
Potential Mechanisms Underlying Resistance to Dementia in Non-Demented Individuals with Alzheimer’s Disease Neuropathology
Abstract
Alzheimer’s disease (AD) is the most common form of dementia and typically characterized by the accumulation of amyloid-β plaques and tau tangles. Intriguingly, there also exists a group of elderly which do not develop dementia during their life, despite the AD neuropathology, the so-called non-demented individuals with AD neuropathology (NDAN). In this review, we provide extensive background on AD pathology and normal aging and discuss potential mechanisms that enable these NDAN individuals to remain cognitively intact. Studies presented in this review show that NDAN subjects are generally higher educated and have a larger cognitive reserve. Furthermore, enhanced neural hypertrophy could compensate for hippocampal and cingulate neural atrophy in NDAN individuals. On a cellular level, these individuals show increased levels of neural stem cells and ‘von Economo neurons’. Furthermore, in NDAN brains, binding of Aβ oligomers to synapses is prevented, resulting in decreased glial activation and reduced neuroinflammation. Overall, the evidence stated here strengthens the idea that some individuals are more resistant to AD pathology, or at least show an elongation of the asymptomatic state of the disease compared to others. Insights into the mechanisms underlying this resistance could provide new insight in understanding normal aging and AD itself. Further research should focus on factors and mechanisms that govern the NDAN cognitive resilience in order to find clues on novel biomarkers, targets, and better treatments of AD.
INTRODUCTION
In the pathology of Alzheimer’s disease (AD), two pivotal key hallmarks have been known and described for decades: amyloid-beta (Aβ) senile plaques and hyperphosphorylated tau proteins that form neurofibrillary tangles (NFT) [1]. Extensive research has shown that abnormal deposition of Aβ plaques and NFTs leads to synaptic damage and eventually to progressive cognitive impairment (e.g., memory loss), associated with dementia [2–5]. So far, drug development has been mainly focused on agents decelerating or preventing the progression of AD; however, most anti-AD therapies have been unsuccessful [6] and many promising findings in AD mouse models failed in translation to the clinical situation [7]. More recently, research is focused on identifying novel biomarkers and developing methods for early detection of AD. Currently, suggested AD biomarkers in the cerebrospinal fluid (CSF) are Aβ42; the Aβ42/40 ratio; phosphorylated tau at threonine residue 181 (pTau181) and tau protein [8].
Intriguingly, a multiplicity of literature originating from a variety of locations of the world has emerged that offers contradicting, yet intriguing, findings on the Aβ- and tau-hypotheses, as these histopathologic changes associated with AD have also been discovered post-mortem in non-demented older individuals. This suggests the existence of a group of non-demented, cognitively normal individuals carrying AD neuropathology.
In different recent studies, several synonyms (Table 1) have been used to describe these non-demented individuals, including but not limited to, non-demented individuals with AD neuropathology (NDAN) [9, 10]; non-demented high potential controls [11]; asymptomatic persons with AD neuropathology [12]; intermediate and high probability mismatches [13]; SuperAgers [14–16]; and asymptomatic AD (ASYMAD) [17–20]. All these studies have investigated older adults (65 + of age) with Aβ- and/or tau-deposition in absence of cognitive decline, dementia (Table 1). In this review, NDAN is defined as individuals that do not show the typical clinical-pathological correlation associated with AD, in other words individuals that carry AD pathology, as assessed post-mortem, but who despite this pathological burden performed cognitive normal antemortem. Important to note is that, in general, normal aging elderly show some degree of AD pathology and of cognitive deterioration, but not to the extent that they identify as ‘NDAN’ or become demented. Looking at the Gaussian distribution of the association between AD pathology and cognitive impairment on a population scale, the NDAN individuals are located on the tail of the Gaussian, as they are relatively rare, with for instance 6 to 15 out of over 3200 volunteers in the Baltimore Longitudinal Study of Aging (BLSA) study [17, 21, 22]. But even though they might be exceptions to the average disease progression, at the same time, this makes them even more interesting to study in the search for a better drug target and treatment for AD.
Table 1
Studies that described non-demented individuals with AD neuropathology (NDAN), using different synonyms. These studies all focus on non-demented individuals with AD neuropathology or preclinical AD, studying brain tissue and/or data on cognitive performance. Cognitive assessment and AD pathology scores are specified in Tables 2 and 3, respectively
| Synonyms of NDAN | Cohort study/Data set | Reference |
| Non-demented individuals with Alzheimer’s Neuropathology (NDAN) | Oregon Brain Bank at Oregon Health and Science University | [9, 10, 223] |
| Nondemented high potential controls (ND-HPC) | Banner Sun Health Research Institute: Brain and Body Donation Program | [11] |
| Asymptomatic person with AD neuropathology | National Alzheimer’s Coordinating Center: Uniform Data Set and Neuropathology Data Set | [12] |
| Intermediate probability mismatches | Massachusetts General Hospital, Mayo Clinic and University of Pittsburgh ADRC Brain Banks | [13, 178] |
| ‘Asymptomatic AD’ (ASYMAD) | The Nun Study/Baltimore Longitudinal Study of Aging (BLSA)/ | [17–20, 179] |
| Cognitively normal with significant AD pathology | Medical Research Council London Neurodegenerative Diseases Brain Bank | |
| Amyloid-PET positive subjective memory complaint seniors (SMCpos) | INveStIGation of AlzHeimer’s PredicTors (INSIGHT) – preAD study | [61, 172, 173, 242] |
| Presymptomatic or incipient AD | Dementia of Alzheimer type | [63] |
| Preclinical AD | – | [64] |
| Pathologically preclinical AD | – | [79] |
| SuperAgers | Australian Imaging, Biomarkers and Lifestyle (AIBL) study of Aging / | [170, 176, 177] |
| 80+-year-olds, protected from Aβ neurodegeneration | Alzheimer’s Disease Neuroimaging Initiative (ADNI) | |
| Cognitive reserve | – | [174, 175] |
To disentangle NDAN from normal aging and AD, different mechanisms have been studied as summarized in Figs. 1 and 2. While some studies suggest that this asymptomatic form of AD is caused by resilience or resistance for Aβ toxicity, others focus on alternative potential mechanisms, such as the status of the risk gene for the apolipoprotein E (APOE), the immune system or neurogenesis.
Fig. 1
Mechanisms in NDAN and AD brain. Top left: Increase of (re)active microglia and astrocytes accompanied with elevated levels of pro-inflammatory cytokines (IL-1, IL-6, TNF-α) in AD. In NDAN brain glial activation is decreased and anti-inflammatory cytokine (IL-4, IL-10) levels are increased. Top right: In AD, proliferation of neural stem cells (NSCs) is decreased and ‘von Economo neurons’ (VENs) are degenerated, while in NDAN NSC proliferation is increased as well as VEN density. Bottom left: Volume of the hippocampus and corpus collosum is increased in NDAN. Bottom right: In AD, Aβ oligomers are able to bind the postsynaptic membrane and induce synaptotoxicity. In NDAN, binding of Aβ oligomers is prevented.
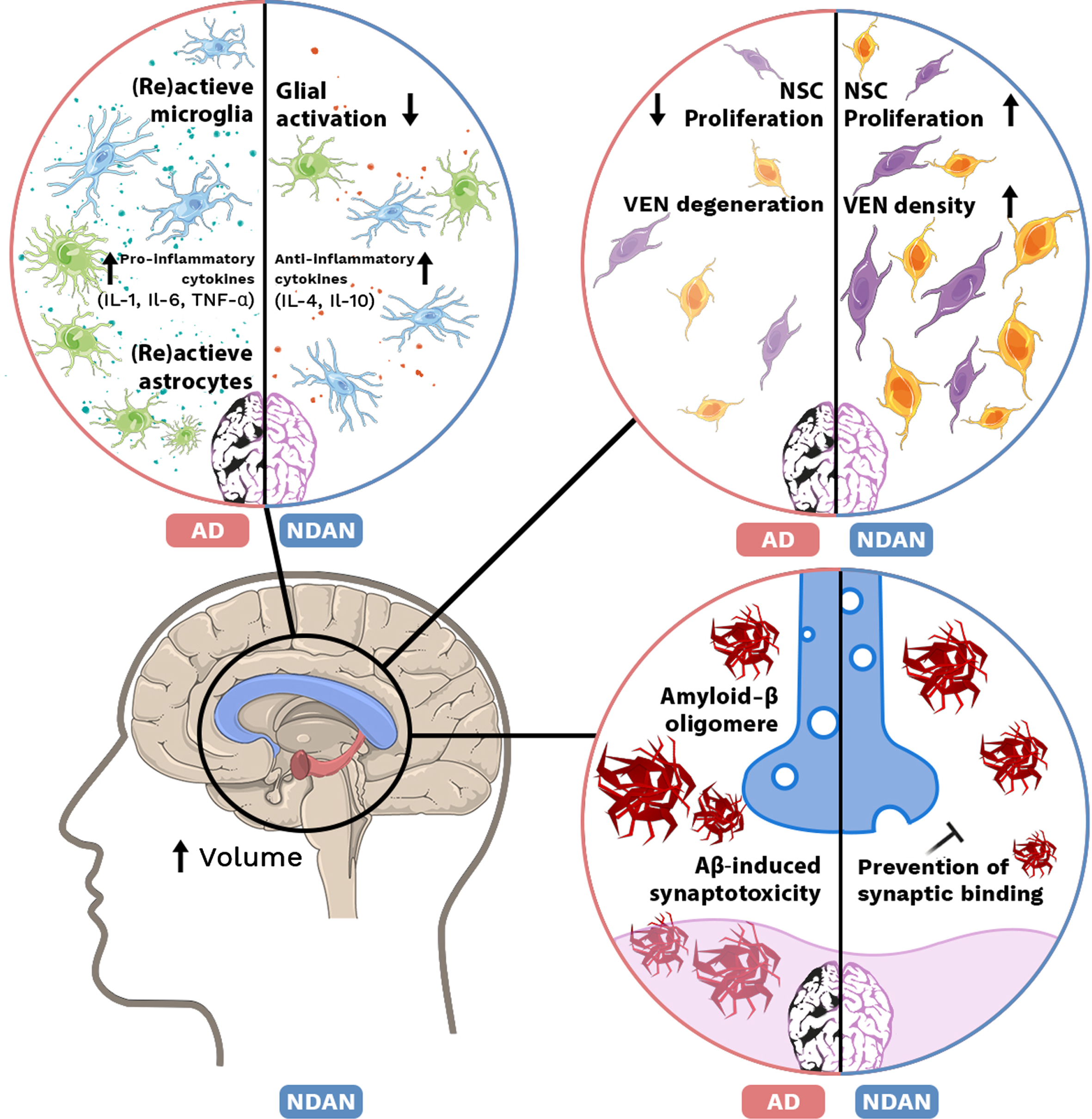
Fig. 2
Mechanisms in “normal” aging, AD and NDAN. Organ level: Shrinkage of grey matter in frontal and parietal volume over time in normal aging. Overall grey matter volume shrinks in AD but increases in NDAN brain. In NDAN the cingulate gyrus is thicker, and the hippocampus is spared, or increased in some cases. Neurons: Neurogenesis is decreased, and brain plasticity is increased in normal aging. In AD, NSCs proliferate less in the subventricular zone and hippocampus and VEN cells degenerate. NSC proliferation in the hippocampus of NDAN brain is increased, as well as VEN density. Inflammation: Microglia and astrocyte response to pro-inflammatory cytokines is increased in normal aging hippocampus. Microglia and astrocytes become more (re)active in AD, due to increased levels of pro-inflammatory cytokines (IL-1, IL-6, and TNF-α). Glial activation is decreased in superior temporal sulcus of NDAN, as anti-inflammatory cytokines (IL-4 and IL-10) are increased. Aβ: The molecular composition of Aβ molecules in normal aging is different than in AD and not distributed to specific areas. In AD, Aβ oligomers bind to synapses and induce synaptotoxicity. In NDAN, postsynaptic binding of Aβ oligomers is prevented by neuromodulators (Zn2+ [230] and Reelin [232]), Insulin [235] and specific APP miRNAs [182]. Tau: NFT distribution in normal aging is restricted to specific areas: entorhinal cortex, olfactory nucleus, and parahippocampal gyrus [49]. NFT levels in these areas are elevated in AD and also spread to the neocortex. In NDAN, NFT formation is inhibited in the temporal cortex, due to lower 0N3R expression [236]. A proposed mechanism for NDAN is to investigate the composition and location of different tau species.
![Mechanisms in “normal” aging, AD and NDAN. Organ level: Shrinkage of grey matter in frontal and parietal volume over time in normal aging. Overall grey matter volume shrinks in AD but increases in NDAN brain. In NDAN the cingulate gyrus is thicker, and the hippocampus is spared, or increased in some cases. Neurons: Neurogenesis is decreased, and brain plasticity is increased in normal aging. In AD, NSCs proliferate less in the subventricular zone and hippocampus and VEN cells degenerate. NSC proliferation in the hippocampus of NDAN brain is increased, as well as VEN density. Inflammation: Microglia and astrocyte response to pro-inflammatory cytokines is increased in normal aging hippocampus. Microglia and astrocytes become more (re)active in AD, due to increased levels of pro-inflammatory cytokines (IL-1, IL-6, and TNF-α). Glial activation is decreased in superior temporal sulcus of NDAN, as anti-inflammatory cytokines (IL-4 and IL-10) are increased. Aβ: The molecular composition of Aβ molecules in normal aging is different than in AD and not distributed to specific areas. In AD, Aβ oligomers bind to synapses and induce synaptotoxicity. In NDAN, postsynaptic binding of Aβ oligomers is prevented by neuromodulators (Zn2+ [230] and Reelin [232]), Insulin [235] and specific APP miRNAs [182]. Tau: NFT distribution in normal aging is restricted to specific areas: entorhinal cortex, olfactory nucleus, and parahippocampal gyrus [49]. NFT levels in these areas are elevated in AD and also spread to the neocortex. In NDAN, NFT formation is inhibited in the temporal cortex, due to lower 0N3R expression [236]. A proposed mechanism for NDAN is to investigate the composition and location of different tau species.](https://content.iospress.com:443/media/jad/2022/87-1/jad-87-1-jad210607/jad-87-jad210607-g002.jpg)
This review provides an overview on literature that focusses on NDAN, referring to all asymptomatic individuals with Aβ- and tau-pathology (Table 1), thereby aiming to provide more insight into potential mechanisms underlying AD pathology in absence of neurodegeneration and cognitive decline. Mechanisms underlying NDAN will also be compared to those underlying “normal” aging and AD in Fig. 2.
PHYSIOLOGY OF THE AGING BRAIN
Normal aging
An important characteristic of AD is cognitive decline, i.e., reduction of memory, attention, processing speed, visuospatial abilities, and executive functions. However, some degree of cognitive decline has also been associated with “normal” aging, as older individuals are often less efficient in cognitive tasks than younger people [23], but, which in general, does not convert into dementia [24]. To gain a better understanding in the process of normal aging, researchers from the Gerontology Branch at the National Institution of Health (NIH) launched the BLSA-study [22]. By following over 3,200 volunteers for more than 60 years, this study shed light on the relationship between aging and cognitive health by looking at occurring changes in brain tissue and cognition over time. Both healthy and demented older volunteers appeared to lose a significant amount of both grey and white matter [25]. In the case of grey matter atrophy, especially the frontal and parietal volume declined over time. And both the ventricles in the brain as well as the volume of ventricular CSF increased over time [25]. These findings suggest that both atrophy and increased ventricles reflect normal aging. However, these physiological changes are brain region dependent, e.g., the mesial temporal and frontal areas are more susceptive to AD pathology, rather than to normal aging [25].
The brain atrophy as seen in the aging brain could either result from neurodegeneration or from alternations in neurogenesis. Neurogenesis (or gliogenesis) is responsible for the production of new brain cells, such as neurons and the glial cells like astrocytes, oligodendrocytes, and ependymal cells [26]. In contrast to neurons which conduct action potentials, glial cells do not, but instead maintain homeostasis, and protect and support neurons. An important type of glial cell involved in the pathology of AD are microglia [27–30]. Unlike other glial cells, microglia do not derive from neurogenesis, but from hematopoietic stem cells [31, 32]. Microglia play an important role in neuroinflammation, functioning as resident macrophages of the central nervous system. In order to understand normal physiological changes during aging, Mayne et al. (2020) discussed functional decline of the adaptive immune system in healthy aging in comparison to several neurodegenerative diseases [33]. They illustrate that the response of microglia and astrocytes to pro-inflammatory cytokines was increased in brains of aging mice indicating higher sensitivity to inflammatory events [33, 34]. Furthermore, the increased secretion of interferon-γ by cytotoxic CD8+ T cells led to decreased neurogenesis in the hippocampus [33, 35]. Overall, their findings clearly indicated a role of the immune system in the reduction in neurogenesis in the aging brain. In addition to T-cells, natural killer cells have been implicated in impaired neurogenesis [35, 36], as well as oxidative stress and impaired DNA repair [37]. Interestingly, while neurogenesis can be reduced due to aging, the brain’s plasticity was found to be enhanced, yet dysregulated in aged rats [38].
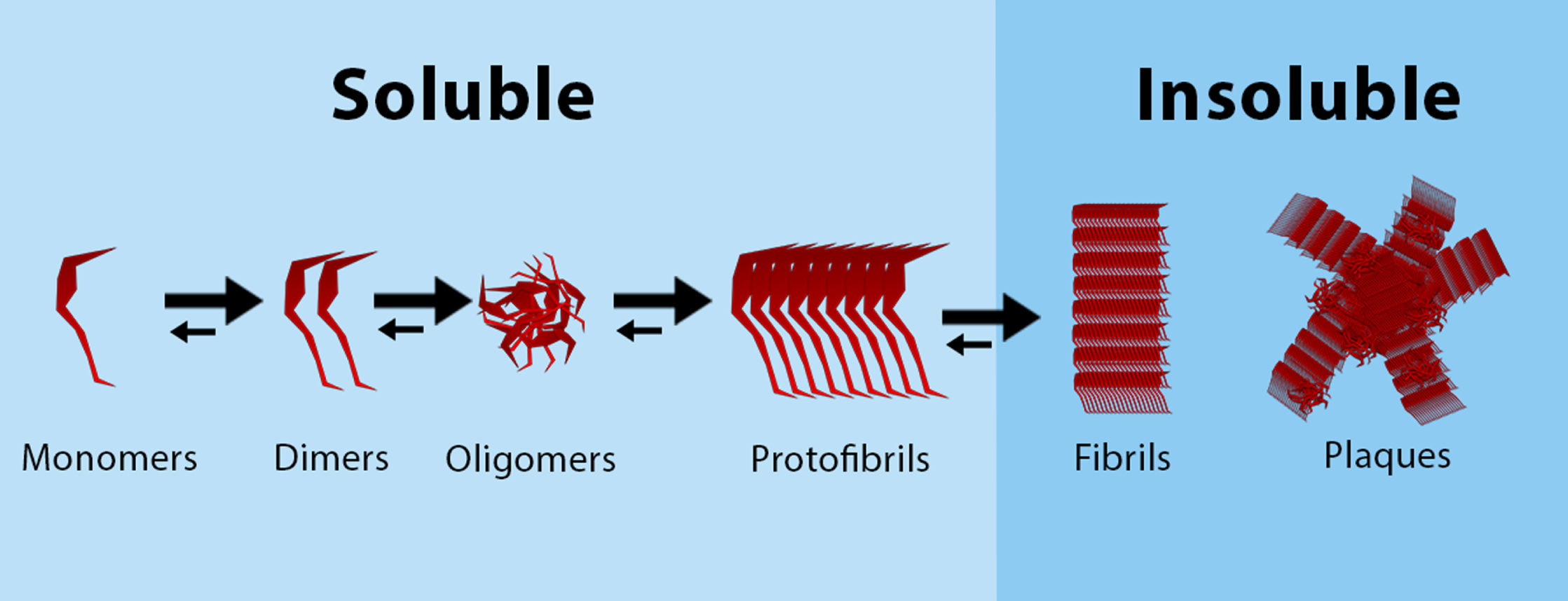
Another feature of “normal” aging is the presence of NFT and Aβ, as found in brains of cognitively normal elderly [39, 40–44]. In “normal” cellular metabolism, the enzymes β- and γ-secretase are responsible for cleavage of the amyloid-β protein precursor (AβPP), yielding Aβ peptides [45]. Aβ peptides can aggregate to form soluble oligomers and insoluble fibrils and plaques (Fig. 3). Perhaps counter intuitively, not the insoluble but the soluble oligomeric Aβ cause more neural damage as it binds easier to synapses, as has been proven extensively [46]. Normal Aβ production and degradation is balanced out by proteolytic degradation, deposition into insoluble plaques and cell-mediated clearance [45]. Tau is “normally” present in the aging brain as a highly soluble protein that primarily maintains microtubules in axons [47]. When tau proteins become hyperphosphorylated, it aggregates and NFTs are formed. As deposition of NFT and Aβ are both associated with AD patients and “normal” aging, an important question is if these NDAN individuals are resilient to tau- and Aβ-pathology. In experiments with virally induced human tau in entorhinal cortex neurons of young and old mice, it was shown that tau spreading was not only age- but also region dependent [48]. More specifically, the human tau expanded from the entorhinal cortex, to the hippocampus and the temporal lobe in old mice. In line with these findings in mice, it was found that NFTs can be distributed to the entorhinal cortex, olfactory nucleus and the parahippocampal gyrus in non-demented aging humans [49]. In mildly demented cases, distribution was similar to non-demented cases, but much more tangles were found in these areas. Only in the severely demented cases, NFTs were found also in the neocortex [49]. Conversely to NFT, consistency in the distribution of Aβ in “normal” aging has not been found yet. In 2005, Piccini et al. did hypothesize that the molecular composition of soluble Aβ aggregates could differ between “normal” aging and AD [50].
Fig. 3
Aggregation of Aβ, from monomers to plaques. Soluble Aβ monomers aggregate together to form dimers and oligomers, that can form soluble protofibrils. These protofibrils aggregate to form insoluble fibrils, that can form amyloid plaques.
MECHANISMS UNDERLYING ALZHEIMER’S DISEASE
AD can be heritable, attributed to mutations in the potential genes that encode for amyloid precursor protein (APP) and presenilins (PSEN) 1 and 2 [51]. Mutations in one of these genes result in increased production of the protein Aβ42. In contrast to the heritable familiar AD cases, in the sporadic AD cases, risk factors consist more of environmental and as well as genetic factors. One well known genetic risk factor is the APOE allele ɛ4, which increases the risk of AD but is not determinative for the disease [52, 53]. Under healthy conditions, ApoE enhances binding to, transportation of, and metabolism of lipids. However, in AD pathology, ApoE binds additionally to Aβ peptides and also regulates Aβ aggregation and clearance [54]. Moreover, the ApoE lipoproteins regulate neuronal signaling, glucose metabolism, and neuroinflammation, all mechanisms implicated with AD, as will be discussed further in this review. Another important causative factor in the pathology of AD is the protein tau. Hyperphosphorylation of tau protein results in the formation of NFTs [55]. Together with Aβ pathology, NFTs are defined as the typical hallmarks of AD.
AD on a body level
Looking at AD on a body level, studies have been focusing on multiple risk factors that are able to modify the onset and the rate of decline seen in AD. Investigated risk factors include age, gender, education, neuropathology, personality, genetics, lifestyle, and cognitive functioning [56–60].
AD symptoms in patients mostly consist of cognitive and functional impairment—dementia. The progressive course of AD is divided in several stages [23]. The first stage, preclinical AD stage, was initially defined as cognitively unimpaired individuals, who contained AD lesions in their brain as identified postmortem [61]. During this stage, clinical symptoms are not yet noticeable [62]. Since individuals in the preclinical stages and NDAN individuals both display AD neuropathology in the brain, but symptoms are not (yet) evident, this preclinical stage provides an interesting perspective for NDAN research [63, 64]. The first clinical stage of cognitive decline starts with subjective cognitive decline (SCD), defined as a very early and subtle cognitive decline which precedes objective cognitive decline [65]. The second stage is called the mild cognitive impairment (MCI) stage [66]. This MCI stage lays between expected cognitive decline associated with normal aging and more severe decline associated with dementia. This MCI stage is marked by symptoms of short term memory loss, especially in retrieving learned facts, and inability to acquire new information [67]. Moreover, deficits occur in planning, executive functions, attentiveness, and impairments in semantic memory (refer to general knowledge). All these MCI symptoms are hard to distinguish from normal aging and can improve over time, remain stable for years, or progress to AD or another type of dementia. During the SCD and MCI stages, diagnoses of patients is difficult, since these first symptoms of AD are hard to distinguish from normal aging. In later stages, the final stage of AD, memory problems become overshadowed by new difficulties: aphasia (difficulty communicating), agnosia (difficulty processing sensory information), and apraxia (inability in executing motor tasks) [68, 69]. During this final AD stage of the disease, it becomes more difficult for patients to execute tasks and therefore, most AD patients eventually need assistance.
To provide good care and disease intervention, a correct diagnosis of the disease is necessary. The current standard to diagnose AD is based on the definition of the Diagnostic and Statistical Manual (DSM)-IIIR for dementia and AD criteria and the diagnostic guidelines from the National Institute on Aging-Alzheimer’s Association (NIA-AA) [70]. This diagnostic method is based on medical history and cognitive testing after ruling out other possible causes. Over the years, these criteria have been specified further to improved accuracy, using imaging and blood tests [62, 66, 71]. Nevertheless, postmortem confirmation of the AD hallmarks is still required for a definitive diagnosis of AD.
With respect to postmortem confirmation, several methods can be used to verify and classify AD. Traditionally, NFT pathology has been assessed using Braak staging system [72, 73] (Fig. 4). The Braak staging system is used to classify the morphological changes that occur over time in AD patients. For long, this system has been used to predict AD, as the latter Braak stages typically indicate a clinical outcome of dementia, even though the system was not developed for clinical-pathological correlation. Nelson et al. (2012) reviewed the validity of the clinical-pathological correlation in AD thereby focusing on neuropathological (Aβ and tau) changes and cognitive impairment using Mini-Mental State Examination (MMSE) scores [74]. The authors state that the cognitive impairment in AD correlates better with neocortical NFT pathology than with Aβ plaque. However, NFT pathology also occurs in normal aging, as described in the previous section. For this reason, it could be questioned if Braak staging provides a valid prediction for AD, and also for NDAN [75, 76]. Especially during the slow-developing pathology of AD, morphological changes in the brain do not always directly translate into behavioral differences. However, at this point the question remains, is this a defect in the pathological evaluation itself, and can Braak staging be used to evaluate AD and NDAN, or is there something else missing? Another neuropathological diagnostic criteria instrument is the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) which evaluates the density of neocortical senile plaques [77–79].
Fig. 4
The Braak staging system adapted from Šimić et al. (2017) [84]. Topographic atrophic progression of AD caused by neurofibrillary degeneration. Initial memory loss correlates with hippocampal atrophy (stages I-II). Followed by disturbance in language & planning function, correlating with atrophy in the temporal, frontal, and parietal cortex (stages III-IV). And finally, impaired perception and movement, as associated with primary sensory and motor cortex (stage V-VI).
![The Braak staging system adapted from Šimić et al. (2017) [84]. Topographic atrophic progression of AD caused by neurofibrillary degeneration. Initial memory loss correlates with hippocampal atrophy (stages I-II). Followed by disturbance in language & planning function, correlating with atrophy in the temporal, frontal, and parietal cortex (stages III-IV). And finally, impaired perception and movement, as associated with primary sensory and motor cortex (stage V-VI).](https://content.iospress.com:443/media/jad/2022/87-1/jad-87-1-jad210607/jad-87-jad210607-g004.jpg)
Altogether, the slow and gradual decline observed in AD ensures the challenges in treatment of this disease. The AD-associated processes will be discussed next on the organ levels, followed by the cellular and biochemical level to gain deeper understanding of AD, and for the ultimate comparison of AD patients and NDAN individuals.
AD on an organ level
AD is characterized by neurodegeneration in specific areas of the brain, causing the cognitive decline as described in the previous section. Typically, histological changes first appear in the limbic system, especially the hippocampus and entorhinal cortex [80]. In particular, atrophy in the CA1 region of the hippocampus is indicative of AD, not of aging [81, 82]. This suggests that memory loss in AD is enhanced by CA1 atrophy, resulting in a greater memory decline than observed in normal aging. After CA1 atrophy, the atrophy affects the temporal and frontal cortex, and parts of the cingulate gyrus and parietal lobe (reviewed in [83, 84]). As a result of this neuronal loss, the ventricular system extends. Meanwhile, the spinal cord and cerebellum seem to stay unaffected [80].
As described in the previous section, the postmortem classification of AD is performed by the Braak staging system (Fig. 4), based on the characteristic distribution pattern of neurofibrillary changes throughout the brain [73]. Initially, the trans entorhinal to the hippocampal region become affected (Stage I & II), thereafter the temporal, frontal, and parietal neocortex (Stage III & IV), and eventually followed by alterations in the primary sensory and motor areas (Stage V & VI). The affected regions correlate with loss of recent memories (stages I-II), impairment of language and planning areas (stages III-IV), and finally malfunctioning of object recognition and motor skills (stages V-VI).
Several AD subtypes have been defined based on the atrophy pattern [85–89]. Ferreira et al. (2017) used magnetic resonance imaging (MRI) to predict and investigate three subtypes of AD: typical AD, limbic-predominant, and hippocampal-sparing forms of AD [89]. In typical AD brains, neurodegeneration appeared in the mesial temporal lobe and in the posterior cortex and/or frontal cortex. In contrast to the typical AD, the limbic-predominant subtype was defined as atrophy in the mesial temporal lobe only, whereas in the hippocampal-sparing AD subtype, this mesial temporal lobe was spared. The latter generally did not experience amnesia. Furthermore, another study identified a subtype of patients with neither hippocampal nor cortical atrophy [90]. This AD subtype did show cognitive impairment, despite their seemingly spared brain areas. This could be explained by possible limitations in the diagnostic accuracy guidelines from the NIA-AA or due to other factors that are not yet identified in the mechanism of AD.
Another aspect of AD is reduction of the cerebral blood flow (CBF). In AD, Aβ peptides indirectly can cause a reduction of CBF due to interference with nitric oxide-mediated vasodilation [91]. Reduction of CBF results in impaired supply of glucose, nutrients, and oxygen, and diminished elimination of toxic products. Therefore, reduction of CBF contributes to neuronal damage. In parallel, Aβ induces reactive oxygen species that activate the release of endothelin-1, which in their turn elicits constriction of pericytes which results in capillary constriction and reduced CBF [92]. Particularly, regional CBF (rCBF) reduction is one of the earlier markers for dysfunction in specific areas associated with AD [93]. This rCBF reduction follows the same trend as the observed areas affected in AD pathology: the posterior cingulate, praecuneus, and mesial temporal lobes. However, rCBF is further reduced in AD patients, namely in the hippocampus, caudate, and thalamus [94]. For this reason, measuring the CBF in these regions, using MRI or positron emission tomography (PET), is a useful method to predict cognitive decline [95, 96].
AD on a cellular level
During AD, neurons and synapses lose their structure and function, and eventually die, resulting in atrophy as described above. Altogether this is referred to as neurodegeneration, hence AD is a neurodegenerative disease. Neurodegeneration as observed in AD results from neurotoxic tau and Aβ, together with aging and inflammation [97].
To get a better idea of neurodegeneration, it is important to not only investigate neuronal death but also neuronal production. Neurons themselves do not proliferate in adult life; however, new neurons can be derived from neuronal stem cells (NSC) by the process of neurogenesis [26]. Adult neurogenesis has been observed in two regions of the brain, namely the mature subventricular zone of the lateral ventricles and the dentate gyrus part of the hippocampus [98, 99]. As indicated previously, mild memory loss is associated with “normal” aging, which could be the result of region-specific neuronal alterations caused by brain plasticity [100] (Fig. 2). Alternatively, neurogenesis in the hippocampus declines with age and therefore causes potential complications in memory function [101]. In AD, neurogenesis is impaired. Studies in AD mice revealed that NSCs in the adult subventricular zone proliferate less and fail to differentiate into healthy mature neurons [102]. Scopa et al. (2020) concluded that neurogenesis in this zone was already impaired in the early stages of AD, due to Aβ oligomers in the endoplasmic reticulum of NSCs. For many years, researchers speculated that AD pathology affected neurogenesis and that neurogenetic misfunction correlated with neuronal loss as seen in AD [103–105]. More recent attention has focused on the groundwork of adult hippocampal neurogenesis in over 90-year-old individuals with MCI and AD [106, 107]. Specifically, MCI patients showed reduced numbers of neuroblasts, premature neurons originating from NSCs [107, 108]. This data suggests that neurogenesis may already decline during MCI. In line with this finding, it is not surprising that adult neurogenesis in the human hippocampus was found to be impaired already in the early stages of AD pathology [109].
In addition to the two hallmarks of AD (Aβ plaques and NFT), neuroinflammation is considered to be the next most important contributor to the progression of AD. Over the last decade, the importance of this process has emerged more and more and in 2020, the link between neuroinflammation and AD has been elaborately discussed in reviews [110, 111]. Neuroinflammation in AD has been observed in human brain tissue and on a molecular imaging level [112–115]. More specifically, on the one hand, astrocytes become more reactive in response to Aβ, releasing pro-inflammatory cytokines [116, 117]. On the other hand, there is increased interest in the brains-resident immune cell, the microglia. As explained earlier, these particular glial cells are the resident immune cells of the brain, that can phagocytose pathogens and display antigens to activate T-lymphocytes [30]. In AD, microglia play a major role in endocytosis and degradation of soluble and fibrillar Aβ species [29]. Initially, this process is beneficial to prevent Aβ accumulation, but as soon as the Aβ levels are chronically elevated, microglia activation will result in prolonged inflammation and enhances AD progression. The impact on AD progression is particularly influenced by the phenotype of microglia (M1 or M2) involved, as type M1 microglia enhance cellular immune responses while type M2 inhibit classic inflammation and promote wound healing [118, 119]. The mixed population of microglia results in a heterogenic neuroinflammatory response that influences AD progression and effectiveness of possible treatments, notably in adults with early AD [112].
Besides (re)active glial cells, several cytokines have been associated with AD. The deposition of Aβ activates astrocytes and microglia that trigger the secretion of several inflammatory cytokines. Firstly, interleukin (IL)-1 can increase AβPP synthesis and secretion, and subsequently generation of Aβ [120]. Furthermore, microglial production of IL-1 activates the p38 mitogen-activated protein kinase (p38-MAPK) pathway and thereby increases the phosphorylation of tau [121]. Secondly, elevated levels of IL-6 have been found in AD brains, which can increase the expression of AβPP as well [122]. Thirdly, IL-10 inhibits the macrophagic function of microglia and thereby indirectly decreases cerebral Aβ clearance [123]. Lastly, tumor necrosis factor (TNF)-α stimulates the activity of γ-secretase, resulting in an increased production of Aβ from AβPP [120]. Increased levels of this pro-inflammatory cytokine were also found in CSF and blood of MCI patients, who later progressed to AD [124]. Moreover, results from a cell culture study indicated that microglia, when activated by Aβ, release reactive oxygen species and also TNF-α, creating a positive feedback loop of enhanced Aβ and microglial induced inflammation [125]. Together, these cytokines affect the proliferation and survival of both neurons and NSCs and enhance the progression of AD. To conclude, these studies clearly indicate that there is a strong relationship between neuroinflammation and the progression of AD, which needs to be considered in the pathology of the disease.
In literature, many different theories of AD etiology have been proposed, including the vascular hypothesis that states a reduction in CBF [126]. The reduction in CBF may result from blood-brain barrier (BBB) dysfunction as indicated by recent studies [127–132], suggesting a potential causal relationship between AD and cardiovascular dysfunction. Under healthy conditions, the BBB protects the brain by preventing access for blood cells, pathogens, and neurotoxic components in the blood [133]. During the early stages of AD pathophysiology, breakdown and dysfunction of the BBB occurs. This BBB breakdown results in leakage throughout the cerebrum, due to dilatation of the BBB’s tight junctions [134]. The integrity of these tight junctions is, among others, regulated by APOE, which is, as mentioned above, an extensively investigated risk gene in AD research [135]. Additionally, another study showed that increased BBB permeability resulted from Aβ-induced pro-inflammatory and cytotoxic events [136]. Together, this indicates that AD is indirectly associated with increased BBB permeability. Furthermore, Montagne et al. (2015) observed a breakdown of the BBB in hippocampal areas, CA1, and dentate gyrus in healthy elderly [127]. These findings suggest that the breakdown of BBB contributes to the cognitive impaired symptoms that are associated with aging and AD.
In short, multiple processes in the brain are affected by AD pathology (Fig. 2). In AD, neurons degenerate, while the formation of new neurons is impaired at the same time. Moreover, astrocytes and microglia become reactive in the presence of Aβ, triggering the release of several pro-inflammatory cytokines. This cytokine release could be associated with the decline observed in BBB tight junction integrity, causing more neurotoxic components to enter the brain and inducing an immune response. Furthermore, CBF is reduced in AD brains, suggesting a decrease in blood supply and decreased synaptic activity. All these mechanisms can be caused by Aβ and NFT pathology of AD, which will be discussed next.
AD on a biochemical level
Amyloid-β pathology
The Aβ molecules exist of peptides derived from AβPP; an integral membrane protein expressed in synapses. It is thought that AβPP functions as a regulator of neural plasticity and synapse formation [137, 138]. Mouse studies suggested that AβPP plays an important factor in neurogenesis, as APP–/– mice showed no increase in neuronal proliferation and reduction in brain weight [139]. Nevertheless, AβPP itself is not responsible for the pathological development occurring in AD. When AβPP is cleaved by β- and γ-secretase, it generates Aβ peptides from different sizes [140–142]. These Aβ isoforms vary from 30 to 51 amino acid residues. The two most prevalent isoforms are Aβ40 and Aβ42, which are produced in both neuronal and nonneuronal cells. The ratio of Aβ42/40 is an important factor to determine fibrillar formation and consequent toxicity of Aβ [143]. Aβ40 is produced in the trans-Golgi network, while Aβ42 is produced in the endoplasmic reticulum (ER) [144]. Especially, the ER-derived isoform, Aβ42, is notorious as it is best at misfolding and forming the toxic Aβ oligomers [145] and consequently, with a higher ratio of Aβ42/40 more toxic oligomers are formed. Neuronal cell culture studies demonstrated that Aβ oligomer exposure induced cell death, which was mediated by acute toxicity triggered by oxidative stress [146, 147].
Not only insoluble Aβ oligomers (which lead to plaque formation), but also soluble Aβ oligomers (Fig. 3) play an important role in the pathology of AD [148] and the presence of high Aβ oligomer levels in CSF of AD patients may indicate an increased risk of AD [149]. In another study, significantly higher levels of Aβ oligomers were found in CSF samples from AD patients compared to controls, as well as an elevated ratio of Aβ oligomers to Aβ42 [150], which, in AβPP-expressing cultured cell lines, were associated with loss of dendritic spines and loss of synaptic function [46]. This observation is crucial because Aβ oligomer-induced synaptic loss results in disruption of neuronal communication.
Shankar et al. (2008) also found that these Aβ oligomers were bound to the synapses and inhibited long-term potentiation, while enhancing long-term depression (LTD). Enhancing LTD in the hippocampus, together with the reduced dendritic spine density, eventually results in interruption of memory and learning ability. Furthermore, Shankar et al. (2008) illustrated the synaptotoxic role of Aβ dimers in this process, as Aβ dimers solubilize amyloid plaques and disrupt glutamatergic synaptic transmission that is necessary for the LTD. It is believed that the misfolded Aβ fulfills a prion function: misfolded proteins that can convey their misfolded shape onto other proteins [151–153]. Not only Aβ, but also tau proteins can be misshaped as a result of these misfolded Aβ [154]. The toxic behavior of misfolding also perturbs when misfolded Aβ and tau enter neurons which contained properly folded and functioning Aβ and tau.
To detect Aβ plaques, a radioactive analogue of thioflavin T, Pittsburgh compound B (PiB) is used. This compound, 2-(4’-[11C]methylaminophenyl)-6-hydroxybenzothiazole (11C-PiB), can bind to Aβ fibrils and is visible through PET imaging [155]. Binding of 11C-PiB to Aβ enables us to diagnose AD pathology and investigate the areas that are affected by Aβ. Yet, Aβ pathology was not significantly associated with cognitive AD symptoms and had, therefore, no predictive advantage, while NFT does [156] (see below). This conclusion contradicts that of Pike et al. (2007) who found a strong correlation between impaired episodic memory and increased binding of PiB [157]. But, as impaired episodic memory is not the only symptom indicative for AD, this approach can be used to fulfil some of the antemortem detection of Aβ, but cannot assess the degree of cognitive impairment.
Tau pathology
The other hallmark of AD are tau proteins, which aggregates into NFTs. Under healthy conditions, tau proteins are highly soluble proteins with different isoforms produced by alternative splicing from the microtubule-associated protein tau (MAPT) gene [158] (Supplementary Figure 1). Primarily, the role of tau in the central nervous system is to stabilize the microtubules in axons; however, these proteins can also be expressed in non-neuronal cells, including astrocytes and oligodendrocytes [159]. Tau proteins can become phosphorylated by different protein kinases on various sites that regulate microtubule binding [160]. In total approximately 45 phosphorylation sites have been reported, either being tyrosine, threonine, or serine residues (Supplementary Figure 2). Phosphorylation of these different residues is regulated by different kinases including GSK-3, PKN, and CDK5 [161, 162]. Phosphorylation interferes with microtubule binding; consequently, the hyperphosphorylated state of tau decreases the solubility of the protein, resulting in its aggregation into bundles of paired helical filaments, the NFTs [163]. During AD, this hyperphosphorylated, insoluble form can no longer bind and stabilize the microtubules, and therefore indirectly demolishes the microtubules, thereby weakening the cytoskeleton and obstructing axonal transport [161]. Inhibition of axonal transport can lead to oxidative stress in mitochondria and eventually cell death [164]. Moreover, tau’s indirect inhibition of axonal transport leads to the accumulation of AβPP, the precursor of the other AD hallmark (see the section on Amyloid-β pathology above). The quantity and distribution pattern of tau correlate with the degree of decreasing cognitive performance and memory in AD. Tau staining is performed using silver and immunohistochemical staining for hyperphosphorylated tau [72, 73]. As discussed previously (see the sections AD on a body & organ level above), the Braak staging system is based on the deposition of tau, the formation of NFT and neuropil threads in neurites [72, 73]. Apart from this system to identify AD stages, Braak et al. pointed out the importance of hyperphosphorylation of tau, as accumulation of tau already starts years before the first signs of AD occur. This highlights the importance of tau as a valuable hallmark for AD. However, NFT distribution was also seen in “normal” aging (Fig. 2); therefore, it is important to also investigate the distribution and composition of tau species in NDAN.
NON-DEMENTED INDIVIDUALS WITH AD NEUROPATHOLOGY
Until the past decade, researchers in the field of AD have been focusing on the link between the clinical symptoms of dementia and the two hallmarks: Aβ and tau. However, several longitudinal, cohort studies have been suggesting that it is not as straightforward as was anticipated. One review discussed demented patients, without amyloid or tau pathology, as well as cognitively intact individuals with severe AD pathology [165]. Nelson et al. (2009) illustrated that individuals without or with mild dementia tend to have less NFT counts in the neocortex, while when NFT counts are over a certain threshold, cognitive impairment is inescapable. Moreover, in cases with many NFT counts, the combined amount neuritic and diffuse amyloid plaques was increased. Therefore, Nelson et al. (2009) strongly agreed with the clinical-pathological correlation of NFT and neuritic and diffuse amyloid plaques to cognitive impairment. In 1958, the BLSA started investigating the normal progression of aging [22]. In this program, the presence of AD neuropathological changes was found in several of the autopsies of individuals that displayed normal, non-demented behavior prior to their death. Another longitudinal aging study, the Nun Study (1986), found that 12% of the cognitively intact participants showed abundant Aβ plaques and NFT during postmortem examination [166–168]. After the successful findings of the Nun Study, the Rush Religious Orders Study (1992) was executed [169]. The Rush Religious Orders researchers, Bennett and Schneider, found that one-third of older people’s brains had serious AD lesions, while these people were not cognitively impaired. Not only in America, but also in Australia an aging and AD study was launched in 2004: the Australian Imaging, Biomarker & Lifestyle Flagship (AIBL) Study [14, 170], and found a significant Aβ deposition in the brains of some non-demented individuals [171]. More recently, a group in Paris investigates the compensation mechanisms in individuals with AD lesions that maintain their cognitive capabilities. To this aim, the “INveStIGation of AlzHeimer’s PredicTors” (INSIGHT-preAD) study was launched in 2013 and is still ongoing [172]. Within the INSIGHT-preAD cohort, positive amyloid deposition was found in asymptomatic individuals [173]. Taken together, these studies indicate the existence of a group of elderly that do not develop dementia during their life, despite the AD neuropathological burden in their brains. Therefore, it is thought that other factors, or maybe even an undetected hallmark in the pathology of AD, play an important role in altering the risk for AD or the progression of AD. So, what distinguishes NDAN individuals from normal AD patients and “normal” aging, and what key features are still missing?
As was pointed out in the introduction of this review, already many different studies have been performed regarding these NDAN cases (Table 1). Although a variety of synonyms have been used, some studies focus on a specific age interval and others investigated the preclinical state of the disease. Moreover, the conditions by which individuals are labeled as NDAN differ between studies. Nevertheless, all these studies have been investigating the potential missing link between the two hallmarks of AD and the cognitive outcome that is expected in this disease. Already at the end of the last century, researchers tried to distinguish normal aging from the preclinical state of AD, in which the quantity of neuropathological changes has not yet affected cognitive behavior. Morris et al. (1996) discussed whether the cerebral deposition of Aβ in nondemented elderly indicated early AD in its presymptomatic phase or simply “normal” aging [63]. Moreover, Schmitt et al. (2000) referred to preclinical AD subjects as having AD-like changes, but exhibiting no cognitive differences [64]. Moreover, they suggested that the aging brain might cope with these changes and remain normal cognitive functioning. Both Morris et al. and Schmitt et al. refer to this phenomenon as the delay in time between the pathological progression and the expression of disease and symptoms. Later researchers started to use the term cognitive reserve (or buffer), in expectance that symptoms will eventually occur [174, 175]. Another synonym that is being used to describe NDAN specifically focusses on non-demented individuals within a certain age range. So did Maarouf et al. (2011) who described a group cognitively intact nonagenarians with high amyloid plaque loads and classified these as non-demented high pathology controls [11]. Similarly, the term ‘SuperAgers’ was used to refer to 80-year-old elderly, with a level of episodic memory that is equal or better than those between 50- to 60-year-old [14, 15, 176, 177]. Alternatively, the group of Gómez-Isla described these non-demented individuals with significant amounts of AD changes as high or intermediate probability mismatches or resilient to the AD pathology [13, 178]. Additionally, scientists even use different terms to refer to a non-symptomatic behavior of AD, dementia, observed in individuals with significant AD neuropathology. The group of Troncoso investigated asymptomatic AD (ASYMAD) in BLSA and conducted their research on a histological, proteomic, and transcriptomic level [21, 179, 180]. Lastly, the synonym that is used in this review, ‘NDAN’, was already described by Zolochevska and Taglialatela [9, 181, 182]. Altogether, these scientists have all contributed to unravel the underlying mechanisms that differentiate NDAN from normal aging and AD. Some of these studies mainly focused on delaying the effects of aging, as aging is an inevitable process that can be seen as a risk factor for AD. At the same time, delaying the effects of aging itself can provide useful insight into the resilience of AD. In this section, these potential mechanisms will be discussed on a body, organ, cellular, and biochemical level aiming to gain a bigger understanding of NDAN.
POTENTIAL MECHANISMS UNDERLYING NDAN
As mentioned earlier, already multiple risk factors for AD have been examined by previous researchers. These factors—age, gender, education, neuropathology, personality, genetics, lifestyle, and cognitive function—can modify the onset and rate of decline of AD and therefore also need to be considered in NDAN research [56]. Morphological changes of the brain and the neurons, inflammation, Aβ, and tau have already been discussed in the context of normal aging and AD (Fig. 2) and will here be discussed in the context of NDAN. First, NDAN is discussed on an organism level from an age, education, and cognitive perspective.
NDAN on a body level
Typically in AD, patients encounter dementia-like symptoms, including memory impairment, difficulties with language, and moving difficulties [183, 184]. Diagnostic classification of AD is initially executed using several cognitive performance tests on, for instance, memory recalling, perceptual speed, and verbal fluency. As explained earlier, pathological diagnoses and cognitive testing is needed to confirm the AD diagnosis, using Braak staging system, CERAD, and NIA-AA neuropathologic criteria for AD. These neuropathologic criteria have also been used to examine cognitive functioning in non-demented elderly [185]. The study by Bennett et al. (2006) found that some of these non-demented individuals were allocated with an intermediate or high likelihood to develop AD, a crucial discovery, as it demonstrates the heterogeneity in the extent to which AD pathology correlates with cognitive impairment.
Several studies performed among different populations worldwide have demonstrated Aβ aggregates and NFT in 20–40% of the cognitively intact elderly [63, 71, 186–190]. Specifically, the study performed by Driscoll et al. (2006) assessed cognitive performances of cognitively normal elderly controls, individuals with MCI or AD, and NDAN individuals over a time span of 16 years or longer [190]. These individuals underwent prospective neurological evaluations on several cognitive domains, including memory, visuospatial abilities, attention, and word knowledge, and eventually underwent autopsy. Interestingly, the authors found that the cognitive trajectories between cognitively normal older individuals and NDAN, declines similarly, whereas the cognitive trajectories of individuals with AD or MCI showed an accelerated decline. Surprisingly, this accelerated decline could be observed already prior to the diagnosis of AD or MCI. In other studies, including the more recent study performed by Dang et al. (2019), the new term ‘SuperAgers’ was introduced which is defined as cognitively strong 80+-year-old NDAN that are resistant to neurodegeneration associated with age and Aβ [14]. In this study, SuperAgers were case-matched to cognitively normal individuals for their age, based on sex and education (control subjects) and their memory performance was investigated over a time span of 90 months. They found that SuperAgers had less Aβ-associated decline compared to control subjects. It can thus be suggested that these individuals are resilient to AD pathology.
Since the discovery of NDAN cases, an Alzheimer’s Association workgroup suggested alterations in the diagnostic guidelines of AD (part of the NIA-AA) which would allow for the separation of NDAN individuals from typical AD patients [71]. Consequently, many researchers started assessing the cognitive function of their subjects using the Clinical Dementia Rating (CDR) score and/or MMSE (Table 2) [191, 192]. Most of the cited studies summarized in Table 2 include only a small amount of individuals with an MMSE score > 27.5, except for the study on SuperAgers and amyloid-PET positive subjective memory complaint seniors (SMCpos) [16, 172], showing that inclusion criteria vary between studies and that the group of NDAN individuals is rare or at least difficult to recruit or study. Some scientists analyzed the clinical-pathological correlation by investigating the correlation between MMSE scores and Braak stages [193]. According to these authors, a “ultimate NDAN”, having a MMSE score of 30 and end-stage tau pathology combined in one individual, did not exist in their analyzed datasets of 3,310 cases. This outcome provides another prospective in the NDAN research. However, the research goal is not to find and then study the “ultimate NDAN”, but to gain insights in the missing link between the currently accepted AD pathology and the actual clinical AD outcome. To achieve this goal, the “ultimate NDAN” cannot and does not have to be the golden standard for NDAN. However, it remains debatable what would be the best cut-off point of the MMSE or CDR score to define individuals as cognitively intact, and should one take into account that an individual with a high score can still be cognitive impaired relative to their own baseline? And what is the threshold that states whether an individual has AD pathology?
Table 2
Selected studies on cognitive diagnostic examination of NDAN individuals
| NDAN or synonym | Number of participants | Age | CDR | MMSE | Time of last evaluation prior to death | References of performed studies |
| NDAN | N = 4 | > 89 | 26–29 | – | [9] | |
| NDAN | N = 8 | 76 – > 89 | 27–30 | Several months | [10] | |
| ND-HPC | N = 8 | 91–100 | 27.7 | – | [11] | |
| Asymptomatic | N = 82 | ≈ 81.3 | 0 | < 1 year | [12] | |
| SA | N = 5 | 87–95 | 29.23±0.88 | – | [16, 177] | |
| ASYMAD | N = 6 | > 75.9 | ≤0.5 | ≈ 11 months | [17] | |
| ASYMAD | N = 13 | ≈ 89 | 28.3±1.1 | ≈ 0.5 year | [18] | |
| ASYMAD | N = 15 | 71–96 | 28–30 | – | [21] | |
| p-preAD | N = 13 + 20 | 53–88 | 0 – 3 | 1–4 weeks | [79] | |
| SMCpos | N = 88 | 70–85 | 0 | ≥27 | – | [172] |
| SA | N = 172 | > 60 | > 24 | – | [176] | |
| High or intermediate likelihood | N = 48 | 28.2 | – | [185] | ||
| Nondemented, meets AD criteria | N = 14 | ≈ 87.62 | – | 23.93 | 1.18 year | [209] |
| NDAN | N = 10 | ≈ 89.5 | 28 | – | [230] |
MMSE, Mini-Mental State Exam; CDR, Clinical Dementia Rating; ND-HPC, non-demented high pathology controls; ASYMAD, Asymptomatic AD; NDAN, non-demented individuals with AD neuropathology; p-preAD, pathologically diagnosed preclinical AD; SA, SuperAgers; SMCpos, amyloid-PET positive subjective memory complaint seniors.
Looking at scoring the cognitive functions of individuals, the CDR is used to distinguish three stages of dementia from healthy individuals, based on their performance in memory, orientation, problem-solving, community affairs, home and hobbies, and personal care [191]. Each category is scored independently as healthy (0), questionable dementia (0.5), mild dementia (1), moderate dementia (2), and severe dementia (3). Unfortunately, not all studies focused on NDAN cases included CDR scoring, but in the studies that did include CDR scores, almost all patients had low scores (between 0 and 0.5) indicating healthy or very questionable dementia (Table 2). The study of Upadhaya et al. (2014) had two exceptions with high scores (2 and 3) that were observed in two ‘pathologically diagnosed preclinical AD’ subjects, specifically a subject with argyrophilic grain disease, another type of dementia and a subject with both corticobasal degeneration and vascular dementia [79].
As an alternative for the CDR scoring, there is the MMSE, a questionnaire to measure cognitive impairment [192]. This test is divided into categories to test the functioning of time and spatial orientation, language, memory, repetition, and attention, resulting in a MMSE score indicating either normal cognition (score 24–30), mild (score 19–23), moderate (score 10–18), or severe cognitive impairment (score≤9). Again, the cognitive function of all non-demented individuals in the NDAN studies was identified with a MMSE score of at least 24, but mostly above 27 (Table 2), indicating strong cognitive functioning. Nevertheless, it is important to keep in mind that even a maximum MMSE score of 30 does not rule out the possibility of developing dementia, and the MMSE score might change over time. Especially in NDAN, it remains uncertain whether the presence of AD pathology in these cognitively high functioning elderlies will still eventually cause dementia had they lived longer. Furthermore, a time delay between the latest cognitive evaluation performed antemortem and the post-mortem analysis of brain tissue is inevitable, for most studies this time delay was less than 1 years (Table 2).
Cognitive “reserve” and level and/or duration of education
One hypothesis that provides an explanation for the dissociation between AD neuropathology and dementia is “reserve”. In 2012, Stern explained that there were two types of reserve: brain reserve and cognitive reserve [175]. Brain reserve refers to physical differences in the brain that could advance tolerability against disease pathology (see the section NDAN on an organ level below). Additionally, cognitive reserve refers to the ‘resistance’ to neuropathological damage. Several studies suggested that cognitive reserve might be involved in NDAN’s resistance to cognitive decline [194–198]. And another study suggested that a higher cognitive reserve enables individuals to elongate the maintenance of their cognitive skills and therefore persist longer in an asymptomatic state [199]. It is hypothesized that individuals with AD pathology that initially performed cognitively well, will eventually develop MCI or AD, regardless of cognitive reserve.
Cognitive reserve refers to flexibility and efficiency in executing tasks and often clusters literacy, IQ, reading, virtue in social networks, lifestyle, and potentially most important, education. Already in 1990, Katzman et al. reported that lower education was indicative for a higher AD prevalence [200]. The Rotterdam Study confirmed these results, as they found as well that an increased risk of AD was associated with a lower educational level [201]. Additionally, a meta-analysis suggested that for every year increase in education, the prevalence of dementia was diminished by 7% [202]. Hence, it could conceivably be hypothesized that NDAN individuals attained a higher education level than AD individuals. In line with this hypothesis, Iacono et al. (2015) investigated the level of education of subjects from the Nun Study [18]. These authors found a significantly higher level of education (Masters or higher) in NDAN cases (61.5%), compared to both MCI (33.3%) and AD (29.3%) subjects. Similarly, in SuperAgers, cognitive reserve was suggested to be mainly developed over years of education [203]. Moreover, this link between level of education and dementia was thought to be caused by the putative unhealthy lifestyles of lower educated individuals. In an attempt to evaluate this hypothesis, Ngandu et al. (2007) demonstrated that unhealthy lifestyles may only independently contribute to reduction of cognitive reserve, not contribute to increased prevalence of AD itself [204]. With respect to the mechanisms underlying education and cognitive function in AD, researchers proposed that this was only associated with amyloid, not NFT [205]. To conclude, considering all this evidence it seems that NDAN individuals generally attended a higher educational level and/or were educated for a longer time, resulting in an increased cognitive reserve.
So higher educated individuals seem to have a greater reserve. However, when individuals with a higher IQ do decline, the terminal cognitive decline is much steeper compared to individuals with a lower IQ, as was concluded by investigating the rate and acceleration of cognitive decline in the final years of disease prior to death [206]. Additionally, another recently published study, which did not specifically look at IQ, but that investigated person-specific cognitive trajectories, also found that terminal decline contributes on average for about 70% to late-life cognitive loss in the investigated cohort [207]. Interestingly, accelerated cognitive decline was also observed in a recent study in individuals who lost their home during the 2011 earthquake in Japan [208]. Even though there was quite some heterogeneity between individuals, the individuals that seemed most vulnerable to post-disaster accelerated cognitive decline tended to be less educated, as well as older, unmarried, not working, living alone, and had also baseline health problems [208].
All these studies provide insights, from different perspective, into the process of resilience to cognitive impairment.
NDAN on an organ level
As was mentioned before, AD is characterized by neuronal atrophy in various regions of the brain (see the section on AD on an organ level). Various subtypes of AD have been described in this section, including cognitively impaired individuals without any hippocampal or cortical atrophy [90]. A possible explanation for these subtypes may be the lack of adequate knowledge in the pathology, one link that is repeatedly observed in NDAN. Are NDAN brains resilient to AD pathology or do they also encounter atrophy?
Hippocampal volume
The volume of the hippocampus is an independent criterion to distinguish between healthy and AD individuals. In one of the investigations from the Nun Study, they found that AD neuropathology was associated with a reduction of the hippocampal volume and that the brains of NDAN individuals did show hippocampal atrophy [209]. In contrast to Gosche et al. (2002), others reported that the volume of both the total brain and hippocampus were elevated in cognitively normal individuals with high AD neuropathology, compared to AD [210, 211]. Furthermore, in baseline measurements of SuperAgers (80+-year-old NDAN group), hippocampal volumes were found to be significantly larger compared to the normal aging controls [176]. These opposing findings suggest that brain and hippocampal volume by itself cannot explain the clinical-pathologic correlation in NDAN.
The genome-wide associated study performed by Guo et al. (2019) associated three small nucleotide polymorphisms in the TOMM40-APOC1 region, located on chromosome 19q13.32, with hippocampal atrophy and cognitive decline in non-demented older adults [212], indicating that genetic background affects hippocampal atrophy. Furthermore, another study indicated that hippocampal atrophy in AD is associated with vascular damage [213]. So far, however, it remains unknown whether the hippocampal and brain atrophy would be directly caused by NFT and Aβ plaques, since NDAN individuals do not necessarily show reduced hippocampal volume and/or brain atrophy despite their AD pathology.
Collectively, in NDAN, multiple studies indicate hippocampal volume changes while one study does not. As far as neurodegeneration is concerned, the most important factor remains age. One study did consider age in their analysis and reported that similar rates of atrophy were observed in both control and Aβ-resilient cases [14], suggesting that larger hippocampal volume could contribute to resilience to Aβ, but that structural decline eventually does occur over time.
Brain area dependency
Next to cognitive reserve (see the section on NDAN on a body level) resiliency to cognitive decline in NDAN could result from brain reserve [174, 175, 214]. Brain reserve refers to the ability to endure age-related changes and pathological damage, without developing symptoms. As mentioned above, the hippocampal volume is one of the mechanisms in brain reserve. Moreover, other structural changes or differences in the brain might be involved in the coping mechanism in NDAN individuals. Research from the AIBL study of Aging group (Table 1) used 11C-PiB PET and MRI brain scans to investigate Aβ deposition in typical AD and NDAN subjects [215]. The authors found a greater temporal grey matter volume in ‘high-PiB healthy controls’, compared to ‘low-PiB healthy controls’. In other words, brain scans of NDAN subjects with high Aβ load showed a larger grey matter volume than healthy controls without Aβ in the temporal lobe.
Typically in AD, the hippocampus, particularly CA1 region, and entorhinal cortex are the first regions that are affected [73]. Afterwards, other parts of the temporal and parietal lobes and some of the frontal cortex and cingulate gyrus are affected. Surprisingly, in the 80+-year-old NDAN group, SuperAgers, a significantly thicker left anterior cingulate gyrus was found in comparison to both other 80+-year-olds and middle-aged controls [216]. Later these findings were confirmed by Gefen et al. (2015) who further reported that this region contains a lower NFT density in SuperAgers compared to other groups [177]. Moreover, they discovered a remarkably high presence of specialized nerve cells in the SuperAgers’ cingulate cortex: ‘von Economo-neurons’ (VEN). These neurons might be important to understand NDAN and will be discussed in the section NDAN on a cellular level. In the BLSA Study, the rCBF, a measure for local neuronal activity, was measured longitudinally up to 10 years prior to death by PET scan of ‘ASYMAD’ individuals, NDAN, and compared to cognitively impaired and cognitively normal older subjects [17]. Their analysis demonstrated that both the NDAN and the cognitively impaired group show similar rCBF declines in the precuneus and mesial temporal cortex over time. These results are consistent with observations seen in subjects with MCI, reflecting the stage before this decline further progresses into AD and before the thalamic regions are affected [94, 217]. Interestingly, only in the NDAN group, not in the cognitively normal, nor in the cognitively impaired group, an increase in rCBF could be observed in the hippocampus, parahippocampal gyrus, and anterior insula over time [17]. Despite the limited sample size in this BLSA study, it can be hypothesized that the resilience observed in NDAN individuals could be mostly contributed to the hippocampal area, the main affected area by AD.
NDAN on a cellular level
Neurons
As was briefly introduced in the previous section, the brains of the group SuperAgers contain a thicker region of the anterior cingulate cortex, which correlated with a higher density of the neuronal subtype VENs [177]. More precisely, Gefen et al. (2015) reported that the VEN density in SuperAgers was approximately 3- to 5-fold higher than in elderly control. VENs are rare, spindle neurons, that are characterized as slim, bipolar neurons located in the anterior cingulate cortex and fronto-insular cortex, and named after Constantin von Economo who first described them [218, 219]. Loss of VENs is associated with frontotemporal dementia, a type of dementia with atrophy in the anterior cingulate cortex early in its progression, resulting in aberrant social behavior [220]. However, the function of these spindle neurons has not yet been completely clarified. Gefen et al. (2018) found no significant association between the density of VENs and Braak stages [16]. However, in the same study, lower VEN density was observed in AD and in amnestic MCI cases, compared to elderly controls. Moreover, they demonstrated the highest density of VENs in SuperAgers, even in comparison with young adults, which was consistent with their previous findings [177]. It would be interesting to investigate whether VENs remain retentive in brains of other NDAN subpopulations.
Neuronal hypertrophy is a hypothesized compensatory mechanism that prevents AD progression. Hypertrophy refers to an increase in size, in this case of neural cells. Studies performed by Iacono et al. reported significant hypertrophy of neuronal cell bodies, nuclei, and nucleoli in NDAN individuals, particularly in the CA1 region of the hippocampus and the anterior cingulate gyrus [197, 221]. This observation was in agreement with findings of the BLSA study, in which neuronal hypertrophy was proposed as a compensatory mechanism for atrophy that prevents AD pathology [222].
As discussed earlier in this review, adult hippocampal neurogenesis is reduced in AD and MCI brains. Moreover, an interesting working hypothesis could be that neurogenesis is preserved in NDAN individuals. In agreement with this hypothesis, a recent study found an elevated amount of NSCs and newly matured neurons in NDAN individuals [9]. Postmortem miRNA analysis of the dentate gyrus in the hippocampi of NDAN revealed that NDAN expressed lower levels of miRNAs known to regulate neurogenesis (miR-9, miR-25, miR-29a, miR-124, miR-132, miR-137) compared to controls, MCI, and AD [9]. Moreover, in the dentate gyrus, immunofluorescence studies for SOX2, a transcription factor crucial for maintenance of neuronal stem cells, also showed a higher amount of SOX2 positive NSCs, compared to AD and MCI controls. Interestingly, a positive correlation was found between the fraction of SOX2 positive cells and cognitive functioning prior to death. Taken together, these findings indicate a role of neurogenesis as a possible mechanism allowing for the resistance of AD pathology [9]. In another study, NSC culture-derived exosomes were investigated for their function in neurogenesis [223]. Exosomes are nano-sized extracellular vesicles, that contain information (e.g., lipids, proteins, RNAs, genetic material) about the cells that secrete them [224]. Moreover, these vesicles have been reported to spread pathological proteins and lipids involved in AD [225]. To investigate whether exosomes originating from neuronal stem cells can prevent synaptic loss and memory impairment, Micci et al., 2019 cultured adult rat hippocampus NSCs and injected their exosomes intracerebroventricularly in healthy mice, and found that the NSC-secreted exosomes showed reduction in Aβ oligomer-binding to synapses, and neutralize memory deficits caused by Aβ oligomers [223]. Surprisingly exosomes originating from mature neuronal hippocampal cell culture did not show these effects, highlighting the importance of NSCs and neurogenesis. miRNA analysis of the exosomes originating from both cell cultures showed an enrichment of miR-485, miR-322, and miR-17 in NSC-derived exosomes. The use of miRNA mimics further validated the role of miR-17 and miR-485 in protecting synapses from Aβ oligomer-binding [223].
The key implication to draw from these two studies is that neurogenesis seems to be preserved in NDAN individuals, resulting from increased levels of hippocampal NSCs, with elevated SOX2 expression and these NSC may secrete exosomes, transporting particular miRNAs that protect synapses against Aβ oligomers. In the following section, other mechanisms will be discussed that prevent Aβ oligomeric toxicity in synapses and preserve cognitive functioning in NDAN individuals.
Taken together, it seems that specialized neurons in the hippocampus, VENs and NSCs, are particularly upregulated in NDAN individuals. This resistance could be parallel to or causal to hippocampal hypertrophy that would increase brain “reserve” in NDAN individuals.
Immune cells
As was pointed out earlier (see the section AD on a cellular level), the important contribution of neuroinflammation in the pathology of AD is becoming more and more acknowledged. Already in the early stages of AD, neuroinflammatory responses are very diverse between individuals [112]. Interestingly, it is believed that inflammation during the early stages of AD contributes to the development of AD and cognitive impairment later in life [112]. Considering NDAN, it could be that inflammation is decreased and therefore inhibits progression of dementia. This could be attributed to the microglial phenotype, M1 and M2, and the ratio in which they have proliferated in the brain after detection of Aβ or tau protein aggregates. Hence, it could be hypothesized that in NDAN the AD pathology does not result in proliferation of, and differentiation into the M1 microglia, but in M2 microglia, showing a stronger phenotype of classic inflammation inhibition. To best of our knowledge to date, however, no single study exists that discriminates between the different microglial phenotypes in NDAN.
The study by Melah et al. (2016) showed that neuroinflammation aggravates neural damage, as they found a correlation between the neuroinflammatory markers, YKL-40 and MCP1, and the neural damage markers, NFL and tau levels, in the CSF in asymptomatic adults with AD pathology carrying the risk gene APOE ɛ4 [226]. However, they did not specifically look at microglia. In another study, microglial activation was evaluated in brains of SuperAgers, to investigate the response to age-related pathology [227]. Older adults in this study showed increased microglial density in white matter regions that correlate with the affected regions in AD as described in the section AD on an organ level. On the other hand, this increased microglial density was not observed in SuperAgers, giving us another interesting possible explanation for the high cognitive functioning in these individuals. These results match those observed in the study of Perez-Nievas et al. (2013), who found a significantly lower amount of microglia, and also of astrocytes, in the superior temporal sulcus of their NDAN group [13]. In this study, the NDAN group was divided into high probability (HP, Braak stage V-VI) to develop AD and intermediate probability (IP, Braak stage III-IV) to develop AD. The lower levels of glial activation in these HP and IP resilient cases indicate that neuroinflammation might be suppressed in NDAN but does not address further mechanistic insights.
A follow-up study performed by the same research group of Perez-Nievas investigated neuroinflammation in NDAN individuals by quantifying cytokines in brain tissue [178]. Using a multiplex immunoassay, increased expression of neurotrophic factors was found in both HP and IP resilient cases, compared to AD cases. Neurotropic factors are beneficial for NDAN cases, since they support neuronal growth, differentiation, and survival [228]. Moreover, both HP and IP resilient cases show upregulated cytokines in the entorhinal cortex, compared to AD cases [178]. Interestingly, in the HP cases IL-1β, IL-6, IL-13, and IL-4 were found to be upregulated, while IL-6 and IL-10 were found to be upregulated in the IP resilient cases. This brings back the hypothesis stated at the beginning of this section, namely that toxicity of AD pathology might depend on the balance between the classic pro-inflammatory M1 microglia population relative to the alternative anti-inflammatory, wound healing M2 microglia population, as classic activation of microglia (M1) is mainly mediated through TNF-α, IL-1, IL-12, IL-23, and interferon-γ, whereas alternative (M2) microglia activation primarily through transforming growth factor-β, IL-4, IL-10, and IL-13 [118, 119]. This combination of findings supports the hypothesis that AD pathology in NDAN results predominantly in activation of the anti-inflammatory M2 microglia and so suppresses neuroinflammation. However, further research is required to investigate the microglial phenotypes during neuroinflammation in NDAN cases.
Collectively, these studies indicate that neuroinflammation is reduced in NDAN cases, as a result of decreased glial density. Furthermore, the different cytokine profiles suggest the hypothetical anti-inflammatory phenotype of microglia.
NDAN on a biochemical level
Multiple mechanism underlying NDAN resistance to AD have already been discussed on an organism, organ, and cellular level. For this section, several biochemical mechanisms will be discussed as potential mechanisms that could explain the resilience to AD in NDAN individuals.
Amyloid-β pathology
One key player in the neuropathology of AD is Aβ. Unfortunately, the amount of amyloid plaques used to identify NDAN individuals differs per author, as is reflected in the different CERAD scores, a semiquantitative measure for neuritic amyloid plaques, in Table 3. Most of these studies investigated NDAN cases around the same age and all have a limited number of subjects. For most articles, only a high CERAD rating was confirmed even though some other articles include CERAD scores lower than moderate.
Table 3
Selected studies on classification of non-demented individuals with AD neuropathology on Braak staging to classify the degree of NFT, and CERAD rating to measure the amyloid plaques
| Synonym | Number of participants | Age | Braak stage | CERAD rating | Reference |
| NDAN | N = 4 | > 89 | IV–VI | – | [9] |
| NDAN | N = 8 | 76– > 89 | IV–VI | – | [10] |
| ND-HPC | N = 8 | 91–100 | III–V | Moderate – frequent | [11] |
| HP | N = 8 | ≈ 88.4 | V–VI | Frequent | [13] |
| IP | N = 12 | ≈ 89.82 | III–IV | Moderate | [13] |
| SA | N = 5 | 87–95 | 0–III | – | [16, 177] |
| ASYMAD | N = 6 | > 75.9 | II–IV | Moderate | [17] |
| ASYMAD | N = 13 | ≈ 89 | I–III | 76.9 % – Moderate | [18] |
| 23.0 % – Frequent | |||||
| AsymAD | N = 33 | 79–96 | I–IV | – | [19] |
| ASYMAD | N = 15 | 71–96 | II–IV | Moderate – frequent | [21] |
| p-preAD | N = 13 + 20 | 53–88 | I–III | None – sparse | [79] |
| HP | N = 12 | ≈ 86.1 | V–VI | Moderate – frequent | [178] |
| IP | N = 21 | ≈ 86.6 | III – IV | Sparse – frequent | [178] |
| AsymAD | N = 21 | ≈ 75.59 | ≥II | Moderate – frequent | [190] |
| NDAN | N = 10 | ≈ 89.5 | IV–VI | Moderate | [230] |
| NDAD | N = 10 | 86.6±5.3 | II–IV | Moderate–frequent | [237] |
CERAD, Consortium to Establish a Registry for Alzheimer’s Disease; ASYMAD, Asymptomatic AD; NDAD, non-demented individuals demonstrating intermediate AD neuropathologies; ND-HPC, non-demented high pathology controls; NDAN, non-demented individuals with AD neuropathology; HP, high probability; IP, intermediate probability; p-preAD, pathologically diagnosed preclinical AD; SA, SuperAgers.
Compared to young healthy adults, cognitively normal aging adults demonstrated increased fibrillar Aβ plaques in the temporal lobe and partially in the parietal region (particularly the precuneus), as visualized by PET using PiB staining [44]. Moreover, tau and Aβ changes in CSF can occur approximately 15 years prior to the clinical onset of AD [229]. Together, these studies highlight that brains of preclinical AD and of normal aging both contain Aβ pathology, which makes it difficult to diagnose AD. Further complicating AD diagnoses are the individuals that remain non-demented, despite the presence of Aβ aggregates in their elderly brain, the individuals we refer to in this paper as NDANs [63, 71, 186–190]. Interestingly, the study by Bjorklund et al. (2012) showed that the levels of Aβ plaques, tau tangles, Aβ42, and low molecular weight Aβ oligomeric species in brains of NDAN were comparable to the levels in AD brains [230]. Despite the similar levels of Aβ species in both AD and NDAN brains, Aβ oligomers did not bind the synapses nor affected the post-synaptic density. Interestingly, using immunohistochemistry and conformation-specific antibodies that recognized Aβ oligomer deposits (NAB61), Perez-Nievas et al. (2013) observed a significant decrease in the load of Aβ oligomeric deposits, in the group they termed ‘high probability mismatches’, NDAN, compared to AD patients [13]. These observations imply that the levels of soluble Aβ species in NDAN are comparable to those in AD, but that Aβ oligomers are less deposited in NDAN. These findings were supported by Bilousova et al. (2016), who found higher levels of soluble oligomeric Aβ surviving within synaptic terminals in demented cases, but not in NDAN [231].
Furthermore, the study of Bjorklund et al. (2012) found that in contrast to AD cases, NDAN individuals did not show Aβ oligomers in the hippocampal postsynapses, even though the Aβ oligomers were present in the ‘soluble fraction’ of the synaptic fractionation performed on the hippocampal tissue [230]. This observation is crucial because it suggests that Aβ oligomeric binding to synapses is needed to maneuver AD pathology into dementia and furthermore suggests a potential safety mechanism in NDAN that prevent binding to synapses. Interestingly, in line with this hypothesis, the same study showed that while the hippocampi of AD cases contained elevated synaptic Zn2+ levels, both in the ‘soluble fraction’ and the synaptic vesicles, NDAN cases only showed elevated levels of Zn2+ at the synaptic vesicles [230]. These findings correlated with regular expression of the synaptic Zn2+ transporter ZnT3, which was impaired in the AD cases [230]. These findings indicate that NDAN cases can better maintain synaptic Zn2+ homeostasis and thereby regulate synaptic binding of Aβ oligomers. This understanding is crucial because it could lead to new therapeutic insights to prevent AD.
As discussed in the section AD on a biochemical level, amyloid-β pathology, soluble Aβ oligomers induce synaptic damage thereby leading to inhibition of long-term potentiation [148]. Interestingly, the neuromodulator Reelin has been suggested to have an Aβ-antagonist-like function at the synapses [232]. Typically, Reelin contributes to the regulation of neuronal migration in the brain development by controlling cell-cell interactions mediated by binding to the postsynaptic APOE receptors VLDLR and APOER2. By binding to VLDLR and APOER2, Reelin is thought to counteract Aβ-induced synaptic suppression [232]. Interestingly, also elevated levels of Reelin were observed in the hippocampal pyramidal neurons of NDAN individuals [233], suggesting a potential compensatory mechanism. However, Reelin was also upregulated in the cortex and CSF of patients with AD [234], and it has been proposed that elevated levels of Reelin do not necessarily contribute to, or are not sufficient for, the resistance of AD pathology [234], but this might depend on other unknown factors.
Interestingly, insulin signaling also seems to fulfill an Aβ-antagonist-like function. Using primary neuronal cell culture models, De Felice et al. (2009) observed that soluble Aβ oligomers induced downregulation of the insulin receptor at the plasma membrane [235]. In fact, adding insulin itself prevented not only binding of soluble Aβ oligomers to the synapses and downregulation of the insulin receptor, but was also able to abolish Aβ-induced oxidative stress and able to prevent Aβ-mediated synapse loss [235]. Interestingly, the protective role of insulin dependent on the kinase activity of the receptor, indicating that insulin signaling is responsible for the protective effect. In a review paper, Taglialatela reported unpublished data on decreased signaling components of the insulin transduction pathway in AD hippocampi compared to controls and NDAN [181]. Furthermore, in the hippocampus of NDAN individuals an even higher pathway activation was observed compared to AD and controls cases [181]. Overall, this suggests that maintenance of the insulin signaling pathway contributes to the preservation of synapses and cognitive integrity.
As described before in the context of neurons in NDAN, NSC-secreted exosomes were able to prevent synaptic degradation through upregulation of specific miRNAs [223]. In line with this finding, the synaptic miRNA expression was investigated in hippocampal tissue of NDAN subjects [182]. Specifically, they identified three miRNAs, miRNA-485, miRNA-4723, and miRNA-149, to be differently expressed in hippocampal tissue of NDAN and AD, compared to controls [182]. All three miRNAs regulate the protein expression of synaptic genes, such as APP and β-secretase 1, and intracerebroventricularly administration to adult wild-type mice resulted in increased synaptic resilience to Aβ oligomers [182]. While a very interesting discovery, focus lied on upregulation of these miRNAs, knockdown studies in cell culture systems and/or animals would provide crucial insights whether the disruption of these miRNAs will lead to or enhance Aβ-toxicity.
Overall, it seems that Aβ oligomers bind to synapses, thereby resulting in synaptic dysfunction in AD. Various observations in NDAN individuals together gave insights on potential mechanisms preventing Aβ-mediated synapse loss: better maintained Zn2+ homeostasis, upregulation of Reelin, increased levels of insulin signaling components, and miRNAs that regulate the protein expression of APP and β-secretase genes in synapses. However, as these findings were done independently of each other, it remains unclear whether they act independently or parallel to each other or even synergistically, and further research is required to understand the full, coherent picture that explains the role of Aβ oligomer binding to preserve cognition and prevent dementia.
Tau pathology
The other key player in the neuropathology of AD is aggregation of the protein tau. Similar to Aβ aggregation, tau aggregation can also be observed in normal aging (section on Normal aging). One of the biochemical mechanisms that could potentially be underlying NDAN resistance to AD pathology is the inhibition of NFT formation resulting from for instance changes in tau isoform expression, as well as changes in activities of tau kinases and phosphatases. One study investigated tau isoforms in brain areas affected and unaffected by NFT, in both AD and control using RT-PCR and they showed that the relative expression of tau isoform 0N3R was higher in the affected temporal cortex than in the unaffected cerebellar cortex, in both AD and control [236]. It would be interesting to investigate the tau isoform expression in NDAN to provide more inside into the NFT formation in NDAN. As was pointed out in the section “normal” aging, tau deposition is generally observed in older adults [44]. This study shows that for in vivo Braak staging, there is a higher within-group variance for cognitively intact elderly, than for young adults and AD patients. This within-group variance could therefore also be observed for NDAN cases. Moreover, between authors, different approaches are used for both Braak stages to identify NDAN cases (Table 3).
So certain amounts of tau species have been found in NDAN brains. What is not yet clear is what type of tau species were found (see also Supplementary Figures 1 and 2), and whether these would differ from the type of tau species found in AD. Perez-Nievas et al. (2013) investigated what tau species could contribute to the resilient mechanism in NDAN, particularly soluble tau versus NFT [13]. In their results, mostly tau monomers were found in controls and NDAN. Compared to healthy controls, only AD cases had an abnormal accumulation of soluble mono- and multimeric species of hyperphosphorylated tau in the synapses, not the high probability mismatch cases [13]. In other words, the accumulation of soluble hyperphosphorylated tau in synapses seems more manageable in NDAN-brains, compared to AD.
Another study investigated the correlation between Braak’s staging system and intact cognitive outcome, using MMSE scores, with data from the Nun Study and the National Alzheimer’s Coordinating Center [12, 193, 197]. This data showed that the cognition of individuals with Braak stages V and VI were significantly different from each other. Therefore it was stated in this paper that these two Braak stages should not be combined in the determination of NDAN individuals [193]. Moreover, there were no cognitive intact individuals found with the highest Braak stage in this study. Therefore, it is necessary to form concrete inclusion criteria to categorize NDAN individuals.
Gene expression
Gene variants and changes in gene expression in NDAN brains can provide insight into the upstream mechanism underlying dementia. Liang et al. (2010) performed postmortem transcriptomics analysis on 6 different brain regions comparing AD individuals, NDAN individuals, and healthy elderly controls [237]. They found that, while both NDAN and AD brains showed similar expression changes in tangle- and plaque related pathways and ubiquitin-proteasomal pathways, NDAN individuals displayed unique expression changes in most brain regions in genes correlating with learning and/or memory processes. More specifically, they found a downregulation several genes, including APOE, while genes such as MAPT and ephrin-B2, involved in cell-cell signaling and mediating neural development, were upregulated [237]. These finding suggest that the transcriptomic changes may represent compensatory mechanisms against cognitive decline. This study stresses the importance of transcriptomic in the quest to unravel underlying mechanisms, focusing on expression of notorious AD-related genes. Nevertheless, recently a study was conducted suggesting another potential component that reduces the risk for AD and other types of dementia. In this study, a genetic variant in the phospholipase Cγ2 (PLCG2) gene was suggested to reduce AD risk [238]. The PLCG2 gene was found to have a role in immune system signaling, suggesting a protective effect against neurodegenerative diseases, including AD. The authors in this study indicate that the genetic variant of this gene is associated with an increased longevity and reduced AD risk [238]. It is therefore not unlikely that a connection exists between this genetic variant and NDAN.
DISCUSSION AND CONCLUSIONS
The aim of this review was to achieve more insight into the mechanism underlying NDAN resistance to dementia. Thus far, we have evaluated various mechanism on a body, organ, cellular, and biochemical level based on a large variety of studies. All the included studies in this review were based on cognitively normal elderly, defined as a MMSE score > 24, and with age 60 years or older, carrying the AD pathology (Table 2). So far in literature, there has not been a concrete definition for NDAN-like individuals, and it can be debated which MMSE cut-off score best reflects cognitively intact elderly. Similarly, the degree of AD pathology in literature, as summarized in Table 3, is inconsistent. Therefore, for future studies, clarity is needed to form concrete inclusion criteria for NDAN, for which Tables 2 and 3 can be used to form an overview.
Key findings that could partly explain why this small group of NDAN individuals, which carry AD pathology but stay cognitively intact, are summarized here. Generally, NDAN individuals have attended higher education, than individuals with MCI or AD, and therefore may possibly have a higher cognitive reserve. Secondly, researchers debate on the relationship between hippocampal volume and NDAN resilience to dementia. However, most of the studies indicate that hippocampal atrophy is compensated for in NDAN, which could result from increased neuronal hypertrophy in NDAN’s hippocampal neurons. Moreover, the cingulate gyrus was found to be thicker and overall the grey volume was larger in NDAN, compared to AD cases. Besides neuronal hypertrophy, two different studies each looking at only one specific cell-type found an elevated presence of specialized neurons, such as VENs and NSCs, in NDAN individuals. Furthermore, adult neurogenesis seems to be preserved in NDAN individuals, which can be attributed to increased levels of hippocampal NSCs and their elevated SOX2-expression allowing for stem-cell renewal. Thirdly, neuroinflammation is suppressed in NDAN as associated with decreased glial activation. Moreover, compared to AD cases, the cytokine signature in NDAN individuals represented more an anti-inflammatory state, with elevated levels IL-4 and IL-13, further thus supporting the hypothesis that neuroinflammation is decreased in NDAN cases. Lastly, looking at the AD hallmarks Aβ and tau pathology, NDAN cases have more tau monomers and less accumulation of soluble hyperphosphorylated tau at the synapses, but further research is needed, including elucidation of the different tau species and their distribution in NDAN cases compared to control and AD cases. Looking at the Aβ pathology, in NDAN several biochemical mechanisms for synaptic resilience to Aβ-oligomeric binding have been suggested, namely preserved synaptic Zn2+ homeostasis, better maintenance of the insulin signaling pathway and increased expression of three miRNAs involved in the regulation of expression of synaptic genes.
Altogether, these different mechanisms discussed in this review shed new light into the underlying mechanism of NDAN cases, “normal” aging, and AD (Fig. 2). It is clear that despite the growing literature regarding NDAN, additional studies will be needed to develop a better picture of NDAN to disentangle NDAN cases from “normal” aging and AD and to gain insights into possible drug targets for AD. Regarding resilience to AD pathology on an organ level, NDAN researchers have been focused primarily on the hippocampus, while cortical regions that are affected in AD, are not taken into account in most investigations in NDAN individuals. Moreover, literature regarding hippocampal volume in NDAN cases show contradicting findings, suggesting that further research is required. Furthermore, other circumstances, including time and aging, that could affect the hippocampal volume and neurogenetic factors will provide further crucial insights. Based on the cytokines profiles of NDAN cases, we hypothesized that NDAN individuals display a different ratio of M1 and M2 microglial phenotypes, specifically we hypothesized that in NDAN, classic inflammation is inhibited by M2-microglial cells for which so far no experimental data is available. On the topic Aβ toxicity, the observation that Aβ-oligomeric binding to synapses is reduced in NDAN, brings new insights into the development of novel therapeutic approaches aiming to improve synaptic resistance to Aβ oligomeric toxicity. Regarding the other AD hallmark, tau, the mechanisms involved in NDAN have to be investigated more extensively, therefore it was proposed to investigate what type of tau species was found in NDAN brain, compared to normal aging and AD, and where it distributed to. What was already found in literature is that NFT formation in the temporal cortex was inhibited, due to lower 0N3R expression.
Various observations in NDAN individuals have tried to provide an explanation how Aβ-synapse binding is mediated: Zn2+ homeostasis, the extracellular matrix protein Reelin, insulin signaling, and miRNAs that regulate APP and β-secretase expression levels in synapses. Unfortunately, these are all different mechanisms studied independently, and a more comprehensive and holistic study looking at these mechanisms all in the same individual, to the best of our knowledge, so far has been lacking. Further a holistic research approach is required to form a coherent entity that explains the role of Aβ oligomer binding to preserve cognition and prevent dementia. The observation that neurogenesis is more pronounced in NDAN individuals than in AD, also provides and interesting therapeutic potential, for instance focused on NSC proliferation in the hippocampus.
Similar to the longitudinal cohort studies that have been performed in America and Australia, among others, more recently, researchers in The Netherlands launched two cohort studies to investigate several types of diseases in elderly [239, 240]. They both investigated the increasing prevalence of dementia with age and investigated the impact of lifestyle factors on AD. However, they did not yet study or publish whether a group of NDAN would exist in this cohort, for the potential reason that they did not yet performed autopsies in this study. Nevertheless, it would be interesting to see also the NDAN individuals in these cohorts.
A major limitation of the field, and thereby as well of this review, is that most research groups use their own synonym to refer to non-demented elderly with AD pathology. Not only do different studies use different synonyms, but also use different measurements and thresholds to classify these subjects. While some studies expect that these subjects will eventually develop dementia, so study them as preclinical, other studies focused only on the individuals with really severe AD pathology. Another issue is the lack of awareness regarding the non-demented individuals in most AD studies. Despite the alterations made by the Alzheimer’s Association to separate the NDAN from AD cases, not all researchers considered NDAN in their study design. Also, we have to consider that NDAN individuals included in the studies generally attended higher education, suggesting that these individuals have already been cognitively stronger than the ones with lower education, creating educational bias. Then, the biggest question in NDAN is time, in particular the contribution of aging in the pathology of AD. In this review, both longitudinal and cross-sectional studies have been presented. In longitudinal studies (e.g., cohort studies), the same subjects are observed at multiple points in time, whereas cross-sectional studies take a snapshot by investigating a population at a fixed time point, for example, postmortem. Looking at the contribution of time in the presented studies, a limitation of postmortem brain analysis in cross-sectional studies is the absence of information on the subjects’ brain during life. The NDAN subjects in the studies described in this review had intermediate to high AD pathology in their brains. However, it could never be confirmed with certainty that these subjects would not eventually have developed dementia and a certain time delay between cognitive testing and death is inevitable. As discussed in the section NDAN on a body level, both Driscoll et al. (2006) and Dang et al. (2019) observed that cognitive functioning of their NDAN group declined over time, comparable to cognitively normal elderly [14, 190]. This suggests that despite their resilience to AD pathology, time remains one of the most important factors in the pathology. Hence, Goldberg (2019) reviews these SuperAger studies, suggesting that age needs to be considered more [241]. Moreover, he implied that SuperAgers are not necessarily superior, but rather “LuckyAgers”, questioning the resilience, cognitive reserve, and neurological changes in these individuals. From this perspective, aging itself seems one of the largest risks for neurodegenerative diseases, as confirmed by the Nun Study. On the other hand, some studies indicated the uniqueness of NDAN individuals, as being cognitively intact despite AD pathology [17, 61]. Especially Dubois et al. (2018), who launched a longitudinal study aiming to discover new insight into the compensation mechanism in cognitively intact individuals with AD pathology [172]. The last issue we would like to raise is that the current literature on NDAN-like individuals implemented their test group by different conditions, therefore comparing different studies could be unjustly or at least requires some caution. So far, no concrete inclusion criteria have been defined to categorize NDAN individuals.
Despite evidence on both sides, resilience to pathology or simply a preclinical indication, it is most important that the contribution of age to AD pathology is not forgotten in any NDAN research, as well as that we would consider NDAN subjects as a unique group that could provide crucial insights into compensation mechanisms in the prevention of dementia. This review intended to form a broad overview into the different aspects that are involved in NDAN and compared these mechanisms to those in “normal” aging and AD itself. The suggestions given in this review lay the groundwork for future research into the mechanism that prevents clinical progression to dementia.
ACKNOWLEDGMENTS
We wish to express appreciation to Christiaan van Leerdam for making all graphical figures that are present in this review.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/21-0607r2).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-210607.
REFERENCES
[1] | Blennow K , de Leon MJ , Zetterberg H ((2006) ) Alzheimer’s disease. Lancet 368: , 387–403. |
[2] | Bejanin A , Schonhaut DR , La Joie R , Kramer JH , Baker SL , Sosa N , Ayakta N , Cantwell A , Janabi M , Lauriola M , O’neil JP , Gorno-Tempini ML , Miller ZA , Rosen HJ , Miller BL , Jagust WJ , Rabinovici GD ((2017) ) Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer’s disease. Brain 140: , 3286–3300. |
[3] | He Z , Guo JL , McBride JD , Narasimhan S , Kim H , Changolkar L , Zhang B , Gathagan RJ , Yue C , Dengler C , Stieber A , Nitla M , Coulter DA , Abel T , Brunden KR , Trojanowski JQ , Lee VMY ((2018) ) Amyloid-β plaques enhance Alzheimer’s brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat Med 24: , 29–38. |
[4] | Irvine GB , El-Agnaf OM , Shankar GM , Walsh DM ((2008) ) Protein aggregation in the brain: The molecular basis for Alzheimer’s andarkinson’s diseases. Mol Med 14: , 451–464. |
[5] | Hardy J , Selkoe DJ ((2002) ) The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 297: , 353–356. |
[6] | Sheinerman KS , Umansky SR ((2013) ) Early detection of neurodegenerative diseases. Cell Cycle 12: , 1–2. |
[7] | Drummond E , Wisniewski T ((2017) ) Alzheimer’s disease: Experimental models and reality. Acta Neuropathol 133: , 155–175. |
[8] | Lewczuk P , Łukaszewicz-Zając M , Mroczko P , Kornhuber J ((2020) ) Clinical significance of fluid biomarkers in Alzheimer’s disease. Pharmacol Rep 72: , 528–542. |
[9] | Briley D , Ghirardi V , Woltjer R , Renck A , Zolochevska O , Taglialatela G , Micci MA ((2016) ) Preserved neurogenesis in non-demented individuals with AD neuropathology. Sci Rep 6: , 27812. |
[10] | Zolochevska O , Bjorklund N , Woltjer R , Wiktorowicz JE , Taglialatela G ((2018) ) Postsynaptic proteome of non-demented individuals with Alzheimer’s disease neuropathology. J Alzheimers Dis 65: , 659–682. |
[11] | Maarouf CL , Daugs ID , Kokjohn TA , Walker DG , Hunter JM , Kruchowsky JC , Woltjer R , Kaye J , Castaño EM , Sabbagh MN , Beach TG , Roher AE ((2011) ) Alzheimer’s disease and non-demented high pathology control nonagenarians: Comparing and contrasting the biochemistry of cognitively successful aging. PLoS One 6: , e27291. |
[12] | Monsell SE , Mock C , Roe CM , Ghoshal N , Morris JC , Cairns NJ , Kukull W ((2013) ) Comparison of symptomatic and asymptomatic persons with Alzheimer disease neuropathology. Neurology 80: , 2121–2129. |
[13] | Perez-Nievas BG , Stein TD , Tai HC , Dols-Icardo O , Scotton TC , Barroeta-Espar I , Fernandez-Carballo L , De Munain EL , Perez J , Marquie M , Serrano-Pozo A , Frosch MP , Lowe V , Parisi JE , Petersen RC , Ikonomovic MD , López OL , Klunk W , Hyman BT , Gómez-Isla T ((2013) ) Dissecting phenotypic traits linked to human resilience to Alzheimer’s pathology. Brain 136: , 2510–2526. |
[14] | Dang C , Harrington KD , Lim YY , Ames D , Hassenstab J , Laws SM , Yassi N , Hickey M , Rainey-Smith SR , Robertson J , Rowe CC , Sohrabi HR ((2019) ) Superior memory reduces 8-year risk of mild cognitive impairment and dementia but not amyloid β-associated cognitive decline in older adults. Arch Clin Neuropsychol 17: , 585–598. |
[15] | Rogalski EJ ((2019) ) Don’t forget—Age is a relevant variable in defining SuperAgers. Alzheimers Dement (Amst) 11: , 560–561. |
[16] | Gefen T , Papastefan ST , Rezvanian A , Bigio EH , Weintraub S , Rogalski E , Mesulam M-M , Geula C ((2018) ) Von economo neurons of the anterior cingulate across the lifespan and in Alzheimer’s disease. Cortex 99: , 69–77. |
[17] | Codispoti KET , Beason-Held LL , Kraut MA , O’Brien RJ , Rudow G , Pletnikova O , Crain B , Troncoso JC , Resnick SM ((2012) ) Longitudinal brain activity changes in asymptomatic Alzheimer disease. Brain Behav 2: , 221–230. |
[18] | Iacono D , Zandi P , Gross M , Markesbery WR , Pletnikova O , Rudow G , Troncoso JC ((2015) ) APOɛ2 and education in cognitively normal older subjects with high levels of AD pathology at autopsy: Findings from the Nun Study. Oncotarget 6: , 14082–14091. |
[19] | Patel H , Hodges AK , Curtis C , Lee SH , Troakes C , Dobson RJB , Newhouse SJ ((2019) ) Transcriptomic analysis of probable asymptomatic and symptomatic alzheimer brains. Brain Behav Immun 80: , 644–656. |
[20] | Snowden SG , Ebshiana AA , Hye A , Pletnikova O , O’Brien R , Yang A , Troncoso J , Legido-Quigley C , Thambisetty M ((2019) ) Neurotransmitter imbalance in the brain and Alzheimer’s disease pathology. J Alzheimers Dis 72: , 35–43. |
[21] | Iacono D , Resnick SM , O’Brien R , Zonderman AB , An Y , Pletnikova O , Rudow G , Crain B , Troncoso JC ((2014) ) Mild cognitive impairment and asymptomatic Alzheimer disease subjects. J Neuropathol Exp Neurol 73: , 295–304. |
[22] | Shock NW , Greulich RC , Costa PT Jr , Andres R , Lakatta EG , Arenberg D , Tobin JD ((1984) ) Normal Human Aging: The Baltimore Longitudinal Study of Aging, US Department of Health and Human Services, Public Health Service, National Institutes of Health, National Institute on Aging, Gerontology Research Center, Washington, DC. |
[23] | Consumer Reports, Evaluating Prescription Drugs Used to Treat: Alzheimer’s Disease Comparing Effectiveness, Safety and Price, Last updated May 2012, http://www.consumerreports.org/health/resources/pdf/best-buy-drugs/AlzheimersFINAL.pdf. Accessed on May 2020. |
[24] | Jessen F , Amariglio RE , Buckley RF , van der Flier WM , Han Y , Molinuevo JL , Rabin L , Rentz DM , Rodriguez-Gomez O , Saykin AJ , Sikkes SAM , Smart CM , Wolfsgruber S , Wagner M ((2020) ) The characterisation of subjective cognitive decline. Lancet Neurol 19: , 271–278. |
[25] | Resnick SM , Pham DL , Kraut MA , Zonderman AB , Davatzikos C ((2003) ) Longitudinal magnetic resonance imaging studies of older adults: A shrinking brain. J Neurosci 23: , 3295–3301. |
[26] | Berninger B , Jessberger S ((2016) ) Engineering of adult neurogenesis and Gliogenesis. Cold Spring Harb Perspect Biol 8: , a018861. |
[27] | Wang W-Y , Tan M-S , Yu J-T , Tan L ((2015) ) Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann Transl Med 3: , 151. |
[28] | Fakhoury M ((2017) ) Microglia and astrocytes in Alzheimer’s disease: Implications for therapy. Curr Neuropharmacol 15: , 508. |
[29] | Lee CYD , Landreth GE ((2010) ) The role of microglia in amyloid clearance from the AD brain. J Neural Transm 117: , 949–960. |
[30] | Colonna M , Butovsky O ((2017) ) Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol 35: , 441–468. |
[31] | Ginhoux F , Prinz M ((2015) ) Origin of microglia: Current concepts and past controversies. Cold Spring Harb Perspect Biol 7: , a020537. |
[32] | Tay TL , Savage JC , Hui CW , Bisht K , Tremblay MÈ ((2017) ) Microglia across the lifespan: From origin to function in brain development, plasticity and cognition. J Physiol 595: , 1929–1945. |
[33] | Mayne K , White JA , McMurran CE , Rivera FJ , de la Fuente AG ((2020) ) Aging and neurodegenerative disease: Is the adaptive immune system a friend or foe? Front Aging Neurosci 12: , 305. |
[34] | Xu YZ , Nygård M , Kristensson K , Bentivoglio M ((2010) ) Regulation of cytokine signaling and T-cell recruitment in the aging mouse brain in response to central inflammatory challenge. Brain Behav Immun 24: , 138–152. |
[35] | Dulken BW , Buckley MT , Negredo PN , Saligrama N , Cayrol R , Leeman DS , George BM , Boutet SC , Hebestreit K , Pluvinage JV , Wyss-Coray T , Weissman IL , Vogel H , Davis MM , Brunet A ((2019) ) Single cell analysis reveals T cell infiltration in old neurogenic niches. Nature 571: , 210. |
[36] | Cuapio A , Ljunggren H-G ((2021) ) Activated natural killer cells hit neurogenesis in the aging brain. Neurosci Bull 37: , 1074. |
[37] | Regnell CE , Hildrestrand GA , Sejersted Y , Medin T , Moldestad O , Rolseth V , Krokeide SZ , Suganthan R , Luna L , Bjørås M , Bergersen LH ((2012) ) Hippocampal adult neurogenesis is maintained by Neil3-dependent repair of oxidative DNA lesions in neural progenitor cells. Cell Rep 2: , 503–510. |
[38] | Cisneros-Franco JM , Ouellet L , Kamal B , De Villers-Sidani E ((2018) ) A brain without brakes: Reduced inhibition is associated with enhanced but dysregulated plasticity in the aged rat auditory cortex. eNeuro 5: , 1–15. |
[39] | Reas ET ((2017) ) Amyloid and tau pathology in normal cognitive aging. J Neurosci 37: , 7561–7563. |
[40] | Troncoso JC , Cataldo AM , Nixon RA , Barnett JL , Lee MK , Checler F , Fowler DR , Smialek JE , Crain B , Martin LJ , Kawas CH ((1998) ) Neuropathology of preclinical and clinical late-onset Alzheimer’s disease. Ann Neurol 43: , 673–676. |
[41] | Troncoso JC , Martin LJ , Dal Forno G , Kawas CH ((1996) ) Neuropathology in controls and demented subjects from the Baltimore longitudinal study of aging. Neurobiol Aging 17: , 365–371. |
[42] | Crystal H , Dickson D , Fuld P , Masur D , Scott R , Mehler M , Masdeu J , Kawas C , Aronson M , Wolfson L ((1988) ) Clinico-pathologic studies in dementia: Nondemented subjects with pathologically confirmed alzheimer’s disease. Neurology 38: , 1682–1687. |
[43] | Knopman DS , Parisi JE , Salviati A , Floriach-Robert M , Boeve BF , Ivnik RJ , Smith GE , Dickson DW , Johnson KA , Petersen LE , McDonald WC , Braak H , Petersen RC ((2003) ) Neuropathology of cognitively normal elderly. J Neuropathol Exp Neurol 62: , 1087–1095. |
[44] | Schöll M , Lockhart SN , Schonhaut DR , O’Neil JP , Janabi M , Ossenkoppele R , Baker SL , Vogel JW , Faria J , Schwimmer HD , Rabinovici GD , Jagust WJ ((2016) ) PET imaging of tau deposition in the aging human brain. Neuron 89: , 971–982. |
[45] | Chen G , Xu T , Yan Y , Zhou Y , Jiang Y , Melcher K , Xu HE ((2017) ) Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol Sin 38: , 1205–1235. |
[46] | Selkoe DJ ((2008) ) Soluble oligomers of the amyloid β-protein impairsynaptic plasticity and behavior. Behav Brain Res 192: , 106–133. |
[47] | Bancher C , Brunner C , Lassmann H , Budka H , Jellinger K , Wiche G , Seitelberger F , Grundke-Iqbal I , Iqbal K , Wisniewski HM ((1989) ) Accumulation of abnormally phosphorylated τ precedes the formation of neurofibrillary tangles in Alzheimer’s disease. Brain Res 477: , 90–99. |
[48] | Wegmann S , Bennett RE , Delorme L , Robbins AB , Hu M , McKenzie D , Kirk MJ , Schiantarelli J , Tunio N , Amaral AC , Fan Z , Nicholls S , Hudry E , Hyman BT ((2019) ) Experimental evidence for the age dependence of tau protein spread in the brain. Sci Adv 5: , eaaw6404. |
[49] | Price JL , Davis PB , Morris JC , White DL ((1991) ) The distribution of tangles, plaques and related immunohistochemical markers in healthy aging and Alzheimer’s disease. Neurobiol Aging 12: , 295–312. |
[50] | Piccini A , Russo C , Gliozzi A , Relini A , Vitali A , Borghi R , Giliberto L , Armirotti A , D’Arrigo C , Bachi A , Cattaneo A , Canale C , Torrassa S , Saido TC , Markesbery W , Gambetti P , Tabaton M ((2005) ) β-amyloid is different in normal aging and in Alzheimer disease. J Biol Chem 280: , 34186–34192. |
[51] | Waring SC , Rosenberg RN ((2008) ) Genome-wide association studies in Alzheimer disease. Arch Neurol 65: , 329–334. |
[52] | Corder EH , Saunders AM , Strittmatter WJ , Schmechel DE , Gaskell PC , Small GW , Roses AD , Haines JL , Pericak-Vance MA ((1993) ) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261: , 921–923. |
[53] | Hauser P , Ryan R ((2013) ) Impact of apolipoprotein E on Alzheimer’s disease. Curr Alzheimer Res 10: , 809–817. |
[54] | Liu CC , Kanekiyo T , Xu H , Bu G ((2013) ) Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat Rev Neurol 9: , 106–118. |
[55] | Goedert M , Spillantini MG , Crowther RA ((1991) ) Tau proteins and neurofibrillary degeneration. Brain Pathol 1: , 279–286. |
[56] | Karr JE , Graham RB , Hofer SM , Muniz-Terrera G ((2018) ) When does cognitive decline begin? A systematic review of change point studies on accelerated decline in cognitive and neurological outcomes preceding mild cognitive impairment, dementia, and death. Psychol Aging 33: , 95–218. |
[57] | Wilson RS , Leurgans SE , Boyle PA , Bennett DA ((2011) ) Cognitive decline in prodromal alzheimer disease and mild cognitive impairment. Arch Neurol 68: , 351–356. |
[58] | Wilson RS , Boyle PA , Yu L , Segawa E , Sytsma J , Bennett DA ((2015) ) Conscientiousness, dementia related pathology, and trajectories of cognitive aging. Psychol Aging 30: , 74–82. |
[59] | Yu L , Boyle P , Schneider JA , Segawa E , Wilson RS , Leurgans S , Bennett DA ((2013) ) APOE ɛ4, Alzheimer’s disease pathology, cerebrovascular disease, and cognitive change over the years prior to death. Psychol Aging 28: , 1015–1023. |
[60] | Livingston G , Huntley J , Sommerlad A , Ames D , Ballard C , Banerjee S , Brayne C , Burns A , Cohen-Mansfield J , Cooper C , Costafreda SG , Dias A , Fox N , Gitlin LN , Howard R , Kales HC , Kivimäki M , Larson EB , Ogunniyi A , Orgeta V , Ritchie K , Rockwood K , Sampson EL , Samus Q , Schneider LS , Selbæk G , Teri L , Mukadam N ((2020) ) Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 396: , 413–446. |
[61] | Dubois B , Hampel H , Feldman HH , Scheltens P , Aisen P , Andrieu S , Bakardjian H , Benali H , Bertram L , Blennow K , Broich K , Cavedo E , Crutch S , Dartigues JF , Duyckaerts C , Epelbaum S , Frisoni GB , Gauthier S , Genthon R , Gouw AA , Habert MO , Holtzman DM , Kivipelto M , Lista S , Molinuevo JL , O’Bryant SE , Rabinovici GD , Rowe C , Salloway S , Schneider LS , Sperling R , Teichmann M , Carrillo MC , Cummings J , Jack CR ((2016) ) Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimers Dement 12: , 292–323. |
[62] | McKhann GM , Knopman DS , Chertkow H , Hyman BT , Jack CR , Kawas CH , Klunk WE , Koroshetz WJ , Manly JJ , Mayeux R , Mohs RC , Morris JC , Rossor MN , Scheltens P , Carrillo MC , Thies B , Weintraub S , Phelps CH ((2011) ) The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7: , 263–269. |
[63] | Morris JC , Storandt M , McKeel DW Jr , Rubin EH , Price JL , Grant EA , Berg L ((1996) ) Cerebral amyloid deposition and diffuse plaques in “normal” aging: Evidence for presymptomatic and very mild Alzheimer’s disease. Neurology 46: , 707–719. |
[64] | Schmitt FA , Davis DG , Wekstein DR , Smith CD , Ashford JW , Markesbery WR ((2000) ) “Preclinical” AD revisited: Neuropathology of cognitively normal older adults. Neurology 55: , 370–376. |
[65] | Rabin LA , Smart CM , Amariglio RE ((2017) ) Subjective cognitive decline in preclinical Alzheimer’s disease. Annu Rev Clin Psychol 13: , 369–396. |
[66] | Albert MS , DeKosky ST , Dickson D , Dubois B , Feldman HH , Fox NC , Gamst A , Holtzman DM , Jagust WJ , Petersen RC , Snyder PJ , Carrillo MC , Thies B , Phelps CH ((2011) ) The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7: , 270–279. |
[67] | Backman L , Jones S , Berger A-K , Laukka EJ , Small BJ ((2004) ) Multiple cognitive deficits during the transition to Alzheimer’s disease. J Intern Med 256: , 195–204. |
[68] | Förstl H , Kurz A ((1999) ) Clinical features of Alzheimer’s disease. Eur Arch Psychiatry Clin Neurosci 249: , 288–290. |
[69] | Taler V , Phillips NA ((2008) ) Language performance in Alzheimer’s disease and mild cognitive impairment: A comparative review. J Clin Exp Neuropsychol 30: , 501–556. |
[70] | McKhann G , Drachman D , Folstein M , Katzman R , Price D , Stadlan EM ((1984) ) Clinical diagnosis of Alzheimer’s Disease: Report of the NINCDS-ADRDA work group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34: , 939–944. |
[71] | Sperling RA , Aisen PS , Beckett LA , Bennett DA , Craft S , Fagan AM , Iwatsubo T , Jack CR , Kaye J , Montine TJ , Park DC , Reiman EM , Rowe CC , Siemers E , Stern Y , Yaffe K , Carrillo MC , Thies B , Morrison-Bogorad M , Wagster MV , Phelps CH ((2011) ) Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7: , 280–292. |
[72] | Braak H , Alafuzoff I , Arzberger T , Kretzschmar H , Tredici K ((2006) ) Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112: , 389–404. |
[73] | Braak H , Braak E ((1991) ) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82: , 239–259. |
[74] | Nelson PT , Alafuzoff I , Bigio EH , Bouras C , Braak H , Cairns NJ , Castellani RJ , Crain BJ , Davies P , Tredici K Del , Duyckaerts C , Frosch MP , Haroutunian V , Hof PR , Hulette CM , Hyman BT , Iwatsubo T , Jellinger KA , Jicha GA , Kövari E , Kukull WA , Leverenz JB , Love S , MacKenzie IR , Mann DM , Masliah E , McKee AC , Montine TJ , Morris JC , Schneider JA , Sonnen JA , Thal DR , Trojanowski JQ , Troncoso JC , Wisniewski T , Woltjer RL , Beach TG ((2012) ) Correlation of Alzheimer disease neuropathologic changes with cognitive status: A review of the literature. J Neuropathol Exp Neurol 71: , 381. |
[75] | Chen SD , Lu JY , Li HQ , Yang YX , Jiang JH , Cui M , Zuo CT , Tan L , Dong Q , Yu JT ; Alzheimer’s Disease Neuroimaging Initiative ((2021) ) Staging tau pathology with tau PET in Alzheimer’s disease: A longitudinal study. Transl Psychiatry 11: , 483. |
[76] | Lowe VJ , Wiste HJ , Senjem ML , Weigand SD , Therneau TM , Boeve BF , Josephs KA , Fang P , Pandey MK , Murray ME , Kantarci K , Jones DT , Vemuri P , Graff-Radford J , Schwarz CG , Machulda MM , Mielke MM , Roberts RO , Knopman DS , Petersen RC , Jack CR ((2018) ) Widespread brain tau and its association with ageing, Braak stage and Alzheimer’s dementia. Brain 141: , 271–287. |
[77] | Murayama S , Saito Y ((2004) ) Symposium: Neuropathological diagnostic criteria and problems of neurodegenerative disorders Neuropathological diagnostic criteria for Alzheimer’s disease. Neuropathology 24: , 254–260. |
[78] | Mirra SS , Heyman A , McKeel D , Sumi SM , Crain BJ , Brownlee LM , Vogel FS , Hughes JP , van Belle G , Berg L , Ball MJ , Bierer LM , Claasen D , Hansen LR , Hart M , Hedreen J , Baltimore B , Hen Derson V , Hyman BT , Joachim C , Mark-Esbery W , Mar Tinez AJ , McKee A , Miller C , Moossy J , Nochlin D , Perl D , Petito C , Rao GR , Schelper RL , Slager U , Terry RD ((1991) ) The consortium to establish a registry for Alzheimer’s disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41: , 479–486. |
[79] | Upadhaya AR , Kosterin I , Kumar S , Von Arnim CAF , Yamaguchi H , Fä M , Walter J , Thal DR , Thal DR ((2014) ) Biochemical stages of amyloid-β peptide aggregation and accumulation in the human brain and their association with symptomatic and pathologically preclinical Alzheimer’s disease. Brain 137: (Pt 3), 887–903. |
[80] | Cross SS ((2013) ) Underwood’s Pathology - a clinical approach, Elsevier Ltd. |
[81] | West MJ , Coleman PD , Flood DG , Troncoso JC ((1994) ) Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer’s disease. Lancet 344: , 769–772. |
[82] | West MJ , Kawas CH , Stewart WF , Rudow GL , Troncoso JC ((2004) ) Hippocampal neurons in pre-clinical Alzheimer’s disease. Neurobiol Aging 25: , 1205–1212. |
[83] | Weiner MW , Veitch DP , Aisen PS , Beckett LA , Cairns NJ , Green RC , Harvey D , Jack CR , Jagust W , Morris JC , Petersen RC , Saykin AJ , Shaw LM , Toga AW , Trojanowski JQ ((2017) ) Recent publications from the Alzheimer’s Disease Neuroimaging Initiative: Reviewing progress toward improved AD clinical trials. Alzheimers Dement 13: , e1–e85. |
[84] | Šimić G , Babić Leko M , Wray S , Harrington CR , Delalle I , Jovanov-Milošević N , Bažadona D , Buée L , de Silva R , Di Giovanni G , Wischik CM , Hof PR ((2017) ) Monoaminergic neuropathology in Alzheimer’s disease. Prog Neurobiol 151: , 101–138. |
[85] | Grothe MJ , Heinsen H , Amaro E , Grinberg LT , Teipel SJ ((2013) ) Cognitive correlates of basal forebrain atrophy and associated cortical hypometabolism in mild cognitive impairment. Psychother Psychosom 26: , 2411–2426. |
[86] | Mah L , Binns MA , Steffens DC ((2015) ) Anxiety symptoms in amnestic mild cognitive impairment are associated with medial temporal atrophy and predict conversion to Alzheimer disease. Am J Geriatr Psychiatry 23: , 466–476. |
[87] | Marshall GA , Lorius N , Locascio JJ , Hyman BT , Rentz DM , Johnson KA , Sperling RA ((2014) ) Regional cortical thinning and cerebrospinal biomarkers predict worsening daily functioning across the Alzheimer’s disease spectrum. J Alzheimers Dis 41: , 719–728. |
[88] | Rafii MS , Taylor CS , Kim HT , Desikan RS , Fleisher AS , Katibian D , Brewer JB , Dale AM , Aisen PS ((2014) ) Neuropsychiatric symptoms and regional neocortical atrophy in mild cognitive impairment and Alzheimer’s disease. Am J Alzheimers Dis Other Demen 29: , 159–165. |
[89] | Ferreira D , Verhagen C , Hernández-Cabrera JA , Cavallin L , Guo CJ , Ekman U , Muehlboeck JS , Simmons A , Barroso J , Wahlund LO , Westman E ((2017) ) Distinct subtypes of Alzheimer’s disease based on patterns of brain atrophy: Longitudinal trajectories and clinical applications. Sci Rep 7: , 46263. |
[90] | Byun MS , Kim SE , Park J , Yi D , Choe YM , Sohn BK , Choi HJ , Baek H , Han JY , Woo JI , Lee DY ((2015) ) Heterogeneity of regional brain atrophy patterns associated with distinct progression rates in Alzheimer’s disease. PLoS One 10: , e0142756. |
[91] | Toda N , Okamura T ((2012) ) Cerebral blood flow regulation by nitric oxide in Alzheimer’s disease. J Alzheimers Dis 32: , 569–578. |
[92] | Nortley R , Korte N , Izquierdo P , Hirunpattarasilp C , Mishra A , Jaunmuktane Z , Kyrargyri V , Pfeiffer T , Khennouf L , Madry C , Gong H , Richard-Loendt A , Huang W , Saito T , Saido TC , Brandner S , Sethi H , Attwell D ((2019) ) Amyloid β oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science 365: , eaav9518. |
[93] | Alsop DC , Dai W , Grossman M , Detre JA ((2013) ) Arterial spin labeling blood flow MRI: Its role in the early characterization of Alzheimer’s disease. J Alzheimers Dis 20: , 871–880. |
[94] | Nation DA , Wierenga CE , Clark LR , Dev SI , Stricker NH , Jak AJ , Salmon DP , Delano-Wood L , Bangen KJ , Rissman RA , Liu TT , Bondi MW ((2013) ) Cortical and subcortical cerebrovascular resistance index in mild cognitive impairment and Alzheimer’s disease. J Alzheimers Dis 36: , 689–698. |
[95] | Wolters FJ , Zonneveld HI , Hofman A , van der Lugt A , Koudstaal PJ , Vernooij MW , Ikram MA ((2017) ) Cerebral perfusion and the risk of dementia. Circulation 136: , 719–728. |
[96] | Benedictus MR , Leeuwis AE , Binnewijzend MAA , Kuijer JPA , Scheltens P , Barkhof F , van der Flier WM , Prins ND ((2017) ) Lower cerebral blood flow is associated with faster cognitive decline in Alzheimer’s disease. Eur Radiol 27: , 1169–1175. |
[97] | Jeong S ((2017) ) Molecular and cellular basis of neurodegeneration in Alzheimer’s disease. Mol Cells 40: , 613–620. |
[98] | Ernst A , Alkass K , Bernard S , Salehpour M , Perl S , Tisdale J , Possnert G , Druid H , Frisén J ((2014) ) Neurogenesis in the striatum of the adult human brain. Cell 156: , 1072–1083. |
[99] | Lim DA , Alvarez-Buylla A ((2016) ) The adult ventricular-subventricular zone (V-SVZ) and olfactory bulb (OB) neurogenesis. Cold Spring Harb Perspect Biol 8: , a018820. |
[100] | Bruke SN , Barnes CA ((2006) ) Neural plasticity in the ageing brain. Nat Rev Neurosci 7: , 30–40. |
[101] | Gemma C , Bachstetter AD , Bickford PC ((2010) ) Neuron-microglia dialogue and hippocampal neurogenesis in the aged brain. Aging Dis 1: , 244. |
[102] | Scopa C , Marrocco F , Latina V , Ruggeri F , Corvaglia V , La Regina F , Ammassari-Teule M , Middei S , Amadoro G , Meli G , Scardigli R , Cattaneo A ((2020) ) Impaired adult neurogenesis is an early event in Alzheimer’s disease neurodegeneration, mediated by intracellular Aβ oligomers. Cell Death Differ 27: , 934–948. |
[103] | Lazarov O , Marr RA ((2010) ) Neurogenesis and Alzheimer’s disease: At the crossroads. Exp Neurol 223: , 267–281. |
[104] | Mu Y , Gage FH ((2011) ) Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Mol Neurodegener 6: , 85. |
[105] | Lucassen PJ , Fitzsimons CP , Salta E , Maletic-Savatic M ((2020) ) Adult neurogenesis, human after all (again): Classic, optimized, and future approaches. Behav Brain Res 381: , 112458. |
[106] | Choi SH , Tanzi RE ((2019) ) Is Alzheimer’s disease a neurogenesis disorder? Cell Stem Cell 25: , 7–8. |
[107] | Tobin MK , Musaraca K , Disouky A , Shetti A , Bheri A , Honer WG , Kim N , Dawe RJ , Bennett DA , Arfanakis K , Lazarov O ((2019) ) Human hippocampal neurogenesis persists in aged adults and Alzheimer’s disease patients HHS Public Access. Cell Stem Cell 24: , 974–982. |
[108] | Maurer MH ((2008) ) Neural stem cells: A functional definition based on proteomics. Electron J Biol 4: , 43–46. |
[109] | Moreno-Jiménez EP , Flor-García M , Terreros-Roncal J , Rábano A , Cafini F , Pallas-Bazarra N , Ávila J , Llorens-Martín M ((2019) ) Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat Med 25: , 554–560. |
[110] | Sung PS , Lin PY , Liu CH , Su HC , Tsai KJ ((2020) ) Neuroinflammation and neurogenesis in alzheimer’s disease and potential therapeutic approaches. Int J Mol Sci 21: , 701. |
[111] | Webers A , Heneka MT , Gleeson PA ((2020) ) The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer’s disease. Immunol Cell Biol 98: , 28–41. |
[112] | Sudduth TL , Schmitt FA , Nelson PT , Wilcock DM ((2013) ) Neuroinflammatory phenotype in early Alzheimer’s disease. Neurobiol Aging 34: , 1051–1059. |
[113] | Gomez-Nicola D , Boche D ((2015) ) Post-mortem analysis of neuroinflammatory changes in human Alzheimer’s disease. Alzheimers Res Ther 7: , 42. |
[114] | Zimmer ER , Leuzy A , Lessa Benedet A , Breitner J , Gauthier S , Rosa-Neto P ((2014) ) Tracking neuroinflammation in Alzheimer’s disease: The role of positron emission tomography imaging. J Neuroinflammation 11: , 120. |
[115] | Chandra A , Valkimadi P , Pagano G , Cousins O , Dervenoulas G , Politis M ((2019) ) Applications of amyloid, tau, and neuroinflammation PET imaging to Alzheimer’s disease and mild cognitive impairment. Hum Brain Mapp 40: , 5424–5442. |
[116] | Rodríguez JJ , Yeh CY , Terzieva S , Olabarria M , Kulijewicz-Nawrot M , Verkhratsky A ((2014) ) Complex and region-specific changes in astroglial markers in the aging brain. Neurobiol Aging 35: , 15–23. |
[117] | Valles SL , Burguet F , Iradi A , Aldasoro M , Vila JM , Aldasoro C , Jordá A ((2020) ) Astrocytes and inflammatory processes in Alzheimer’s disease. In Glia in Health and Disease. IntechOpen. https://doi.org/10.5772/intechopen.88701 |
[118] | Mosser DM , Edwards JP ((2008) ) Exploring the full spectrum of macrophage activation. Nat Rev Immunol 8: , 958–969. |
[119] | Martinez FO , Helming L , Gordon S ((2009) ) Alternative activation of macrophages: An immunologic functional perspective. Annu Rev Immunol 27: , 451–483. |
[120] | Liaoi YF , Wang BJ , Cheng HT , Kuo LH , Wolfe MS ((2004) ) Tumor necrosis factor-α, interleukin-1β, and interferon-γ stimulate γ-secretase-mediated cleavage of amyloid precursor protein through a JNK-dependent MAPK pathway. J Biol Chem 279: , 49523–49532. |
[121] | Li Y , Liu L , Barger SW , Griffin WST ((2003) ) Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J Neurosci 23: , 1605–1611. |
[122] | Ringheim GE , Szczepanik AM , Petko W , Burgher KL , Zhu SZ , Chao CC ((1998) ) Enhancement of beta-amyloid precursor protein transcription and expression by the soluble interleukin-6 receptor/interleukin-6 complex. Mol Brain Res 55: , 35–44. |
[123] | Guillot-Sestier MV , Doty KR , Gate D , Rodriguez J , Leung BP , Rezai-Zadeh K , Town T ((2015) ) Il10 deficiency rebalances innate immunity to mitigate Alzheimer-like pathology. Neuron 85: , 534–548. |
[124] | Tarkowski E , Andreasen N , Tarkowski A , Blennow K ((2003) ) Intrathecal inflammation precedes development of Alzheimer’s disease. J Neurol Neurosurg Psychiatry 74: , 1200–1205. |
[125] | Combs CK , Colleen Karlo J , Kao SC , Landreth GE ((2001) ) β-amyloid stimulation of microglia anti monocytes results in TNFα-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J Neurosci 21: , 1179–1188. |
[126] | Kelleher RJ , Soiza RL ((2013) ) Evidence of endothelial dysfunction in the development of Alzheimer’s disease: Is Alzheimer’s a vascular disorder? Am J Cardiovasc Dis 3: , 226. |
[127] | Montagne A , Barnes SR , Sweeney MD , Halliday MR , Sagare AP , Zhao Z , Toga AW , Jacobs RE , Liu CY , Amezcua L , Harrington MG , Chui HC , Law M , Zlokovic BV ((2015) ) Blood-brain barrier breakdown in the aging human hippocampus. Neuron 85: , 296–302. |
[128] | Montagne A , Nation DA , Pa J , Sweeney MD , Toga AW , Zlokovic BV ((2016) ) Brain imaging of neurovascular dysfunction in Alzheimer’s disease. Acta Neuropathol 131: , 687–707. |
[129] | Kisler K , Nelson AR , Montagne A , Zlokovic BV ((2017) ) Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat Rev Neurosci 18: , 419–434. |
[130] | Sweeney MD , Kisler K , Montagne A , Toga AW , Zlokovic BV ((2018) ) The role of brain vasculature in neurodegenerative disorders. Nat Neurosci 21: , 1318–1331. |
[131] | Wong KH , Riaz MK , Xie Y , Zhang X , Liu Q , Chen H , Bian Z , Chen X , Lu A , Yang Z ((2019) ) Review of current strategies for delivering Alzheimer’s disease drugs across the blood-brain barrier. Int J Mol Sci 20: , 381. |
[132] | Sweeney MD , Montagne A , Sagare AP , Nation DA , Schneider LS , Chui HC , Harrington MG , Pa J , Law M , Wang DJJ , Jacobs RE , Doubal FN , Ramirez J , Black SE , Nedergaard M , Benveniste H , Dichgans M , Iadecola C , Love S , Bath PM , Markus HS , Salman RA , Allan SM , Quinn TJ , Kalaria RN , Werring DJ , Carare RO , Touyz RM , Williams SCR , Moskowitz MA , Katusic ZS , Lutz SE , Lazarov O , Minshall RD , Rehman J , Davis TP , Wellington CL , González HM , Yuan C , Lockhart SN , Hughes TM , Chen CLH , Sachdev P , O’Brien JT , Skoog I , Pantoni L , Gustafson DR , Biessels GJ , Wallin A , Smith EE , Mok V , Wong A , Passmore P , Barkof F , Muller M , Breteler MMB , Román GC , Hamel E , Seshadri S , Gottesman RF , van Buchem MA , Arvanitakis Z , Schneider JA , Drewes LR , Hachinski V , Finch CE , Toga AW , Wardlaw JM , Zlokovic BV ((2019) ) Vascular dysfunction—The disregarded partner of Alzheimer’s disease. Alzheimers Dement 15: , 158–167. |
[133] | Montagne A , Zhao Z , Zlokovic BV ((2017) ) Alzheimer’s disease: A matter of blood-brain barrier dysfunction? J Exp Med 214: , 3151–3169. |
[134] | Van De Haar HJ , Burgmans S , Jansen JFA , Van Osch MJP , Van Buchem MA , Muller M , Hofman PAM , Verhey FRJ , Backes WH ((2016) ) Blood-brain barrier leakage in patients with early Alzheimer disease. Radiology 281: , 527–535. |
[135] | Nishitsuji K , Hosono T , Nakamura T , Bu G , Michikawa M ((2011) ) Apolipoprotein E regulates the integrity of tight junctions in an isoform-dependent manner in an in vitro blood-brain barrier model. J Biol Chem 286: , 17536–17542. |
[136] | Erickson MA , Banks WA ((2013) ) Blood-brain barrier dysfunction as a cause and consequence of Alzheimer’s disease. J Cereb Blood Flow Metab 33: , 1500–1513. |
[137] | Priller C , Bauer T , Mitteregger G , Krebs B , Kretzschmar HA , Herms J ((2006) ) Synapse formation and function is modulated by the amyloid precursor protein. J Neurosci 20: , 7951–7963. |
[138] | Turner PR , O’Connor K , Tate WP , Abraham WC ((2003) ) Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Prog Neurobiol 70: , 1–32. |
[139] | Magara F , Müller U , Li ZW , Lipp HP , Weissmann C , Stagljar M , Wolfer DP ((1999) ) Genetic background changes the pattern of forebrain commissure defects in transgenic mice underexpressing the β-amyloid-precursor protein. Proc Natl Acad Sci U S A 96: , 4656–4661. |
[140] | Vassar R , Bennett BD , Babu-Khan S , Kahn S , Mendiaz EA , Denis P , Teplow DB , Ross S , Amarante P , Loeloff R , Luo Y , Fisher S , Fuller J , Edenson S , Lile J , Jarosinski MA , Biere AL , Curran E , Burgess T , Louis JC , Collins F , Treanor J , Rogers G , Citron M ((1999) ) β-Secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286: , 735–741. |
[141] | Vassar R ((2002) ) β-secretase (BACE) as a drug target for Alzheimer’s disease. Adv Drug Deliv Rev 54: , 1589–1602. |
[142] | Roberts SB ((2002) ) γ-secretase inhibitors and Alzheimer’s disease. Adv Drug Deliv Rev 54: , 1579–1588. |
[143] | Jan A , Gokce O , Luthi-Carter R , Lashuel HA ((2008) ) The ratio of monomeric to aggregated forms of Aβ40 and Aβ42 is an important determinant of amyloid-β aggregation, fibrillogenesis, and toxicity. J Biol Chem 283: , 28176–28189. |
[144] | Hartmann T , Bieger SC , Brühl B , Tienari PJ , Ida N , Allsop D , Roberts GW , Masters CL , Dotti CG , Unsicker K , Beyreuther K ((1997) ) Distinct sites of intracellular production for Alzheimer’s disease Aβ40/42 amyloid peptides. Nat Med 3: , 1016–1020. |
[145] | Bitan G , Kirkitadze MD , Lomakin A , Vollers SS , Benedek GB , Teplow DB ((2003) ) Amyloid β-protein (Aβ) assembly: Aβ40 and Aβ42 oligomerize through distinct pathways. Proc Natl Acad Sci U S A 100: , 330–335. |
[146] | Yankner BA , Dawes LR , Fisher S , Villa-Komaroff L , Oster-Granite ML , Neve RL ((1989) ) Neurotoxicity of a fragment of the amyloid precursor associated with Alzheimer’s disease. Science 245: , 417–420. |
[147] | Deshpande A , Mina E , Glabe C , Busciglio J ((2006) ) Different conformations of amyloid β induce neurotoxicity by distinct mechanisms in human cortical neurons. J Neurosci 26: , 6011–6018. |
[148] | Shankar GM , Li S , Mehta TH , Garcia-Munoz A , Shepardson NE , Smith I , Brett FM , Farrell MA , Rowan MJ , Lemere CA , Regan CM , Walsh DM , Sabatini BL , Selkoe DJ ((2008) ) Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med 14: , 837–842. |
[149] | Hölttä M , Hansson O , Andreasson U , Hertze J , Minthon L , Nägga K , Andreasen N , Zetterberg H , Blennow K ((2013) ) Evaluating amyloid-β oligomers in cerebrospinal fluid as a biomarker for Alzheimer’s disease. PLoS One 8: , e67898. |
[150] | Herskovits AZ , Locascio JJ , Peskind ER , Li G , Hyman BT ((2013) ) A Luminex assay detects amyloid β oligomers in Alzheimer’s disease cerebrospinal fluid. PLoS One 8: , e67898. |
[151] | Haass C , Selkoe DJ ((2007) ) Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat Rev Mol Cell Biol 8: , 101–112. |
[152] | Condello C , Stöehr J ((2018) ) Aβ propagation and strains: Implications for the phenotypic diversity in Alzheimer’s disease. Neurobiol Dis 109: , 191–200. |
[153] | Liyanage SI , Weaver DF ((2019) ) Misfolded proteins as a therapeutic target in Alzheimer’s disease. Adv Protein Chem Struct Biol 118: , 371–411. |
[154] | Nussbaum JM , Seward ME , Bloom GS ((2013) ) Alzheimer disease - A tale of two prions. Prion 7: , 14–19. |
[155] | Klunk WE , Engler H , Nordberg A , Wang Y , Blomqvist G , Holt DP , Bergström M , Savitcheva I , Huang GF , Estrada S , Ausén B , Debnath ML , Barletta J , Price JC , Sandell J , Lopresti BJ , Wall A , Koivisto P , Antoni G , Mathis CA , Långström B ((2004) ) Imaging brain amyloid in Alzheimer’s disease with Pittsburgh compound-B. Ann Neurol 55: , 306–319. |
[156] | Giannakopoulos P , Herrmann FR , Bussière T , Bouras C , Kövari E , Perl DP , Morrison JH , Gold G , Hof PR ((2003) ) Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 60: , 1495–1500. |
[157] | Pike KE , Savage G , Villemagne VL , Ng S , Moss SA , Maruff P , Mathis CA , Klunk WE , Masters CL , Rowe CC , Rowe C ((2007) ) β-amyloid imaging and memory in non-demented individuals: Evidence for preclinical Alzheimer’s disease. Brain 130: , 2837–2844. |
[158] | Goedert M , Spillantini MG , Jakes R , Rutherford D , Crowther RA ((1989) ) Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 3: , 519–526. |
[159] | Gu Y , Oyama F , Ihara Y ((1996) ) τ is widely expressed in rat tissues. J Neurochem 67: , 1235–1244. |
[160] | Pierre M , Nunez J ((1983) ) Multisite phosphorylation of tau proteins from rat brain. Biochem Biophys Res Commun 115: , 212–219. |
[161] | Mudher A , Lovestone S ((2002) ) Alzheimer’s disease - Do tauists and baptists finally shake hands? Trends Neurosci 25: , 22–26. |
[162] | Rad SK , Arya A , Karimian H , Madhavan P , Rizwan F , Koshy S , Prabhu G ((2018) ) Mechanism involved in insulin resistance via accumulation of β-amyloid and neurofibrillary tangles: Link between type 2 diabetes and Alzheimer’s disease. Drug Des Devel Ther 12: , 3999–4021. |
[163] | Alonso ADC , Grundke-Iqbal I , Barra HS , Iqbal K ((1997) ) Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: Sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc Natl Acad Sci U S A 94: , 298–303. |
[164] | Mandelkow EM , Stamer K , Vogel R , Thies E , Mandelkow E ((2003) ) Clogging of axons by tau, inhibition of axonal traffic and starvation of synapses. Neurobiol Aging 24: , 1079–1085. |
[165] | Nelson PT , Braak H , Markesbery WR ((2009) ) Neuropathology and cognitive impairment in Alzheimer disease: A complex but coherent relationship. J Neuropathol Exp Neurol 68: , 14. |
[166] | Snowdon DA ((1997) ) Aging and Alzheimer’s disease: Lessons from the Nun Study. Gerontologist 37: , 150–156. |
[167] | Snowdon DA , Nun Study ((2003) ) Healthy aging and dementia: Findings from the Nun Study. Ann Intern Med 139(5 Pt 2): , 450–454. |
[168] | Riley KP , Snowdon DA , Desrosiers MF , Markesbery WR ((2005) ) Early life linguistic ability, late life cognitive function, and neuropathology: Findings from the Nun Study. Neurobiol Aging 26: , 341–347. |
[169] | Bennett DA , Schneider JA , Arvanitakis Z , Wilson RS ((2013) ) Overview and findings from the Religious Orders Study. Curr Alzheimer Res 9: , 628–645. |
[170] | Ellis KA , Bush AI , Darby D , De Fazio D , Foster J , Hudson P , Lautenschlager NT , Lenzo N , Martins RN , Maruff P , Masters C , Milner A , Pike K , Rowe C , Savage G , Szoeke C , Taddel K , Villemagne V , Woodward M , Ames D ((2009) ) The Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging: Methodology and baseline characteristics of 1112 individuals recruited for a longitudinal study of Alzheimer’s disease. Int Psychogeriatr 21: , 672–687. |
[171] | Clark CM , Davatzikos C , Borthakur A , Newberg A , Leight S , Lee M-Y , Trojanowski JQ ((2008) ) Biomarkers for early detection of Alzheimer pathology. Neurosignals 16: , 11–18. |
[172] | Dubois B , Epelbaum S , Nyasse F , Bakardjian H , Gagliardi G , Uspenskaya O , Houot M , Lista S , Cacciamani F , Potier M-C , Bertrand A , Lamari F , Benali H , Mangin J-F , Colliot O , Genthon R , Habert M-O , Hampel H ((2018) ) Cognitive and neuroimaging features and brain β-amyloidosis in individuals at risk of Alzheimer’s disease (INSIGHT-preAD): A longitudinal observational study. Lancet Neurol 17: , 335–346. |
[173] | Xicota L , Ichou F , Lejeune FX , Colsch B , Tenenhaus A , Leroy I , Fontaine G , Lhomme M , Bertin H , Habert MO , Epelbaum S , Dubois B , Mochel F , Potier MC ((2019) ) Multi-omics signature of brain amyloid deposition in asymptomatic individuals at-risk for Alzheimer’s disease: The INSIGHT-preAD study. EBioMedicine 47: , 518–528. |
[174] | Rossini PM , Miraglia F , Alù F , Cotelli M , Ferreri F , Iorio R Di , Iodice F , Vecchio F ((2020) ) Neurophysiological hallmarks of neurodegenerative cognitive decline: The study of brain connectivity as a biomarker of early dementia. J Pers Med 10: , 34. |
[175] | Stern Y ((2012) ) Cognitive reserve in ageing and Alzheimer’s disease. Lancet Neurol 11: , 1006–1012. |
[176] | Dang C , Yassi N , Harrington KD , Xia Y , Lim YY , Ames D , Laws SM , Hickey M , Rainey-Smith S , Sohrabi HR , Doecke JD , Fripp J , Salvado O , Snyder PJ , Weinborn M , Villemagne VL , Rowe CC , Masters CL , Maruff P , Research Group A ((2019) ) Special Section: Are the rates of age-and amyloid β-associated cortical atrophy influenced by sustained exceptional cognitive functioning in older adults? Rates of age-and amyloid b-associated cortical atrophy in older adults with superior memory performa. Alzheimers Dement (Amst) 11: , 566–575. |
[177] | Gefen T , Peterson M , Papastefan ST , Martersteck A , Whitney K , Rademaker A , Bigio EH , Weintraub S , Rogalski E , Mesulam M-M , Geula C ((2015) ) Morphometric and histologic substrates of cingulate integrity in elders with exceptional memory capacity. J Neurosci 35: , 1781–1797. |
[178] | Barroeta-Espar I , Weinstock LD , Perez-Nievas BG , Meltzer AC , Siao Tick Chong M , Amaral AC , Murray ME , Moulder KL , Morris JC , Cairns NJ , Parisi JE , Lowe VJ , Petersen RC , Kofler J , Ikonomovic MD , López O , Klunk WE , Mayeux RP , Frosch MP , Wood LB , Gomez-Isla T ((2019) ) Distinct cytokine profiles in human brains resilient to Alzheimer’s pathology. Neurobiol Dis 121: , 327–337. |
[179] | Driscoll I , Troncoso J ((2011) ) Asymptomatic Alzheimer’s disease: A prodrome or a state of resilience? Curr Alzheimer Res 8: , 330–5. |
[180] | Seyfried NT , Dammer EB , Swarup V , Nandakumar D , Duong DM , Yin L , Deng Q , Nguyen T , Hales CM , Wingo T , Glass J , Gearing M , Thambisetty M , Troncoso JC , Geschwind DH , Lah JJ , Levey AI ((2017) ) A multi-network approach identifies protein-specific co-expression in asymptomatic and symptomatic Alzheimer’s disease. Cell Syst 4: , 60–72.e4. |
[181] | Zolochevska O , Taglialatela G ((2016) ) Non-demented individuals with Alzheimer’s disease neuropathology: Resistance to cognitive decline may reveal new treatment strategies. Curr Pharm Des 22: , 4063–4068. |
[182] | Zolochevska O , Taglialatela G ((2020) ) Selected microRNAs increase synaptic resilience to the damaging binding of the Alzheimer’s disease amyloid beta oligomers. Mol Neurobiol 57: , 2232–2243. |
[183] | Galvin JE , Sadowsky CH ((2012) ) Practical guidelines for the recognition and diagnosis of dementia. J Am Board Fam Med 25: , 367–382. |
[184] | Ljubenkov PA , Geschwind MD ((2016) ) Dementia. Semin Neurol 36: , 397–404. |
[185] | Bennett DA , Schneider JA , Arvanitakis Z , Kelly JF , Aggarwal NT , Shah RC , Wilson RS ((2006) ) Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology 66: , 1837–1844. |
[186] | Sheline YI , Raichle ME , Snyder AZ , Morris JC , Head D , Wang S , Mintun MA ((2010) ) Amyloid plaques disrupt resting state default mode network connectivity in cognitively normal elderly. Biol Psychiatry 67: , 584–587. |
[187] | De Meyer G , Shapiro F , Vanderstichele H , Vanmechelen E , Engelborghs S , De Deyn PP , Coart E , Hansson O , Minthon L , Zetterberg H , Blennow K , Shaw L , Trojanowski JQ ((2010) ) Diagnosis-independent Alzheimer disease biomarker signature in cognitively normal elderly people. Arch Neurol 67: , 949–956. |
[188] | Rowe CC , Ellis KA , Rimajova M , Bourgeat P , Pike KE , Jones G , Fripp J , Tochon-Danguy H , Morandeau L , O’Keefe G , Price R , Raniga P , Robins P , Acosta O , Lenzo N , Szoeke C , Salvado O , Head R , Martins R , Masters CL , Ames D , Villemagne VL ((2010) ) Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging 31: , 1275–1283. |
[189] | O’brien RJ , Resnick SM , Zonderman AB , Ferrucci L , Crain BJ , Pletnikova O , Rudow G , Iacono D , Riudavets MA , Driscoll I , Price DL , Martin LJ , Troncoso JC ((2009) ) Neuropathologic studies of the Baltimore Longitudinal Study of Aging (BLSA). J Alzheimers Dis 18: , 665–675. |
[190] | Driscoll I , Resnick SM , Troncoso JC , An Y , O’brien R , Zonderman AB ((2006) ) Impact of Alzheimer’s pathology on cognitive trajectories in nondemented elderly. Ann Neurol 60: , 688–695. |
[191] | Hughes CP , Berg L , Danziger WL , Coben L , Martin RL ((1982) ) A new clinical scale for the staging of dementia. Br J Psychiatry 140: , 566–572. |
[192] | Folstein MF , Folstein SE , McHugh PR ((1975) ) “Mini-mental state”: A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12: , 189–198. |
[193] | Abner EL , Kryscio RJ , Schmitt FA , Santacruz KS , Jicha GA , Lin Y , Neltner JM , Smith CD , Van Eldik LJ , Nelson PT ((2011) ) “End-stage” neurofibrillary tangle pathology in preclinical Alzheimer’s disease: Fact or fiction? J Alzheimers Dis 25: , 453. |
[194] | Bennett DA , Wilson RS , Schneider JA , Evans DA , Mendes de Leon CF , Arnold SE , Barnes LL , Bienias JL ((2003) ) Education modifies the relation of AD pathology to level of cognitive function in older persons. Neurology 60: , 1909–1915. |
[195] | Coffey CE , Saxton JA , Ratcliff G , Bryan RN , Lucke JF ((1999) ) Relation of education to brain size in normal aging: Implications for the reserve hypothesis. Neurology 53: , 189–196. |
[196] | Groot C , Van Loenhoud AC , Barkhof F , Van Berckel BNM , Koene T , Teunissen CC , Scheltens P , Van Der Flier WM , Ossenkoppele R ((2018) ) Differential effects of cognitive reserve and brain reserve on cognition in Alzheimer disease. Neurology 90: , e149–e156. |
[197] | Iacono D , Markesbery WR , Gross M , Pletnikova O , Rudow G , Zandi BP , Troncoso JC ((2009) ) The Nun Study: Clinically silent AD, neuronal hypertrophy, and linguistic skills in early life. Neurology 73: , 665–673. |
[198] | Roe CM , Xiong C , Miller JP , Morris JC ((2007) ) Education and Alzheimer disease without dementia: Support for the cognitive reserve hypothesis. Neurology 68: , 223–228. |
[199] | Soldan A , Pettigrew C , Cai Q , Wang J , Wang MC , Moghekar A , Miller MI , Albert M ((2017) ) Cognitive reserve and long-term change in cognition in aging and preclinical Alzheimer’s disease. Neurobiol Aging 60: , 164–172. |
[200] | Zhang M , Katzman R , Salmon D , Jin H , Cai G , Wang Z , Qu G , Grant I , Yu E , Levy P , Klauber MR , Liu WT ((1990) ) The prevalence of dementia and Alzheimer’s disease in Shanghai, China: Impact of age, gender, and education. Ann Neurol 27: , 428–437. |
[201] | Ott A , Breteler MM , Van Harskamp F , Claus jules J , Van Der Cammen TJM , Grobbee DE , Hofman A ((1995) ) Prevalence of Alzheimer’s disease and vascular dementia: Association With education. The Rotterdam study. BMJ 310: , 970. |
[202] | Xu W , Tan L , Wang HF , Tan MS , Tan L , Li JQ , Zhao QF , Yu JT ((2016) ) Education and risk of dementia: Dose-response meta-analysis of prospective cohort studies. Mol Neurobiol 53: , 3113–3123. |
[203] | Balduino E , Ribeiro De Melo BA , De Sousa Mota Da Silva L , Martinelli JE , Cecato JF ((2019) ) The “SuperAgers” construct in clinical practice: Neuropsychological assessment of illiterate and educated elderly. Int Psychogeriatr 32: , 191–198. |
[204] | Ngandu T , Von Strauss E , Helkala EL , Winblad B , Nissinen A , Tuomilehto J , Soininen H , Kivipelto M ((2007) ) Education and dementia: What lies behind the association? Neurology 69: , 1442–1450. |
[205] | Bennett DA , Schneider JA , Wilson RS , Bienias JL , Arnold SE ((2005) ) Education modifies the association of amyloid but not tangles with cognitive function. Neurology 65: , 953–955. |
[206] | Thorvaldsson V , Skoog I , Johansson B ((2017) ) IQ as moderator of terminal decline in perceptual and motor speed, spatial, and verbal ability: Testing the cognitive reserve hypothesis in a population-based sample followed from age 70 until death. Psychol Aging 32: , 148–157. |
[207] | Wilson RS , Yu L , Leurgans SE , Bennett DA , Boyle PA ((2020) ) Proportion of cognitive loss attributable to terminal decline. Neurology 94: , e42–e50. |
[208] | Shiba K , Daoud A , Hikichi H , Yazawa A , Aida J , Kondo K , Kawachi I ((2021) ) Heterogeneity in cognitive disability after a major disaster: A natural experiment study. Sci Adv 7: , eabj2610. |
[209] | Gosche KM , Mortimer JA , Smith CD , Markesbery WR , Snowdon DA ((2002) ) Hippocampal volume as an index of Alzheimer neuropathology: Findings from the Nun study. Neurology 58: , 1476–1482. |
[210] | Erten-Lyons D , Woltjer RL , Dodge H , Nixon R , Vorobik R , Calvert BJF , Leahy M , Montine T , Kaye J ((2009) ) Factors associated with resistance to dementia despite high Alzheimer disease pathology. Neurology 72: , 354–360. |
[211] | Vandenberghe R ((2014) ) The relationship between amyloid deposition, neurodegeneration, and cognitive decline in dementia topical collection on neuroimaging. Curr Neurol Neurosci Rep 14: , 498. |
[212] | Guo Y , Xu W , Li JQ , Ou YN , Shen XN , Huang YY , Dong Q , Tan L , Yu JT ((2019) ) Genome-wide association study of hippocampal atrophy rate in non-demented elders. Aging (Albany NY) 11: , 10468–10484. |
[213] | Fiford CM , Manning EN , Bartlett JW , Cash DM , Malone IB , Ridgway GR , Lehmann M , Leung KK , Sudre CH , Ourselin S , Biessels GJ , Carmichael OT , Fox NC , Cardoso MJ , Barnes J ((2017) ) White matter hyperintensities are associated with disproportionate progressive hippocampal atrophy. Hippocampus 27: , 249–262. |
[214] | Fratiglioni L , Wang H-X ((2007) ) Brain Reserve Hypothesis in Dementia, IOS Press, Stockholm. |
[215] | Chételat G , Villemagne VL , Pike KE , Baron J-C , Bourgeat P , Jones G , Faux NG , Ellis KA , Salvado O , Szoeke C , Martins RN , Ames D , Masters CL , Rowe CC ((2010) ) Larger temporal volume in elderly with high versus low beta-amyloid deposition. Brain 133: , 3349–3358. |
[216] | Harrison TM , Weintraub S , Mesulam M-M , Rogalski E ((2012) ) Superior memory and higher cortical volumes in unusually successful cognitive aging. J Int Neuropsychol Soc 18: , 1081–1085. |
[217] | Clark LR , Nation DA , Wierenga CE , Bangen KJ , Dev SI , Shin DD , Delano-Wood L , Liu TT , Rissman RA , Bondi MW ((2015) ) Elevated cerebrovascular resistance index is associated with cognitive dysfunction in the very-old. Alzheimers Res Ther 7: , 3. |
[218] | Watson KK , Jones TK , Allman JM ((2006) ) Dendritic architecture of the von Economo neurons. Neuroscience 141: , 1107–1112. |
[219] | von Economo CF , Koskinas GN ((1925) ) Die cytoarchitektonik der hirnrinde des erwachsenen menschen, Springer. |
[220] | Tan RH , Yang Y , Mccann H , Shepherd C , Halliday GM ((2019) ) von Economo neurons in behavioral variant frontotemporal dementia with underlying Alzheimer’s disease. J Alzheimers Dis 69: , 963–967. |
[221] | Iacono D , O’Brien R , Resnick SM , Zonderman AB , Pletnikova O , Rudow G , An Y , West MJ , Crain B , Troncoso JC ((2008) ) Neuronal hypertrophy in asymptomatic Alzheimer disease. J Neuropathol Exp Neurol 67: , 578–589. |
[222] | Riudavets MA , Iacono D , Resnick SM , O’Brien R , Zonderman AB , Martin LJ , Rudow G , Pletnikova O , Troncoso JC ((2007) ) Resistance to Alzheimer’s pathology is associated with nuclear hypertrophy in neurons. Neurobiol Aging 28: , 1484–1492. |
[223] | Micci M-A , Krishnan B , Bishop E , Zhang W-R , Guptarak J , Grant A , Zolochevska O , Tumurbaatar B , Franklin W , Marino C , Widen SG , Luthra A , Kernie SG , Taglialatela G ((2019) ) Hippocampal stem cells promotes synaptic resistance to the dysfunctional impact of amyloid beta oligomers via secreted exosomes. Mol Neurodegener 14: , 25. |
[224] | Vlassov AV , Magdaleno S , Setterquist R , Conrad R ((2012) ) Exosomes: Current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochim Biophys Acta 1820: , 940–948. |
[225] | Xiao T , Zhang W , Jiao B , Pan C-Z , Liu X , Shen L ((2017) ) The role of exosomes in the pathogenesis of Alzheimer’ disease. Transl Neurodegener 6: , 3. |
[226] | Melah KE , Yuan-Fu Lu S , Hoscheidt SM , Alexander AL , Adluru N , Destiche DJ , Carlsson CM , Zetterberg H , Blennow K , Okonkwo OC , Gleason CE , Maritza Dowling N , Bratzke LC , Rowley HA , Sager MA , Asthana S , Johnson SC , Bendlin BB ((2016) ) CSF markers of Alzheimer’s pathology and microglial activation are associated with altered white matter microstructure in asymptomatic adults at risk for Alzheimer’s disease HHS Public Access. J Alzheimers Dis 50: , 873–886. |
[227] | Gefen T , Kim G , Bolbolan K , Geoly A , Ohm D , Oboudiyat C , Shahidehpour R , Rademaker A , Weintraub S , Bigio EH , Mesulam MM , Rogalski E , Geula C ((2019) ) Activated microglia in cortical white matter across cognitive aging trajectories. Front Aging Neurosci 11: , 94. |
[228] | Richardson PM ((1991) ) Neurotrophic factors in regeneration. Curr Opin Neurobiol 1: , 401–406. |
[229] | Fagan AM , Xiong C , Jasielec MS , Bateman RJ , Goate AM , Benzinger TLS , Ghetti B , Martins RN , Masters CL , Mayeux R , Ringman JM , Rossor MN , Salloway S , Schofield PR , Sperling RA , Marcus D , Cairns NJ , Buckles VD , Ladenson JH , Morris JC , Holtzman DM , Dominantly Inherited Alzheimer Network ((2014) ) Longitudinal change in CSF biomarkers in autosomal-dominant Alzheimer disease. Sci Transl Med 6: , 226ra30. |
[230] | Bjorklund NL , Reese LC , Sadagoparamanujam V-M , Ghirardi V , Woltjer RL , Taglialatela G ((2012) ) Absence of amyloid β oligomers at the postsynapse and regulated synaptic Zn2+in cognitively intact aged individuals with Alzheimer’s disease neuropathology. Mol Neurodegener 7: , 23. |
[231] | Bilousova T , Miller CA , Poon WW , Vinters HV , Corrada M , Kawas C , Hayden EY , Teplow DB , Glabe C , Albay R , Cole GM , Teng E , Gylys KH ((2016) ) Synaptic amyloid-β oligomers precede p-Tau and differentiate high pathology control cases. Am J Pathol 186: , 185–198. |
[232] | Durakoglugil MS , Chen Y , White CL , Kavalali ET , Herz J ((2009) ) Reelin signaling antagonizes β-amyloid at the synapse. Proc Natl Acad Sci U S A 106: , 15938–15943. |
[233] | Kramer PL , Xu H , Woltjer RL , Westaway SK , Clark D , Erten-Lyons D , Kaye JA , Welsh-Bohmer KA , Troncoso JC , Markesbery WR , Petersen RC , Turner RS , Kukull WA , Bennett DA , Galasko D , Morris JC , Ott J ((2011) ) Alzheimer disease pathology in cognitively healthy elderly: A genome-wide study. Neurobiol Aging 32: , 2113–2122. |
[234] | Botella-López A , Burgaya F , Gavín R , García-Ayllón MS , Gómez-Tortosa E , Peña-Casanova J , Ureña JM , Del Río JA , Blesa R , Soriano E , Sáez-Valero J ((2006) ) Reelin expression and glycosylation patterns are altered in Alzheimer’s disease. Proc Natl Acad Sci U S A 103: , 5573–5578. |
[235] | De Felice FG , Vieira MNN , Bomfim TR , Decker H , Velasco PT , Lambert MP , Viola KL , Zhao WQ , Ferreira ST , Klein WL ((2009) ) Protection of synapses against Alzheimer’s-linked toxins: Insulin signaling prevents the pathogenic binding of Aβ oligomers. Proc Natl Acad Sci U S A 106: , 1971–1976. |
[236] | Boutajangout A , Boom A , Leroy K , Brion JP ((2004) ) Expression of tau mRNA and soluble tau isoforms in affected and non-affected brain areas in Alzheimer’s disease. FEBS Lett 576: , 183–189. |
[237] | Liang WS , Dunckley T , Beach TG , Grover A , Mastroeni D , Ramsey K , Caselli RJ , Kukull WA , McKeel D , Morris JC , Hulette CM , Schmechel D , Reiman EM , Rogers J , Stephan DA ((2010) ) Neuronal gene expression in non-demented individuals with intermediate Alzheimer’s disease neuropathology. Neurobiol Aging 31: , 549–566. |
[238] | van der Lee SJ , Conway OJ , Jansen I , Carrasquillo MM , Kleineidam L , van den Akker E , Hernández I , van Eijk KR , Stringa N , Chen JA , Zettergren A , Andlauer TFM , Diez-Fairen M , Simon-Sanchez J , Lleó A , Zetterberg H , Nygaard M , Blauwendraat C , Savage JE , Mengel-From J , Moreno-Grau S , Wagner M , Fortea J , Keogh MJ , Blennow K , Skoog I , Friese MA , Pletnikova O , Zulaica M , Lage C , de Rojas I , Riedel-Heller S , Illán-Gala I , Wei W , Jeune B , Orellana A , Then Bergh F , Wang X , Hulsman M , Beker N , Tesi N , Morris CM , Indakoetxea B , Collij LE , Scherer M , Morenas-Rodríguez E , Ironside JW , van Berckel BNM , Alcolea D , Wiendl H , Strickland SL , Pastor P , Rodríguez Rodríguez E , Boeve BF , Petersen RC , Ferman TJ , van Gerpen JA , Reinders MJT , Uitti RJ , Tárraga L , Maier W , Dols-Icardo O , Kawalia A , Dalmasso MC , Boada M , Zettl UK , van Schoor NM , Beekman M , Allen M , Masliah E , de Munain AL , Pantelyat A , Wszolek ZK , Ross OA , Dickson DW , Graff-Radford NR , Knopman D , Rademakers R , Lemstra AW , Pijnenburg YAL , Scheltens P , Gasser T , Chinnery PF , Hemmer B , Huisman MA , Troncoso J , Moreno F , Nohr EA , Sørensen TIA , Heutink P , Sánchez-Juan P , Posthuma D , Coppola G , Karydas AM , Varpetian A , Foroud TM , Levey AI , Kukull WA , Mendez MF , Ringman J , Chui H , Cotman C , DeCarli C , Miller BL , Geschwind DH , Clarimón J , Christensen K , Ertekin-Taner N , Scholz SW , Ramirez A , Ruiz A , Slagboom E , van der Flier WM , Holstege H ((2019) ) A nonsynonymous mutation in PLCG2 reduces the risk of Alzheimer’s disease, dementia with Lewy bodies and frontotemporal dementia, and increases the likelihood of longevity. Acta Neuropathol 138: , 237–250. |
[239] | Hofman A , Murad SD , Van Duijn CM , Franco OH , Goedegebure A , Arfan Ikram M , Klaver CCW , Nijsten TEC , Peeters RP , Stricker BHC , Tiemeier HW , Uitterlinden AG , Vernooij MW ((2013) ) The Rotterdam Study: 2014 objectives and design update. Eur J Epidemiol 28: , 889–926. |
[240] | Van Der Flier WM , Scheltens P ((2018) ) Amsterdam dementia cohort: Performing research to optimize care. J Alzheimers Dis 62: , 1091–1111. |
[241] | Goldberg TE ((2019) ) Comments about SuperAging and SuperAgers. Alzheimers Dement (Amst) 11: , 564–565. |
[242] | Babiloni C , Lopez S , Del Percio C , Noce G , Pascarelli MT , Lizio R , Teipel SJ , González-Escamilla G , Bakardjian H , George N , Cavedo E , Lista S , Chiesa PA , Vergallo A , Lemercier P , Spinelli G , Grothe MJ , Potier MC , Stocchi F , Ferri R , Habert MO , Fraga FJ , Dubois B , Hampel H ((2020) ) Resting-state posterior alpha rhythms are abnormal in subjective memory complaint seniors with preclinical Alzheimer’s neuropathology and high education level: The INSIGHT-preAD study. Neurobiol Aging 90: , 43–59. |


