
Oxysterols and Oxysterol Sulfates in Alzheimer’s Disease Brain and Cerebrospinal Fluid
Abstract
Background:
Brain cholesterol levels are tightly regulated but increasing evidence indicates that cholesterol metabolism may drive Alzheimer’s disease (AD)-associated pathological changes. Recent advances in understanding of mitochondrial dysfunction in AD brain have presented a vital role played by mitochondria in oxysterol biosynthesis and their involvement in pathophysiology. Oxysterol accumulation in brain is controlled by various enzymatic pathways including sulfation. While research into oxysterol is under the areas of active investigation, there is less evidence for oxysterol sulfate levels in human brain.
Objective:
This study investigates the hypothesis that AD brain oxysterol detoxification via sulfation is impaired in later stages of disease resulting in oxysterol accumulation.
Methods:
Lipids were extracted from postmortem frozen brain tissue and cerebrospinal (CSF) from late- (Braak stage III-IV) and early- (Braak stage I-II) stage AD patients. Samples were spiked with internal standards prior to lipid extraction. Oxysterols were enriched with a two-step solid phase extraction using a polymeric SPE column and further separation was achieved by LC-MS/MS.
Results:
Oxysterols, 26-hydroxycholesterol (26-OHC), 25-hydroxycholesterol (25-OHC), and 7-oxycholesterol levels were higher in brain tissue and mitochondria extracted from late-stage AD brain tissue except for 24S-hydroxycholesterol, which was decreased in late AD. However, oxysterol sulfates are significantly lower in the AD frontal cortex. Oxysterols, 25-OHC, and 7-oxocholesterol was higher is CSF but 26-OHC and oxysterol sulfate levels were not changed.
Conclusion:
Our results show oxysterol metabolism is altered in AD brain mitochondria, favoring synthesis of 26-OHC, 25-OHC, and 7-oxocholesterol, and this may influence brain mitochondrial function and acceleration of the disease.
INTRODUCTION
Alzheimer’s disease (AD) is the most common neurodegenerative disorder worldwide. It represents 70% of total dementia cases and clinically presented as a progressive loss of cognitive abilities and functional independence [1]. Pathophysiologically, AD is characterized by the presence of intracellular neurofibrillary tangles and extracellular deposition of amyloid plaques, resulting in neuronal dysfunction and neuronal loss [2]. Although the link between these two AD pathological hallmarks and their involvement in neuronal synaptic dysfunction is unclear, abundant evidence support a strong link to oxidative stress mediated assaults in the brain tissues [3]. Of particular significance, the role of cholesterol homeostasis was investigated heavily in the past decades [4, 5] with its links to apolipoprotein E (APOE) type 4 allele, which presents as the most robust genetic risk factor for late-onset AD [6].
Under normal physiological conditions brain cholesterol is produced and metabolized in situ by the glial cells [7] independent of peripheral cholesterol levels [8]. To maintain normal lipids hemostasis, 70% of the brain cholesterol remains in its non-esterified form while excess cholesterol is oxidized enzymatically by the cytochrome p450 family, forming oxysterols [7]. At least 40% of the brain cholesterol is converted into 24S-hydroxycholesterol (24S-OHC, also known as cerebrosterol) by the neuron-specific enzyme CYP46A1 [9]. This enzyme is highly expressed in pyramidal cells of the cortex and hippocampus, granule cells of the dentate gyrus and Purkinje cells of the cerebellum [10]. Brain 24-OHC levels has been shown to reflect neuronal dysfunction during late stage of AD based on the Braak staging system of neurofibrillary pathology [5, 9, 11]. Polymorphisms in the CYP46 gene were found to associate with increased amyloid-β (Aβ) load in the brain, as well as increased cerebrospinal fluid (CSF) levels of Aβ and phosphorylated tau [12]. Beside 24S-OHC, brain cells also synthesize other oxysterols such as 25-hydroxycholesterol (25-OHC) and (25R)26-hydroxycholesterol (26-OHC; also known as 27-hydroxycholesterol) via the actions of cholesterol 25-hydroxylase (CH25H) and cholesterol 27-hydroxylase (CYP27A1).
Mitochondria play an important role in the synthesis of oxysterols via cytochrome P450 enzymes [13]. The mitochondrial inner membrane enzyme CYP27 initiates the acidic pathway of oxysterol synthesis to form monohydroxy oxysterols 26-OHC and 25-OHC followed by 7α-hydroxylation via CYP7B1 to form dihydroxy oxysterols: 7α,26-dihydroxycholesterol and 7α,25-dihydroxycholesterol. Even though mitochondrial involvement in oxysterol biosynthesis is well defined, it is not clear if this pathway is impaired in AD brain. In addition to enzymatic production, oxysterols can be also generated non-enzymatically through free radical mediated reactions specially during inflammation. Free radical derived 7-oxycholesterols [7β hydroxy cholesterol (7β-OHC) and 7-keto cholesterol (7-KC)] have been found in brain, CSF [14], and plasma from AD patients [15]. Since oxysterols are important mediators in variety of cell functions including intracellular signaling [4], cell death [16], cell-cell communications [17], and inflammation [15], alteration to oxysterol homeostasis affects cellular health.
Another regulatory pathway of cholesterol metabolism is sulfation. Cholesterol and oxysterols can be sulfated by sulfotransferases (SULT) at the 3 position of ring A of cholesterol to form cholesterol sulfate or oxysterol 3-sulfates [18]. Sulfotransferases, SULT2B1b, SULT2B1a, and SULT2A1 produce several oxysterol 3-sulfates including 7-ketocholesterol 3-sulfate, 24(S)-OHC-3-sulfate, or 25-OHC -3-sulfate. Cholesterol sulfate is the most abundant sterol sulfate in human plasma [19] and in the brain, cholesterol sulfate is a substrate for the synthesis of neurosteroids which display neuroprotective properties [20]. New evidence suggests that oxysterol sulfates are biologically active metabolites and not merely a detoxification end-products of the sterol metabolism [21, 22]. Oxysterol sulfates have been shown to be involve in lipid metabolism, inflammatory responses, and hepatic cell proliferation [21].
Our understanding of oxysterols and oxysterol sulfates, including their levels in brain, is emerging with the aid of quantitative lipidomics [23–25]. However, it is not clear the levels of oxysterol sulfates in AD brain tissue or their physiological and pathophysiological roles in AD. In this study we hypothesize that as AD develops, brain mitochondria contribute to the altered oxysterol metabolism, and this is partly through decreased levels of oxysterol sulfation. To investigate this hypothesis, we adopted a high-sensitive mass spectrometry approach to measure low abundant oxysterol and sulfated oxysterol metabolites in the frontal cortex, generally vulnerable in AD, of postmortem brain samples and CSF.
MATERIALS AND METHODS
Chemicals
Authentic standards (24(S)-hydroxycholesterol, 26-hydroxycholesterol, 25-hydroxycholesterol, 7β-hydroxycholesterol) and deuterated (24(R/S)-hydroxycholesterol-d7, 25-hydroxycholesterol-d6, 26-hydroxycholesterol-d6, 7β-hydroxycholesterol-d7, 7-ketocholesterol-d5) were purchased from Avanti polar lipids, Alabama. Authentic standard 7 keto cholesterol was purchased from Cayman chemicals, MI, USA. Butyl acetate, hexane, isopropanol, methanol, and formic acid (HPLC/MS grade) were purchased from Fisher Scientific, UK. Butylated hydroxytoluene (BHT) was from Sigma-Aldrich, UK. Oasis HLB Prime cartridges were purchased from Waters.
Tissue samples
Primary frontal cortex tissue samples from individuals diagnosed with AD or age-sex-matched controls were obtained from the Brains for Dementia Research (BDR), London Brain Bank. BDR (brainsfordementiaresearch.org.uk) project is a growing longitudinal cohort of controls and dementia samples. Twenty frozen brain tissue samples (0.5 mg) and matching CSF were obtained from brains for dementia with (n = 10, 74–89 years old, mean age 82.4 years) and without (n = 10, 72–91 years old, mean age 81 years) AD. Donors had provided written informed consent for brain donation and the use of the material and clinical information for research purposes under Research Ethics Committee approval (REC 15/SC/0639, HTA license 12217). The genotype data for the BDR cohort is available on the Dementia Platform UK upon request (https://www.dementiasplatform.uk/).
Enriched mitochondrial fractions
Enriched mitochondrial fractions were separated by differential centrifugation according to our previously published protocols [26]. Briefly, previously flash frozen cerebellar samples were placed in GentleMACS C tubes with mitochondria extraction buffer (50 mM Tris-HCl pH 7.4, 100 mM KCl, 1.5 mM MgCl2, 1 mM EGTA, 50 mM HEPES and 100 mM sucrose; all sourced from Sigma-Aldrich, UK) and homogenized using a GentleMACS Dissociator (Miltenyi Biotec). The resulting homogenates were spun at 4°C in an Eppendorf Model 5417R Microcentrifuge (Fisher Scientific); first at 850 x g for 10 min, then the supernatant obtained was centrifuged separately at 1000 x g for 10 min to yield a nuclear pellet and a final spin at 10000 x g for 30 min to produce the mitochondrial pellet; the remaining supernatant contained the cytosolic fraction. Fractions were stored at –80°C.
Extraction of free oxysterols from tissue, CSF, and mitochondria
Frozen tissues (50 mg) spiked with internal standards (1 ng of 24OHC-d7, 25OHC-d6, 26OHC-d6, 7βOHC-d7, 7-keto-OHC-d5) were homogenized with a Jencons-PLS T8.01, IKA® homogenizer in 500μL of ice-cold methanol with 4 mg/ml BHT. Human CSF samples (400μl) spiked with internal standards was mixed with 1,600μL ice-cold methanol containing 4 mg/ml BHT as we described before [14]. Enriched mitochondrial fractions (1 mg/ml) were incubated in 100μL of ice-cold methanol with 4 mg/ml BHT. All samples were incubated in ice for 10 min before centrifugation at 14,000 × g for 10 min. The methanolic supernatants were diluted with acidified water up to 12.5% of methanol for loading on to a solid phase extraction (SPE) cartridge. Oxysterols were enriched using two-step SPE using a polymeric SPE column (HLB PRiME, Waters) as described by Dias et al. (2018) [27].
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis of oxysterols
The oxysterol analysis was done using liquid chromatography (LC, DIONEX UltiMate 3000, Thermo Scientific UK Ltd., Hemel Hempstead) on-line coupled to the ESI-QqLIT-MS/MS (QTRAP 5500, AB Sciex UK Ltd., Warrington) as previously described by Dias et al. (2018) [27]. Multiple reaction monitoring with transitions of 367.2/161 for 24S-OHC, 367.4/147 for 25-OHC, 385.4/161 for 26-OHC, 385.4/81 for 7β-OHC, and 401.4/95 for 7-KC were used to collect data. Data were examined using Analyst Software 1.7.2 (AB Sciex, Warrington, UK).
Semi-quantification of oxysterol sulfates in brain tissue and CSF
Lipids from frozen tissues (10 mg) and CSF (100μL) were extracted by the Folch protocol [28]. Extraction was done in glass vials and repeated twice, the organic layer from each was combined and evaporated to dry under nitrogen stream in an ice bath. Phospholipids were quantified by spectrophotometry measurement of inorganic phosphorous as described before [29]. Lipid extracts were resuspended in chloroform: methanol (1:1, v/v) and normalized to a final concentration of 25 ng phospholipid per microliter in 100% methanol. Oxysterol sulfates were analyzed by mass spectrometry in a 5500 QTrap instrument (ABSciex, Warrington, UK) operating in the negative ion detection mode over the mass range of 350–1000 Da with direct infusion at a flow rate of 10μL min–1 as we described before [29]. Detection of oxysterol sulfates in lipid extracts was achieved by precursor ion scanning (PIS) at m/z 97.0 and confirmed by PIS at m/z 80 collected at 1000 Da/s scan speed with step size of 0.1 Da. Oxysterol PIS mass spectrum of samples at m/z 481.4 was used to calculate the levels of oxysterol sulfates.
Statistical analysis
All analyses were performed using SPSS® software (IBM®, Version 25, USA). Kolmogorov–Smirnov test was used to determine if the data set was well-modelled by a normal distribution prior to statistical analysis. Means of continuous variables were compared by independent t-test. Bivariate Pearson correlation was used to test the degree of association between the variables. A p value of < 0.05 was considered statistically significant in all the performed analyses.
RESULTS
Oxysterol levels are altered in AD brain tissue and CSF
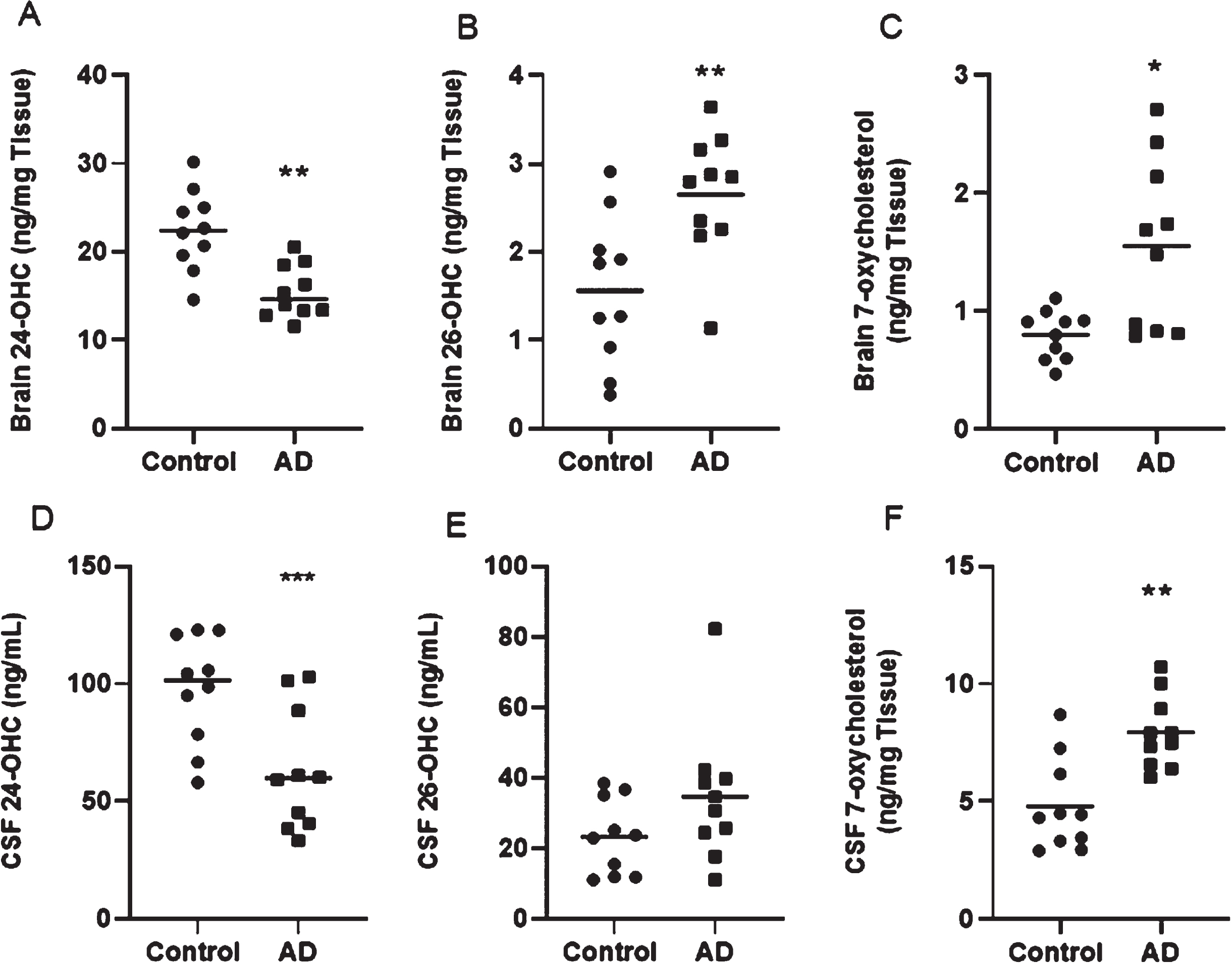
The study comprised postmortem tissue sample from the frontal cortex of AD brains and CSF, classified as early (control) or late AD based on the Braak staging system of neurofibrillary pathology (early AD: stages I and II; late AD: stages IV–VI) [30]. No significant differences between the patients and control group were observed with regard to age (82.4±4.45 years versus 81.00±7.28 years), postmortem delay (47.05±23.28 hours versus 53.20±26.21 hours), brain pH (6.31±0.29 versus 6.56±0.23), and Thal amyloid phase (3.14±1.46 versus 1.5±0.707), except for the Braak tangle classification. Control brains did not report senile plaques and tau pathology and aging changes were consistent with Braak stage II. Enzymatic origin 24S-OHC was significantly decreased in both AD brain (Fig. 1A) and CSF (Fig. 1D) (p < 0.001). Enzymatically generated 26-OHC and non-enzymatically generated 7-oxycholesterols (7β-OHC and 7-KC) were significantly elevated in AD brain tissue (Fig. 1B, C) and CSF (Fig. 1E, F).
Fig. 1
Oxysterol analysis in brain tissue and CSF. *Significant p-values are indicated where p < 0.05 was considered significant.
The relationship of oxysterols to APOE, Thal phase, and Braak stage
Postmortem delay and APOE polymorphism did not correlate to the oxysterol concentrations in the brain tissues and CSF p > 0.05 (Table 1). Thal amyloid phase was positively corelated to the 26-OHC concentration in the brain and the CSF (p = 0.044, r = 0.679 and p = 0.026, r = 0.727 respectively). Braak tangle staging was positively correlated to 7-oxycholesterols in the brain and the CSF (p = 0.025, r = 0.527; p = < 0.0001, r = 0.787) and 26-OHC in the brain (p = 0.022, r = 0.535), and negatively to 24S-OHC in the brain (p = 0.003, r = –0.662) (Table 1).
Table 1
Correlation between oxysterols, APOE, Thal amyloid phase and Braak tangle stage
| Postmortem Delay | APOE | Thal amyloid phase | Braak tangle staging | |
| Brain | ||||
| 24S-OHC (ng/mg tissue) | p = 0.356 | p = 0.293 | p = 0.173 | p = 0.003* |
| r = 0.218 | r = 0.247 | r = –0.498 | r = –0.662 | |
| 26-OHC (ng/mg tissue) | p = 0.978 | p = 0.560 | p = 0.044* | p = 0.022* |
| r = 0.007 | r = 0.138 | r = 0.679 | r = 0.535 | |
| 7-oxycholesterols (ng/mg tissue) | p = 0.466 | p = 0.987 | p = 0.10 | p = 0.025* |
| r = –0.178 | r = 0.004 | r = 0.799 | r = 0.527 | |
| CSF | ||||
| 24S-OHC (ng/mL) | p = 0.243 | p = 0.212 | p = 0.162 | p = 0.052 |
| r = 0.301 | r = 0.292 | r = 0.509 | r = –0.466 | |
| 26-OHC (ng/mL) | p = 0.301 | p = 0.674 | p = 0.026* | p = 0.368 |
| r = 0.893 | r = –0.10 | r = 0.727 | r = 0.225 | |
| 7-oxycholesterols (ng/mL) | p = 0.469 | p = 0.827 | p = 0.094 | p≤0.0001* |
| r = –0.172 | r = –0.052 | r = 0.591 | r = 0.787 | |
APOE, Apolipoprotein E; 24S-OHC, 24S-hydroxycholesterol; 26-OHC, 26-hydroxycholesterol; 7-oxycholesterols, 7β cholesterol and 7-Ketocholesterol; CSF, cerebrospinal fluid. *Significant p-values are indicated where p < 0.05 was considered significant.
Oxysterols in brain mitochondria
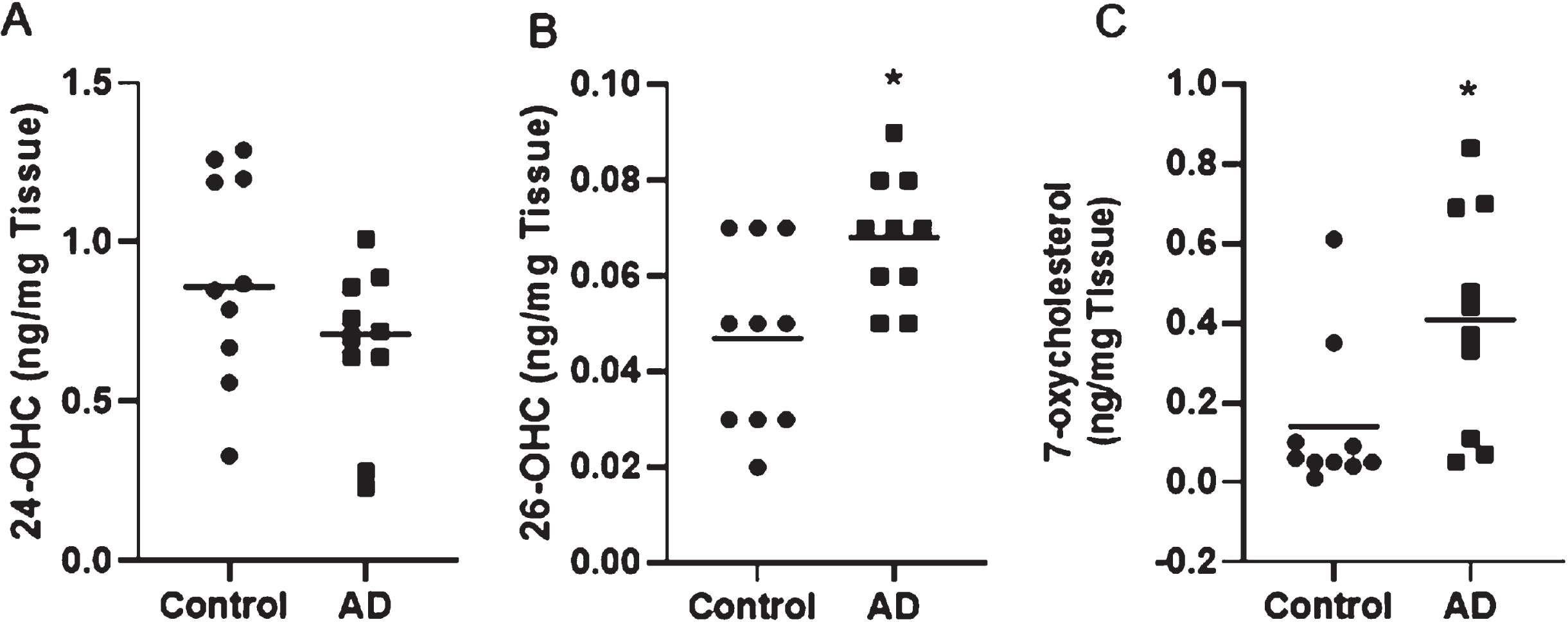
Since the acidic pathway of oxysterol synthesis is catalyzed by mitochondrial sterol hydroxylases, we investigated the distribution of oxysterols in mitochondria isolated from brain tissue. Similar to oxysterols levels in the brain tissue, 24S-OHC levels were significantly lower in brain mitochondria (Fig. 2A). The levels of 26-OHC, 7β-OHC, and 7-KC was significantly upregulated in AD brain mitochondria (Fig. 2B, C, D, respectively). Positive correlation was found between mitochondria 26-OHC and Braak tangle staging (p = 0.029, r = 0.513).
Fig. 2
Oxysterol analysis in brain mitochondria. *Significant p-values are indicated where p < 0.05 was considered significant.
Oxysterol sulfate levels are reduced in AD brain
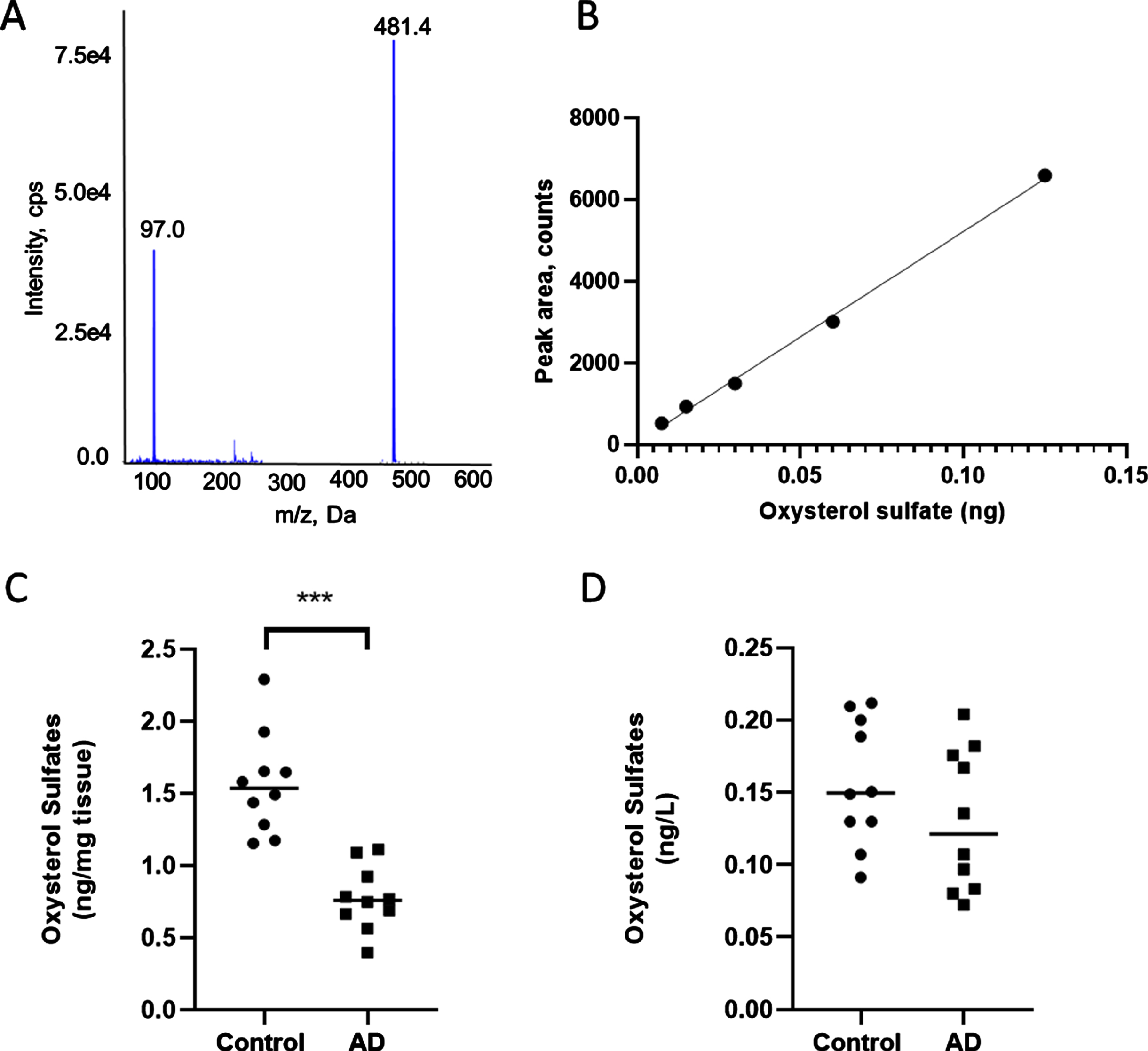
Lipid sulfation is known as a detoxifying mechanism to remove oxidized lipids [25]. In order to understand the level of oxysterol sulfate levels in AD brain, we applied recently developed mass spectrometry methods for semi-quantification of sulfate-based lipids [29]. Direct injection of 25HC3S standard was analyzed by PIS 97 and PIS 80 targeted method (Fig. 3A). After fragmentation at 30 eV, the major ions detected were the parent ion 25-OHC (m/z 481.4 Da) and the sulfate moiety (m/z 97 Da). Figure 3B confirms a linear dynamic range between 7.5 pg to 125 pg for 25HC3S. The limit of detection and the limit of quantification were 5 pg and 6.5 pg, respectively. Analyte peaks for the parent ion 25-OHC were higher than the limit of detection. 25HC3S levels were significantly reduced in the AD brain tissue compared to the control (0.77±0.07 ng/mg tissue versus 1.56±0.11 ng/mg tissue; p < 0.001) (Fig. 3C). However, 25HC3S levels in AD CSF were not significantly lower compared to control (0.13±0.02 ng/L versus 0.16±0.01 ng/L respectively) (Fig. 3D).
Fig. 3
Oxysterol sulfate analysis by MS. A) Identification of 25-hydroxycholesterol-3-sulfate by MS analysis. B) Linear dynamic range of oxysterol sulfate. C) Scatter plot showing oxysterol sulfate levels in control and AD brain tissue (***p < 0.001). D) scatter plot showing oxysterol sulfate levels in control and AD CSF.
DISCUSSION
This study analyzed free, non-esterified oxysterols in the tissue, mitochondria, and CSF of AD patients and controls. Even though these free, non-esterified molecules make up only a small proportion of the total oxysterols, they are biologically active metabolites [17, 31–33]. This study shows oxysterol levels for 26-OHC, 25-OHC, 7β-OHC, and 7-KC were raised in the late stage of AD frontal cortex with the exception to 24S-OHC, which was decreased. Our observations agree with previously published data for distribution of oxysterols in late AD brain tissue [11]. Here we show for the first time that mitochondria isolated from AD frontal cortex also contain increased levels of 26-OHC, 25-OHC, 7β-OHC, and 7-KC, and decreased 24S-OHC levels even after correcting to total protein.
For decades, Aβ and neurofibrillary tangles were considered the primary cause of AD [34, 35] and main disease staging systems such as the Braak tangle and Thal amyloid were developed depending on the location of amyloid lesion and the severity of the pathological changes [30]. To date, a large body of research has shown mitochondrial dysfunction in the brain of AD patients [36]. Since mitochondria play an important role in steroidogenesis, perturbed cholesterol metabolism and mitochondrial dysfunction has been suggested as contributors of AD. This study shows a significantly lower levels of 24S-OHC in the frontal cortex of AD brains and in isolated mitochondria compared to the study controls. Since 24S-OHC is mainly synthesized by neuronal cells, reduction of 24S-OHC could suggest the loss of neuronal mass in AD. However, low levels in mitochondrial 24S-OHC and increased levels of 26-OHC, 25-OHC, 7β-OHC, and 7-KC suggest that mitochondrial oxysterol pathway is also altered in AD. Previous reports demonstrated beneficial roles played by 24S-OHC [37]. For example, 24S-OHC favors α-secretase activity with subsequent increased levels of soluble amyloid protein precursor (sAPP) that favors safer removal of sAPP compared to oligomeric Aβ formation [37], 24S-OHC could selectively modulate the main memory controlling receptor in the human brain N-methyl-D-aspartate (NMDA) [38] and in vivo studies reported a potential improvement of memory by the overexpression of CYP46A enzyme and the modulation of its main metabolite 24S-OHC [39, 40]. Our analysis revealed a direct correlation between brain tissues 24S-OHC and the Braak stages of neurofibrillary tangles. Although this remains to be confirmed in more definitive experiments, our results support the preclinical evidence for 24S-OHC as a potent AβPP modulator [41]. Since 24S-OHC has neuronal origin and more than 60% of cholesterol removal from brain is achieved via oxidation to 24S-OHC [42], which traverses the blood-brain barrier, there was much interest to investigate it as a biomarker in circulation [9, 43]. However, we did not observe changes to 24S-OHC levels in CSF between control and AD.
Oxysterol homeostasis is maintained in the brain by both biosynthesis and efflux. Unlike cholesterol, excess oxysterols can be exported from the brain across blood-brain barrier or CSF. Likewise, some peripheral-derived oxysterols (e.g., 26-OHC) have been shown to be imported into the brain across the blood-brain barrier [44]. However, excess 26-OHC is suggested to be metabolized and subsequently eliminated from the brain as 7α-hydroxy-3-oxo-4-cholestenoic acid by neuronal cells [45]. During AD, the shrinking neuronal mass could negatively affect this process. Therefore, 26-OHC could act as an important marker for blood-brain barrier integrity where compromised blood-brain and blood-CSF barrier integrity may allow peripheral oxysterols to be transported to brain tissue [46] subsequently mediating negative effects on neuronal functions [33, 47, 48]. Our results show increased 26-OHC levels in AD frontal cortex, mitochondria, and CSF. Correlation analysis also revealed for the first time a positive clinical correlation between phases of amyloid deposition (Thal amyloid) and 26-OHC concentration in the brain and the CSF. Likewise, neurofibrillary pathology staging (Braak tangle) was affected positively by 26-OHC concentration in the brain and the mitochondria which adds to the growing evidence proposing the mitochondria as a key organelle in AD etiology. It may be possible that oxysterols in the brain is fundamental to maintain neuronal health thus, altered brain oxysterol concentrations could be a key to counter the detrimental effects of AD pathology.
Aside from enzymatically generated oxysterols, free radical generated 7-oxycholesterols were significantly increased in AD brain tissue, mitochondria, and CSF and correlated with Braak tangle staging. The enzymes 11β-hydroxysteroid dehydrogenase (11β-HSD) type 1 and type 2 are responsible for the interconversion of 7β-OHC and 7-KC [49]. 11β-HSD1 was reported to catalyze the reduction of 7KC to 7βOHC [49] and 11β-HSD2 was found to catalyze the oxidation of 7βOHC to 7KC [50]. 7-oxycholesterols have been shown to cytotoxic to the neural cells via multiple stress-response pathways. For example, 7-oxycholesterols increases the production of reactive oxygen species and triggers an apoptotic stress response [51]. 7-KC induced cell death found to be associated with mitochondrial dysfunctions, including changes to oxidative phosphorylation resulting energy imbalance in oligodendrocytes [52]. CSF 7-KC levels in cognitively healthy adults were associated with Aβ levels and white matter microstructure indicating the potential effect of 7-KC in the Aβ aggregation at early stage of the disease [14]. Collectively, this work shows important correlations between enzymatically produced and free radical generated oxysterols in AD brain. By emphasizing the role of mitochondrial cholesterol metabolism in AD, this study shows the importance of targeting brain mitochondria in AD.
Based on these measures, next we sought to investigate whether conversion of oxysterols to oxysterol sulfate for removal is altered in AD. This paper presents, for the first time, that oxysterol sulfate 25HC3S is significantly lower in AD frontal cortex. Sulfate-based lipids have increased water solubility than the parent oxidized form. Therefore, SULTs are known to be involved in detoxification of cytotoxic oxidized lipids [25]. Previous reports suggest a significantly lower copy number of SULT genes in AD [53] and lower SULT enzymatic activity compared to non-AD controls [54]. Therefore, it is possible that this pathway is impaired in AD and oxysterols may accumulate in the brain. Even though 25HC3S levels are lower in AD CSF, they were not statistically significant. Further experiments will be needed to confirm if this is due to sample number or due to another mechanism. Apart of detoxification, recent studies have shown that 25HC3S regulates important cellular events, including responses to stress signals via epigenetic modification, lipid hemostasis, regulating cellular inflammatory responses, and cell proliferation via the regulation of the activity of nuclear receptors.
It is interesting to compare the distribution of oxysterols in mitochondria with oxysterol sulfates. The key steps of this mechanism are depicted in Fig. 4. However, there were some limitations in this study: 1) sample numbers, 2) stages of AD development, and 3) access to different brain regions. Addressing above limitations would provide further insight into the interplay between oxysterols and oxysterol sulfates in the AD brain. In summary, this work suggest that cytotoxic oxysterols are accumulated in AD brain in the absence of SULT detoxification systems and open a new avenue to improve our understanding of the pathophysiological effects of oxysterol sulfates in AD.
Fig. 4
A schematic representing the key steps of mitochondrial oxysterol synthesis and conversion to oxysterol sulfates in cytosol. AD patients are reported to have low levels of brain sulfotransferase (SULT) genes and enzymatic activity [53, 54]. This may result oxysterols accumulation in AD brain cells and mitochondria.
![A schematic representing the key steps of mitochondrial oxysterol synthesis and conversion to oxysterol sulfates in cytosol. AD patients are reported to have low levels of brain sulfotransferase (SULT) genes and enzymatic activity [53, 54]. This may result oxysterols accumulation in AD brain cells and mitochondria.](https://content.iospress.com:443/media/jad/2022/87-4/jad-87-4-jad220083/jad-87-jad220083-g004.jpg)
ACKNOWLEDGMENTS
The authors acknowledge the MRC London Brain Bank for Neurodegenerative Diseases for providing samples. IHKD and LC would like to thank funding received by Alzheimer’s Research UK Midlands network.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/22-0083r1).
REFERENCES
[1] | Mucke L ((2009) ) Alzheimer’s disease. Nature 461: , 895–897. |
[2] | DeTure MA , Dickson DW ((2019) ) The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener 14: , 32. |
[3] | Chauhan V , Chauhan A ((2006) ) Oxidative stress in Alzheimer’s disease. Pathophysiology 13: , 195–208. |
[4] | Mutemberezi V , Guillemot-Legris O , Muccioli GG ((2016) ) Oxysterols: From cholesterol metabolites to key mediators. Prog Lipid Res 64: , 152–169. |
[5] | Zarrouk A , Vejux A , Mackrill J , O’Callaghan Y , Hammami M , O’Brien N , Lizard G ((2014) ) Involvement of oxysterols in age-related diseases and ageing processes. Ageing Res Rev 18: , 148–162. |
[6] | Stocker H , Möllers T , Perna L , Brenner H ((2018) ) The genetic risk of Alzheimer’s disease beyond APOE ɛ4: Systematic review of Alzheimer’s genetic risk scores. Transl Psychiatry 8: , 166–166. |
[7] | Björkhem I , Meaney S ((2004) ) Brain cholesterol: Long secret life behind a barrier. Arterioscler Thromb Vasc Biol 24: , 806–815. |
[8] | Dias Irundika HK , Polidori Maria C , Griffiths Helen R ((2014) ) Hypercholesterolaemia-induced oxidative stress at the blood–brain barrier. Biochem Soc Trans 42: , 1001–1005. |
[9] | Gamba P , Giannelli S , Staurenghi E , Testa G , Sottero B , Biasi F , Poli G , Leonarduzzi G ((2021) ) The controversial role of 24-S-hydroxycholesterol in Alzheimer’s disease. Antioxidants (Basel) 10: , 740. |
[10] | Björkhem I ((2006) ) Crossing the barrier: Oxysterols as cholesterol transporters and metabolic modulators in the brain. J Intern Med 260: , 493–508. |
[11] | Testa G , Staurenghi E , Zerbinati C , Gargiulo S , Iuliano L , Giaccone G , Fantò F , Poli G , Leonarduzzi G , Gamba P ((2016) ) Changes in brain oxysterols at different stages of Alzheimer’s disease: Their involvement in neuroinflammation. Redox Biol 10: , 24–33. |
[12] | Kölsch H , Lütjohann D , Ludwig M , Schulte A , Ptok U , Jessen F , von Bergmann K , Rao ML , Maier W , Heun R ((2002) ) Polymorphism in the cholesterol 24S-hydroxylase gene is associated with Alzheimer’s disease. Mol Psychiatry 7: , 899–902. |
[13] | Papadopoulos V , Miller WL ((2012) ) Role of mitochondria in steroidogenesis. Best Pract Res Clin Endocrinol Metab 26: , 771–790. |
[14] | Iriondo A , García-Sebastian M , Arrospide A , Arriba M , Aurtenetxe S , Barandiaran M , Clerigue M , Ecay-Torres M , Estanga A , Gabilondo A , Izagirre A , Saldias J , Tainta M , Villanua J , Blennow K , Zetterberg H , Mar J , Abad-García B , Dias IHK , Goñi FM , Martínez-Lage P ((2020) ) Cerebrospinal fluid 7-ketocholesterol level is associated with amyloid-β 42 and white matter microstructure in cognitively healthy adults. J Alzheimers Dis 76: , 643–656. |
[15] | Anderson A , Campo A , Fulton E , Corwin A , Jerome WG , O’Connor MS ((2020) ) 7-Ketocholesterol in disease and aging. Redox Biol 29: , 101380. |
[16] | Lütjohann D , Papassotiropoulos A , Björkhem I , Locatelli S , Bagli M , Oehring RD , Schlegel U , Jessen F , Rao ML , von Bergmann K , Heun R ((2000) ) Plasma 24S-hydroxycholesterol (cerebrosterol) is increased in Alzheimer and vascular demented patients. J Lipid Res 41: , 195–198. |
[17] | de Freitas FA , Levy D , Zarrouk A , Lizard G , Bydlowski SP ((2021) ) Impact of oxysterols on cell death, proliferation, and differentiation induction: Current status. Cells 10: , 2301. |
[18] | Javitt NB , Lee YC , Shimizu C , Fuda H , Strott CA ((2001) ) Cholesterol and hydroxycholesterol sulfotransferases: Identification, distinction from dehydroepiandrosterone sulfotransferase, and differential tissue expression. Endocrinology 142: , 2978–2984. |
[19] | Strott CA , Higashi Y ((2003) ) Cholesterol sulfate in human physiology: What’s it all about? J Lipid Res 44: , 1268–1278. |
[20] | Borowicz KK , Piskorska B , Banach M , Czuczwar SJ ((2011) ) Neuroprotective actions of neurosteroids. Front Endocrinol 2: , 50–50. |
[21] | Ren S , Ning Y ((2014) ) Sulfation of 25-hydroxycholesterol regulates lipid metabolism, inflammatory responses, and cell proliferation. Am J Physiol Endocrinol Metab 306: , E123–E130. |
[22] | Wang Y , Li X , Ren S ((2021) ) Cholesterol metabolites 25-hydroxycholesterol and 25-hydroxycholesterol 3-sulfate are potent paired regulators: From discovery to clinical usage. Metabolites 11: , 9. |
[23] | Sánchez-Guijo A , Oji V , Hartmann MF , Traupe H , Wudy SA ((2015) ) Simultaneous quantification of cholesterol sulfate, androgen sulfates, and progestagen sulfates in human serum by LC-MS/MS. J Lipid Res 56: , 1843–1851. |
[24] | Sánchez-Guijo A , Oji V , Hartmann MF , Schuppe HC , Traupe H , Wudy SA , ((2015) ) High levels of oxysterol sulfates in serum of patients with steroid sulfatase deficiency. J Lipid Res 56: , 403–412. |
[25] | Fuda H , Javitt NB , Mitamura K , Ikegawa S , Strott CA ((2007) ) Oxysterols are substrates for cholesterol sulfotransferase. J Lipid Res 48: , 1343–1352. |
[26] | Ingram TL , Shephard F , Sarmad S , Ortori CA , Barrett DA , Chakrabarti L ((2020) ) Sex specific inflammatory profiles of cerebellar mitochondria are attenuated in Parkinson’s disease. Aging 12: , 17713–17737. |
[27] | Dias IHK , Milic I , Lip GYH , Devitt A , Polidori MC , Griffiths HR ((2018) ) Simvastatin reduces circulating oxysterol levels in men with hypercholesterolaemia. Redox Biol 16: , 139–145. |
[28] | Folch J , Lees M , Sloane Stanley GH ((1957) ) A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem 226: , 497–509. |
[29] | Dias IHK , Ferreira R , Gruber F , Vitorino R , Rivas-Urbina A , Sanchez-Quesada JL , Vieira Silva J , Fardilha M , de Freitas V , Reis A ((2019) ) Sulfate-based lipids: Analysis of healthy human fluids and cell extracts. Chem Phys Lipids 221: , 53–64. |
[30] | Braak H , Alafuzoff I , Arzberger T , Kretzschmar H , Del Tredici K ((2006) ) Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112: , 389–404. |
[31] | Dias IHK , Mistry J , Fell S , Reis A , Spickett CM , Polidori MC , Lip GYH , Griffiths HR ((2014) ) Oxidized LDL lipids increase β-amyloid production by SH-SY5Y cells through glutathione depletion and lipid raft formation. Free Radic Biol Med 75: , 48–59. |
[32] | Gamba P , Guglielmotto M , Testa G , Monteleone D , Zerbinati C , Gargiulo S , Biasi F , Iuliano L , Giaccone G , Mauro A , Poli G , Tamagno E , Leonarduzzi G ((2014) ) Up-regulation of β-amyloidogenesis in neuron-like human cells by both 24- and 27-hydroxycholesterol: Protective effect of N-acetyl-cysteine. Aging Cell 13: , 561–572. |
[33] | Mateos L , Ismail MA , Gil-Bea FJ , Leoni V , Winblad B , Björkhem I , Cedazo-Mínguez A ((2011) ) Upregulation of brain renin angiotensin system by 27-hydroxycholesterol in Alzheimer’s disease. J Alzheimers Dis 24: , 669–679. |
[34] | Hyman BT , Phelps CH , Beach TG , Bigio EH , Cairns NJ , Carrillo MC , Dickson DW , Duyckaerts C , Frosch MP , Masliah E , Mirra SS , Nelson PT , Schneider JA , Thal DR , Thies B , Trojanowski JQ , Vinters HV , Montine TJ ((2012) ) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement 8: , 1–13. |
[35] | Montine TJ , Phelps CH , Beach TG , Bigio EH , Cairns NJ , Dickson DW , Duyckaerts C , Frosch MP , Masliah E , Mirra SS , Nelson PT , Schneider JA , Thal DR , Trojanowski JQ , Vinters HV , Hyman BT , National Institute on A Alzheimer’s A ((2012) ) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: A practical approach. Acta Neuropathol 123: , 1–11. |
[36] | Wang W , Zhao F , Ma X , Perry G , Zhu X ((2020) ) Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol Neurodegener 15: , 30. |
[37] | Famer D , Meaney S , Mousavi M , Nordberg A , Björkhem I , Crisby M ((2007) ) Regulation of α- and β-secretase activity by oxysterols: Cerebrosterol stimulates processing of APP via the α-secretase pathway. Biochem Biophys Res Commun 359: , 46–50. |
[38] | Paul SM , Doherty JJ , Robichaud AJ , Belfort GM , Chow BY , Hammond RS , Crawford DC , Linsenbardt AJ , Shu HJ , Izumi Y , Mennerick SJ , Zorumski CF ((2013) ) The major brain cholesterol metabolite 24(S)-hydroxycholesterol is a potent allosteric modulator of N-methyl-D-aspartate receptors. J Neurosci 33: , 17290–17300. |
[39] | Maioli S , Båvner A , Ali Z , Heverin M , Ismail MA , Puerta E , Olin M , Saeed A , Shafaati M , Parini P , Cedazo-Minguez A , Björkhem I ((2013) ) Is it possible to improve memory function by upregulation of the cholesterol 24S-hydroxylase (CYP46A1) in the brain? PLoS One 8: , e68534. |
[40] | Kotti T , Head DD , McKenna CE , Russell DW ((2008) ) Biphasic requirement for geranylgeraniol in hippocampal long-term potentiation. Proc Natl Acad Sci 105: , 11394–11399. |
[41] | Loera-Valencia R , Goikolea J , Parrado-Fernandez C , Merino-Serrais P , Maioli S ((2019) ) Alterations in cholesterol metabolism as a risk factor for developing Alzheimer’s disease: Potential novel targets for treatment. J Steroid Biochem Mol Biol 190: , 104–114. |
[42] | Vaya J , Schipper HM ((2007) ) Oxysterols, cholesterol homeostasis, and Alzheimer disease. J Neurochem 102: , 1727–1737. |
[43] | Chiappelli J , Quinton MS , Volfson D , Cwik M , Marshall W , Bruce H , Goldwaser E , Kvarta M , Summerfelt A , Kochunov P , O’Donnell P , Hong LE ((2020) ) Assessment of brain cholesterol metabolism biomarker 24S-hydroxycholesterol in schizophrenia. NPJ Schizophrenia 6: , 34. |
[44] | Björkhem I , Cedazo-Minguez A , Leoni V , Meaney S ((2009) ) Oxysterols and neurodegenerative diseases. Mol Aspects Med 30: , 171–179. |
[45] | Meaney S , Heverin M , Panzenboeck U , Ekström L , Axelsson M , Andersson U , Diczfalusy U , Pikuleva I , Wahren J , Sattler W , Björkhem I ((2007) ) Novel route for elimination of brain oxysterols across the blood-brain barrier: Conversion into 7alpha-hydroxy-3-oxo-4-cholestenoic acid. J Lipid Res 48: , 944–951. |
[46] | Leoni V , Masterman T , Patel P , Meaney S , Diczfalusy U , Björkhem I ((2003) ) Side chain oxidized oxysterols in cerebrospinal fluid and the integrity of blood-brain and blood-cerebrospinal fluid barriers. J Lipid Res 44: , 793–799. |
[47] | Leoni V , Masterman T , Mousavi FS , Wretlind B , Wahlund LO , Diczfalusy U , Hillert J , Björkhem I ((2004) ) Diagnostic use of cerebral and extracerebral oxysterols. Clin Chem Lab Med 42: , 186–191. |
[48] | Ismail MA , Mateos L , Maioli S , Merino-Serrais P , Ali Z , Lodeiro M , Westman E , Leitersdorf E , Gulyás B , Olof-Wahlund L , Winblad B , Savitcheva I , Björkhem I , Cedazo-Mínguez A ((2017) ) 27-Hydroxycholesterol impairs neuronal glucose uptake through an IRAP/GLUT4 system dysregulation. J Exp Med 214: , 699–717. |
[49] | Schweizer RAS , Zürcher M , Balazs Z , Dick B , Odermatt A ((2004) ) Rapid hepatic metabolism of 7-ketocholesterol by 11beta-hydroxysteroid dehydrogenase type 1: Species-specific differences between the rat, human, and hamster enzyme. J Biol Chem 279: , 18415–18424. |
[50] | Raleigh DR , Sever N , Choksi PK , Sigg MA , Hines KM , Thompson BM , Elnatan D , Jaishankar P , Bisignano P , Garcia-Gonzalo FR , Krup AL , Eberl M , Byrne EFX , Siebold C , Wong SY , Renslo AR , Grabe M , McDonald JG , Xu L , Beachy PA , Reiter JF ((2018) ) Cilia-associated oxysterols activate smoothened. Mol Cell 72: , 316–327.e315. |
[51] | Leonarduzzi G , Vizio B , Sottero B , Verde V , Gamba P , Mascia C , Chiarpotto E , Poli G , Biasi F ((2006) ) Early involvement of ROS overproduction in apoptosis induced by 7-ketocholesterol. Antioxid Redox Signal 8: , 375–380. |
[52] | Leoni V , Nury T , Vejux A , Zarrouk A , Caccia C , Debbabi M , Fromont A , Sghaier R , Moreau T , Lizard G ((2017) ) Mitochondrial dysfunctions in 7-ketocholesterol-treated 158N oligodendrocytes without or with α-tocopherol: Impacts on the cellular profil of tricarboxylic cycle-associated organic acids, long chain saturated and unsaturated fatty acids, oxysterols, cholesterol and cholesterol precursors. J Steroid Biochem Mol Biol 169: , 96–110. |
[53] | Butcher NJ , Horne MK , Mellick GD , Fowler CJ , Masters CL , Minchin RF , group Ar ((2018) ) Sulfotransferase 1A3/4 copy number variation is associated with neurodegenerative disease. Pharmacogenomics J 18: , 209–214. |
[54] | Vanková M , Hill M , Velíková M , Vcelák J , Vacínová G , Lukásová P , Vejrazková D , Dvoráková K , Rusina R , Holmerová I , Jarolímová E , Vanková H , Bendlová B ((2015) ) Reduced sulfotransferase SULT2A1 activity in patients with Alzheimer’s disease. Physiol Res 64: , S265–273. |


