
Cognitive Effects of the BET Protein Inhibitor Apabetalone: A Prespecified Montreal Cognitive Assessment Analysis Nested in the BETonMACE Randomized Controlled Trial
Abstract
Background:
Epigenetic changes may contribute importantly to cognitive decline in late life including Alzheimer’s disease (AD) and vascular dementia (VaD). Bromodomain and extra-terminal (BET) proteins are epigenetic “readers” that may distort normal gene expression and contribute to chronic disorders.
Objective:
To assess the effects of apabetalone, a small molecule BET protein inhibitor, on cognitive performance of patients 70 years or older participating in a randomized trial of patients at high risk for major cardiovascular events (MACE).
Methods:
The Montreal Cognitive Assessment (MoCA) was performed on all patients 70 years or older at the time of randomization. 464 participants were randomized to apabetalone or placebo in the cognition sub-study. In a prespecified analysis, participants were assigned to one of three groups: MoCA score≥26 (normal performance), MoCA score 25–22 (mild cognitive impairment), and MoCA score≤21 (dementia). Exposure to apabetalone was equivalent in the treatment groups in each MoCA-defined group.
Results:
Apabetalone was associated with an increased total MoCA score in participants with baseline MoCA score of≤21 (p = 0.02). There was no significant difference in change from baseline in the treatment groups with higher MoCA scores. In the cognition study, more patients randomized to apabetalone discontinued study drug for adverse effects (11.3% versus 7.9%).
Conclusion:
In this randomized controlled study, apabetalone was associated with improved cognition as measured by MoCA scores in those with baseline scores of 21 or less. BET protein inhibitors warrant further investigation for late life cognitive disorders.
INTRODUCTION
Neurodegenerative disorders such as Alzheimer’s disease (AD) and vascular disorders including vascular dementia (VaD) are major causes of late-life cognitive decline. AD and related disorders (ADRD) including VaD are a significant burden to global healthcare systems and there is a large unmet need for effective therapies to prevent or retard ADRD progression [1]. There is considerable evidence that the risk of AD and VaD increases with age and the presence of cardiovascular disease, diabetes, and chronic kidney disease [2–6]. Populations with diabetes and cardiovascular disease (CVD) typically have cerebrovascular pathology at autopsy [7]. Accordingly, the focus of drug development has recently expanded to include processes common to these disorders, including vascular inflammation and calcification, neuroprotection, lipid metabolism, protein degradation and clearance, and glucose metabolism [8]. There is growing appreciation that these processes are regulated by epigenetic transcriptional controls.
Bromodomain and extra-terminal (BET) proteins are epigenetic ‘readers’ that recognize and bind to acetylated lysine residues on histone tails and other nuclear proteins [9]. These interactions localize BET proteins to discrete locations along chromatin strands, where they recruit and facilitate assembly of factors that control gene expression [10, 11]. Through this mechanism BET proteins can play a role in maladaptive gene expression leading to chronic disease states including CVD [9–11]. Recently a role for BET protein-dependent effects in central nervous system (CNS) disorders, including memory disorders and AD, has been recognized [12–15].
Apabetalone is an orally bioavailable, small molecule BET protein inhibitor (BETi) that was recently assessed for secondary prevention of CVD events in 2,425 patients with diabetes and recent acute coronary syndrome (ACS) in the randomized, placebo controlled BETonMACE trial [16]. Apabetalone-treated participants experienced an 18% favorable trend in the incidence of the primary composite endpoint of major adverse cardiovascular events (MACE, comprising cardiovascular death, non-fatal myocardial infarction, and stroke (p = 0.11)) and a 41% reduction in hospitalization for congestive heart failure (p = 0.03) [17–19].
To explore the effect of apabetalone on cognition in this population with risk factors for AD and VaD, we collected performance on the Montreal Cognitive Assessment (MoCA) in BETonMACE participants 70 or more years of age. MoCA is a brief, 30-point screening instrument for cognitive dysfunction [20]. The MoCA assesses eight cognitive domains including attention and concentration, executive function, memory, language, visuoconstructional skills, conceptual thinking, calculations, and orientation. MoCA is not a diagnostic tool and will capture cognitive impairment from any cause. The MoCA has been shown to be sensitive to change in AD, VaD, and post-stroke patients [21–23]. In this nested prespecified analysis of the BETonMACE trial, we examined the effects of apabetalone on cognition using the MoCA.
A secondary aim was to determine if serum alkaline phosphatase level was a biomarker associated with cognitive change in the MoCA analysis cohort. Serum alkaline phosphatase is increased in the brain and plasma in AD [24]. Alkaline phosphatase is elevated in stroke and may be a biomarker for VaD or mixed dementia [24]. Apabetalone consistently lowers serum alkaline phosphatase [25].
MATERIALS AND METHODS
Study design
The design and primary CVD results of the phase III BETonMACE trial have been reported [16–19]. The protocol was approved by the responsible institutional review board or ethics committee at each participating site. In brief, eligible participants had an ACS within 7–90 days prior to randomization, a low high-density lipoprotein cholesterol (HDL-C) level, and a diagnosis of type 2 diabetes. Among exclusion criteria was any condition which, in the opinion of the investigator, was likely to prevent the subject from complying with the requirements of or completing the study. Qualifying patients who provided written, informed consent were randomized in a 1:1 ratio to receive apabetalone 100 mg orally twice daily or matching placebo. In addition, participants received high-intensity statin therapy with atorvastatin or rosuvastatin and other clinically defined standard of care. The primary outcome was time to the first occurrence of MACE.
Cognitive function and alkaline phosphatase measurements
Cognitive evaluation with the MoCA in patients 70 years and older at time of randomization was included as a prespecified endpoint in the supplemental statistical analysis plan (included in the Supplementary Material). Patients were classified into three mutually exclusive categories according to a baseline MoCA score of 30–26, 25–22, or≤21. The cut-off score of 26 is regarded as the threshold separating cognitively normal individuals from those with cognitive impairment [23]. Scores of 25–22 typically reflect mild cognitive impairment (MCI), and scores of 21 and below are consistent with dementia [20]. MoCA was collected at baseline, 12 months, and 24 months, as well as at the last visit on treatment (LVT). Biochemical measurements were obtained at baseline, week 24, week 52, and every 24 weeks until LVT. Liver function biochemical parameters including alkaline phosphatase (ALP) were obtained at baseline and every 2 weeks until week 12, then every 4 weeks until week 28, then every 12 weeks until LVT. For each individual patient, median biochemical parameter values were calculated across all collected time points after baseline until LVT. All parameter measurements were performed by a central laboratory (ICON, Farmingdale, New York). The normal range for serum ALP was 40–150 U/L.
Statistical analysis
Baseline characteristics were summarized for all patients who completed a baseline MoCA and for the MoCA subgroups by treatment arm. Data were expressed as mean (standard deviation (SD)) or median (interquartile range (IQR)) for continuous variables and counts and percentages for categorical variables. Change in MoCA score from baseline to last observation captured (LOC) was evaluated in the MoCA subgroups using an analysis of variance model (ANOVA). Changes in MoCA were analyzed using analysis of covariance models (ANCOVA) with baseline MoCA score, statin, age, sex, race, years of education, duration between baseline and LOC, and baseline clinical chemistry measurements as covariates. Results from the ANCOVA analyses are reported as least squares (LS) means. Treatment duration between baseline and LOC was evaluated as mean (standard deviation [SD]) with comparison between treatment groups using Student’s t-test. For biochemical parameters, a median was calculated across all post-randomization time points for each patient until LOC to calculate change from baseline and percent change from baseline. The analysis approach for change in biochemical parameters used a Wilcoxon test between treatment groups with results summarized as median (IQR). Analyses were performed with R software, version 3.5.1 or higher (R Foundation for Statistical Computing). p-values less than 0.05 were considered statistically significant without adjustment for multiplicity in this prespecified exploratory analysis.
RESULTS
Of a total of 2,425 randomized participants in the BETonMACE trial at 190 sites in 13 countries, 1,215 were assigned to apabetalone and 1,210 to placebo. A total of 485 participants (20%) 70 years of age or older were eligible for the cognitive sub-study. Of these, two did not receive apabetalone or placebo and 19 did not have a baseline MoCA measurement and were excluded from analyses, 464 (95.7%) had a baseline MoCA measurement and comprised the cognition subgroup; 212 were assigned to apabetalone and 252 to placebo. The aggregate cognition subgroup included 223 participants (48%) with baseline MoCA≥26, 144 participants (31%) with baseline MoCA 25–22, and 97 participants (21%) with baseline MoCA≤21. A comparison non-cognition subgroup comprised all patients < 70 years of age.
Figure 1 shows the patient flow diagram for the cognition subgroup. Table 1 shows baseline demographic and clinical characteristics of the trial participants according to baseline MoCA category. The average age of the cognition subgroup was 73 years, compared to 59 years in the non-cognition subgroup. Consistent with this age difference, participants in the cognition subgroup were more likely to be female, to have a history of hypertension and a longer duration of diabetes, to have higher systolic and lower diastolic blood pressures, to be a current smoker, and to have had non-ST elevation MI as the qualifying ACS. Irrespective of cognition status, more than 90% of participants received inhibitors of the renin-angiotensin pathway, beta-blockers, and anti-platelet agents. However, fewer participants in the cognition subgroup were treated with high-intensity statins, metformin, insulin, sodium-glucose cotransporter-2 (SGLT2) inhibitors, or glucagon-like peptide 1 (GLP-1) receptor agonists. Levels of glucose, cholesterol (total and low-density lipoprotein (LDL), triglycerides, high-sensitivity C-reactive protein, and alkaline phosphatases were similar in cognition and non-cognition subgroups while hemoglobin A1c (HbA1c) and alanine aminotransferase were lower and HDL-C, total bilirubin, and the neutrophil to lymphocyte ratio were higher in the cognition subgroup. In each baseline MoCA category, baseline characteristics and time from baseline to LOC were well-balanced among treatment groups. The treatment duration from baseline to LOC was balanced between apabetalone and placebo irrespective of baseline MoCA. Among participants with baseline MoCA score≥26, treatment duration was 729 (529–951) days in the placebo-treated group compared to 699 (541–909) days in the apabetalone-treated group; in participants with baseline MoCA score 25–22, 694 (513–937), and 731 (548–939) days in placebo-treated and apabetalone-treated groups, respectively; and, in participants with baseline MoCA score≤21, 702 (512–912) and 701 (493–778) days in placebo-treated and apabetalone-treated groups, respectively (p > 0.05 for all three comparisons).
Fig. 1
Patient flow diagram for the cognition subgroup of the BETonMACE trial of apabetalone.
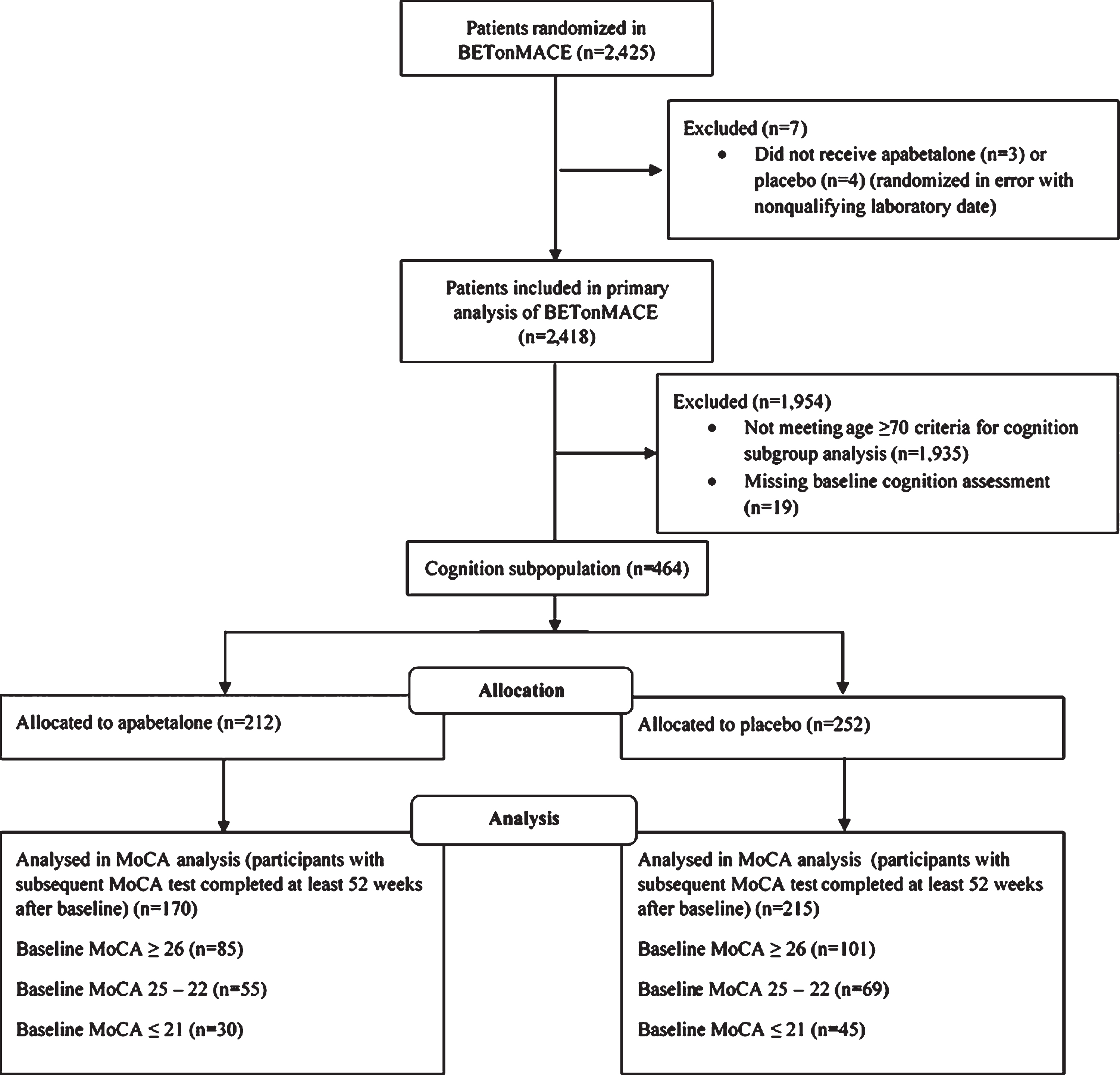
Table 1
Demographics, clinical, pharmacologic and laboratory characteristics of the BETonMACE trial participants at baseline according to MoCA subgroup and assigned treatment group
| Full study cohort according to cognition subgroup | Patients with MoCA≥26 by assigned treatment group | Patients with MoCA 25–22 by assigned treatment group | Patients with MoCA≤21 by assigned treatment group | |||||
| Non-cognition subgroup | Cognition subgroup | Placebo | Apabetalone | Placebo | Apabetalone | Placebo | Apabetalone | |
| Number of participants | 1,935 | 464 | 119 | 104 | 80 | 64 | 53 | 44 |
| Age, y | 59 (53–64) | 73 (71–76) | 73 (71–76) | 73 (71–76) | 73.5 (71–76) | 73 (71–77) | 75 (72–77) | 73.5 (71–76) |
| Female, n (%) | 446 (23.0%) | 167 (36.0%) | 47 (39.5%) | 32 (30.8%) | 27 (33.8%) | 19 (29.7%) | 23 (43.4%) | 19 (43.2%) |
| White, n (%) | 1,684 (87.0%) | 416 (89.7%) | 113 (95.0%) | 98 (94.2%) | 71 (88.8%) | 57 (89.1%) | 47 (88.7%) | 30 (68.2%) |
| Asian, n (%) | 30 (1.6%) | 9 (1.9%) | 2 (1.7%) | 2 (1.9%) | 1 (1.3%) | 0 (0.0%) | 2 (3.8%) | 2 (4.5%) |
| Other race, n (%) | 221 (11.4%) | 39 (8.4%) | 4 (3.4%) | 4 (3.8%) | 8 (10%) | 7 (10.9%) | 4 (7.5%) | 12 (27.3%) |
| Body mass index, kg/m2 | 30.5 (5.0) | 29.3 (4.6) | 29.5 (4.3) | 29.5 (4.5) | 29.2 (5.2) | 29.4 (4.2) | 29.0 (4.8) | 28.4 (4.9) |
| Hypertension history, n (%) | 1,684 (87.0%) | 440 (94.8%) | 117 (98.3%) | 100 (96.2%) | 71 (88.8%) | 60 (93.8%) | 49 (92.5%) | 43 (97.7%) |
| Smoking status, n (%) | 245 (12.7%) | 27 (5.8%) | 4 (3.4%) | 6 (5.8%) | 7 (8.8%) | 6 (9.4%) | 1 (1.9%) | 3 (6.8%) |
| Diabetes duration, years | 8.0 (7.3) | 10.9 (8.7) | 10.9 (9.1) | 10.9 (8.7) | 10.1 (6.8) | 10.2 (8.4) | 11.0 (9.5) | 13.3 (9.7) |
| Index ACS | ||||||||
| Myocardial infarction, n (%) | 1,444 (75.1%) | 322 (70.0%) | 81 (69.8%) | 69 (66.3%) | 59 (74.7%) | 42 (65.6%) | 38 (71.7%) | 33 (75.0%) |
| NSTEMI, n (%) | 649 (45.1%) | 183 (57.5%) | 52 (65.0%) | 39 (58.2%) | 35 (59.3%) | 27 (64.3%) | 16 (43.2%) | 14 (42.4%) |
| STEMI, n (%) | 789 (54.9%) | 135 (42.5%) | 28 (35.0%) | 28 (41.8%) | 24 (40.7%) | 15 (35.7%) | 21 (56.8%) | 19 (57.6%) |
| Unstable angina, n (%) | 480 (24.9%) | 138 (30.0%) | 35 (30.2%) | 35 (33.7%) | 20 (25.3%) | 22 (34.4%) | 15 (28.3%) | 11 (25.0%) |
| Time from index ACS, days | 39 (25–62) | 33 (24–60) | 30 (23–52) | 31 (23–63) | 39 (27–59) | 31 (24–62) | 37 (25–62) | 41 (28–67) |
| Cardiovascular medications | ||||||||
| Atorvastatin, n (%) | 1,003 (51.8%) | 231 (49.8%) | 61 (51.3%) | 50 (48.1%) | 40 (50.0%) | 34 (53.1%) | 24 (45.3%) | 22 (50.0%) |
| Rosuvastatin, n (%) | 932 (48.2%) | 233 (50.2%) | 58 (48.7%) | 54 (51.9%) | 40 (50.0%) | 30 (46.9%) | 29 (54.7%) | 22 (50.0%) |
| Intensive statin therapy, n (%) | 1,783 (92.1%) | 394 (84.9%) | 99 (83.2%) | 89 (85.6%) | 65 (81.3%) | 56 (87.5%) | 47 (88.7%) | 38 (86.4%) |
| Ezetimibe, n (%) | 55 (2.8%) | 10 (2.2%) | 6 (5.0%) | 2 (1.9%) | 1 (1.3%) | 1 (1.6%) | 0 (0.0%) | 0 (0.0%) |
| ACE inhibitors or ARB, n (%) | 1,782 (92.1%) | 431 (92.9%) | 112 (94.1%) | 99 (95.2%) | 74 (92.5%) | 57 (89.1%) | 48 (90.6%) | 41 (93.2%) |
| Beta-blockers, n (%) | 1,750 (90.4%) | 423 (91.2%) | 107 (89.9%) | 96 (92.3%) | 72 (90.0%) | 62 (96.9%) | 47 (88.7%) | 39 (88.6%) |
| Antiplatelet agents, n (%) | 1,917 (99.1%) | 455 (98.1%) | 117 (98.3%) | 100 (96.2%) | 79 (98.8%) | 63 (98.4%) | 52 (98.1%) | 44 (100.0%) |
| Diabetes medications | ||||||||
| Metformin, n (%) | 1,620 (83.7%) | 359 (77.4%) | 97 (81.5%) | 78 (75.0%) | 52 (65.0%) | 52 (81.3%) | 42 (79.2%) | 38 (86.4%) |
| Insulin, n (%) | 749 (38.7%) | 156 (33.6%) | 38 (31.9%) | 30 (28.8%) | 25 (31.3%) | 26 (40.6%) | 17 (32.1%) | 20 (45.5%) |
| Sulfonylureas, n (%) | 552 (28.5%) | 150 (32.3%) | 33 (27.7%) | 35 (33.7%) | 20 (25.0%) | 26 (40.6%) | 19 (35.8%) | 17 (38.6%) |
| DPP4 inhibitors, n (%) | 286 (14.8%) | 70 (15.1%) | 19 (16.0%) | 13 (12.5%) | 14 (17.5%) | 13 (20.3%) | 5 (9.4%) | 6 (13.6%) |
| SGLT2 inhibitors, n (%) | 267 (13.8%) | 27 (5.8%) | 9 (7.6%) | 6 (5.8%) | 3 (3.8%) | 6 (9.4%) | 3 (5.7%) | 0 (0.0%) |
| GLP1 receptor agonists, n (%) | 78 (4.0%) | 6 (1.3%) | 2 (1.7%) | 0 (0.0%) | 1 (1.3%) | 1 (1.6%) | 2 (3.8%) | 0 (0.0%) |
| Biochemical parameters | ||||||||
| HbA1c, % | 7.4 (6.5–8.8) | 7.0 (6.3–8.1) | 7.1 (6.3–8.2) | 6.9 (6.3–7.9) | 7.0 (6.2–7.9) | 7.3 (6.5–8.1) | 7.0 (6.4–8.5) | 7.3 (6.5–8.9) |
| Serum glucose, mg/dL | 136.0 | 132.4 | 129.7 | 134.5 | 127.2 | 137.3 | 130.6 | 140.1 |
| (110.4–176.5) | (109.9–167.7) | (107.2–161.1) | (116.9–157.9) | (109.9–167.7) | (112.3–182.7) | (100.2–183.0) | (106.8–179.5) | |
| Total cholesterol, mg/dL | 129.9 | 128.0 | 129.2 | 126.1 | 129.9 | 123.4 | 133.8 | 117.2 |
| (111.4–156.8) | (107.8–151.2) | (113.3–155.5) | (108.2–149.6) | (109.0–149.7) | (102.4–143.8) | (110.6–150.8) | (103.3–140) | |
| LDL cholesterol, mg/dL | 65.4 (49.5–85.5) | 63.8 (48.0–83.1) | 63.4 (50.9–81.0) | 62.8 (46–85) | 66.3 (50.1–85.0) | 66.3 (46.5–81.7) | 64.0 (52.7–82.4) | 57.0 (45.5–77.3) |
| HDL cholesterol, mg/dL | 33.3 (29.8–36.7) | 34.0 (30.5–37.5) | 34.8 (31.7–38.7) | 33.3 (29.8–37.1) | 34.0 (31.3–37.5) | 32.9 (30.1–37.1) | 34.8 (31.3–37.9) | 33.6 (29.3–37.3) |
| Triglycerides, mg/dL | 148.8 | 146.1 | 158.5 | 146.6 | 143.0 | 139.9 | 146.1 | 130.6 |
| (114.3–204.6) | (111.4–190.7) | (128.0–205.0) | (118.9–192.2) | (104.1–174.5) | (108.9–184.2) | (128.4–191.3) | (103.2–179.8) | |
| Alkaline phosphatase, U/L | 78.0 (64.0–95.0) | 76.5 (62.0–92.0) | 77.0 (61.0–91.0) | 76 (63–92.3) | 76.5 (61.0–93.3) | 74.0 (61.8–84.0) | 77.0 (66.0–92.0) | 81.0 (59.8–101.3) |
| Alanine aminotransferase, U/L | 23.0 (17.0–31.0) | 19.0 (14.0–26.0) | 20.0 (15.3–27.8) | 19 (14.5–26) | 19.0 (15.0–27.0) | 18.0 (14.0–23.3) | 19.0 (15.0–25.0) | 19.0 (13.5–25.5) |
| Systolic BP (mmHg) | 129.0 | 130.0 | 132.0 | 131 | 131.5 | 130.0 | 129.0 | 135.5 |
| (120.0–139.0) | (122.0–140.0) | (122.0–140.0) | (122.8–140) | (121.8–140.0) | (125.0–136.0) | (120.0–137.0) | (128.0–145.0) | |
| Diastolic BP (mmHg) | 78.0 (70.0–82.0) | 75.5 (70.0–80.0) | 78.0 (70.5–83.0) | 77 (70–80) | 73.5 (67.8–80.0) | 75.5 (70.0–80.0) | 74.0 (70.0–80.0) | 77.0 (68.0–83.0) |
| Total bilirubin, umol/L | 9.0 (6.7–11.8) | 9.6 (7.5–13.0) | 9.9 (7.6–13.0) | 9.6 (7.3–12.6) | 9.3 (7.2–13.4) | 9.5 (8.0–11.8) | 9.8 (8.5–13.8) | 9.6 (7.2–14.1) |
| hsCRP, mg/L | 2.9 (1.2–6.1) | 2.4 (1.1–6.2) | 1.3 (0.8–2.3) | 3.5 (1.7–6) | 1.7 (0.4–5.5) | 2.1 (1.1–3.7) | 5.2 (3.3–10.3) | 2.2 (0.8–5.1) |
| [n = 390] | [n = 89] | [n = 23] | [n = 23] | [n = 10] | [n = 16] | [n = 13] | [n = 4] | |
| NLR, ratio | 2.5 (1.9–3.3) | 2.8 (2.2–3.7) | 2.5 (2.1–3.5) | 2.8 (2.1–3.7) | 3.0 (2.2–3.8) | 3.0 (2.2–3.9) | 2.5 (2.2–3.6) | 2.9 (2.1–3.8) |
| MoCA characteristics | ||||||||
| Total MoCA score | n/a | 25 (22–27) | 28 (26–29) | 27 (26–29) | 24 (23–25) | 24 (23–25) | 18 (16–20) | 18.5 (16–20) |
| > 12 years of education, n (%) | n/a | 139 (30%) | 47 (39.5%) | 33 (31.7%) | 23 (28.8%) | 19 (29.7%) | 8 (15.1%) | 9 (20.5%) |
| Post randomization | ||||||||
| Time from baseline to last | n/a | 701 (523–912) | 729 (529–951) | 699 (541–909) | 694 (513–937) | 731 (548–939) | 702 (512–912) | 701 (493–778) |
| observation captured, days | ||||||||
MoCA, Montreal Cognitive Assessment; ACS, acute coronary syndrome; NSTEMI, non-ST segment elevation myocardial infarction; STEMI, ST-segment elevation myocardial infarction; ACE, angiotensin-converting enzyme; ARB, Angiotensin II receptor blocker; DPP4, dipeptidyl peptidase; SGLT2, sodium-glucose cotransporter 2; GLP1, glucagon-like peptide 1; HbA1c, hemoglobin A1C; LDL, low-density lipoprotein; HDL, high-density lipoprotein; BP, blood pressure; hsCRP, high-sensitivity C-reactive protein; NLR, neutrophil-lymphocyte ratio. Continuous variables are presented as mean (SD) or median (quartile 1−quartile 3). Categorical variables are presented as n (%). p-values comparing groups at baseline were calculated using z-test or Wilcoxon tests for continuous variables and chi-square tests for categorical variables. p-values of < 0.05 are considered statistically significant and are highlighted in bold. †There was no significant difference in proportion of MI versus unstable angina as an index event (p = 0.61), but among those with MI as the index event, there were significant differences in proportion of STEMI versus non-STEMI (p = 0.025).
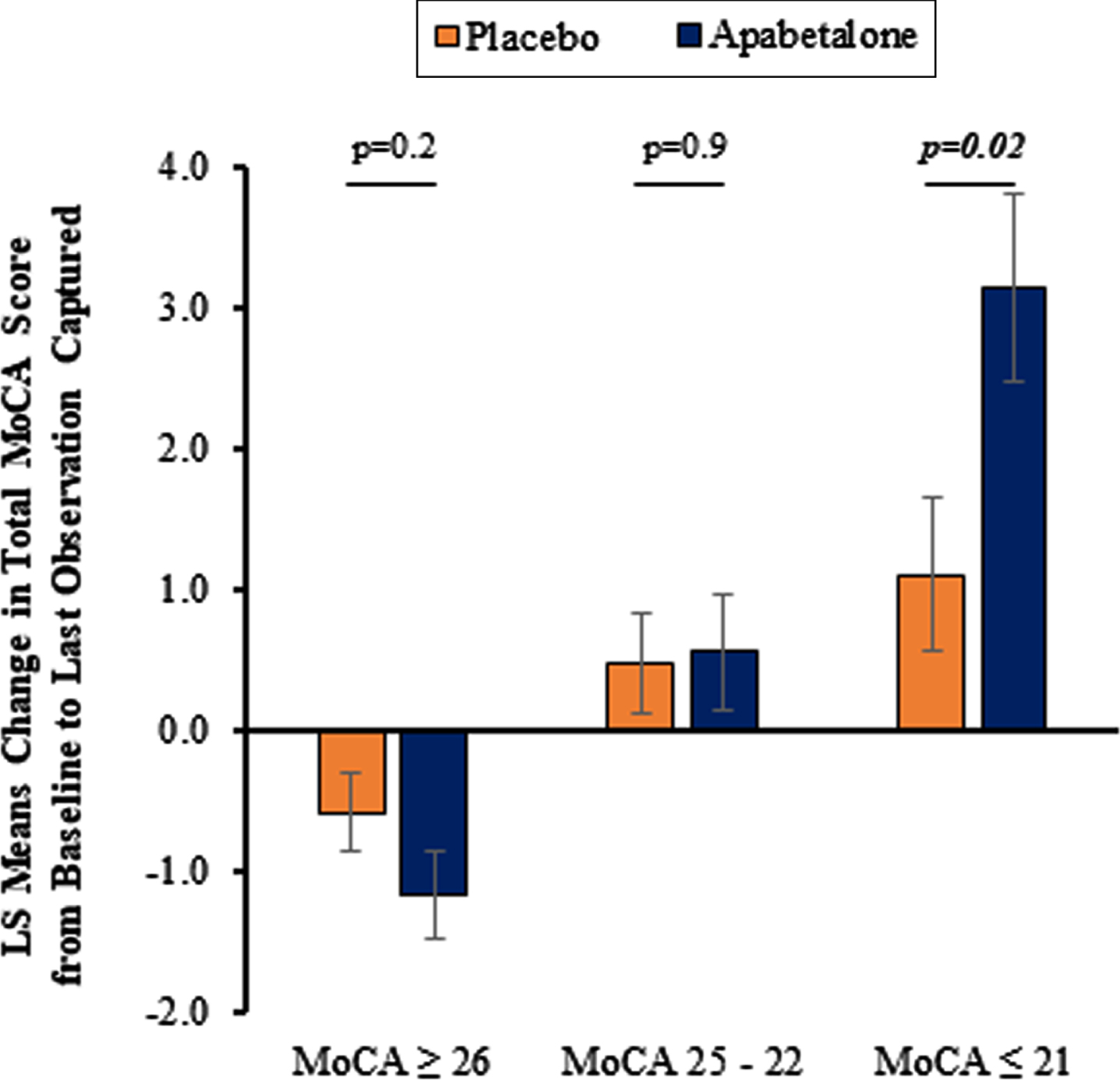
Apabetalone treatment was associated with a significantly increased total MoCA score compared to placebo among participants with baseline MoCA≤21 (p = 0.02). There was no difference between treatment groups in total MoCA score among participants with baseline MoCA 25–22 or MoCA≥26. Changes in MoCA score over the course of the study from baseline to LOC for the prespecified baseline MoCA score ranges and treatment groups are shown in Fig. 2. Supplementary Table 1 shows that in the participants with baseline MoCA≤21 placebo treatment shifted 16 (35%) participants to the 25–22 strata, and 1 (2%) to the≥26 strata, which compared to 14 (46%) and 3 (10%) for the apabetalone group, consistent with an apabetalone treatment effect. Shifts for the participants with baseline MoCA 25–22 and≥26 differed minimally between placebo and apabetalone.
Fig. 2
Least Squares (LS) mean change in total Montreal Cognitive Assessment (MoCA) score from baseline to last observation captured according to MoCA subgroup and assigned treatment group. Error bars represent standard error of the LS means. p-values were calculated using ANCOVA statistical analysis to compare change in total MoCA from baseline to last observation captured between apabetalone-treated patients and placebo with baseline total MoCA serving as a covariate and treatment arm as a factor. p-values of < 0.05 are considered statistically significant.
Supplementary Table 2 shows the analysis of changes in MoCA from baseline to LOC across the MoCA domains in both treatment groups. There was a trend (p = 0.099) for greater improvement in delayed recall in the most severely impaired group compared to placebo; this trend was not seen in the higher functioning patients. The analysis indicated an association between MoCA change from baseline to LOC and baseline MoCA category in the apabetalone-treatment group but not in placebo-treatment group.
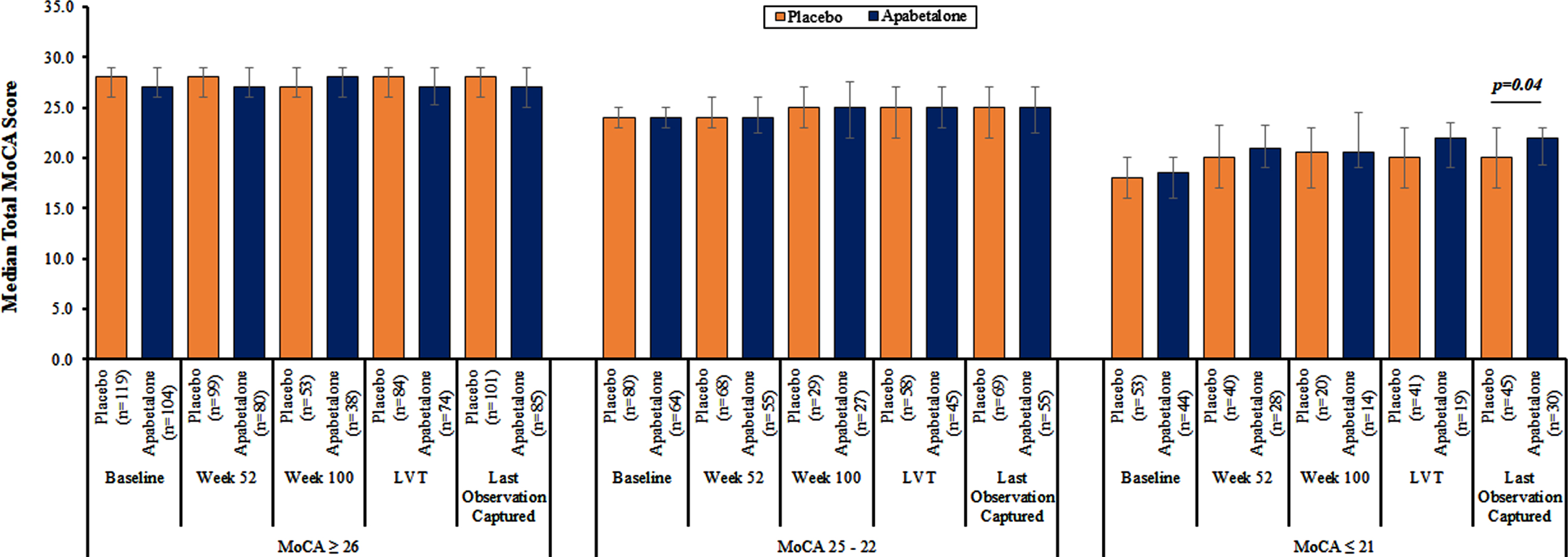
Table 2 shows the results of ANCOVA models of change from baseline to LOC in total MoCA scores between treatment groups. Apabetalone treatment was associated with a significantly increased total MoCA score compared to placebo among participants with baseline MoCA≤21 after adjustment for demographic, clinical, and treatment variables. Figure 3 shows that assessing treatment effects on the MoCA at 52 and 100 weeks and last value treated provided lower numbers of participants compared to LOC. LOC provided the highest number of participants and thereby best power to detect treatment effects.
Table 2
Change in total MoCA score from baseline to last observation captured according to MoCA subgroup and assigned treatment group. Data are presented as Least Squares (LS) means with standard error
| Model | Model covariates | Patients with MoCA ≥26 by assigned treatment group | Patients with MoCA 25–22 by assigned treatment group | Patients with MoCA ≤21 by assigned treatment group | ||||||
| Placebo (n = 101) | Apabetalone (n = 85) | p | Placebo (n = 69) | Apabetalone (n = 55) | p | Placebo (n = 45) | Apabetalone (n = 30) | p | ||
| 1 | Baseline total MoCA score | –0.6 (0.3) | –1.2 (0.3) | 0.2 | 0.5 (0.4) | 0.6 (0.4) | 0.9 | 1.1 (0.5) | 3.1 (0.7) | 0.03 |
| 2 | Model 1 plus adjustment for statin, age, sex, race, years of education, duration between baseline and last observation captured, baseline ALP, HDL and HbA1c | –0.6 (0.3) | –1.4 (0.3) | 0.2 | 0.4 (0.4) | 0.4 (0.5) | 0.8 | 1.7 (0.7) | 3.8 (0.8) | 0.02 |
MoCA, Montreal Cognitive Assessment; ALP; alkaline phosphatase; HbA1c, hemoglobin A1C; HDL, high-density lipoprotein. p-values were calculated using ANCOVA statistical analysis to compare change in total MoCA from baseline to last observation captured between apabetalone-treated patients and placebo. p-values of < 0.05 are considered statistically significant.
Fig.3
Median total Montreal Cognitive Assessment (MoCA) score from baseline to last observation captured according to MoCA subgroup and assigned treatment group. Error bars represent the interquartile ranges. p-values were calculated using Wilcoxon tests for continuous variables. p-values of < 0.05 are considered statistically significant and are highlighted. All other p-values were not significant. LVT, last visit on treatment.
Biochemical parameters and their change across all time points following treatment with placebo and apabetalone are summarized in Supplementary Table 3. Apabetalone treatment resulted in decreased alkaline phosphatase levels irrespective of baseline MoCA category. Among participants with baseline MoCA≥26, median change from baseline of alkaline phosphatase was 0.0 U/L in the placebo-treated group compared to –8.5 U/L in the apabetalone-treated group (p < 0.0001); in participants with baseline MoCA 25–22, –1.0 U/L in placebo-treated and –9.0 U/L in apabetalone-treated groups (p = 0.003); and in participants with baseline MoCA≤21, –3.0 U/L in placebo-treated and –8.5 U/L in apabetalone-treated groups (p = 0.03). No correlation was found between change in MoCA scores and change in alkaline phosphatase levels.
In the cognition subgroup, more participants allocated to apabetalone than placebo discontinued study drug [24 (11.3%) versus 20 (7.9%)] for adverse events including elevated liver function tests (LFT). Fewer participants in the cognition subgroup allocated to apabetalone than placebo died during the study [8 (3.8%) versus 13 (5.2%)]. Table 3 shows that among participants in the cognition subgroup, similar numbers of participants in the apabetalone and placebo groups experienced adverse events (150 [71%] versus 171 [68%] and serious adverse events (36% versus 35%).
Table 3
Adverse Events (AEs) according to assigned treatment group1
| Non-cognition subgroup by assigned treatment group | Cognition subgroup by assigned treatment group | |||
| Placebo (n = 943) | Apabetalone (n = 992) | Placebo (n = 252) | Apabetalone (n = 212) | |
| Patients with at least 1 adverse event2 (%) | 641 (68%) | 675 (68%) | 171 (68%) | 150 (71%) |
| Patients with at least 1 adverse event leading to study drug discontinuation (%)2 | 56 (6%) | 85 (9%) | 20 (8%) | 22 (10%) |
| Patients with at least 1 serious adverse event (%)2 | 246 (26%) | 278 (28%) | 90 (36%) | 75 (35%) |
| Frequent adverse events2,3 | ||||
| Alanine aminotransferase increased | 14 (1%) | 52 (5%) | 4 (2%) | 12 (6%) |
| Acute myocardial infarction | 37 (4%) | 33 (3%) | 13 (5%) | 8 (4%) |
| Angina | 59 (6%) | 58 (6%) | 17 (7%) | 15 (7%) |
| Unstable angina | 32 (3%) | 53 (5%) | 9 (4%) | 5 (2%) |
| Bronchitis | 17 (2%) | 22 (2%) | 15 (6%) | 3 (1%) |
| Cardiac failure | 19 (2%) | 17 (2%) | 18 (7%) | 5 (2%) |
| Diabetes mellitus | 56 (6%) | 68 (7%) | 6 (2%) | 8 (4%) |
| Hypertension | 59 (6%) | 63 (6%) | 12 (5%) | 9 (4%) |
| Influenza | 38 (4%) | 30 (3%) | 9 (4%) | 12 (6%) |
| Nasopharyngitis | 48 (5%) | 43 (4%) | 8 (3%) | 3 (1%) |
| Urinary tract infection | 29 (3%) | 42 (4%) | 11 (4%) | 16 (8%) |
1Safety population includes all patients who received at least 1 dose of study medication. 2Includes treatment-emergent adverse events only, defined as those occurring after the first dose and within 14 days of the last dose of the study drug. 3Defined as occurring with a frequency of 5% or more in any of the cognition or treatment groups.
DISCUSSION
Therapeutic intervention through epigenetic approaches, and specifically BET inhibition, is a novel area of study in CNS diseases and in late-life cognitive disorders such as VaD and AD [12, 27, 28]. Apabetalone is a small molecule BETi and in the BETonMACE trial, was associated with an 18% favorable trend towards a lower risk of the composite MACE endpoint (p = 0.11) and a 41% reduction in hospitalization for congestive heart failure (p = 0.03) [17–19]. These observations suggest that apabetalone-mediated BET protein inhibition may have beneficial cardiovascular effects in patients with diabetes who are at elevated risk for further cardiovascular events.
Meta-analyses indicate that diabetes increases the risk of all-cause dementia by a factor of 1.7, VaD by a factor of 2.27 [29] and AD by a factor of 1.36 [7]. At autopsy, patients with diabetes have increased cerebrovascular pathology compared to non-diabetic patients [30]. When diabetes, cerebrovascular disease and AD-related pathology occur concomitantly, they interact to worsen cognition [31–33]. Converging evidence that BET proteins play a role in AD, neurodegeneration, and cerebrovascular disease in the setting of diabetes provided the rationale for assessing the effects of apabetalone on cognitive outcomes in older participants in the BETonMACE trial [12–15, 27, 28, 34]. Age of at least 70 years rendered the analysis subgroup at high risk for VaD and AD and defined a subgroup in which a cognitive effect of apabetalone might be more readily identified [35]. Results of this nested, prespecified analysis suggest that in those participants with cognitive impairment at baseline, apabetalone treatment resulted in cognitive improvement over the ensuing 2 years. A likely reason is that the post-ACS, type 2 diabetes population with MoCA≤21 had a more advanced cerebral vascular and circulatory deficit, and thus were more likely to have benefited from the proposed mechanism of apabetalone.
The treatment-placebo difference on the MoCA for those with baseline scores of 21 or less was 2.1 (1.7 for the placebo group; 3.8 for the treatment group; p = 0.03). This 2.1-point difference is in the upper range of the minimum clinically important difference (MCID) of MoCA scores which varies between 1.22 and 2.15 in a similar (post-stroke) population treated with rehabilitation [21]. This outcome is sufficiently promising to warrant further investigation into the possible beneficial effects of apabetalone on cognition in patients with dementia of AD, vascular, or mixed etiology.
Mechanisms underlying an effect of apabetalone on cognitive function cannot be determined in a large, multicenter clinical trial but can be hypothesis-generating. Effects may include reduction of vascular inflammation and calcification, modulation of the complement and coagulation cascades, decrease in lipid levels, and reduced systemic inflammation, as have previously been reported for apabetalone [36, 37]. Improved cerebral blood flow [38, 39] and a reduction in inflammation-induced dysfunction of the blood-brain barrier (BBB) may contribute to cognitive improvement [40–45]. Systemic inflammation activates BBB endothelial cells, heightening the surface abundance and release of inflammatory molecules into the brain and into circulating blood, increasing BBB permeability to peripheral leukocytes. Vascular inflammation and BBB breakdown are both causative factors of neuroinflammatory and neurodegenerative processes [46]. Apabetalone suppresses BBB endothelial cell secretion of cytokines and chemokines and reduces the abundance of surface adhesion molecules in vitro. It lowers monocyte chemokine receptor expression, and monocyte chemoattraction [47]. As a result, apabetalone prevents the binding of monocytes to BBB endothelial cells stimulated by cytokines [47]. In a mouse model of systemic inflammation, administration of oral apabetalone decreases brain expression of pro-inflammatory mediators, despite being largely BBB impermeant [47]. Apabetalone is proposed to protect the BBB by reducing systemic and local inflammation, resulting in less neuroinflammation and decreased neurodegeneration.
The search for blood-based biomarkers for cognitive dysfunction is an area of active research with hope of identifying biomarkers that are diagnostic, prognostic, and predictive of therapeutic outcomes [48, 49]. Serum alkaline phosphatase has recently been shown to be associated with AD diagnosis and pathophysiology and to predict cognitive decline [50–52]. Alkaline phosphatase has previously been shown to decrease in apabetalone-treated patients in studies of CVD or CKD patients [25]. In this cognition sub-study, alkaline phosphatase levels decreased significantly in the apabetalone-treatment subgroup compared to the placebo group across all pre-defined cognitive subgroups. Apabetalone exhibited direct effects on the expression of alkaline phosphatase in cellular models [53]. It is unknown whether modulation of alkaline phosphatase levels reflects an attenuation of underlying inflammation [54]. No relationship between MoCA change and change in alkaline phosphatase was observed in this study.
This study has important limitations. Dichotomization of the study population at age 70 years was arbitrary but intended to focus on patients at high risk for cognitive decline. Effects of apabetalone on cognition in younger patients are unknown. The MoCA is a brief instrument that was feasible to administer in a large, multicenter cardiovascular clinical trial. More comprehensive cognitive assessments are required to better define the effects of apabetalone. Neither the duration of treatment to achieve optimal effects on cognition nor the persistence of effects on cognition after discontinuation of treatment could be determined in the BETonMACE trial. In BETonMACE the cause of the cognitive impairment in the participants with MoCA scores of 21 or less is unknown, and we cannot deduce whether the treatment effects occurred in participants with AD, VaD, or both. MoCA scores increased directionally from baseline in the treatment group with the lowest baseline MoCA score category and decreased directionally from baseline in both treatment groups of the highest baseline MoCA score category. These observations are consistent with regression to the mean in both treatment groups; however, this would not account for the significant differences between treatment groups in the evolution of MoCA scores over time. Notwithstanding these acknowledged limitations, the data justify further study of apabetalone’s effects on cognition.
In conclusion, the results of the cognitive sub-study of the BETonMACE trial hold promise that BET protein inhibition with apabetalone may provide a novel therapeutic approach for patients with concurrent CVD and cognitive impairment. New treatments for AD and VaD are US and global research priorities [55]. The findings of this sub-study of BETonMACE suggest epigenetic interventions may be a safe and efficacious therapeutic approach to augment cognitive capacity and warrant further study [56]. More specifically, the current findings provide impetus for a dedicated, placebo-controlled trial to test the efficacy of apabetalone on cognitive function with assessment instruments, outcome measures, and trial design strategies suited to investigate cognitive effects.
ACKNOWLEDGMENTS
The BETonMACE trial was funded by Resverlogix Corp.
Dr. Ray acknowledges support from the NIHR Imperial BRC.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/21-0570r2).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-210570.
REFERENCES
[1] | GBD 2016 Dementia Collaborators ((2019) ) Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 18: , 88–106. |
[2] | Song R , Xu H , Dintica CS , Pan KY , Qi X , Buchman AS , Bennett DA , Xu W ((2020) ) Associations between cardiovascular risk, structural brain changes, and cognitive decline. J Am Coll Cardiol 75: , 2525–2534. |
[3] | de Brujin RFAG , Ikram MA ((2014) ) Cardiovascular risk factors and future risk of Alzheimer’s disease. BMC Med 12: , 130. |
[4] | Cholerton B , Baker LD , Montine TJ , Craft S ((2016) ) Type 2 diabetes, cognition, and dementia in older adults: Toward a precision health approach. Diabetes Spectrum 29: , 210–219. |
[5] | Schilling MA ((2016) ) Unraveling Alzheimer’s: Making sense of the relationship between diabetes and Alzheimer’s disease. J Alzheimers Dis 51: , 961–977. |
[6] | Zhang CY , He FF , Su H , Zhang C , Meng XF ((2020) ) Association between chronic kidney disease and Alzheimer’s disease: An update. Metab Brain Dis 35: , 883–894. |
[7] | Zhang J , Chen C , Hua S , Liao H , Wang M , Xiong Y , Cao F ((2017) ) An updated meta-analysis of cohort studies: Diabetes and risk of Alzheimer’s disease. Diabetes Res Clin Pract 124: , 41–47. |
[8] | Scheltens P , De Strooper B , Kivipelto M , Holstege H , Chételat G , Teunissen CE , Cummings J , van der Flier WM ((2021) ) Alzheimer’s disease. Lancet 397: , 1577–1590. |
[9] | Yang XJ ((2004) ) Lysine acetylation and the bromodomain: A new partnership for signaling. BioEssays 26: , 1076–1087. |
[10] | Borck PC , Guo LW , Plutzky J ((2020) ) BET epigenetic reader proteins in cardiovascular transcriptional programs. Circ Res 126: , 1190–1208. |
[11] | Kulikowski E , Rakai BD , Wong N ((2021) ) Inhibitors of bromodomain and extra-terminal proteins for treating multiple human diseases. Med Res Rev 41: , 223–245. |
[12] | Singh MB , Sartor GC ((2020) ) BET bromodomains as novel epigenetic targets for brain health and disease. Neuropharmacology 181: , 108306. |
[13] | Benito E , Ramachandran B , Schroeder H , Schmidt G , Urbanke H , Burkhardt S , Capece V , Dean C , Fischer A ((2017) ) The BET/BRD inhibitor JQ1 improves brain plasticity in WT and APP mice. Transl Psychiatry 7: , e1239. |
[14] | Magistri M , Velmeshev D , Makhmutova M , Patel P , Sartor GC , Volmar CH , Wahlestedt C , Faghihi MA ((2016) ) The BET-bromodomain inhibitor JQ1 reduces inflammation and tau phosphorylation at Ser396 in the brain of the 3xTg model of Alzheimer’s disease. Cur Alzheimer Res 13: , 985–995. |
[15] | Korb E , Herre M , Zucker-Scharff I , Darnell RB , Allis CD ((2015) ) BET protein Brd4 activates transcription in neurons and BET inhibitor Jq1 blocks memory in mice. Nat Neurosci 18: , 1464–1473. |
[16] | Ray KK , Nicholls SJ , Ginsberg HD , Johansson JO , Kalantar-Zadeh K , Kulikowski E , Toth PP , Wong N , Cummings JL , Sweeney M , Schwartz GG ((2019) ) Effect of selective BET protein inhibitor apabetalone on cardiovascular outcomes in patients with acute coronary syndrome and diabetes: Rationale, design, and baseline characteristics of the BETonMACE trial. Am Heart J 217: , 72–83. |
[17] | Ray KK , Nicholls SJ , Buhr KA , Ginsberg HN , Johansson JO , Kalantar-Zadeh K , Kulikowski E , Toth PP , Wong N , Sweeney M , Schwartz GG , BETonMACE Investigators and Committees ((2020) ) Effect of apabetalone added to standard therapy on major adverse cardiovascular events in patients with recent acute coronary syndrome and type 2 diabetes: A randomized clinical trial. JAMA 323: , 1565–1573. |
[18] | Nicholls SJ , Schwartz GG , Buhr KA , Ginsberg HN , Johansson JO , Kalantar-Zadeh K , Kulikowski E , Toth PP , Wong N , Sweeney M , Ray KK , BETonMACE Investigators ((2021) ) Apabetalone and hospitalization for heart failure in patients following an acute coronary syndrome: A prespecified analysis of the BETonMACE study. Cardiovasc Diabetol 20: , 13–. |
[19] | Kalantar-Zadeh K , Schwartz GG , Nicholls SJ , Buhr KA , Ginsberg HN , Johansson JO , Kulikowski E , Lebioda K , Toth PP , Wong N , Sweeney M , Ray KK , BETonMACE Investigators ((2021) ) Effect of apabetalone on cardiovascular events in diabetes, CKD, and recent acute coronary syndrome: Results from the BETonMACE randomized controlled trial. Clin J Am Soc Nephrol 16: , 705–716. |
[20] | Nasreddine ZS , Phillips NA , Bédirian V , Charbonneau S , Whitehead V , Collin I , Cummings JL , Chertkow H ((2005) ) The Montreal Cognitive Assessment, MoCA: A brief screening tool for mild cognitive impairment. J Am Geriatr Soc 53: , 695–699. |
[21] | Wu CY , Hung SJ , Lin KC , Chen KH , Chen P , Tsay PK ((2019) ) Responsiveness, minimal clinically important difference, and validity of the MoCA in stroke rehabilitation. Occup Ther Int 2019: , 2517658. |
[22] | Costa AS , Reich A , Fimm B , Ketteler ST , Schulz JB , Reetz K ((2014) ) Evidence of the sensitivity of the MoCA alternate forms in monitoring cognitive change in early Alzheimer’s disease. Dement Geriatr Cogn Disord 37: , 95–103. |
[23] | Freitas S , Simões MR , Alves L , Vicente M , Santana I ((2012) ) Montreal Cognitive Assessment (MoCA): Validation study for vascular dementia. J Int Neuropsychol Soc 18: , 1031–1040. |
[24] | Vardy ER , Kellett KA , Cocklin SL , Hooper NM ((2012) ) Alkaline phosphatase is increased in both brain and plasma in Alzheimer’s disease. Neurodegenerative Dis 9: , 31–37. |
[25] | Haarhaus M , Ray KK , Nicholls SJ , Schwartz GG , Kulikowski E , Johansson JO , Sweeney M , Halliday C , Lebioda K , Wong N , Brandenburg V , Beddhu S , Tonelli M , Zoccali C , Kalantar-Zadeh K ((2019) ) Apabetalone lowers serum alkaline phosphatase and improves cardiovascular risk in patients with cardiovascular disease. Atherosclerosis 290: , 59–65. |
[26] | Rossetti HC , Lacritz LH , Cullum CM , Weiner MF ((2011) ) Normative data for the Montreal Cognitive Assessment (MoCA) in a population-based sample. Neurology 77: , 1272–1275. |
[27] | Ciceri F , Rotllant D , Maes T ((2017) ) Understanding epigenetic alterations in Alzheimer’s and Parkinson’s disease: Towards targeted biomarkers and therapies. Curr Pharm Des 23: , 839–857. |
[28] | Wang J , Yu JT , Tan MS , Jiang T , Tan L ((2013) ) Epigenetic mechanisms in Alzheimer’s disease: Implications for pathogenesis and therapy. Ageing Res Rev 12: , 1024–1041. |
[29] | Biessels GJ , Despa F ((2018) ) Cognitive decline and dementia in diabetes mellitus: Mechanisms and clinical implications. Nat Rev Endocrinol 14: , 591–604. |
[30] | Ahtiluoto S , Polvikoski T , Peltonen M , Solomon A , Tuomilehto J , Winblad B , Sulkava R , Kivipelto M ((2010) ) Diabetes, Alzheimer disease, and vascular dementia: A population-based neuropathologic study. Neurology 75: , 1195–1202. |
[31] | Abner EL , Nelson PT , Kryscio RJ , Schmitt FA , Fardo DW , Woltjer RL , Cairns NJ , Yu L , Dodge HH , Xiong C , Masaki K , Tyas SL , Bennett DA , Schneider JA , Arvanitakis Z ((2016) ) Diabetes is associated with cerebrovascular but not Alzheimer’s disease neuropathology. Alzheimers Dement 12: , 882–889. |
[32] | Kivipelto M , Ngandu T , Laatikainen T , Winblad B , Soininen H , Tuomilehto J ((2006) ) Risk score for the prediction of dementia risk in 20 years among middle aged people: A longitudinal, population-based study. Lancet Neurol 5: , 735–741. |
[33] | Snowdon DA , Greiner LH , Mortimer JA , Riley KP , Greiner PA , Markesbery WR ((1997) ) Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA 277: , 813–817. |
[34] | DeMars KM , Yang C , Candelario-Jalil E ((2019) ) Neuroprotective effects of targeting BET proteins for degradation with dBET1 in aged mice subjected to ischemic stroke. Neurochem Int 127: , 94–102. |
[35] | Brookmeyer R , Abdalla N , Kawas CH , Corrada MM ((2018) ) Forecasting the prevalence of preclinical and clinical Alzheimer’s disease in the United States. Alzheimers Dement 14: , 121–129. |
[36] | Wasiak S , Dzobo KE , Rakai BD , Kaiser Y , Versloot M , Bahjat M , Stotz SC , Fu L , Sweeney M , Johansson JO , Wong NCW , Stroes ESG , Kroon J , Kulikowski E ((2020) ) BET protein inhibitor apabetalone (RVX-208) suppresses pro-inflammatory hyper-activation of monocytes from patients with cardiovascular disease and type 2 diabetes. Clin Epigenet 12: , 166. |
[37] | Tsujikawa LM , Fu L , Das S , Halliday C , Rakai BD , Stotz SC , Sarsons CD , Gilham D , Daze E , Wasiak S , Studer D , Rinker KD , Sweeney M , Johansson JO , Wong NCW , Kulikowski E ((2019) ) Apabetalone (RVX-208) reduces vascular inflammation in vitro and in CVD patients by a BET-dependent epigenetic mechanism. Clin Epigenet 11: , 102. |
[38] | Korte N , Nortley R , Attwell D ((2020) ) Cerebral blood flow decrease as an early pathological mechanism in Alzheimer’s disease. Acta Neuropathol 140: , 793–810. |
[39] | Bracko O , Njiru BN , Swallow M , Ali M , Haft-Javaherian M , Schaffer CB ((2019) ) Increasing cerebral blood flow improves cognition into late stages in Alzheimer’s disease mice. J Cereb Blood Flow Metab 40: , 1441–1452. |
[40] | Noe CR , Noe-Letschnig M , Handschuh P , Noe CA , Lanzenberger R ((2020) ) Dysfunction of the blood-brain barrier—a key step in neurodegeneration and dementia. Front Aging Neurosci 12: , 185. |
[41] | Yang AC , Stevens MY , Chen MB , Lee DP , Stähli D , Gate D , Contrepois K , Chen W , Iram T , Zhang L , Vest RT , Chaney A , Lehallier B , Olsson N , du Bois H , Hsieh R , Cropper HC , Berdnik D , Li L , Wang EY , Traber GM , Bertozzi CR , Luo J , Snyder MP , Elias JE , Quake SR , James ML , Wyss-Coray T ((2020) ) Physiological blood-brain transport is impaired with age by a shift in transcytosis. Nature 583: , 425–430. |
[42] | Sweeney MD , Sagare AP , Zlokovic BV ((2018) ) Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol 14: , 133–150. |
[43] | Varatharaj A , Galea I ((2017) ) The blood-brain barrier in systemic inflammation. Brain Behav Immun 60: , 1–12. |
[44] | Zenaro E , Piacentino G , Constantin G ((2017) ) The blood-brain barrier in Alzheimer’s disease. Neurobiol Dis 107: , 41–56. |
[45] | Takeda S , Sato N , Morishita R ((2014) ) Systemic inflammation, blood-brain barrier vulnerability and cognitive/non-cognitive symptoms in Alzheimer disease: Relevance to pathogenesis and therapy. Front Aging Neurosci 6: , 171. |
[46] | Gorelick PB , Counts SE , Nyenhuis D ((2016) ) Vascular cognitive impairment and dementia. Biochim Biophys Acta 1862: , 860–868. |
[47] | Wasiak S , Fu L , Daze E , Gilham D , Rakai BD , Stotz SC , Tsujikawa LM , Sarsons CD , Jahagirdar R , Wong NCW , Sweeney M , Johansson JO , Kulikowski E ((2021) ) The epigenetic modulator apabetalone decreases neuroinflammation in blood brain barrier cell models and LPS-treated mice. CTAD 2021 (abstract accepted for oral presentation). |
[48] | Cummings J ((2019) ) The role of biomarkers in Alzheimer’s disease drug development. Adv Exp Med Biol 1118: , 29–61. |
[49] | Cummings J , Feldman HH , Scheltens P ((2019) ) The “rights” of precision drug development for Alzheimer’s disease. Alz Res Therapy 11: , 76. |
[50] | Kellett KA , Williams J , Vardy ER , Smith AD , Hooper NM ((2011) ) Plasma alkaline phosphatase is elevated in Alzheimer’s disease and inversely correlates with cognitive function. Int J Mol Epidemiol Genet 2: , 114–121. |
[51] | Boccardi V , Bubba V , Murasecco I , Pigliautile M , Monastero R , Cecchetti R , Scamosci M , Bastiani P , Mecocci P , ReGAL 2.0 Study Group ((2021) ) Serum alkaline phosphatase is elevated and inversely correlated with cognitive functions in subjective cognitive decline: Results from the ReGAl 2.0 project. Aging Clin Exp Res 33: , 603–609. |
[52] | Nho K , Kueider-Paisley A , Ahmad S , MahmoudianDehkordi S , Arnold M , Risacher SL , Louie G , Blach C , Baillie R , Han X , Kastenmüller G , Trojanowski JQ , Shaw LM , Weiner MW , Doraiswamy PM , van Duijn C , Saykin AJ , Kaddurah-Daouk R , Alzheimer’s Disease Neuroimaging Initiative and the Alzheimer Disease Metabolomics Consortium ((2019) ) Association of altered liver enzymes with Alzheimer disease diagnosis, cognition, neuroimaging measures, and cerebrospinal fluid biomarkers. JAMA Network Open 2: , e197978. |
[53] | Gilham D , Tsujikawa LM , Sarsons CD , Halliday C , Wasiak S , Stotz SC , Jahagirdar R , Sweeney M , Johansson JO , Wong NCW , Kalantar-Zadeh K , Kulikowski E ((2019) ) Apabetalone downregulates factors and pathways associated with vascular calcification. Atherosclerosis 280: , 75–84. |
[54] | Nicholas DA , Andrieu G , Strissel KJ , Nikolajczyk BS , Denis GV ((2017) ) BET bromodomain proteins and epigenetic regulation of inflammation: Implications for type 2 diabetes and breast cancer. Cell Mol Life Sci 74: , 231–243. |
[55] | Corriveau RA , Koroshetz WJ , Gladman JT , Jeon S , Babcock D , Bennett DA , Carmichael ST , Dickinson SL , Dickson DW , Emr M , Fillit H , Greenberg SM , Hutton ML , Knopman DS , Manly JJ , Marder KS , Moy CS , Phelps CH , Scott PA , Seeley WW , Sieber BA , Silverberg NB , Sutherland ML , Taylor A , Torborg CL , Waddy SP , Gubitz AK , Holtzman DM ((2017) ) Alzheimer’s Disease-Related Dementias Summit 2016: National research priorities. Neurology 89: , 2381–2391. |
[56] | Strafella C , Caputo V , Galota MR , Zampatti S , Marella G , Mauriello S , Cascella R , Giardina E ((2018) ) Application of precision medicine in neurodegenerative diseases. Front Neurol 9: , 701. |


