-synuclein (α-Syn) phosphorylation at serine-129, which may increase the risk of development of Lewy body disease [100]. Indeed, ischemic brain damage is known to be the defining pathological process in vascular dementia, and stroke is a major risk factor for dementia [101, 102]. It is therefore possible that acute large cerebral vascular occlusion associated with hypercoagulability in severely affected COVID-19 patients will increase the risk of having dementia to some extent [100]. Hyperphosphorylation of tau was reported to increase as well after cerebral hypoperfusion [103]. Indeed, murine studies have shown that short-term mechanical ventilations may cause AD neuropathologies, such as Aβ deposition, systemic and neurologic inflammation, and disruption of the BBB. Furthermore, another plausible possibility is that SARS-CoV-2 could accelerate the generation of Aβ since this latter has a potent antimicrobial activity as part of the immune response leading to the initiation of the amyloid cascade [66]. Decrease in the level of total tau, a biomarker of neuronal death, was reported in one study that investigated severe cases of COVID-19 patients. Neurofilaments are also known to be released following axonal damage and neuronal death. The level of the light chain neurofilament subunit was found to be elevated in the serum and the CSF of COVID-19 patients [66, 104– 108]. The dysregulation of autophagy has long been reported in AD patients during the progress of the disease and is now being reported as a mechanism by which COVID-19 acts after infection of target cells. In fact, ORF3a of the COVID-19 virus SARS-CoV-2 hinders autophagy activity by blocking fusion of autophagosomes/amphisomes with lysosomes [109].
The hippocampus appears to be the target for the respiratory non-coronavirus infections. Indeed, in mice infected with the influenza virus, the morphology and function of the hippocampus, a short-term deterioration in the hippocampus-dependent learning and a reduction in the long-term potentiation associated impairment in spatial memory were reported [110]. This may raise the question about the role of the infection in the acceleration of the hippocampal related degeneration such as in AD and in disease onset in the previously asymptomatic individuals. Mice studies emphasized that viral infection could affect the acceleration of AD-related tau pathology and consequently the impairment of spatial memory [111] which is one of the major features of AD [112]. An overlap in the regional vulnerability of the temporal lobe between AD/PD and SARS-COV-2 infection may cause long-term cognitive decline that can be associated with the acceleration of the onset of the neurodegenerative dementia akin to other neurotropic viruses such as HIV and herpes viruses. In fact, these two viruses were reported to accelerate amyloid-β and tau pathologies [112]. This suggests that AD patients who have COVID-19 may have an acceleration of Alzheimer-like symptoms and pathology [98]. In a recent study, Reiken et al. (2022) showed evidence that links SARS-Cov-2 infection to activation of TGF-β signaling and oxidative stress and an activation of the neuropathological pathways that cause tau hyperphosphorylation which are typically associated with AD. They further reported a leakage of ryanodine receptors (RyR 2) that may promote behavioral and cognitive defects in COVID-19 patients [113].
Potential role of dementia in increasing the vulnerability to SARS-CoV-2 infection
Pre-existing AD was reported to predict a high risk of mortality in elderly individuals with COVID-19 [65, 79, 80, 114]. PD and vascular dementia, however, predict a high risk of infection but not mortality from COVID-19 [78]. The most frequent symptoms that accompany the critical cases are hypoactive delirium and functional status worsening with delirium being the major symptom in the intensive care unit [80]. Besides, the nature of the disease by itself could increase the vulnerability to the infection. The social and cognitive behavior of AD patients may increase the risk of contagion and the need for cares since they cannot follow the recommendations of public health authorities [115]. Motor agitation, intrusiveness, or wandering could make the matter more complicated which may further hamper the isolation procedure during the pandemic for individuals with dementia [78].
A new finding that reinforces the link between AD and COVID-19 susceptibility is the increased prevalence of the APOE4 allele which is the most important susceptibility gene to AD [116].
APOE4 genotype and COVID-19
Apolipoprotein E is a lipid binding protein, and its gene encodes three different major isoforms, APOE2, APOE3, and APOE4, which have allele frequency of 8.4%, 77.9%, and 13.7%, respectively in the general population [117, 118]. In AD patients, the frequency of APOE4 allele is nearly 40% [117, 118] and is associated with a 14-fold increased risk in people with European ancestry compared to APOE3 allele which is considered neutral and APOE2 allele which is considered protective for AD [118, 119]. In the CNS, the APOE gene is highly expressed in astrocytes and to a lower extent in neurons [117]. Astrocytes transport cholesterol to neurons via ApoE receptors [117]. Recently, an original study using data from the UK data bank of 451,367 people with European ancestry showed that there is a strong positive association (OR > 2.3, 95% CI) between APOE4/4 and the susceptibility to severe COVID-19 [67, 120]. Indeed, the APOE4/4 genotype was associated with a 4-fold increase in mortality after testing positive in the UK biobank. This increased susceptibility to the severity of COVID-19 was independent of preexisting dementia, cardiovascular disease, and type-2 diabetes [12, 67]. Following this study, another group of researchers using ApoE4 neurons showed that they are more prone to infection compared to ApoE3 neurons [121]. ApoE4 neurons co-cultured with astrocytes are more prone to a higher infection rate by SARS-CoV-2 compared to neurons cultured alone, indicating that astrocytes could exacerbate and enhance SARS-CoV-2 infection in neurons [121]. The same study showed that SARS-CoV-2 infection could result in neurite degeneration in both ApoE3 and ApoE4 neurons with a more dramatic deteriorative effect on ApoE4 neurons than ApoE3 neurons. Furthermore, astrocytes showed an exacerbated cellular response with signs of astrocytes reactivity and fragmentation of nucleus as a sign of cell death [121].
Of interest to note that the APOE4/4 gene was also associated with a higher susceptibility to the herpes simplex virus type I (HSV-1) latent infection and higher viral load than to APOE3 in murine studies [122]. Furthermore, the same gene was associated with higher susceptibility to Human Immunodeficiency Virus Type I (HIV-1), acceleration of disease progression, and more severe cases compared to the APOE3/3 genotype [121, 123]. Hence, besides its role as a major risk factor for developing AD, APOE4 is further identified as a major risk factor for the severity of COVID-19 and the acceleration of neurodegeneration which widen its role in infectious diseases. The findings that APOE4/4 was associated with increased risk of COVID-19 infection and that in human induced pluripotent stem cell models astrocytes and neurons that express APOE4/4 alleles are more prone to infection compared to those that express APOE3/3 and that the rate of infection increases in neuron-astrocyte co-cultures further show that a potential link between AD and COVID-19 may exist [67, 120, 121]. Besides its well-known role in influencing Aβ pathology and lipid homeostasis, APOE4/4 was recently reported to exacerbate neuronal inflammation in the brain [12]. APOE4/4 carriers were found to display reduced expression of antiviral defense genes compared to APOE3/3 individuals, and this may explain the susceptibility of some people to COVID-19 [80]. The APOE4 genotype is thus a risk factor not only for dementia and AD but also for COVID-19.
COVID-19 and PD
Potential role of COVID-19 in the initiation and acceleration of PD
More than 80% of COVID-19 cases presented to hospitals were reported to manifest hyposmia or ageusia as a prognosis for the infection [66, 124]. Hyposmia is very well known as a prodromal feature of PD as well as AD [125– 127]. Hence, patients that develop hyposmia may be are at increased risk of developing NDs or that hyposmia is a sign of inflammatory reaction in the olfactory mucosa. Furthermore, since the olfactory bulb is a selective target for α-Syn pathology deposition and that hyposmia is a common feature to both PD and COVID-19, it may not be just a coincidence. Maybe hyposmia development in COVID-19 cases is a harbinger for future development of PD [20].
Furthermore, the presence of antibodies against coronaviruses that cause the common cold, coronavirus OC43 and 229E, were reported in the CSF of PD patients [21]. In addition, an association between influenza A virus infection and development of transient parkinsonism was reported. Besides, many avian flu survivors developed parkinsonism and other infections were associated with a transient or permanent parkinsonism such as Epstein-Barr, Japanese encephalitis, Coxsackie, West Nile, Western equine encephalomyelitis, and human immunodeficiency virus [126, 128– 131]. Hence, it is very important to follow up the COVID-19 survivors that have developed permanent hyposmia, syncope, and persistent confusion given the importance of these conditions to AD and PD [126]. Parkinsonism in these infections was mostly developed because of induction of neuroinflammation and/or hypoxia with structural/functional damage in basal ganglia [128].
A number of murine preclinical studies have addressed the link between viral infection and PD. Jang et al. (2009) reported the potential of neurotropic Type A influenza virus (A/Vietnam, 1203/04, H5N1, a.k.a. bird flu) to induce Parkinsonism; these authors found that this virus was able to directly infect neurons with high affinity to circuits involved in PD; following this infection, mice exhibited ataxia tremor and bradykinesia [132], which was concomitant with a transient but significant loss of dopaminergic neurons phenotype, an early neuroinflammatory program, long-lasting microgliosis and an increase in α-Syn expression [133]. Furthermore, although the 2009 H1N1 influenza pandemic (CA/09) virus was not neurotropic, it does indeed induce a significant inflammatory response in the CNS, including the substantia niagra Pars compacta (SNpc) [126].
Currently there is no strong evidence that supports that PD patients are at greater risk for having SARS-CoV-2 infection or that SARS-CoV-2 infection may predispose survivors to PD. Nevertheless, some cases of worsening of PD symptoms in COVID-19 were reported in old people and one case of an acute hypokinetic syndrome with hyposmia was reported following COVID-19 infection [126]. Furthermore, developing PD could certainly be a possibility since H1N1 replicates in neuronal cells and could induce seeds of aggregated α-Syn, key factors in the pathogenesis of synucleinopathies [134, 135]. Besides, the ACE-2 receptor of the S1 subunit of the COVID-19 implicated in the angiotensin system was shown to be implicated in the neuroinflammatory response and the neurodegenerative mechanisms observed in PD. Indeed, an increase in the levels of pro-inflammatory cytokines, such as tumor necrosis factor and interleukin 1 beta (IL-1β), were linked to an increased risk of PD [136]. Hence, the presence of coronaviruses antibodies in the CSF of PD patients and the ability of the virus to enter through the nasal cavity to cause anosmia/hyposmia are consolidated evidence that may link COVID-19 to movement disorders, especially PD [20].
Potential role of PD in increasing the vulnerability to COVID-19
Some studies proposed that the nature of the disease may have a protective effect against COVID-19 since ACE-2 is highly expressed in the dopaminergic neurons which are decreased due to the disease progression and that α-Syn was reported to play an anti-viral role during West Nile virus infection [78]. However, it was reported that COVID-19 invade the brains of PD patients and can worsen motor and non-motor symptoms [126]. In fact, COVID-19 infection can cause chronic intermittent damage to the dopamine-producing neurons of the substantia nigra in humans due to chronic inflammation. The accumulation of the α-Syn in the patient’s brain may further activate microglia and worsen the neuroinflammatory process which may warrant for an effect of the infection on PD progression [137]. Other indirect factors that may have contributed to the susceptibility to the infection could be the COVID-19 lockdowns that increased stress, self-isolation, and anxiety as well as prolonged immobility which could further worsen the outcomes of PD patients infected with the virus [78]. Several studies have reported that PD is associated with a high risk of infectivity with the COVID-19 but not with the mortality level. However, some studies have reported COVID associated increase in mortality in PD cases. The mortality rate was 20% in PD patients in Italy which is higher than the general population [78]. Another study reported that PD patients with older age (mean, 78.3 years) and longer disease duration (mean, 12.7 years) were particularly susceptible to COVID-19, with a case fatality rate as high as 40% [138]. Infectious disease including viral infections were reported to increase the risk of developing PD by 20% [139]. Hence, viral infection might target pathways essential for PD and induce neuronal injury leading to loss of dopaminergic neurons and probably initiating the disease [139].
COVID-19 and aging
It is now becoming evident after the surge of the pandemic and from the available data across the UK, Italy, US, and several other countries that SARS-CoV-2 affects mostly aged people with a high mortality rate especially in residential home patients. Aged people with pre-existing co-morbidities such as chronic lung disease, cardiovascular disease, and diabetes are even at higher risk of deterioration and poor outcomes [140, 141]. In fact, the estimated median age for all COVID-19 related death is 81 years with a case fatality rate in patients aged≥80 years of > 20% [20, 142]. In the US, the fatality rate for COVID-19 increased with age from 3 to 5% between 65 and 74 years, 4 to 11% between 75 and 84 years, and 10 to 27% above 85 years. Among COVID-19 patients over 65 years, there were 45% hospitalizations, 53% of intensive care unit admissions, and 80% of death [143]. Furthermore, in China, the fatality rate was 0.1% among children and 14.8% in older individuals [144].
Several reasons could be behind the susceptibility of aged people to COVID-19 infection, it could be due to a gradual loss of BBB integrity because of the effect of aging [66, 145], the deterioration of the immune system [146, 147] or decrease in the capacity of viral clearance and recognition. Indeed, an evident decrease in the respiratory immune barrier was reported in people aged > 60 years especially alveolar phagocytosis, ventilation, tracheal epithelial ciliary movement, and cough reflex, resulting in poor virus clearance [140, 148]. Besides, the process of aging could lead to an abnormal differentiation of T cells, phagocyte viability, and the secretion of natural killer cells because of the degeneration of the thymus glands, the spleen, and lymph nodes [140, 149, 150].
The presence of co-morbidities such as diabetes and hypertension lead to a reduction in the expression of ACE-2 receptor and an upregulation of the angiotensin II proinflammatory signaling [151, 152]. Furthermore, the ageing process is usually accompanied with a state of low-grade inflammation [140, 153]. Infection with SARS-CoV-2 may accelerate this process and induce the cytokine storm. Hence, pro-inflammatory cytokines can be actively transported through the BBB and activate glia cells leading to the induction of a neuroinflammatory response and increasing oxidative stress. The body’s immune system fails to clear these inflammatory factors in a timely manner [154], leading to damaging cell structures and functions resulting in excitotoxicity [151].
VIRAL INFECTION CAN ACCELERATE TAU PATHOLOGY IN SEVERAL TAUOPATHIES
Tauopathies are divided into two different categories: primary and secondary. In the primary category, tau pathology is the major pathological characteristic like frontotemporal dementia with Parkinsonism linked to chromosome 17 and Pick’s disease. In the second category, tau pathology is usually found along with another driving force contributing to the disease like amyloid-β peptide, prion protein, and 34-mer amyloid Bri. This group includes familial and sporadic AD, familial Gerstmann-Sträussler– Scheinker disease, and familial British dementia [155]. In these diseases, the propagation of tau between brain cells follows a prion-like spreading which is essential to the ND process. Although tau is mainly intracellular and can be released by dying neurons in the extracellular space, it has been reported that tau can be also actively released into the extracellular space both in vivo and in vitro. Because tau is an intracellular protein, its propagation requires seeding, aggregation, uptake, and release [155]. Phosphorylation of tau by certain kinases such as CDK-5 and GSK-3β can cause its hyperphosphorylation, leading to its dissociation from the microtubules, misfolding, and becoming toxic seeds which are then released from the cells [156]. Tau released by a donor cell then undergoes aggregation either before or after release, then it is taken up by a recipient cell. Finally, tau aggregation is induced in the recipient cell so that the propagation can occur [157].
Protein aggregates in ND diseases can transmit to unaffected cells, hence causing templation of the normal soluble proteins leading to their aberrant conformation. These proteinopathic seeds could be released extracellularly, released in association with extracellular vesicles or exchanged by direct cell-to-cell contact. A recent study suggested that enabling the interaction between viral ligands and host cells could contribute to and accelerate the intercellular proteinopathic seed transmission [25] (Fig. 3). Indeed, using different cellular models propagating prions or pathogenic tau aggregates, it was shown that vesicular stomatitis virus glycoprotein (VSVG) and SARS-CoV-2 spike S were able to accelerate the induction of the seeding via cell contact or ligand-decorated extracellular vesicles. In fact, coculture of VSV-G-expressing donor cells with recipient cells greatly enhanced protein aggregate induction in the latter. Likewise, SARS-CoV-2 spike S protein and its receptor ACE2 contributed to the spreading of cytosolic prions and tau aggregates. These results may suggest that viral infection could facilitate the intercellular spreading of the misfolded proteins. Glycoproteins that are especially expressed during the acute phase of infection could have an essential role in the spreading of the proteins seeding in vivo. This intercellular transmission of proteinopathic seeds is suggested as a common mechanism of ND. This then further suggests that certain viral infections could modulate the spread of the proteinopathic seeds leading to the acceleration of the disease [25].
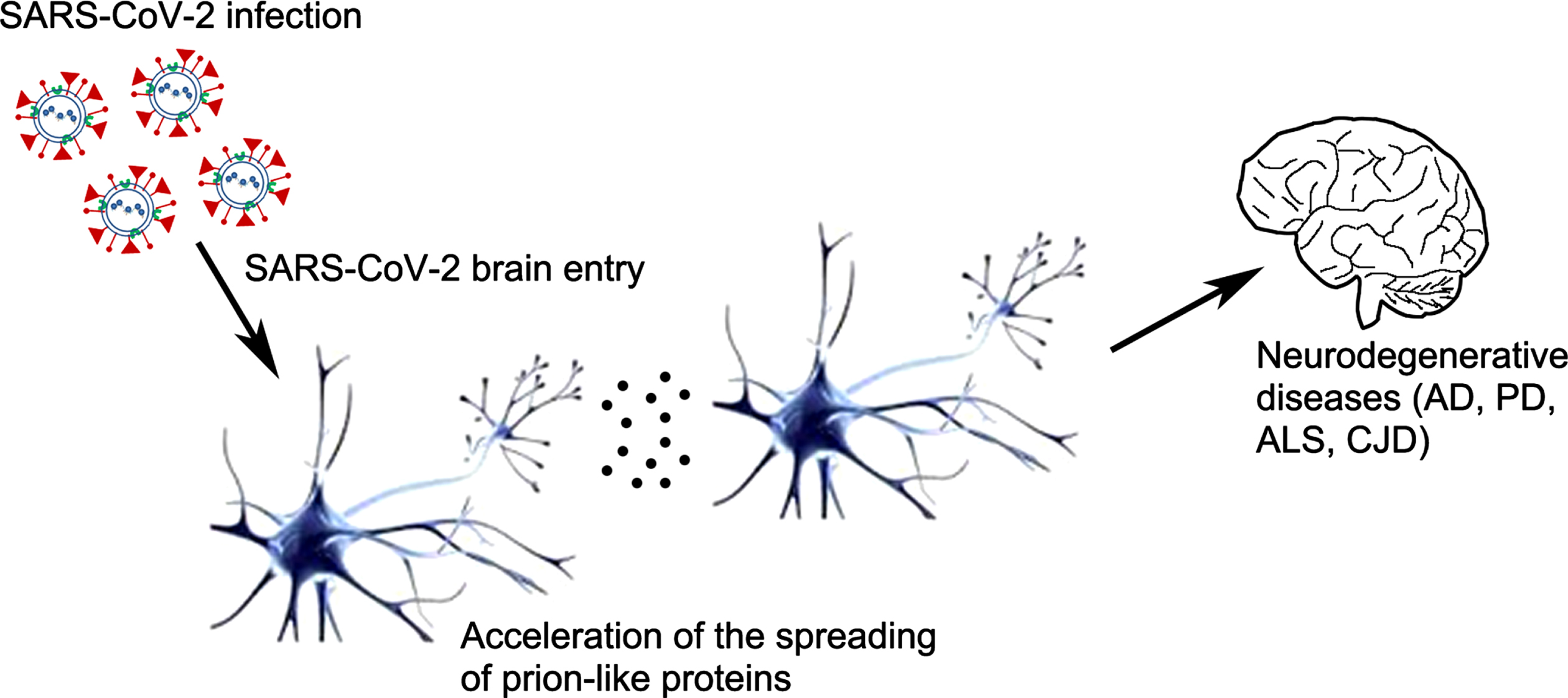
Fig. 3
Possible role of SARS-CoV-2 infection in the acceleration of the spread of prion-like proteins. The interaction between viral ligands and host cells could contribute to and accelerate the intercellular proteinopathic seed transmission through the intercellular spreading of the misfolded proteins which is suggested as a common mechanism of neurodegenerative diseases (AD, PD, ALS, CJD). This then further suggests that certain viral infections could modulate the spread of the proteinopathic seeds leading to the acceleration of the disease which could be the case for the SARS-CoV-2 virus. AD, Alzheimer’s disease; PD, Parkinson’s disease, ALS, amyotrophic lateral sclerosis; CJD, Creutzfeldt-Jakob disease.
Microbial brain infection could play a role in the development of ND. It was found that 25% of HIV patients that do not take antiretroviral therapy develop neurological disorders associated with diffuse Aβ plaque deposition and tau neurofibrillary tangles [158]. Viral infections could also affect the processing, deposition, and clearance of ND-related proteins [25, 159]. Co-infection of fibroblasts with scrapie and Moloney leukemia retrovirus caused an increase in the protein aggregates spreading [25, 160]. Furthermore, as mentioned above in this article, certain viruses like herpes, HIV, West Nile virus, and H1N1 influenza A virus were reported to be associated with several ND such AD, PD, or Alzheimer-like symptoms [24, 161]. Some viruses were even used as seeds to accelerate the spread of brain aggregation-prone proteins that were implicated in the development of ND. Two main mechanisms were proposed to be involved in the aggregation promotion: 1) Aggregation of the prion proteins on the viral surface [162] and 2) cross seeding with functional amyloid which are viral derived peptides that can promote seeding of the pathologic brain proteins [24].
SARS-COV-2 has recently been shown to bind to heparin and heparin-binding proteins via its spike protein. This binding mechanism is known to be used in pathological amyloid proteins where heparin acts as an accelerator of their aggregation [163]. Hence, heparin may serve as the recognition binding site for the aggregation-prone proteins to their initial binding which finally end up into fibrillar processes. Other viruses could infect the host cells though the use of the heparan sulfate proteoglycan-endocytosis [163]. One of the heparin-binding viruses HSV-1 was reported to promote Aβ42 aggregation both in vivo and in vitro. Therefore, since SARS-CoV-2 also has several heparin-binding sites within the spike S1 subunit [163], a similar mechanism of the acceleration of the seeding of the aggregation-prone proteins such as Aβ, α-Syn, tau, and prions could be present [24].
The cleavage of the SARS-COV-2 virus via its spike protein at the S1/S2 site and the S2’ results in a cleavage of a peptide (∼150 Aas) the pathological function of which is still unknown. If this peptide is dissociated and released into the intracellular or the extracellular space it might induce some immunological reactions or act by itself as a functional amyloid. Hence, this peptide could seed aggregation of brain proteins as a possible pathological mechanism like some viral and bacterial-derived functional amyloids. Hence, in a predictive study the self-aggregation properties of this peptide were measured and compared to other aggregation-prone proteins and the results of the AGGRESCAN revealed that S-CoV-peptide has the highest aggregation propensity and that α-Syn has the lowest propensity and the range was as follow: S-CoV-peptide >Aβ40 >M45 > α-Syn [24]. α-Syn was also proposed to become prion proteins decades ago when Lewy bodies in grafted brain cells were transplanted to PD patients [164, 165]. Furthermore, previous studies reported that recombinant α-Syn assembled into fibrils that induce the seeding of α-Syn prions both in vitro and in vivo [166]. Prions were also suspected to be the cause of amyotrophic lateral sclerosis via the aggregation of a mutant human superoxide dismutase (SOD1) that acts as a prion and in the pathogenesis of Huntington’s disease, expanded polyglutamine repeats in a huntingtin protein fragment showed self-propagation features hence acting as prions [166, 167].
Going back to history, in the last 10 years there was three coronavirus epidemics (SARS, MERS, and COVID-19), hence this epidemic is unlikely to be the last one. It is, therefore, mandatory for the scientific researchers to focus more on the long-term consequences of this infection on the neurological sequelae and the development of ND [112].
CONCLUSIONS
In the present review, we have investigated a potential relationship between COVID-19 and several NDs such as AD, PD, and aging. From the limited current data available in the literature about SARS-CoV-2 infection, we can speculate about the future development of ND as an aftermath of the epidemic. Prion-like spread is a common feature between these diseases and was recently proposed to accelerate in the presence of COVID-19 infection which may further increase the risk of ND. Indeed, viral infection was recently suggested to facilitate the intercellular spread of the misfolded proteins and several recent studies have linked the acceleration of the seeding properties of several prion-like proteins tested in response to coronaviruses infection. Hence, certainly this mutualistic relationship between, ND, neuroinflammation, prion-like protein spread, and SARS-CoV-2 infection needs further investigation.
ACKNOWLEDGMENTS
The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University for funding this work through the Small Research group project under grant number (R.G.P.1/295/43).
We are very grateful to Dr. Wen Hu, Head of Neuro-regeneration Lab. at New York State Institute for Basic Research, Staten Island for his help in the preparation of figures.
All authors are funded through the Small Research group project from the Deanship of Scientific Research at King Khalid University under research grant number (R.G.P.1/295/43).
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/22-0105r1).
REFERENCES
[1] | World Health Organization ((2020) ) https://www.who.int/news/item/27-04-2020-who-timeline---covid-19 |
[2] | McIntosh K , Chao RK , Krause HE , Wasil R , Mocega HE , Mufson MA ((1974) ) Coronavirus infection in acute lower respiratory tract disease of infants. J Infect Dis. 130: , 502–507. |
[3] | Centers for Disease Control and Prevention ((2020) ) Severe acute respiratory syndrome (SARS). https://www.cdc.gov/sars/index.html, Last updated December 6, 2017. |
[4] | World Health Organization ((2019) ) WHO MERS Global Summary and Assessment of Risk. https://apps.who.int/iris/rest/bitstreams/1239951/retrieve |
[5] | Berger JR ((2020) ) COVID-19 and the nervous system. J Neurovirol. 26: , 143–148. |
[6] | Zhang Q , Xiang R , Huo S , Zhou Y , Jiang S , Wang Q , Yu F ((2021) ) Molecular mechanism of interaction between SARS-CoV-2 and host cells and interventional therapy. Signal Transduct Target Ther. 6: , 233. |
[7] | Lu R , Zhao X , Li J , Niu P , Yang B , Wu H , Wang W , Song H , Huang B , Zhu N , Bi Y , Ma X , Zhan F , Wang L , Hu T , Zhou H , Hu Z , Zhou W , Zhao L , Chen J , Meng Y , Wang J , Lin Y , Yuan J , Xie Z , Ma J , Liu WJ , Wang D , Xu W , Holmes EC , Gao GF , Wu G , Chen W , Shi W , Tan W ((2020) ) Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet. 395: , 565–574. |
[8] | Naqvi AAT , Fatima K , Mohammad T , Fatima U , Singh IK , Singh A , Atif SM , Hariprasad G , Hasan GM , Hassan MI ((2020) ) Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochim Biophys Acta Mol Basis Dis. 1866: , 165878. |
[9] | Fehr AR , Perlman S ((2015) ) Coronaviruses: An overview of their replication and pathogenesis. Methods Mol Biol. 1282: , 1–23. |
[10] | Hamming I , Timens W , Bulthuis MLC , Lely AT , Navis GJ , van Goor H ((2004) ) Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 203: , 631–637. |
[11] | Allen AM , O’Callaghan EL , Mendelsohn FAO , Chai SY ((2009) ) Neuronal angiotensin. In Encyclopedia of Neuroscience., pp. 697–702. |
[12] | Abate G , Memo M , Uberti D ((2020) ) Impact of COVID-19 on Alzheimer’s disease risk: Viewpoint for research action. Healthcare. 8: , 286. |
[13] | Matschke J , Lütgehetmann M , Hagel C , Sperhake JP , Schröder AS , Edler C , Mushumba H , Fitzek A , Allweiss L , Dandri M , Dottermusch M , Heinemann A , Pfefferle S , Schwabenland M , Sumner Magruder D , Bonn S , Prinz M , Gerloff C , Püschel K , Krasemann S , Aepfelbacher M , Glatzel M ((2020) ) Neuropathology of patients with COVID-19 in Germany: A post-mortem case series. Lancet Neurol. 19: , 919–929. |
[14] | Ellul MA , Benjamin L , Singh B , Lant S , Michael BD , Easton A , Kneen R , Defres S , Sejvar J , Solomon T ((2020) ) Neurological associations of COVID-19. Lancet Neurol. 19: , 767–783. |
[15] | Liotta EM , Batra A , Clark JR , Shlobin NA , Hoffman SC , Orban ZS , Koralnik IJ ((2020) ) Frequent neurologic manifestations and encephalopathy-associated morbidity in Covid-19 patients. Ann Clin Transl Neurol. 7: , 2221–2230. |
[16] | Nasiri E , Naseri A , Yazdchi M , Talebi M ((2021) ) Is there a link between COVID-19 and Creutzfeldt-Jakob Disease? A case report. J Res Clin Med. 9: , 26. |
[17] | Mao L , Jin H , Wang M , Hu Y , Chen S , He Q , Chang J , Hong C , Zhou Y , Wang D , Miao X , Li Y , Hu B ((2020) ) Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurol. 77: , 683–690. |
[18] | Ding Q , Shults NV , Harris BT , Suzuki YJ ((2020) ) Angiotensin-converting enzyme 2 (ACE2) is upregulated in Alzheimer’s disease brain. bioRxiv [Preprint]., doi: 10.1101/2020.10.08.331157. |
[19] | Matías-Guiu J , Gomez-Pinedo U , Montero-Escribano P , Gomez-Iglesias P , Porta-Etessam J , Matias-Guiu JA ((2020) ) Should we expect neurological symptoms in the SARS-CoV-2 epidemic? Neurologia. 35: , 170–175. |
[20] | Ferini-Strambi L , Salsone M ((2020) ) COVID-19 and neurological disorders: Are neurodegenerative or neuroimmunological diseases more vulnerable? J Neurol. 268: , 409–419. |
[21] | Fazzini E , Fleming J , Fahn S ((1992) ) Cerebrospinal fluid antibodies to coronavirus in patients with Parkinson’s disease. Mov Disord. 7: , 153–158. |
[22] | Gatto EM , Fernandez Boccazzi J ((2020) ) COVID-19 and neurodegeneration: What can we learn from the past? Eur J Neurol. 27: , e45. |
[23] | Serrano-Castro PJ , Estivill-Torrús G , Cabezudo-García P , Reyes-Bueno JA , Ciano Petersen N , Aguilar-Castillo MJ , Suárez-Pérez J , Jiménez-Hernández MD , Moya-Molina M , Oliver-Martos B , Arrabal-Gómez C , Rodríguez de Fonseca F ((2020) ) Impact of SARS-CoV-2 infection on neurodegenerative and neuropsychiatric diseases: A delayed pandemic? Neurologia. 35: , 245–251. |
[24] | Tavassoly O , Safavi F , Tavassoly I ((2020) ) Seeding brain protein aggregation by SARS-CoV-2 as a possible long-term complication of COVID-19 infection. ACS Chem Neurosci. 11: , 3704–3706. |
[25] | Liu S , Hossinger A , Heumüller S-E , Hornberger A , Buravlova O , Konstantoulea K , Müller SA , Paulsen L , Rousseau F , Schymkowitz J , Lichtenthaler SF , Neumann M , Denner P , Vorberg IM ((2021) ) Highly efficient intercellular spreading of protein misfolding mediated by viral ligand-receptor interactions. Nat Commun. 12: , 5739. |
[26] | Li YC , Bai WZ , Hirano N , Hayashida T , Hashikawa T ((2012) ) Coronavirus infection of rat dorsal root ganglia: Ultrastructural characterization of viral replication, transfer, and the early response of satellite cells. Virus Res. 163: , 628–635. |
[27] | Li YC , Bai WZ , Hirano N , Hayashida T , Taniguchi T , Sugita Y , Tohyama K , Hashikawa T ((2013) ) Neurotropic virus tracing suggests a membranous-coating-mediated mechanism for transsynaptic communication. J Comp Neurol. 521: , 203–212. |
[28] | Matsuda K , Park CH , Sunden Y , Kimura T , Ochiai K , Kida H , Umemura T ((2004) ) The vagus nerve is one route of transneural invasion for intranasally inoculated influenza A virus in mice. Vet Pathol. 41: , 101–107. |
[29] | Swanson PA , McGavern DB ((2015) ) Viral diseases of the central nervous system. Curr Opin Virol. 11: , 44–54. |
[30] | Berth SH , Leopold PL , Morfini G ((2009) ) Virus-induced neuronal dysfunction and degeneration. Front Biosci. 14: , 5239–5259. |
[31] | Bohmwald K , Gálvez NMS , Ríos M , Kalergis AM ((2018) ) Neurologic alterations due to respiratory virus infections. Front Cell Neurosci. 12: , 386. |
[32] | Desforges M , Le Coupanec A , Stodola JK , Meessen-Pinard M , Talbot PJ ((2014) ) Human coronaviruses: Viral and cellular factors involved in neuroinvasiveness and neuropathogenesis. Virus Res. 194: , 145–158. |
[33] | Lima M , Siokas V , Aloizou AM , Liampas I , Mentis AFA , Tsouris Z , Papadimitriou A , Mitsias PD , Tsatsakis A , Bogdanos DP , Baloyannis SJ , Dardiotis E ((2020) ) Unraveling the possible routes of SARS-COV-2 invasion into the central nervous system. Curr Treat Options Neurol. 22: , 37. |
[34] | Mengeling WL , Boothe AD , Ritchie AE ((1972) ) Characteristics of a coronavirus (strain 67N) of pigs. Am J Vet Res. 33: , 297–308. |
[35] | Andries K , Pensaert MB ((1980) ) Immunofluorescence studies on the pathogenesis of hemagglutinating encephalomyelitis virus infection in pigs after oronasal inoculation. Am J Vet Res. 41: , 1372–1378. |
[36] | Dubé M , Le Coupanec A , Wong AHM , Rini JM , Desforges M , Talbot PJ ((2018) ) Axonal transport enables neuron-to-neuron propagation of human coronavirus OC43. J Virol. 97: , e00404–18. |
[37] | Koyuncu OO , Hogue IB , Enquist LW ((2013) ) Virus infections in the nervous system. Cell Host Microbe. 13: , 379–393. |
[38] | Netland J , Meyerholz DK , Moore S , Cassell M , Perlman S ((2008) ) Severe acute respiratory syndrome coronavirus infection causes neuronal death in the absence of encephalitis in mice transgenic for human ACE2. J Virol. 82: , 7264–7275. |
[39] | Li K , Wohlford-Lenane C , Perlman S , Zhao J , Jewell AK , Reznikov LR , Gibson-Corley KN , Meyerholz DK , McCray PB ((2015) ) Middle east respiratory syndrome coronavirus causes multiple organ damage and lethal disease in mice transgenic for human dipeptidyl peptidase 4. J Infect Dis. 213: , 712–722. |
[40] | Lechien JR , Chiesa-Estomba CM , De Siati DR , Horoi M , Le Bon SD , Rodriguez A , Dequanter D , Blecic S , El Afia F , Distinguin L , Chekkoury-Idrissi Y , Hans S , Delgado IL , Calvo-Henriquez C , Lavigne P , Falanga C , Barillari MR , Cammaroto G , Khalife M , Leich P , Souchay C , Rossi C , Journe F , Hsieh J , Edjlali M , Carlier R , Ris L , Lovato A , De Filippis C , Coppee F , Fakhry N , Ayad T , Saussez S ((2020) ) Olfactory and gustatory dysfunctions as a clinical presentation of mild-to-moderate forms of the coronavirus disease (COVID-19): A multicenter European study. Eur Arch Otorhinolaryngol. 277: , 2251–2261. |
[41] | Sungnak W , Huang N , Bécavin C , Berg M , Queen R , Litvinukova M , Talavera-López C , Maatz H , Reichart D , Sampaziotis F , Worlock KB , Yoshida M , Barnes JL , Banovich NE , Barbry P , Brazma A , Collin J , Desai TJ , Duong TE , Eickelberg O , Falk C , Farzan M , Glass I , Gupta RK , Haniffa M , Horvath P , Hubner N , Hung D , Kaminski N , Krasnow M , Kropski JA , Kuhnemund M , Lako M , Lee H , Leroy S , Linnarson S , Lundeberg J , Meyer KB , Miao Z , Misharin AV , Nawijn MC , Nikolic MZ , Noseda M , Ordovas-Montanes J , Oudit GY , Pe’er D , Powell J , Quake S , Rajagopal J , Tata PR , Rawlins EL , Regev A , Reyfman PA , Rozenblatt-Rosen O , Saeb-Parsy K , Samakovlis C , Schiller HB , Schultze JL , Seibold MA , Seidman CE , Seidman JG , Shalek AK , Shepherd D , Spence J , Spira A , Sun X , Teichmann SA , Theis FJ , Tsankov AM , Vallier L , van den Berge M , Whitsett J , Xavier R , Xu Y , Zaragosi LE , Zerti D , Zhang H , Zhang K , Rojas M , Figueiredo F ((2020) ) SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat Med. 26: , 681–687. |
[42] | Moriguchi T , Harii N , Goto J , Harada D , Sugawara H , Takamino J , Ueno M , Sakata H , Kondo K , Myose N , Nakao A , Takeda M , Haro H , Inoue O , Suzuki-Inoue K , Kubokawa K , Ogihara S , Sasaki T , Kinouchi H , Kojin H , Ito M , Onishi H , Shimizu T , Sasaki Y , Enomoto N , Ishihara H , Furuya S , Yamamoto T , Shimada S ((2020) ) A first case of meningitis/encephalitis associated with SARS-Coronavirus-2. Int J Infect Dis. 94: , 55–58. |
[43] | Rhea EM , Logsdon AF , Hansen KM , Williams LM , Reed MJ , Baumann KK , Holden SJ , Raber J , Banks WA , Erickson MA ((2020) ) The S1 protein of SARS-CoV-2 crosses the blood-brain barrier in mice. Nat Neurosci. 24: , 386–378. |
[44] | Saleki K , Banazadeh M , Saghazadeh A , Rezaei N ((2020) ) The involvement of the central nervous system in patients with COVID-19. Rev Neurosci. 31: , 453–456. |
[45] | Li Z , Liu T , Yang N , Han D , Mi X , Li Y , Liu K , Vuylsteke A , Xiang H , Guo X ((2020) ) Neurological manifestations of patients with COVID-19: Potential routes of SARS-CoV-2 neuroinvasion from the periphery to the brain. Front Med. 14: , 533–541. |
[46] | McCray PB , Pewe L , Wohlford-Lenane C , Hickey M , Manzel L , Shi L , Netland J , Jia HP , Halabi C , Sigmund CD , Meyerholz DK , Kirby P , Look DC , Perlman S ((2007) ) Lethal infection of K18-hACE2 mice infected with severe acute respiratory syndrome coronavirus. J Virol. 81: , 813–821. |
[47] | Wohleb ES , McKim DB , Sheridan JF , Godbout JP ((2015) ) Monocyte trafficking to the brain with stress and inflammation: A novel axis of immune-to-brain communication that influences mood and behavior. Front Neurosci. 8: , 447. |
[48] | Zhang X , Chen X , Chen L , Deng C , Zou X , Liu W , Yu H , Chen B , Sun X ((2020) ) The infection evidence of SARS-COV-2 in ocular surface: A single-center cross-sectional study. medRxiv, doi: https://doi.org/10.1101/2020.02.26.20027938 |
[49] | Khelashvili G , Plante A , Doktorova M , Weinstein H ((2021) ) Ca2+-dependent mechanism of membrane insertion and destabilization by the SARS-CoV-2 fusion peptide. Biophys J. 120: , 1105–1119. |
[50] | Tang T , Bidon M , Jaimes JA , Whittaker GR , Daniel S ((2020) ) Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antiviral Res. 178: , 104792. |
[51] | Marsh M ((2005) ) Membrane trafficking in viral replication, Springer, Berlin New York. |
[52] | Eckert DM , Kim PS ((2001) ) Mechanisms of viral membrane fusion and its inhibition. Annu Rev Biochem. 70: , 777–810. |
[53] | Skehel JJ , Wiley DC ((2000) ) Receptor binding and membrane fusion in virus entry: The influenza hemagglutinin. Annu Rev Biochem. 69: , 531–569. |
[54] | Tamm LK , Han X ((2000) ) Viral fusion peptides: A tool set to disrupt and connect biological membranes. Biosci Rep. 20: , 501–518. |
[55] | Gorgun D , Lihan M , Kapoor K , Tajkhorshid E ((2020) ) Binding mode of SARS-CoV2 fusion peptide to human cellular membrane. Biophys J. 120: , 2914–2926. |
[56] | Hassanzadeh K , Perez Pena H , Dragotto J , Buccarello L , Iorio F , Pieraccini S , Sancini G , Feligioni M ((2020) ) Considerations around the SARS-CoV-2 spike protein with particular attention to COVID-19 brain infection and neurological symptoms. ACS Chem Neurosci. 11: , 2361–2369. |
[57] | Amin M , Sorour MK , Kasry A ((2020) ) Comparing the binding interactions in the receptor binding domains of SARS-CoV-2 and SARS-CoV. J Phys Chem Lett. 11: , 4897–4900. |
[58] | Gussow AB , Auslander N , Faure G , Wolf YI , Zhang F , Koonin EV ((2020) ) Genomic determinants of pathogenicity in SARS-CoV-2 and other human coronaviruses. Proc Natl Acad Sci U S A. 117: , 15193–15199. |
[59] | Yan R , Zhang Y , Li Y , Xia L , Guo Y , Zhou Q ((2020) ) Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 367: , 1444–1448. |
[60] | Reza-Zaldívar EE , Hernández-Sapiéns MA , Minjarez B , Gómez-Pinedo U , Márquez-Aguirre AL , Mateos-Díaz JC , Matias-Guiu J , Canales-Aguirre AA ((2021) ) Infection mechanism of SARS-COV-2 and its implication on the nervous system. Front Immunol. 11: , 621735. |
[61] | Heneka MT , Golenbock D , Latz E , Morgan D , Brown R ((2020) ) Immediate and long-term consequences of COVID-19 infections for the development of neurological disease. Alzheimers Res Ther. 12: , 69. |
[62] | Helms J , Kremer S , Merdji H , Clere-Jehl R , Schenck M , Kummerlen C , Collange O , Boulay C , Fafi-Kremer S , Ohana M , Anheim M , Meziani F ((2020) ) Neurologic features in severe SARS-CoV-2 infection. N Engl J Med. 382: , 2268–2270. |
[63] | Sfera A , Bullock K , Price A , Inderias L , Osorio C ((2018) ) Ferrosenescence: The iron age of neurodegeneration? Mech Ageing Dev. 174: , 63–75. |
[64] | Gomez-Pinedo U , Matias-Guiu J , Sanclemente-Alaman I , Moreno-Jimenez L , Montero-Escribano P , Matias-Guiu JA ((2020) ) Is the brain a reservoir organ for SARS-CoV2? J Med Virol. 92: , 2354–2355. |
[65] | Bianchetti A , Rozzini R , Guerini F , Boffelli S , Ranieri P , Minelli G , Bianchetti L , Trabucchi M ((2020) ) Clinical presentation of COVID19 in dementia patients. J Nutr Health Aging. 24: , 560–562. |
[66] | Ciaccio M , Sasso BL , Scazzone C , Gambino CM , Ciaccio AM , Bivona G , Piccoli T , Giglio RV , Agnello L ((2021) ) COVID-19 and Alzheimer’s disease. Brain Sci. 11: , 305. |
[67] | Kuo CL , Pilling LC , Atkins JL , Masoli JAH , Delgado J , Kuchel GA , Melzer D ((2020) ) APOE E4 Genotype predicts severe COVID-19 in the UK biobank community cohort. J Gerontol A Biol Sci Med Sci. 75: , 2231–2232. |
[68] | Wang QQ , Xu R , Volkow ND ((2021) ) Increased risk of COVID-19 infection and mortality in people with mental disorders: Analysis from electronic health records in the United States. World Psychiatry. 20: , 124–130. |
[69] | Wang R , Simoneau CR , Kulsuptrakul J , Bouhaddou M , Travisano KA , Hayashi JM , Carlson-Stevermer J , Zengel JR , Richards CM , Fozouni P , Oki J , Rodriguez L , Joehnk B , Walcott K , Holden K , Sil A , Carette JE , Krogan NJ , Ott M , Puschnik AS ((2021) ) Genetic screens identify host factors for SARS-CoV-2 and common cold coronaviruses. Cell. 184: , 106–119.e14. |
[70] | Baggen J , Persoons L , Vanstreels E , Jansen S , Van Looveren D , Boeckx B , Geudens V , De Man J , Jochmans D , Wauters J , Wauters E , Vanaudenaerde BM , Lambrechts D , Neyts J , Dallmeier K , Thibaut HJ , Jacquemyn M , Maes P , Daelemans D ((2021) ) Genome-wide CRISPR screening identifies TMEM106B as a proviral host factor for SARS-CoV-2. Nat Genet. 53: , 435–444. |
[71] | Schneider WM , Luna JM , Hoffmann HH , Sánchez-Rivera FJ , Leal AA , Ashbrook AW , Le Pen J , Ricardo-Lax I , Michailidis E , Peace A , Stenzel AF , Lowe SW , MacDonald MR , Rice CM , Poirier JT ((2021) ) Genome-scale identification of SARS-CoV-2 and pan-coronavirus host factor networks. Cell. 184: , 120–132.e14. |
[72] | Yang AC , Kern F , Losada PM , Agam MR , Maat CA , Schmartz GP , Fehlmann T , Stein JA , Schaum N , Lee DP , Calcuttawala K , Vest RT , Berdnik D , Lu N , Hahn O , Gate D , McNerney MW , Channappa D , Cobos I , Ludwig N , Schulz-Schaeffer WJ , Keller A , Wyss-Coray T ((2021) ) Dysregulation of brain and choroid plexus cell types in severe COVID-19. Nature. 595: , 565–571. |
[73] | Hampshire A , Trender W , Chamberlain SR , Jolly AE , Grant JE , Patrick F , Mazibuko N , Williams SC , Barnby JM , Hellyer P , Mehta MA ((2021) ) Cognitive deficits in people who have recovered from COVID-19. EClinicalMedicine. 39: , 101044. |
[74] | Cedars-Sinai Staff ((2021) ) How COVID-19 Compromises Brain Function. https://www.cedars-sinai.org/blog/covid-19-brain-fog.html |
[75] | Gavidia M ((2021) ) Latest News in Parkinson Disease: COVID-19 Neurological Complications, Technological Innovations, and More. https://www.ajmc.com/view/latest-news-in-parkinson-disease-covid-19-neurological-complications-technological-innovations-and-more |
[76] | Jaywant A , Vanderlind WM , Alexopoulos GS , Fridman CB , Perlis RH , Gunning FM ((2021) ) Frequency and profile of objective cognitive deficits in hospitalized patients recovering from COVID-19. Neuropsychopharmacology. 46: , 2235–2240. |
[77] | GlobalData Healthcare ((2021) ) Covid-19 may result in brain damage and increase the risk of dementia. https://www.clinicaltrialsarena.com/comment/covid-19-brain-damage-dementia-risk/ |
[78] | Yu Y , Travaglio M , Popovic R , Leal NS , Martins LM ((2021) ) Alzheimer’s and Parkinson’s diseases predict different COVID-19 outcomes: A UK biobank study. Geriatrics. 6: , 10. |
[79] | Wang Q , Davis PB , Gurney ME , Xu R ((2021) ) COVID-19 and dementia: Analyses of risk, disparity, and outcomes from electronic health records in the US. Alzheimers Dement. 17: , 1297–1306. |
[80] | Zhou Y , Xu J , Hou Y , Leverenz JB , Kallianpur A , Mehra R , Liu Y , Yu H , Pieper AA , Jehi L , Cheng F ((2021) ) Network medicine links SARS-CoV-2/COVID-19 infection to brain microvascular injury and neuroinflammation in dementia-like cognitive impairment. Alzheimers Res Ther. 13: , 110. |
[81] | Méndez R , Balanzá-Martínez V , Luperdi SC , Estrada I , Latorre A , González-Jiménez P , Feced L , Bouzas L , Yépez K , Ferrando A , Hervás D , Zaldívar E , Reyes S , Berk M , Menéndez R ((2021) ) Short-term neuropsychiatric outcomes and quality of life in COVID-19 survivors. J Intern Med. 290: , 621–631. |
[82] | Toubasi AA , AbuAnzeh RB , Tawileh HBA , Aldebei RH , Alryalat SAS ((2021) ) A meta-analysis: The mortality and severity of COVID-19 among patients with mental disorders. Psychiatry Res. 299: , 113856. |
[83] | Fong TG , Davis D , Growdon ME , Albuquerque A , Inouye SK ((2015) ) The interface between delirium and dementia in elderly adults. Lancet Neurol. 14: , 823–832. |
[84] | Fong TG , Jones RN , Shi P , Marcantonio ER , Yap L , Rudolph JL , Yang FM , Kiely DK , Inouye SK ((2009) ) Delirium accelerates cognitive decline in Alzheimer disease. Neurology. 72: , 1570–1575. |
[85] | Arnold C ((2020) ) Could COVID delirium bring on dementia? Nature. 588: , 22–24. |
[86] | Goldberg TE , Chen C , Wang Y , Jung E , Swanson A , Ing C , Garcia PS , Whittington RA , Moitra V ((2020) ) Association of delirium with long-term cognitive decline: A meta-analysis. JAMA Neurol. 77: , 1373. |
[87] | Tejera D , Mercan D , Sanchez-Caro JM , Hanan M , Greenberg D , Soreq H , Latz E , Golenbock D , Heneka MT ((2019) ) Systemic inflammation impairs microglial Aβ clearance through NLRP 3 inflammasome. EMBO J. 38: , e101064. |
[88] | Venegas C , Kumar S , Franklin BS , Dierkes T , Brinkschulte R , Tejera D , Vieira-Saecker A , Schwartz S , Santarelli F , Kummer MP , Griep A , Gelpi E , Beilharz M , Riedel D , Golenbock DT , Geyer M , Walter J , Latz E , Heneka MT ((2017) ) Microglia-derived ASC specks crossseed amyloid-β in Alzheimer’s disease. Nature. 552: , 355–361. |
[89] | Young MJ , O’Hare M , Matiello M , Schmahmann JD ((2020) ) Creutzfeldt-Jakob disease in a man with COVID-19: SARS-CoV-2-accelerated neurodegeneration? Brain Behav Immun. 89: , 601–603. |
[90] | Merad M , Martin JC ((2020) ) Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat Rev Immunol. 20: , 355–362. |
[91] | Cavalli G , De Luca G , Campochiaro C , Della-Torre E , Ripa M , Canetti D , Oltolini C , Castiglioni B , Tassan Din C , Boffini N , Tomelleri A , Farina N , Ruggeri A , Rovere-Querini P , Di Lucca G , Martinenghi S , Scotti R , Tresoldi M , Ciceri F , Landoni G , Zangrillo A , Scarpellini P , Dagna L ((2020) ) Interleukin-1 blockade with high-dose anakinra in patients with COVID-19, acute respiratory distress syndrome, and hyperinflammation: A retrospective cohort study. Lancet Rheumatol. 2: , e325–e331. |
[92] | de Erausquin GA , Snyder H , Carrillo M , Hosseini AA , Brugha TS , Seshadri S ((2021) ) The chronic neuropsychiatric sequelae of COVID-19: The need for a prospective study of viral impact on brain functioning. Alzheimers Dement. 17: , 1056–1065. |
[93] | Rahman MA , Islam K , Rahman S , Alamin M ((2021) ) Neurobiochemical cross-talk between COVID-19 and Alzheimer’s disease. Mol Neurobiol. 58: , 1017–1023. |
[94] | Cojocaru IM , Cojocaru M , Miu G , Sapira V ((2011) ) Study of interleukin-6 production in Alzheimer’s disease. Romanian J Intern Med Rev Roum Med Interne. 49: , 55–58. |
[95] | Huang W-J , Zhang X , Chen W-W ((2016) ) Role of oxidative stress in Alzheimer’s disease. Biomed Rep. 4: , 519–522. |
[96] | Pereira A ((2020) ) Long-term neurological threats of COVID-19: A call to update the thinking about the outcomes of the coronavirus pandemic. Front Neurol. 11: , 308. |
[97] | Lu Y , Li X , Geng D , Mei N , Wu P-Y , Huang C-C , Jia T , Zhao Y , Wang D , Xiao A , Yin B ((2020) ) Cerebral micro-structural changes in COVID-19 patients - an MRI-based 3-month follow-up study. EClinicalMedicine. 25: , 100484. |
[98] | Daroische R , Hemminghyth MS , Eilertsen TH , Breitve MH , Chwiszczuk LJ ((2021) ) Cognitive impairment after COVID-19— a review on objective test data. Front Neurol. 12: , 699582. |
[99] | Lee S , Viqar F , Zimmerman ME , Narkhede A , Tosto G , Benzinger TLS , Marcus DS , Fagan AM , Goate A , Fox NC , Cairns NJ , Holtzman DM , Buckles V , Ghetti B , McDade E , Martins RN , Saykin AJ , Masters CL , Ringman JM , Ryan NS , Förster S , Laske C , Schofield PR , Sperling RA , Salloway S , Correia S , Jack C , Weiner M , Bateman RJ , Morris JC , Mayeux R , Brickman AM , for the Dominantly Inherited Alzheimer Network ((2016) ) White matter hyperintensities are a core feature of Alzheimer’s disease: Evidence from the dominantly inherited Alzheimer network: White Matter Hyperintensities in Familial AD. Ann Neurol. 79: , 929–939. |
[100] | Love S , Miners JS ((2016) ) Cerebrovascular disease in ageing and Alzheimer’s disease. Acta Neuropathol (Berl). 131: , 645–658. |
[101] | Helzner EP , Luchsinger JA , Scarmeas N , Cosentino S , Brickman AM , Glymour MM , Stern Y ((2009) ) Contribution of vascular risk factors to the progression in Alzheimer disease. Arch Neurol. 66: , 343–348. |
[102] | Hemming ML , Selkoe DJ ((2005) ) Amyloid β-protein is degraded by cellular angiotensin-converting enzyme (ACE) and elevated by an ACE inhibitor. J Biol Chem. 280: , 37644–37650. |
[103] | Wen Y , Yang S , Liu R , Simpkins JW ((2004) ) Transient cerebral ischemia induces site-specific hyperphosphorylation of tau protein. Brain Res. 1022: , 30–38. |
[104] | Virhammar J , Nääs A , Fällmar D , Cunningham JL , Klang A , Ashton NJ , Jackmann S , Westman G , Frithiof R , Blennow K , Zetterberg H , Kumlien E , Rostami E ((2021) ) Biomarkers for central nervous system injury in cerebrospinal fluid are elevated in COVID-19 and associated with neurological symptoms and disease severity. Eur J Neurol. 28: , 3324–3331. |
[105] | Ameres M , Brandstetter S , Toncheva AA , Kabesch M , Leppert D , Kuhle J , Wellmann S ((2020) ) Association of neuronal injury blood marker neurofilament light chain with mild-to-moderate COVID-19. J Neurol. 267: , 3476–3478. |
[106] | Edén A , Kanberg N , Gostner J , Fuchs D , Hagberg L , Andersson L-M , Lindh M , Price RW , Zetterberg H , Gisslén M ((2020) ) CSF biomarkers in patients with COVID-19 and neurological symptoms: A case series. Neurology. 96: , e294–e300. |
[107] | Espíndola OM , Brandão CO , Gomes YCP , Siqueira M , Soares CN , Lima MASD , Leite ACCB , Torezani G , Araujo AQC , Silva MTT ((2021) ) Cerebrospinal fluid findings in neurological diseases associated with COVID-19 and insights into mechanisms of disease development. Int J Infect Dis. 102: , 155–162. |
[108] | Sutter R , Hert L , De Marchis GM , Twerenbold R , Kappos L , Naegelin Y , Kuster GM , Benkert P , Jost J , Maceski AM , Rüegg S , Siegemund M , Leppert D , Tschudin-Sutter S , Kuhle J ((2021) ) Serum neurofilament light chain levels in the intensive care unit: Comparison between severely ill patients with and without coronavirus disease 2019. Ann Neurol. 89: , 610–616. |
[109] | Miao G , Zhao H , Li Y , Ji M , Chen Y , Shi Y , Bi Y , Wang P , Zhang H ((2021) ) ORF3a of the COVID-19 virus SARS-CoV-2 blocks HOPS complex-mediated assembly of the SNARE complex required for autolysosome formation. Dev Cell. 56: , 427–442. |
[110] | Hosseini S , Wilk E , Michaelsen-Preusse K , Gerhauser I , Baumgärtner W , Geffers R , Schughart K , Korte M ((2018) ) Long-term neuroinflammation induced by influenza A virus infection and the impact on hippocampal neuron morphology and function. J Neurosci. 38: , 3060–3080. |
[111] | Sy M , Kitazawa M , Medeiros R , Whitman L , Cheng D , Lane TE , LaFerla FM ((2011) ) Inflammation induced by infection potentiates tau pathological features in transgenic mice. Am J Pathol. 178: , 2811–2822. |
[112] | Ritchie K , Chan D , Watermeyer T ((2020) ) The cognitive consequences of the COVID-19 epidemic: Collateral damage? Brain Commun. 2: , fcaa069. |
[113] | Reiken S , Sittenfeld L , Dridi H , Liu Y , Liu X , Marks AR ((2022) ) Alzheimer’s-like signaling in brains of COVID-19 patients. Alzheimers Dement. 18: , 955–965. |
[114] | Atkins JL , Masoli JAH , Delgado J , Pilling LC , Kuo C-L , Kuchel GA , Melzer D ((2020) ) Preexisting comorbidities predicting COVID-19 and mortality in the UK Biobank Community Cohort. J Gerontol Ser A. 75: , 2224–2230. |
[115] | Brown EE , Kumar S , Rajji TK , Pollock BG , Mulsant BH ((2020) ) Anticipating and mitigating the impact of the COVID-19 pandemic on Alzheimer’s disease and related dementias. Am J Geriatr Psychiatry. 28: , 712–721. |
[116] | Perry G ((2020) ) Alzheimer’s disease patients in the crosshairs of COVID-19. J Alzheimers Dis. 76: , 1. |
[117] | Liu CC , Kanekiyo T , Xu H , Bu G ((2013) ) Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat Rev Neurol. 9: , 106–118. |
[118] | Farrer LA , Cupples LA , Haines JL , Hyman B , Kukull WA , Mayeux R , Myers RH , Pericak-Vance MA , Risch N , Van Duijn CM ((1997) ) Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease: A meta-analysis. JAMA. 278: , 1349–1356. |
[119] | Bertram L , Tanzi RE ((2008) ) Thirty years of Alzheimer’s disease genetics: The implications of systematic meta-analyses. Nat Rev Neurosci. 9: , 768–778. |
[120] | Kuo CL , Pilling LC , Atkins JL , Masoli JAH , Delgado J , Kuchel GA , Melzer D , Newman AB ((2020) ) ApoE e4e4 genotype and mortality with COVID-19 in UK Biobank. J Gerontol Ser A. 75: , 1801–1803. |
[121] | Wang C , Zhang M , Garcia G , Tian E , Cui Q , Chen X , Sun G , Wang J , Arumugaswami V , Shi Y ((2021) ) ApoE-isoform-dependent SARS-CoV-2 neurotropism and cellular response. Cell Stem Cell. 28: , 331–342.e5. |
[122] | Burgos JS , Ramirez C , Sastre I , Valdivieso F ((2006) ) Effect of Apolipoprotein E on the cerebral load of latent herpes simplex virus type 1 DNA. J Virol. 80: , 5383–5387. |
[123] | Burt TD , Agan BK , Marconi VC , He W , Kulkarni H , Mold JE , Cavrois M , Huang Y , Mahley RW , Dolan MJ , McCune JM , Ahuja SK ((2008) ) Apolipoprotein (apo) E4 enhances HIV-1 cell entry in vitro, and the APOE 4/4 genotype accelerates HIV disease progression. Proc Natl Acad Sci U S A. 105: , 8718–8723. |
[124] | Jiménez-Ruiz A , García-Grimshaw M , Ruiz-Sandoval JL ((2020) ) Manifestaciones neurológicas por COVID-19 neurologic manifestations. Gac Med Mex. 156: , 257. |
[125] | Kotecha AM , Corrêa ADC , Fisher KM , Rushworth JV ((2018) ) Olfactory dysfunction as a global biomarker for sniffing out Alzheimer’s disease: A meta-analysis. Biosensors. 8: , E41. |
[126] | Sulzer D , Antonini A , Leta V , Nordvig A , Smeyne RJ , Goldman JE , Al-Dalahmah O , Zecca L , Sette A , Bubacco L , Meucci O , Moro E , Harms AS , Xu Y , Fahn S , Ray Chaudhuri K ((2020) ) COVID-19 and possible links with Parkinson’s disease and parkinsonism: From bench to bedside. NPJ Parkinsons Dis. 6: , 18. |
[127] | Heinzel S , Berg D , Gasser T , Chen H , Yao C , Postuma RB , the MDS Task Force on the Definition of Parkinson’s Disease ((2019) ) Update of the MDS research criteria for prodromal Parkinson’s disease. Mov Disord. 34: , 1464–1470. |
[128] | Limphaibool N , Iwanowski P , Holstad MJV , Kobylarek D , Kozubski W ((2019) ) Infectious etiologies of Parkinsonism: Pathomechanisms and clinical implications. Front Neurol. 10: , 652. |
[129] | Henry J , Smeyne RJ , Jang H , Miller B , Okun MS ((2010) ) Parkinsonism and neurological manifestations of influenza throughout the 20th and 21st centuries. Parkinsonism Relat Disord. 16: , 566–571. |
[130] | Arabi YM , Harthi A , Hussein J , Bouchama A , Johani S , Hajeer AH , Saeed BT , Wahbi A , Saedy A , AlDabbagh T , Okaili R , Sadat M , Balkhy H ((2015) ) Severe neurologic syndrome associated with Middle East respiratory syndrome corona virus (MERS-CoV). Infection. 43: , 495–501. |
[131] | Jang H , Boltz DA , Webster RG , Smeyne RJ ((2009) ) Viral parkinsonism. Biochim Biophys Acta. 1792: , 714–721. |
[132] | Jang H , Boltz D , Sturm-Ramirez K , Shepherd KR , Jiao Y , Webster R , Smeyne RJ ((2009) ) Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc Natl Acad Sci U S A. 106: , 14063–14068. |
[133] | Jang H , Boltz D , McClaren J , Pani AK , Smeyne M , Korff A , Webster R , Smeyne RJ ((2012) ) Inflammatory effects of highly pathogenic H5N1 influenza virus infection in the CNS of mice. J Neurosci. 32: , 1545–1559. |
[134] | Calderón-Garcidueñas L , Torres-Jardón R , Franco-Lira M , Kulesza R , González-Maciel A , Reynoso-Robles R , Brito-Aguilar R , García-Arreola B , Revueltas-Ficachi P , Barrera-Velázquez JA , García-Alonso G , García-Rojas E , Mukherjee PS , Delgado-Chávez R ((2020) ) Environmental nanoparticles, SARS-CoV-2 brain involvement, and potential acceleration of Alzheimer’s and Parkinson’s diseases in young urbanites exposed to air pollution. J Alzheimers Dis. 78: , 479–503. |
[135] | Marreiros R , Müller-Schiffmann A , Trossbach SV , Prikulis I , Hänsch S , Weidtkamp-Peters S , Moreira AR , Sahu S , Soloviev I , Selvarajah S , Lingappa VR , Korth C ((2020) ) Disruption of cellular proteostasis by H1N1 influenza A virus causes α-synuclein aggregation. Proc Natl Acad Sci U S A. 117: , 6741–6751. |
[136] | Disdier C , Chalansonnet M , Gagnaire F , Gaté L , Cosnier F , Devoy J , Saba W , Lund AK , Brun E , Mabondzo A ((2017) ) Brain Inflammation, blood brain barrier dysfunction and neuronal synaptophysin decrease after inhalation exposure to titanium dioxide nano-aerosol in aging rats. Sci Rep. 7: , 12196. |
[137] | Achbani A , Sine H , Naciri A , Baba MA , Kharbach A , Bouchriti Y , Nejmeddine M ((2020) ) Can the 2019 novel coronavirus cause Parkinson’s disease? Mov Disord. 35: , 1102–1103. |
[138] | Hu C , Chen C , Dong X-P ((2021) ) Impact of COVID-19 pandemic on patients with neurodegenerative diseases. Front Aging Neurosci. 13: , 664965. |
[139] | Schirinzi T , Landi D , Liguori C ((2021) ) COVID-19: Dealing with a potential risk factor for chronic neurological disorders. J Neurol. 268: , 1171–1178. |
[140] | Pang L , Liu Y , Shen M , Ye J , Chen R , Lan Z , Wu Z , Guo Y , Zhang P ((2020) ) Influence of aging on deterioration of patients with COVID-19. Aging. 12: , 26248–26262. |
[141] | Ashapkin VV , Kutueva LI , Kurchashova SY , Kireev II ((2019) ) Are there common mechanisms between the Hutchinson-Gilford progeria syndrome and natural aging? Front Genet. 10: , 455. |
[142] | Onder G , Rezza G , Brusaferro S ((2020) ) Case-fatality rate and characteristics of patients dying in relation to COVID-19 in Italy. JAMA. 323: , 1775–1776. |
[143] | Le Couteur DG , Anderson RM , Newman AB ((2020) ) COVID-19 through the lens of gerontology. J Gerontol A Biol Sci Med Sci. 75: , e119–e120. |
[144] | Promislow DEL , Anderson R ((2020) ) A geroscience perspective on COVID-19 mortality. J Gerontol A Biol Sci Med Sci. 75: , e30–e33. |
[145] | Montagne A , Barnes SR , Sweeney MD , Halliday MR , Sagare AP , Zhao Z , Toga AW , Jacobs RE , Liu CY , Amezcua L , Harrington MG , Chui HC , Law M , Zlokovic BV ((2015) ) Blood-brain barrier breakdown in the aging human hippocampus. Neuron. 85: , 296–302. |
[146] | Karuppan MKM , Devadoss D , Nair M , Chand HS , Lakshmana MK ((2021) ) SARS-CoV-2 infection in the central and peripheral nervous system-associated morbidities and their potential mechanism. Mol Neurobiol. 58: , 2465–2480. |
[147] | Wu C , Chen X , Cai Y , Xia J , Zhou X , Xu S , Huang H , Zhang L , Zhou X , Du C , Zhang Y , Song J , Wang S , Chao Y , Yang Z , Xu J , Zhou X , Chen D , Xiong W , Xu L , Zhou F , Jiang J , Bai C , Zheng J , Song Y ((2020) ) Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med. 180: , 934–943. |
[148] | Boe DM , Boule LA , Kovacs EJ ((2017) ) Innate immune responses in the ageing lung. Clin Exp Immunol. 187: , 16–25. |
[149] | Liberale L , Montecucco F , Tardif JC , Libby P , Camici GG ((2020) ) Inflamm-ageing: The role of inflammation in age-dependent cardiovascular disease. Eur Heart J. 41: , 2974–2982. |
[150] | Mueller AL , Mcnamara MS , Sinclair DA ((2020) ) Why does COVID-19 disproportionately affect older people? Aging. 12: , 9959–9981. |
[151] | Baig AM , Sanders EC ((2020) ) Potential neuroinvasive pathways of SARS-CoV-2: Deciphering the spectrum of neurological deficit seen in coronavirus disease-2019 (COVID-19). J Med Virol. 92: , 1845–1857. |
[152] | AlGhatrif M , Cingolani O , Lakatta EG ((2020) ) The dilemma of coronavirus disease 2019, aging, and cardiovascular disease. JAMA Cardiol. 5: , 747–748. |
[153] | Calder PC , Bosco N , Bourdet-Sicard R , Capuron L , Delzenne N , Doré J , Franceschi C , Lehtinen MJ , Recker T , Salvioli S , Visioli F ((2017) ) Health relevance of the modification of low grade inflammation in ageing (inflammageing) and the role of nutrition. Ageing Res Rev. 40: , 95–119. |
[154] | Harenberg J , Favaloro E ((2020) ) COVID-19: Progression of disease and intravascular coagulation-present status and future perspectives. Clin Chem Lab Med. 58: , 1029–1036. |
[155] | Fuster-Matanzo A , Hernández F , Ávila J ((2018) ) Tau spreading mechanisms; implications for dysfunctional tauopathies. Int J Mol Sci. 19: , 645. |
[156] | Hanger DP , Anderton BH , Noble W ((2009) ) Tau phosphorylation: The therapeutic challenge for neurodegenerative disease. Trends Mol Med. 15: , 112–119. |
[157] | Kanmert D , Cantlon A , Muratore CR , Jin M , O’Malley TT , Lee G , Young-Pearse TL , Selkoe DJ , Walsh DM ((2015) ) C-terminally truncated forms of tau, but not full-length tau or its C-terminal fragments, are released from neurons independently of cell death. J Neurosci. 35: , 10851–10865. |
[158] | Hategan A , Masliah E , Nath A ((2019) ) HIV and Alzheimer’s disease: Complex interactions of HIV-Tat with amyloid β peptide and Tau protein. J Neurovirol. 25: , 648–660. |
[159] | Kodidela S , Gerth K , Haque S , Gong Y , Ismael S , Singh A , Ishrat T , Kumar S ((2019) ) Extracellular vesicles: A possible link between HIV and Alzheimer’s disease-like pathology in HIV subjects? Cells. 8: , 968. |
[160] | Leblanc P , Alais S , Porto-Carreiro I , Lehmann S , Grassi J , Raposo G , Darlix JL ((2006) ) Retrovirus infection strongly enhances scrapie infectivity release in cell culture. EMBO J. 25: , 2674–2685. |
[161] | Zhou L , Miranda-Saksena M , Saksena NK ((2013) ) Viruses and neurodegeneration. Virol J. 10: , 172. |
[162] | Ezzat K , Pernemalm M , Pålsson S , Roberts TC , Järver P , Dondalska A , Bestas B , Sobkowiak MJ , Levänen B , Sköld M , Thompson EA , Saher O , Kari OK , Lajunen T , Sverremark Ekström E , Nilsson C , Ishchenko Y , Malm T , Wood MJA , Power UF , Masich S , Lindén A , Sandberg JK , Lehtiö J , Spetz A-L, EL , Andaloussi S ((2019) ) The viral protein corona directs viral pathogenesis and amyloid aggregation. Nat Commun. 10: , 2331. |
[163] | Tavassoly O , Safavi F , Tavassoly I ((2020) ) Heparin-binding peptides as novel therapies to stop SARS-CoV-2 cellular entry and infection. Mol Pharmacol. 98: , 612–619. |
[164] | Kordower JH , Chu Y , Hauser RA , Freeman TB , Olanow CW ((2008) ) Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med. 14: , 504–506. |
[165] | Li J-Y , Englund E , Holton JL , Soulet D , Hagell P , Lees AJ , Lashley T , Quinn NP , Rehncrona S , Björklund A , Widner H , Revesz T , Lindvall O , Brundin P ((2008) ) Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med. 14: , 501–503. |
[166] | Prusiner SB ((2013) ) Biology and genetics of prions causing neurodegeneration. Annu Rev Genet. 47: , 601–623. |
[167] | Chien P , Weissman JS , DePace AH ((2004) ) Emerging principles of conformation-based prion inheritance. Annu Rev Biochem. 73: , 617–656. |


