
Porphyromonas gingivalis Outer Membrane Vesicles as the Major Driver of and Explanation for Neuropathogenesis, the Cholinergic Hypothesis, Iron Dyshomeostasis, and Salivary Lactoferrin in Alzheimer’s Disease
Abstract
Porphyromonas gingivalis (Pg) is a primary oral pathogen in the widespread biofilm-induced “chronic” multi-systems inflammatory disease(s) including Alzheimer’s disease (AD). It is possibly the only second identified unique example of a biological extremophile in the human body. Having a better understanding of the key microbiological and genetic mechanisms of its pathogenesis and disease induction are central to its future diagnosis, treatment, and possible prevention. The published literature around the role of Pg in AD highlights the bacteria’s direct role within the brain to cause disease. The available evidence, although somewhat adopted, does not fully support this as the major process. There are alternative pathogenic/virulence features associated with Pg that have been overlooked and may better explain the pathogenic processes found in the “infection hypothesis” of AD. A better explanation is offered here for the discrepancy in the relatively low amounts of “Pg bacteria” residing in the brain compared to the rather florid amounts and broad distribution of one or more of its major bacterial protein toxins. Related to this, the “Gingipains Hypothesis”, AD-related iron dyshomeostasis, and the early reduced salivary lactoferrin, along with the resurrection of the Cholinergic Hypothesis may now be integrated into one working model. The current paper suggests the highly evolved and developed Type IX secretory cargo system of Pg producing outer membrane vesicles may better explain the observed diseases. Thus it is hoped this paper can provide a unifying model for the sporadic form of AD and guide the direction of research, treatment, and possible prevention.
PORPHYROMONAS GINGIVALIS: MEET THE BIOLOGICAL EXTREMOPHILE
Extremophile, an organism that is tolerant to environmental extremes and has evolved to grow optimally under one or more of these extreme conditions, hence the suffix phile, meaning “one who loves.” Extremophiles are of biotechnological interest, as they produce extremozymes, defined as enzymes that are functional under extreme conditions [1]. It seems that they should be also of medical interest as they may well represent an important discovery in our understanding of complex and seemingly less obvious disease processes. The term “Biological Extremophile” is being coined here for the first time that we know. Its point is to bring to view what is known by many in environmental microbiology, where there exists as the definition states, bacteria that have solved incredible local conditions for colonization growth and survival with the evolution of very unique metabolic/survival pathways. This type of evolution in biology is more rarely observed and by nature should put these organisms in a unique, extremely small class of their own. Porphyromonas gingivalis (Pg) is a microbiological enigma. In some ways resembling the distant Halophiles (salt-loving), as a Gram-negative non-spore forming, non-motile, pigment forming bacteria that reproduce by binary fission.
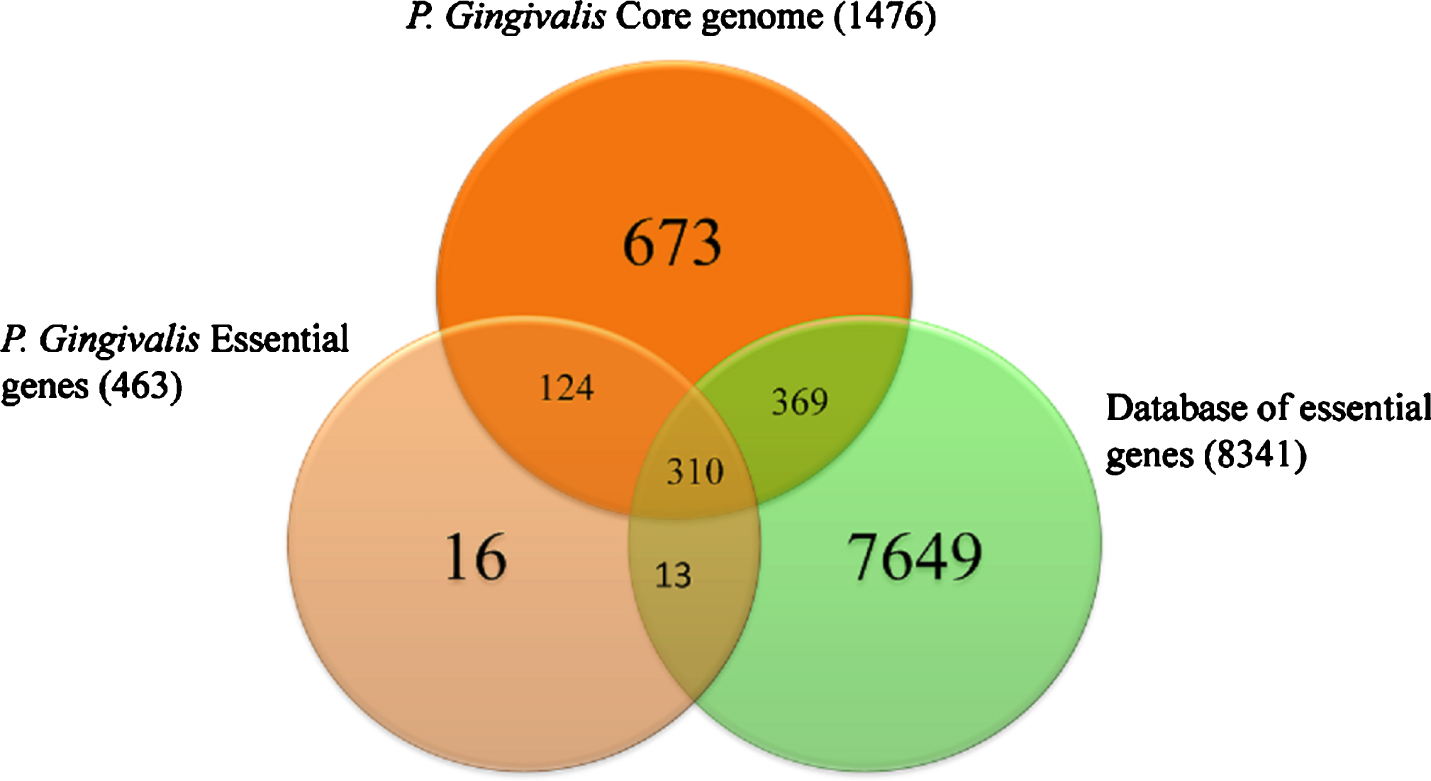
Pg belongs to the phylum Bacteroidetes and is a nonmotile, Gram-negative, black-pigmented, assaccharolytic, oligotrophic, acidophilic, alkaliphilic, poly-extremophile rod-/-coccobacilli shaped, obligately anaerobic, non-spore-forming pathogenic bacterium. It has to overcome significant changes in pH (from acidic pH 5.5 to basic pH 8.5, high to low oxygen tension, temperature deviations of up to 4°C (35–39°C), numerous anti-bacterial peptides from resident commensal bacteria, numerous bacteriophages (of which none have been found in Pg—the only one to date) and a wide range of numerous anti-microbial proteins and peptides in saliva (∼1,256) and a rapidly changing nutritional condition, often described as “feast or famine”. The whole genome sequence of Pg type strain ATCC 33227 was determined in 2008 by Naito et al. [2], deposited as Genbank accession AP009380. The genome comprises a single circular chromosome of 2.35 million bp in size, averaging 48.4% G + C content, and is without a plasmid. Coding sequence coverage is 86.1%. The 2,090 protein coding sequences identified have an average length of 970 bp. Mutagenesis experiments have identified 463 genes essential for in vitro viability for strain ATCC 33227, and of those, 424 are contained within its core genome of 1,476 genes (Fig. 1) [3, 4]. Sequence alignment shows that extensive rearrangements have taken place between the two strains to produce strain-specific coding sequences (CDSs) 60% specific to ATCC 33277, 68% specific to W83 [5]. Gene similarities to other bacterial species such as Bacteroides fragilis suggests that horizontal transfers have taken place.
Fig. 1
Comparisons and overlap of essential genes and the core genome of P. gingivalis type ATCC 33227 together with the entire curated database of essential genes. Modified with permission from Klein BA, Tenorio EL, Lazinski DW, Camilli A, Duncan MJ, Hu LT (2012) Identification of essential genes of the periodontal pathogen Porphyromonas gingivalis. BMC Genomics 13, 578, under the terms of the Creative Commons CC BY license.
More recent work shows the ability of Pg to alter culture medium pH by fermenting amino acids into ammonia and weak acids such as butyric acid, acetic acid, propionic acid, and succinic acid [6]. Nelson et al. (2003) back this discovery through examination of the complete Pg genome sequence, finding multiple amino acid catabolic pathways ultimately leading to the production of ammonia [6]. Base creation, specifically synthesis of ammonia, appears to be the response Pg enacts to overcome acidic pH stress. Pg is unable to use carbohydrates for growth. Instead, Pg must rely on acquisition of peptides in order to provide essential nutrients, such as carbon and nitrogen. This study examined Pg by comparing bacteria shocked at three acidic to neutral pH conditions (5.5, 6.5, and 7.0) to bacteria vii shocked at pH 8.5. Using microarray to examine global gene expression, differential gene expression was identified in all conditions, with total genes differentially regulated ranging from 30 to almost 500 genes. Among these, genes for ammonia production, and cation gradients were found significantly upregulated in acidic conditions, indicating a possible role in base creation and cation transport for survival of Pg. Interestingly, in the stomach, Heliobacter pylori (H. pylori) is the only other exceedingly rare example of a biological extremophile in the human body and the only other bacteria in the medical microbiology literature to exhibit and break the rule of “one pathogen one disease” paradigm (to be discussed later in the paper) having evolved the ability to survive in extremely acidic conditions with periods of rapid pH changes through another novel system of ammonia synthesis with the production of urease [7].
Pg is known to constitute up to 5% of the oral bacteria population in healthy individuals, but as periodontal disease progresses, a shift is seen toward a greater percentage of Pg. Its specific niche is the periodontal pocket or sulcus below the gumline, the crevice between the tooth and the gingiva (gums) [8]. It, however, can be found in many locations in the oral cavity. Pg relies on the presence of “primary colonizers” to establish an early biofilm [9]. Typically, these are Gram-positive streptococci, which express receptors specific to the acquired pellicles that cover the tooth surface. Secondary colonizers such as Pg then colonize that existing biofilm in various spatial and temporal manners which are often co-dependent. For Pg, a key role is played by the long rod-like Fusobacterium nucleatum (F. nucleatum) which acts as a “bridge” between the early and late colonizers such as Pg which have receptors that attach to F. nucleatum [10]. In many cases, Pg is found as the minority bacterial species in the polymicrobial biofilm.
THE PRIMARY ORAL-SYSTEMIC PATHOGEN
Periodontal disease, including Pg infection, has been implicated in various conditions, disorders or diseases including, without limitation: vascular disease (e.g., cardiovascular disease, atherosclerosis, coronary artery disease, myocardial infarction, stroke and post event myocardial hypertrophy), systemic cardio-metabolic syndrome disease (e.g., type II diabetes, insulin resistance and metabolic syndrome related obesity), rheumatoid arthritis, cancer (e.g., oral-esophageal, gastrointestinal, or pancreatic cancer), renal disease, gut microbiome-related disorder (e.g., inflammatory bowel disease, irritable bowel syndrome, celiac disease), non-alcoholic fatty liver disease, non-alcoholic steatohepatitis, allergy, asthma, wound closure, age-related macular degeneration, cerebral and abdominal aneurysms, glioma, large vessel stroke C-IMT, microvascular defects and associated dementias (e.g., Parkinson’s disease), peri-implantitis and/or periodontal disease and/or associated bone loss, cognitive disorders (e.g., early, middle, and/or late dementia, Alzheimer’s disease (AD)), and longevity or age-related disorders, regenerative and stem cell dysfunction [11–14]. Within a relatively small group of oral bacterial pathogens, Pg known as a member of the Red/Orange Complex, are involved in chronic gingivitis and periodontitis. Pg was given the name “Keystone Pathogen” by Hajishengallis, to what has become identified over the years as the major driver of both the local and systemic diseases associated with an oral dysbiosis of the microbiome of the mouth and gums [15].
VIRULENCE OF Pg CELLS
Just the listing of so many systemic inflammatory diseases, that are so anatomically distant from the primary site of infection in the oral cavity, suggests that either: 1) a rather regular/progressive seeding of the body via systemic planktonic bacterial spread of the bacteria is occurring and establishing other major/minor reservoirs of infection-causing many end organ pathologies directly and/or 2) the orally localized Pg infection is producing multiple systems diseases from afar. If the latter is the case, what would that mechanism(s) be and what is the evidence for one mechanism over the other or both? Since its first discovery in the 1950s by H. Werner in Germany [16], Pg and its over 10,000 peer reviewed publications (and counting) have roared into the field of medical microbiology and suggests a very active interest in many aspects of the organism and suggests a further source of discovery for a highly complex set of evolved processes ranging from infection, to chronic colonization, host evasion, metabolism, genomics, biofilm formation, its poly-microbial interactions and overall survival.
Gram-negative bacteria like Pg have evolved numerous cellular and extracellular mechanisms for accomplishing these feats [17]. In this hypothesis, the extracellular mechanism(s) will be discussed and integrated into a primary model of Pg disease pathogenesis. It is hoped that the information here will open thought, dialogue, and research into a different direction than is the currently held dogma of Pg—that of the bacterial cells being the focus of distant site organ disease. The extracellular mechanisms of bacterial pathogenesis, known to exist for a number of other Gram-negative bacteria, fits the existing data better and makes a more than plausible explanation how Pg alters host cell biology, functions of innate and acquired immune responses, avoidance and deregulation, nutrient acquisition, pathogenesis, and biofilm development and enables chronic (lifelong in the case of Pg) bacterial colonization.
Pg: AVOIDING LAND MINES IN THE ORAL CAVITY-ORAL BACTERIOPHAGES
Pg exists in an environment loaded with symbiotic commensal bacterial communities. In addition to a number of mechanisms to protect their community, there exist large numbers of bacteriophages that regulate and lyse invading opportunistic bacteria. Bacteria have developed a sort of primitive immune system to address the infection with these bacteriophages. The oral cavity contains approximately 700 prokaryote species harboring known CRISPR Cas systems, including type I, type II, type III, type V, and type VI, and unidentified CRISPR Cas systems. Moreover, recently, new functions, other than defense, have been attributed to CRISPR systems of the oral microbiome [18] compared to healthy sites in the same individual; it would be expected that under both conditions at both sites, oral phages would be present. There is increasing evidence to suggest that different CRISPR Cas systems in the human oral microbiome can affect bacterial physiology through different mechanisms including defense against the invasion of foreign mobile elements, biofilm formation, acquisition of resistance genes, DNA repair, regulation of interspecific competition and intraspecific diversification, stress responses, and gene expression regulation [19]. The mechanism of the CRISPR-Cas system was first demonstrated in 2007 in Streptococcus thermophilus [20]. Clustered regularly interspaced short palindromic repeats CRISPRs belong to a family of DNA sequences characterized by short direct repeats that are separated by spacers [21]. Almost half (45%) of the species in Bacteria and the majority (80%) in Archaea contain CRISPR-Cas systems [22]. Through an adaptation process, the system is able to store memories of previous exposure to foreign DNA in the spacer sequences across generations of progenies [22–24]. Hence, the spacers serve as the ‘acquired memory’ of this adaptive immunization system and are at the center of CRISPR defense providing immunity against specific phages or plasmids. Cas proteins are encoded by the cas genes that are located proximal to the CRISPR array.
Most amazingly even though bacteriophages have been isolated from many oral bacteria, a bacteriophage for Pg has yet to be isolated despite the efforts in this area [25, 26]. This is a significant finding in the bacterial community among the medically important bacteria. The fact that it sits at the top of the bacterial phage food chain gives Pg a sort of bacterial immortality. ISPR-Cas systems may defend against oral phages; there are studies and clinical data that do not fully support the notion that oral phages alone are responsible for the changed expression occurring mainly in the context of the transition from periodontal health to disease. CRISPR-Cas genes were highly up-regulated at sites with clinical evidence of transition to disease.
Pg FLYING BELOW THE HOST RADAR-INFLAMMATION ENIGMA
Host recognition of Gram-negative bacteria occurs via detection of LPS expressed on the bacterial membrane by the innate immune receptor, TLR4 [27]. This initial recognition is critical on the host side for instructing host immunity and promoting an inflammatory response that eradicates the pathogen from the host [28, 29]. However, on the pathogen side a number of Gram-negative organisms have evolved mechanisms to modify their lipid A species, the component of bacterial LPS that directly activates the TLR4 complex (Fig. 2), as a strategy to evade immune detection and establish infection [30]. Lipid A is initially synthesized as a b-19, 6-linked disaccharide of glucosamine that is phosphorylated and fatty acylated [31]. An unmodified version of this lipid A structure is typically expressed by E. coli and induces a robust inflammatory response as is when in septic shock [32]. Modifications to this basic lipid A structure are observed in alterations to acyl chains or terminal phosphate groups [33]. H. pylori [34], Legionella pneumophila [35], Yersinia pestis [36], and Francisella novicida [37] express under-acylated lipid A moieties, in comparison to the canonical LPS expressed by E. coli, and are poorly recognized by TLR4. Yersinia pestis [38] and Francisella tularensis [39] expression of structurally divergent forms of lipid A is highly regulated by local environmental conditions such as temperature, allowing these pathogens to adapt to harsh environmental conditions in the host.
Fig. 2
P. gingivalis dysregulates host cell immune activation facilitating systemic inflammation. The host predominantly senses P. gingivalis infection through engagement of TLR2 while the involvement of TLR4-dependent recognition is significantly impaired. Expression of antagonistic or immunologically inert lipid A by P. gingivalis attenuates production of proinflammatory mediators and prevents activation of the inflammasome (potentially through evasion of an unknown sensor) that facilitates intracellular survival. These events allow the pathogen to disseminate and to exacerbate systemic inflammation. In contrast, increased immunostimulatory potential at TLR4, through expression of an agonistic lipid A moiety, results in increased production of proinflammatory mediators, inflammasome activation, and reduced survival of the bacterium in macrophages leading to attenuated systemic inflammation. Reprinted with permission from Slocum C, Coats SR, Hua N, Kramer C, Papadopoulos G, Weinberg EO, Gudino CV, Hamilton JA, Darveau RP, Genco CA (2014) Distinct lipid A moieties contribute to pathogen-induced site-specific vascular inflammation. PLoS Pathog 10, e1004215, under the terms of the Creative Commons Attribution License.
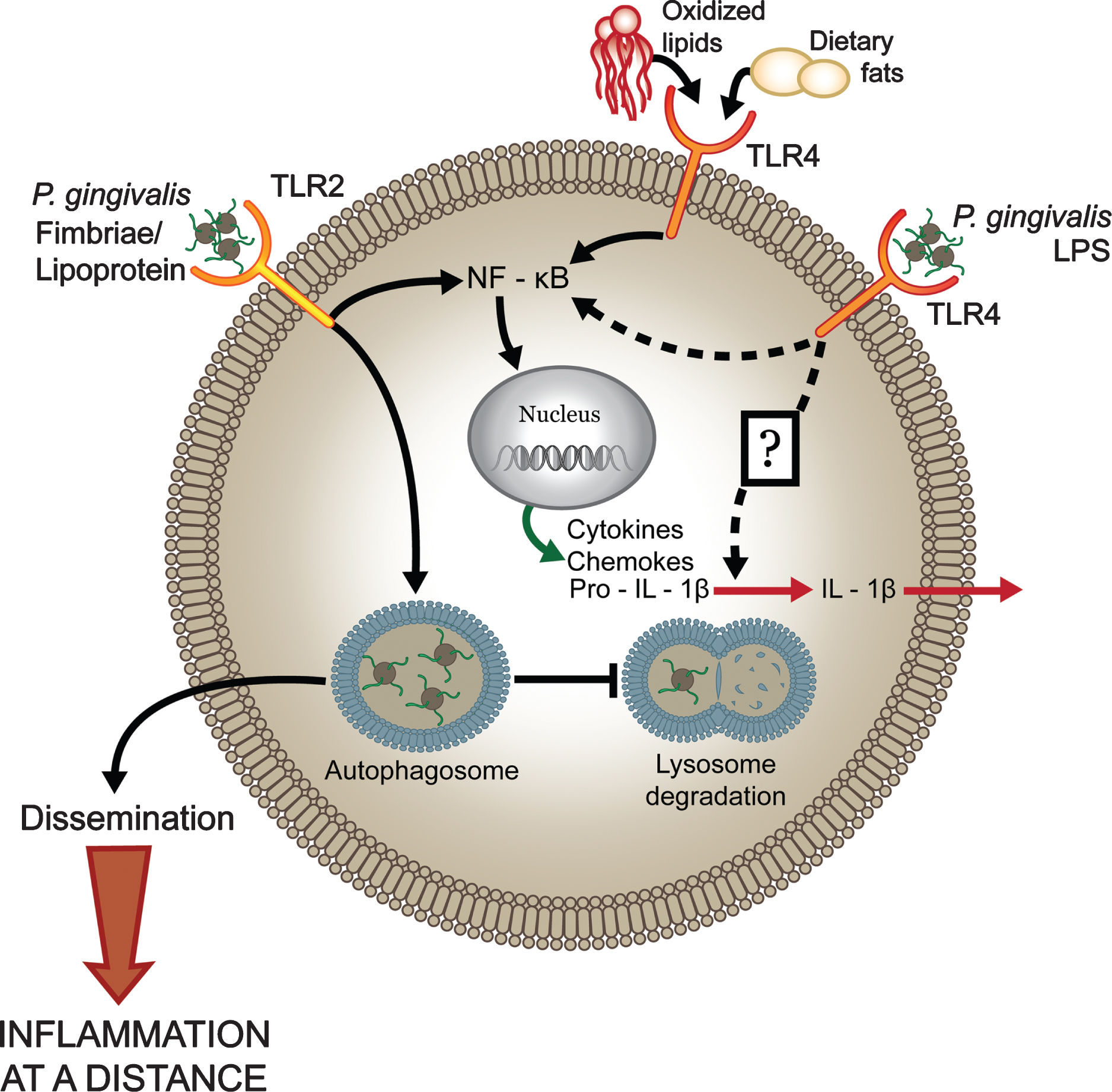
It has been postulated that the ability of these types of pathogens to cause persistent infection and severe disease is partially due to evasion of host immune detection at TLR4 [40]. Recently, it has been revealed that in addition to evasion of TLR4 signaling, lipid A modifications promote evasion of the noncanonical inflammasome by preventing activation of caspase-11 [41]. Activation of the inflammasome is characterized by the production of the prion-flammatory mediators IL-1b and IL-18 and is associated with downstream events such as pyroptosis [42]. Due to its role in host innate defense, a number of pathogens have evolved strategies to evade activation of this complex [43]. Pg expression of antagonist lipid A species results in attenuated production of proinflammatory mediators and evasion of non-canonical inflammasome activation, facilitating bacterial survival in the macrophage. Infection of atherosclerosis-prone ApoE2/2 mice with this strain resulted in progression of chronic inflammation in the vasculature. Notably, and most importantly, the ability of Pg to induce local inflammatory bone loss was independent of lipid A modifications, supporting distinct mechanisms for induction of local versus systemic inflammation.
Evasion of immune detection at TLR4 contributes to pathogen persistence and facilitates low grade chronic inflammation. Pathogen evasion of inflammasome activation has been proposed to serve a dual role: to dampen cytokine production and to prevent host cell death in order to provide an intracellular niche for the pathogen to survive [44]. One pathogen that has successfully adapted to evade the inflammasome is H. pylori, through expression of its tetra-acylated lipid A [41]. An important observation from this study was that H. pylori failed to activate the inflammasome. It has recently been reported that Gram-negative bacteria utilize a non-canonical pathway for inflammasome activation that is mediated by caspase-11 [45, 46].
Kayagaki et al. [41] have shown that H. pylori, again the only other known bacteria than Pg in the human host to exist as a chronic pathogen in extreme environment like the gastric mucosa and oral submucosa respectively and to cause multiple serious diseases and whose tetraacylated lipid A also poorly activates the TLR4 complex and fail to trigger the non-canonical inflammasome. Although TLR4 signaling correlates with non-canonical inflammasome activity, this group reported that activation of the non-canonical inflammasome is independent of TLR4 signaling, further suggesting an unknown sensor in the cytosol that detects modified lipid A (Fig. 3). It was found that caspase-11 expression was essential for IL-1b production elicited by Pg strain PG1587381, pointing to a pivotal role for activation of the non-canonical inflammasome in Pg infection. In all of these cases, it appears again that OMVs play an important role in delivering toxins such as the various LPS moieties, thus allowing for their translocation into various host immune cell types.
Fig. 3
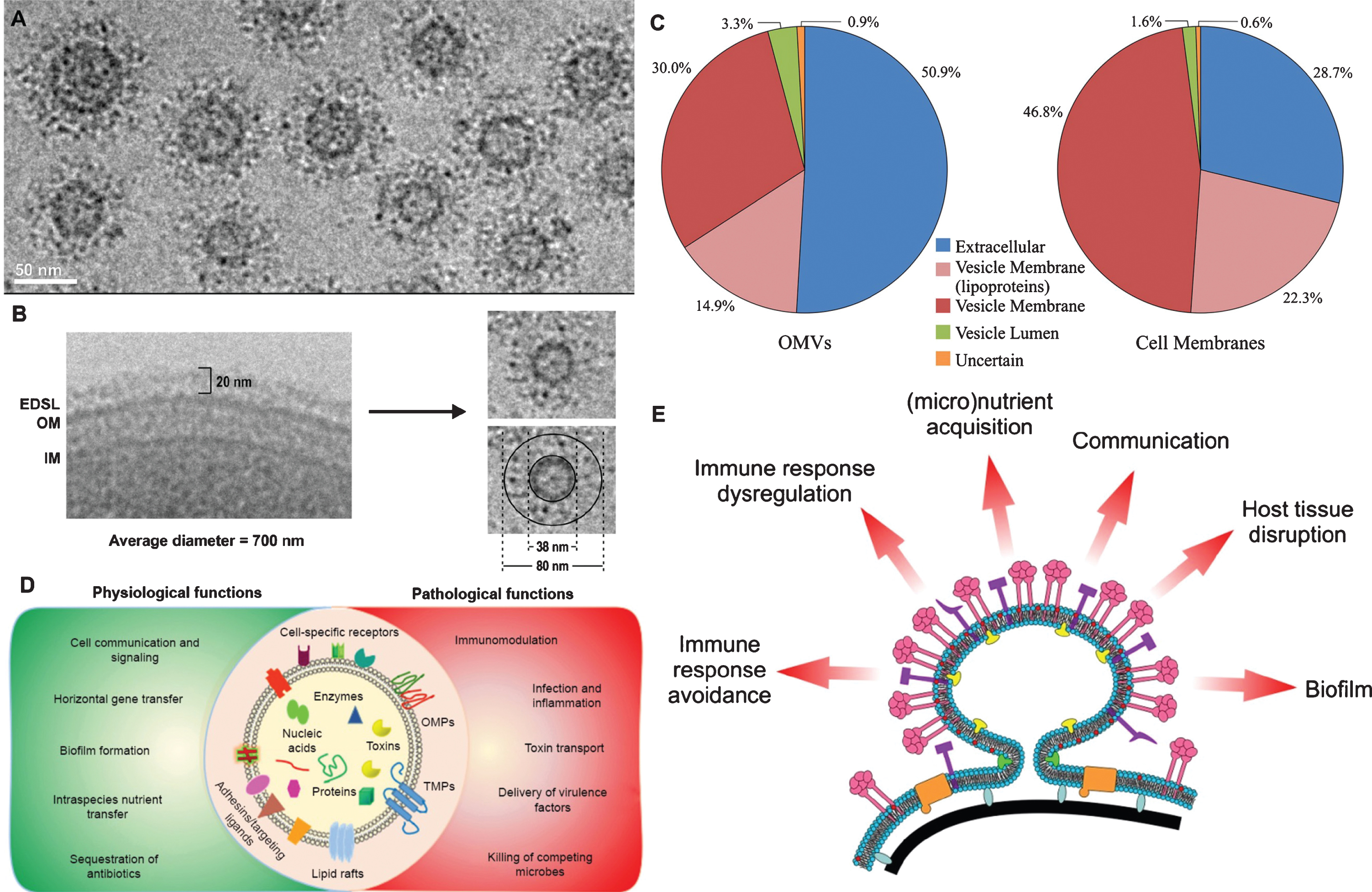
Outer membrane vesicles (OMVs) have an increased amount of EDSL relative to cells. A) A cryo-TEM image of several OMVs showing the presence of a thick EDSL surrounding the membrane of each vesicle. B) A cryo-TEM image of a typical P. gingivalis cell and a typical vesicle with a diameter of 80 nm, with dimensions of the membrane and EDSL shown. The increased curvature of OMVs allows for an increased amount of surface layer (EDSL) relative to the OM. C) CTD Proteins are enriched in OMVs. The average total Mascot scores obtained for proteins within each OMV localization were summed to give an estimate of the total protein content of that locale. The corresponding scores for these proteins obtained from the cell membranes sample were similarly summed. D) It can be seen that the extracellular proteins (CTD proteins) are highly enriched in the OMVs relative to the outer membrane of the cell. E) A schematic of P. gingivalis OMV biogenesis and their putative functions. Pink symbols represent the RgpA/Kgp proteinase adhesin complexes and other type IX secretion system substrates. Purple symbols represent adhesive lipoproteins including HmuY and IhtB. Tan symbols represent TonB-linked transporters. Red symbols denote the abundance of anionic lipopolysaccharide (A-LPS) in the OMV membrane while yellow and green symbols indicate hypothetical proteins/compounds involved in biogenesis of the OMVs and the curvature of the membrane. Black represents the cell wall and blue/grey indicates Brauns proteins linking the cell wall to the outer membrane. Panels A-C are reprinted with permission from Veith PD, Chen YY, Gorasia DG, Chen D, Glew MD, O’Brien-Simpson NM, Cecil JD, Holden JA, Reynolds EC (2014) Porphyromonas gingivalis outer membrane vesicles exclusively contain outer membrane and periplasmic proteins and carry a cargo enriched with virulence factors. J Proteome Res 13, 2420–2432. Copyright (2014) American Chemical Society. Panel D is reprinted with permission from Jain S, Pillai J (2017) Bacterial membrane vesicles as novel nanosystems for drug delivery. Int J Nanomedicine 12, 6329–6341, under the Creative Commons Attribution - Non Commercial (unported, v3.0) License. Panel E is reprinted with permission from Gui MJ, Dashper SG, Slakeski N, Chen YY, Reynolds EC (2016) Spheres of influence: Porphyromonas gingivalis outer membrane vesicles. Mol Oral Microbiol 31, 365–378, with permission the Wiley online library under the Creative Commons Attribution (CC BY) license.
It is well established that atherosclerosis progression is due to excessive production of proinflammatory mediators [30]. Although lipid deposition is considered a leading contributor to the inflammation, additional stimuli, such as infectious agents, have been considered as sources for the continuous inflammation. Infection with Pg induced low levels of proinflammatory mediators but accelerated chronic distant site inflammatory atherosclerosis. Thus, the question arises as to why a pathogen that induces low levels of inflammatory mediators would accelerate chronic inflammatory atherosclerosis (to be discussed later).
OMVs VERSUS BACTERIAL CELLS
Outer membrane vesicles
There is good evidence that some oral bacterial pathogens are implicated in neuroinflammatory diseases and can be localized in the brain [47, 48], but if and how these microbes cross the blood-brain barrier (BBB) is still very debatable. The numbers overall found in the brain are very few and limited in number, making an etiology associated linkage difficult. It has been shown that extracellular vesicles can among many things facilitate the inter-kingdom exchange of RNAs as communication molecules both within and outside of the biofilm [49, 50]. Host cells take up the RNA and vast protein cargo of OMVs and along with the OMVs themselves in toto [51]. OMVs were first discovered in Vibrio cholerae in 1967 [52] but have not received much attention in disease causation until more recently. Recent findings suggest that OMVs are utilized by bacterial pathogens and commensal bacteria to manipulate the host innate and acquired immune responses and communicate with host cells in preparation of invasion and or to interfere with local cellular host defenses. In general, OMVs were reported earlier to consist of a proto-liposome membrane with microbe-associated molecular pattern recognition molecules, exo-endotoxins, e.g., LPS, and peptidoglycans. This limited view, however, has changed greatly [53, 54] and will be discussed.
In Pg, the major and biologically significant virulence factor(s) are the production of small (20–500 nm) proteo-liposomal membrane vesicles (OMVs) via the Type IX cargo secretion system that organizes and distributes macro and micro molecules through its cell membrane and into specific protein-lipo-protein structures [55] (Fig. 3). Through three major mechanisms, this protein-protein ligand, protein-RNA, RNA-mediated and DNA-mediated signal and manipulates a multitude of cell types in a wide variety of ways.
Pg OMVs vary significantly in their biogenesis and protein cargos [56]. Some strains of Pg OMVs have greatly enhanced and different cargos with extraordinarily large numbers (>100) of concentrated molecular components mostly derived from that of the bacterial cell membranes and its outer membrane constituents including all the known virulence proteins, lipoproteins-LPS and cell signaling factors, miRNAs, msRNA, sRNAs, exRNAs, and DNA. The Pg OMV proteome consists of 30 C-terminal secretion signals known as CTD proteins which localize to the EDSL, 79 vesicle membrane proteins, 27 vesicle lumen proteins, and 15 others less defined for a total of 151. An abbreviated list is found in Table 1. A search of the literature for recent studies on Pg and other Gram-negative OMV proteomes revealed eight studies that reported the identification of at least 100 proteins [56]. A major conclusion from this work is the wholesale concentration of Pg CTD proteins onto/into the surface of OMVs. The total Mascot score of all CTD proteins from a OMV sample accounted for 51% of the total, approximately half of which was contributed by the endo-peptidases known as the lysine and arginine gingipains. In contrast, only 3.3% of the total Mascot score was contributed by proteins localized to the lumen. The LptO protein, having a role in the attachment of CTD proteins to the cell surface, was one of the proteins most enriched in the OMVs. The other two proteins of known function that were highly enriched in the vesicle membrane were IhtB and HmuY, both of which are surface-exposed lipoproteins involved in iron acquisition (to be discussed later). The overall picture is that OMVs are highly virulent particles, equipped with the tools to invade host tissues, dysregulate and modulate various cellular functions, avoid the adaptive and acquired immune responses, and degrade protein such as hemoglobin for the acquisition of both peptides and heme. Gingipains consist of arginine-specific (Rgp [two subtypes RgpA and RgpB]) and lysine-specific (Kgp) protease types which have been implicated in a wide range of both pathological and physiological processes of the Pg. This includes degradation of collagen and fibronectin, degradation of various cytokines, resulting in a disturbance of the host cytokine network, disruption of host defense mechanisms, inhibition of interactions between host cells and the extracellular matrix and loss of viability in host cells, such as fibroblasts and endothelial cells [57, 58]. Evidence of their importance is observed in (Fig. 3), and Table 1 demonstrates the large amounts found in Pg OMVs.
Table 1
Comparison of the top 20 most abundant proteins identified in Porphyromonas gingivalis 33277 and W83 vesicles. Reprinted with permission from Mantri CK, Chen CH, Dong X, Goodwin JS, Pratap S, Paromov V, Xie H (2015) Fimbriae-mediated outer membrane vesicle production and invasion of Porphyromonas gingivalis. Microbiology Open 4, 53–65, under the terms of the Creative Commons CC BY license
| Rank | 33277 | W83 |
| 1 | Lys-gingipain, kgp | Arg-gingipain, RgpA |
| 2 | Arg-gingipain, RgpA | Receptor antigen A, RagA |
| 3 | Por secretion system protein PorV | Por secretion system protein PorV |
| 4 | Arg-gingipain, RgpB | Arg-gingipain, RgpB |
| 5 | Receptor antigen A, RagA | Receptor antigen B, RagB |
| 6 | Peptidylarginine deiminase | Peptidylarginine deiminase |
| 7 | Hemagglutinin protein, HagA | Hemagglutinin protein HagA |
| 8 | Major fimbrial subunit protein type-1 FimA | Immunoreactive 61 kDa antigen |
| 9 | Receptor antigen B, RagB2 | Uncharacterized protein (PG_1823, PGN_1744) |
| 10 | Immunoreactive 61 kDa antigen | Zinc carboxypeptidase, putative |
| 11 | Uncharacterized protein (PGN_1744) | Uncharacterized protein |
| 12 | Mfa1 fimbrilin, Mfa1 | Uncharacterized protein (PG_1626, PGN_0477) |
| 13 | Putative lipoprotein | Putative lipoprotein |
| 14 | Zinc carboxypeptidase, putative | 35 kDa hemin binding protein |
| 15 | Immunoreactive 23 kDa antigen | Extracellular protease, putative |
| 16 | Uncharacterized protein (PGN_0477) | Immunoreactive 47 kDa antigen |
| 17 | Immunoreactive 47 kDa antigen | Uncharacterized protein |
| 18 | 35 kDa hemin binding protein | Uncharacterized protein |
| 19 | Putative uncharacterized protein | Heme-binding protein FetB |
| 20 | Extracellular protease, putative | Outer membrane protein 41 |
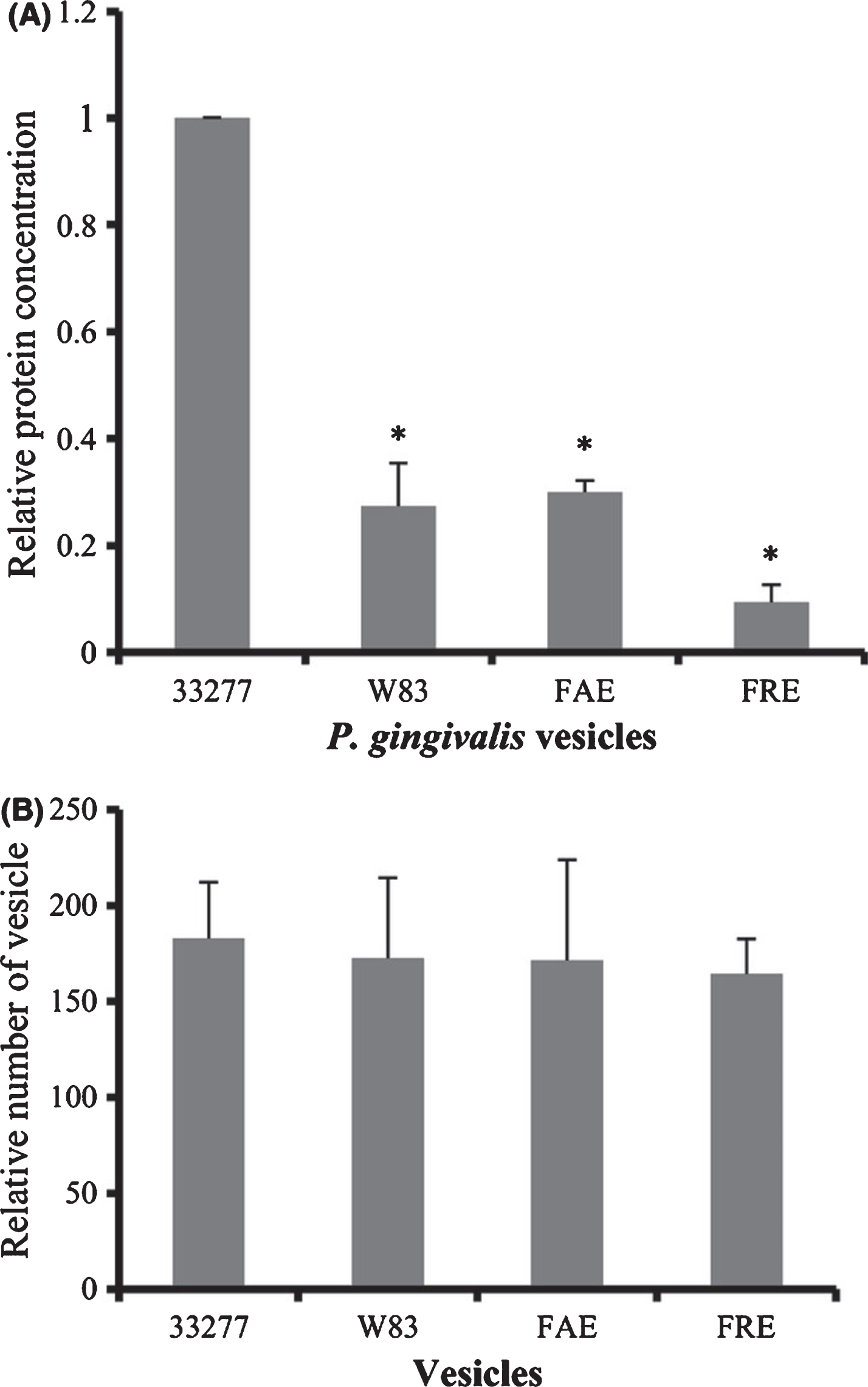
The fimbriae are proteinaceous filamentous projections on the Pg bacterial cell surface that mediate bacterial adherence to and entry into periodontal cells, are essential to plaque formation [59] and incorporated into OMVs. The fimA gene is classified into six types (genotypes I, Ib, II, III, IV, and V) on the basis of different nucleotide sequences, with fimA genotypes II and IV being associated with increased virulence and prevalent in isolates from patients with periodontitis and other systemic cardiovascular inflammatory diseases. Together, fimbriae and gingipains are responsible for the adhesive, proteolytic, and OMV production capacity. Mantri et al. [60] demonstrated a higher level of vesiculation was found in the growth media of strain 33277 when compared to strain W83 media (Fig. 4). An intriguing finding was that two a fimbriated strains, the fimA mutant and the fimR mutant, produced significantly fewer vesicles than its parent strain 33277. A recent study by Kerr et al. [61] using transmission electron microscopy showed a copious number of OMVs on the surfaces of many Pg strains including 33277, while fewer vesicles were observed on the surface of W83. Another interesting finding was the very different levels of OMV proteins that were found in the two different strains of Pg (Fig. 4).These results suggest that FimA production or its regulatory system is likely involved in vesicle biogenesis and their proteome in Pg. These findings strongly suggest that strains of Pg with specific fim A subtypes have very different qualitative and quantitative changes in their OMV related biochemical and biological properties and suggests that a better understanding of these characteristics may shed light in terms of the ability and types of clinical disease they may producing (Fig. 5).
Fig. 4
Comparison of vesicle production from P. gingivalis strains. A) Protein concentrations of vesicle suspensions. B) Equal amounts of vesicle suspensions were determined by averaging the number of vesicles in 10 electron microscopic images. C) Different strain of P. gingivalis. Reprinted with permission from Mantri CK, Chen CH, Dong X, Goodwin JS, Pratap S, Paromov V, Xie H (2015) Fimbriae-mediated outer membrane vesicle production and invasion of Porphyromonas gingivalis. Microbiology Open 4, 53–65, under the terms of the Creative Commons CC BY license.
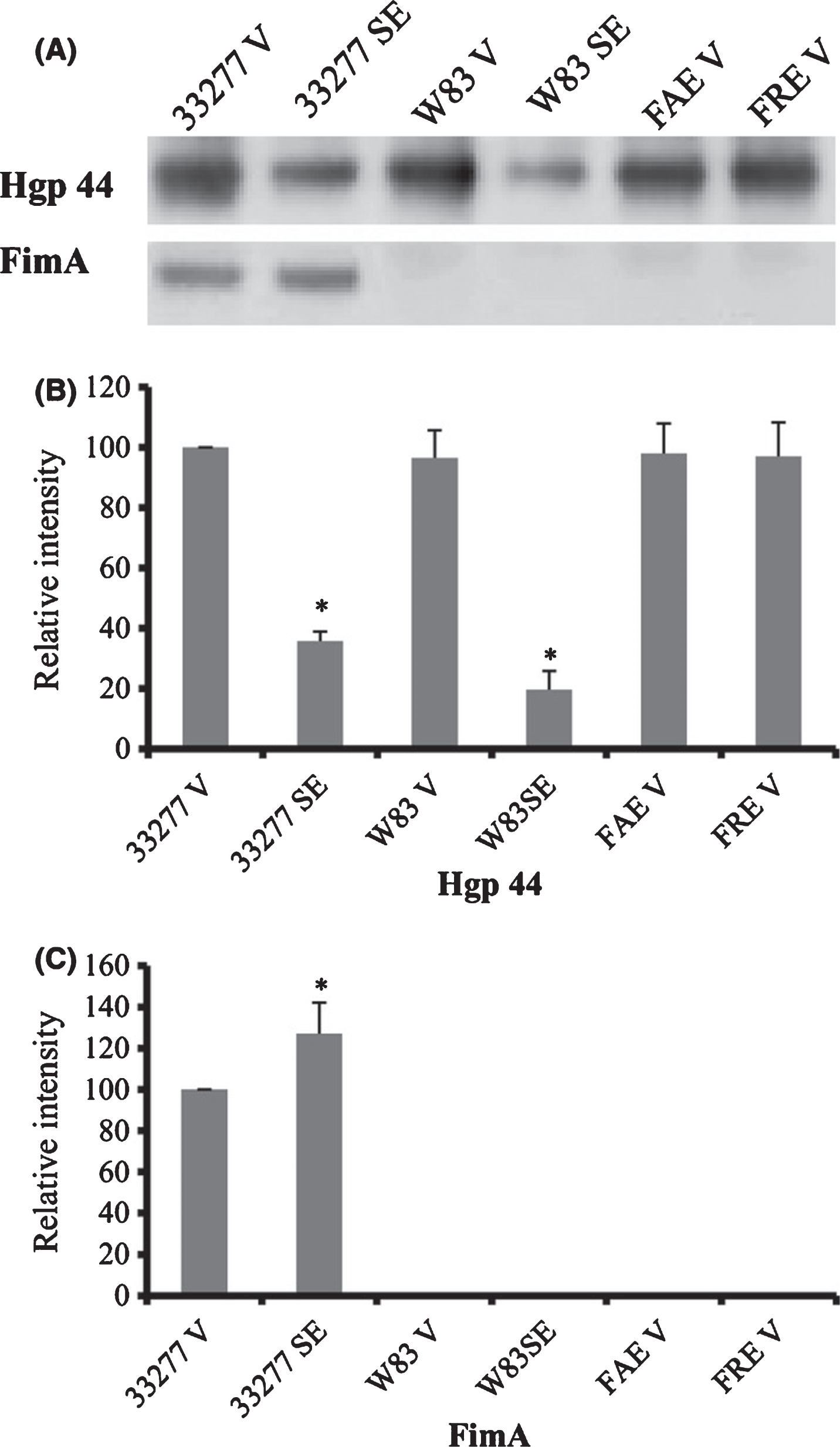
Fig. 5
Expression of Hgp44 and FimA protein in P. gingivalis vesicles. A) Levels of FimA and Hgp44 of gingipains in surface extracts (SE) of Pg33277 or an afimbriated strain (W83), and in vesicles (V) of 33277, W83, fimAmutant (FAE), and fimRmutant (FRE) were analyzed using western blot analysis with rabbit anti-FimA or rabbit anti-Hgp44 sera. Reprinted with permission from Mantri CK, Chen CH, Dong X, Goodwin JS, Pratap S, Paromov V, Xie H (2015) Fimbriae-mediated outer membrane vesicle production and invasion of Porphyromonas gingivalis. Microbiology Open 4, 53–65, under the terms of the Creative Commons CC BY license.
Following entry of OMV into host cells, OMV-associated gingipains degrade cellular functional molecules resulting in cellular impairment. Via OMV, Pg virulence factors can penetrate an intact mucosa and enter underlying host tissues [62], ultimately resulting in periodontitis. Finally, the lipopolysaccharide of Pg (LPS-PG) is integrated into and transported via OMVs and known to be involved in the etiology of distant organ diseases-for example, chronic systemic exposure to the lipopolysaccharide of Pg induces the accumulation of amyloid-β (Aβ) in the brain of middle-aged mice an abnormal accumulation of which is the most significant pathological hallmark of AD [63]. These data imply that vesicles production and assembly are regulated in Pg.
OMVs: By the numbers
Understanding how Pg bacterial cells and OMVs spatially exist in situ and interact with host tissues in a setting of health and disease is important. This includes to what extent they co-localize or not within the oral tissues they inhabit, and where they mostly reside in their poly-microbial biofilm as well as to what extent they invade and can inhabit the epithelia cells of the mouth and gums. Although both Pg and its OMVs have been found to invade multiple cell types both in vivo and in vitro, existing studies demonstrating their localization, especially in the primary source of the mouth remains an important unmet issue and pertains to how the bacteria may be mediated local versus systemic disease [64]. Surprisingly to date, there are very few studies specifically addressing this issue; however, as will be now discussed, there are some relevant studies that begin to address this important topic. We have seen that Pg can secrete large numbers and amounts (protein concentrations) via OMVs.
Stepping back for a moment, what evidence is there in the literature for OMV specific production in the setting of disease? As seen in Fig. 6, there are some reported studies directly demonstrating bacteria in situ and their secreted OMVs in various diseases, demonstrating that OMVs are in fact released in the actual diseased tissues and not just a tissue culture phenomena by Gram-negative bacteria.
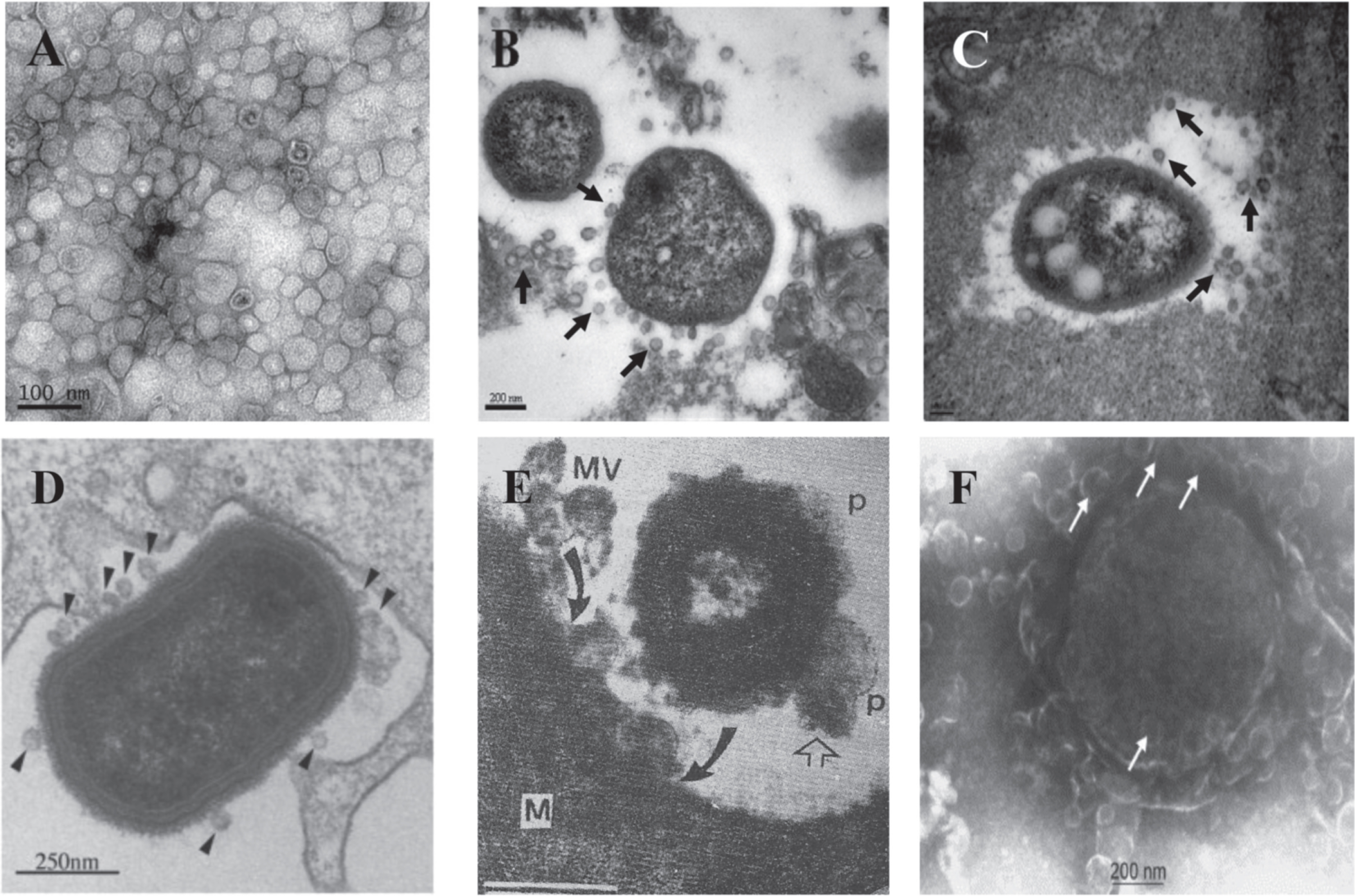
Fig. 6
Secretion of OMVs in situ from A. baumanniiduring in vitro culture and in vivo infection. A) Transmission electron micrograph of OMVs prepared from A. baumannii ATCC 19606T cultured in LB broth for 24 h. B, C) Secretion of OMVs from A. baumannii ATCC 19606T in a murine pneumonia model. Mice were infected with 16107 CFU of bacteria intratracheally and sacrificed 48 h after bacterial injection. Arrows indicate the OMVs secreted from A. baumannii. D) TEM image of P. gingivalis entering human epithelial cell and producing numerous OMVs (arrowheads). E) TEM of human Salmonella organism bearing periplasmic organelles (p, line arrow) on its surface and releasing bacterial outer membrane vesicles (MV) being endocytosed (curved arrow) by macrophage cell (M) in chicken ileum in vivo. F) Meningococcus cell in plasma of acute meningitis patient showing blebbing of numerous outer membrane vesicles with a blood level of LPS of 1700 endotoxin units/mL or approx. 170 ng/ml of purified E. coli LPS (65,000X mag.) Panels A-C are reprinted with permission from Jin JS, Kwon SO, Moon DC, Gurung M, Lee JH, Kim SI, Lee JC (2011) Acinetobacter baumannii secretes cytotoxic outer membrane protein A via outer membrane vesicles. PLoS One 6, e17027, under the Creative Commons Attribution License. Panel D is reprinted from Amano A, Takeuchi H, Furuta N (2010) Outer membrane vesicles function as offensive weapons in host-parasite interactions. Microbes Infect 12, 791–798, with permission Elsevier. Panel E is reprinted from Wikiwant (https://www.wikiwand.com/en/Membrane_vesicle_trafficking) under the Creative Commons Attribution-ShareAlike 4.0 International License (CC BY-SA). Panel F is reprinted from Namork E, Brandtzaeg P (2002) Fatal meningococcal septicaemia with “blebbing” meningococcus. Lancet 360, 1741, with permission from Elsevier.
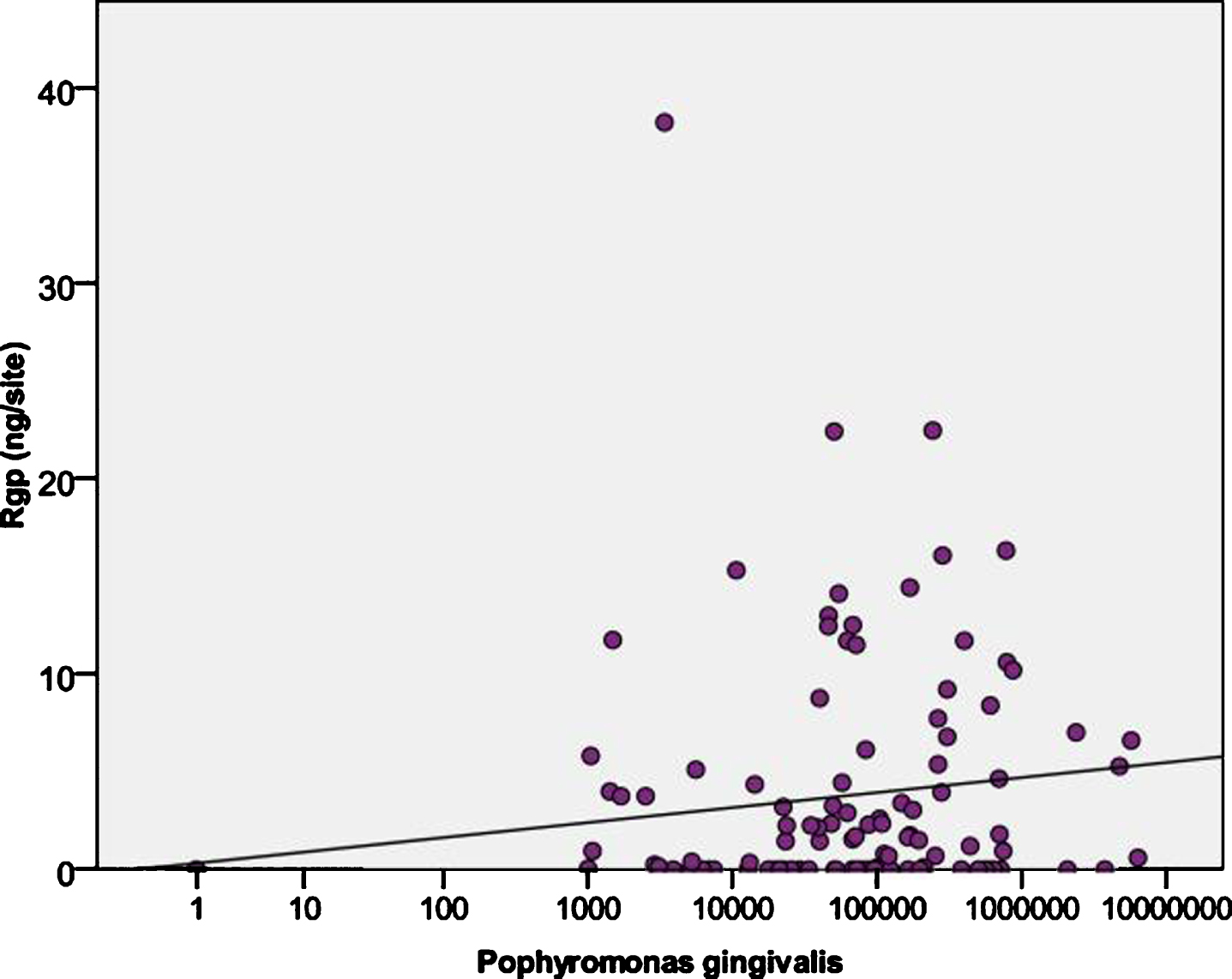
In our own studies of Pg in culture and as mentioned depending on the strains, hundreds of OMVs can be observed emerging from the cell membrane at the same time (Nara et al., unpublished studies) and on most if not all cells suggesting that at any relative time point 1.0×10(9) CFUs of Pg can produce 1.0×10 (11) or greater OMVs, which over time on a continuing basis could produce a significant amount of material (Nara et al., unpublished studies). In vivo data is lacking on the amounts and conditions whereby OMVs can be produced from many Gram-negative bacteria. Mantri et al. [60] demonstrated using Pg OMVs the detachment of human gingival fibroblasts (HGFs) or human oral keratinocytes (HOKs) cells in vitro was not observed with a protein concentration of vesicles (0.5μg/mL), but resulted in detachment when HGFs were exposed to a higher protein concentration of vesicles (>3μg/mL) from both 33277 and W83 (data not shown). Therefore, it is conceivable that production of Pg OMVs may be a major mechanism for release and delivery of gingipains/LPS and many other virulence factors to local and distant tissue sites throughout the body. In the only in vivo study to date, Guentsch et al. [65] in Pg infected patients demonstrated by ELISA, using a specific anti-gingipains antibody, that gingipains levels taken from Pg positive patients by paper points resulted in 64% of the 109 samples being positive for both Pg bacteria and gingipains in the GCF per gum/tooth site. The levels ranged from 0–38.2 ng with a median of 1.45 ng/site in a GCF volume of 1-2μl. This suggests that gingipains in GCF could be on the order of 1.5 ng/μl or 0.001μg/ml or 1.0 mg/L, approaching normal levels of serum CRP. Thus, it appears that under clinical conditions, relatively high amounts of the major cell free Pg gingipains toxins are being released (Fig. 7) and available to the GCF/lymphatic and micro-vascular systems. As the majority of Pg gingipains are known to be packaged and associated with OMVs, this represents the only direct measurement to date in the literature. Currently we are conducting studies to measure Pg OMVs in blood of Pg infected patients (Nara et al., unpublished studies).
Fig. 7
Levels of arginine-specific cysteine protease (Rgps from P. gingivalis) in GCF fluids in relation to the load of P. gingivalis. Reprinted from Guentsch A, Kramesberger M, Sroka A, Pfister W, Potempa J, Eick S (2011) Comparison of gingival crevicular fluid sampling methods in patients with severe chronic periodontitis. J Periodontol 82, 1051–1060, with permission from John Wiley and Sons.
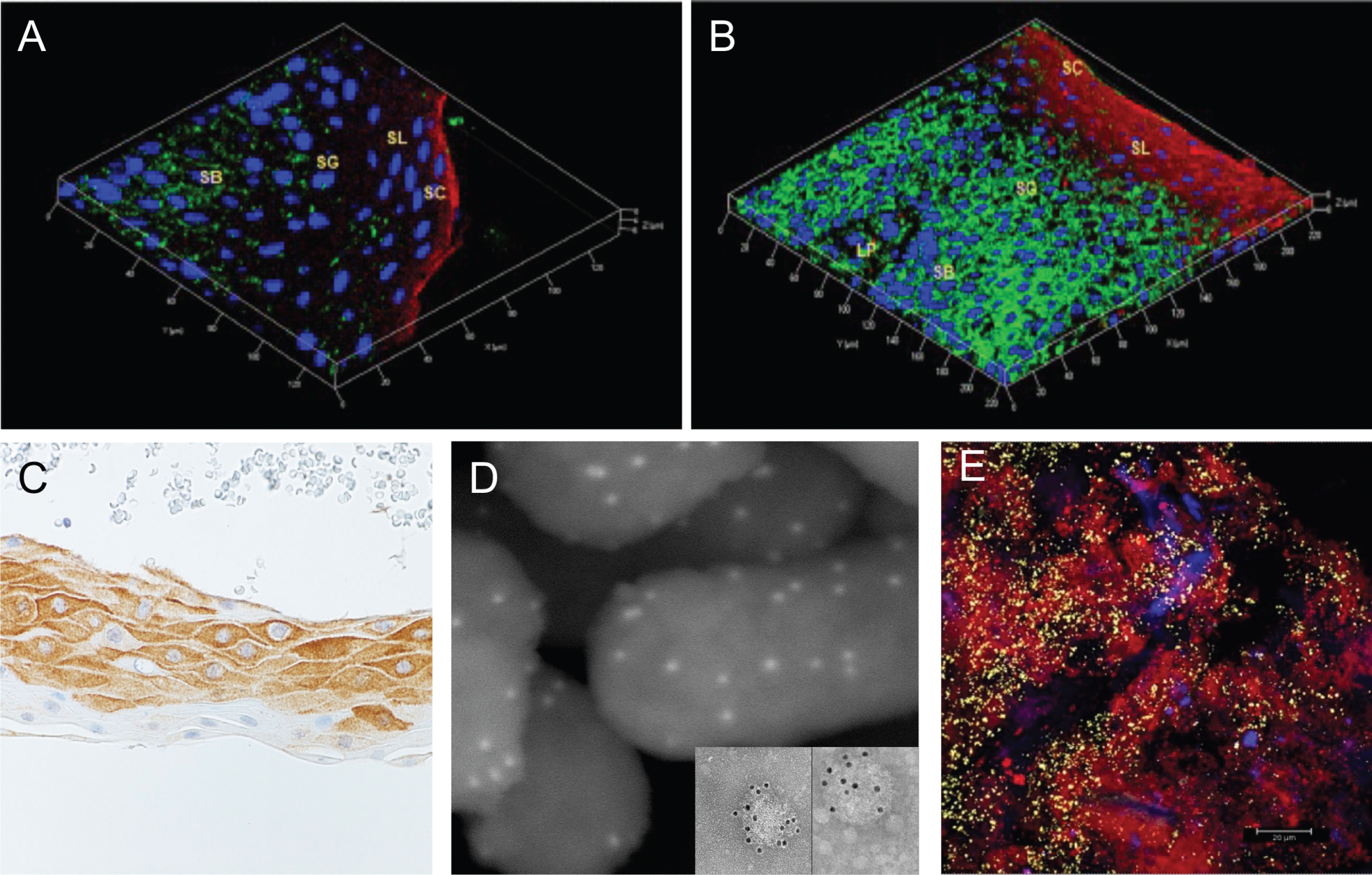
To assign the local production of OMVs being responsible for systemic diseases from Pg colonies found in the mouth requires some direct evidence whereby the bacteria are producing measurable amounts of OMVs in tissues. Evolutionarily speaking, keeping the vascular system sterile especially from oral bacteria while having the oral mucosal epithelium serve as a physical barrier with a variety of antibacterial host defense machineries should stop most large amounts of bacteria from getting into systemic circulation thus causing septic shock. An elegant study by Lee et al. [66] looked at an the in situ intraepithelial localization of Pg and Fili factor alocis, using clinically derived human gingival samples. Gingival tissues from these patients reveal increased formation of gingival microvasculature associated gingival epithelium under chronic inflammation and the attending clinical sign of bleeding on probing. Interestingly, when using a polyclonal, anti-Pg antibody, which was not well characterized for the target Pg antigen, a rather broad and homogenously diffuse staining pattern was observed concentrated and localized near the basement membrane or external basal lamina of the gingival epithelium (Fig. 8B) suggesting that a florid number of bacterial cells must be throughout the tissues. However, when FISH-RNA analysis/overlay (that detects metabolically active bacteria) was performed on these same sections (Fig. 8AA), significantly reduced and focal smaller well delineated clumping/bacterial colonies were observed. This result suggested smaller numbers of bacteria were actually in the tissue which- consistent with multiple other studies demonstrating that Pg in the oral cavity resides in very low amounts relative to other opportunistic bacterial sp. Using our own highly specific mouse monoclonal antibody KB-001, that is directed to a unique epitope of the HagA/gingipains protein and decorates both emerging Pg OMVs on the bacterial surface and after release (Fig. 8C,D), resulted in a diffuse fine homogeneous granular staining pattern throughout the gingival epithelium in a pattern not resembling bacterial cells/colonies but more distributed solubilized bacterial proteins. A similar result was reported by O’Brien-Simpson et al. [67] using a similar antibody to the gingipains antigen complex demonstrated a similar diffuse staining. Hence, they concluded the staining pattern was indicative of a concentration gradient of the RgpA-Kgp antigen in the gingival connective tissue, as would be expected from diffusion of the antigen from subgingival plaque into the subjacent tissue. It is significant that the adhesins associated with the RgpA-Kgp proteinase/adhesin complexes have been shown to bind to various connective tissue proteins and proteoglycans such as fibronectin, laminin, and collagen type V [67, 68]. The result is also consistent with binding of RgpA-Kgp to endothelial cells [69]. Targeting of the RgpA-Kgp complexes to vessels and subsequent vascular disruption may be highly significant in the virulence of Pg in periodontitis [69].
Fig. 8
In situ and IHC analysis of P. gingivalis (Pg)-infected gum tissue. A) The metabolically active Pg cells (FISH-labeling) are demonstrated. B) In same section, the overall demonstration of the Pg/bacterial antigens using polyclonal anti-Pg antisera. C) Immuno-histochemical staining of Pg infected gum tissues using OMVanti-HagA/gingipain Mab. D) Immuno-gold SEM of W83 Pg cells showing formation of outer membrane vesicles and localization of the anti-HagA MAb and reactivity to purified W83 OMVs. E) Gingival acellular epithelial biofilm from a Pg positive patient. CLSM image: differentiation of a specific bacterial group. 2D overlay of 3D CLSM stack data. Biofilm stained with EUBmix (red, all bacteria), Bac303 (blue, Bacteroidetes), and POGI (yellow, Porphyromonas gingivalis). Panels A and B are adapted with permission from Lee JS, Spooner R, Chowdhury N, Pandey V, Wellslager B, Atanasova KR, Evans Z, Yilmaz Ö (2020) In situ intraepithelial localizations of opportunistic pathogens, Porphyromonas gingivalis and Filifactor alocis, in human gingiva. Curr Res Microbial Sci 1, 7–17, under the Creative Commons CC-BY-NC-ND license. Panel E is adapted with permission from Klug B, Rodler C, Koller M, Wimmer G, Kessler HH, Grube M, Santigli E (2011) Oral biofilm analysis of palatal expanders by fluorescence in-situ hybridization and confocal laser scanning microscopy. J Vis Exp (56), e2967, from JOVE.
Pg bacterial OMVs, based on their small size and their adhesive and proteolytic activities, are known to spread more readily in tissues than in bacterial cells [53]. Pg vesicles were shown to penetrate more rapidly through a monolayer of HOKs and then invade HGFs than whole bacterial cells. According to the current state of knowledge, Pg cells spread in periodontal tissues via intercellular translocation established by actin-dependent membrane protrusions or through intercellular junctions, which require gingipain activity [70–72]. Takeuchi et al. [72] also showed an epithelial translocation of Pg cells that was mediated by an endocytotic recycling pathway (Fig. 9). Although Pg vesicles were able to enter human epithelial cells by an endocytic pathway and were sorted to lysosomal compartments [62], there was no evidence of the vesicles trafficking out the cells through recycling endosomes. Further evidence for this was reported by Kumar et al. that showed Pg was not detectable in stable/healthy clinical states and comprised only 0.8% of total clones in active human periodontitis [73], although its presence has been significantly correlated with progressive bone loss [74]. Similarly low levels of Pg have been reported in another human study (predominantly found at≤0.03 of the total microbiota) [75] and in the mouse periodontitis model [76] was detected in the murine periodontal tissue at least 4 log10 units lower than the total oral bacteria. The anti-Pg sera again appeared to be reacting either with a florid intracellular accumulation of dead/inert bacteria or more likely to a bacterial-associated antigen that would be more consistent with the production and spread of the OMVs/solubilized gingipains/LPS protein of the bacteria. Because DNA specific staining was not done it cannot be definitely ruled out that the observed staining with the Pg anti-sera was not due to inert or dead bacteria in the tissue, but highly unlikely.
Fig. 9
Time course study using labeled P. gingivalis OMVs (lime green circles) in human HOK epithelial cells. All OMVs were found within cells by 30 minutes. Reprinted from Olsen I, Amano A (2015) Outer membrane vesicles – offensive weapons or good Samaritans? J Oral Microbiol 7, 1, with permission by Taylor & Francis Ltd (http://www.tandfonline.com).
The collective evidence in the oral cavity strongly suggests that regularly distributed bio-polyclonal biofilm colonies of Pg are deeper in the secular tissues and extracellular while the OMVs produced by them are more diffusely spread to surrounding tissues and in the GCF/lymph and micro-vascular systems.
IN SITU OBSERVATION OF OMVs IN DISEASE
Vesicle productions during infection from both intracellular and extracellular bacterial pathogens have been identified in diverse host tissues, revealing the ability of vesicles to access a variety of environments within the host (Fig. 6). Because of their small size and the lack of known unique biological markers, OMVs are difficult to detect in biopsies and host specimens. Vesicle production and composition are influenced by environmental factors that pathogens experience inside the host. Not surprisingly, vesicles are often observed near colonizing bacteria. Chronic H. pylori colonization of the stomach leads to peptic ulcers and, in some cases, cancer. Vesiculating H. pylori and vesicle binding to gastric cells have been detected in gastric biopsies and with tissue culture cells [77, 78]. Vesicles containing vacuolating cytotoxin observed in H. pylori-colonized human gastric epithelium biopsies were similar in appearance and composition to H. pylori vesicles made in vitro. Chlamydial OMVs were apparent in McCoy mouse fibroblast cells infected with either Chlamydia trachomatis or Chlamydia psittaci [79, 80]. A B. burgdorferi isolate that causes Lyme disease was incubated with human skin for 24 h and was found to produce vesicles after invading in the dermis [81]. Vesicles have also been detected in the fluids from infected hosts, demonstrating their ability to disseminate distant from the site of infection. Both vesiculating Neisseria meningitidis and freely diffused vesicles were detected in asymptomatic carriers and symptomatic patients, including in the cerebrospinal fluid from a patient with meningitis and blood from a patient who died from meningitis [80–85]. Similarly, vesiculating Borrelia and free vesicles were detected in the urine and blood of B. burgdorferi-infected mice as well as in infected ticks [86].
DIRECT EVIDENCE FOR OMVs VERSUS Pg CELLS IN AD
The relationship between AD and periodontitis has mostly focused on Pg LPS and other bacterial products that can cause local host responses and regionally generated inflammatory cytokines, which might gain access to the brain [87]. Furthermore, although local host response and regionally generated inflammatory cytokines seem to be the main cause of AD, with little to no evidence that bacterium is the cause of AD until recently. Recent studies in both rodent animal models and human tissues have shown the putative presence of the periodontal pathogen Pg in the brains of individuals with AD [88–93]. It remains unclear, however, how a periodontopathogen such as Pg crosses the BBB to infect and possibly colonize the brain. Direct evidence for this however in the limited studies done to date is not as convincing as has been adopted in the literature and popular press and will be reviewed here.
Although a study has indicated that bacterial OMVs increase BBB permeability [94], the question of whether they can cross the BBB has not been resolved. It seems likely now that if the distribution of Pg cells to OMV material in the oral cavity where the bulk of the infection is known to occur in the body is as summarized above that a similar situation would exist in the brain. To produce the amount and distribution of the HagA/gingipains antigen/protein in AD brain tissues as we have observed would have to be the result of a significant amount of Pg bacteria in and around the tissues such as the brain. The argument this happens by a slow process of a few bacteria spreading slowing throughout the brain although hypothetical is problematic from the position that the brain is a very poor environment for growing metabolically active Gram-negative, anerobic, assacryoltic organism like Pg. The brain has one of the highest oxygen tension environments in the body and uses glucose as its main energy source. In addition, Pg flourishes in a polymicrobial biofilm in a noticeable colony. Nothing like this has ever been reported nor observed. Furthermore, if the human oral cavity of a Pg-infected person is used as a comparison, it can be seen that the gingival submucosa and associated epithelia cells are for the most part uniformly and diffusely filled with HagA/gingipains antigens (see Fig. 8B, C). Pg-positive people, depending on many factors including stage of diseases, have a wide range of Pg CFU/ml, from 0 to 10(4–6) CFU/ml. The older the person, the greater the prevalence and higher the titer.
As mentioned, Pg is a keystone pathogen in chronic periodontitis [15, 95]. That is to say, it modulates the size and composition of the local microbial community to promote periodontitis and an inflammatory milieu. Moreover, it is capable of escaping into the bloodstream and colonizing extraoral tissues [96–99]. Recent studies identified Pg lipopolysaccharide [89] and DNA [47] in autopsy specimens from the brains of people who had AD. Moreover, Pg DNA was present in the cerebrospinal fluid and saliva of individuals with mild to moderate cognitive impairment, clinically diagnosed as having probable AD [89]. In terms of direct evidence for Pg bacteria being in the brains of AD tissues, there are none to date. There are no reports of direct visualization of bacteria in the brain, just 16s rRNA. In general, issues with these studies include the cohort assessed was not selected based on periodontal or any other disease; future studies would require specific cohorts selected for the presence and absence of periodontal or other disease. Additionally, any infection, which initiates the neuropathology of AD, may occur 15–20 years pre-mortem and therefore the bacteria identified here may be due to prolonged secondary infection after BBB breakdown.
IN VITRO INTRACELLULAR STUDIES OF Pg
Pg has been reported following intracellular invasion to exhibit a range of behaviors from a cellular stasis and apoptosis to very limited division. Whole-cell quantitative proteomic analyses were conducted to investigate the change from an extracellular to intracellular lifestyle for Pg. Global protein abundance data for Pg strain ATCC 33277 internalized for 18 hours within human gingival epithelial cells and controls exposed to gingival cell culture medium were obtained at sufficient coverage to provide strong evidence that these changes are profound [100]. A total of 385 proteins were overexpressed in internalized Pg relative to controls; 240 proteins were shown to be underexpressed. This represented in total about 28% of the protein encoding ORFs annotated for this organism, and slightly less than half of the proteins that were observed experimentally [100]. Importantly, for the issue of PG bacterial cells in the brain reported to be responsible for producing the broadly diffuse and copious amounts of gingipains in the brain (Fig. 10) [47], production of several proteases, including the classical virulence factors RgpA, RgpB, and Kgp, were decreased. What emerges from these studies and measurements is a biologically consistent picture of an organism that down-regulates production of destructive proteases, coordinates responses to elevated oxygen tension and iron availability, dispenses with production of energetically costly surface molecules that have outlived their use, and ramps up protein production to thrive in a nutritionally rich intracellular environment. Unless Pg possesses a mechanism to restrain growth and division, in the long-term the increase in bacterial numbers may tip the ecological balance away from health and toward cell and tissue destruction. There is one report that HCAE endothelial intracellular Pg cells could increase 2.7 X in 8 hours [100]. No evidence of infectivity or cell to cell transmission was reported. Pg could be detected intracellularly at 48-hours post invasion in large numbers by immunofluorescent microscopy. While different entry pathways and intracellular trafficking in various cell types have been proposed [101–104] and observed at a relatively high multiplicity of infection (MOI) of 100, virtually all host cells in culture contained several bacterial cells, and that recovery of viable intracellular Pg was relatively high a few hours post-invasion. Over a period of 48 hours, however, while the level of intracellular localization did not change, the recovery of viable microbes on blood agar plates (BAP) diminished to virtually zero for smooth muscle cells and endothelial cells (and very low numbers for KB, a continuous epithelial cell line). Most of the Pg bacteria in all three cell types are perinuclear at this stage, similarly to what has been demonstrated in primary oral epithelial cells [105, 106]. It is unclear why Pg could not be recovered on BAP at this stage. The inability to recover Pg on BAP has been reported before. For example, high amount of Pg genomic DNA in atheromas using Q-PCR but no bacteria could be directly recovered on BAP, as previously observed. One intriguing possibility again is that OMVs secreted from Pg in the oral cavity and containing various RNAs and DNAs were responsible for the PCR positive intracellular results.
Fig. 10
Immunohistochemical localization of gingipains in human gum tissues, AD brain tissues. Adapted with permission from Dominy SS, Lynch C, Ermini F, et al. (2019) Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv 5, eaau3333, under the Creative Commons Attribution license (CC BY 4.0).
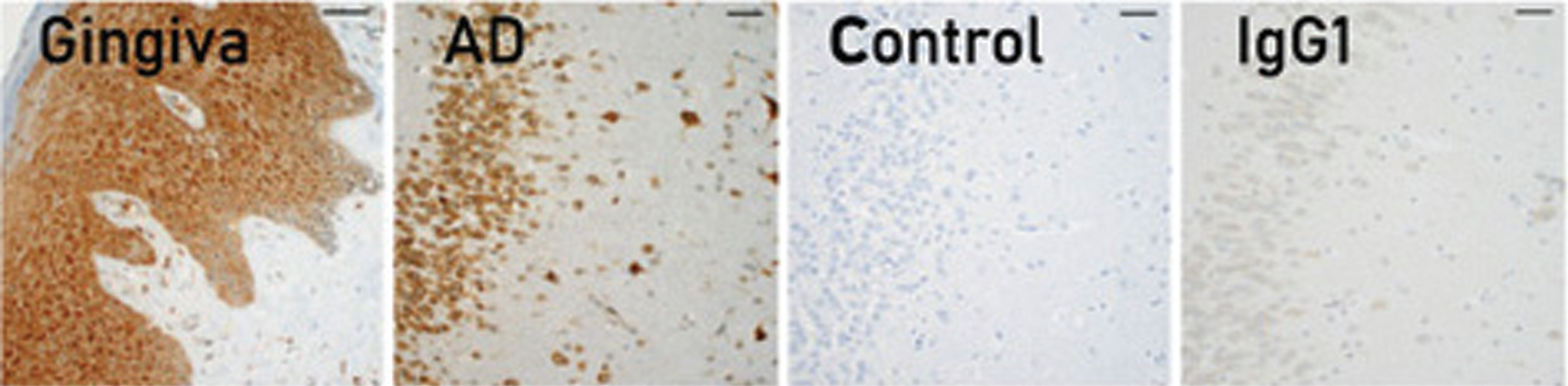
It could not be ruled out that Pg may actually have entered a latent state to reside intracellularly, resulting in inability to be cultivated on BAP, and that it may leave such a state upon contact with fresh host cells.
There is only one study in the literature addressing the infection of neural cells with Pg [107]. In this study Pg, at a high MOI was found to infect limited numbers of neurons/axons with but a few bacterial cells and express gingipains and a neuropathological phenotype which included autophagic vacuoles in neurites, an accumulation of multivesicular bodies in the perikaryon, degradation and phosphorylation of tau, and the loss of synapses after 48 hours. Pg cells exhibited increasing association with lysosomal compartments suggesting their inactivation by the cell. In addition, it was demonstrated that after 72 hours of infection, Pg is in a viable but non-culturable state and expresses both active Rgp and Kgp. Larger aggregates of bacteria appeared partially engulfed by neuronal membrane and produced focal amounts of gingipain detectable by immunohistochemistry. The authors suggested “these Pg aggregates appear to be producing gingipains and bacteria and continuously infect neurons over longer time periods.” No evidence of this, however, was demonstrated in this or any other study to date. Given that such few bacteria were able to infect neural cells at such a high MOI and produce such few gingipains, it seems hard to therefore assign a direct intracellular bacterial source for the repeatedly observed diffuse cytoplasmic immunoreactive HagA/gingipains staining of nerve cells in AD brain sections as seen in Fig. 10.
DIRECT EVIDENCE FOR OMV UPTAKE INTO THE BRAIN
András et al. hypothesized in 2015 that bacterial OMVs may cross the BBB and affect AD pathology [108], as exosomes from various cell types have been associated with AD pathology [109]. The relationship between AD and periodontitis has mostly focused on Pg and E. coli LPS and other bacterial endo-and exo-toxins products that can cause local host responses and regionally generated inflammatory cytokines, which might gain access to the brain. In the case of LPS, Pg’s unique LPS was found to be more frequently found in AD brain tissues. Furthermore, although local host response and regionally generated inflammatory cytokines seem to be at the center of current dogma and the main cause of AD, with no evidence that Pg bacteria itself is the cause of AD until recently. Some recent studies have shown the presence of periodontal pathogen Pg in the brains of Pg-infected mouse models and individuals with AD [47]. It remains unclear, however, how the periodontopathogen crosses the BBB to colonize the brain (to be discussed later). Although a study has indicated that bacterial OMVs increase BBB permeability [108], the question of whether they can cross the BBB has not been resolved.
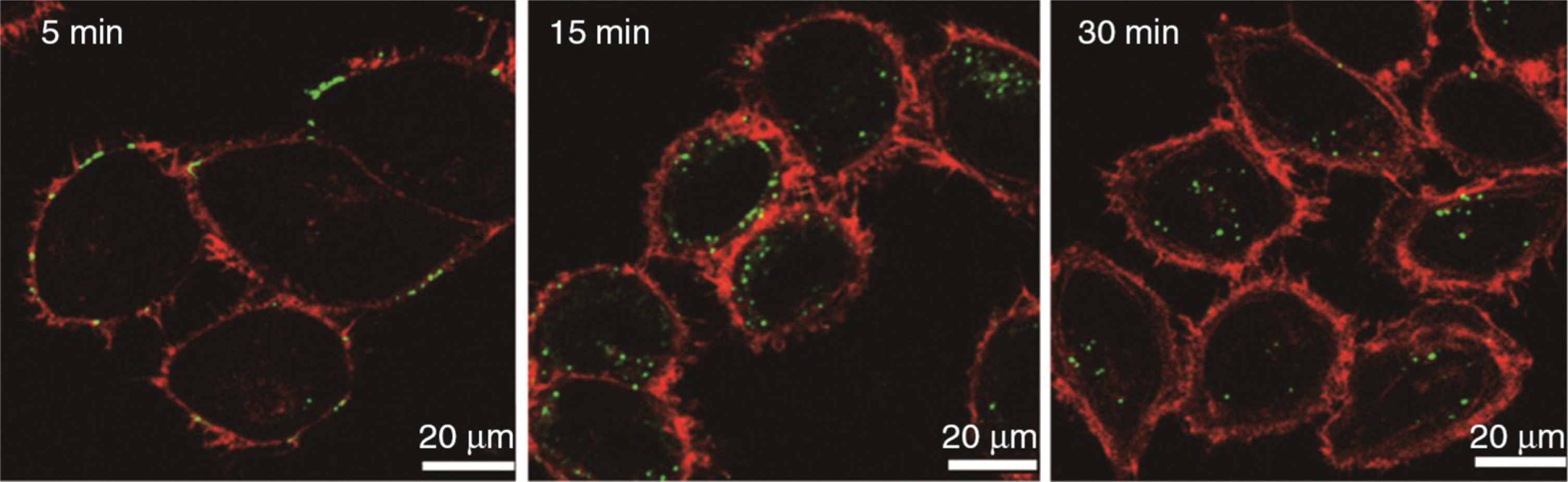
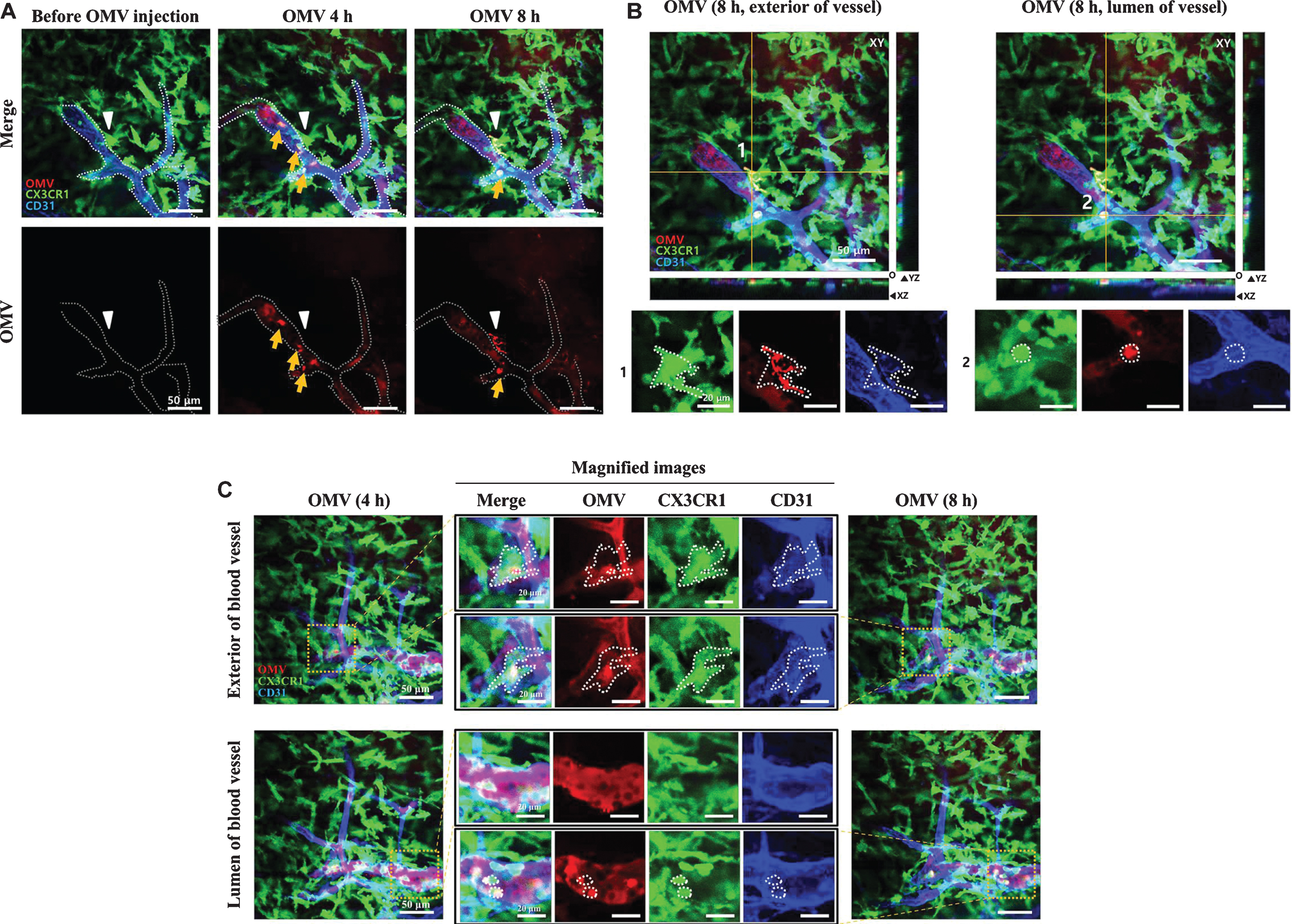
In a landmark study by Han et al., it was shown for the first time that oral bacterial OMVs can systemically cross a normal C57BL6 mouse BBB 24 hours after intracardiac injection in a dose dependent manner (Fig. 11) [94]. In addition, it was reported suggesting another mechanism, whereby exRNA in OMV stimulates the host response. Because OMVs contain numerous various bioactive molecules, such as bacterial proteins and RNAs, OMVs from commensal bacteria may regularly affect brain function. Because the majority of EV RNA consists of sRNAs such as miRNAs and msRNAs [110, 111], it would not be surprising if commensal bacterial exRNAs affected the brain immune response via microbe associated molecular patterns pattern recognition receptor signaling pathways or by exogenous miRNAs incorporating into host RISC. Although the role of exogenous miRNAs needs further investigation, it was demonstrated in this same report that Aa exRNAs can promote the production of brain TNF α. An association between AD and TNF-α has been repeatedly suggested [112, 113], and patients with periodontitis have been shown to have an elevated risk of AD [65]. These results imply that OMVs from periodontal pathogens cause AD via leaky gum. Additional studies demonstrated that injected labeled OMVs were taken up into meningeal macrophages by 8 hours and cortical microglial cells by 24–48 hours (see Figs. 12 and 13) [114]. These studies demonstrate the dynamic nature and uptake of these virulence factors into the first order peripheral neuro-cellular types and proceeding further into the micro-glial cells in the brain parenchyma. Bacterial OMV-derived microbe-host genetic communication requires more extensive research not only at the site of symptoms and in the brain but also from a systemic perspective.
Fig. 11
Aa OMVs cross the BBB of mice. Two-dimensional light sheet fluorescence microscopy analysis of the mouse brain (cortex). A) Lipid tracer dye was treated to OMV or PBS (control) then ultracentrifuged to remove unincorporated dye. The mouse brain was perfused 24 h after cardiac injection of stained OMV or PBS and cleared using the brain clearing (clarity) technique. Lectin was injected to present the brain vessels (green); OMVs are shown in red due to the lipid tracer dye DiD (red). Scale bar, 100 mm. B) Aa OMVs were pre-stained with lipid tracer dye DiD (red) and RNA-specific dye Syto RNA-Select (green) before injection in mice. The mouse brain was perfused 4 and 24 h after cardiac injection of stained OMVs. The 24 h cortex region shows red spots (OMV) and green spots (RNA inside OMVs), suggesting OMVs containing RNA can cross the BBB, whereas 4 h OMVs remain in brain vessels. Scale bars, 50 mm. Reprinted from Han EC, Choi SY, Lee Y, Park JW, Hong SH, Lee HJ (2019) Extracellular RNAs in periodontopathogenic outer membrane vesicles promote TNF-α production in human macrophages and cross the blood-brain barrier in mice. FASEB J 33, 13412–13422, with permission from John Wiley and Sons.
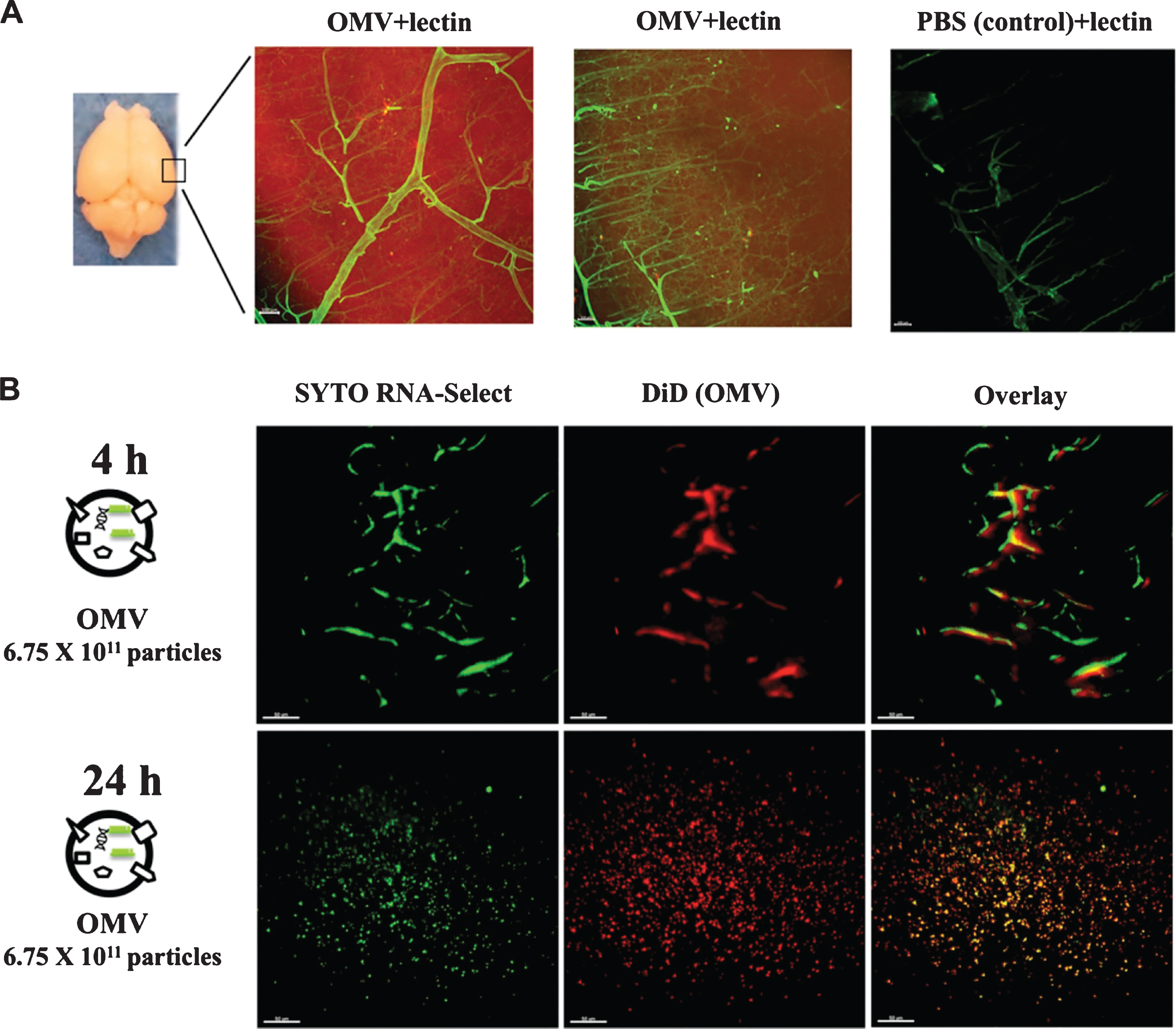
Fig. 12
Delivery of Aa OMVs into microglial cells. Intravital image was captured 24 h after Aa OMV i.v. injection. OMVs were colocalized with GFP-positive microglial cells. Scale bar: 50μm. Reprinted with permission from Ha JY, Choi SY, Lee JH, Hong SH, Lee HJ (2020) Delivery of periodontopathogenic extracellular vesicles to brain monocytes and microglial IL-6 promotion by RNA Cargo. Front Mol Biosci 7, 596366, under the Creative Commons Attribution License (CC BY).
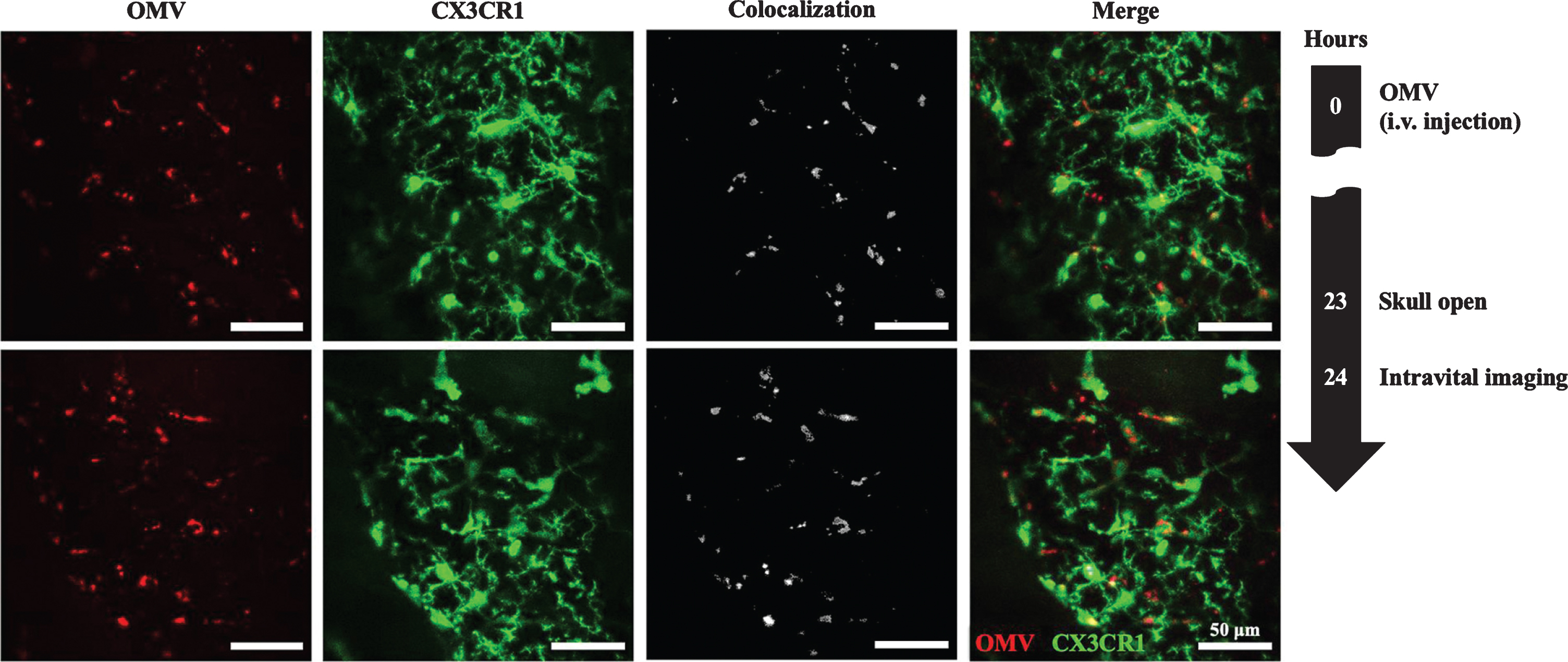
Fig. 13
Visualization of tail-vein-injected OMVs and meningeal macrophages of live CX3CR1-GFP mice. A,B) OMVs taken up by monocytes were compared between 4 and 8 h after OMV injection. Confocal microscopic acquisition results show the OMV distribution before and 4 and 8 h after OMV i.v. injection of mice. A) The white dotted line delineates the lumen of blood vessels. White arrowheads indicate Aa OMV-colocalized meningeal macrophages, and yellow arrows indicate OMV-colocalized monocytes/macrophages in the lumen of vessel. Scale bar: 50 mm. B) Representative image stacks (5 mm intervals) acquired during confocal imaging of mice with GFP-positive microglial cells for OMVs injected (8 h) shown in (A). The white dotted area refers to the colocalization of OMVs (red) and CX3CR1-GFP-positive monocytes (green). Scale bar: 50 mm; magnified scale: 20 mm. C) Colocalized monocytes/macrophages (dotted areas in the magnified images) in the exterior and lumen of the meningeal blood vessel were compared. Magnified images (scale bar: 20 mm) of colocalized monocytes (shown in yellow in the merged images) in the dotted area are clearly seen in both the exterior and lumen of the meningeal blood vessel. Reprinted with permission from Ha JY, Choi SY, Lee JH, Hong SH, Lee HJ (2020) Delivery of periodontopathogenic extracellular vesicles to brain monocytes and microglial IL-6 promotion by RNA Cargo. Front Mol Biosci 7, 596366, under the Creative Commons Attribution License (CC BY).
ANIMAL MODELS OF Pg INFECTION
Regarding AD, once the oral cavity is infected, Pg bacteria may hypothetically access the brain and spread via a number of pathways including 1) infection of monocytes followed by brain recruitment, 2) direct infection and damage to endothelial cells protecting the BBB, and/or 3) infection and spreading through cranial nerves (e.g., olfactory or trigeminal) to the brain. After entering the brain, it is suggested that Pg spreads slowly over many years from neuron to neuron (trojan horse mechanism) along anatomically connected pathways, similar to what has been demonstrated for endothelial vascular cell-to-cell transmission of Pg [47]. Tau pathology has also been suggested to spread from neuron to neuron, with a pattern resembling an infectious process with tau being a target of gingipain proteolysis, and the tau pathology seen in AD brains being due to the trans-neuronal spread of Pg, with direct damage of tau by gingipain proteolysis [47].
Direct evidence for Pg bacteria colonizing the brain and being responsible for the AD-like brain pathology in mice is limited to a few rodent models in which multiple, regular, chronically re-infected over many weeks (e.g., 6–22 weeks) and with large amounts of bacteria: for example, the oral dosing of live Pg (W83), serotype 1 encapsulated, bacterial mono-infection using 1×10(9) CFU, repeated every other day of every week for 22 weeks. These studies used both various genetic lines of mice and wild type mice and demonstrated in some cases the presence of Pg DNA in the brain parenchyma. In none of these experimental mouse models did the authors show they could recover infectious bacteria from the tissues nor demonstrate metabolically viable cells by FISH staining or demonstrating any visually intact bacterial cells from the brain tissues. Also, the relative amount of bacterial DNA and its exact cell distribution were never fully discussed leaving the question, was there a functional infection in the brain tissue and could that infection be enough to induce the broader observed pathology. The study by Ilievski et al. using 3x a week for 22 weeks of 10(9) CFUs reported up to 12 bacterial cells per field and 6pg copy numbers of 16S RNA per 5 FFPE [88]. There was no discussion of the relationship of these bacterial cells and copy number to the distribution of the neuropathology and inflammation reported in specific regions of the brain. By the authors own admission, the status of these bacterial cells as being metabolically viable and/or and bacterial RNA copy numbers being of a level to suggest local active colonization was not done.
Since it takes a metabolically active bacterial cell to produce the gingipains and LPS in amounts to diffuse out of brains cells and throughout the brain parenchyma, it seems less likely that there was ever a productive infection of the brain. The other problems with these studies are the vast amounts of Pg culture material > 10(9) CFUs/ml used on a daily weekly basis for out to 28 weeks and tested soon after terminating the infections. No authors described perfusing the brains before doing any PCR DNA studies thus leaving in doubt that the signal may have come from Pg cells and/or DNA in the blood, not to mention that without knowing which specific primers were used there could have been Pg OMVs in the blood or the brain tissue which are known to contain coding RNAs and DNAs for multiple gene products including hmuY gene. Furthermore, the Pg cultures were not separated into OMVs and whole PG bacterial cells so as to delineate the source of the PCR signals. Without these two simple yet important modifications in the study design, no conclusion can be made that the Pg DNA brain signal seen in mouse models were due a specific bacterial infection in the mouse brains and more likely being due to the much larger numbers of OMVs over bacteria that were inoculated orally into the mice. Additionally, cofounding these studies is the excess volume of Pg culture given to the animals that could have resulted in their swallowing of the Pg cells and OMVs. This is known to cause a dysbiosis of the GI tract in as little as few weeks [115].
GINGIPAINS HYPOTHESIS
The Gingipain Hypothesis proposes that a productive Pg infection of brain tissue thus producing gingipains and causing AD [47]. In infected human cells, Pg is found in vacuoles that contain undegraded ribosomes [116]. Pg ferments amino acids as an energy source [6]. Co-localized ribosomes may provide a particularly digestible source of amino acids because of their enrichment for the positively charged residues that gingipains cleave. Cytosolically free Pg quickly localizes to the rough endoplasmic reticulum (ER) to form autophagosome-like vacuoles [117]. However, it is not known how Pg traffics to the ER and thus it is speculated that ER translocation processes may be exploited by Pg to reach the endoplasmic reticulum. Sequestration of ribosomes in autophagosome-like vacuoles by Pg may cause increased transcription of cytosolic ribosomal protein genes because ribosome biogenesis is highly regulated [118]. As stated earlier, intracellular Pg cells down regulate gingipains synthesis and the type IX cargo secretion system. Further study is needed to experimentally test if Pg exploits cytosolic ribosomes and ER translocation pathways. Our model rather suggests that Pg OMVs entering the brain through the BBB are the more likely source of this diffuse toxic insult to the brain and not a direct infection by Pg.
EARLIER THEORY OF AD: THE CHOLINERGIC HYPOTHESIS
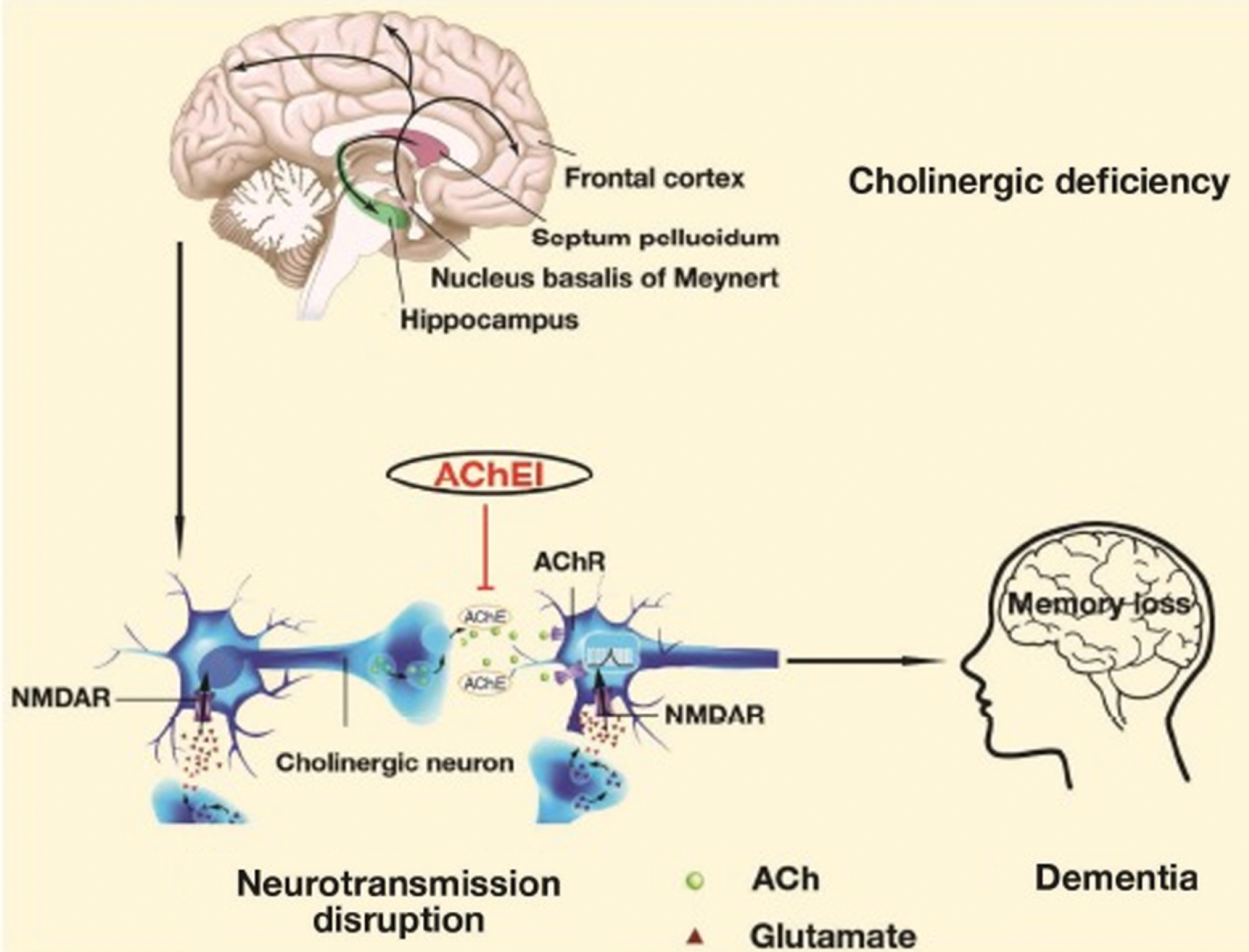
In the 1960s and 1970s, psychologists gave young healthy subjects cholinergic inhibitors (which prevent acetylcholine from signaling to post-synaptic cells) [119]. These young healthy subjects had memory problems and cognitive problems that resembled patients with AD, showing that acetylcholine was important for memory. Most of the brain’s supply of acetylcholine derives from the nucleus basalis of Meynert in the basal forebrain, so doctors investigated this area during the autopsies of patients with AD and showed that the nucleus basalis seems to be particularly vulnerable to the neurodegeneration seen in AD [120, 121]. Patients showed decreased numbers of healthy neurons in this area, and decreased cholinergic projections to the hippocampus and entorhinal cortex (areas we also knew were important for memory), suggesting that patients with AD may have decreased acetylcholine (Fig. 14) [119–121].
Fig. 14
Simplified drawing of the Cholinergic Hypothesis. Reprinted from Wang Y, Wang H, Chen HZ (2016) AChE inhibition-based multi-target-directed ligands, a novel pharmacological approach for the symptomatic and disease-modifying therapy of Alzheimer’s disease. Curr Neuropharmacol 14, 364–375, under the Creative Commons Attribution-Non-Commercial 4.0 International Public License (CC BY-NC 4.0).
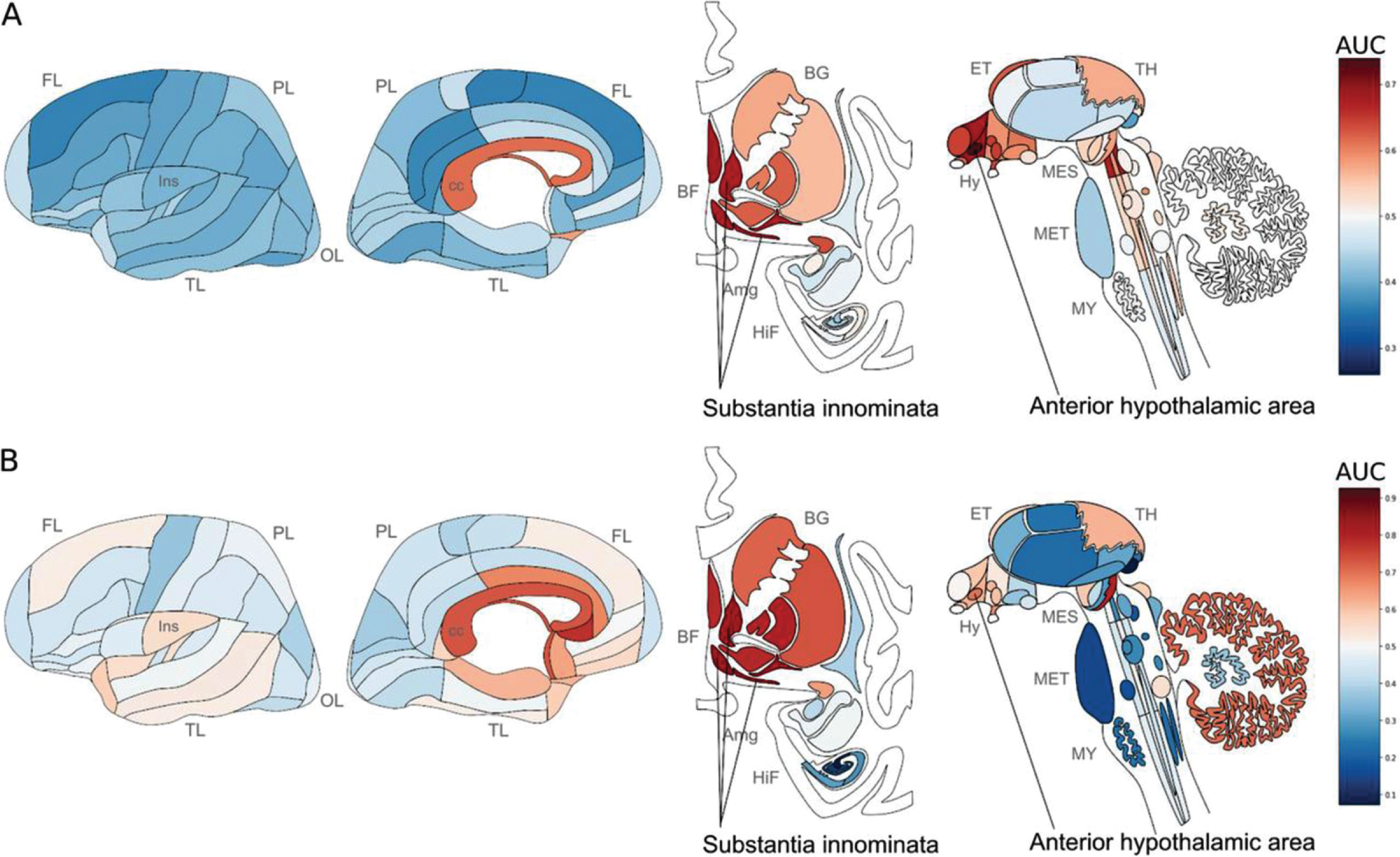
This observation in fact is now more consistent with some studies looking at the neuroanatomical analysis of Pg associated genes (gingipains) which mark cholinergic neurons, basal forebrain and anterior hypothalamic regions, and regions near ventricles and peripheral neurons are also enriched, suggesting relevance to Pg brain entry (Fig. 15) [122]. Pg OMVs mediating these activities since the majority of gingipains are loaded into OMVs and not the small soluble fraction coming off the bacterial cell membrane. While ribosomal genes are enriched for these residues, a specific set which function in SRP dependent ER co-translational translocation has been previously associated with periodontitis and AD [123, 124]. These genes are also upregulated in brain tissue with detected Pg RNA, suggesting a transcriptional response to gingipain driven degradation of ER localized ribosomes (Fig. 15). At the mechanistic level, the highlighted translocation genes may explain how Pg transits to the ER upon cell entry. Also, downregulation of synapse and cell-cell contact genes support findings of synapse loss upon Pg infection [107], thus suggesting in total Pg’s early response and susceptibility at the molecular and anatomical level, new associations that may be relevant to the early stages for onset AD pathogenesis. Proteins that are enriched for arginine and lysine residues, which are potential gingipain cleavage sites bind RNA and DNA. These results can modify and now link parts of the “gingipain” and “cholinergic hypotheses.”
Fig. 15
Neuroanatomical heatmaps marking specific expression of the genes upregulated in samples with P. gingivalis reads (A) and ER translocation genes (B). AUC values range from depleted expression in dark blue to enriched in dark red. Brain region abbreviations: FL, frontal lobe; PL, parietal lobe; TL, temporal lobe; OL, occipital lobe; BF, basal forebrain; BG, basal ganglia; AmG, amygdala; HiF, hippocampal formation; EP, epithalamus; TH, thalamus; Hy, hypothalamus; MES, mesencephalon; MET, metencephalon; MY, myelencephalon. Reprinted with permission from Patel S, Howard D, Chowdhury N, Derieux C, Wellslager B, Yilmaz Ö, French L (2021) Characterization of human genes modulated by Porphyromonas gingivalis highlights the ribosome, hypothalamus, and cholinergic neurons. Front Immunol 12, 2165, under the Creative Commons Attribution License (CC BY).
POSSIBLE RELATIONSHIP BETWEEN Pg AND DECREASED SALIVARY LACTOFERRIN LEVELS IN AD
The above discussion and association of Pg OMVs either targeting and/or seeking out tissues higher in arginine and lysine bearing amino acids, especially in the brain is intriguing on many levels. Should the neuroanatomical analysis of Pg gingipains mark cholinergic neurons, basal forebrain and anterior hypothalamic regions, and regions near ventricles and peripheral neurons, an early pathway to Pg OMV to brain entry may now be identified. These results link Pg gingipains and therefore most likely OMVs and cholinergic hypotheses. Extending this mechanism forward would mean ultimately interference with the cholinergic input to the salivary glands. The salivary glands including the submandibular gland, one of the major salivary glands, together with the sublingual and the parotid glands make up the oral host defense apparatus providing a vast array of anti-microbial effector functions to the oral cavity. The secretion of saliva is controlled by the autonomic nervous system. It is known that pilocarpine, a muscarinic agonist, injected intracerebroventricularly induced salivary secretion by activating central autonomic efferent fibers have demonstrated that an important center for eliciting salivary secretion exists in the hypothalamus and involves cholinergic pathways [125]. Numerous projections exist from the lateral hypothalamus to salivary nuclei located in the brain stem. Thus, lesions of the lateral or ventromedial hypothalamus impair salivary secretion.
There are recent reports demonstrating that decreased salivary levels of lactoferrin (Lf) are found as excellent early diagnostic performance markers to detect prodromal AD and AD dementia and actually distinguishing them from other dementia as frontotemporal dementia (FTD), with sensitivities and specificities over 87% and 91% [126]. Therefore, it is remarkable that salivary Lf levels do not decrease in some other dementias-such as FTD. A possible explanation could be that the hypothalamic region, described above due to the HagA/gingipains/OMV driven neuropathology of Pg that controls body innate immunity, is affected in the earliest pro-domal stages of AD, but not in FTD [127]. In addition to amyloid plaques and neurofibrillary tangles, functional studies suggest that hypothalamic dysfunction is a common event in AD, often early in the course of disease [128]. Although there are evidences indicating that certain hypothalamic regions are also affected in FTD, specifically those that correlate with abnormal eating behaviors [129], they are different to those affected in AD. The apparently AD-specific salivary Lf reduction may thus not only be useful in the differential diagnosis but could also provide important insights into selective immune vulnerability in neurodegenerative diseases. As mentioned above the secretion of salivary proteins is controlled by cholinergic parasympathetic nerves that release acetylcholine, evoking the secretion of saliva by acinar cells in the salivary gland. These parasympathetic nerves are connected with the hypothalamus [128–130]. We propose that early hypothalamic Aβ accumulation is associated with Pg OMVs gingipains deposition found in postmortem brain tissue with the upregulation of ER translocation genes in the context of AD. This could be an early switch that begins the loss of control and disrupt hypothalamic function affecting salivary gland regulation that ultimately results in reduced salivary Lf secretion. Pg is known to degrade Lf for its major early iron source in oral cavity. Should neural based impairment of the salivary glands produce a decline in the steady-state level of Lf, a major switch in an otherwise delicate balance between Pg and the oral cavity may ensue. More specifically, the diminishing oral salivary iron source would further signal to Pg the need for new iron source.
Pg AND THE RUSTY BRAIN: IRON DYSHOMEOSTASIS
Pg is a heme auxotroph, and many studies have highlighted the major influence the environmental concentration of heme has on Pg gene and protein expression as well as the growth and virulence capacity of the microorganism Heme can be derived from host hemoproteins present in the saliva, gingival crevicular fluid, and erythrocytes in the oral cavity. In vivo concentrations of free heme have been found to be too low (10–24 M) to support bacterial growth without the help of specialized heme acquisition systems produced by the bacteria themselves. Normal human saliva, however, has been demonstrated to support the complex poly-microbial biofilms associated with Pg [131]. Thus at least initially it seems that Pg in the oral cavity would exist in a heme normo/excess environment. This condition promotes OMVs to inhereit more cargo proteins associated with increased amounts of adhesive molecules. This promotes a biofilm formation and maintenance situation. OMVs under these conditions when in lymphatic and or blood vessels may now target specific endothelial cell markers throughout the body including the brain. Heme-limited conditions to some extent mimic periodontically healthy tissues, where there is little to no bleeding, whereas heme-excess conditions mimic the disease state where damage to the microvasculature and consequent bleeding is common. The importance of acquiring this essential micronutrient is highlighted by the number of heme acquisition systems identified in Pg to date.
Under the diminishing levels of Lf described above and the signaling to Pg, an iron limiting environment would lead to a well-known mechanism for upregulation of its OMV production and an increase in specific scavenging proteins and loading of iron (Fig. 16). Thus, more iron laden OMVs will be created, heralding again another shift in the exiting balance. These iron-ladened OMVs from the oral cavity may now when passing through the brain start bringing in more iron and heralding the next shift in neuropathogenesis of AD. Thus, an additional insult from the earlier low grade neural inflammation in the cholinergic neurons, basal forebrain and anterior hypothalamic regions, and regions near ventricles may now shift and expand into those areas of the brain so typically involved in AD (hippocampus and temporal lobes).
Fig. 16
Enrichment of proteins onto OMVs. The average LFQ values obtained for each of the 112 proteins common to OMVs and WCLs are plotted against each other on a log scale. A) Heme-excess and B) heme-limited. Proteins above the green line are considered enriched in OMVs, whereas proteins below the red line are considered depleted in OMVs relative to the WCLs. Panels Reprinted with permission from Veith PD, Chen YY, Gorasia DG, Chen D, Glew MD, O’Brien-Simpson NM, Cecil JD, Holden JA, Reynolds EC (2014) Porphyromonas gingivalis outer membrane vesicles exclusively contain outer membrane and periplasmic proteins and carry a cargo enriched with virulence factors. J Proteome Res 13, 2420–2432. Copyright (2014) American Chemical Society.
The extent to which lower amounts of non-iron containing OMVs verses higher containing iron OMVs may be involved in switching the early cognitive-decline form of AD into a more aggressive form of neuropathology and progressing dementia is not known. However, it is not unreasonable to think the shift now to a greater deposition of higher iron into the deep gray matter and total neocortex, and regionally in temporal and occipital lobes would not be seen as a poorer prognostic indicator for AD disease progression. It is well documented that brain iron accumulation is associated with cognitive deterioration earlier and independently of brain volume loss. Moreover, those iron concentrating neuro-anatomic locations over time coincide with the insults mapped for Pg gingipains (Figs. 15 and 17). Changes in iron levels over time in the hippocampus, thalamus, putamen, and caudate nucleus correlated with cognitive decline in individuals with Parkinson’s disease (PD). In this light, it is interesting to consider the pathogenesis of sporadic AD or PD being highly complex, sharing several cardinal cellular and molecular mechanisms, such as proteotoxicity, mitochondrial dysfunction, and oxidative stress. All of these have been suggested, based on postmortem studies in patients and evidence acquired from various experimental models—as the abnormal accumulation of proteins of altered conformation is considered as the most characteristic molecular signature of AD or PD, it is highly unlikely that only proteinopathy acts as the driving force of the pathology, while others such as oxidative stress and mitochondrial dysfunction are secondary consequences. Instead, it is plausible that they represent interacting damage pathways that culminate in neuronal death and degeneration. Thus, the cross talk among proteinopathy, oxidative stress, and mitochondrial dysfunctions on the one hand and the genetic or nongenetic factors that trigger these damage mechanisms on the other have to be analyzed critically to obtain a deeper understanding of the pathogenesis of AD and PD. Pg OMVs again given the neuroanatomical locations seems a common denominator. The literature is replete with studies that demonstrate the varied aspects of proteotoxicity culminating in neurodegeneration in AD or PD, as well as the detailed biophysical aspects of altered protein conformation and aggregation in these disorders.
Fig. 17
QSM covarying with lower cognitive performance (MoCA) reveals increased brain tissue iron in hippocampus, thalamus, putamen and caudate nucleus in Parkinson’s disease. Whole brain results are overlaid onto the study-wise QSM template in the MNI coordinate system. Red/yellow clusters represent statistical significance at PFDR < 0.05 as indicated by the color bar. Numbers represent slice position in millimeters in MNI coordinate space. FDR, false discovery rate; MNI, Montreal Neurological Institute; MoCA, Montreal Cognitive Assessment; QSM, quantitative susceptibility mapping. Reprinted with permission from Thomas GEC, Leyland LA, Schrag AE, Lees AJ, Acosta-Cabronero J, Weil RS (2020) Brain iron deposition is linked with cognitive severity in Parkinson’s disease. J Neurol Neurosurg Psychiatry 91, 418–425, under the Creative Commons Attribution 4.0 Unported (CC BY 4.0) license.
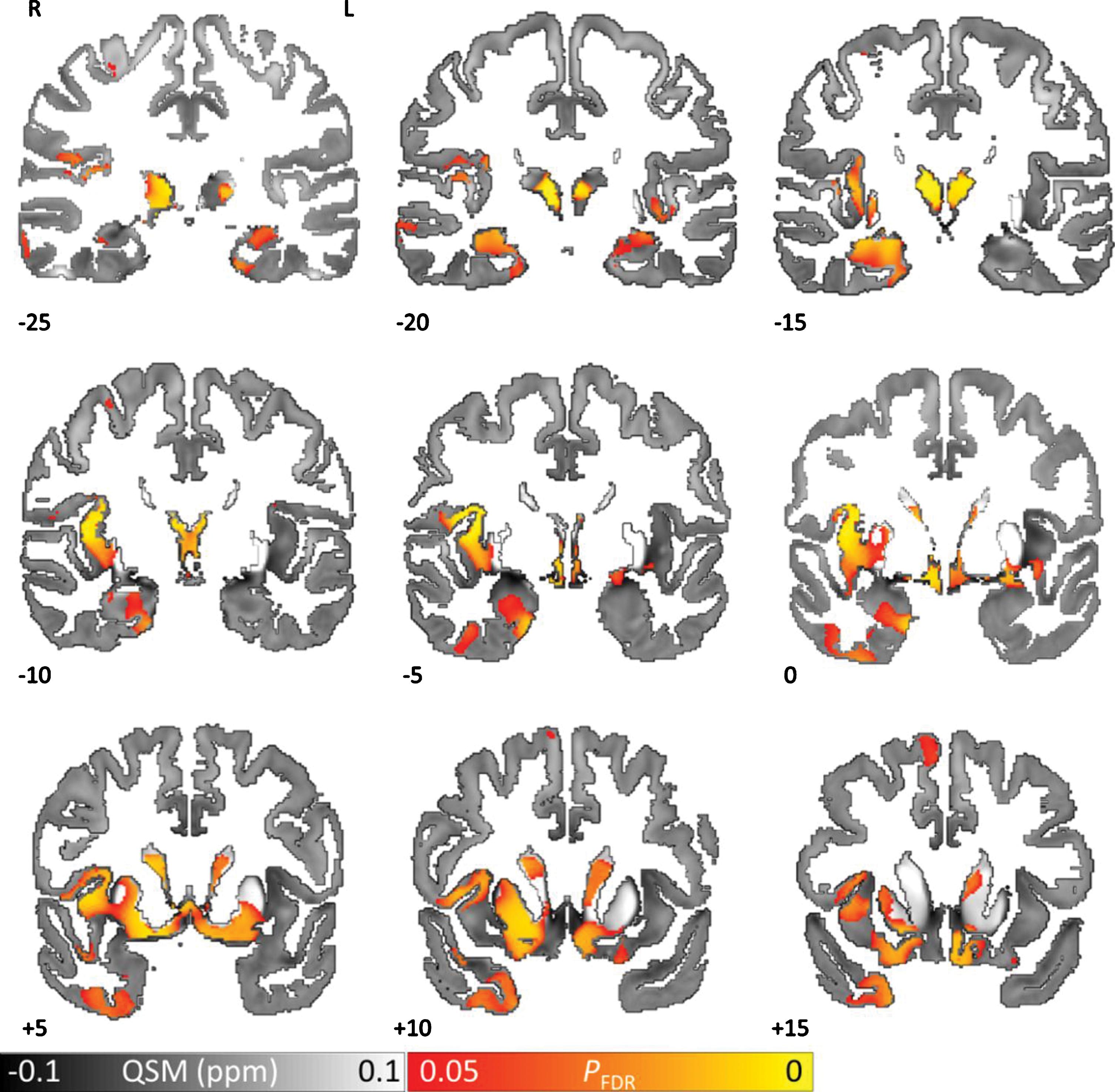
Pg OMVs DUAL EDGE SWORDS FOR THE BRAIN
As stated above, the shift of OMVs from low to high iron would then contribute to the deposition of Aβ and the formation of neurofibrillary tangles, which in turn, promotes the development of AD [132]. To add insult to injury, Lf is known to block Aβ aggregation, tauopathy spread, and neuronal damage [132]. It also acts as an iron-binding protein and is strongly upregulated in the brains of patients with AD. Lf readily crosses to the BBB, and has been shown to ameliorate mitochondrial dysfunction and reduce cellular damage by scavenging reactive oxygen species (ROS) in animal models of AD [133, 134]. At increasing and high levels, iron is inherently toxic to neuronal cells and generates ROS directly or through iron-chelated enzymes [135]. In accordance with this, increased iron deposition upregulated expression of the ROS-generating enzymes P22 phox, Gp91phox, and Xdh (increased by 63.45%, 112.27%, and 183.75%, respectively) in the brains of aged mice. Thus another mechanism associated with ROS accumulation as seen in the aging brain is yet another low grade chronic state of inflammation, contributing to malfunctions of the brain and impaired cognition.
In transgenic mice with AD, double-immunofluorescence labeling with antibodies directed against Aβ and Lf was used and Lf depositions localized to Aβ plaques and regions of amyloid angiopathy were found [136]. Both the intensity and number of Lf-positive depositions increased with age. The upregulation of Lf in the brains of both AD patients and transgenic mice with AD indicated an important protective role for Lf in infected AD-brain tissue. It is tempting to speculate that the high consumption of Lf in AD could lead to reduced Lf levels over time, particularly when AD is promoted by long-term chronic infection. Interestingly, it is suggested that Lf was down regulated in the saliva of AD patients like several other factors of systemic immunity [137]. If Lf decline, again as described above as a trigger of the upstream shift in oral dysbiosis, more specifically Pg and its regulation of the oral environment via OMVs, it is completely plausible that it could also be an important factor in the etiology or pathophysiology of AD. It is remarkable that the levels of Lf are increased in the brains of AD patients, at least initially, and also reduced in their whole saliva. The latter scenario could aggravate the BBB and setup the brain for additional less invasive, oro-dontophlic bacteria and other non-specific microbial/viral infections. It is also possible that low levels of Lf in the whole saliva of AD patients may affect the Lf concentration in the brain since salivary Lf is transferred into the brain via the sublingual route.
One final intriguing aspect of Pg and AD was that it was the most powerful Lf-degrading bacterium of several periodontal pathogens tested in vitro. It is plausible that the effect of Pg on Lf could be added to its wide capacity of immune suppression, acting both in the periodontal pocket and in the AD brain. There are also other proteins and peptides in saliva but their functions and interactions with the oral microbiome remain to be determined. Clearly, when the level of whole saliva Lf is reduced, its composition is changed and this could promote dysbiosis and the risk of associated oral diseases such as caries, gingivitis, periodontitis and fungal infections, and possibly AD.
THE PRIMARY ORAL-NEUROGENIC Pg OMV MODEL
The working model for Pg OMV regulated chronic oral-neurogenic disease outlines the primary bacterial player and its major virulence effector, the OMV. This is somewhat of a departure from the current and ongoing studies for the “Gingipains Hypothesis” put forth by Dominy et al. that offered some evidence that Pg and gingipains in the brain play a central role in the pathogenesis of AD and providing a new conceptual framework for the AD infection model of disease treatment. There are very few examples in the peer reviewed literature of where bacteria and its major virulence factors are found in large amounts intra- and intercellularly both locally and translocated into a given tissue such as the brain and attempts to ignore their primary role in the disease processes. We are now preparing a manuscript demonstrating significant amounts of specific Pg OMV related virulence factors in human AD brains (Nara et al., unpublished study). The issue here may well be that we forgot some very important past bacterial pathogenesis discoveries that exist as rare occurrences/reporting? in the medical literature. Typically bacterially mediated disease such as gramnegative endo-toxemia mediated by LPS and Staphlolococcal endo-toxemia present as hyper-acute to acute to others that are sub-acute in timing. The possibly forgotten dogma, however, is that of the pivotal work described in 1982 by Drs. Barry J. Marshall and Robin Warren, two Australian researchers who discovered the bacterium H. pylori and deciphered its role in gastritis, peptic ulcer disease, and later gastric cancer, and were awarded the Nobel Prize in Physiology or Medicine in 2005 [138].
Digressing a bit, the H. pylori example above is worth re-stating and represents at the time a very unexpected but paradigm shifting discovery in medicine that now in the context of this hypothesis paper specific to another biological extremophile (Pg) may be even more relevant in connecting the Pg oral infection and its associated systemic toxic OMVs into a more clear example for direct causality in these neurogenerative diseases. As is well known now, it was demonstrated that gastritis, and ulceration of the stomach or duodenum, and later gastric cancer were the direct causality and result of infection with a curved, Gram-negative bacilli and furthermore that infection with 1 bacterial organism can and does lead to multiple disease conditions. At the time when Warren and Marshall announced their findings, it was a long-standing belief in medical teaching and practice the high acid content of the stomach would preclude any organisms from colonizing, thus stress and lifestyle factors were promulgated as the major causes of peptic ulcer disease. Warren and Marshall rebutted that dogma, and it was soon clear that H. pylori causes more than 90% of duodenal ulcers, up to 80% of gastric ulcers, and a 2- to 8-fold increased risk of developing gastric cancer and mucosal-associated-lymphoid-type lymphoma. Interestingly, some H. pylori bacterial strains use a needle-like appendage to inject a toxin produced by a gene called cytotoxin-associated gene A (CagA) into the junctions where cells of the stomach lining meet. CagA alters the structure of stomach cells and allows the bacteria to attach to them more easily [139]. Long-term exposure to the toxin causes chronic inflammation mediated by IL-1β. However, not all strains of H. pylori carry the CagA gene and are less virulent. It is interesting to note that CagA is loaded into its OMVs and secreted from the outer membrane exactly like that of HagA/gingipains/hemagglutinin/adhesin complex of Pg (Fig. 18).
Fig. 18
Localization of CagA outer membrane in H. pylori. CagA (green) was detected using an anti-CagA antibody and analyzed by STED microscopy. Note the surface vesicles containing CagA. Bacterial DNA was stained using DAPI (blue). Bar, 1μM. Note vesicular appearances of protein. Reprinted with permission from Jarzab M, Posselt G, Meisner-Kober N, Wessler S (2020) Helicobacter pylori-derived outer membrane vesicles (OMVs): role in bacterial pathogenesis? Microorganisms 8, 1328, under the Creative Commons Attribution License.
Again, recent work on H. pylori beyond targeting the epithelium as it does, the bio-distribution of H. pylori OMVs due to the induction of mucosal barrier instability they create, their small size and penetration of deeper layers of the gastric mucosa leads to their entry into the circulation [139]. It was shown that CagA is present in vesicles derived from the serum of patients infected with cagA positive strains [140], demonstrating that either OMVs themselves or secondary exosomes secreted from mucosal cells targeted by OMVs might enter the blood stream. Just like H. pylori’s examples given, the Pg OMVs in similar sub-acute to chronic processes impact innocent bystander organs like the vasculature, heart, and brain. The authors state “An unprecedented mechanism for the delivery of microbial virulence factors to sites distant from the primary infection site, which may provoke more systemic clinical effects or symptoms that are seemingly unrelated to the primary infection” [140]. It is also interesting to note that Pg OMVs contain transforming protein factors similar to CagA and found in close associations with multiple cancers of the oral-esophageal, gastro-intestinal, pulmonary, and pancreatic origins.
The authors in our working model believe Pg and its OMVs represent another and even possibly more extreme case of the H. pylori example. In this case, instead of the unique, hostile, gastric mucosal epithelial stomach lining, it is on and in the gum and oral cavity lining and may well represent another major case of where a Gram-negative bacteria as evolved into a highly specialized and public access point into the body having one of the body’s most robust and complex set of host defenses. Pg can and does exist in the oral cavity in what appears in some cases as oral cavity/gum disease free state. This may well be due to the existence of multiple strains and genotypes of the organism. As we know from the biofilm associated Distributed genome/Supra-genome hypothesis of bacterial pathogenesis, a large number of virulence genes are “shared” in the biofilm thereby transferring new properties to the acquiring bacteria. Once this is done, major new phenotypes emerge, and disease processes begin. Ongoing Pg genotyping work (Immoratii et al., unpublished studies) and others [141] have demonstrated and suggest that specific sets of virulence genes of Pg exist (fim A). Assuming that when this transformation occurs or the direct infection with virulent genotypes of Pg occurs, the entirety of the processes described in this paper ensue. More specifically, as is the case for Pg, in such a hostile, microbe-eliminating/killing environment as is found in the oral cavity, has it that these nanoscale, chemically dynamic, proteo-liposomes that are released from the bacterial cell and into surrounding tissues, and are rapidly internalized into the surrounding epithelial, fibroblasts and immune cells can and do have off site un-expected pathogenic consequences.
As depicted in (Figs. 18–20 and Table 2), the work presented here of Pg OMV-mediated sporadic AD in some or all forms an oral-neurogenic driven disease process is believed to be the major driving processes for this and the multiple other systemic inflammatory diseases listed in the beginning. Pg exists initially and possibly ultimately as a small population poly-microbial infection. Depending on environmental signaling, more specifically iron mostly coming from salivary Lf provide a heme excess environment for some time (Phase 1). It is hypothesized that Pg OMVs at this stage have a unique molecular signature that is enriched in various adhesion molecules. These find their way through and around the interstitial spaces (lymphatics) and epithelium/basement membrane to nearby microvascular networks (Fig. 19). Once there, they circulate to the brain and bind to yet unknown endothelia extravasation signaling molecules and though the BBB/meningeal lining cells and finally into adjacent neural parenchymal cells. These are to possibly exist and explain the early localization to the cholinergic neurons, basal forebrain, and anterior hypothalamic regions and regions near ventricles and peripheral neurons, an early pathway to Pg OMV entry to brain (Beginning of Phase 2) (Table 2). Ultimately the brain inflammation in this region leads to a shift in the delicate balance of salivary Lf coming from the decreased production of the salivary glands leading ultimately to shift in biofilm sensing system to a heme limited environment. Phase 3 begins the shift now to Pg OMVs enriching their protein cargo for increased iron scavenging. OMVs now entering the brain bring in iron with them and possibly through other unknown endothelial signaling and or now a general breakdown of the BBB, these Fe-loaded OMVs target the hippocampus and frontal-temporal lobes and neo-cortex. This is a more pathogenic period for the brain with the loss of the Lf protein protection system of the brain and the more incessant loading of iron a more advanced stage of AD occurs. Sometime between Phases 2-3, there is a greater chance for the entry of either more Pg bacterial cells other non-specific bacteria, viruses and fungi to locate in the parenchyma. This being due to both the loss of BBB integrity and innate and acquired immune suppression. The early cognitive decline seen in the prodromal period is most likely occurring in Phase 2. The more progressive cognition and memory losses coming in the Phase 3 period when both the Lf protection system is failing and the iron dyshomeostasis is occurring through the iron loaded OMV mediated period.
Fig. 19
Schematic for the working model of P. gingivalis (Pg) OMV-mediated local and systemic disease production. Pg colonies in green circles form poly-microbial biofilm in the gingival sulcus and tissues in Phases of chronicity. Released Pg OMVs depending on quorum sensing mechanisms are released with various protein/lipo-protein cargos through local and distant site trans-cellular and direct migration resulting in systemic organ/cellular modulation/inflammation and degeneration (cardio-vascular, neuro-vascular/anatomic). Top panel is from https://www.hygiene-in-practice.com/media/biofilm-development/ (Adapted © Legger | Dreamstime.com). Bottom panel is reprinted from Cecil JD, Sirisaengtaksin N, O’Brien-Simpson NM, Krachler AM (2019) Outer membrane vesicle-host cell interactions. Microbiol Spectr 7, 10.1128/microbiolspec.PSIB-0001-2018, with permission from ASM Press.
Fig. 20
P. gingivalis (Pg) OMV model of blood-brain barrier (BBB)/ventricle entry. A) Schematic of BBB/ventricle flow showing Pg OMVs (blue circles). OMVs released mainly from the oral cavity circulate in blood and bind to or trans-migrate neuro-vascular endothelial cells of the BBB. OMVs in the CSF have deeper and different access to neuro-anatomic locations associated with AD and PD. B) IHC of Pg HagA/gingipains antigen in choroid plexus in ventricles of hippocampus. C) Cross sectional drawing for enlarging ventricles and AD stage. Panel A is reprinted from Nau R, Sörgel F, Eiffert H (2010) Penetration of drugs through the blood-cerebrospinal fluid/blood-brain barrier for treatment of central nervous system infections. Clin Microbiol Rev 23, 858–883, with permission from the American Society for Microbiology. Panel C is from https://wiki.mcmaster.ca/LIFESCI_4M03/_detail/brain-w550.jpg?id=group_4_presentation_2_-_alzheimers.
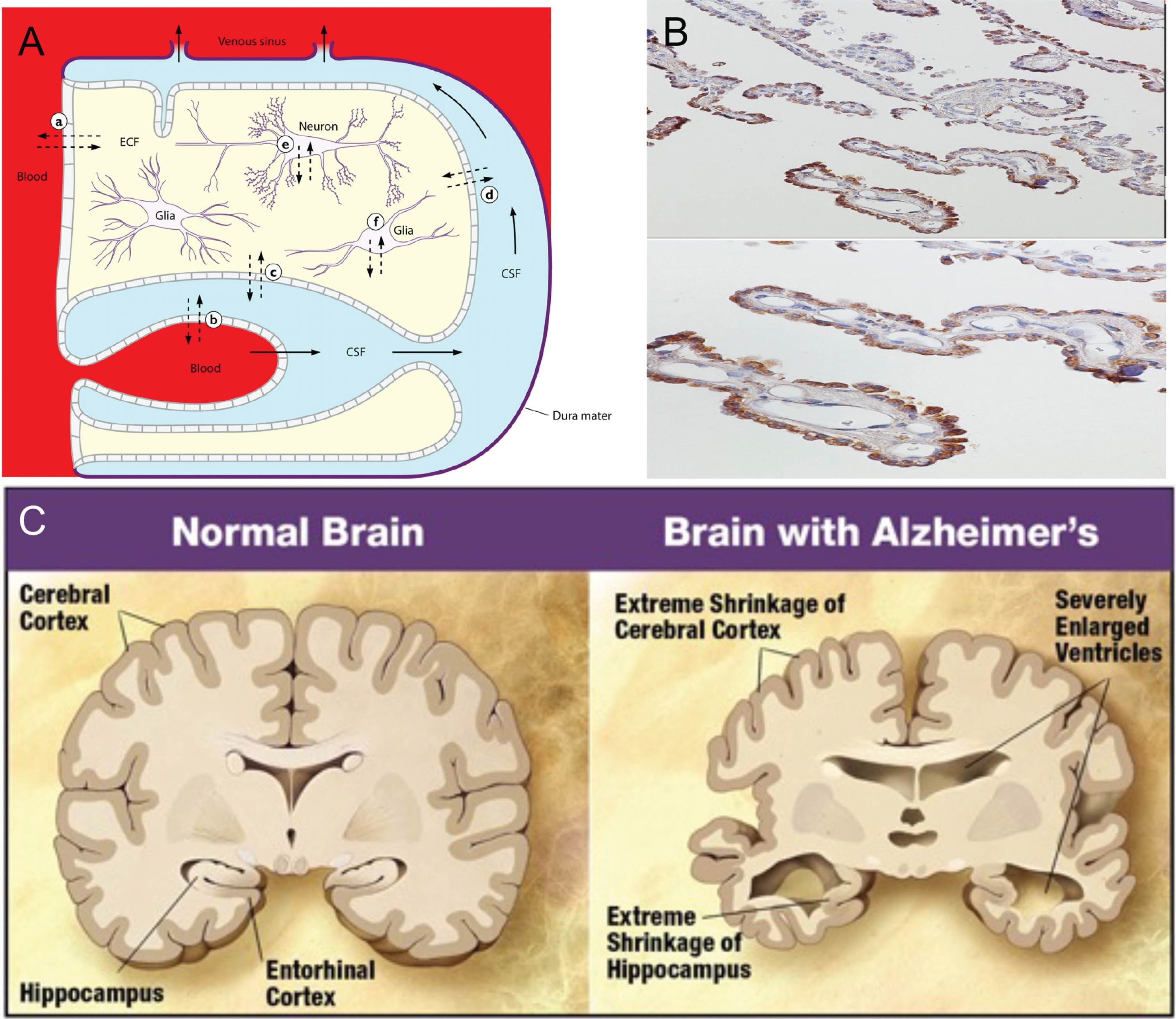
Table 2
Pg OMV mediated Oro-neuro-pathogenesis in AD
| Phase -1 | Oral/gingival colonization- Initial small avirulent Pg colony formation with virulent genotype transformation via horizontal gene transfer by type IV pili mediated distribute gene network poly-microbial biofilm matrix. No clinical brain disease. Time: 1 year-initial onset however could be highly variable from 20–40 years of age. No Stage. |
| Phase-2 | Formation of complex poly-microbial biofilm:- with salivary lactoferrin iron acquisition mediated through fimA virulence type IX cargo system. (Heme excess period-iron equilibrium). Secretion of adhesin enriched OMV cargo in the oral biofilm with vascular/lymphatic dissemination via BBB to arginine and lysine enriched tissues of to cholinergic neurons, basal forebrain and anterior hypothalamic regions and regions near ventricles. Brain lactoferrin protection system intact. Start of vascular insult, brain inflammation and amyloid-β formation in vessel walls and amyloid precursor protein. Beginning of OMV accumulation of the brain-No clinical disease. Stage 1; Time 2–5 years. Ages 30–50. |
| Phase-3 | Deterioration of cholinergic centers- to support salivary gland lactoferrin secretion into oral cavity. Pg biofilm senses heme limiting environment due to lactoferrin sources being consumed and switches to heme scavenging protein, complex cargo on OMVs. Brain lactoferrin system begins to slow down and fail. BBB beginning to become leaky in various deep locations. Brain inflammation present and expanding. Amyloid-β 42 and neurofibrillary tangles. Stage 2–4; Time 5–10 years. Ages 50–60. |
| Phase-4 | Iron Dyshomostasis period- with heme limited iron-rich OMVs switch iron sources now taking heme from red blood cells and continuing dissemination to and expanding on the degeneration of the deeper cholinergic, basal forebrain and anterior hypothalamic centers to expand into hippocampus. Breakdown of BBB and onset of some non-specific microbial and viral opportunistic infection and or innocent by-stander localizations-mistaken for primary pathogens of disease. All of Phase 4 pathology plus plaques forming and the beginning loss of supporting cells and neuronal dysfunction. Stage 5-6; Ages 60–80. |
| Phase-5 | Collapse of the Brain- OMVs regularly delivering toxic levels of iron to brain regularly coupled to HagA/gingipain/hemagginitin/adhesin load, total loss of brain lactoferrin system advanced neuropathology. Loss of neuronal synapse and cell death shrinkage. Stage 6-7; Ages 60–80 |
In summary, a working model for explaining the evidence of the relatively low amounts of Pg bacteria residing in the AD brain as compared to the rather florid and broad systemic distribution of Pg specific virulence factors is now presented. The multiple number of earlier, somewhat desparted disparate in nature and other AD inter-related findings such as the Gingipains Hypothesis, AD related iron dyshomeostasis, early reduced salivary Lf, along with resurrecting the Cholinergic Hypothesis may now further be explained. Furthermore, this review suggests that the highly developed and complex regulated Type IX secretory cargo system of Pg producing OMVs and/or as soluble molecules becoming systemic may now better explain a more unifying model for the sporadic form of AD disease and guide the direction of research, treatment and possible prevention.
CONCLUSION
Through long-distance transmission, Pg OMVs can reach distant target organs and induce related systemic diseases. A recent review of this subject [142] indicates the correlation between Pg OMVs and systemic system diseases. First, in cardio-metabolic diseases, Pg OMVs attenuate insulin-induced Akt/GSK-3β signaling in hepatic HepG2 cells, thereby causing changes in glucose metabolism in the liver and promoting the development of diabetes. Second, Pg OMVs activate Rho-associated protein kinase (ROCK), which is a kinase belonging to the AGC family of serine-threonine kinases. It is involved mainly in regulating the shape and movement of cells by acting on the cytoskeleton. Pg-mediated ROCK activation of human umbilical vein endothelial cells through ERK1/2 and p38 MAPK-dependent mechanisms promote endothelial dysfunction, promote vascular smooth muscle cell calcification through ERK1/2-RUNX2, and increase vascular permeability by cleaving endothelial cell connexins such as PECAM-1, thereby promoting cardiovascular disease. Third, Pg OMVs contain peptidylarginine deiminase (PPAD), which is closely related to rheumatoid arthritis, and are modified by A-LPS to protect PPAD from proteolytic degradation. However, the relationship between Pg OMVs and other systemic diseases including non-alcoholic fatty liver disease and carcinoma is still in early stages of research, and further exploration is needed. The transforming factor Cag of H. pylori, however, serves notice of how Pg OMVs could participate in cellular transformation as well. In summary, compared to its parent bacteria, OMVs contain a higher concentration of virulence factors, and a thin membrane is used to protect their smaller structures and insulate the toxic factors from systemic inflammatory cascades until these toxic payloads are delivered to various organ and vascular based tissue targets. These characteristics enable OMVs to achieve long-distance transmission and reach locations where the parent bacteria cannot access, amplifying the pathogenic effects of Pg in the oral environment and even in various parts of the body.
ACKNOWLEDGMENTS
Peter L. Nara and Daniel Sindelar are full time employees and Marc Penn is a part-time employee of Keystone Bio during the writing of and review of this manuscript. Jan Potempa was supported via a grant entitled “The link between PD and AD” - grant 2019/01/Y/NZ1/00016 from the Polish National Science Center (NCN) within the EC program ERA-NET_JPND / JPco-fuND2. Sue Griffin was supported by NIH/NIA AG012411. The authors additionally thank Ann Progulsky-Fox and Sasanka Chuckkaalli (Center for Molecular Biology, University of Florida) for fruitful discussions.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/21-0448).
REFERENCES
[1] | Merino N , Aronson HS , Bojanova DP , Feyhl-Buska J , Wong ML , Zhang S , Giovannelli D ((2019) ) Living at the extremes: Extremophiles and the limits of life in a planetary context. Front Microbiol 10: , 780. |
[2] | Naito M , Hirakawa H , Yamashita A , Ohara N , Shoji M , Yukitake H , Nakayama K , Toh H , Yoshimura F , Kuhara S , Hattori M , Hayashi T , Nakayama K ((2008) ) Determination of the genome sequence of Porphyromonas gingivalis strain ATCC 33277 and genomic comparison with strain W83 revealed extensive genome rearrangements in P. gingivalis. DNA Res 15: , 215–225. |
[3] | Takahashi N ((2003) ) Acid-neutralizing activity during amino acid fermentation by Porphyromonas gingivalis, Prevotella intermedia and Fusobacterium nucleatum. Oral Microbiol Immunol 18: , 109–113. |
[4] | Klein BA , Tenorio EL , Lazinski DW , Camilli A , Duncan MJ , Hu LT ((2012) ) Identification of essential genes of the periodontal pathogen Porphyromonas gingivalis. BMC Genomics 13: , 578. |
[5] | Acuña-Amador L , Primot A , Cadieu E , Roulet A , Barloy-Hubler F ((2018) ) Genomic repeats, misassembly and reannotation: A case study with long-read resequencing of Porphyromonas gingivalis reference strains. BMC Genomics 19: , 54. |
[6] | Nelson KE , Fleischmann RD , DeBoy RT , Paulsen IT , Fouts DE , Eisen JA , Daugherty SC , Dodson RJ , Durkin AS , Gwinn M , Haft DH , Kolonay JF , Nelson WC , Mason T , Tallon L , Gray J , Granger D , Tettelin H , Dong H , Galvin JL , Duncan MJ , Dewhirst FE , Fraser CM ((2004) ) Complete genome sequence of the oral pathogenic bacterium Porphyromonas gingivalis strain W83. J Bacteriol 186: , 593–593. |
[7] | Merrell DS , Goodrich ML , Otto G , Tompkins LS , Falkow S ((2003) ) pH-regulated gene expression of the gastric pathogen Helicobacter pylori. Infect Immun 71: , 3529–3539. |
[8] | Tanner ACR ((2015) ) Anaerobic culture to detect periodontal and caries pathogens. J Oral Biosci 57: , 18–26. |
[9] | Bakaletz LO ((2004) ) Developing animal models for polymicrobial diseases. Nat Rev Microbiol 2: , 552–568. |
[10] | Sbordone L , Bortolaia C ((2003) ) Oral microbial biofilms and plaque-related diseases: Microbial communities and their role in the shift from oral health to disease. Clin Oral Investig 7: , 181–188. |
[11] | Mei F , Xie M , Huang X , Long Y , Lu X , Wang X , Chen L ((2020) ) Porphyromonas gingivalis and its systemic impact: Current status. Pathogens 9: , 944. |
[12] | Fiorillo L , Cervino G , Laino L , D’Amico C , Mauceri R , Tozum TF , Gaeta M , Cicciù M ((2019) ) Porphyromonas gingivalis, periodontal and systemic implications: A systematic review. Dent J 7: , 114. |
[13] | Gholizadeh P , Eslami H , Yousefi M , Asgharzadeh M , Aghazadeh M , Kafil HS ((2016) ) Role of oral microbiome on oral cancers, a review. Biomed Pharmacother 84: , 552–558. |
[14] | Mulhall H , Huck O , Amar S ((2020) ) Porphyromonas gingivalis, a long-range pathogen: Systemic impact and therapeutic implications. Microorganisms 8: , 1–15. |
[15] | Hajishengallis G , Darveau RP , Curtis MA ((2012) ) The keystone-pathogen hypothesis. Nat Rev Microbiol 10: , 717–725. |
[16] | Loos BG , Dyer DW , Whittam TS , Selander RK ((1993) ) Genetic structure of populations of Porphyromonas gingivalis associated with periodontitis and other oral infections. Infect Immun 61: , 204–212. |
[17] | Gupta RS ((2011) ) Origin of diderm (Gram-negative) bacteria: Antibiotic selection pressure rather than endosymbiosis likely led to the evolution of bacterial cells with two membranes. Antonie Van Leeuwenhoek 100: , 171–182. |
[18] | Gong T , Zeng J , Tang B , Zhou X , Li Y ((2020) ) CRISPR-Cas systems in oral microbiome: From immune defense to physiological regulation. Mol Oral Microbiol 35: , 41–48. |
[19] | Sampson TR , Saroj SD , Llewellyn AC , Tzeng YL , Weiss DS ((2013) ) A CRISPR/Cas system mediates bacterial innate immune evasion and virulence. Nature 497: , 254–257. |
[20] | Makarova KS , Wolf YI , Iranzo J , Shmakov SA , Alkhnbashi OS , Brouns SJJ , Charpentier E , Cheng D , Haft DH , Horvath P , Moineau S , Mojica FJM , Scott D , Shah SA , Siksnys V , Terns MP , Venclovas Č , White MF , Yakunin AF , Yan W , Zhang F , Garrett RA , Backofen R , van der Oost J , Barrangou R , Koonin EV ((2020) ) Evolutionary classification of CRISPR-Cas systems: A burst of class 2 and derived variants. Nat Rev Microbiol 18: , 67–83. |
[21] | Jansen R , Embden JD , Gaastra W , Schouls LM ((2002) ) Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol 43: , 1565–1575. |
[22] | Sampson TR , Weiss DS ((2014) ) CRISPR-Cas systems: New players in gene regulation and bacterial physiology. Front Cell Infect Microbiol 4: , 37. |
[23] | Marraffini LA ((2015) ) CRISPR-Cas immunity in prokaryotes. Nature 526: , 55–61. |
[24] | Sternberg SH , Richter H , Charpentier E , Qimron U ((2016) ) Adaptation in CRISPR-Cas Systems. Mol Cell 61: , 797–808. |
[25] | Hitch G , Pratten J , Taylor PW ((2004) ) Isolation of bacteriophages from the oral cavity. Lett Appl Microbiol 39: , 215–219. |
[26] | Solbiati J , Duran-Pinedo A , Godoy Rocha F , Gibson FC , Frias-Lopez J ((2020) ) Virulence of the pathogen Porphyromonas gingivalis is controlled by the CRISPR-Cas protein Cas3. mSystems 5: , e00852–20. |
[27] | Rosenfeld Y , Shai Y ((2006) ) Lipopolysaccharide (Endotoxin)-host defense antibacterial peptides interactions: Role in bacterial resistance and prevention of sepsis. Biochim Biophys Acta 1758: , 1513–1522. |
[28] | Zamyatina A , Heine H ((2020) ) Lipopolysaccharide recognition in the crossroads of TLR4 and Caspase-4/11 mediated inflammatory pathways. Front Immunol 11: , 585146. |
[29] | Ishii KJ , Koyama S , Nakagawa A , Coban C , Akira S ((2008) ) Host innate immune receptors and beyond: Making sense of microbial infections. Cell Host Microbe 3: , 352–363. |
[30] | Slocum C , Coats SR , Hua N , Kramer C , Papadopoulos G , Weinberg EO , Gudino C V. , Hamilton JA , Darveau RP , Genco CA ((2014) ) Distinct lipid A moieties contribute to pathogen-induced site-specific vascular inflammation. PLoS Pathog 10: , e1004215. |
[31] | Needham BD , Trent MS ((2013) ) Fortifying the barrier: The impact of lipid A remodelling on bacterial pathogenesis. Nat Rev Microbiol 11: , 467–481. |
[32] | Raetz CRH , Whitfield C ((2002) ) Lipopolysaccharide endotoxins. Annu Rev Biochem 71: , 635–700. |
[33] | Coats SR , Berezow AB , To TT , Jain S , Bainbridge BW , Banani KP , Darveau RP ((2011) ) The lipid A phosphate position determines differential host toll-like receptor 4 responses to phylogenetically related symbiotic and pathogenic bacteria. Infect Immun 79: , 203–210. |
[34] | Cullen TW , Giles DK , Wolf LN , Ecobichon C , Boneca IG , Trent MS ((2011) ) Helicobacter pylori versus the host: Remodeling of the bacterial outer membrane is required for survival in the gastric mucosa. PLoS Pathog 7: , e1002454. |
[35] | Neumeister B , Faigle M , Sommer M , Zähringer U , Stelter F , Menzel R , Schütt C , Northoff H ((1998) ) Low endotoxic potential of Legionella pneumophila lipopolysaccharide due to failure of interaction with the monocyte lipopolysaccharide receptor CD14. Infect Immun 66: , 4151–4157. |
[36] | Kawahara K , Tsukano H , Watanabe H , Lindner B , Matsuura M ((2002) ) Modification of the structure and activity of lipid A in Yersinia pestis lipopolysaccharide by growth temperature. Infect Immun 70: , 4092–4098. |
[37] | Hajjar AM , Harvey MD , Shaffer SA , Goodlett DR , Sjöstedt A , Edebro H , Forsman M , Byström M , Pelletier M , Wilson CB , Miller SI , Skerrett SJ , Ernst RK ((2006) ) Lack of in vitro and in vivo recognition of Francisella tularensis subspecies lipopolysaccharide by toll-like receptors. Infect Immun 74: , 6730–6738. |
[38] | Rebeil R , Ernst RK , Jarrett CO , Adams KN , Miller SI , Hinnebusch BJ ((2006) ) Characterization of late acyltransferase genes of Yersinia pestis and their role in temperature-dependent lipid A variation. J Bacteriol 188: , 1381–1388. |
[39] | Li Y , Powell DA , Shaffer SA , Rasko DA , Pelletier MR , Leszyk JD , Scott AJ , Masoudi A , Goodlett DR , Wang X , Raetz CRH , Ernst RK ((2012) ) LPS remodeling is an evolved survival strategy for bacteria. Proc Natl Acad Sci U S A 109: , 13877–13877. |
[40] | Maeshima N , Fernandez RC ((2013) ) Recognition of lipid A variants by the TLR4-MD-2 receptor complex. Front Cell Infect Microbiol 3: , 3. |
[41] | Kayagaki N , Wong MT , Stowe IB , Ramani SR , Gonzalez LC , Akashi-Takamura S , Miyake K , Zhang J , Lee WP , Muszynski A , Forsberg LS , Carlson RW , Dixit VM ((2013) ) Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341: , 1246–1249. |
[42] | Franchi L , Eigenbrod T , Muñoz-Planillo R , Nuñez G ((2009) ) The inflammasome: A caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol 10: , 241–247. |
[43] | Taxman DJ , Huang MTH , Ting JPY ((2010) ) Inflammasome inhibition as a pathogenic stealth mechanism. Cell Host Microbe 8: , 7–11. |
[44] | Shimada K , Crother TR , Karlin J , Dagvadorj J , Chiba N , Chen S , Ramanujan VK , Wolf AJ , Vergnes L , Ojcius DM , Rentsendorj A , Vargas M , Guerrero C , Wang Y , Fitzgerald KA , Underhill DM , Town T , Arditi M ((2012) ) Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36: , 401–414. |
[45] | Kayagaki N , Warming S , Lamkanfi M , Walle Vande L , Louie S , Dong J , Newton K , Qu Y , Liu J , Heldens S , Zhang J , Lee WP , Roose-Girma M , Dixit VM ((2011) ) Non-canonical inflammasome activation targets caspase-11. Nature 479: , 117–121. |
[46] | Rathinam VAK , Vanaja SK , Waggoner L , Sokolovska A , Becker C , Stuart LM , Leong JM , Fitzgerald KA ((2012) ) TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by Gram-negative bacteria. Cell 150: , 606–619. |
[47] | Dominy SS , Lynch C , Ermini F , Benedyk M , Marczyk A , Konradi A , Nguyen M , Haditsch U , Raha D , Griffin C , Holsinger LJ , Arastu-Kapur S , Kaba S , Lee A , Ryder MI , Potempa B , Mydel P , Hellvard A , Adamowicz K , Hasturk H , Walker GD , Reynolds EC , Faull RLMM , Curtis MA , Dragunow M , Potempa J ((2019) ) Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv 5: , eaau3333. |
[48] | Wu Y , Du S , Johnson JL , Tung H-Y , Landers CT , Liu Y , Seman BG , Wheeler RT , Costa-Mattioli M , Kheradmand F , Zheng H , Corry DB ((2019) ) Microglia and amyloid precursor protein coordinate control of transient Candida cerebritis with memory deficits. Nat Commun 10: , 58. |
[49] | Choi J-W , Um J-H , Cho J-H , Lee H-J ((2017) ) Tiny RNAs and their voyage via extracellular vesicles: Secretion of bacterial small RNA and eukaryotic microRNA. Exp Biol Med 242: , 1475–1481. |
[50] | Lee H-J ((2019) ) Microbe-host communication by small RNAs in extracellular vesicles: Vehicles for transkingdom RNA Transportation. Int J Mol Sci 20: , 1487. |
[51] | Choi J-W , Kim S-C , Hong S-H , Lee H-J ((2017) ) Secretable small RNAs via outer membrane vesicles in periodontal pathogens. J Dent Res 96: , 458–466. |
[52] | Chatterjee SN , Das J ((1967) ) Electron microscopic observations on the excretion of cell-wall material by Vibrio cholerae. J Gen Microbiol 49: , 1–11. |
[53] | Kuehn MJ ((2005) ) Bacterial outer membrane vesicles and the host-pathogen interaction. Genes Dev 19: , 2645–2655. |
[54] | Bomberger JM , MacEachran DP , Coutermarsh BA , Ye S , O’Toole GA , Stanton BA ((2009) ) Long-distance delivery of bacterial virulence factors by Pseudomonas aeruginosa outer membrane vesicles. PLoS Pathog 5: , e1000382. |
[55] | Nonaka S , Nakanishi H ((2020) ) Secreted gingipains from Porphyromonas gingivalis induce microglia migration through endosomal signaling by protease-activated receptor 2. Neurochem Int 140: , 104840. |
[56] | Veith PD , Chen YY , Gorasia DG , Chen D , Glew MD , O’Brien-Simpson NM , Cecil JD , Holden JA , Reynolds EC ((2014) ) Porphyromonas gingivalis outer membrane vesicles exclusively contain outer membrane and periplasmic proteins and carry a cargo enriched with virulence factors. J Proteome Res 13: , 2420–2432. |
[57] | Lamont RJ , Jenkinson HF ((1998) ) Life below the gum line: Pathogenic mechanisms of Porphyromonas gingivalis. Microbiol Mol Biol Rev 62: , 1244–1263. |
[58] | Bostanci N , Belibasakis GN ((2012) ) Porphyromonas gingivalis: An invasive and evasive opportunistic oral pathogen. FEMS Microbiol Lett 333: , 1–9. |
[59] | Kebschull M , Demmer RT , Papapanou PN ((2010) ) “Gum bug, leave my heart alone!”—Epidemiologic and mechanistic evidence linking periodontal infections and atherosclerosis. J Dent Res 89: , 879–902. |
[60] | Mantri CK , Chen CH , Dong X , Goodwin JS , Pratap S , Paromov V , Xie H ((2015) ) Fimbriae-mediated outer membrane vesicle production and invasion of Porphyromonas gingivalis. Microbiologyopen 4: , 53–65. |
[61] | Kerr JE , Abramian JR , Dao D-H V. , Rigney TW , Fritz J , Pham T , Gay I , Parthasarathy K , Wang B , Zhang W , Tribble GD ((2014) ) Genetic exchange of fimbrial alleles exemplifies the adaptive virulence strategy of Porphyromonas gingivalis. PLoS One 9: , e91696. |
[62] | Furuta N , Tsuda K , Omori H , Yoshimori T , Yoshimura F , Amano A ((2009) ) Porphyromonas gingivalis outer membrane vesicles enter human epithelial cells via an endocytic pathway and are sorted to lysosomal compartments. Infect Immun 77: , 4187–4196. |
[63] | Minter MR , Taylor JM , Crack PJ ((2016) ) The contribution of neuroinflammation to amyloid toxicity in Alzheimer’s disease. J Neurochem 136: , 457–474. |
[64] | Jan AT ((2017) ) Outer membrane vesicles (OMVs) of Gram-negative bacteria: A perspective update. Front Microbiol 8: , 1053. |
[65] | Guentsch A , Kramesberger M , Sroka A , Pfister W , Potempa J , Eick S ((2011) ) Comparison of gingival crevicular fluid sampling methods in patients with severe chronic periodontitis. J Periodontol 82: , 1051–1060. |
[66] | Lee JS , Spooner R , Chowdhury N , Pandey V , Wellslager B , Atanasova KR , Evans Z , Yilmaz Ö ((2020) ) In situ intraepithelial localizations of opportunistic pathogens, Porphyromonas gingivalis and Filifactor alocis, in human gingiva. Curr Res Microb Sci 1: , 7–17. |
[67] | O’Brien-Simpson NM , Pathirana RD , Walker GD , Reynolds EC ((2009) ) Porphyromonas gingivalis RgpA-Kgp proteinase-adhesin complexes penetrate gingival tissue and induce proinflammatory cytokines or apoptosis in a concentration-dependent manner. Infect Immun 77: , 1246–1261. |
[68] | Pathirana RD , O’Brien-Simpson NM , Veith PD , Riley PF , Reynolds EC ((2006) ) Characterization of proteinase-adhesin complexes of Porphyromonas gingivalis. Microbiology 152: , 2381–2394. |
[69] | Potempa J , Banbula A , Travis J ((2000) ) Role of bacterial proteinases in matrix destruction and modulation of host responses. Periodontol 2000 24: , 153–192. |
[70] | Andrian E , Grenier D , Rouabhia M ((2004) ) In vitro models of tissue penetration and destruction by Porphyromonas gingivalis. Infect Immun 72: , 4689–4698. |
[71] | Yilmaz O , Verbeke P , Lamont RJ , Ojcius DM ((2006) ) Intercellular spreading of Porphyromonas gingivalis infection in primary gingival epithelial cells. Infect Immun 74: , 703–710. |
[72] | Takeuchi H , Furuta N , Morisaki I , Amano A ((2011) ) Exit of intracellular Porphyromonas gingivalis from gingival epithelial cells is mediated by endocytic recycling pathway. Cell Microbiol 13: , 677–691. |
[73] | Kumar PS , Leys EJ , Bryk JM , Martinez FJ , Moeschberger ML , Griffen AL ((2006) ) Changes in periodontal health status are associated with bacterial community shifts as assessed by quantitative 16S cloning and sequencing. J Clin Microbiol 44: , 3665–3673. |
[74] | Chaves ES , Jeffcoat MK , Ryerson CC , Snyder B ((2000) ) Persistent bacterial colonization of Porphyromonas gingivalis, Prevotella intermedia, and Actinobacillus actinomycetemcomitans in periodontitis and its association with alveolar bone loss after 6 months of therapy. J Clin Periodontol 27: , 897–903. |
[75] | Doungudomdacha S , Rawlinson A , Douglas CWI ((2000) ) Enumeration of Porphyromonas gingivalis, Prevotella intermedia and Actinobacillus actinomycetemcomitans in subgingival plaque samples by a quantitative-competitive PCR method. J Med Microbiol 49: , 861–874. |
[76] | Pathirana RD , O’Brien-Simpson NM , Brammar GC , Slakeski N , Reynolds EC ((2007) ) Kgp and RgpB, but not RgpA, are important for Porphyromonas gingivalis virulence in the murine periodontitis model. Infect Immun 75: , 1436–1442. |
[77] | Fiocca R , Necchi V , Sommi P , Ricci V , Telford J , Cover TL , Solcia E ((1999) ) Release ofHelicobacter pylori vacuolating cytotoxin by both a specific secretion pathway and budding of outer membrane vesicles. Uptake of released toxin and vesicles by gastric epithelium. J Pathol 188: , 220–226. |
[78] | Keenan J ((2000) ) A role for the bacterial outer membrane in the pathogenesis of Helicobacter pylori infection. FEMS Microbiol Lett 182: , 259–264. |
[79] | Frohlich KM , Hua Z , Quayle AJ , Wang J , Lewis ME , Chou C , Luo M , Buckner LR , Shen L ((2014) ) Membrane vesicle production by Chlamydia trachomatis as an adaptive response. Front Cell Infect Microbiol 4: , 73. |
[80] | Stirling P , Richmond SJ ((1980) ) Production of outer membrane blebs during chlamydial replication. FEMS Microbiol Lett 9: , 103–105. |
[81] | Beermann C , Groscurth P , Filgueira L , Wunderli-Allenspach H , Groscurth P , Filgueira L ((2000) ) Lipoproteins from Borrelia burgdorferi applied in liposomes and presented by dendritic cells induce CD8+T-lymphocytes in vitro. Cell Immunol 201: , 124–131. |
[82] | Craven DE , Peppler MS , Frasch CE , Mocca LF , McGrath PP , Washington G ((1980) ) Adherence of isolates of Neisseria meningitidis from patients and carriers to human buccal epithelial cells. J Infect Dis 142: , 556–568. |
[83] | Stephens DS , Edwards KM , Morris F , McGee ZA ((1982) ) Pili and outer membrane appendages on Neisseria meningitidis in the cerebrospinal fluid of an infant. J Infect Dis 146: , 568–568. |
[84] | Brandtzaeg P , Bryn K , Kierulf P , Ovstebø R , Namork E , Aase B , Jantzen E ((1992) ) Meningococcal endotoxin in lethal septic shock plasma studied by gas chromatography, mass-spectrometry, ultracentrifugation, and electron microscopy. J Clin Invest 89: , 816–823. |
[85] | Namork E , Brandtzaeg P ((2002) ) Fatal meningococcal septicaemia with “blebbing” meningococcus. Lancet 360: , 1741. |
[86] | Dorward DW , Garon CF ((1989) ) DNA-binding proteins in cells and membrane blebs of Neisseria gonorrhoeae. J Bacteriol 171: , 4196–4201. |
[87] | Lee J , Lee Y , Yuk D , Choi D , Ban S , Oh K , Hong J ((2008) ) Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J Neuroinflammation 5: , 37. |
[88] | Ilievski V , Zuchowska PK , Green SJ , Toth PT , Ragozzino ME , Le K , Aljewari HW , O’Brien-Simpson NM , Reynolds EC , Watanabe K ((2018) ) Chronic oral application of a periodontal pathogen results in brain inflammation, neurodegeneration and amyloid beta production in wild type mice. PLoS One 13: , e0204941. |
[89] | Poole S , Singhrao SK , Kesavalu L , Curtis MA , Crean S ((2013) ) Determining the presence of periodontopathic virulence factors in short-term postmortem Alzheimer’s disease brain tissue. J Alzheimers Dis 36: , 665–677. |
[90] | Poole S , Singhrao SK , Chukkapalli S , Rivera M , Velsko I , Kesavalu L , Crean S ((2014) ) Active invasion of Porphyromonas gingivalis and infection-induced complement activation in ApoE-/- mice brains. J Alzheimers Dis 43: , 67–80. |
[91] | Singhrao SK , Chukkapalli S , Poole S , Velsko I , Crean SJ , Kesavalu L ((2017) ) Chronic Porphyromonas gingivalis infection accelerates the occurrence of age-related granules in ApoE - / - mice brains. J Oral Microbiol 9: , 1270602. |
[92] | Ishida N , Ishihara Y , Ishida K , Tada H , Kato Y , Isoda R , Hagiwara M , Michikawa M , Noguchi T , Matsushita K ((2013) ) Periodontitis induced by bacterial infection exacerbates features of Alzheimer’s disease in transgenic mice. Alzheimers Dement 9: , P851–P851. |
[93] | Liu Y , Wu Z , Nakanishi Y , Ni J , Hayashi Y , Takayama F , Zhou Y , Kadowaki T , Nakanishi H ((2017) ) Infection of microglia with Porphyromonas gingivalis promotes cell migration and an inflammatory response through the gingipain-mediated activation of protease-activated receptor-2 in mice. Sci Rep 7: , 11759. |
[94] | Han EC , Choi SY , Lee Y , Park JW , Hong SH , Lee HJ ((2019) ) Extracellular RNAs in periodontopathogenic outer membrane vesicles promote TNF-α production in human macrophages and cross the blood-brain barrier in mice. FASEB J 33: , 13412–13422. |
[95] | Darveau RP , Hajishengallis G , Curtis MA ((2012) ) Porphyromonas gingivalis as a potential community activist for disease. J Dent Res 91: , 816–820. |
[96] | Katz J , Chegini N , Shiverick KT , Lamont RJ ((2009) ) Localization of P. gingivalis in preterm delivery placenta. J Dent Res 88: , 575–578. |
[97] | Mahendra J , Mahendra L , Kurian VM , Jaishankar K , Mythilli R ((2009) ) Prevalence of periodontal pathogens in coronary atherosclerotic plaque of patients undergoing coronary artery bypass graft surgery. J Maxillofac Oral Surg 8: , 108–113. |
[98] | Mougeot J-LC , Stevens CB , Paster BJ , Brennan MT , Lockhart PB , Mougeot FKB ((2017) ) Porphyromonas gingivalis is the most abundant species detected in coronary and femoral arteries. J Oral Microbiol 9: , 1281562. |
[99] | Ishikawa M , Yoshida K , Okamura H , Ochiai K , Takamura H , Fujiwara N , Ozaki K ((2013) ) Oral Porphyromonas gingivalis translocates to the liver and regulates hepatic glycogen synthesis through the Akt/GSK-3β signaling pathway. Biochim Biophys Acta 1832: , 2035–2043. |
[100] | Xia Q , Wang T , Taub F , Park Y , Capestany CA , Lamont RJ , Hackett M ((2007) ) Quantitative proteomics of intracellular Porphyromonas gingivalis. Proteomics 7: , 4323–4337. |
[101] | Li L , Michel R , Cohen J , DeCarlo A , Kozarov E ((2008) ) Intracellular survival and vascular cell-to-cell transmission of Porphyromonas gingivalis. BMC Microbiol 8: , 26. |
[102] | Deshpande RG , Khan M , Genco CA ((1998) ) Invasion strategies of the oral pathogen Porphyromonas gingivalis: Implications for cardiovascular disease. Invasion Metastasis 18: , 57–69. |
[103] | Deshpande RG , Khan MB , Attardo Genco C ((1998) ) Invasion of aortic and heart endothelial cells by Porphyromonas gingivalis. Infect Immun 66: , 5337–5343. |
[104] | Duncan MJ , Nakao S , Skobe Z , Xie H ((1993) ) Interactions of Porphyromonas gingivalis with epithelial cells. Infect Immun 61: , 2260–2265. |
[105] | Yilmaz Ö , Young PA , Lamont RJ , Kenny GE ((2003) ) Gingival epithelial cell signalling and cytoskeletal responses to Porphyromonas gingivalis invasion. Microbiology 149: , 2417–2426. |
[106] | Belton CM , Izutsu KT , Goodwin PC , Park Y , Lamont RJ ((1999) ) Fluorescence image analysis of the association between Porphyromonas gingivalis and gingival epithelial cells. Cell Microbiol 1: , 215–223. |
[107] | Haditsch U , Roth T , Rodriguez L , Hancock S , Cecere T , Nguyen M , Arastu-Kapur S , Broce S , Raha D , Lynch CC , Holsinger LJ , Dominy SS , Ermini F ((2020) ) Alzheimer’s disease-like neurodegeneration in Porphyromonas gingivalis infected neurons with persistent expression of active gingipains. J Alzheimers Dis 75: , 1361–1376. |
[108] | András IE , Toborek M ((2016) ) Extracellular vesicles of the blood-brain barrier. Tissue Barriers 4: , e1131804. |
[109] | Jiang L , Dong H , Cao H , Ji X , Luan S , Liu J ((2019) ) Exosomes in pathogenesis, diagnosis, and treatment of Alzheimer’s disease. Med Sci Monit 25: , 3329–3335. |
[110] | Garcia-Contreras M , Shah SH , Tamayo A , Robbins PD , Golberg RB , Mendez AJ , Ricordi C ((2017) ) Plasma-derived exosome characterization reveals a distinct microRNA signature in long duration Type 1 diabetes. Sci Rep 7: , 5998. |
[111] | Ghosal A , Upadhyaya BB , Fritz J V. , Heintz-Buschart A , Desai MS , Yusuf D , Huang D , Baumuratov A , Wang K , Galas D , Wilmes P ((2015) ) The extracellular RNA complement of Escherichia coli. Microbiologyopen 4: , 252–266. |
[112] | Choi S , Kim K , Chang J , Kim SM , Kim SJ , Cho HJ , Park SM ((2019) ) Association of chronic periodontitis on Alzheimer’s disease or vascular dementia. J Am Geriatr Soc 67: , 1234–1239. |
[113] | Kamer AR , Craig RG , Pirraglia E , Dasanayake AP , Norman RG , Boylan RJ , Nehorayoff A , Glodzik L , Brys M , de Leon MJ ((2009) ) TNF-α and antibodies to periodontal bacteria discriminate between Alzheimer’s disease patients and normal subjects. J Neuroimmunol 216: , 92–97. |
[114] | Ha JY , Choi SY , Lee JH , Hong SH , Lee HJ ((2020) ) Delivery of periodontopathogenic extracellular vesicles to brain monocytes and microglial IL-6 promotion by RNA cargo. Front Mol Biosci 7: , 596366. |
[115] | Kobayashi R , Ogawa Y , Hashizume-Takizawa T , Kurita-Ochiai T ((2020) ) Oral bacteria affect the gut microbiome and intestinal immunity. Pathog Dis 78: , ftaa024. |
[116] | Dorn BR , Dunn WA , Progulske-Fox A ((2001) ) Porphyromonas gingivalis traffics to autophagosomes in human coronary artery endothelial cells. Infect Immun 69: , 5698–5708. |
[117] | Lee K , Roberts JS , Choi CH , Atanasova KR , Yilmaz Ö ((2018) ) Porphyromonas gingivalis traffics into endoplasmic reticulum-rich-autophagosomes for successful survival in human gingival epithelial cells. Virulence 9: , 845–859. |
[118] | Lempiäinen H , Shore D ((2009) ) Growth control and ribosome biogenesis. Curr Opin Cell Biol 21: , 855–863. |
[119] | Francis PT , Palmer AM , Snape M , Wilcock GK ((1999) ) The cholinergic hypothesis of Alzheimer’s disease: A review of progress. J Neurol Neurosurg Psychiatry 66: , 137–147. |
[120] | Drachman DA ((1974) ) Human memory and the cholinergic system. Arch Neurol 30: , 113. |
[121] | Rønneberg A , Nordgarden H , Skaare AB , Willumsen T ((2019) ) Selective loss of central cholinergic neurons in Alzheimer’s disease. Int J Paediatr Dent 29: , 683–683. |
[122] | Patel S , Howard D , Chowdhury N , Derieux C , Wellslager B , Yilmaz Ö , French L ((2021) ) Characterization of human genes modulated by Porphyromonas gingivalis highlights the ribosome, hypothalamus, and cholinergic neurons. Front Immunol 12: , 2165. |
[123] | Patel S , Howard D , Man A , Schwartz D , Jee J , Felsky D , Pausova Z , Paus T , French L ((2020) ) Donor-specific transcriptomic analysis of Alzheimer’s disease-associated hypometabolism highlights a unique donor, ribosomal proteins and microglia. eneuro 7: , 0255–20.2020. |
[124] | Felsky D , Sariya S , Schneider JA , Bennett DA , Mayeux R , De Jager PL , Tosto G ((2020) ) The Caribbean-Hispanic Alzheimer’s brain transcriptome reveals ancestry-specific disease mechanisms. Alzheimers Dement 16: , 43068. |
[125] | Proctor GB , Carpenter GH ((2014) ) Salivary secretion: Mechanism and neural regulation. Monogr Oral Sci 24: , 14–29. |
[126] | González-Sánchez M , Bartolome F , Antequera D , Puertas-Martín V , González P , Gómez-Grande A , Llamas-Velasco S , Herrero-San Martín A , Pérez-Martínez D , Villarejo-Galende A , Atienza M , Palomar-Bonet M , Cantero JL , Perry G , Orive G , Ibañez B , Bueno H , Fuster V , Carro E ((2020) ) Decreased salivary lactoferrin levels are specific to Alzheimer’s disease. EBioMedicine 57: , 102834. |
[127] | Wang J , Bi M , Liu H , Song N , Xie J ((2015) ) The protective effect of lactoferrin on ventral mesencephalon neurons against MPP+is not connected with its iron binding ability. Sci Rep 5: , 10729. |
[128] | Baloyannis SJ , Mavroudis I , Mitilineos D , Baloyannis IS , Costa VG ((2015) ) The hypothalamus in Alzheimer’s disease. Am J Alzheimers Dis Other Demen 30: , 478–487. |
[129] | Piguet O , Petersén Å , Yin Ka Lam B , Gabery S , Murphy K , Hodges JR , Halliday GM ((2011) ) Eating and hypothalamus changes in behavioral-variant frontotemporal dementia. Ann Neurol 69: , 312–319. |
[130] | Ishii M , Iadecola C ((2015) ) Metabolic and non-cognitive manifestations of Alzheimer’s disease: The hypothalamus as both culprit and target of pathology. Cell Metab 22: , 761–776. |
[131] | Jiang Y , Brandt BW , Buijs MJ , Cheng L , Exterkate RAM , Crielaard W , Deng DM ((2021) ) Manipulation of saliva-derived microcosm biofilms to resemble dysbiotic subgingival microbiota. Appl Environ Microbiol 87: , 1–14. |
[132] | Myhre O , Utkilen H , Duale N , Brunborg G , Hofer T ((2013) ) Metal dyshomeostasis and inflammation in Alzheimer’s and Parkinson’s diseases: Possible impact of environmental exposures. Oxid Med Cell Longev 2013: , 726954. |
[133] | Park Y-G , Jeong J-K , Lee J-H , Lee Y-J , Seol J-W , Kim S-J , HUR T-Y , Jung Y-H , Kang S-J , Park S-Y ((2013) ) Lactoferrin protects against prion protein-induced cell death in neuronal cells by preventing mitochondrial dysfunction. Int J Mol Med 31: , 325–330. |
[134] | Guo C , Yang ZH , Zhang S , Chai R , Xue H , Zhang YH , Li JY , Wang ZY ((2017) ) Intranasal lactoferrin enhances α-secretase-dependent amyloid precursor protein processing via the ERK1/2-CREB and HIF-1α pathways in an Alzheimer’s disease mouse model. Neuropsychopharmacology 42: , 2504–2515. |
[135] | Muñoz Y , Carrasco CM , Campos JD , Aguirre P , Núñez MT ((2016) ) Parkinson’s disease: The mitochondria-iron link. Parkinsons Dis 2016: , 7049108. |
[136] | Wang L , Sato H , Zhao S , Tooyama I ((2010) ) Deposition of lactoferrin in fibrillar-type senile plaques in the brains of transgenic mouse models of Alzheimer’s disease. Neurosci Lett 481: , 164–167. |
[137] | Olsen I , Singhrao SK ((2021) ) Low levels of salivary lactoferrin may affect oral dysbiosis and contribute to Alzheimer’s disease: A hypothesis. Med Hypotheses 146: , 110393. |
[138] | Robinwarren J ((1983) ) Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet 321: , 1273–1275. |
[139] | Jarzab M , Posselt G , Meisner-Kober N , Wessler S ((2020) ) Helicobacter pylori-derived outer membrane vesicles (OMVs): Role in bacterial pathogenesis? Microorganisms 8: , 1328. |
[140] | Shimoda A , Ueda K , Nishiumi S , Murata-Kamiya N , Mukai S , Sawada S , Azuma T , Hatakeyama M , Akiyoshi K ((2016) ) Exosomes as nanocarriers for systemic delivery of the Helicobacter pylori virulence factor CagA. Sci Rep 6: , 18346. |
[141] | Kugaji M , Muddapur U , Bhat K , Joshi V , Manubolu M , Pathakoti K , Peram MR , Kumbar V ((2020) ) Variation in the occurrence of fima genotypes of porphyromonas gingivalis in periodontal health and disease. Int J Environ Res Public Health 17: , 1826. |
[142] | Zhang Z , Liu D , Liu S , Zhang S , Pan Y ((2021) ) The role of Porphyromonas gingivalis outer membrane vesicles in periodontal disease and related systemic diseases. Front Cell Infect Microbiol 10: , 585917. |


