
Cerebral Amyloid Angiopathy in a Mouse Model of Alzheimer’s Disease Associates with Upregulated Angiopoietin and Downregulated Hypoxia-Inducible Factor
Abstract
Background:
Vascular pathology is a common feature in patients with advanced Alzheimer’s disease, with cerebral amyloid angiopathy (CAA) and microvascular changes commonly observed at autopsies and in genetic mouse models. However, despite a plethora of studies addressing the possible impact of CAA on brain vasculature, results have remained contradictory, showing reduced, unchanged, or even increased capillary densities in human and rodent brains overexpressing amyloid-β in Alzheimer’s disease and Down’s syndrome.
Objective:
We asked if CAA is associated with changes in angiogenetic factors or receptors and if so, whether this would translate into morphological alterations in pericyte coverage and vessel density.
Methods:
We utilized the transgenic mice carrying the Arctic (E693G) and Swedish (KM670/6701NL) amyloid precursor protein which develop severe CAA in addition to parenchymal plaques.
Results:
The main finding of the present study was that CAA in Tg-ArcSwe mice is associated with upregulated angiopoietin and downregulated hypoxia-inducible factor. In the same mice, we combined immunohistochemistry and electron microscopy to quantify the extent of CAA and investigate to which degree vessels associated with amyloid plaques were pathologically affected. We found that despite a severe amount of CAA and alterations in several angiogenetic factors in Tg-ArcSwe mice, this was not translated into significant morphological alterations like changes in pericyte coverage or vessel density.
Conclusion:
Our data suggest that CAA does not impact vascular density but might affect capillary turnover by causing changes in the expression levels of angiogenetic factors.
INTRODUCTION
Alzheimer’s disease (AD) is the leading cause of dementia, currently estimated to affect more than 50 million people worldwide, with numbers rapidly increasing [1]. AD is characterized by an insidious onset and neuropathological features developing decades before symptoms of memory loss, cognitive decline, and behavioral changes occur [2]. The amyloid cascade hypothesis, dominating the field since its proposal in 1992 [3], holds that cleavage of amyloid-β protein precursor (AβPP) into amyloid-β (Aβ) initiates a pathological cascade, where Aβ aggregates into Aβ plaques in the brain parenchyma, in turn causing extensive loss of neurons, and eventually of cognitive function. Even though numerous studies point to a strong involvement of Aβ in the development of AD, alternative pathophysiological mechanisms must be considered. Since vascular abnormalities are commonly observed in AD brains [4, 5] several studies have explored the vascular components of the disease. Early anatomical studies in human AD cases report abnormal tortuous, kinking, looping, twisting, spiraling, bundle forming, and knob-like structures in vessels [6–10]. Cerebral amyloid angiopathy (CAA), characterized by accumulation of Aβ around cerebral blood vessels, is reported in approximately 90%of AD patients [11, 12]. Epidemiological data suggest that vascular factors like mid-life hypertension, atherosclerosis, and diabetes may increase AD risk, and several reports indicate that the first change in AD is a decrease of cerebral blood flow [13–15]. Angiogenetic changes, aberrant vascular cell function, reduced vascular coverage, and a loss of blood-brain barrier integrity may be consequences of amyloid toxicity, but may also contribute to increased Aβ deposition in the parenchyma and around vessels in a vicious cycle that enhances disease progression [13, 16]. However, attempts to study these vascular changes in both rodents and humans have yielded contradictory results, in particular with respect to angiogenesis (reviewed in [13]).
Angiogenesis is a complex process involving both hypoxia-inducible factor (HIF)-dependent angiogenetic factors (e.g., vascular endothelial growth factor (VEGF) and its tyrosine kinase receptor; VEGFR-1, angiopoietins (ANGPT-1 and ANGPT-2), and the endothelial tyrosine kinase receptor, TIE-2 and the VEGF co-receptor neuropilin; NRP-1) and HIF-independent angiogenetic factors (e.g., vascular endothelial growth factor receptor (VEGFR-2)). These factors are crucial in controlling and timing regenerative angiogenesis in the adult rodent brain [17–19]. Ubiquitously expressed hypoxia-inducible factor-1, alpha subunit (HIF- 1α) is involved in the cellular adaptation to low oxygen tension and has evoked interest for its potential neuroprotective role [16]. HIF- 1α seems to be a crucial compensatory mechanism against overexpression of AβPP and diminished clearance of Aβ. While recent experimental studies suggest a neuroprotective effect of stabilizing HIFs, there is limited knowledge of their role in AD [16].
AD has been associated with pericyte degeneration, which might impact important brain functions and cause blood-brain barrier leakage, reduced blood flow, and aberrant angiogenesis [20, 21]. Pericytes play an important dynamic and at times opposing role in angiogenesis [20], while reduced blood flow is an early marker of AD [13, 22]. Recent studies have indicated that this is caused by constriction of capillaries by contractile pericytes, probably evoked by oligomeric Aβ [14, 23].
The Tg-ArcSwe mouse model carries a human AβPP cDNA with the Arctic (p. E693G) and Swedish (p. KM670/671NL) mutations (Tg(Thy1-APPSweArc)BLngn; MGI: 4420229), and develops severe CAA from around 8 months in addition to parenchymal plaques (between the age 5-6 months) [24, 25]. The Swedish mutation is located immediately adjacent to the β-secretase site in AβPP [26], while the Arctic mutation is located within the Aβ sequence. A pathological mutation within the Aβ sequence, like the Dutch (E693Q) and Flemish (A692G) mutations, generally causes a phenotype slightly different from AD, with intracerebral hemorrhages in addition to massive amyloid accumulation in cerebral blood vessel walls and parenchymal amyloid plaques [27]. However, despite a vast amount of CAA being observed in the Tg-ArcSwe model [28], intracerebral hemorrhages, are not observed [27]. This makes the Tg-ArcSwe model a promising tool for assessing the impact of CAA on angiogenesis and vascular density and turnover.
With the aim of arriving at a better understanding of AD, we set out to test if CAA in Tg-ArcSwe is associated with changes in angiogenetic factors or receptors and morphological changes in pericyte coverage and vascular density. To address these questions, we utilized immunohistochemistry at the light microscopic and electron microscopic level combined with unbiased stereology and qPCR analysis methods.
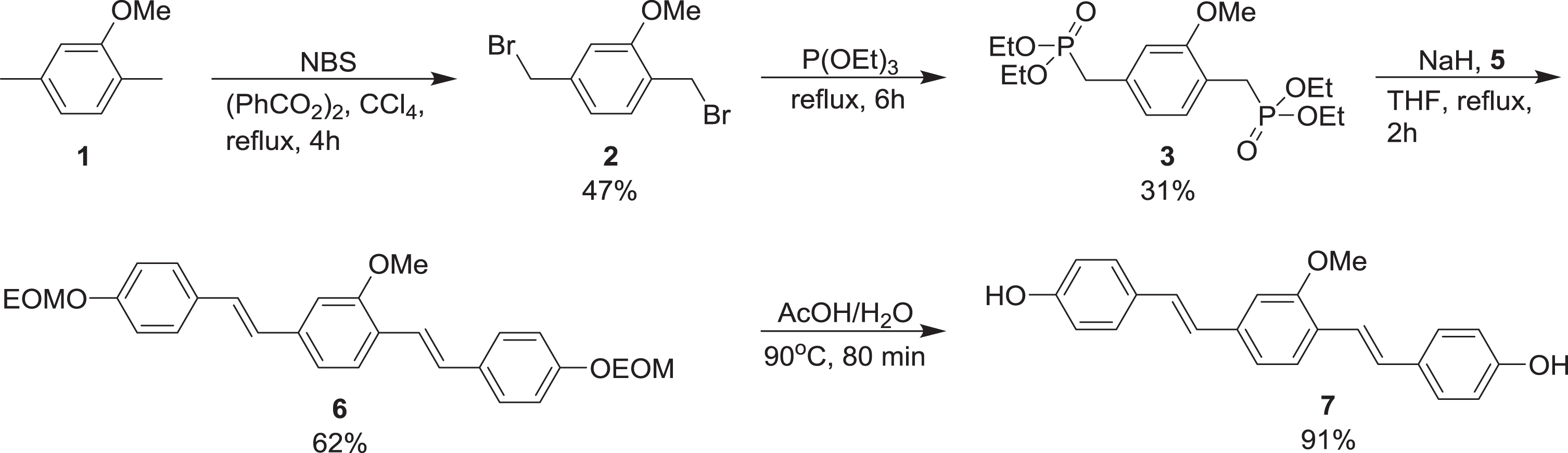
Scheme 1
Synthesis of Methoxy-X04. 5, 4-(ethoxymethoxy)benzaldehyde; EOM, ethoxymethyl
MATERIALS AND METHODS
Animals
Thirty mice carrying a human AβPP cDNA with the Arctic (p. E693G) and Swedish (p. KM670/671NL) mutation (Tg-ArcSwe), and twenty-eight wild type littermate controls (WT) were used for different analyses as indicated in Table 1. The animals were housed under standard conditions with 12-h dark-light cycles and unrestricted access to food and water. All animal procedures were in accordance with European (2010/63/EU and ETS123) guidelines for the care and use of laboratory animals and approved by the Norwegian Food Safety Authority (FOTS id 5693).
Table 1
Animals used in the study
| Data analysis | Genotype | Age (mo) | Number of animals | |||
| Quantitative-real time | WT littermates | 11 – 13 | 8 | |||
| polymerase chain reaction | Tg-ArcSwe | 11 – 13 | 8 | |||
| Stereology: Area fraction | WT littermates | 11 – 13 | 12 | |||
| fractionator quantification | Tg-ArcSwe | 11 –13 | 14 | |||
| Electron microscopy | WT littermates | 4 | 16 | 4 | 4 | |
| quantification | Tg-ArcSwe | 4 | 16 | 4 | 4 | |
Synthesis of Methoxy-X04
Methoxy-X04 [29] was synthesized largely according to published methods. First, 1,4-bis(bromomethyl)-2-methoxybenzene 2, obtained by dibromination of 2,5-dimethylanisole 1, was converted to the corresponding bisphosphonate 3 in a Michaelis-Arbuzov reaction (Scheme 1) [30]. Next, this was deprotonated with NaH and allowed to react with two equivalents of 4-(ethoxymethoxy)benzaldehyde 5 in a double Wittig-Horner/Horner-Wadsworth-Emmons reaction, giving the diene 6 [30, 31]. Finally, deprotection of 6 under acidic conditions afforded Methoxy-X04 7 [29].
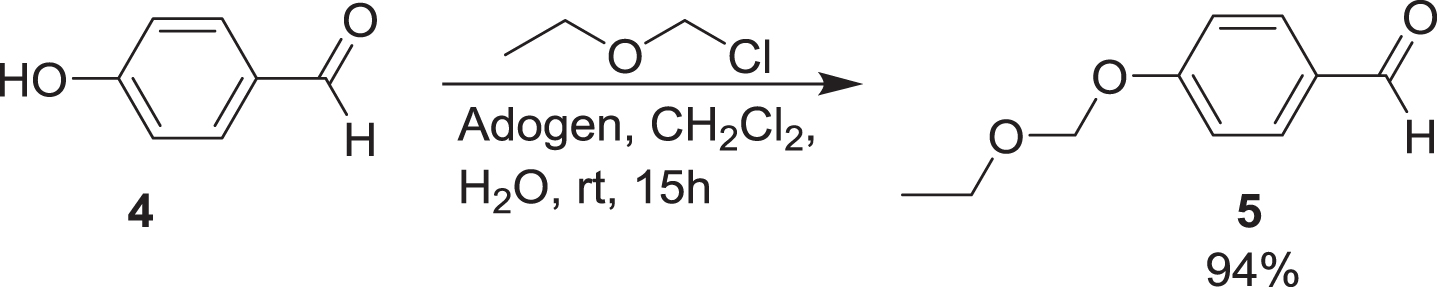
Some minor adjustments were made to the published methods, one being the replacement of methoxymethyl (MOM) with ethoxymethyl (EOM) as protecting group for the phenolic hydroxy group of 4-hydroxybenzaldehyde 4 (Scheme 2) [32].
Scheme 2
Protection of the phenolic hydroxy group of 4.
Experimental details and spectroscopic data for the intermediates and Methoxy-X04 are provided in the Supplementary Material. The large coupling constants observed for the olefinic protons (J = 17 Hz) strongly suggest that the configuration at both double bonds is (E).
Tissue processing for light microscopic immunohistochemistry and qPCR analysis
The animals were weighed and injected intraperitoneally with 7 mg/g Methoxy-X04 dissolved in 0.1 M phosphate buffer saline (PBS) 24 h before they were sacrificed. In previous experiments, we have injected several wild type mice to ensure no unspecific binding of Methoxy-X04. Animals (Tg-ArcSwe and WT littermates) used for qPCR analysis was not injected with Methoxy-X04.
Prior to sacrifice, the mice were anesthetized with Isofluran (Isoflo, Abbot Laboratories, Abbot Park, IL, USA) and ZRF mixture (Zolazepam 3.8 mg/ml, Tiletamin 3.8 mg/ml Xylazine 0.45 mg/ml, and Fentanyl 2.6μg/ml; single intraperitoneal injection) and transcardially injected with 100μl Texas red-labeled dextran (molecular weight 70 kDa; lysine fixable; 25 mg/ml stock solution diluted to 5 mg/ml in PBS; cat# D1830; Thermo Fisher Scientific) for 5 min before decapitation, using a protocol adapted from Wälchli et al. [33]. The lysine-fixable dextran used in this study is a hydrophilic polysaccharide with low toxicity that has covalently bound lysine residues. This permits the dextran tracer to be conjugated to surrounding biomolecules by aldehyde-mediated fixation for subsequent detection by immunohistochemical and ultrastructural techniques [34].
Following decapitation, the brains were dissected out. For 16 of the brains (8 Tg-ArcSwe and 8 WT littermates), the right hemisphere was immersion fixed in freshly dissolved 4%formaldehyde in 0.1 M phosphate buffer (PB) for 3 days before being transferred to 0.1%formaldehyde in 0.1 M PB at 4°C until further usage. For these animals the left hemisphere was subdivided into tissue blocks including frontal cortex, hippocampus, cerebellum, and the rest of the brain, which were frozen and stored in –80°C pending analysis.
Cryoprotective steps in graded sucrose solution (10%, 20%, and 30%sucrose in PB) were performed on the immersion fixed material, before cutting each hemisphere in serial 40μm thick sagittal free-floating sections on a freeze microtome (Thermo Scientific™ Microm KS 34). Sections were stored in 0.1%formaldehyde in PB at 4°C, protected from light, until further use. Every 12th section from the right hemisphere were mounted on glass slides with ProLong Gold Antifade Mountant (Invitrogen, Thermo Fisher Scientific) and stored at –20°C and protected from light.
Stereological evaluation of vessel area fraction in cerebral cortex
To estimate the area fraction of cerebral vessels, unbiased stereology of Texas red-labeled blood vessels was performed in cerebral cortex of Tg-ArcSwe mice and WT littermates. Animals with insufficient Texas red-labeled dextran labelling were excluded from the analysis, and 14 Tg-ArcSwe animals and 12 WT littermates were chosen for final analysis. The analyst was blinded for the genotype. For stereological quantification, each 12th section, starting randomly at section number 4, 8, or 12 in the series were chosen. This yielded 8–11 sections for analysis of the entire cerebral cortex per animal. The sections were examined by an Olympus Bx52 microscope (Olympus, Tokyo, Japan) equipped with a motorized stage (LEP MAC5000, LUDL Electronic Products Ltd., Hawthorne, NY, USA), and Optronics MicroFire digital camera (Optronics Goleta CA, USA) running the Stereo Investigator software (version 2017.02.2; MBF Bioscience, Chicago, IL, USA).
Regions of interest were manually drawn using a 4×objective (Olympus UPlanApo, NA 0.16) in bright field mode. The cerebral cortex was delineated anteriorly by a line connecting the rhinal fissure and corpus callosum, ventrally by the outer boundary of corpus callosum, posteriorly by the dorsal subiculum and dorsally by the external surface of the brain. Texas red –labeled blood vessels were identified and marked through a 40×objective (Olympus UPlanApo, NA 0.85) using the Area Fraction Fractionator approach [35]. A 300×300μm counting frame were positioned within the region of interest on an isotropic sampling grid (850×850μm) with gridlines spaced at 50μm. Texas red - labeled blood vessels were counted if their outer boarder fell within the intersection.
Immunohistochemistry for light microscopy
A subset of sections was washed in PBS 0.01 M for 10 min, followed by two times in 0.1%TritonX100 in PBS (PBST) for 5 min. The PBST was removed and blocking (10%normal donkey serum (NDS), 1%bovine serum albumin (BSA), 0.5 %Triton X100 in PBS) was performed for 1 h at room temperature. This was directly followed by incubation overnight at room temperature with primary antibody (PDGFRβ; host: rabbit; diluted 1:100; Abcam; cat# ab32570) diluted in antibody solution (ABS; 3%NDS, 1 %BSA, 0.1%Triton X100 in PBS). The following day the sections were rinsed in 0.1%PBST two times for 1 min and three times for 10 min. Secondary antibody (CY2-conjugated donkey anti-rabbit; diluted 1:500; Jackson ImmunoResearch Labs.; cat# 711-225-152) was diluted in ABS, and the sections were incubated for 1 h at room temperature. After the second incubation, the sections were washed in PBS for 10 min, three times. All sections were transferred to ddH2O and mounted with ProLong Gold antifade reagent (Thermo Fisher Scientific; cat# P36934). Z-stack images were acquired using a Zeiss LSM 710 confocal microscope, at 20×, 40×, or 63×magnification at an optical thickness of 1μm. The stacks obtained were 3D rendered using ImageJ software, FIJI [36]. The images were used for qualitative purposes.
Tissue processing for electron microscopy
16 animals (8 WT littermates and 8 Tg-ArcSwe) were used for electron microscopy analysis and sacrificed either at 4 months or 16 months of age. These animals were anesthetized with 0.3 mL ZRF mixture intraperitoneally and checked for absence of reflexes before they were transcardially perfused with ice cold 2%dextran (freshly dissolved in 0.1 M PB) for 20–30 s, followed by 4%formaldehyde and 0.1%glutaraldehyde in 0.1 M PB (pH 7.4) for 10 min. The brains were extracted and postfixed in the same solution for 72 h and stored in 1/10 fixative at 4°C until further preparation. Pieces from cerebral cortex were dissected and embedded in Lowicryl HM20 following our laboratory’s standard protocol [37].
Briefly, the tissue pieces were cryoprotected in 4%glucose dissolved in 0.1 M PB for 2 h followed by increasing concentrations (10%, 20%, and 30%) of glycerol before plunging the tissue specimens into liquid propane at –170°C in a liquid nitrogen-cooled cryofixation unit KF80 (Reichert, Vienna, Austria). Freeze substitution was undertaken in 0.5%uranyl acetate in anhydrous methanol at –90°C for 48 h in a freeze substitution unit (Leica EM AFS, Leica Microsystems GmbH, Wien, Austria). The temperature was gradually increased to –45°C, and Lowicryl HM20 was gradually substituted for methanol for embedding the tissue. The specimens were polymerized under ultraviolet light for 48 h at –45°C. Ultrathin sections were cut at 80–100 nm and placed on Formvar-coated single-hole grids. The grids were incubated in 2%uranyl acetate followed by 0.3%lead citrate for 90 s each to enhance the contrast for imaging.
Several sections, chosen randomly, from the frontal cortex of each animal were investigated, and all capillaries deemed to be intact and without severe artefacts were imaged. The frontal cortex was chosen for analysis as we have previously shown that this is a region severely affected by CAA in Tg-ArcSwe mice [28]. For 4-month-old Tg-ArcSwe mice and WT littermates this yielded a total number of 184 and 141 capillaries, respectively, while for the 16-month-old animals, 196 capillaries were included from the Tg-ArcSwe mice and 168 from the WT littermates.
Electron micrographs were obtained digitally from a transmission electron microscope (Tecnai 12, Philips Electron Optics BV, Eindhoven, The Netherlands). The images were exported at 11500x in tiff format and blinded to the analyst.
On selected sections, postembedding immunogold labeling with antibody Aβx-40 [38] was performed to identify amyloid deposit for qualitative purposes. The procedure has previously been described [39].
Electron microscopic image analysis
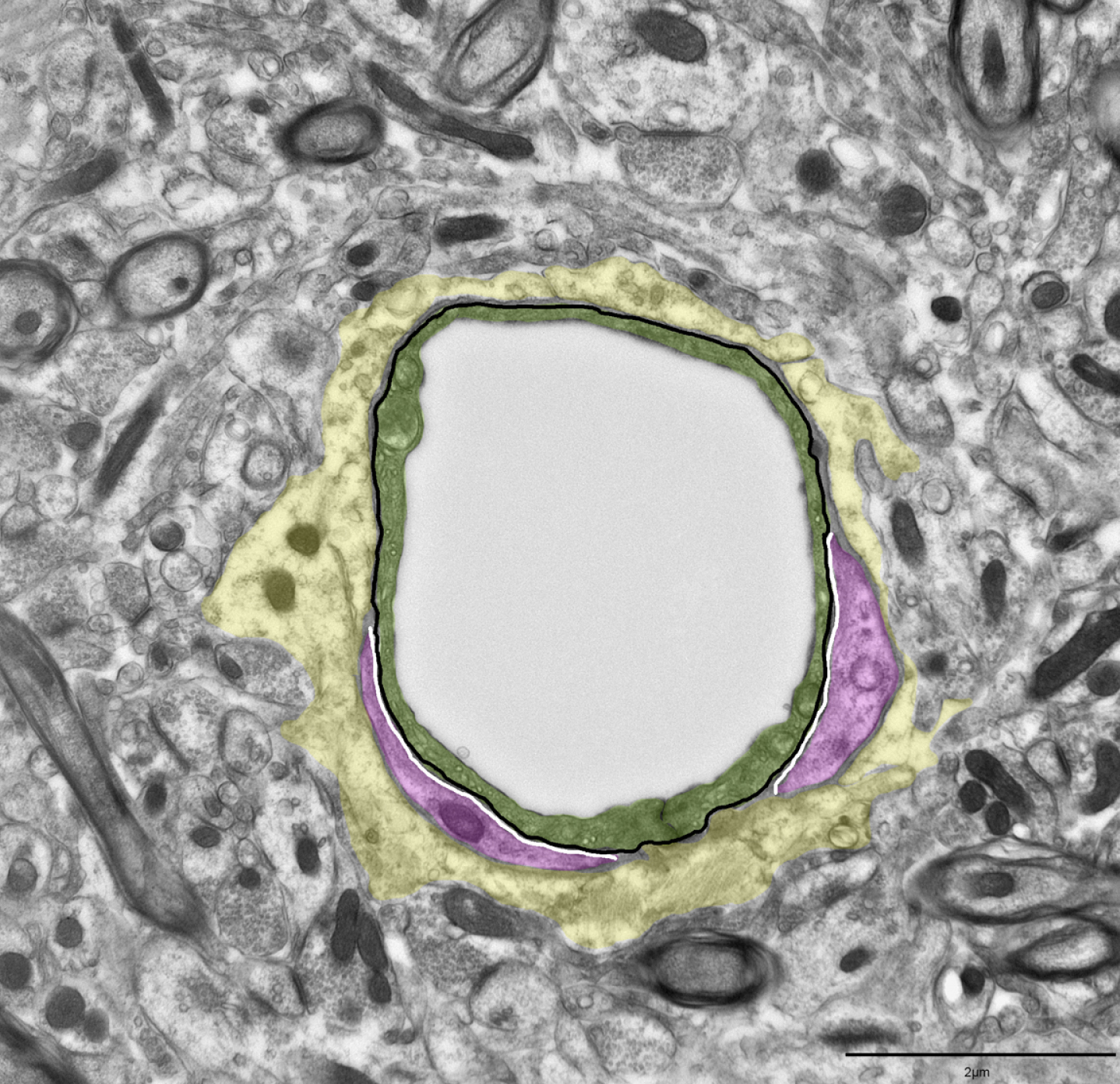
Pericytes were defined as cells located within the perivascular basal membrane, recognized by the splitting of the basal membrane on opposite sides of the cells. Pericyte coverage was measured as percentage of endothelial membrane covered by pericytes by measuring the total perimeter length of the capillary endothelium and the parts of it lined by pericytes (Fig. 1) [40]. The image analysis were performed in MATLAB R2017a® (The MathWorks, Inc., Natick, MA, USA) using software developed for automatic detection of immunogold particles [41]. The resolution was set at 11500x, while the nm per pixel was set at 4.3228.
Fig. 1
Procedure for quantification of pericyte coverage. Cross section of a capillary in cortex. Pericyte coverage was measured as the percentage of the endothelial cell perimeter covered by pericytes. The black line outlines the endothelial cell perimeter, and the white lines indicate endothelial cell membrane domains covered by a pericyte. Green overlay shows the endothelium and magenta identify pericytes while yellow outlines the astrocyte endfeet. Scale bar 2μm.
RNA isolation and real time PCR
Prior to RNA extraction, tissues were suspended in RNAlater™-ICE (Ambion; cat#: AM7030) for 48 h and then proceeded with extraction. The RNeasy Mini Kit (QIAGEN, Hilden, Germany), including the on column DNase digestion, was used to isolate total RNA from the frontal cortex tissue samples. The RNA concentration and integrity were determined using NanoDrop 2000c spectrophotometer (ThermoFisher Scientific) and ethidium bromide visualization after agarose gel electrophoresis, respectively. 1μg of total RNA was reverse-transcribed into cDNA with Oligo (dT)15 using the GoScript Reverse Transcription System (Promega, Madison, WI, USA, cat#: A5001) following manufacturer’s protocol. All the cDNA samples were diluted in Tris-EDTA buffer (pH 8.0) to a final concentration of 2.5 ng/μl. Real-Time PCR was carried out in a total volume of 20μl, containing 2x AB Power SYBR® Green PCR Master Mix (ThermoFisher Scientific) with gene specific primers (at a final concentration of 200 nM) and 2μl cDNA samples. Amplification was performed on the StepOnePlus system (Applied Biosystems) with the following conditions: 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min, followed by melting curve analysis to check for unspecific secondary products. Each sample was run in duplicates. Using the NormFinder software [42], HPRT1 was determined as an internal control for normalization of the gene expression. The primers were designed online using Primer-BLAST and setting the amplicon size to a maximum of 200 bp. All the primers designed spans exon-exon junction and standards were prepared for each primer pair as previously described [43]. List of primers used in the study are presented in Table 2.
Table 2
Primer list
| Gene | Protein names | Accession | Forward (5’) | Reverse (3’) |
| Hif1a | Hypoxia inducible factor 1, alpha subunit | NM_010431.2 | TTTTGGCAGCGATGACACAG | AAGTGGCTTTGGAGTTTCCG |
| Vegfa | Vascular endothelial growth factor A | NM_001025250.3 | AAAACACAGACTCGCGTTGC | TGGCTTGTCACATCTGCAAG |
| Vegfb transcript variant 1 | Vascular endothelial growth factor B | NM_011697.3 | CTCCCCAGCTGACATCATCC | CTTGTCACTTTCGCGGCTTC |
| Vegfr1/mFlt1 transcript variant 1 | Vascular endothelial growth factor receptor 1 | NM_010228.4 | TGTTTGAGGACTATCAGCTGGAC | TGCACGGGCCTGATGTG |
| Vegfr1/mFLT1 transcript variant 2 | Vascular endothelial growth factor receptor 1 | NM_001363135.1 | GAGAGCATCTCTCAGCGCAT | CATTCGGCACATCTGTGACAT |
| Vegfr2/Kdr transcript variant 1 | Vascular endothelial growth factor receptor 2 | NM_010612.3 | ATAGTGTGAAGAGCTGGGAGCA | GCTGAAAGCCTTGTAATCTCTCTA |
| Vegfr2/Kdr transcript variant 2 | Vascular endothelial growth factor receptor 1 | NM_001363216.1 | AGATTACTTGCAGGGGACAGC | CCGGTACGAGCACTTGTAGG |
| Angpt1 | Angiopoietin 1 | NM_009640.3 | TACAACACCGGGAAGATGGAAG | AATGAACTCGTTCCCAAGCC |
| Angpt2 | Angiopoietin 2 | NM_007426.4 | ACCGCGGGCAAAATAAGTAG | AAGGACCACATGCGTCAAAC |
| Tie1 | Tyrosine kinase with immunoglobulin-like and EGF-like domains 1 | NM_011587.2 | GGTCTGCCATTGTGGTGGAT | CATAAGGGCAGAAGGTCCCC |
| Tie2 | TEK receptor tyrosine kinase | NM_013690.3 | CTTCATCCACTCAGTGCCCC | GTACCTGGCCGAGTACACAC |
Statistical analysis
For qPCR analysis, mean copy number per ng of total RNA was compared between genotypes by Mann-Whitney U test in SPSS Statistics 26 (SPSS, Chicago, IL, USA). To evaluate vessel density unpaired two-sided Student’s t-test was performed using GraphPad Prism version 8.0.1 for Windows (GraphPad Software, San Diego, CA USA, http://www.graphpad.com). Pericyte coverage was evaluated by Student’s t-test in MATLAB R2017a®. A p-value≤0.05 was considered significant. The data are presented as mean with SEM.
RESULTS
Amyloid-β accumulates in vascular smooth muscle cells and around capillaries
In Tg-ArcSwe mice, Aβ accumulates around leptomeningeal vessels, penetrating arterioles, capillaries and less frequently around veins. At approximately 12 months of age 13.66%±5.16 of the vessels in cerebral cortex possess CAA (Fig. 2). In the leptomeningeal arteries and arterioles, Aβ is deposited in smooth muscle cells, while around capillaries it accumulates on the outside and inside of astrocytic endfeet (Fig. 2). This is described in detail in a 3D reconstruction of capillaries in our previous paper [25].
Fig. 2
Aβ accumulates in leptomeningeal and penetrating arterioles and around capillaries in Tg-ArcSwe. A) Light microscopic image of a penetrating arteriole with CAA from the upper layer of cortex. Red, Texas red conjugated dextran; Cyan, Methoxy-X04. Scale bar 20μm. B) Light microscopic image of an arteriole (big arrow) and capillary (small arrow) in cerebral cortex in Tg-ArcSwe. The image demonstrates the difference in appearance of Aβ depositions. C) Electron micrograph of a leptomeningeal artery with Aβ accumulating in the vascular smooth muscle cells. Scale bar 5μm. D) Enlargement of marked area demonstrated in C. The image shows in detail how the Aβ fibrils deposits in the vSMCs. The Aβ fibrils are marked with immunogold histochemistry (black dots). Scale bar 500 nm. V, vessel; vSMC, vascular smooth muscle cell. E) An electron micrograph of a cortical capillary with Aβ accumulation marked with immunogold histochemistry. The dotted line marks the outer boundary of the Aβ fibrils. The fibrils are so densely packed near the endothelial cell making it difficult to define other cell types of the neurovascular unit. Scale bar 2μm. F) Quantitative assessment by unbiased stereology of CAA burden in cerebral cortex in 11–13-month-old Tg-ArcSwe animals demonstrated by area fraction. In these animals (n = 5) 13.66%±5.16% of the total vessels in cortex are affected by CAA. The data are presented as mean with SEM.

Angiogenetic factors are altered in Tg-ArcSwe animals with no changes in blood vessel density
In our study, we performed quantitative PCR on RNA samples extracted from cerebral cortex of 11–13-month-old Tg-ArcSwe mice (n = 8) and compared with WT littermates (n = 8). Strikingly, Hif-1 α is strongly downregulated in the transgenic animals compared to the WT littermates (p < 0.001). We also found a significant downregulation of vascular endothelial growth factor receptor 2 variant 2 (Vegfr2-var 2) in Tg-ArcSwe compared to WT littermates (p = 0.038). However, angiopoietin 1 (Angpt1) and angiopoietin 2 (Angpt2) were both significantly upregulated in Tg-ArcSwe compared to WT littermates (p = 0.038 and 0.05), so was Tie2, the most important angiopoietin receptor (p = 0.015). Vegfa, Vegfb, Vegfr1 variant 1 and 2 and Vegfr 2 variant 1 remained unchanged. Mean copy numbers with SEM are demonstrated in Fig. 3 and Supplementary Table 1.
Fig. 3
Genes involved in angiogenesis and stereological quantification of vascular density in cerebral cortex. A) Representative confocal micrograph of Texas red-labeled blood vessels in the cerebral cortex of a wild type littermate animal. Scale bar 20μm. B) Representative confocal micrograph of Texas red-labeled blood vessels in the cerebral cortex of a Tg-ArcSwe animal. A penetrating arteriole with Aβ deposits and a parenchymal Aβ-plaque visualized by Methoxy-X04 can also be seen. C,D) Various genes of importance for angiogenesis were tested by qPCR. There is a significant downregulation of Hif1a (p < 0.001) and Vegfr2 transcript variant 2 (p = 0.038) in Tg-ArcSwe compared to WT littermates (n = 8 in both groups), while Angpt1 (p = 0.038), Angpt2 (p = 0.05), and the most important receptor for the angiopoietins, Tie2 (p = 0.015), are all upregulated in Tg-ArcSwe (as seen in F). E) No significant difference in vessel density in the cerebral cortex of 11–13-month-old WT littermates (17.23%±1.01%, n = 12) compared to Tg-ArcSwe littermates (15.45%±0.74%; p = 0,44, n = 14). Data are presented as mean with SEM.
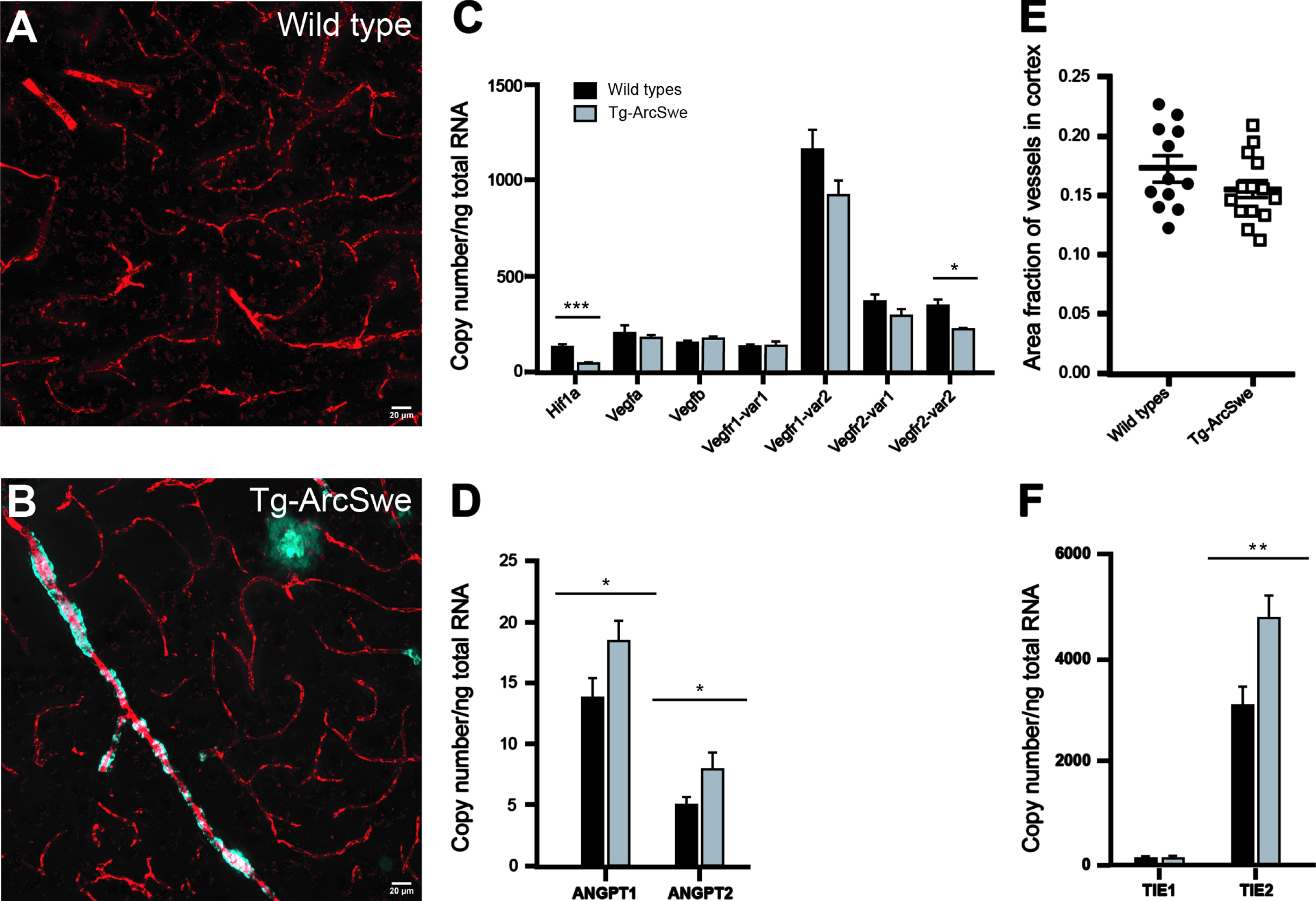
Based on our qPCR results, it was reasonable to expect that changes in angiogenetic factors could influence vessel density. However, unbiased stereological quantification showed no significant difference in vessel density between Tg-ArcSwe animals and WT littermates (p = 0.44), as demonstrated in Fig. 3E.
No changes in pericyte coverage of aged Tg-ArcSwe animals
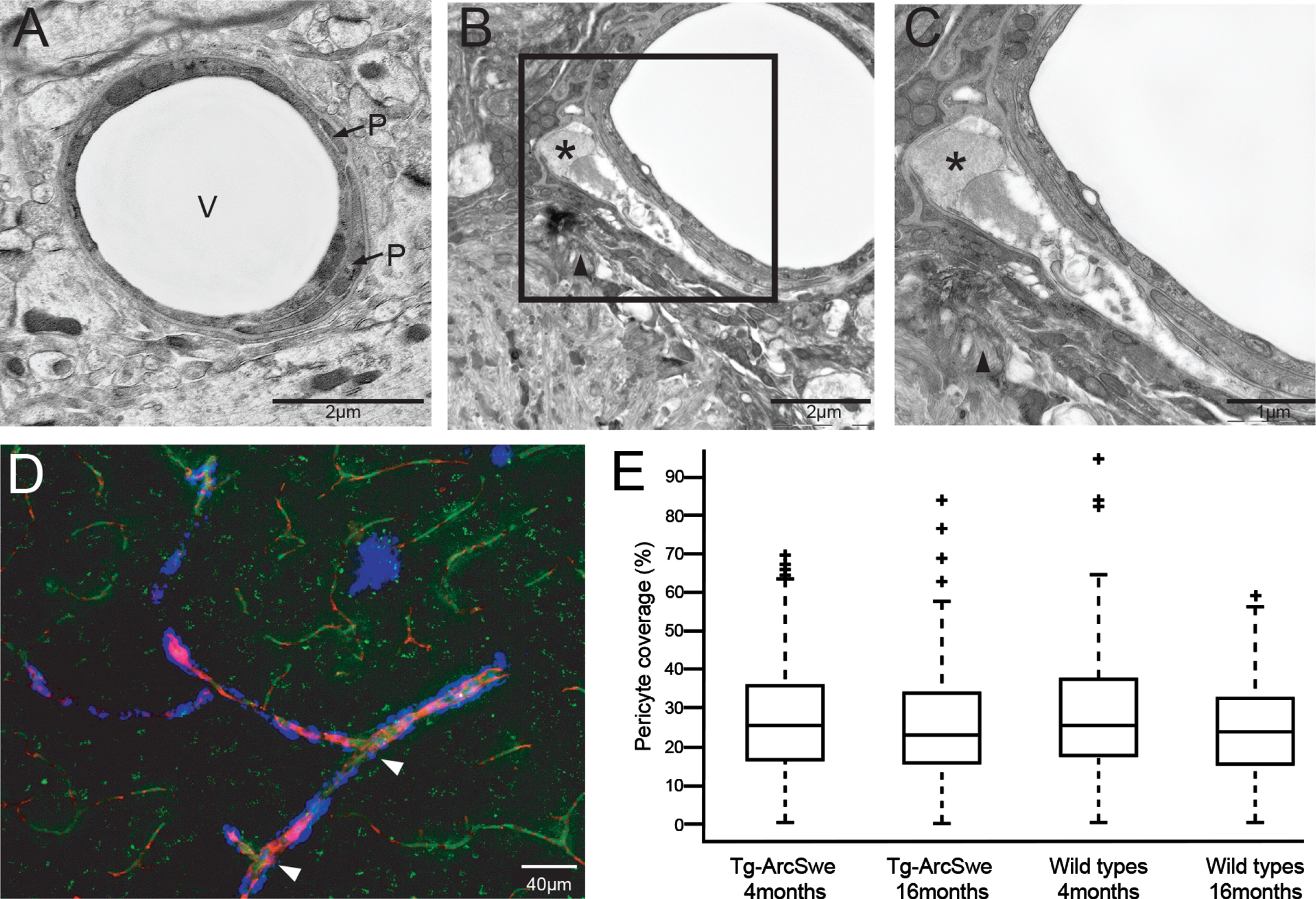
Analysis of electron micrographs showed numerous profiles of pericytes displaying distinct pathological changes, ranging from swelling to outright degeneration (Fig. 4A-C and Supplementary Figure 1). Many pericytes showed accumulation of inclusion bodies. The pathologically altered pericytes were found to be in direct contact with Aβ fibrils (Fig. 4 and Supplementary Figure 1), while pericytes not associated with Aβ fibrils did not display any overt pathology. The identity of Aβ fibrils was confirmed by immunogold cytochemistry (Supplementary Figure 1). However, we did not observe any significant differences in pericyte coverage between Tg-ArcSwe and WT littermates, neither at 4 months (p = 0.38) of age (before the onset of parenchymal Aβ plaques and CAA) nor at 16 months (p = 0.77) (Fig. 4).
Fig. 4
Degenerative pericytes in a terminal arteriole and capillary with Aβ. The figure demonstrates how Aβ affects pericyte morphology. A) Electron micrograph of a vessel found in a wild type littermate mouse demonstrating normal vessel morphology. Scale bar: 2μm. B) Pericytes in close proximity to a parenchymal Aβ plaque (arrow head). The outer pericyte (marked with asterisk) is swollen with large inclusions. Scale bar: 2μm. C) Enlargement of framed area in B to demonstrate a pathological swollen pericyte with large inclusions (marked with asterisk) in detail. The arrowhead points to Aβ fibrils. Scale bar: 1μm. D) Confocal image in a Tg-ArcSwe mouse stained with PDGFRβ (green), Methoxy-X04 (blue), and blood vessels visualized by Texas red conjugated dextran (red). Vessels without Aβ shows no sign of degenerated pericytes with a continuous and homogenous PDGFRβ-immuno-staining. In presence of Aβ, PDGFRβ-immunostaining is confluence or absent (indicated by arrows). Scale bar 40μm. V, vessel; P, pericyte. E) Quantitative assessment of pericyte coverage in cerebral capillaries. The box plot demonstrates no significant difference between Tg-ArcSwe and WT littermates (n = 4 in both groups), neither at 4 months (26.84%±14.73%in Tg-ArcSwe and 28.33%±15.50%in wild types; p = 0.38) or at 16 months (25.86%±15.04%in Tg-ArcSwe and 25.44%±12.72%in wild types; p = 0.77).
DISCUSSION
We have demonstrated by stereological quantification that a substantial fraction of cerebral vessels are affected by CAA at 11–13 months of age. Our qPCR investigations showed that important angiogenetic factors were significantly changed in Tg-ArcSwe mice, with a downregulation of Hif1a and Vegfr2 var 2 mRNA, and an upregulation of Angpt1, Angpt2, and Tie2 receptor mRNA. However, despite substantial CAA, altered angiogenetic regulating factors, and morphologically damaged pericytes, we found no significant changes in pericyte coverage or vessel density.
So far, research on angiogenesis in AD has provided conflicting results. Previous studies on postmortem human brains and AD mice models reported reductions in vascular density in cortex, hippocampus, and basal forebrain [6, 10, 44, 45], but there is also considerable evidence of increased angiogenesis in both human and AD mice model brains [46–49]. Further, a recent study by Xu et al. demonstrates a decrease in vascular density in aged APP23 mice, with a transient increase in density at 9 months [50]. It is worth noting that some studies and particularly older ones fail to take possible cortical atrophy into account (reviewed in [13]). Our use of intravasal fluorescent labelling allowed us to restrict our analysis to perfused and thereby functional vessels in cerebral cortex. By contrast, postmortem labeling of all blood vessels, as used in several previous studies [44–47, 49, 51–53], includes non-perfused as well as aberrant vessels that may have contributed to inconsistent results.
Pericytes play an important effect in angiogenesis. Early in the process, pericytes secrete VEGF-A to stimulate sprouting, endothelial proliferation and survival, but later pericytes inhibit endothelial proliferation [20]. Although we did not detect any global difference in pericyte coverage or vessel density in cerebral cortex, the considerable variance in vessel density among animals may have precluded detection of local changes. Interestingly, the substantial pathological alterations of pericytes in direct contact with Aβ fibrils did not correlate with reduced pericyte coverage or changed vessel density. Whether vascular pathology increases and correlates with vascular dysfunction at older age in Tg-ArcSwe mice is unknown.
A healthy vascular network is of paramount importance for the continuous supply of oxygen and glucose required for brain function. Several studies report that hypometabolism, characterized by decreased brain glucose consumption, may cause an imbalance in the production and clearance of Aβ by altering the AβPP expression and turnover [54–57]. The expression of HIFs is enhanced as a compensatory mechanism that alters the expression of genes involved in glycolysis, angiogenesis, and cell survival [16]. Hence, we particularly focused on HIF-1 α and HIF-dependent angiogenetic factors. HIF-independent factors, like VEGFR2, are also relevant as they are important in promoting capillary recanalization and minimizing subsequent pruning [58]. The most salient observation in our study is a substantial decrease in mRNA levels of Hif1a in 11–13-month-old Tg-ArcSwe mice. HIF-1α induces transcriptions of more than 60 proteins involved in inflammation and angiogenesis, including VEGF, VEGFR-1, ANGPT-1, ANGPT-2, NRP-1, and TIE-1 [16]. Our finding is consistent with previous studies indicating a role of HIF-1α in neurodegenerative diseases, including AD [16, 59, 60]. Notably, postmortem analyses of AD brains have shown reduced levels of HIF-1, particularly HIF-1α [61]. Our finding is, however, at odds with that of De Lemos et al. who reported no significant difference in Hif1a mRNA levels in 3–12-month-old APP/PS1 mice [62].
Schubert et al. [63] have previously demonstrated that Aβ-dependent astrocyte activation was implicated in the long-term decrease in HIF-1α expression and reduction in the rate of glycolysis. Activated astrocytes are a well-known feature of AD [64, 65] and occur in Tg-ArcSwe mice as well [25, 66]. Several studies have reported a possible correlation between AD and ANG-TIE signaling, but its exact role remains unclear [67]. Our present finding that the Angpt1/Tie2 system is transcriptionally upregulated in Tg-ArcSwe mice might reflect dysregulation of vascular repair and vessel maturation, as also suggested in several studies demonstrating that ANGPT-2 inhibits the protective role of the ANGPT-1/TIE-2-system [19, 68–70].
Having revealed significant changes in the levels of angiogenetic factors—most notably Hif1a mRNA—we went on to investigate whether these changes are translated into an alteration in vessel density or pericyte coverage. Pericytes are instrumental in vessel neogenesis [71–73] and also directly involved in the regulation of blood flow through their contractile properties [14, 74–77]. However, we found no evidence of a significant change in vessel density, nor in the extent of pericyte coverage. Despite that recent studies have reported pericyte loss in association with AD [78, 79], our results regarding pericyte coverage are in line with that of Fernandez-Klett et al. [80], who did not discover any difference in pericyte coverage between human AD cases and controls. Obviously, if the observed changes in angiogenetic factors are upstream of vascular pathologies their effects must be more subtle. Future studies are needed to clarify to which extent these factors affect vessel turnover or vessel dynamics.
Even if pericyte coverage was unchanged in the present model, pericyte pathology was rampant and often extensive. Nortley et al. [14] demonstrated that pericytes exposed to Aβ caused a constriction of the capillaries, and even a 30% reduction in the capillary diameter was enough to reduce the blood flow by 58%. In our study, pathological changes were restricted to pericytes closely associated with amyloid deposits while pericytes distant from amyloid deposits generally displayed normal morphology. The present study thus clearly identifies pericyte pathology as a prevalent component of CAA.
CONCLUSION AND PERSPECTIVES
Cerebrovascular changes have been implicated in the pathophysiology of AD and are potential targets for treatment. In this paper we utilized the Tg-ArcSwe mouse model to investigate angiogenetic markers and vascular density in the presence of marked CAA. We observe a pronounced change in mRNA expression level of Hif1a, as well as a significant downregulation in Vegfr2 var 2 mRNA but an upregulation of Angpt1, Angpt2, and Tie2 receptor mRNA. Our morphological analyses indicate that these changes are not coupled to any alteration in vascular density or pericyte coverage. However, vessels with CAA show extensive pathology in pericytes. Our observations in the present AD model suggest that CAA is associated with aberrant vessel turnover with no changes in vascular density. Vessel turnover and pericyte pathology stand out as an important issue for further studies on the pathophysiology of AD.
ACKNOWLEDGMENTS
This work was supported by Norwegian Health Association, Alzheimer fond, Norwegian Civitan, Alzheimer fond and grant from UNIFOR. The authors thank Bjørg Riber, Grazyna Babinska and Hong Qu for expert histotechnical assistance and Carina Knudsen for aiding in final preparation of figures. Prof. Lars Lannfelt at the Uppsala University Transgenic Facility (UUTF) and Prof. Lars N. G. Nilsson at the University of Oslo are gratefully acknowledged for their support in developing the Tg-ArcSwe mouse model. We also thank Prof. Lars N. G. Nilsson for aiding in genotyping the animals.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/21-0571r1).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-210571.
REFERENCES
[1] | Alzheimer’s Disease International ((2019) ) World Alzheimer Report 2019. Attitudes to Dementia. London. |
[2] | Alzheimer’s Association ((2019) ) 2019 Alzheimer’s disease facts and figures. Alzheimers Dement 15: , 321–387. |
[3] | Hardy JA , Higgins GA ((1992) ) Alzheimer’s disease: The amyloid cascade hypothesis. Science 256: , 184. |
[4] | Ellis RJ , Olichney JM , Thal LJ , Mirra SS , Morris JC , Beekly D , Heyman A ((1996) ) Cerebral amyloid angiopathy in the brains of patients with Alzheimer’s disease: The CERAD experience, part XV. Neurology 46: , 1592–1596. |
[5] | Klohs J , Rudin M , Shimshek DR , Beckmann N ((2014) ) Imaging of cerebrovascular pathology in animal models of Alzheimer’s disease. Front Aging Neurosci 6: , 32. |
[6] | Fischer VW , Siddiqi A , Yusufaly Y ((1990) ) Altered angioarchitecture in selected areas of brains with Alzheimer’s disease. Acta Neuropathol 79: , 672–679. |
[7] | Baloyannis SJ , Baloyannis IS ((2012) ) The vascular factor in Alzheimer’s disease: A study in Golgi technique and electron microscopy. J Neurol Sci 322: , 117–121. |
[8] | Hassler O ((1965) ) Vascular changes in senile brains - A micro-angiographic study. Acta Neuropathol 5: , 40–53. |
[9] | Beskow J , Hassler O , Ottosson JO ((1971) ) Cerebral arterial deformities in relation to senile deterioration. Acta Psychiatr Scand 47: , 111–119. |
[10] | Kalaria RN , Kroon SN ((1992) ) Expression of leukocyte antigen CD34 by brain capillaries in Alzheimer’s disease and neurologically normal subjects. Acta Neuropathol 84: , 606–612. |
[11] | Arvanitakis Z , Leurgans SE , Wang Z , Wilson RS , Bennett DA , Schneider JA ((2011) ) Cerebral amyloid angiopathy pathology and cognitive domains in older persons. Ann Neurol 69: , 320–327. |
[12] | Jellinger KA ((2002) ) Alzheimer disease and cerebrovascular pathology: An update. J Neural Transm 109: , 813–836. |
[13] | Govindpani K , McNamara LG , Smith NR , Vinnakota C , Waldvogel HJ , Faull RL , Kwakowsky A ((2019) ) Vascular dysfunction in Alzheimer’s disease: A prelude to the pathological process or a consequence of it? J Clin Med 8: , 651. |
[14] | Nortley R , Korte N , Izquierdo P , Hirunpattarasilp C , Mishra A , Jaunmuktane Z , Kyrargyri V , Pfeiffer T , Khennouf L , Madry C , Gong H , Richard-Loendt A , Huang W , Saito T , Saido TC , Brandner S , Sethi H , Attwell D ((2019) ) Amyloid β oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science 365: , eaav9518. |
[15] | Korte N , Nortley R , Attwell D ((2020) ) Cerebral blood flow decrease as an early pathological mechanism in Alzheimer’s disease. Acta Neuropathol 1: , 3. |
[16] | Ashok BS , Ajith TA , Sivanesan S ((2017) ) Hypoxia-inducible factors as neuroprotective agent in Alzheimer’s disease. Clin Exp Pharmacol Physiol 44: , 327–334. |
[17] | Beck H , Plate KH ((2009) ) Angiogenesis after cerebral ischemia. Acta Neuropathol 117: , 481–496. |
[18] | Benderro GF , Lamanna JC ((2014) ) HIF-1α/COX-2 expression and mouse brain capillary remodeling during prolonged moderate hypoxia and subsequent re-oxygenation. Brain Res 1569: , 41–47. |
[19] | Trollmann R , Mühlberger T , Richter M , Boie G , Feigenspan A , Brackmann F , Jung S ((2018) ) Differential regulation of angiogenesis in the developing mouse brain in response to exogenous activation of the hypoxia-inducible transcription factor system. Brain Res 1688: , 91–102. |
[20] | Winkler EA , Sagare AP , Zlokovic BV ((2014) ) The pericyte: A forgotten cell type with important implications for Alzheimer’s disease? Brain Pathol 24: , 371–386. |
[21] | Lendahl U , Nilsson P , Betsholtz C ((2019) ) Emerging links between cerebrovascular and neurodegenerative diseases—a special role for pericytes. EMBO Rep 20: , e48070. |
[22] | Iturria-Medina Y , Sotero RC , Toussaint PJ , Mateos-Pérez JM , Evans AC ; Alzheimer’s Disease Neuroimaging Initiative ((2016) ) Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat Commun 7: , 1934. |
[23] | Miners JS , Schulz I , Love S ((2018) ) Differing associations between Aβ accumulation, hypoperfusion, blood-brain barrier dysfunction and loss of PDGFRB pericyte marker in the precuneus and parietal white matter in Alzheimer’s disease. J Cereb Blood Flow Metab 38: , 103–115. |
[24] | Lord A , Kalimo H , Eckman C , Zhang XQ , Lannfelt L , Nilsson LNG ((2006) ) The Arctic Alzheimer mutation facilitates early intraneuronal Aβ aggregation and senile plaque formation in transgenic mice. Neurobiol Aging 27: , 67–77. |
[25] | Yang J , Lunde LK , Nuntagij P , Oguchi T , Camassa LMA , Nilsson LNG , Lannfelt L , Xu Y , Amiry-Moghaddam M , Ottersen OP , Torp R ((2011) ) Loss of astrocyte polarization in the Tg-ArcSwe mouse model of Alzheimer’s disease. J Alzheimers Dis 27: , 711–722. |
[26] | Mullan M , Crawford F , Axelman K , Houlden H , Lilius L , Winblad B , Lannfelt L ((1992) ) A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N-terminus of β-amyloid. Nat Genet 1: , 345–347. |
[27] | Nilsberth C , Westlind-Danielsson A , Eckman CB , Condron MM , Axelman K , Forsell C , Stenh C , Luthman J , Teplow DB , Younkin SG , Näslund J , Lannfelt L ((2001) ) The “Arctic” APP mutation (E693G) causes Alzheimer’s disease by enhanced Aβ protofibril formation. Nat Neurosci 4: , 887–893. |
[28] | Lillehaug S , Syverstad GH , Nilsson LNG , Bjaalie JG , Leergaard TB , Torp R ((2014) ) Brainwide distribution and variance of amyloid-beta deposits in tg-ArcSwe mice. Neurobiol Aging 35: , 556–564. |
[29] | Klunk WE , Bacskai BJ , Mathis CA , Kajdasz ST , McLellan ME , Frosch MP , Debnath ML , Holt DP , Wang Y , Hyman BT ((2002) ) Imaging Abeta plaques in living transgenic mice with multiphoton microscopy and methoxy-X04, a systemically administered Congo red derivative. J Neuropathol Exp Neurol 61: , 797–805. |
[30] | Stankoff B , Wang Y , Bottlaender M , Aigrot MS , Dolle F , Wu C , Feinstein D , Huang GF , Semah F , Mathis CA , Klunk W , Gould RM , Lubetzki C , Zalc B ((2006) ) Imaging of CNS myelin by positron-emission tomography. Proc Natl Acad Sci U S A 103: , 9304–9309. |
[31] | Wang Y , Mathis CA , Huang GF , Holt DP , Debnath ML , Klunk WE ((2002) ) Synthesis and 11c-labelling of (E,E)-1-(3′,4′-dihydroxystyryl)-4-(3′-methoxy-4 ′-hydroxystyryl) benzene for PET imaging of amyloid deposits. J Label Compd Radiopharm 45: , 647–664. |
[32] | Sebhat IK , Tan YL , Widdowson DA , Wilhelm R , White AJP , Williams DJ ((2000) ) Directed lithiation in arenetricarbonylchromium(0) complexes: Assessment of some directing group specificities and of electrophilic quench efficacies. Tetrahedron 56: , 6121–6134. |
[33] | Wälchli T , Mateos JM , Weinman O , Babic D , Regli L , Hoerstrup SP , Gerhardt H , Schwab ME , Vogel J ((2015) ) Quantitative assessment of angiogenesis, perfused blood vessels and endothelial tip cells in the postnatal mouse brain. Nat Protoc 10: , 53–74. |
[34] | Fluorescent and Biotinylated Dextrans—Section 14.5. Thermo Fisher Scientific, NO. |
[35] | Nagelhus EA , Amiry-Moghaddam M , Bergersen LH , Bjaalie JG , Eriksson J , Gundersen V , Leergaard TB , Morth JP , Storm-Mathisen J , Torp R , Walhovd KB , Tønjum T ((2013) ) The glia doctrine: Addressing the role of glial cells in healthy brain ageing. Mech Ageing Dev 134: , 449–459. |
[36] | Schindelin J , Arganda-Carreras I , Frise E , Kaynig V , Longair M , Pietzsch T , Preibisch S , Rueden C , Saalfeld S , Schmid B , Tinevez JY , White DJ , Hartenstein V , Eliceiri K , Tomancak P , Cardona A ((2012) ) Fiji: An open-source platform for biological-image analysis. Nat Methods 9: , 676–682. |
[37] | Takumi Y , Ramírez-León V , Laake P , Rinvik E , Ottersen OP ((1999) ) Different modes of expression of AMPA and NMDA receptors in hippocampal synapses. Nat Neurosci 2: , 618–624. |
[38] | Philipson O , Lord A , Lalowski M , Soliymani R , Baumann M , Thyberg J , Bogdanovic N , Olofsson T , Tjernberg LO , Ingelsson M ((2012) ) The Arctic amyloid-β precursor protein (AβPP) mutation results in distinct plaques and accumulation of N-and C-truncated Aβ. Neurobiol Aging 33: , 1010. e1𠄣 1010. e13. |
[39] | Hoddevik EH , Khan FH , Rahmani S , Ottersen OP , Boldt HB , Amiry-Moghaddam M ((2017) ) Factors determining the density of AQP4 water channel molecules at the brain–blood interface. Brain Struct Funct 222: , 1753–1766. |
[40] | Eidsvaag VA , Hansson HA , Heuser K , Nagelhus EA , Eide PK ((2017) ) Brain capillary ultrastructure in idiopathic normal pressure hydrocephalus: Relationship with static and pulsatile intracranial pressure. J Neuropathol Exp Neurol 76: , 1034–1045. |
[41] | Enger R ((2017) ) Automated gold particle quantification of immunogold labeled micrographs. J Neurosci Methods 286: , 31–37. |
[42] | Andersen CL , Jensen JL , Ørntoft TF ((2004) ) Normalization of real-time quantitative reverse transcription-PCR data: A model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res 64: , 5245–5250. |
[43] | Rao SB , Skauli N , Jovanovic N , Katoozi S , Frigeri A , Froehner SC , Adams ME , Ottersen OP , Amiry-Moghaddam M ((2021) ) Orchestrating aquaporin-4 and connexin-43 expression in brain: Differential roles of α1- and β1- syntrophin. Biochim Biophys Acta Biomembr 1863: , 183616. |
[44] | Buée L , Hof PR , Bouras C , Delacourte A , Perl DP , Morrison JH , Fillit HM ((1994) ) Pathological alterations of the cerebral microvasculature in Alzheimer’s disease and related dementing disorders. Acta Neuropathol 87: , 469–480. |
[45] | Suter O-C , Sunthorn T , Kraftsik R , Straubel J , Darekar P , Khalili K , Miklossy J ((2002) ) Cerebral hypoperfusion generates cortical watershed microinfarcts in Alzheimer disease. Stroke 33: , 1986–1992. |
[46] | Desai BS , Schneider JA , Li JL , Carvey PM , Hendey B ((2009) ) Evidence of angiogenic vessels in Alzheimer’s disease. J Neural Transm 116: , 587–597. |
[47] | Biron KE , Dickstein DL , Gopaul R , Jefferies WA ((2011) ) Amyloid triggers extensive cerebral angiogenesis causing blood brain barrier permeability and hypervascularity in Alzheimer’s disease. PLoS One 6: , 23789. |
[48] | Meyer EP , Ulmann-Schuler A , Staufenbiel M , Krucker T ((2008) ) Altered morphology and 3D architecture of brain vasculature in a mouse model for Alzheimer’s disease. Proc Natl Acad Sci U S A 105: , 3587–3592. |
[49] | Bell MA , Ball MJ ((1981) ) Morphometric comparison of hippocampal microvasculature in ageing and demented people: Diameters and densities. Acta Neuropathol 53: , 299–318. |
[50] | Xu X , Meng T , Wen Q , Tao M , Wang P , Zhong K , Shen Y ((2020) ) Dynamic changes in vascular size and density in transgenic mice with Alzheimer’s disease. Aging (Albany NY) 12: , 17224–17234. |
[51] | Lee GD , Aruna JH , Barrett PM , Lei DL , Ingram DK , Mouton PR ((2005) ) Stereological analysis of microvascular parameters in a double transgenic model of Alzheimer’s disease. Brain Res Bull 65: , 317–322. |
[52] | Wang P , Xie ZH , Guo YJ , Zhao CP , Jiang H , Song Y , Zhu ZY , Lai C , Xu SL , Bi JZ ((2011) ) VEGF-induced angiogenesis ameliorates the memory impairment in APP transgenic mouse model of Alzheimer’s disease. Biochem Biophys Res Commun 411: , 620–626. |
[53] | Daschil N , Kniewallner KM , Obermair GJ , Hutter-Paier B , Windisch M , Marksteiner J , Humpel C ((2015) ) L-type calcium channel blockers and substance P induce angiogenesis of cortical vessels associated with beta-amyloid plaques in an Alzheimer mouse model. Neurobiol Aging 36: , 1333–1341. |
[54] | Gabuzda D , Busciglio J , Chen LB , Matsudaira P , Yankner BA ((1994) ) Inhibition of energy metabolism alters the processing of amyloid precursor protein and induces a potentially amyloidogenic derivative. J Biol Chem 269: , 13623–13628. |
[55] | Ferreira L.I , Resende R , Ferreiro E , Rego C.A , Pereira F.C ((2012) ) Multiple defects in energy metabolism in Alzheimer’s disease. Curr Drug Targets 11: , 1193–1206. |
[56] | Shoffner JM ((1997) ) Oxidative phosphorylation defects and Alzheimer’s disease. Neurogenetics 1: , 13–19. |
[57] | Cunnane S , Nugent S , Roy M , Courchesne-Loyer A , Croteau E , Tremblay S , Castellano A , Pifferi F , Bocti C , Paquet N , Begdouri H , Bentourkia M , Turcotte E , Allard M , Barberger-Gateau P , Fulop T , Rapoport SI ((2011) ) Brain fuel metabolism, aging, and Alzheimer’s disease. Nutrition 27: , 3–20. |
[58] | Reeson P , Choi K , Brown CE ((2018) ) VEGF signaling regulates the fate of obstructed capillaries in mouse cortex. Elife 7: , e33670. |
[59] | Merelli A , Rodríguez JCG , Folch J , Regueiro MR , Camins A , Lazarowski A ((2018) ) Understanding the role of hypoxia inducible factor during neurodegeneration for new therapeutics opportunities. Curr Neuropharmacol 16: , 1484–1498. |
[60] | Correia SC , Moreira PI ((2010) ) Hypoxia-inducible factor 1: A new hope to counteract neurodegeneration? J Neurochem 112: , 1–12. |
[61] | Liu Y , Liu F , Iqbal K , Grundke-Iqbal I , Gong CX ((2008) ) Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Lett 582: , 359–364. |
[62] | De Lemos ML , De La Torre AV , Petrov D , Brox S , Folch J , Pallás M , Lazarowski A , Beas-Zarate C , Auladell C , Camins A ((2013) ) Evaluation of hypoxia inducible factor expression in inflammatory and neurodegenerative brain models. Int J Biochem Cell Biol 45: , 1377–1388. |
[63] | Schubert D , Soucek T , Blouw B ((2009) ) The induction of HIF-1 reduces astrocyte activation by amyloid beta peptide. Eur J Neurosci 29: , 1323–1334. |
[64] | Olabarria M , Noristani HN , Verkhratsky A , Rodríguez JJ ((2010) ) Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer’s disease. Glia 58: , 831–838. |
[65] | Pekny M , Pekna M , Messing A , Steinhäuser C , Lee JM , Parpura V , Hol EM , Sofroniew MV , Verkhratsky A ((2016) ) Astrocytes: A central element in neurological diseases. Acta Neuropathol 131: , 323–345. |
[66] | Yang J , Zhang R , Shi C , Mao C , Yang Z , Suo Z , Torp R , Xu Y ((2017) ) AQP4 association with amyloid deposition and astrocyte pathology in the Tg-ArcSwe mouse model of Alzheimer’s disease. J Alzheimers Dis 57: , 157–169. |
[67] | Hayashi SI , Rakugi H , Morishita R ((2020) ) Insight into the role of angiopoietins in ageing-associated diseases. Cells 9: , 2636. |
[68] | Du Z , Zheng JW , Zhang ZY , Wang YA ((2017) ) Review of the endothelial pathogenic mechanism of TIE2-related venous malformation. J Vasc Surg Venous Lymphat Disord 5: , 740–748. |
[69] | Nag S , Manias JL , Kapadia A , Stewart DJ ((2017) ) Molecular changes associated with the protective effects of angiopoietin-1 during blood-brain barrier breakdown post-injury. Mol Neurobiol 54: , 4232–4242. |
[70] | Nätynki M , Kangas J , Miinalainen I , Sormunen R , Pietilä R , Soblet J , Boon LM , Vikkula M , Limaye N , Eklund L ((2015) ) Common and specific effects of TIE2 mutations causing venous malformations. Hum Mol Genet 24: , 6374–6389. |
[71] | Winkler EA , Bell RD , Zlokovic BV ((2011) ) Central nervous system pericytes in health and disease. Nat Neurosci 14: , 1398–1405. |
[72] | Armulik A , Genové G , Betsholtz C ((2011) ) Pericytes: Developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell 21: , 193–215. |
[73] | Arnold TD , Niaudet C , Pang MF , Siegenthaler J , Gaengel K , Jung B , Ferrero GM , Mukouyama YS , Fuxe J , Akhurst R , Betsholtz C , Sheppard D , Reichardt LF ((2014) ) Excessive vascular sprouting underlies cerebral hemorrhage in mice lacking αVβ8-TGFβ signaling in the brain. Development 141: , 4489–4499. |
[74] | Peppiatt CM , Howarth C , Mobbs P , Attwell D ((2006) ) Bidirectional control of CNS capillary diameter by pericytes. Nature 443: , 700–704. |
[75] | Hall CN , Reynell C , Gesslein B , Hamilton NB , Mishra A , Sutherland BA , OÂ Farrell FM , Buchan AM , Lauritzen M , Attwell D ((2014) ) Capillary pericytes regulate cerebral blood flow in health and disease. Nature 508: , 55–60. |
[76] | Attwell D , Mishra A , Hall CN , O’Farrell FM , Dalkara T ((2016) ) What is a pericyte? J Cereb Blood Flow Metab 36: , 451–455. |
[77] | Sweeney MD , Kisler K , Montagne A , Toga AW , Zlokovic BV ((2018) ) The role of brain vasculature in neurodegenerative disorders. Nat Neurosci 21: , 1318–1331. |
[78] | Halliday MR , Rege SV , Ma Q , Zhao Z , Miller CA , Winkler EA , Zlokovic BV ((2016) ) Accelerated pericyte degeneration and blood-brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. J Cereb Blood Flow Metab 36: , 216–227. |
[79] | Sengillo JD , Winkler EA , Walker CT , Sullivan JS , Johnson M , Zlokovic BV ((2013) ) Deficiency in mural vascular cells coincides with blood-brain barrier disruption in alzheimer’s disease. Brain Pathol 23: , 303–310. |
[80] | Fernandez-Klett F , Brandt L , Fernández-Zapata C , Abuelnor B , Middeldorp J , Sluijs JA , Curtis M , Faull R , Harris LW , Bahn S , Hol EM , Priller J ((2020) ) Denser brain capillary network with preserved pericytes in Alzheimer’s disease. Brain Pathol 30: , 1071–1086. |


