
A Novel Approach to the Treatment and Prevention of Alzheimer’s Disease Based on the Pathology and Microbiology
Abstract
Utilizing the pathology and microbiology found in tissue from patients with documented Alzheimer’s disease (AD), the pathogenesis of this fateful disorder has been made clear. Borrelia burgdorferi and Treponema denticola spirochetes enter the brain, mostly via neuronal pathways and the entorhinal circulation. These organisms easily pass through the blood-brain barrier and have an affinity for neural tissue. Once in the brain, the spirochetes make intra- and extracellular biofilms, and it is the biofilms that create the pathology. Specifically, it is the intracellular biofilms that are ultimately responsible for neurofibrillary tangles and dendritic disintegration. The extracellular biofilms are responsible for the inflammation that initially is generated by the first responder, Toll-like receptor 2. The hypothesis that arises from this work is two-pronged: one is related to prevention; the other to treatment. Regarding prevention, it is very likely possible that AD could be prevented by periodic administration of penicillin (PCN), which would kill the spirochetes before they made biofilms; this would prevent the disease and would not allow any of the above deleterious changes generated by the biofilms to occur. As regards treatment, it may be possible to slow or prevent further decline in early AD by administration of PCN together with a biofilm disperser. The disperser would disrupt the biofilm coating and enable the PCN to kill the spirochetes. This protocol could be administered in a trial with the control arm utilizing the current treatment. The progress of the treatment could be evaluated by one of the current blood tests that is semi-quantitative. The specific protocols are listed.
DEFINITIONS
Histologic stains and staining patterns in Alzheimer’s disease
The pathology stains included for study of these postmortem, fixed tissue, Alzheimer’s disease (AD) hippocampal specimens were: hematoxylin and eosin, periodic acid Schiff (PAS), Congo red (CR), spirochete immunostains, and amyloid-β (Aβ) immunostains. This paper utilizes the PAS and CR extensively. The PAS stains polysaccharides that make up the bulk of spirochetal biofilms, and the CR stains amyloid fibrils that make up much of the infrastructure of the biofilms. Microbial biofilms and their staining patterns have been documented in many prior works.
Microbiome versus pathobiome
The microbiome refers to all the organisms found in a location while the pathobiome refers only to the organisms involved in creating or leading to disease. In atopic dermatitis, there are thousands of organisms in the skin microbiome, but the pathobiome consists only of normal flora staphylococci that are capable of making biofilms that occlude the eccrine ducts. In psoriasis, there are hundreds of organisms in the nasopharynx microbiome, but only the streptococci that make biofilms (principally in the tonsils) is in the pathobiome.
Antibiotics
Penicillin (PCN) or semi synthetic penicillins (azithromycin for those who are allergic) were chosen as antibiotics for AD prevention for many reasons. They are bactericidal, achieve an effective minimum inhibitory concentration (MIC) at ordinary doses, and easily cross the blood-brain barrier and neuronal cell membranes. AD has a superb prototype in syphilitic dementia that is responsive to PCN. A short course of PCN would eliminate the spirochetes as it does in latent syphilis; and, as preventive therapy, one would expect minimal resistance to develop. In early AD, the longer course, together with a biofilm disperser, disrupts the horizontal transfer of resistance genes.
Doxycycline and minocycline have been ineffective at clearing other spirochetal infections most likely because they do not achieve an effective MIC at ordinary doses.
Scores of attempts have been made to treat AD, but none has been successful. These attempts have mostly been focused on clearing or decreasing production of Aβ and/or hyperphosphorylated tau protein (p-tau) from those affected because these molecules are abundant in AD brains. One of the reasons offered by various investigators for failure in these efforts has been administration of the potential treatment modalities “too late” in the disease [1]. This seems plausible, but it does not account for the possibility that the selected targets of the disease were not what was actually causing the disease. Recent studies highlight a “pathogen” that leads to both Aβ and p-tau; this pathogen also leads to “inflammation” which has recently been included as a possible target along with Aβ and p-tau [2].
The pathogens that are ultimately responsible for all these injurious phenomena (especially the production of biofilms) are spirochetes; and there are three spirochetes involved in producing dementia. These are Borrelia burgdorferi (BB) [3, 4], Treponema denticola (TD) (spirochete representing many dental treponemata such as T. pectinovorum, T. Maltophilum, T socranskii, and others) [5], and Treponema pallidum (TP) (the spirochete that causes syphilitic dementia, general paresis [GP]) (Fig. 1). This triumvirate makes up the “pathobiome” of AD; much has recently been made of the “microbiome” affecting AD, but it seems that the pathobiome has signal importance [3]. All three of these microbes have been found in brains of patients with dementia: BB by culture, Treponema denticola by PCR, and TP by pathology and experience, not all of which has been upstanding. The fact that microbes likely create AD becomes a “tacit” component of the hypothesis regarding treatment and prevention.
Fig. 1
Brain tissue from GP (left) and AD (right); arrows show helical (coiled) nature of the spirochetes in each specimen. TP immunostain (L) and BB immunostain (R) 100X (from Miklossy et al. [14]).
![Brain tissue from GP (left) and AD (right); arrows show helical (coiled) nature of the spirochetes in each specimen. TP immunostain (L) and BB immunostain (R) 100X (from Miklossy et al. [14]).](https://content.iospress.com:443/media/jad/2021/84-1/jad-84-1-jad210429/jad-84-jad210429-g001.jpg)
These organisms, however, do not create AD in their planktonic state: it is only when they create biofilms that they initiate disease. They do this so slowly that it takes up to two years to create a single biofilm [6]; further, the organisms need to be fully formed and not an “L-form or round-body” to be part of the quorum necessary to create the biofilm [7, 8]. The biofilms that are created are found both intra- and extracellularly, and they have different roles in each location [9] (Fig. 2). Inside the neuron, simultaneously with making biofilm, they create Aβ [6, 10]. By known mechanisms, the intracellular Aβ then interacts with ordinary tau protein, and this interaction leads to p-tau which leads to disintegration of dendrites [11] (Fig. 3). This is likely the single most important occurrence in the pathogenesis of AD and is clearly demonstrated in the figure and the “schematic” (Fig. 4).
Fig. 2
Hippocampal specimen from AD demonstrating extracellular biofilms (senile plaques-black arrow), intracellular biofilms (red arrows), neurofibrillary tangles with biofilm (blue arrow). PAS stain 40X.
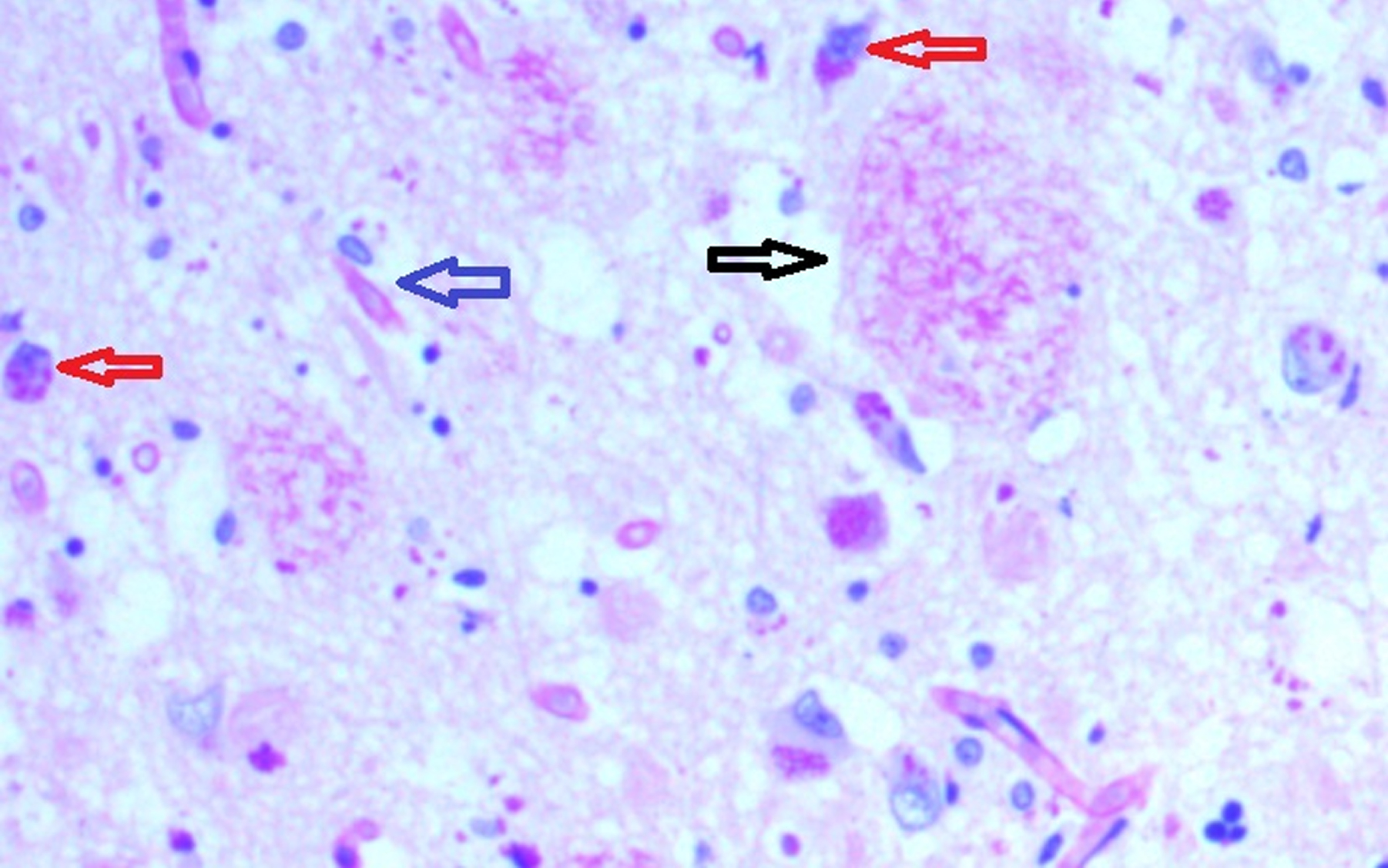
Fig. 3
Neuron (L) containing intracellular biofilm (Congo red positive) with intact dendrites and Neuron (R) showing deformed, blunted dendrites. Arrows show uninvolved dendrites. Congo red 100X.
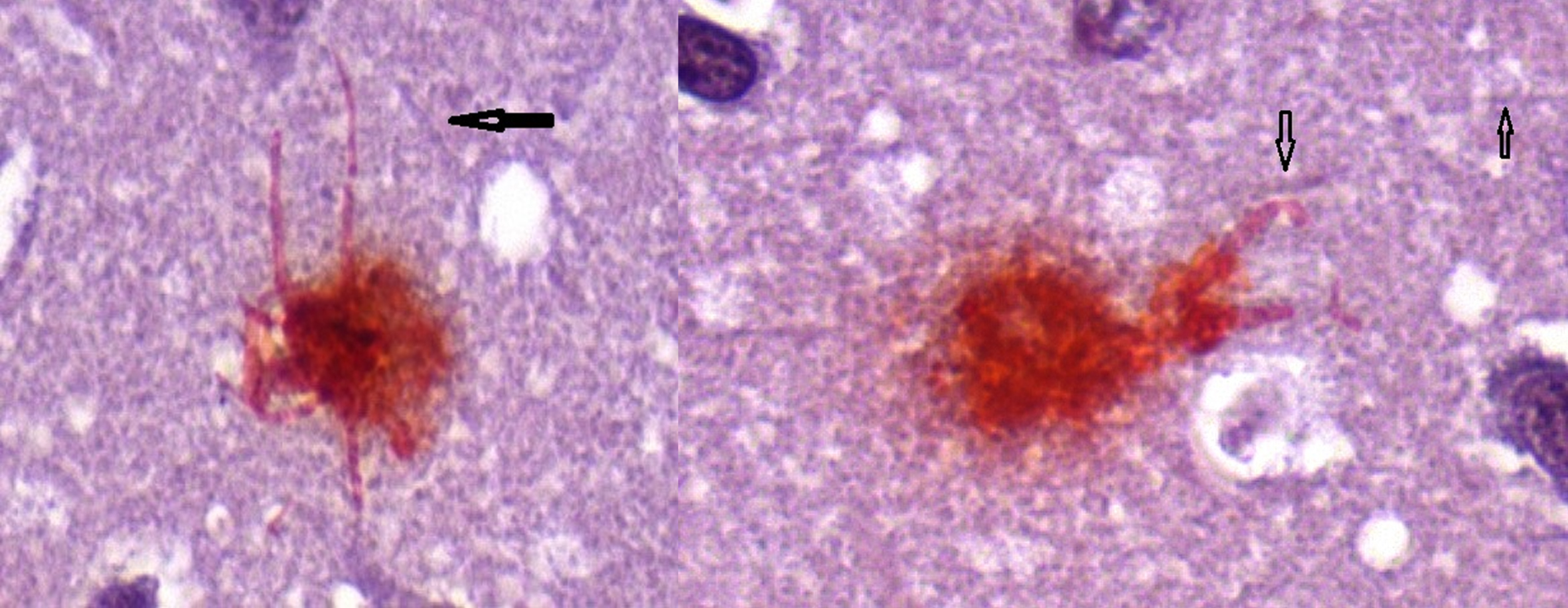
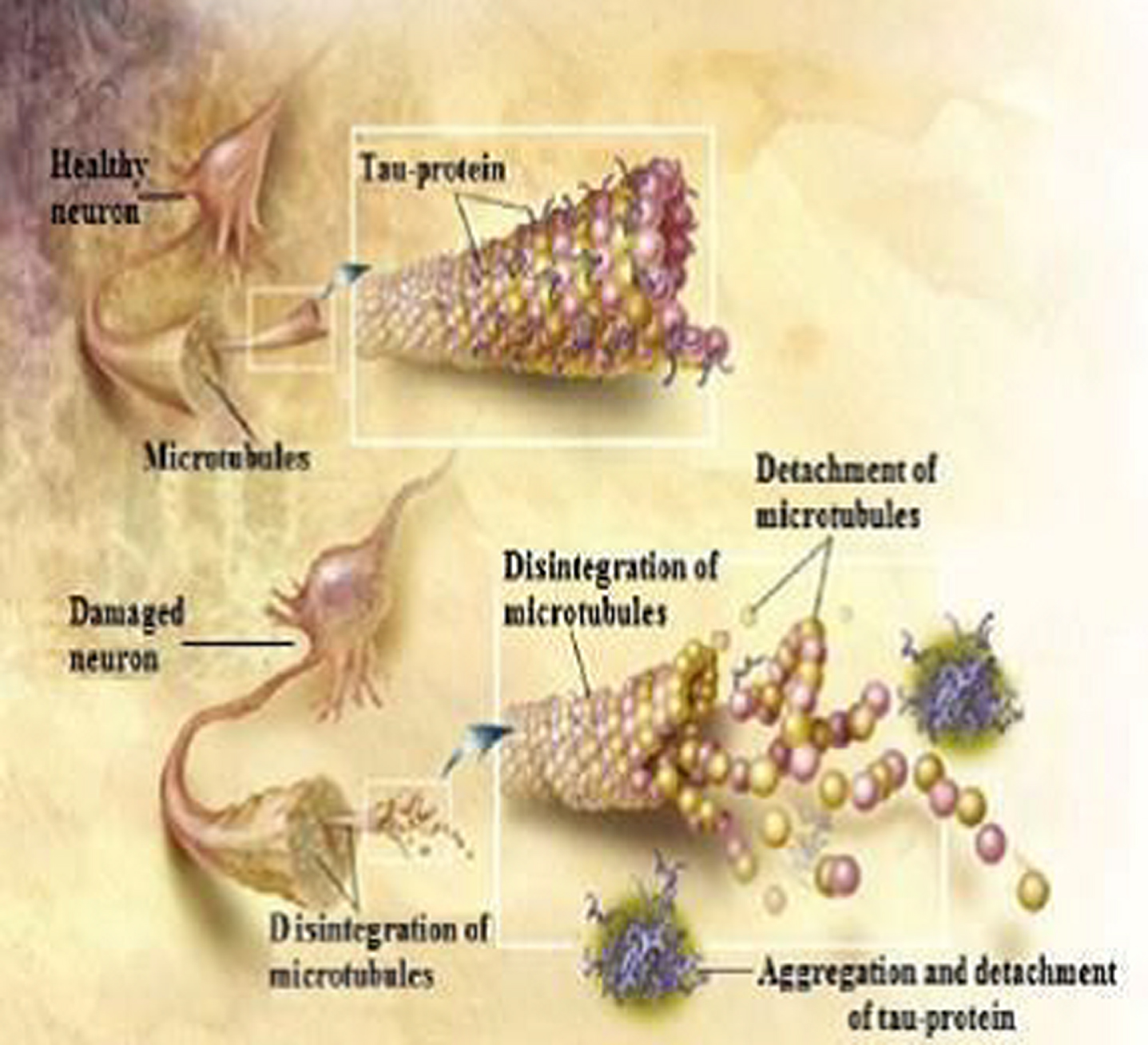
Fig. 4
Tau protein when phosphorylated causes disruption of the neuronal dendrite (from ADEAR/Wikimedia Commons).
The extracellular, senile plaques are made up of spirochetal biofilms coated with Aβ (Fig. 5). The biofilms in these plaques have attachment sites for the innate immune system molecule Toll-like receptor 2 (TLR2), and TLR2, by known pathways, leads to the production of Aβ [6, 12]. Thus, the microbes create the Aβ intracellularly while they are making biofilms, and, extracellularly, the spirochetal biofilms together with the immune system create Aβ. Consequently, the spirochete/biofilm complex creates and results in both the Aβ and p-tau, whose attempted elimination has failed. The protocols for removing Aβ and p-tau are truly “too late”, but they seem misdirected toward the real cause of the disease. Moreover, all the trials with antibiotics were done without the addition of biofilm dispersers, thus rendering them completely ineffective. This includes the recent report on Lewy body dementia where BB were still present after many courses of antibiotics [13].
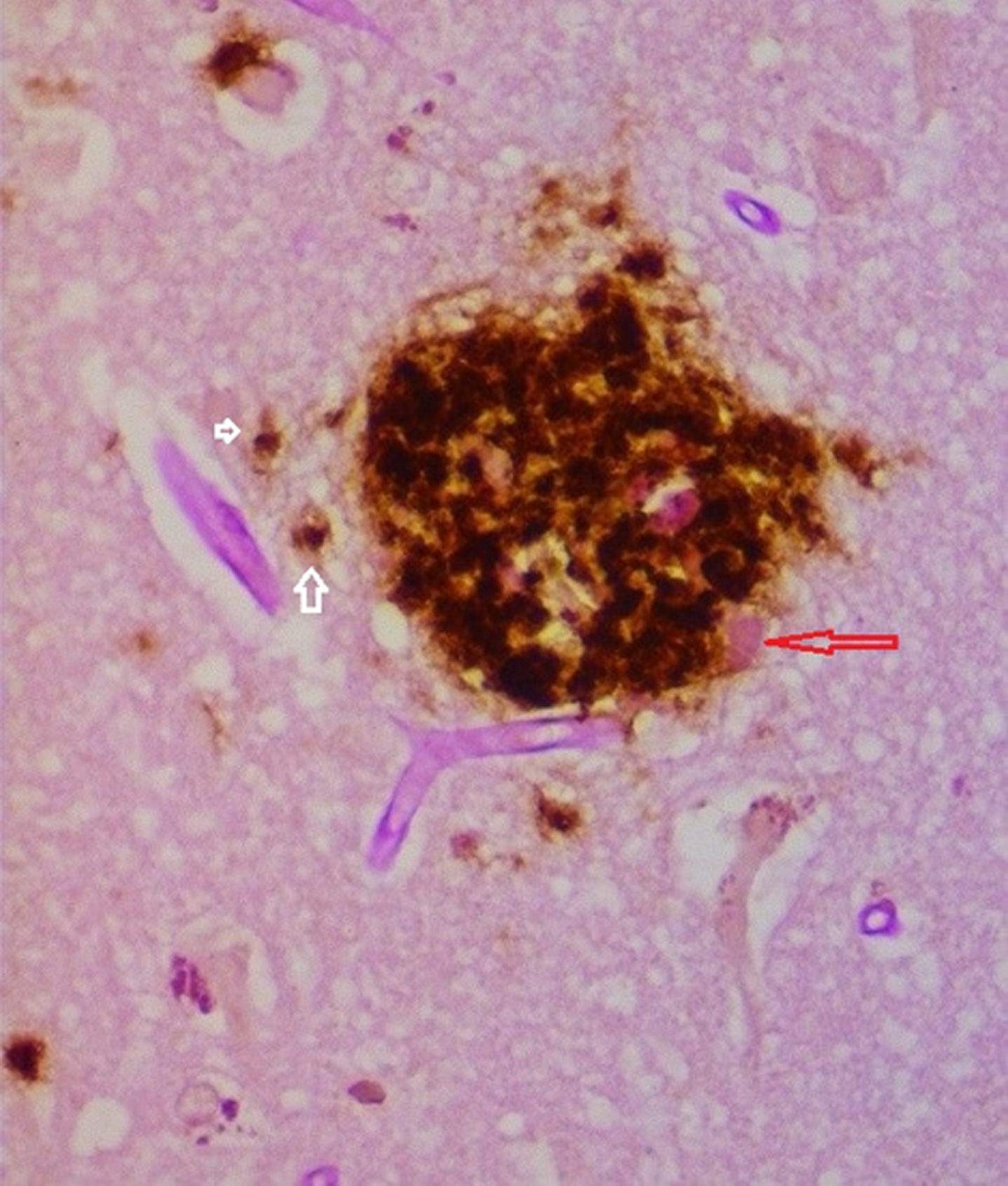
Fig. 5
Senile plaque demonstrating biofilm (pink material red arrow) coated by Aβ. Intracellular Aβ noted in cells to the left of the plaque (white arrows). Combined PAS and Aβ immunostain 40X.
Foremost among the considerations of microbiome versus pathobiome is the pathology, which is helical (Fig. 1) in all cases and not coccoid, rod-like, fungal, or viral. In other words, it is spirochetal. Further, the pathology illuminates the role of biofilms, and, even further, the pathology shows that the tissue findings of GP and AD are similar in all respects [14]. The organisms that have previously been considered in the AD microbiome (herpes simplex (HSV), Chlamydia pneumoniae, Porphyromonas gingivalis (PG), and others) are excluded by the observations that they are not helical or may not form biofilms. Examples would include C hlamydia pneumoniae that is not helical and does not form biofilms, PG that forms biofilms but is not helical, and HSV that is neither helical, nor has it been shown to form biofilms [15–17]. As a gram-negative rod, PG can make biofilms in minutes, not years; this also would deny it a lead role in this slowly developing disease. In the future, HSV may be shown to form biofilms, but these would only be intracellular. Viruses require the genetic material inside cells (which they hi-jack) to form biofilms [18]. All these various organisms may be found in AD brains because the biofilms made by one organism have attachment sites for other organisms and may easily incorporate them into a multiorganism community [19]. Thus, the other organisms, while they are not primary, can, and very likely do, join the community; this finding has recently been demonstrated [20]. The pathway of spirochetes leading to AD is sequentially documented in the Supplementary Material.
Two blood tests have recently shown positivity in AD. The results of the C2N test reveal three groupings: one is true positivity for dementia, another is positive for mild cognitive impairment, and the third is no positivity for AD [21]. The other blood test is a microarray test for AD that is 100%positive in early disease [22]. Trials could be initiated with the C2N test, confirmed by the microarray test with the endpoint of the trial aimed at moving to a lower group. This would be like the situation in syphilis where effective treatment lowers the rapid plasma reagin (RPR) blood test by two dilutions or more. One other test, very recently introduced, utilizes saliva to measure lactoferrin [23]. If the lactoferrin is lower than expected, it is an excellent measure for AD [23, 24]. As with the microarray test, this test would be useful for the confirmation of the disease.
To date, there is no test that shows pre-AD. This group (which would likely include seniors and younger patients with a family history of early onset AD) could be treated like latent syphilis with a course of PCN or azithromycin (for PCN allergic). The reason for treating pre-AD differently from latent syphilis is related to the microbiology: 75%or more of AD is caused by dental spirochetes that cause constant seeding of the brain [5]. This differs from the one time seeding of TP (with syphilis) or BB (from a bite of an infected tick). Because of this, pre-AD would require a course of PCN periodically (perhaps yearly) that would make it similar to a PCN vaccine. In this prevention effort, it does not seem ethically possible to do a trial for pre-AD where the patients in the placebo arm could get AD, similar to Oslo and Tuskegee where there was no treatment arm [25].
Specifically, the treatment of early AD or mild cognitive impairment while utilizing the C2N test as above, would be PCN or azithromycin accompanied by a biofilm disperser for a considerable interval (perhaps 3 months) [26]. This is based on preliminary trials at varying lengths (4, 8, or 12 weeks) where some benefit (clinically) was observed. The shorter intervals were not as successful as the longer.
Many AD patients are already on biofilm dispersers, i.e., donepezil, a chemical piperidine, or citalopram, a chemical furan, are administered for different reasons; nonetheless, each is a biofilm disperser [26, 28]. Each disperser likely works too slowly to recognize a change in the downward clinical course of the disease. Oral and topical biofilm dispersers have been tabulated [26]. If not already taking a disperser, rifampin could be utilized in the treatment arm, as its mechanism for biofilm dispersion is known [29]. Together with Amoxicillin, rifampin has been shown to treat tertiary Lyme arthritis unresponsive to full dose (100 mg bid) doxycycline (which equals 1/2 the MIC for BB) [26]. Minocycline would likely be similar in inefficacy (or lack thereof) to doxycycline [13, 30]. Round-body or granulovacuoles (until they revert to their helical form) should they arise from PCN administration have no observable clinical effect in syphilis or neuroborreliosis (where they have been found) [8]. Also, periodic administration of PCN may kill the organisms when they revert to their helical form. Certain biofilm dispersers work too effectively: haloperidol, a chemical piperidine, is responsible for a 200%increase in death when given to AD patients [31].
PCN may be given as 500 mg of Pen-V-K q 6 h or amoxicillin 500 mg TID for 2 weeks. (This is the equivalent of an IM injection of benzathine PCN.) Azithromycin 500 mg bid for 7 days followed by 500 mg daily would achieve maximal levels (because of the 6-day half-life). If a higher serum level of PCN is required, probenecid may be administered; this doubles the serum concentration because it inhibits the renal excretion of the drug. Benzathine PCN IM, which could be used in place of oral PCN (and reduce any concerns of compliance), is in such short supply, that it is currently recommended for use only in pregnant women with syphilis.
The first prong of the hypothesis that spirochetes lead to AD is likely to be met because of the similarity of the pathology and microbiology in GP and AD. GP becomes a superb prototype for AD, and the treatment of pre-AD should respond similarly to the treatment of latent syphilis with PCN with the eradication of the involved spirochetes. The second prong could be confirmed by a clinical trial as above.
There are more concerns with the preventive arm of the hypothesis because it is not known when to begin the antibiotic, to whom to administer it, or how to judge efficacy. One might consider for inclusion: patients 65 or older with a family history of AD, patients with a stroke, patients with a history of Lyme disease treated with tetracycline or similar, and patients with severe periodontal disease. Patients with secondary or tertiary Lyme disease could consider the mild cognitive impairment treatment protocol. Until a serologic test (like the RPR in latent syphilis) is developed, evidence for efficacy will be elusive. The absence of disease would be the indicator the treatment works. This would be similar to treating and preventing tertiary disease in a patient known to have had primary or secondary syphilis without a known positive serology.
Another concern is the development of resistance, not in the spirochetes (as has been discussed) but in commensal bacterial species because of the administration of PCN. This is mitigated by the short, pulsed course of administration [32, 33]. It is further mitigated by long term administration of PCN for psoriasis with minimal resistance and maximal benefit noted [34]. A further concern is the impact of stressors on biofilms. Increased numbers of extracellular biofilms would lead to increased TLR2 immunologic activity while fewer biofilms would lead to less immunologic stress [6, 12]. Diabetes and nicotine have opposite effects [36]: diabetes, with hyperosmolar serum, causes more biofilms to form; nicotine, a biofilm disperser, eventually causes more biofilms because dispersal is equivalent to the release of “exporter” cells.
Treatment of early AD is perhaps less controversial because the patient population is well-defined and the efficacy of treatment can be measured. Other concerns in both arms of the hypothesis relate to the impact of other organisms in the disease. Does the mixed community create more disease than a biofilm made only by a spirochete? This hypothesis is largely based on the prototype of tertiary syphilis being prevented by a single dose of benzathine PCN given during the latent phase of the disease. As mentioned previously, AD differs from GP because of the constant seeding by oral spirochetes necessitating periodic treatment. The entire hypothesis lends substance to the recent discussion of antimicrobials for AD [36].
ACKNOWLEDGMENTS
Much of this work was presented at “Prevention Alzheimer” conference in Crans Montana, Switzerland in 2018.
The author’s disclosure is available online (https://www.j-alz.com/manuscript-disclosures/21-0429r2).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-210429.
REFERENCES
[1] | Yiannopolou KG , Anastasiou AI , Zachariou V , Pelidou SH ((2019) ) Reasons for failed trials of disease-modifying treatments for Alzheimer disease and their contribution in recent research. Biomedicines 7: , 97. |
[2] | Lee YJ , Han SB , Nam SY , Oh KW , Hong JT ((2010) ) Inflammation and Alzheimer’s disease. Arch Pharm Res 33: , 1539–1556 |
[3] | Miklossy J ((2011) ) Alzheimer’s disease - a neurospirochetosis. Analysis of the evidence following Koch’s and Hill’s criteria. J Neuroinflammation 8: , 90. |
[4] | MacDonald AB ((1986) ) Borrelia in the brains of patients dying with dementia. JAMA 256: , 2195–2196. |
[5] | Riviere GR , Riviere GH , Smith KS ((2002) ) Molecular and immunological evidence of oral treponemes in the human brain and their association with Alzheimer’s disease. Oral Microbiol Immunol 17: , 113–118. |
[6] | Allen HB ((2016) ) Alzheimer’s disease: Assessing the role of spirochetes, biofilms, the immune system, and beta amyloid with regard to potential treatment and prevention. J Alzheimers Dis 53: , 1271–1276. |
[7] | MacDonald AB ((2006) ) Plaques of Alzheimer’s disease originate from cysts of Borrelia burgdorferi, the Lyme disease spirochete. Med Hypotheses 67: , 592–600. |
[8] | Miklossy J , Kasas S , Zurn AD , McCall S , Yu S , McGeer PP ((2008) ) Persisting atypical and cystic forms of Borrelia burgdorferi and local inflammation in Lyme neuroborreliosis. J Neuroinflammation 5: , 40. |
[9] | Allen HB , Allawh R , Touati A , Katsetos C , Joshi SG ((2017) ) Alzheimer’s disease: The novel finding of intracellular biofilms. J Neuroinfect Dis 8: , 247. |
[10] | Miklossy J ((2016) ) Bacterial amyloid and DNA are important constituents of senile plaques: Further evidence of the spirochetal and biofilm nature of senile plaques. J Alzheimers Dis 53: , 1459–1473. |
[11] | Iqbal K , Alonso AC , Chen S , Chohan MO , El-Akkad E , Gong C , Khatoon S , Liu F , Rahman A , Tanimukai H , Grundke-Iqbal I ((2005) ) Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta 1739: , 198–210. |
[12] | Tukel C , Wilson RP , Nishimori M , Pezeshki M , Chromy BA , Baumier AG ((2009) ) Responses to amyloids of microbial and host origin are mediated through toll-like receptor 2. Cell Host Microbe 6: , 45–53. |
[13] | Gadila SKG , Roslklija G , Dwork AJ , Fallon BA , Embers ME ((2021) ) Detecting Borrelia spirochetes: A case study with validation among autopsy specimens. Front Neurol 12: , 628045. |
[14] | Miklossy J ((2018) ) Historic evidence to support a causal relationship between spirochetal infections and Alzheimer’s disease. Front Aging Neurosci 7: , 46. |
[15] | Itzhaki RF , Wozniak MA ((2008) ) Herpes simplex virus type 1 in Alzheimer’s disease: The enemy within. J Alzheimers Dis 13: , 393–405. |
[16] | Balin BJ , Little CS , Hammond CJ , Appelt DM , Whittum-Hudson JA , Gérard HC , Hudson AP ((2008) ) Chlamydophila pneumoniae and the etiology of late-onset Alzheimer’s disease. J Alzheimers Dis 13: , 371–380. |
[17] | Singhrao SK , Harding A , Poole S , Kesavalu L , Crean S ((2015) ) Porphyromonas gingivalis periodontal infection and its putative links with Alzheimer’s disease. Mediators Inflamm 2015: , 137357. |
[18] | Allen HB , Allawh RM , Ballal S ((2017) ) Virally-induced, intracellular biofilms; novel findings in. Clin Microbiol 6: , 302. |
[19] | Stewart PS , Camper AK , Handran SD , Huang C-T , Warnecke M ((1997) ) Spatial distribution and coexistence of Klebsiella pneumoniae and Pseudomonas aeruginosa in biofilms. Microb Ecol 33: , 2–10. |
[20] | Sapi E , Gupta K , Wawrzeniak K , Gaur G , Torres J , Filush K , Melillo A , Zelger B ((2019) ) Borrelia and Chlamydia can form mixed biofilms in infected human skin tissues. Eur J Microbiol Immunol 9: , 46–55. |
[21] | Coghlan A (2017) Blood test detects Alzheimer’s plaques building up in brain. NewScientist.com. https://www.newscientist.com/article/2141198-blood-test-detects-alzheimes-plaques-building-up-in-brain/, Accessed 1/12/2021. |
[22] | De Marshall C , Nagele EP , Sarker A , Goldwaser EL , Kosciuk M , Goldwaser EL , Kosciuk M , Thayasivam U , Han M , Belinka B , Nagele RG. ((2016) ) Detection of Alzheimer’s disease at mild cognitive impairment and disease progression on using autoantibodies as blood-based biomarkers. Alzheimers Dement 3: , 51–62. |
[23] | González-Sánchez M , Bartolome F , Antequera D , González-Sánchez M , Bartolome F , Antequera D , Puertas-Martín V , González P , Gómez-Grande A , Llamas-Velasco S , Herrero-San Martín A , Pérez-Martínez D , Villarejo-Galende A , Atienza M , Palomar-Bonet M , Cantero JL , Perry G , Orive G , Ibañez B , Bueno H , Fuster V , Carro E ((2020) ) Decreased salivary lactoferrin levels are specific to Alzheimer’s disease. E Bio Medicine 57: , 102834. |
[24] | Dashper SG , Pan Y , Veith PD , Toh EC , Liu SW , Cross KJ , Reynolds EC ((2012) ) Lactoferrin inhibits Porphyromonas gingivalis proteinases and has sustained biofilm inhibitory activity. Antimicrob Agents Chemother 56: , 1548–1556. |
[25] | Allen HB , Allawh RM , Ilyas EN , Joshi SG ((2018) ) Bioethical challenges arising from the microbiology and pathology of Alzheimer’s disease. Curr Neurobiol 9: , 26. |
[26] | Allen HB , Hossain C , Abidi N , Larijani M , Joshi SG ((2017) ) Penicillin: The old/new wonder drug. Adv Tech Biol Med 5: , 197. |
[27] | Ghanwate NA ((2012) ) Biofilm eradication studies on uropathogenicusing ciprofloxacin and nitrofurantoin. Int J Pharm 3: , 127–131. |
[28] | Kagan S , Jabbour A , Sionov E , Alquntar AA , Steinberg D , Srebnik M , Nir-Paz R , Weiss A , Polacheck I ((2014) ) Anti-Candida albicans biofilm effect of novel heterocyclic compounds. J Antimicrob Chemother 69: , 416–427. |
[29] | Zheng Z , Stewart PS ((2002) ) Penetration of rifampin through Staphylococcus epidermidis biofilms. Antimicrob Agents Chemother 46: , 900–903. |
[30] | Allen HB , Allawh RM , Gresham K , Donnelly K , Goyal K ((2018) ) Tertiary Lyme disease. Clin Microbiol 7: , 309. |
[31] | Huybrechts KF , Gerhard T , Crystal S , Olfson M , Avorn J , Levin R , Lucas JA , Schneeweiss S ((2012) ) Differential risk of death in older residents in nursing homes prescribed specific antipsychotic drugs: Population based cohort study. BMJ 344: , e977. |
[32] | Raymond B ((2019) ) Five rues for resistance management in the antibiotic apocalypse, a road map for integrated microbial management. Evol Appl 12: , 1079–1091. |
[33] | Baker CM , Ferrari MJ , Shea K ((2018) ) Beyond dose: Pulsed antibiotic treatment schedules can maintain individual benefit while reducing resistance. Sci Rep 8: , 5866. |
[34] | Saxena VN , Dogra J ((2005) ) Long-term use of penicillin for the treatment of chronic plaque psoriasis. Eur J Dermatol 15: , 359–362. |
[35] | Allen HB , Allawh RM , Ilyas EN , Miklossy J and Joshi SG ((2018) ) Alzheimer’s disease: Possible mechanisms for worsening of the disease. J Curr Neurobiol 9: , 59–65. |
[36] | Norins LC ((2021) ) Licensed anti-microbial drugs logical for clinical trials against pathogens currently suspected in Alzheimer’s disease. Antibiotics 10: , 327. |


