
Tau Protein Phosphorylated at Threonine-231 is Expressed Abundantly in the Cerebellum in Prion Encephalopathies
Abstract
Background:
Transmissible spongiform encephalopathies (TSEs) are rare neurodegenerative disorders that affect animals and humans. Bovine spongiform encephalopathy (BSE) in cattle, and Creutzfeld-Jakob Disease (CJD) in humans belong to this group. The causative agent of TSEs is called “prion”, which corresponds to a pathological form (PrPSc) of a normal cellular protein (PrPC) expressed in nerve cells. PrPSc is resistant to degradation and can induce abnormal folding of PrPC, and TSEs are characterized by extensive spongiosis and gliosis and the presence of PrPSc amyloid plaques. CJD presents initially with clinical symptoms similar to Alzheimer’s disease (AD). In AD, tau aggregates and amyloid-β protein plaques are associated with memory loss and cognitive impairment in patients.
Objective:
In this work, we study the role of tau and its relationship with PrPSc plaques in CJD.
Methods:
Multiple immunostainings with specific antibodies were carried out and analyzed by confocal microscopy.
Results:
We found increased expression of the glial fibrillary acidic protein (GFAP) and matrix metalloproteinase (MMP-9), and an exacerbated apoptosis in the granular layer in cases with prion disease. In these cases, tau protein phosphorylated at Thr-231 was overexpressed in the axons and dendrites of Purkinje cells and the extensions of parallel fibers in the cerebellum.
Conclusion:
We conclude that phosphorylation of tau may be a response to a toxic and inflammatory environment generated by the pathological form of prion.
INTRODUCTION
Transmissible spongiform encephalopathies (TSEs) form a group of rare and incurable neurodegenerative diseases. Although TSEs have long incubation episodes, disease progression is inexorable and rapid. The initial clinical manifestations are heterogeneous, with disturbances in memory, gait coordination, personality, and behavior [1–3]. Once the clinical symptoms appear [1], patients die within 6–9 months for sporadic cases, and up to 10 years for genetic cases [4, 5]. The etiologic agent for TSE is termed “prion” (the word being derived from “proteinaceous infectious particle”), according to the nature and ability of an altered protein species (scrapie-associated prion protein, PrPSc) to induce a change in the conformational structure of PrPC (cellular prion protein,) [6, 7]. Physiological roles for PrPC includes copper homeostasis, protection against stress, cell adhesion, and neuronal excitability [6, 8]. PrPSc can cause TSEs and is extremely resistant to thermal decontamination and sterilization methods that are effective against viruses, bacteria, and fungi [9]. Once PrPSc has entered the central nervous system (CNS), progressive neurological signs and degenerative morphological changes (spongiosis, gliosis, and PrPSc-amyloid plaques) appear [1], invariably culminating in rapid death [1, 10]. Human TSEs can be grouped into three classes: I) genetic cases associated with mutations in the gene encoding PrPC, including Familial Creutzfeld-Jakob Disease (fCJD), Gertsmann-Straussler-Scheinker Syndrome (GSS), and Fatal Familial Insomnia (FFI) [5]; II) sporadic cases of CJD (sCJD) and FFI (sFFI) [11]; and III) cases caused by the accidental transmission of a pathogenic prion by surgical procedures such as iatrogenic CJD (iCJD) or “new variant” CJD (nvCJD) caused by oral ingestion of bovine spongiform encephalopathy (BSE) [4]. Approximately 350 new CJD cases (1 per million people) are reported annually in the United States, comprising 1% iatrogenic, 14% genetic, and 85% sporadic forms [12]. Although cerebellar dysfunction is the prominent symptom of TSEs [1, 11], clinical signs indicate that the brainstem, middle brain, hypothalamus, hippocampus, and cerebral cortex may also be affected [2, 3, 5, 11]. Pre-mortem diagnosis consists of encephalograms (EEG), magnetic resonance imaging (MRI), and positron emission tomography (PET) [5]. However, a definitive diagnosis is only confirmed through immunohistochemical evidence of PrPSc lesions [1, 5].
In contrast, neurodegenerative disorders termed “tauopathies” are a group of sporadic or familial neurodegenerative diseases characterized by the accumulation of fibrillar and hyperphosphorylated tau in neurons and glial cells [13]. Extracellular plaques composed of an amyloid-β (Aβ) core surrounded by dystrophic neurites [14] are observed in AD, the most common tauopathy in which pathological tau protein and Aβ plaques are associated with cognitive dysfunction [15].
Differential diagnosis between the early stages of prion disease and AD is not well established as very similar symptoms appear in both conditions, with the primary difference being that dementia occurs rapidly in prion disease [16, 17]. In this study, we analyzed the expression of PrPSc and tau in the cerebellar tissue of prion disease, AD, and healthy patients. In prion disease, PrPSc deposits aggregate and accumulate as prion plaques. Astrogliosis favors an aggravated brain inflammation and neuronal death of the cerebellar granular layer (GL). The presence of hyperphosphorylated tau has been reported in cases of human TSEs [16–20]. Here, we observed the presence of tau phosphorylated at Thr-231 in the axons and dendrites of Purkinje cells and in parallel fibres of the surviving granular cells. This suggests that phosphorylation may be increased by the exacerbated inflammatory activity caused by astrogliosis in prion diseases.
MATERIALS AND METHODS
Brain tissue
This study examined the brain tissue of seven patients diagnosed with prion disease (Table 1). All cerebellar sections included representative cell areas of the molecular layer (ML), Purkinje and granular cells, and white matter (Wm). The tissues were obtained from the National Dementia BioBank, FESC, UNAM, according to the institutional guidelines of bioethics. The cerebellum of patients with prion disease, AD, and those without neurodegenerative disease was maintained in a 10% formalin solution. The fragments were submerged in 10% sucrose in PBS for 24 h and subsequently cut into 50-μm thick coronal slices on a sliding microphone (Leica, Heidelberg. Germany).
Table 1
Characterisation of cases
| Sex | Age at death (y) | Presenting symptoms | Disease duration (mo) | Diagnosis |
| Male | 57 | Mental disorders and rapid progression of gait disorders | 15 | sCJD |
| Female | 65 | Progressive ataxia for gait | 8 | sCJD |
| Male | 76 | Dementia syndrome of rapid evolution with gait apraxia | 15 | sCJD |
| Female | 33 | Alterations in gait and progressive dementia | 10 | sCJD |
| Female | 55 | Throbbing headache followed by episodes of sudden forgetfulness | 26 | fCJD |
| Female | 43 | Rapidly evolving memory loss | 12 | fCJD |
| Female | 47 | Sensory neuropathy and cerebellar syndrome | 72 | GSS |
| Male | 47 | Dementia | severe | AD |
| Female | 80 | Dementia | severe | AD |
| Male | 80 | Dementia | severe | AD |
| Male | 47 | Pneumonia | CONTROL | |
| Male | 79 | Pneumonia | CONTROL | |
| Female | 57 | Pneumonia | CONTROL |
sCJD, sporadic Creutzfeld-Jakob disease; fCJD, familial Creutzfeld-Jakob disease; GSS, Gertsmann-Straussler-Scheinker syndrome; AD, Alzheimer’s disease; CONTROL, brains without neurodegenerative disease.
Immunohistochemistry
Free-floating tissue sections were pretreated with H2O2 (0.3% in PBS), to block endogenous peroxidase activity, for 10 min. Non-specific antibody binding sites were blocked with an IgG free albumin solution (0.2% in PBS). Tissues were incubated with the primary antibody cocktail (Table 2) diluted in 0.2% Triton X-100 in PBS (Sigma-Aldrich Co.) overnight at 4°C. Immunoreactivity was visualized using horseradish peroxidase (HRP) and 3-3-diaminobenzidine (DAB, Sigma-Aldrich Co.) as chromogen. Sections were counterstained with hematoxylin.
Table 2
List of primary antibodies used for immunohistochemistry and immunofluorescence studies
| Antibody | Protein / Epitope | Source |
| 3F4 | PrPC and PrPSc: aa109–112 | BioLegend |
| pTau231 | Tau: Phospho-Thr231 | Invitrogen |
| pTau396 | Tau: Phospho-Ser396 | Invitrogen |
| pTau416 | Tau: Phospho-Ser416 | Abcam |
| CP13 | Tau: Phospho-Ser202 | [21] |
| TG3 | Tau: Regional conformational change with phosphorylation at Thr231 and Ser235 | [22] |
| PHF-1 | Tau: Phospho–396/404 | Invitrogen |
| Tau-C3 | Tau: Truncation at Asp421 | [23] |
| 423 | Tau: Truncation at Glu391 | [24] |
| NF-L | Neurofilament - light | Abcam |
| GFAP | Reactive astroglia (glial fibrillary acidic protein) | GeneTex |
| MMP-9 | Active matrix metalloprotease 9 | Invitrogen |
| ssDNA F7-26 | Fragmented DNA | Chemicon |
Immunofluorescence
Antigenic epitopes were retrieved using a citrate buffer (0.1 M citric acid, 0.1 M sodium citrate, pH 6.0) at 100°C for 30 min. Lipofuscin autofluorescence was removed with Sudan B Black (SBB) solution (SPI® Supplies, West Chester, PA) (0.1% SBB in 70% ethanol) for 10 min. Non-specific sites were blocked with IgG-free albumin solution (0.2% Sigma-Aldrich Co.) in PBS at room temperature for 20 min. Tissues were then incubated with the primary antibody cocktail (Table 2) diluted in 0.2% Triton X-100 (Sigma-Aldrich Co.) in PBS overnight at 4°C. Primary antibodies were revealed with the appropriate secondary antibody coupled to different fluorochromes: Dylight®-488 (Vector Laboratories, CA, USA) and rhodamine isothiocyanate-TRITC (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA). Sections were counterstained with TO-PRO®-3 (Thermo Fisher Scientific) and mounted on slides with anti-photobleaching medium Vectashield (Vector Laboratories).
Confocal microscopy
Double and triple-immunolabeled sections were examined using a confocal laser scanning microscope (TCS-SP8, Leica, Heidelberg, Germany) using the 20x, 40x, and 100x lenses. Ten to fifteen consecutive individual sections were sequentially scanned at 0.8–1.0μm intervals for two or three channels throughout the z-axis of the sample. The collected images were projected and analyzed onto the two-dimensional plane using a pseudocolor display of green (FITC, excitation spectrum: 488 nm), red (TRITC, excitation spectrum: 540 nm), and blue (TO-PRO®-3, spectrum excitation: 650 nm).
RESULTS
Presence of prion plaques in human cerebellum
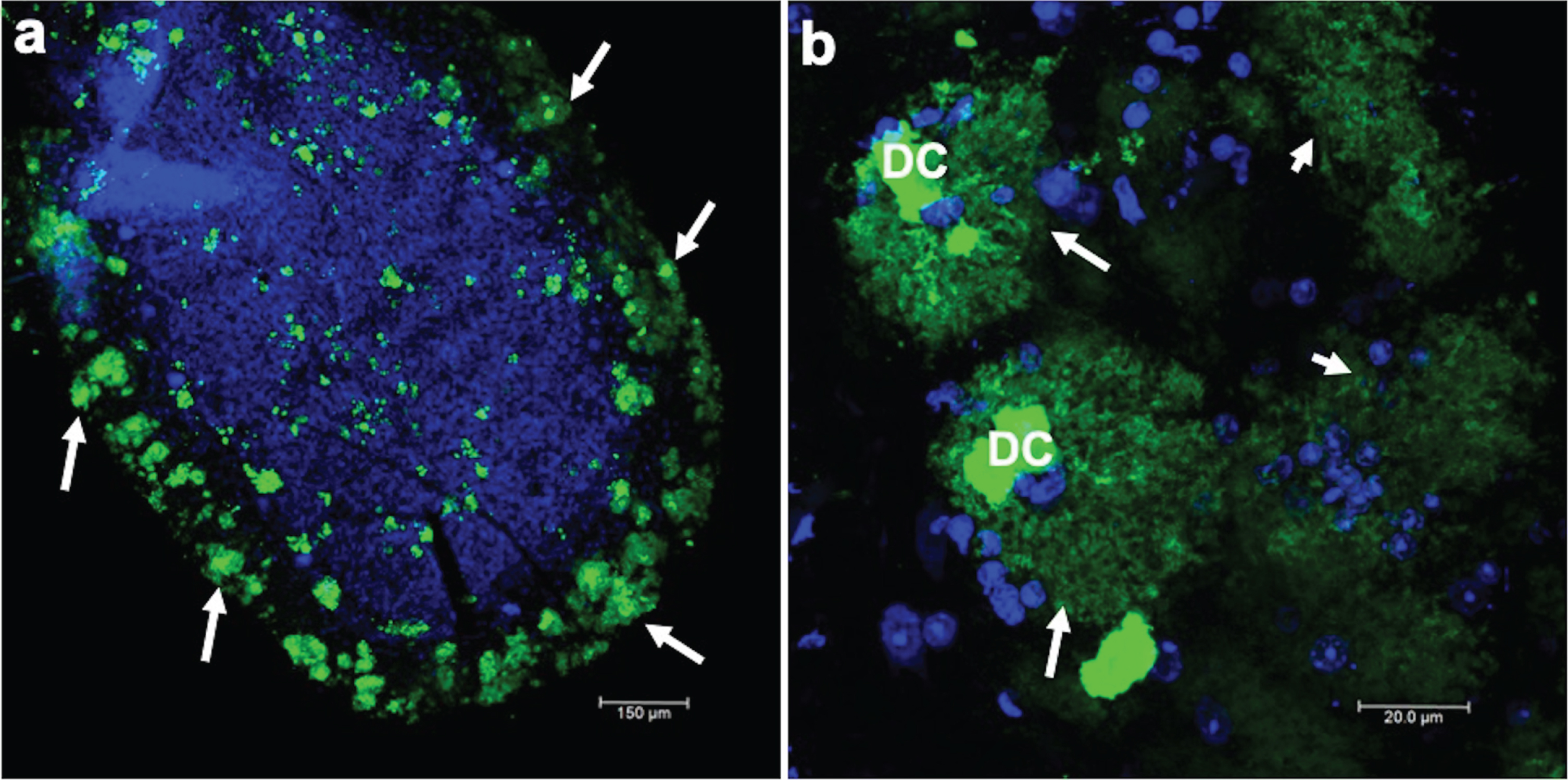
Prion encephalopathies are fatal progressive neurodegenerative diseases. These diseases affect both animals and humans. In humans, only 10% of cases are of genetic etiology; the rest are of either sporadic or infectious origin. The clinical diagnosis of these diseases is based on accelerated and short-term cognitive impairment. Histopathologically, the nervous tissue has cavities and prion amyloidogenic plaques throughout the brain parenchyma. Plaques are revealed using specific markers, such as the anti-prion 3F4 antibody. Thus, a confirmatory diagnosis was performed by staining against this prion protein in cerebellar slices. There were a large number of 3F4-immunoreactive plaques in the cerebellum (Fig. 1a) for all prion disease cases analyzed. In Fig. 1a, a cerebellar folia with many prion plaques (green channel, arrows), counterstained with TO-PRO(R)-3 to show nuclei, is shown at low magnification. Prion plaques were characterized by a dense core (DC), highly immunoreactive to the 3F4 antibody, surrounded by a fuzzy crown (Fig. 1b, arrows). The DC was made up of several small spheres joined together. Diffuse plaques were also present (Fig. 1b, short arrows).
Fig. 1
Immunofluorescent staining of the prion protein. a) Low magnification where a folia of the cerebellum is observed, in which there is strong 3F4 immunoreactivity. The nuclei are stained blue with TO-PRO(R)-3. b) Amplification of classic prion plaques that are characterized by a dense core (DC) and, in the periphery, diffuse staining in the shape of a crown (arrows) is observed, whereas in other structures (short arrows) a dense core is not observed.
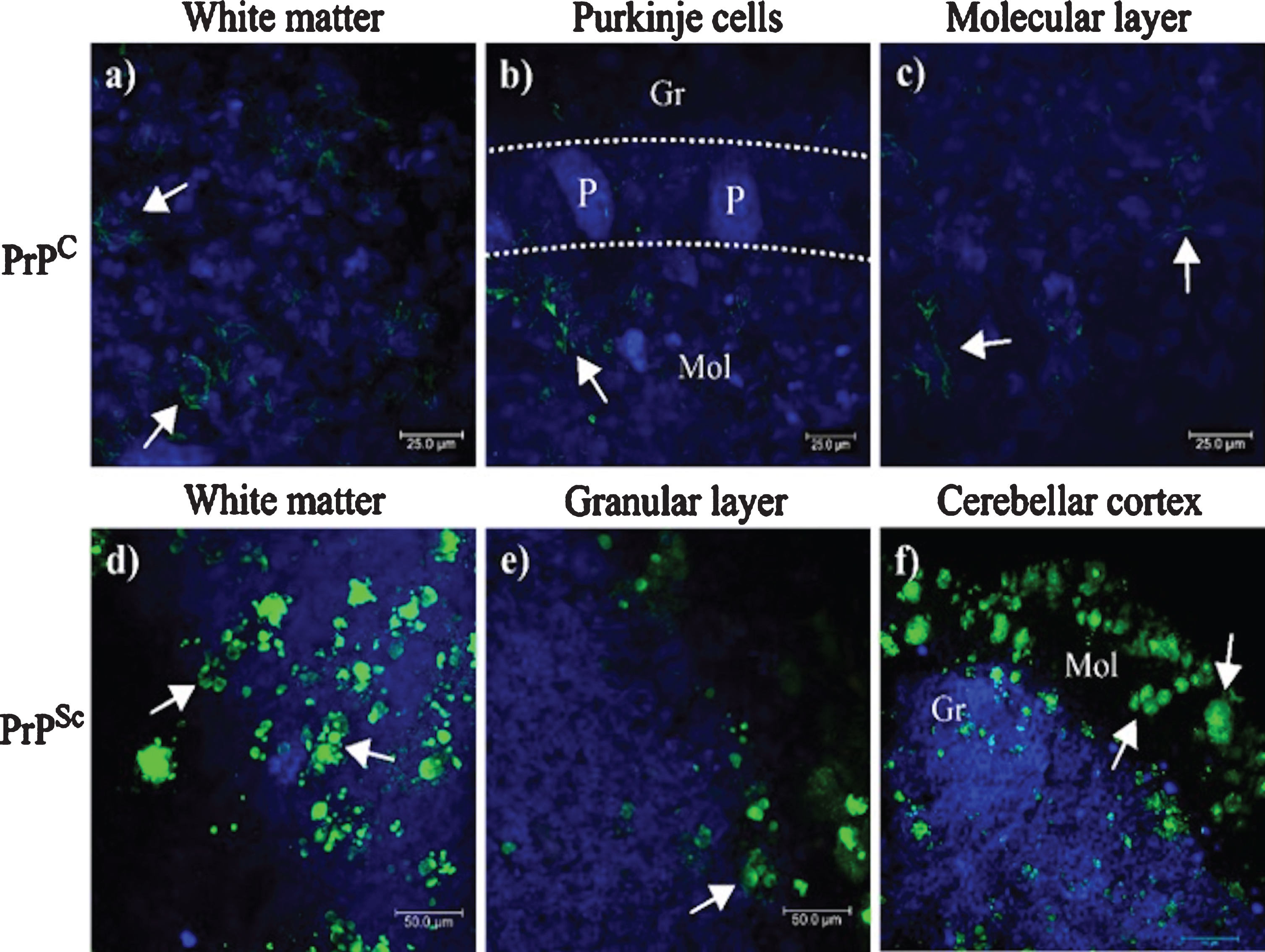
PrPC distribution
In the cerebellum, PrPC expression pattern in the Wm (Fig. 2a, arrows) and in the ML (Fig. 1c, arrows) suggest that PrPSc aggregates, expressed probably in the fibrous astrocytes of the Wm and protoplasmic of the neuropil of the GL and ML, originated from PrPC. 3F4 immunoreactivity was located in the cell cytoplasm in both GL and ML (Fig. 2b, c, arrows). No prion plaques were observed in the Wm (Fig. 2a). The structures observed corresponded to small lump-like cell aggregates and residual cell granules of approximately 5–10μm in length (Fig. 2d, e), suggesting that they may be the minimal nuclei that eventually form the prion plaques in the ML (Fig. 2f).
Fig. 2
Location of PrPC and PrPSc in the cerebellum. 3F4 reactivity shows the cytoplasmic localization of PrPC in Wm (a), in GL (b), and ML (c). Cells with PrPC cytoplasmic expression appear to correspond in shape, size, and location to astrocytes. PrPSc aggregates increase in size and density of Wm (d), GL (e), and ML (f). GL, Granular layer; ML, Molecular layer; Wm, white matter; P, Purkinje cells. Images obtained using a Leica SP8 confocal microscope.
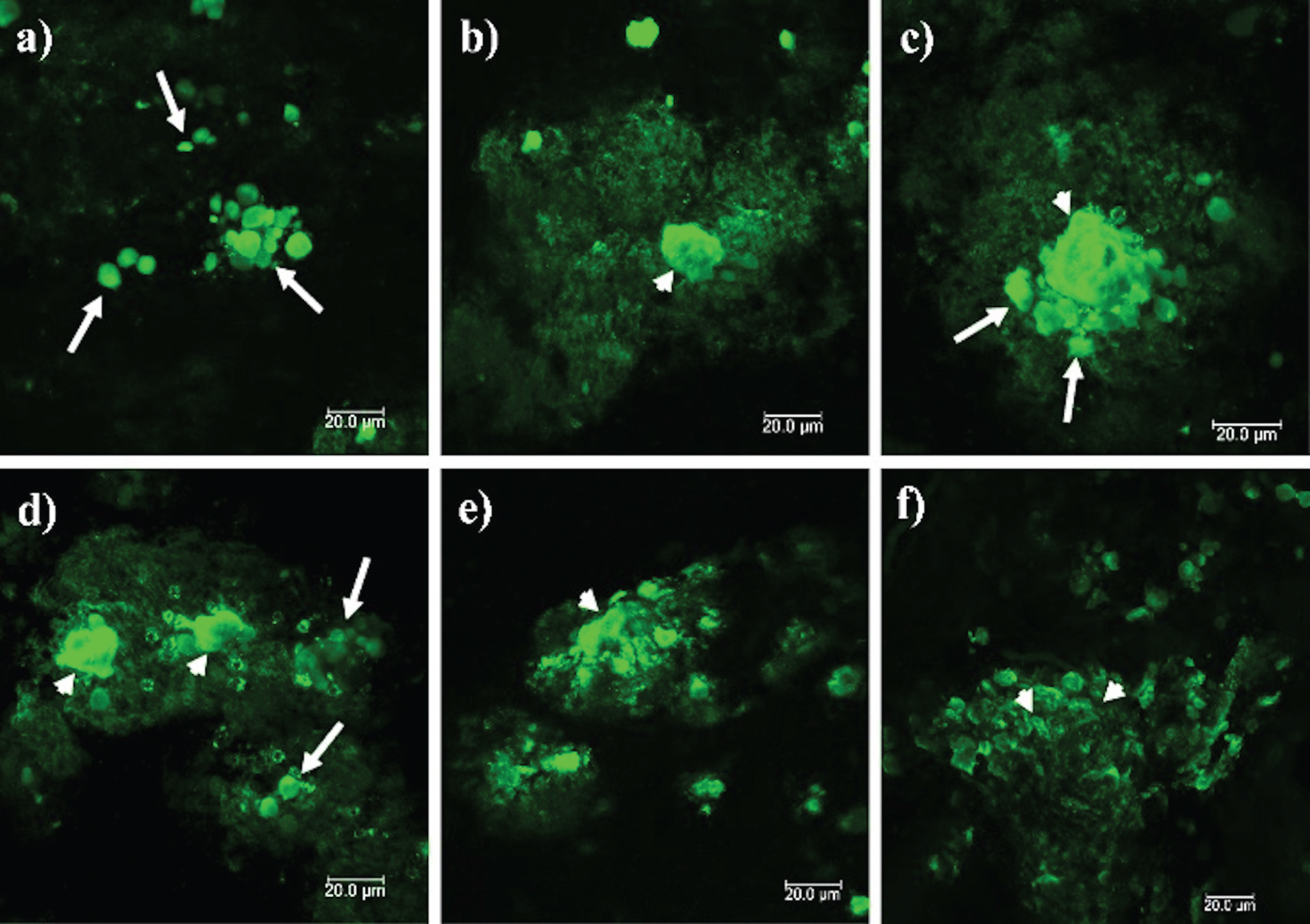
Aggregation of PrPSc in the cerebellum
The pattern of PrPSc aggregates observed in Fig. 2(d-f) was observed in all of the prion disease cases. Figure 3 corresponds to the stages of prion particle aggregation, suggesting that prion plaque formation begins with aggregation of cellular components. These structures are packed in a membrane (Fig. 3a). Diffuse staining around the plaques suggests that free PrPSc induces the replication and conversion of PrPC into PrPSc (Fig. 3b). The accumulation of packed cellular structures gives rise to dense aggregates (Fig. 3c, d), which increase in size until generating the insoluble plaques observed in the ML of the cerebellum (Fig. 3e, f).
Fig. 3
PrPSc deposits in the different cerebellar layers. Immunofluorescence was performed with the anti-PrP 3F4 monoclonal antibody. Small arrows show prominent PrPSc plaques and long arrows show cell aggregates. a) Aggregates of 5–10μm in diameter are observed in the Wm. b) PrPSc aggregates become denser and seem to attract others towards the GL (c). d, e) Dense aggregates interact with others, giving rise to prion plaques with a size larger than 50μm (f). GL: granular layer. ML, molecular layer; Wm, white matter. Images obtained using a Leica SP8 confocal microscope.
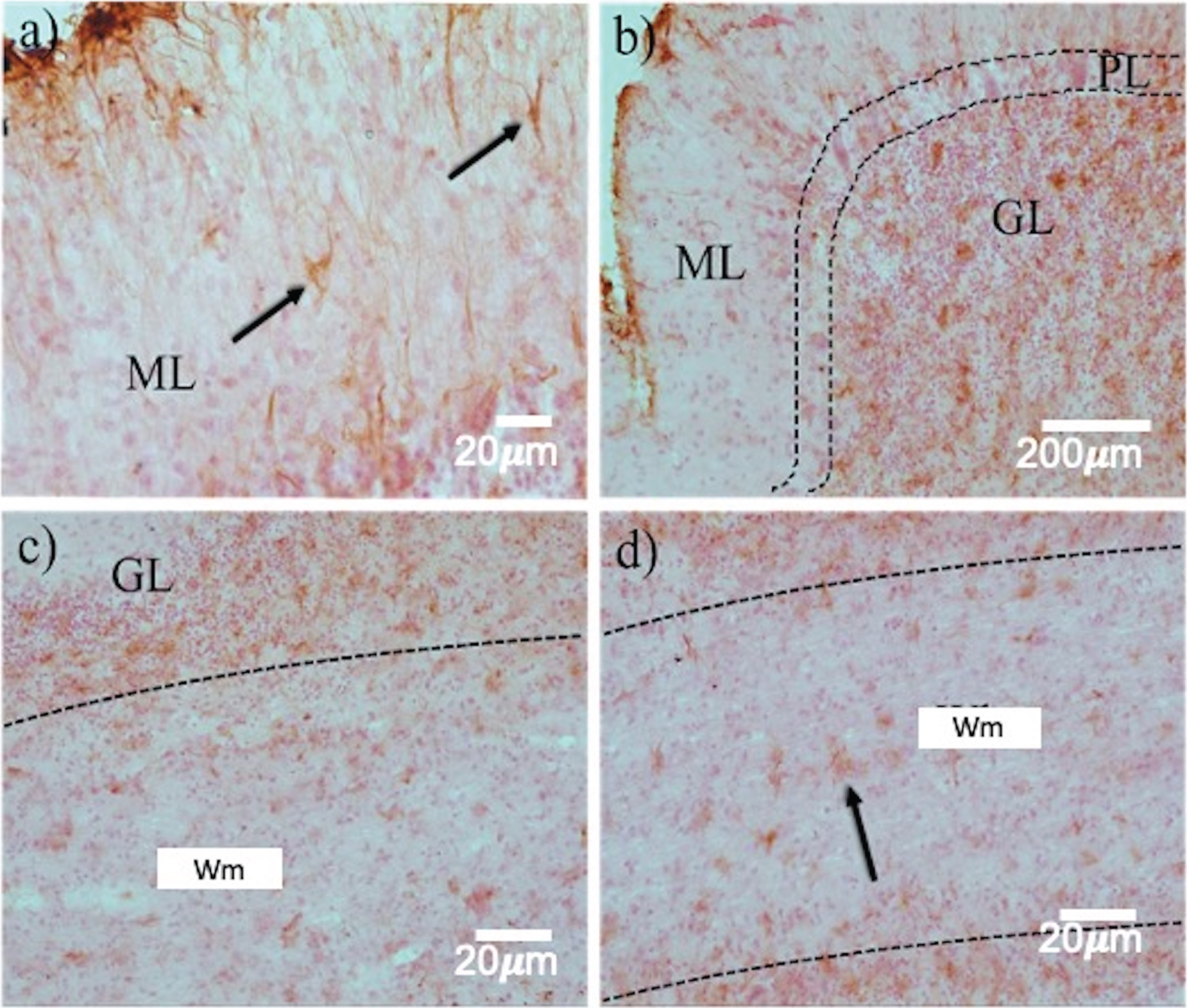
Mechanism of cell death in prion infection
The expression of reactive astrocytes, matrix metalloproteinase-9 (MMP-9), and apoptotic bodies were analyzed to determine the type of neuronal death and those cells affected in prion disease. Reactive astrocytes were distinguished in the ML (Fig. 4a, arrows), GL (Fig. 4b, c), and Wm (Fig. 4d, arrows). Although astrogliosis was evident in all layers of the cerebellum, GFAP staining was most robust in GL. This implied damage, cellular hypertrophy, and destruction of nearby neurons in this region (Fig. 4b, c, arrows).
Fig. 4
Astrogliosis in the layers of the cerebellum in prion disease. GFAP immunohistochemistry revealed a highly variable expression pattern in the different cell layers. Between ML a) and GL (b, c), a dramatic increase in reactive astrocytes (arrows) was observed. d) In the Wm, astrocytosis is kept in a smaller quantity. ML, Molecular layer; GL, Granular layer; Wm, white matter. White arrows point out Purkinje neurons.
In contrast, it has been reported that MMP-9 is highly expressed in the brains of AD patients, associated with a neuroprotective role in response to neuronal death caused by Aβ plaques [25, 26]. The ML (outermost layer) contains the parallel fibers that run through the cortical layer of the cerebellum, the dendritic projections of the Purkinje cells, interneurons (superficial stellate cells and deep basket cells) that establish GABAergic synapses on Purkinje neurons, and neuroglia. The GL is composed of granular cells, Golgi type II cells, cerebellar glomeruli, and protoplasmic astrocytes. In ML, MMP-9 expression was located in structures that morphologically correspond to glial structures (Fig. 5a). MMP-9 was not evident in the Purkinje cell layer (Fig. 5b). MMP-9 immunoreactivity was more marked in GL than in ML (Fig. 5b, c). Finally, abundant MMP-9 was also observed in the Wm, suggesting that astrocytes are expressing this protein in response to PrPSc aggregation, similar to what occurs with plaques (Fig. 5d).
Fig. 5
MMP-9 expression in the different layers of the cerebellum. From the outermost layer to the Wm, in a) abundant MMP-9 expression (arrows) is observed in the ML, decreasing the expression towards the Purkinje cell layer in b), in c) MMP-9 increases considerably in the GL. In d) MMP-9 is observed in the Wm. The dotted lines show the point of intersection between one neural layer and another. P, Purkinje cells; ML, Molecular layer; GL, Granular layer; Wm, White matter. Images obtained using a Leica SP8 confocal microscope.
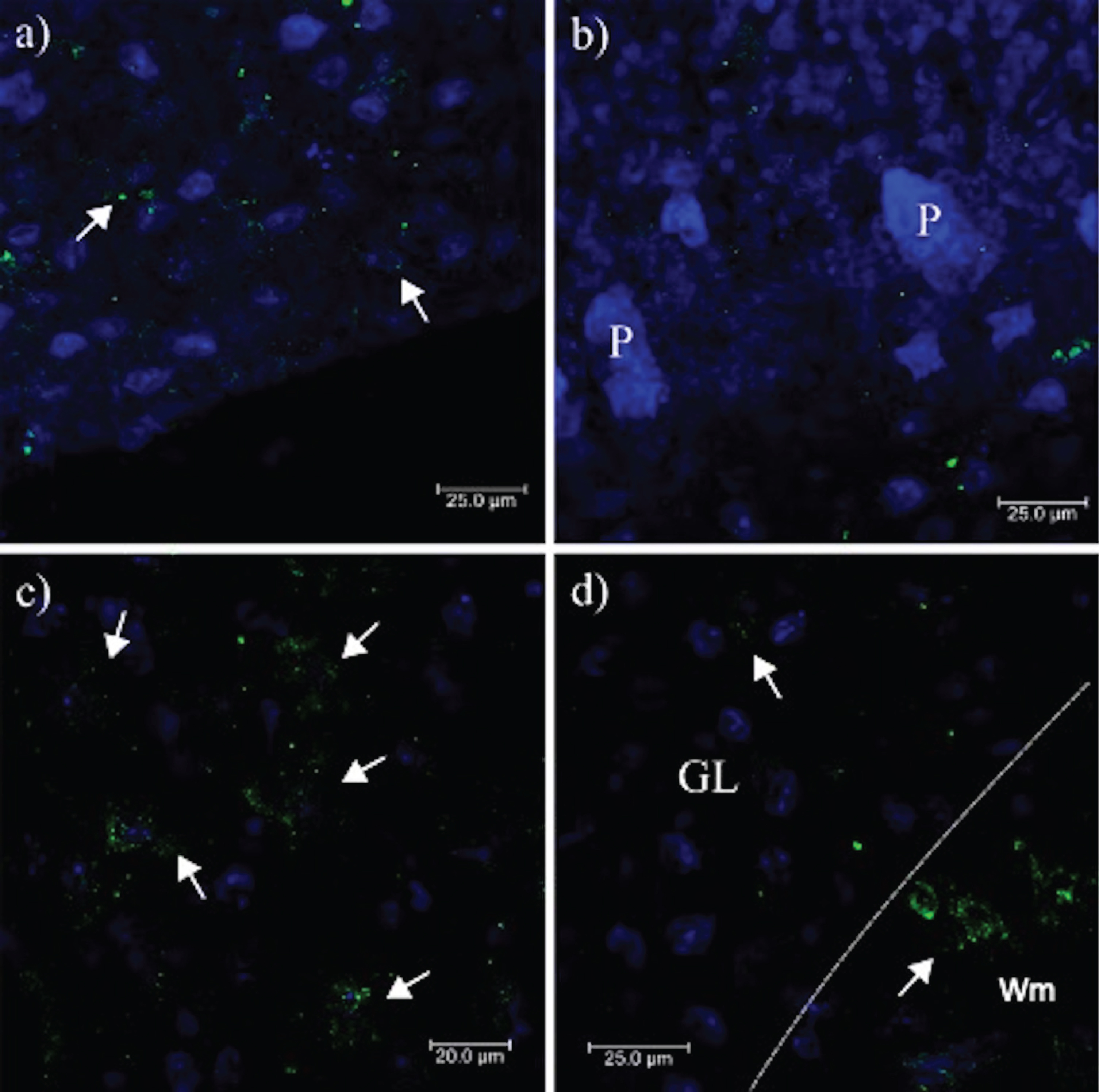
To demonstrate the phenomenon of apoptosis in the cerebellum, the ssDNA antibody F7–26 was used. Apoptotic cells were distributed in the cerebellar cortex, mainly in the GL (Fig. 6a, b), with much less reactivity towards the ML (Fig. 6a, b). The Wm is the region with intrinsic fibers that connect the cerebellar hemispheres; the afferents of the spinal cord and the cerebral cortex and the efferents consisting mainly of the axons of Purkinje cells). In Wm, apoptotic bodies were not observed (Fig. 6c). These structures were associated with an increased GFAP-positive astrocytic activity (Fig. 4), indicating that the type of cells that die in prion infection was granular (Fig. 6d). Purkinje cells and other cells located in the ML were not affected. The distribution of small PrPSc-aggregates (Fig. 2d, e, f, and Fig. 3) and the expression of MMP-9 (Fig. 5) in the Wm and the neuropil of the cerebellar cortex suggest that the conversion of the PrPC in PrPSc, and the loss of protective function of astrocytes in the recapture of neurotransmitters, generate a toxic environment that induces the granular cell death. Cell death triggers the expression of tau phosphorylated at Thr-231, avoiding the collapse of the neuronal circuit of the afferent fibers (corresponding to the mossy fibers (coming from the nuclei of the bridge and the spinal cord), climbing (somatosensory cortex and the lower olive), serotonergic (Rafe nuclei) and noradrenergic (locus coeruleus) systems), axons of the Purkinje cells and the deep nuclei projections. The collapse of this cytoarchitecture could be due to the cytotoxic environment produced more by oxidative glutamate toxicity than by PrPSc plaques.
Fig. 6
Staining of apoptotic cells in the cerebellum. a, b) Marked immunostaining of cell apoptosis (green) in the cerebellar folia. c) There is no reactivity in the Wm. d) The nuclei of the intact granular cells (blue) and the damaged ones (green) in the GL are shown at 100X magnification. P, Purkinje cells; ML, Molecular layer; GL, Granular layer; Wm, White matter. Images obtained using a Leica SP8 confocal microscope.
Tau phosphorylation in prion disease
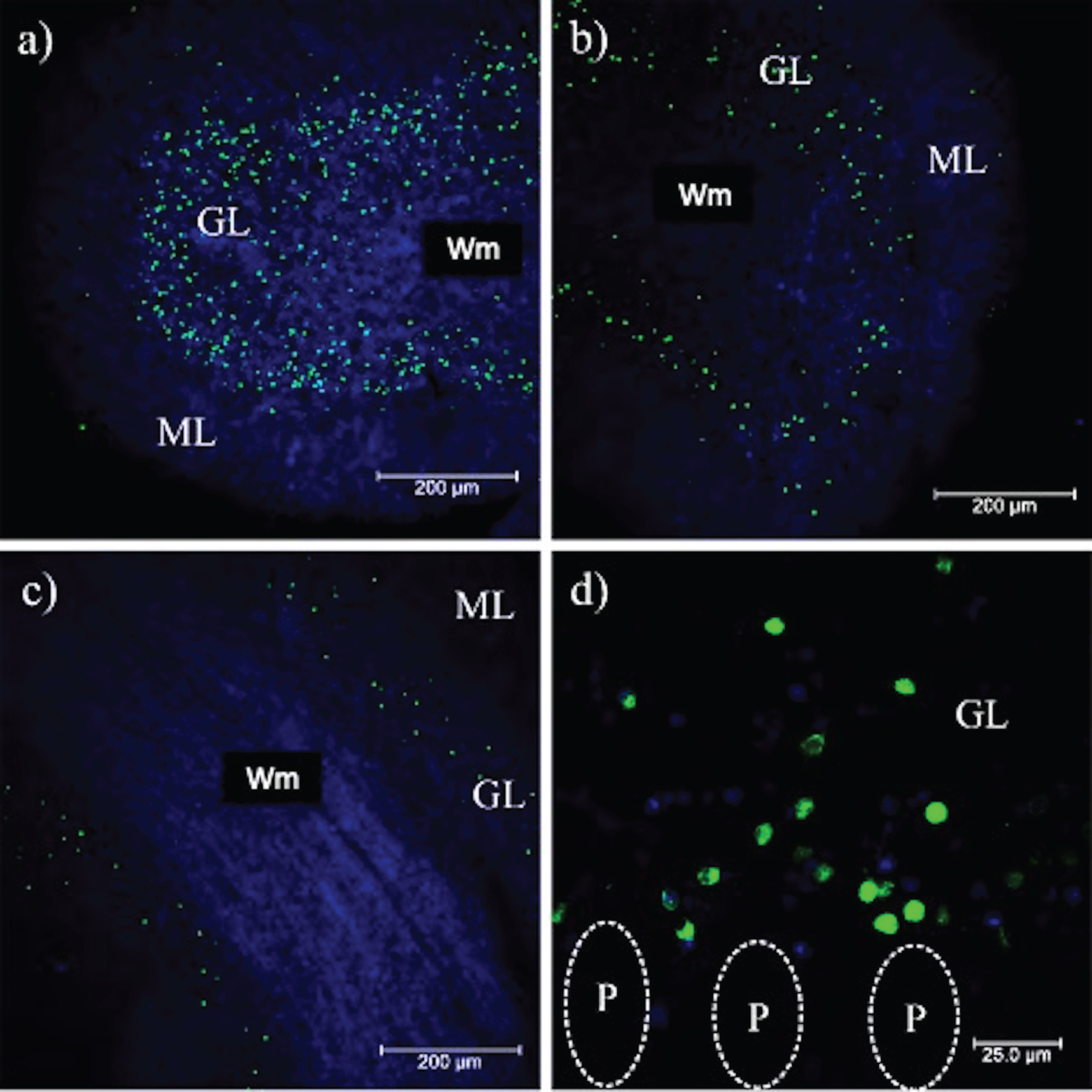
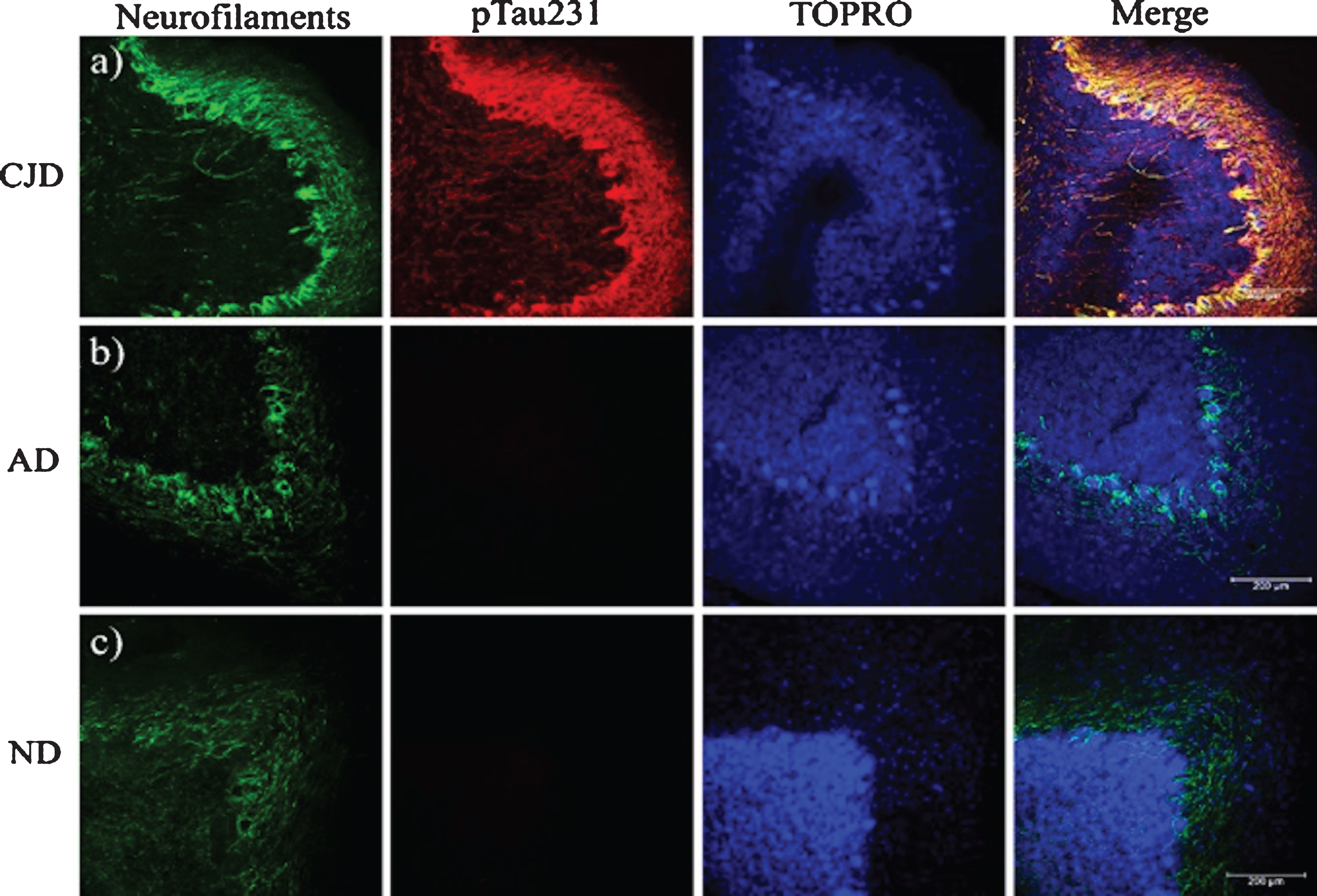
PrPSc plaques were abundant in the ML (Figs. 1 and 2). 3F4-reactive plaques ap-peared to have a uniform distribution both on the outermost (Fig. 7a) and innermost foliae, closest to the deep nuclei (Fig. 7b). The size and number of PrPSc plaques were also significant, demonstrating that PrPC could interact with PrPSc in the ML. Cell integrity could be noted in the parallel fibers (Fig. 7c) with the pTau231 antibody. Axonal torpedoes, formed in response to cerebellar lesions, increased in number according to the state of atrophy of the cerebellum [27]. There was no immunoreactivity with the pTau396 antibody, which reacts with tau phosphorylated at Ser396 (Fig. 7d). The other tau antibodies used in this study against tau post-translational changes (pTau416, CP13, TG3, PHF1, TauC3, 423) did not show any immunoreactivity in the cerebellum (data not shown). The absence of colocalization between the PrP plaques and pTau231 indicated that there was no direct interaction between these structures (Fig. 7a). Axons and dendrites of the Purkinje cells were intact, thus the neurotoxic response produced by the prion plaques near the parallel fibers (which regulate the cells in the basket, the stellate cells, and the Purkinje cells) did not interrupt their excitatory activity. Plaques increased tau phosphorylation at Thr-231. Phosphorylation at this site may act as a neuroprotective signal in response to the neurotoxic environment (produced by prion plaques, located in the ML, converting PrPC to PrPSc (Figs. 2 and 3), and the collapse of neuronal cytoarchitecture due to the granular cell death (Fig. 7a). To identify a possible neuroprotective role for tau, we analyzed different cases according to disease duration with the levels of expression of pTau231. Tau expression was similar for eCJD, fCJD, and GSS patients (Fig.7a, c). In cases of eCJD (Fig. 7a), fCJD (Fig. 7b), and GSS (Fig. 7c), the extent of tau phosphorylation at pTau231 was similar. The levels of expression of tau phosphorylated at Thr-231 produced by the neurotoxic environment, plaques, and free PrPSc were increased in TSEs. Tau phosphorylation at Thr-231 is a neuroprotective response to neurodegeneration caused by the collapse of the neuronal cytoarchitecture, loss of function of astrocytes, and massive granular cell death. It supports the idea that tau aggregates do not contribute to the clinico-pathological characteristics in the terminal stages of prion diseases. To demonstrate this, we analyzed and compared the expression of neurofilaments between prion disease, AD (Fig. 8b), and healthy cases (Fig. 8c). Tau phosphorylation at Thr-231 and overexpression of neurofilaments modulated the axonal cytoskeleton in response to the prion (merged image, Fig. 8a).
Fig. 7
Phosphorylated tau in the cerebellum. In a) and b) pT231 reactivity (red) demonstrates phosphorylation of tau at Thr-231 in both external and internal folia, respectively. Aggregated PrPSc (green) and nuclei (blue) are shown. c) Parallel fibers stained with pT231 in the ML. d) No reactivity is observed with the pT396 antibody. In the magnification of b), axons of the Purkinje cells recognized with the pT231 antibody are indicated. ML, molecular layer; Wm, white matter; P, Purkinje cells. Images obtained with the Leica SP8 confocal microscope.
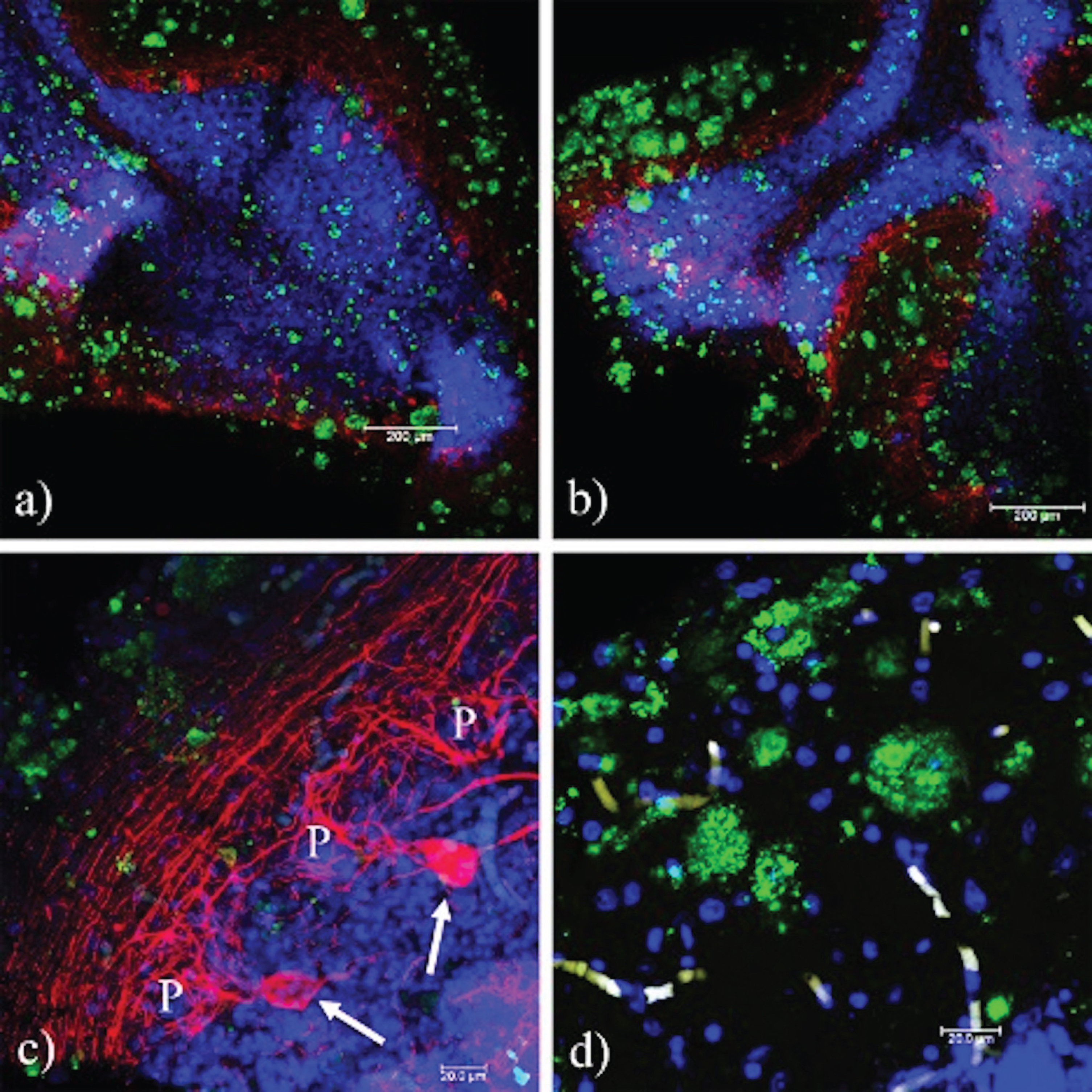
Fig. 8
The pTau231 immunoreactivity pattern is similar in the cerebellum of familial and sporadic TSEs. a) Absence of interaction between PrPSc and pTau. A positive reaction to pTau231 (red) in the axons of the Purkinje cells, and the projections of the parallel fibers from the granular cells is observed, (b) F33-10 (eECJ), (c) F55-26 (fECJ), and (d) F47–72 (GSS). The identifier for cases corresponds to the sex, age (yrs), and duration of the disease (months). P, Purkinje cell.
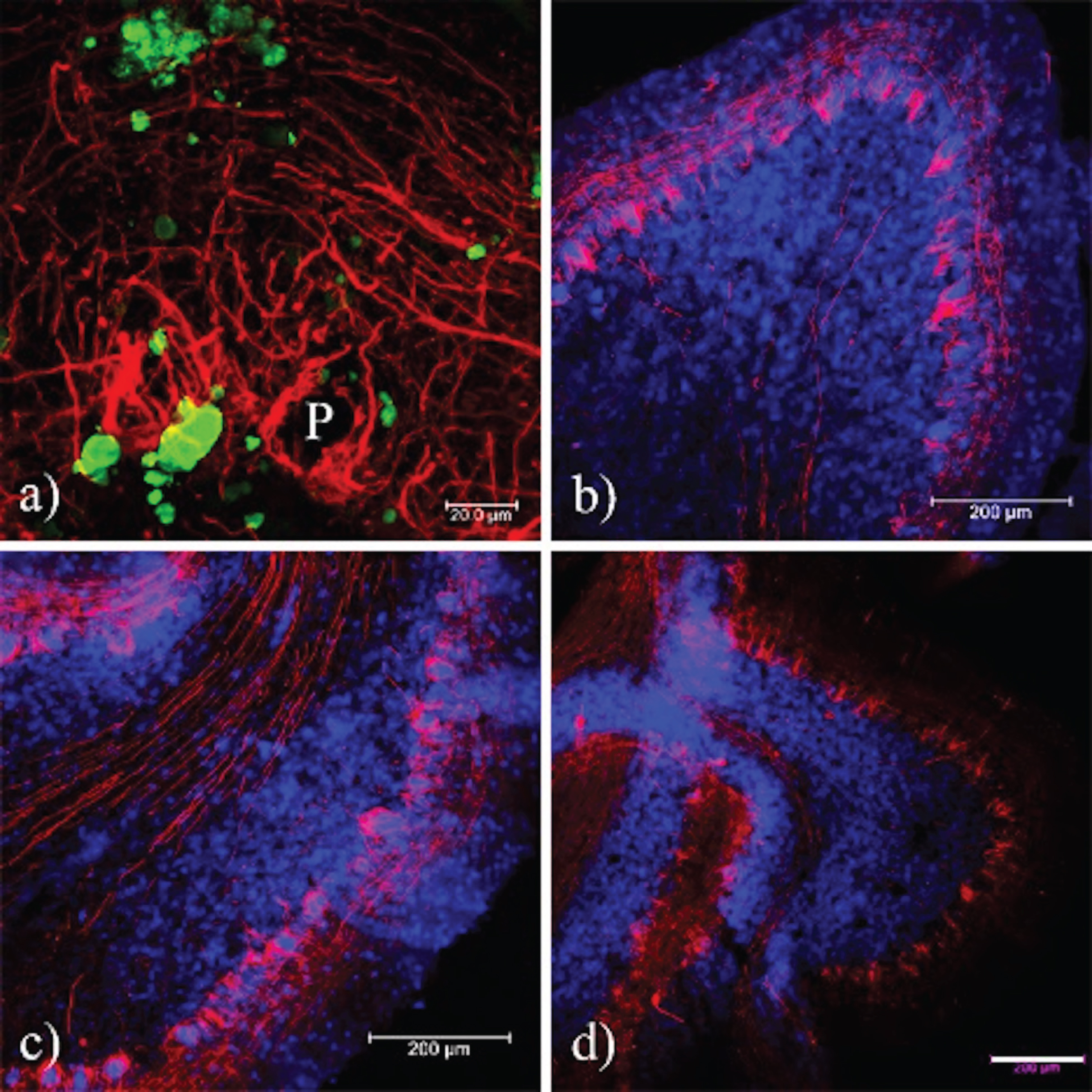
Fig. 9
Presence of neurofilaments in cases of prion disease. a) In prion diseases, the expression of Nf-L is increased and co-localizes with the tau phosphorylated at Thr-231. b, c) In AD and healthy brains, the expression of neurofilaments is similar, whereas the pTau231 antibody does not exhibit any signal. Sections were contrasted with TO-PRO(R)-3 as a background control. CJD, Creutzfeld-Jakob disease; AD, Alzheimer’s disease; ND, Non-dementia. Images obtained using a Leica SP8 confocal microscope.
DISCUSSION
The intricate pattern of tau phosphorylation and its relationship to the prion aggregation is still unknown. Even the mechanism that initiates the conversion of PrPC to PrPSc remains poorly understood. It has been suggested that alteration of PrPC to PrPSc may take place in the endoplasmic reticulum, where proteins appear defective after synthesis and are translocated to the cytosol for degradation in the proteasome via the endoplasmic-reticulum-associated protein degradation (ERAD) pathway [28, 29]. Our observations in Fig. 2a, b, and c are consistent with this mechanism. PrPC in the cytoplasm undergoes the conformational change to PrPSc (Fig. 2d-f) which accumulates as aggregates in the cerebellum (Fig. 1d, e). These aggregates, which vary in size, appear to be the initiators of the aggregation events that ultimately form the extracellular prion protein plaques observed in the ML (Fig. 1f and Fig. 2d-f). Prion plaques would not be the cause of the replication of the agent, but rather the free PrPSc, which is seen as diffuse staining around the aggregates (Fig. 3a-d). The PrPSc is likely to arise from astrocytes, interneurons, and granular cells since the 3F4 immunoreactivity is localized in regions where these cells are most abundant. Previous studies have shown that PrP knockout astrocytes influence the neuronal survival in the regulation of glutamatergic neurotransmission [30]. Both astrocytes and microglia are harmful in CJD [31–34]. According to our observations we suggest that, in astrocytes, the glutamatergic transmission is made via the synapses of mossy fibers (in GL) and granular cells with their lateral fibers on Purkinje neurons (in ML) (Fig. 10). Phosphorylation of tau decreases MT binding, and is observed during neurodegeneration in AD [35–38]. This post-translational modification has been reported in sporadic, familial, and acquired forms of prion diseases. Paired helical filaments appear in rare genetic prion diseases, such as neocortical areas in GSS [29] and thalamus in FFI [18]. Neurofibrillary tangles have been found in the frontal cortex, cerebellum, and hippocampus in familial and sporadic CJD [17, 19]. In TSE, dystrophic neurites associated with prion plaques were observed; these prion plaques were negative for Aβ antibodies and positive for PrPSc [39]. Tau-laden dystrophic neurites are commonly associated with Aβ plaques in AD [40]. Hyperphosphorylated tau protein has also been detected in neurons and glial cells of transgenic mice infected with BSE [41]. Previous studies have documented that PrPSc induces tubulin oligomerization and that the presence of the tau protein could be capable of protecting the microtubule from the effect of the PrP protein [42]. Furthermore, the accumulation and replication of PrPSc disrupts biological processes, distinctly in different cell types [43]. In our study, prion plaques have a heterogeneous pattern of aggregation and localization depending on the type of TSE [39], the polymorphism involved, and duration of the disease [11].
Fig. 10
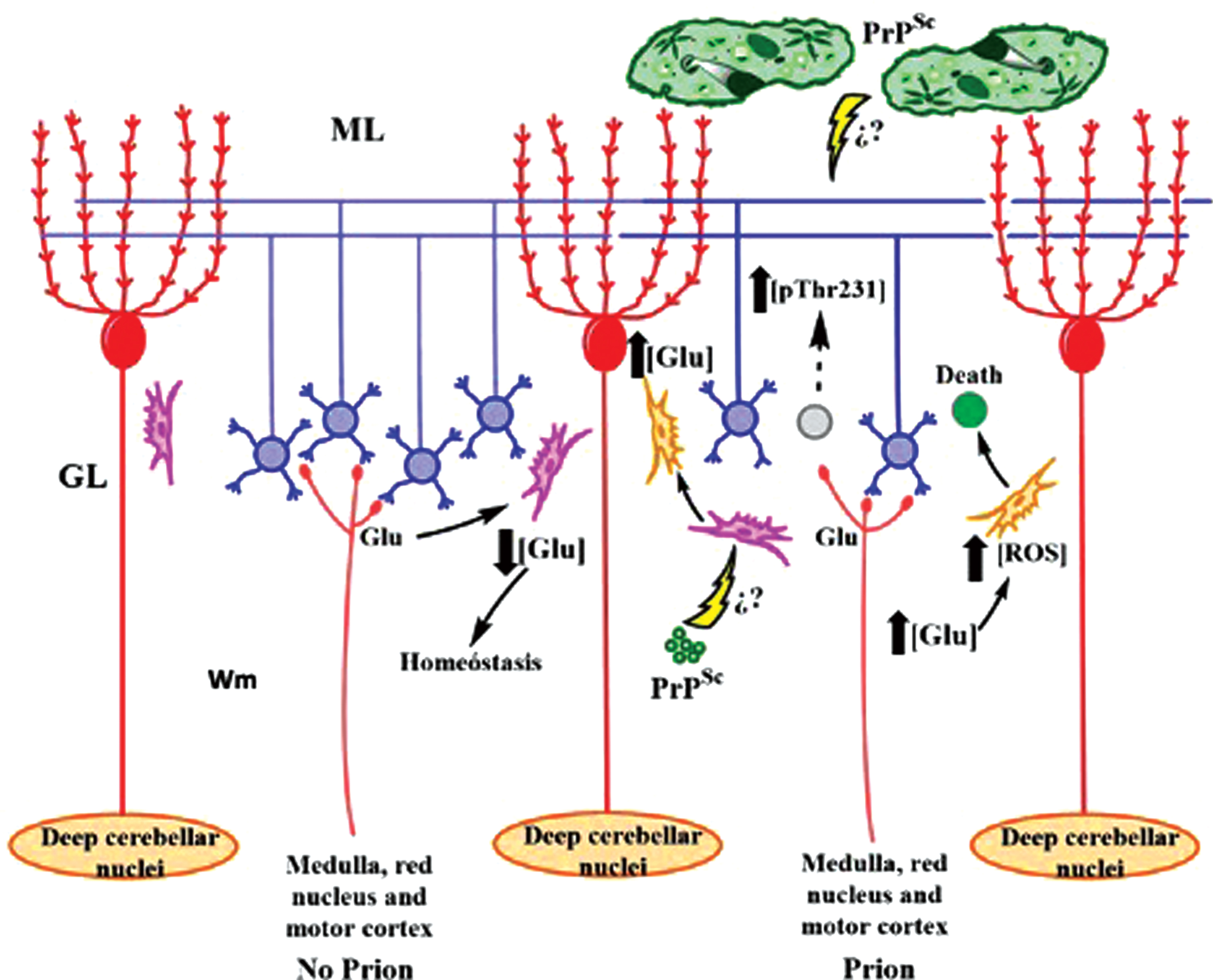
Dynamics of cerebellar dysfunction in TSEs. (Left) Astrocytes regulate the glutamatergic transmission of granular cells (blue) in healthy cerebellum. (Right) PrPSc causes protoplasmic astrocyte dysfunction (pink) and reactive astrocyte activation (orange). It increases the cytotoxic Glu concentration, causes neuronal death, the CG (gray) of the cerebellar glomeruli, and activates the phosphorylation of tau to prevent the depolymerization of the parallel fibers that innervate the Purkinje cells (red). ML, molecular layer; GL, granular layer; Wm, white matter. Question marks represent pathways that have not yet been demonstrated in the literature.
Phosphorylation of tau at Thr231 in the axons and dendrites of the Purkinje cells, and parallel fibers from granule cells, is a response to neuronal loss in the GL and prion aggregates (Figs. 7 and 8). These observations suggest that phosphorylation at Thr231 could prevent microtubule depolymerization and regulate the neurotoxic effect of the prion protein. Neurofilaments provide the structural support for myelinated axons, essential for effective nerve conduction, organelles and synaptic vesicles dynamic [44]. Figures 7 and 8 showed that pTau231 overexpression is linked to its neuroprotective role against prion plaques (and represented schematically in Fig. 10). This suggests that PrPSc deposits do not induce the neurofibrillary changes observed in the cerebellum. The integrity of Purkinje cells may indicate that early tau phosphorylation may prevent breakdown of the microtubule network and the loss of the cerebellar neuronal circuit.
Taking into account the differences in PrPSc plaque aggregation (Fig. 3) and the role of astrocytes in the neuroinflammatory response, what is the role of PrPSc in the loss and death of granule cells in the cerebellum? Considering that reactive astrocytes respond to pathological brain situations, such as ischemia, infection, and neurodegenerative diseases [31], we hypothesize that the loss in function of astrocytes contributes to neurodegeneration in TSEs. In this work, a detailed examination was performed to confirm the role of astrocytes in the granular cell apoptosis. The temporal and anatomical correlation between the PrPSc deposition, reactive astrogliosis located mainly in GL (Fig. 4), and neuropathological damage evidenced by MMP-9 (Fig. 5), suggest that PrPSc has an increased tendency to aggregate from the cellular debris observed in the Wm (Fig. 2d). Astrogliosis has been shown as a hallmark of prion diseases, being induced by PrPSc [7, 21, 32, 33, 45, 46]. MMP-9, expressed in response to stimuli such as pro-inflammatory peptides and cytokines caused by the prion particle [26, 47, 48] and activated microglia, are present both in sporadic and genetic TSEs, and animal models infected with prions [49]. Microglia release interleukin IL1-β in response to neurotoxicity caused by PrPSc aggregates, similar to that which occurs in AD with Aβ plaques [13] and, in turn, promotes the secretion of MMP-9 by reactive astrocytes [26, 47]. On the other hand, PrPC is known to modulate NMDA-type glutamate receptors by mediating plasticity and synaptic transmission [8], and acting as a divalent ion sensor, regulating the balance of Cu2+ and Zn2+ via AMPA receptors [50, 51]. We hypothesize that astrogliosis increases the concentration of extracellular glutamate, leading to hyperexcitability of NMDA receptors. This causes increased intracellular Ca2+ and NOS and the release of Cyt-C via calpain [52], leading to apoptosis due to oxidative stress. AMPA receptors may be related to superoxide dismutase (SOD) dysfunction, due to (a) decrease in the concentration of enzymatic cofactors (Cu2+ and Zn2+); (b) loss of PrPC function (by its conversion to PrPSc); and (c) its proteolysis by the activity of MMP-9 (Fig. 11). The distribution of reactive astrocytes was observed in parallel with the PrPSc topography. DNA fragmentation, however, was exclusive to the GL (Fig. 6) suggesting that the damage is produced by free radicals due to astrocytic activity and excess extracellular glutamate [30]. Previous immunocytochemical and expression studies of Bax, Bcl-2, Fas, and Casp-3, have shown inconsistencies, since only Bax and not Casp-3 expression was reported [53, 54]. Furthermore, the anti-apoptotic activity of Bcl-2 was reported to reduce neuronal loss in the cerebellum of transgenic mice [55]. A toxic PrP form could be produced directly from PrPC expressed in cells of the GL, discarding a direct neurotoxic role for PrPSc plaques. Our findings support apoptosis as the relevant cell death pathway in TSEs. However, this pathway may be activated, in turn, by a further pathway.
Fig. 11
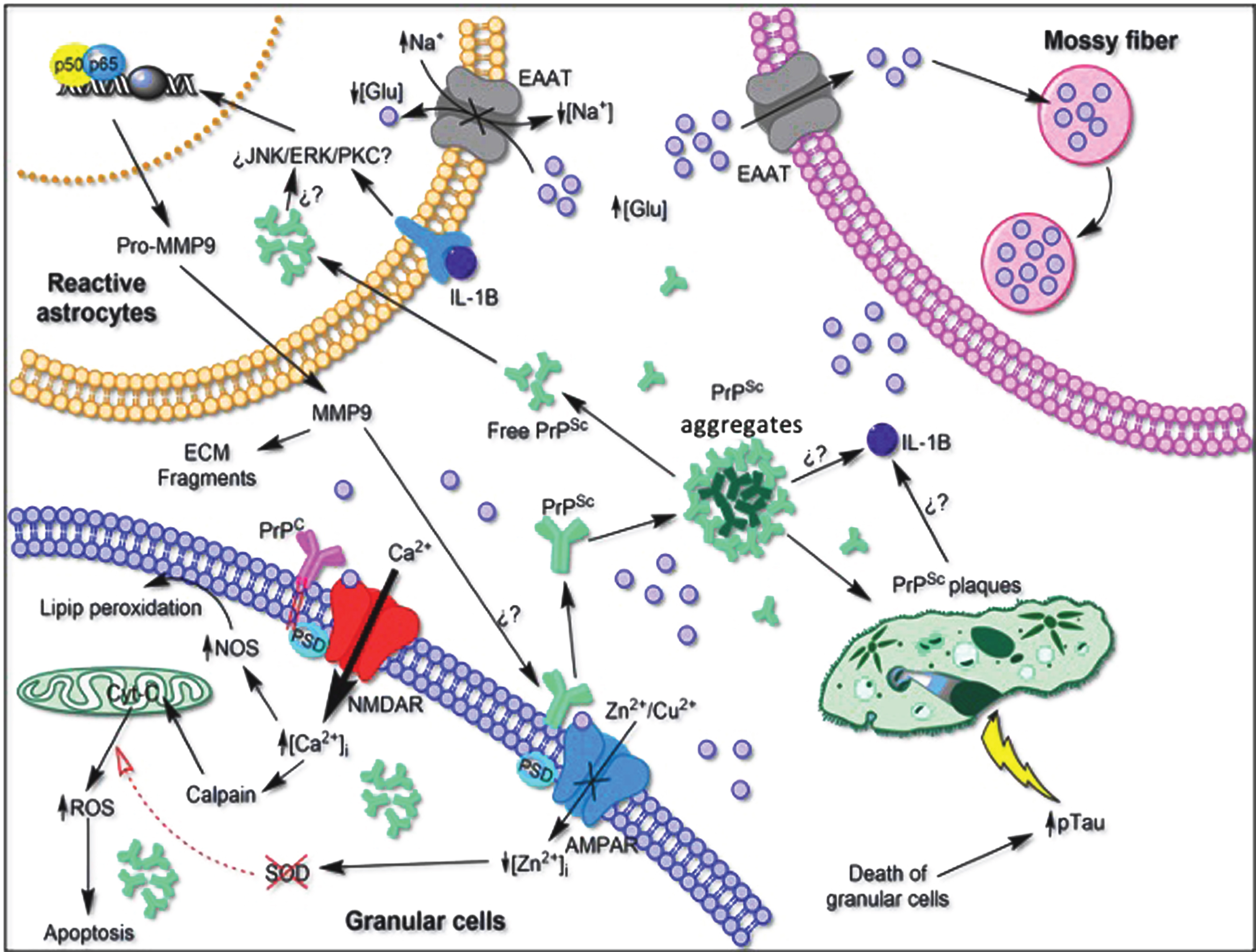
Hypothetical scheme of the neurotoxic environment produced by the proliferation of reactive astrocytes and its relationship with the loss of function of PrPC in the cerebellum. Astrocytes can be stimulated by signaling cascades that result in detrimental effects on the neural environment. Hyperexcitability of NMDA receptors and loss of function of AMPA receptors to maintain the balance of Zn2+ and Cu2+ ions can lead to increased oxidative stress and cell death. The conformational change from PrPC to PrPSc initiates the aggregation of the insoluble plaques, producing a neuroinflammatory response of the microglia, releasing pro-inflammatory cytokines that activate astrogliosis. Both the death of granule cells and the toxic environment produced by prion plaques could initiate the phosphorylation of tau to maintain innervation on Purkinje cells. EAAT, Excitatory amino acid transporter. Question marks represent pathways that have not yet been demonstrated in the literature. NOS: nitric-oxide synthase; AMPAR: AMPA receptor; ROS: reactive oxygen species; ECM: extracellular matrix; MMP-9: matrix metalloproteinase-9, IL-1B: interleukin 1 beta; Glu: glutamate, PSD: postsynaptic density protein; NMDAR: N-methyl-D-aspartate receptor; JNK: c-Jun N-terminal kinase; ERK: extracellular signal-regulated kinase; PKC: protein kinase C.
CONCLUSION
Our study suggests that an early event that prevents MT destabilization in prion disease is the phosphorylation of tau at Thr-231. Reactive astrocytes and the loss of homeostasis in the interstitium lead to death of granular cells and the development of prion pathology. This reactive astrocytosis would be a key event in the pathological mechanisms related to prion disease (as depicted in Fig. 11). Consequent tissue damage could arise via the following pathways: 1) intracellular accumulation of PrPc forms affects cellular function and leads to neuronal death; free PrPSc observed around the dense plaques acts as the cytotoxic factor as opposed to the extracellular deposits of PrPSc within plaques; 2) tau protein responds to the presence of prion plaques; and 3) astrogliosis and elevated MMP-9 expression appear to lead to neuronal death. Since the mechanisms of degeneration in prion diseases may be multiple and complex, our findings contribute to an understanding of the molecular mechanisms associated with the development of prion pathogenesis and a role for phospho-tau protein in this process.
ACKNOWLEDGMENTS
The authors want to express their gratitude to the following: Dr. P. Davies† (Albert Einstein College of Medicine, Bronx, NY, USA) and Lester I. Binder† (North Western, Chicago, IL, USA) for the generous gift of mAbs TG-3 and Alz-50, and Tau-1, Tau-5 and Tau-7, respectively; Tec. Amparo Viramontes Pintos for the handling of the brain tissue; support in the confocal microscopy unit of CIIDIR Durango, Instituto Politécnico Nacional; Union Medica University Clinic, Dominican Republic, for their support and collaboration in the development of this research project. We also want to express our gratitude to the Mexican families who have donated the brain of their loved ones affected with Alzheimer's disease and made our research possible. This work is dedicated to the memory of Professor Dr. José Raúl Mena López†. †Deceased.
This work was supported by Fondo Nacional de Ciencia y Tecnologia, FONDOCyT, from the Ministry of Higher Education, Science and Technology, Dominican Republic (2015-3A2-127 to MP-H) and (2018-2019-2A3-208 to JL-M and MP-H).
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/20-1308r2).
REFERENCES
[1] | du Plessis DG ((2008) ) Prion protein disease and neuropathology of prion disease. Neuroimaging Clin N Am 18: , 163–182. |
[2] | Kaneko M , Sugiyama N , Sasayama D , Yamaoka K , Miya-kawa T , Arima K , Tsuchiya K , Hasegawa K , Washizuka S , Hanihara T , Amano N , Yagishita S ((2008) ) Prion disease causes less severe lesions in human hippocampus than other parts of brain. Psychiatry Clin Neurosci 62: , 264–270. |
[3] | Kim MO , Geschwind MD ((2015) ) Clinical update of Jakob-Creutzfeldt disease. Curr Opin Neurol 28: , 302–310. |
[4] | Imran M , Mahmood S ((2011) ) An overview of animal prion diseases. Virol J 8: , 493. |
[5] | Mastrianni JA ((2010) ) The genetics of prion diseases. Genet Med 12: , 187–195. |
[6] | Castle AR , Gill AC ((2017) ) Physiological functions of the cellular prion protein. Front Mol Biosci 4: , 19. |
[7] | Hetz C , Maundrell K , Soto C ((2003) ) Is loss of function of the prion protein the cause of prion disorders? Trends Mol Med 9: , 237–243. |
[8] | Biasini E , Turnbaugh JA , Unterberger U , Harris DA ((2012) ) Prion protein at the crossroads of physiology and disease. Trends Neurosci 35: , 92–103. |
[9] | Prusiner SB ((1982) ) Novel proteinaceous infectious particles cause scrapie. Science 216: , 136–144. |
[10] | Jeffrey M , Gonzalez L ((2007) ) Classical sheep transmissible spongiform encephalopathies: Pathogenesis, pathological phenotypes and clinical disease. Neuropathol Appl Neurobiol 33: , 373–394. |
[11] | Gambetti P , Cali I , Notari S , Kong Q , Zou WQ , Surewicz WK ((2011) ) Molecular biology and pathology of prion strains in sporadic human prion diseases. Acta Neuropathol 121: , 79–90. |
[12] | Maddox RA , Person MK , Blevins JE , Abrams JY , Appleby BS , Schonberger LB , Belay ED ((2020) ) Prion disease incidence in the United States: 2003-2015. Neurology 94: , e153–e157. |
[13] | Arendt T , Stieler JT , Holzer M ((2016) ) Tau and tauopathies. Brain Res Bull 126: , 238–292. |
[14] | Hansra GK , Popov G , Banaczek PO , Vogiatzis M , Jegathees T , Goldbury CS , Cullen KM ((2019) ) The neuritic plaque in Alzheimer’s disease: Perivascular degeneration of neuronal processes. Neurobiol Aging 82: , 88–101. |
[15] | Martin L , Latypova X , Terro F ((2011) ) Post-translational modifications of tau protein: Implications for Alzheimer’s disease. Neurochem Int 58: , 458–471. |
[16] | Chi NF , Lee YC , Lu YC , Wu HM , Soong BW ((2010) ) Transmissible spongiform encephalopathies with P102L mutation of PRNP manifesting different phenotypes: Clinical, neuroimaging, and electrophysiological studies in Chinese kindred in Taiwan. J Neurol 257: , 191–197. |
[17] | Jayadev S , Nochlin D , Poorkaj P , Steinbart EJ , Mastrianni JA , Montine TJ , Ghetti B , Schellenberg GD , Bird TD , Leverenz JB ((2011) ) Familial prion disease with Alzheimer disease-like tau pathology and clinical phenotype. Ann Neurol 69: , 712–720. |
[18] | Jansen C , Parchi P , Jelles B , Gouw AA , Beunders G , van Spaendonk RM , van de Kamp JM , Lemstra AW , Capellari S , Rozemuller AJ ((2011) ) The first case of fatal familial insomnia (FFI) in the Netherlands: A patient from Egyptian descent with concurrent four repeat tau deposits. Neuropathol Appl Neurobiol 37: , 549–553. |
[19] | Reiniger L , Lukic A , Linehan J , Rudge P , Collinge J , Mead S , Brandner S ((2011) ) Tau, prions and Aβ: The triad of neurodegeneration. Acta Neuropathol 121: , 5–20. |
[20] | Tranchant C , Sergeant N , Wattez A , Mohr M , Warter JM , Delacourte A ((1997) ) Neurofibrillary tangles in Gerstmann-Straussler-Scheinker syndrome with the A117V prion gene mutation. J Neurol Neurosurg Psychiatry 63: , 240–246. |
[21] | Asuni AA , Perry VH , O’Connor V ((2010) ) Change in tau phosphorylation associated with neurodegeneration in the ME7 model of prion disease. Biochem Soc Trans 38: , 545–551. |
[22] | Jicha GA , Lane E , Vincent I , Otvos L Jr. , Hoffmann R , Davies P ((1997) ) A conformation- and phosphorylation-dependent antibody recognizing the paired helical filaments of Alzheimer’s disease. J Neurochem 69: , 2087–2095. |
[23] | Gamblin TC , Chen F , Zambrano A , Abraha A , Lagalwar S , Guillozet AL , Lu M , Fu Y , Garcia-Sierra F , LaPointe N , Miller R , Berry RW , Binder LI , Cryns VL ((2003) ) Caspase cleavage of tau: Linking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proc Natl Acad Sci U S A 100: , 10032–10037. |
[24] | Novak M , Wischik CM , Edwards P , Pannell R , Milstein C ((1989) ) Characterisation of the first monoclonal antibody against the pronase resistant core of the Alzheimer PHF. Prog Clin Biol Res 317: , 755–761. |
[25] | Fragkouli A , Tsilibary EC , Tzinia AK ((2014) ) Neuroprotective role of MMP-9 overexpression in the brain of Alzheimer’s 5xFAD mice. Neurobiol Dis 70: , 179–189. |
[26] | Wang XX , Tan MS , Yu JT , Tan L ((2014) ) Matrix metalloproteinases and their multiple roles in Alzheimer’s disease. Biomed Res Int 2014: , 908636. |
[27] | Louis ED , Kuo SH , Vonsattel JP , Faust PL ((2014) ) Torpedo formation and Purkinje cell loss: Modeling their relationship in cerebellar disease. Cerebellum 13: , 433–439. |
[28] | Beranger F , Mange A , Goud B , Lehmann S ((2002) ) Stimulation of PrPC retrograde transport toward the endoplasmic reticulum increases accumulation of PrPSc in prion-infected cells. J Biol Chem 277: , 38972–38977. |
[29] | Ma J , Wollmann R , Lindquist S ((2002) ) Neurotoxicity and neurodegeneration when PrP accumulates in the cytosol. Science 298: , 1781–1785. |
[30] | Pathmajeyan MS , Patel SA , Carroll JA , Seib T , Striebel JF , Bridges RJ , Chesebro B ((2011) ) Increased excitatory amino acid transport into murine prion protein knockout astrocytes cultured in vitro. Glia 59: , 1684–1694. |
[31] | Ben Haim L , Carrillo-de Sauvage MA , Ceyzeriat K , Escartin C ((2015) ) Elusive roles for reactive astrocytes in neurodegenerative diseases. Front Cell Neurosci 9: , 278. |
[32] | Garces M , Toledano A , Badiola JJ , Monzón M ((2016) ) Morphological change of glia in prion and a prion-like disorder. J Alzheimers Neurodegener Dis 2: , 005. |
[33] | Monzon M , Hernandez RS , Garces M , Sarasa R , Badiola JJ ((2018) ) Glial alterations in human prion diseases: A correlative study of astroglia, reactive microglia, protein deposition, and neuropathological lesions. Medicine (Baltimore) 97: , e0320. |
[34] | Schmitz M , Dittmar K , Llorens F , Gelpi E , Ferrer I , Schulz-Schaeffer WJ , Zerr I ((2017) ) Hereditary human prion diseases: An update. Mol Neurobiol 54: , 4138–4149. |
[35] | Goedert M , Spillantini MG , Potier MC , Ulrich J , Crowther RA ((1989) ) Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: Differential expression of tau protein mRNAs in human brain. EMBO J 8: , 393–399. |
[36] | Goode BL , Denis PE , Panda D , Radeke MJ , Miller HP , Wilson L , Feinstein SC ((1997) ) Functional interactions between the proline-rich and repeat regions of tau enhance microtubule binding and assembly. Mol Biol Cell 8: , 353–365. |
[37] | Iqbal K , Novak M ((2006) ) From tangles to tau protein. Bratisl Lek Listy 107: , 341–342. |
[38] | Jakes R , Novak M , Davison M , Wischik CM ((1991) ) Identification of 3- and 4-repeat tau isoforms within the PHF in Alzheimer’s disease. EMBO J 10: , 2725–2729. |
[39] | Sikorska B , Liberski PP , Sobow T , Budka H , Ironside JW ((2009) ) Ultrastructural study of florid plaques in variant Creutzfeldt-Jakob disease: A comparison with amyloid plaques in kuru, sporadic Creutzfeldt-Jakob disease and Gerstmann-Straussler-Scheinker disease. Neuropathol Appl Neurobiol 35: , 46–59. |
[40] | Mena R , Edwards P , Perez-Olvera O , Wischik CM ((1995) ) Monitoring pathological assembly of tau and β-amyloid proteins in Alzheimer’s disease. Acta Neuropathol 89: , 50–56. |
[41] | Bautista MJ , Gutierrez J , Salguero FJ , Fernandez de Marco MM , Romero-Trevejo JL , Gomez-Villamandos JC ((2006) ) BSE infection in bovine PrP transgenic mice leads to hyperphosphorylation of tau-protein. Vet Microbiol 115: , 293–301. |
[42] | Osiecka KM , Nieznanska H , Skowronek KJ , Jozwiak J , Nieznanski K ((2011) ) Tau inhibits tubulin oligomerization induced by prion protein. Biochim Biophys Acta 1813: , 1845–1853. |
[43] | Brambilla L , Martorana F , Rossi D ((2013) ) Astrocyte signaling and neurodegeneration: New insights into CNS disorders. Prion 7: , 28–36. |
[44] | Yuan A , Rao MV , Veeranna , Nixon RA ((2012) ) Neurofilaments at a glance. J Cell Sci 125: , 3257–3263. |
[45] | Acevedo-Morantes CY , Wille H ((2014) ) The structure of human prions: From biology to structural models-considerations and pitfalls. Viruses 6: , 3875–3892. |
[46] | Garces M , Guijarro MI , Vargas A , Badiola JJ , Monzon M ((2019) ) Neuroglial patterns are shared by cerebella from prion and prion-like disorder affected patients. Mech Ageing Dev 184: , 111176. |
[47] | Hsi-Lung H , Chuen-Mao Y ((2014) ) The role of matrix meta-lloproteinase-9 in pro-inflammatory factors-induced brain inflammation and neurodegenerative diseases. Inflamm Cell Signal 1: , e124. |
[48] | Wang X , Li Y , Chen J , Guo X , Guan H , Li C ((2014) ) Differential expression profiling of matrix metalloproteinases and tissue inhibitors of metalloproteinases in females with or without pelvic organ prolapse. Mol Med Rep 10: , 2004–2008. |
[49] | Aguzzi A , Nuvolone M , Zhu C ((2013) ) The immunobiology of prion diseases. Nat Rev Immunol 13: , 888–902. |
[50] | Kovacs GG , Budka H ((2008) ) Prion diseases: From protein to cell pathology. Am J Pathol 172: , 555–565. |
[51] | Watt NT , Griffiths HH , Hooper NM ((2013) ) Neuronal zinc regulation and the prion protein. Prion 7: , 203–208. |
[52] | Llorens F , Thune K , Sikorska B , Schmitz M , Tahir W , Fernandez-Borges N , Cramm M , Gotzmann N , Carmona M , Streichenberger N , Michel U , Zafar S , Schuetz AL , Rajput A , Andreoletti O , Bonn S , Fischer A , Liberski PP , Torres JM , Ferrer I , Zerr I ((2017) ) Altered Ca2+ homeostasis induces Calpain-Cathepsin axis activation in sporadic Creutzfeldt-Jakob disease. Acta Neuropathol Commun 5: , 35. |
[53] | Kovacs GG , Laszlo L , Bakos A , Minarovits J , Bishop MT , Strobel T , Vajna B , Mitrova E , Majtenyi K ((2005) ) Increased incidence of genetic human prion disease in Hungary. Neurology 65: , 1666–1669. |
[54] | Jodoin J , Misiewicz M , Makhijani P , Giannopoulos PN , Hammond J , Goodyer CG , LeBlanc AC , Loss of anti-Bax function in Gerstmann-Straussler-Scheinker syndrome-associated prion protein mutants. PLoS One 4: , e6647. |
[55] | Nicolas O , Gavin R , Braun N , Urena JM , Fontana X , Soriano E , Aguzzi A , del Rio JA ((2007) ) Bcl-2 overexpression delays caspase-3 activation and rescues cerebellar degeneration in prion-deficient mice that overexpress amino-terminally truncated prion. FASEB J 21: , 3107–3117. |


