
Broader Insights into Understanding Tumor Necrosis Factor and Neurodegenerative Disease Pathogenesis Infer New Therapeutic Approaches
Abstract
Proinflammatory cytokines such as tumor necrosis factor (TNF), with its now appreciated key roles in neurophysiology as well as neuropathophysiology, are sufficiently well-documented to be useful tools for enquiry into the natural history of neurodegenerative diseases. We review the broader literature on TNF to rationalize why abruptly-acquired neurodegenerative states do not exhibit the remorseless clinical progression seen in those states with gradual onsets. We propose that the three typically non-worsening neurodegenerative syndromes, post-stroke, post-traumatic brain injury (TBI), and post cardiac arrest, usually become and remain static because of excess cerebral TNF induced by the initial dramatic peak keeping microglia chronically activated through an autocrine loop of microglial activation through excess cerebral TNF. The existence of this autocrine loop rationalizes post-damage repair with perispinal etanercept and proposes a treatment for cerebral aspects of COVID-19 chronicity. Another insufficiently considered aspect of cerebral proinflammatory cytokines is the fitness of the endogenous cerebral anti-TNF system provided by norepinephrine (NE), generated and distributed throughout the brain from the locus coeruleus (LC). We propose that an intact LC, and therefore an intact NE-mediated endogenous anti-cerebral TNF system, plus the DAMP (damage or danger-associated molecular pattern) input having diminished, is what allows post-stroke, post-TBI, and post cardiac arrest patients a strong long-term survival advantage over Alzheimer’s disease and Parkinson’s disease sufferers. In contrast, Alzheimer’s disease and Parkinson’s disease patients remorselessly worsen, being handicapped by sustained, accumulating, DAMP and PAMP (pathogen-associated molecular patterns) input, as well as loss of the LC-origin, NE-mediated, endogenous anti-cerebral TNF system. Adrenergic receptor agonists may counter this.
INTRODUCTION
Despite possessing a predisposition for developing Alzheimer’s disease (AD) or Parkinson’s disease (PD), as discussed later, survivors of stroke, traumatic brain injury (TBI), and cardiac arrest have a much better long-term prognosis than do individuals primarily developing one of this pair of gradual onset states. Nevertheless all five conditions have much in common, including certain symptoms and signs, changes in laboratory or tissue markers, and therapeutic response, in either experimental models or patients, to anti-tumor necrosis factor (TNF) biologicals [1]. What they do not share are their speed of onset, this being abrupt in stroke, TBI, and cardiac arrest—albeit stepwise with the accumulating consequences of chronic traumatic encephalopathy arising from repeated head injuries in contact sports—while profoundly gradual in AD and PD. The onset in the former group is self-evidently a consequence of an abruptly-acquired brain catastrophe arising from some sudden combination of hypoxia, ischemia, and tissue injury in a hitherto normal brain, implying a normally functional locus coeruleus (LC), and thus, as discussed later, an intact norepinephrine (NE)-mediated endogenous anti-cerebral TNF. This rapidity is clearly very different from the pernicious onset of AD and PD, in which cerebral change may well have been present long before its clinical manifestation. The other shared distinctive feature of this abrupt-onset trio is their consequences for the rest of the patient’s life. Typically, should the initial few weeks severe of illness be weathered, the degree of clinical severity lessens and becomes relatively stable, albeit often very limiting, for many years. This is quite distinct from the gradual but remorseless worsening that characterizes AD and PD, as well as other, rarer, neurodegenerative conditions. These group distinctions are accepted as common knowledge, but, so far as we are aware, their origin has not been formally addressed. Here we propose that the logic of the inflammatory theory of the pathogenesis of these disease states now makes it possible to compare them more accurately than was possible before.
Moreover, we argue that understanding the pathogenesis of post-stroke, TBI, and cardiac arrest syndromes in these terms has wide ramifications. For example, sufficient time has now passed since the onset of the COVID-19 pandemic for several long-term debilitating cerebral effects of the virus to have been clearly identified and described. On the basis of the reasoning put forward in the present text, we propose below the case for etanercept, the specific anti-TNF biological, administered via the perispinal route, a method to get large molecules around the blood-brain barrier, [2–4] being a logical treatment for neurological aspects of COVID-19, as it is proving to be for post-stroke syndromes.
RAPID-ONSET DISEASE STATES PREDISPOSE TO THOSE WITH GRADUAL ONSETS
The shared basic biology across these neurodegenerative conditions can be inferred from reports that affliction by one of these abrupt-onset states predispose to subsequently developing other from this group with a slow onset. Examples include TBI making patients more likely to be subsequently diagnosed with AD [5–7] or with PD [7–9]. Stroke predisposing to AD [10], and PD to stroke [11], are also in the literature. Despite previous arguments to the contrary, the point has been recently made, from two independent sources [12, 13], that chronic traumatic encephalopathy may, rather than inevitably will, lead to one of the terminal neurodegenerative states. The case has also been made that cardiac arrest predisposes to subsequent onset of AD [14]. These observations are consistent with successful experimental therapeutic outcomes on each side of this rapid/gradual onset rate divide, with anti-TNF agents successfully treating experimental stroke [15, 16] and TBI [17] as well as reducing the burden in AD [18] and PD [19] models. This adds credence to earlier post-stroke perispinal etanercept case reports [20–22]. The mouse AD data [18] is consistent with earlier etanercept reports in the form of a six months open trial [23], and long-term subcutaneous use for rheumatoid arthritis improving cognition in older patients [24] and reducing AD incidence [25], and Pfizer (unpublished, https://www.washingtonpost.com/business/economy/pfizer-had-clues-its-blockbuster-drug-could-prevent-alzheimers-why-didnt-it-tell-the-world/2019/06/04/9092e08a-7a61-11e9-8bb7-0fc796cf2ec0_story.html). Similarly, the anti-TNF effects of XPro1595 noted above in a mouse PD model [19] is consistent with a report of anti-TNF treatment for inflammatory bowel disease patients reducing their incidence of PD [26]. One obvious interpretation of these data is, as discussed later, that essentially the same pathophysiology is at work in both the rapid and slow onset groups, despite their superficial differences in outcome. Indeed, a TBI/AD sequence of drug development has been proposed [27, 28] on the argument that any agent that ameliorates TBI is very likely to improve AD.
Even so, none of the above, or anything else so far as we are aware, addresses why, once afflicted, most patients who survive any of these three acquired brain injuries should, after a degree of improvement over the first months, remain stable, whereas those suffering from AD or PD remorselessly worsen. Although this pair of conditions is evidently more complex in their pathophysiology than that of the others, there are, as summarized above, similarities in pathophysiology as well as shared responses to anti-TNF biologicals improving AD [18] and PD [19] models. The large number of anti-TNF agents and intense competition between patent extension attempts and approved biosimilars [29] provides evidence of the widescale awareness and perceived importance of this cytokine in the pathogenesis of neurodegenerative diseases.
THE KEY RELEVANCE OF TNF IN ALL THESE CONDITIONS
Despite their striking differences in onset and course, many characteristics of these two distinctive groups of neurodegenerative diseases overlap closely. Inflammation involving activated microglia driving chronic disease progression is a common theme [30]. We and others had already reviewed how much effort had already been put into understanding the roles of TNF in the physiological, and, should homeostatic levels be exceeded, pathophysiological cerebral consequences in the brain [31]. Similarly broad set of roles for this cytokine has long been acknowledged outside the blood-brain barrier [32]. More recently, the first double-blind randomized controlled clinical trial of perispinal etanercept for chronic stroke has been published [33]. We have also discussed what we regard as the unusual phenomenon of agents, well-represented in the literature as possessing anti-TNF activity, having this attribute ignored in new therapeutic research directed towards mouse models of (slow-onset) PD [34] and (abrupt-onset) stroke and TBI [35]. Specifically, given the extensive links in the MCC950 and maroviroc literatures to their anti-TNF activity (see [1]), although perhaps advantageous from a patent-holder perspective, it seems scientifically indefensible not to have used specific anti-TNF biologicals such as etanercept as a positive control in these studies.
Studies on the roles of the delta family of phosphoinosositide 3-kinases (PI3Ks) of cytokine signaling pathways independently demonstrate the centrality of TNF in models of stroke [36] and AD [37], the most common members of the rapid and slow onset groups respectively. This has been achieved in a stroke model by inhibiting one of this family of kinases, which reduced TNF trafficking and secretion of lipopolysaccharide-induced TNF in macrophages [38]. The same researchers subsequently showed, through a series of in vitro approaches using the PI3Kδ inhibitor CAL-101 [39], that secretion of TNF and other biomarkers of neuroinflammation could be reduced in a mouse cerebral stroke model [36]. Although essentially unable to cross the blood-brain barrier [36], this inhibitor nevertheless exhibited in vitro outcomes consistent with the ability, should it be administered perispinally to ensure it reaches the brain [3] in order to reduce TNF and thus neuroinflammation in a mouse model of AD. As might be expected, genetically inactivating this kinase produced the same TNF and neuroinflammation outcomes in vivo as the inhibitor did in vitro, as well as reducing cognitive decline [37]. Notably, a range of other anti- or proinflammatory cytokines were unaffected. This is consistent with a new study that demonstrates that TNF, but not interleukin-1β, entirely controls metaplastic inhibition of long-term potentiation within the CA-1 area of the hippocampus in an AD model [40].
Changes in Triggering Receptor Expressed on Myeloid cells 2 (TREM2) and Apolipoprotein epsilon 4 (APOE4) positivity, both consistent with higher levels of TNF, are associated with both the rapid and slow onset conditions discussed here. At least one from each onset rate category, stroke and AD, are exacerbated when TREM2 is low [41, 42]. Similarly, stroke [43], TBI [44], AD [45], and PD [46] have the same association with APOE4 status, and the same changes in cerebral biomarkers, including increased proinflammatory cytokines levels, are present. APOE4 individuals have an enhanced innate immunity response by various criteria, including increased TNF generation, after intravenous infusion with a TLR4 ligand, bacterial lipopolysaccharide (LPS) [47]. Other elements of this cytokine framework, such as those entailing Interleukin-1β, Interleukin-17, Interleukin-23, and inflammasomes, which can be activated by TNF [48], are omitted here for brevity.
PAMPs, DAMPs, AND THE TNF CASCADE RESPONSE IN INNATE IMMUNITY AND DISEASE
What leads to the same array of functionally related primitive cytokines, dominated by TNF, being generated in strikingly different circumstances, and inducing surprisingly similar disease outcomes? Since this question is central to this article, the sense of the terms PAMP (pathogen-associated molecular patterns) and DAMP (damage or danger-associated molecular pattern) warrant summarizing here. These molecular patterns are on the surface of the molecules that trigger the inflammatory signaling pathways that are implicated. Others [49] have coined the overarching term alarmins for these two classes of receptor ligands to highlight their essentially similar consequences. In brief, the concept began with PAMPs being an explanation of broad-band pathogen recognition that triggers innate immunity against infectious organisms [50]. The term has evolved to encompass the pathogenesis, through excess TNF, of the infectious disease caused by the pathogen involved [51, 52]. The concept of DAMPs arose through others [53–55] proposing a novel rationalization of the danger that arises from destructive consequences of a self/non-self immunological interaction. With these ideas expanding beyond infectious disease, the implications of tissue damage generating functionally identical triggers for the TNF cascade through the effects of trauma or hypoxia has led to the D in DAMP standing, nowadays, for damage more frequently than for danger. In practice, the two are interchangeable. Activation of TNF-producing cells (mostly microglia and astrocytes in a cerebral context) occurs when DAMPs and PAMPs are recognized by, and activate, pattern recognition receptors (PRRs) [50]. The toll-like receptors (TLRs) [56] discussed here are one of the best described families of PRRs.
Driven by hypoxia and tissue damage, the three abruptly-acquired brain injuries we have been discussing cause release of DAMPs. Mitochondrial DNA (mtDNA) and high mobility group box B-1 (HMGB1) protein escaping from the mitochondria [57] and cell nuclei [58] respectively, of damaged cells, are the dominant known DAMPs that trigger functional and cellular loss after tissue injury and hypoxia, in this case in the brain. Persistent HMGB1 release has been confirmed in patients post-stroke [59], post-TBI [60], and after cardiac arrest [61].
AMYLOID, SYNUCLEIN, AND TAU ASSOCIATIONS ACROSS THE BOARD
Recalling that amyloid-β (Aβ), alpha-synuclein (α−Syn), and tau (i.e., hyperphosphorylated tau, or p-tau) are the stalwarts of the AD and PD literatures, we note here that the promotor region of the amyloid precursor protein gene is controlled by TNF [62], and currently emerging work links them more closely [63] [64]. Moreover, as discussed earlier, both mtDNA and HMGB1 are the DAMPs most likely to cause enhance TNF production in TBI and stroke [65, 66]. Aβ [67] and p-tau [68] are DAMPs, as is α−Syn [69, 70]. This is consistent with increases in p-tau being induced by microglial proinflammatory cytokines [71] in stroke [72, 73], as are α-Syn [74, 75] and Aβ [76, 77]. Indeed, one commentator [27] refers to some 70% of 55 patients who died within 24 hours of head injury already exhibiting increased amyloid-β protein precursor (AβPP) in brain sections, with the earliest seen after two hours of survival. In addition, inflammasome activation occurs in all three of these conditions [78–80]. This can be expected to have induced the IL-1β, via caspase-1 activation, generated downstream from TNF.
Brain injury observed after cardiac arrest, the third of these abrupt-onset neurodegenerative states, is clearly a consequence of a rapid-onset generalized cerebral ischemic hypoxia. As with stroke and TBI survival, it exhibits histologically detectable rapid increases in Aβ [81] and p-tau [82, 83]. The cerebral effects of this hypoxia can be expected to be diffuse rather than focused on some particular brain region as in stroke and TBI, but the fundamentals can be expected to be the same. Survivable cardiac arrest causes a rapid increase in brain TNF in a rat model [84], a finding consistent with hypoxia, the hallmark of cardiac arrest, inducing release of the DAMP, HMGB1 [85]. Raised transactive response DNA-binding protein with a molecular weight of 43 kDa (TDP-43), a DAMP first identified in frontotemporal dementia [86], has recently been recorded in TBI, but as yet only in a single case [87]. Increased levels of HMGB1 and S100B, known biomarkers for slow-onset neurodegenerative states such as AD and PD [88, 89], are also documented after cardiac arrest [90–92]. In a model of post-ischemic brain damage, brain microinjection of HMGB1 increased the transcription levels of pro-inflammatory mediators [93]. This is consistent with brain injury in cardiac arrest rats being attenuated by blocking HMGB1 activity [91]. These associations of biomarkers and mediators in AD and PD are consistent with the therapeutic implications of recent detailed analyses of global inflammation in experimental stroke [94], intracerebral hemorrhage [95], and TBI [96]. The relevant biomarkers are also present in cardiac arrest survivors and animal models of this condition [83, 90, 97, 98].
DAMPs AND PAMPs THAT CAN CONTRIBUTE TO THE GRADUAL ONSET CEREBRAL SYNDROMES
From the above evidence AD and PD can, in essence, be viewed as infinitely less acute, but much more insidious, versions of the altered physiology and subsequent syndromes seen in survivors of stroke, TBI, or cardiac arrest. Most importantly in generating the syndromes that ensue, the DAMPs and PAMPs that instigate AD and PD, unnoticed in minimal quantities, demonstrate their effects slowly and gradually, implying their correspondingly gradual appearance. This is a far cry from the rapid rise, but non-persistence, seen with mtDNA and HMGB1, the DAMPs acutely released after trauma and acute hypoxia. Moreover, gradual onset DAMPs are typically persistent, and often act cumulatively. We use as examples a DAMP generator, lead (Pb), and air pollutant particles that function as DAMPs directly, and PAMPs arising from pathogens chronically activating TLRs on or in brain cells. As we have reviewed in a brain context previously [99], in practice both intracerebral DAMPs and PAMPs thereby chronically generate a cascade of excess proinflammatory cytokines, the functionally dominant one being TNF.
LEAD (Pb) AS A GRADUAL AND CUMULATIVE DAMP GENERATOR
Ingested lead (Pb), long appreciated to be toxic, has a half-life of decades in bone. Its mining, smelting, and presence in the human environment, mainly from old paint, has been recognized for nearly 50 years to lead to an AD-like syndrome [100]. In our view it deserves a wider press in the neurodegenerative disease literature than it currently receives. The harmful effects of Pb on intelligence, learning, memory, executive function, processing speed, language, visuospatial skills and affect, glutamate release, long-term potentiation and synaptic plasticity, increased generation of AβPP, and increased Aβ deposition [101–103] are now documented in some detail. So too are Pb-induced α-Syn accumulation [104] and increased tau phosphorylation [104–106]. Pb has also been linked to increased TNF generation, including by microglia [107–109]. As we have previously summarized [110], the DNA hypomethylation literature on Pb toxicity documents how this metal generates DAMP activity. Epigenetic changes in the gene for Dnmt3a2, a DNA methyltransferase, occur in the hippocampus of young animals exposed to lead [111, 112], rationalizing the epigenetic changes seen in the aging primate brain developing AD after exposure to Pb when young [113]. The key concept here is that hypomethylated DNA, whether in lead-poisoned brains or innately in bacterial DNA [114] or mtDNA [115], is a DAMP, i.e., an agonist for TLR9, which generates a TNF-initiated proinflammatory cytokine cascade. When generated by lead poisoning [111, 112, 116], it does so cumulatively and chronically because of the long half-life of this element in bone, as noted earlier. Tellingly, chronic Pb toxicity is also associated with PD [117–119].
AIR POLLUTION PARTICLES AS GRADUAL AND CUMULATIVE DAMPs
An interest in the harmful effects of air pollution on health began some 90 years ago, with a focus on excess production of cytokines such as TNF for the past 20 years, with tobacco smoke and airborne bacteria initially falling under suspicion. Curiously, actual studies of small particles in this context began with work on fine material worn from hip protheses harming joint tissue by causing macrophages to generate this cytokine [120]. By this time air pollution had already been linked, in a histological study comparing street dogs in Mexico City with controls from less polluted locations, with AD-like changes in the brain [121]. This was extended to human tissues in 2008 [122], and has been much refined since by the same authors [123]. Increases in downstream DAMPs such as Aβ and α-Syn have been documented in these studies. Others have focused on the effects on brain histology and function through studying diesel exhaust particles specifically [124] and air pollution in general [125] on cognitive performance. Notably, air-borne fine particulate matter has been correlated with incidence of dementia [126].
PAMPs RELEASED FROM QUITE DIFFERENT INFECTIOUS ORGANISMS CONTRIBUTING TO GENERATING THE SAME DISEASE PROCESSES
The concept that different triggers from widely different origins can generate essentially identical syndromes is not at all new. For example, LPS, much later shown to be the archetypal TLR4 agonist (and indeed the original TNF inducer [127]), had, as we reviewed [128], been reasoned in the 1940s to generate an acute human syndrome indistinguishable from Plasmodium falciparum malaria. Nowadays this clinical mimicry can be rationalized as different PAMPs generating excessive output of the same inflammatory cytokines. For example, this protozoan produces a PAMP, an agonist of TLR9 [129], that, in a more recent blinded study, is agreed to induce a disease virtually indistinguishable from HINI pandemic influenza [130]. This is despite PAMPs on this virus being quite unrelated to those of malaria and activating PRRs that are quite different from TLRs.
This principle also applies to the chronic neurodegenerative state, AD, for which a polymicrobial infection concept has been described in detail [131]. The possibility of an infectious etiology for AD has repeatedly been proposed for decades, as reviewed in 2014 [132] and 2016 [133] editorials. Constructing a model has been a challenge, with competing cases having been made for chronic cerebral infections with viruses, bacteria, spirochetes, chlamydias, fungi, and periodontal disease initiated by any pathogen. Notably, within the bacterial perspective, LPS, clearly not a component of viruses, has recently been considered in detail across the pathogenesis of neurodegenerative diseases in general [134]. Its capacity to induce TNF is mentioned, but unfortunately the pivotal roles of this cytokine, and the importance of keeping it in homeostasis in these conditions, were omitted. Nevertheless, an earlier report that etanercept attenuates such changes [135] leaves us with negligible doubt that the harmful effects of effects of LPS on brain are mediated by TNF.
Perhaps the strongest single-organism argument has been made for Herpes Simplex Virus 1 infection, with recently reviewed [136] population-based evidence for a causal link to AD [137]. Studies on Helicobacter pylori [138], including the reported beneficial effects of its eradication on five year survival in AD [139], provide a parallel example. As acknowledged in most of the current literature, however, negligible evidence exists that a particular pathogen, or indeed any, is necessarily required for AD to develop. Nor, so far as we are aware, has anyone presenting the virus argument for an infectious origin for AD done so in terms of the PAMPs concept, or considered the possibility of the presence of chronic excessive DAMPs, or a mixture of the two, generating the same outcome. Nevertheless, a logic emerges that certain infectious agents can act alone, in that specifically treating chronic cerebral infections where possible has the potential to reduce PAMP load, and therefore dementia incidence, in certain populations [137]. Even so, persistent activities of DAMPs and PAMPs acting simultaneously are easily overlooked. Consider the cumulative influences of living in a historic Pb mining or smelting district with thick diesel-fueled traffic and having a chronic dormant cerebral viral infection of which one is unaware. In some individuals, any one of these influences, alone, in adequate amounts, may be sufficient to induce enough neurodegeneration to produce functional change. Plausibly a critical mass of persistent DAMP(s) and often PAMP(s), particularly when influenced by a known genetic predisposition [123], may well, once the total TLR agonist load reaches a critical level, be the most frequent way that AD and PD are induced. From first principles, such a mass of persistent TLR agonist from a chronic subclinical cerebral infection could be predicted to exacerbate the influence of a given degree of hypoxia in stroke, TBI, or cardiac arrest.
EVIDENCE FOR HARMFUL EFFECT OF EXCESS TNF ON THE LC
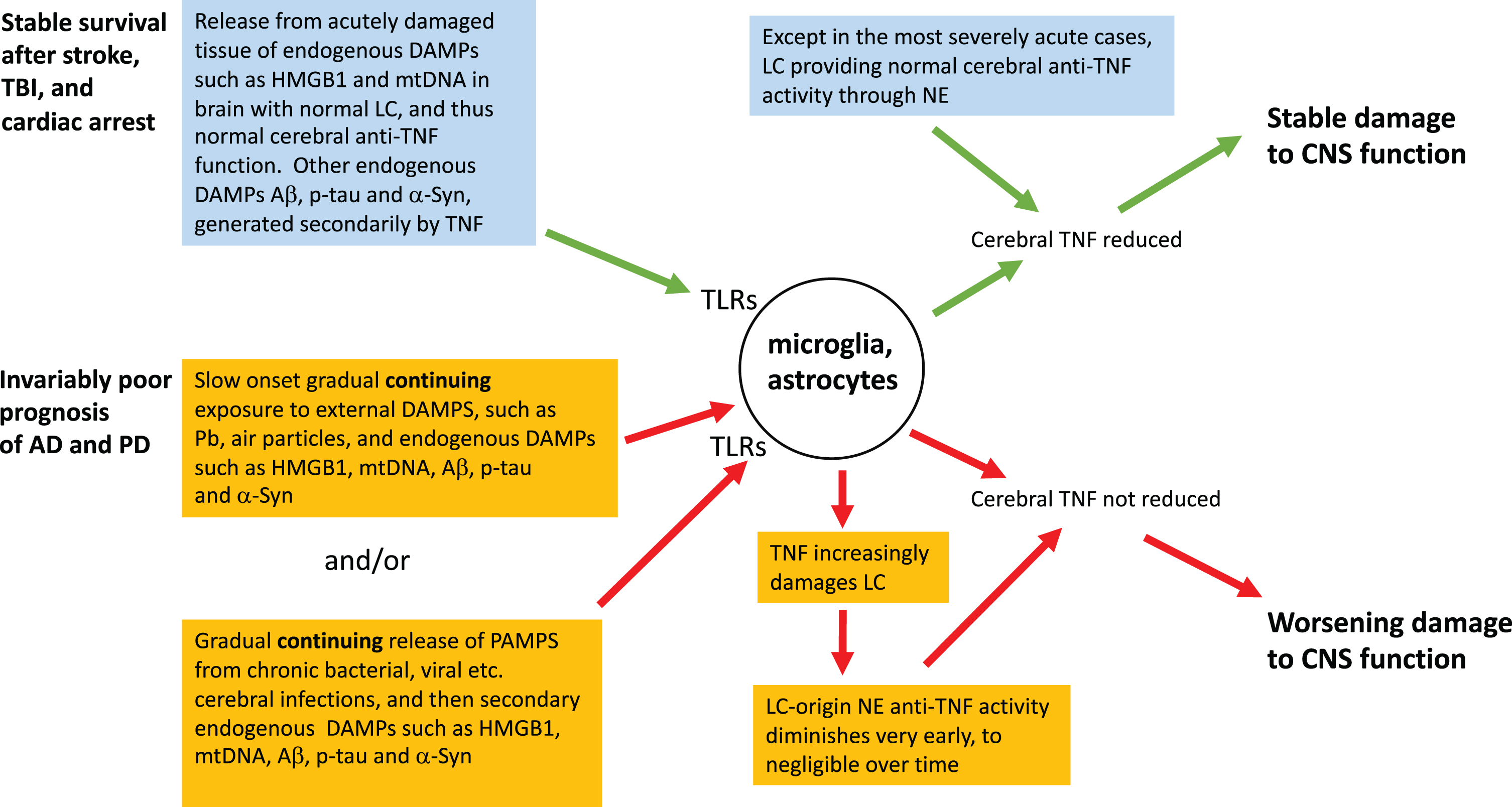
In simple TLR agonist terms, the fundamental difference between the abrupt-onset trio and AD or PD is the rate of production, dose size, and persistence of a patient’s cerebral exposure to DAMPs and PAMPs (Fig. 1). Arising from the abrupt onset of cataclysmic tissue damage and hypoxia, the immediate triggers for stroke, TBI, and the cerebral consequences of cardiac arrest are striking, and typically unprecedented in the prior experience of the individual. DAMPs, particularly mtDNA and HMGB1, are acutely released in survivors’ brains, and a locally severe cytokine storm establishes a new homeostatic baseline associated with chronically activated microglia, and consequently altered clinical function, in survivors. Nevertheless, mtDNA release and hypoxia, and therefore the secondary DAMPs they have induced, are long gone. Thus therapeutically deactivating the microglia by removing the excess TNF they are generating [140] can be expected to produce long-lasting clinical improvement [20, 21], as discussed earlier. This static state can be expected to be established post stroke, post TBI and post cardiac arrest, but not in AD or PD, for reasons that include the abrupt-onset trio brain usually having a healthy LC (Fig. 1). Thus, as documented and expanded upon in the next section, adequate NE, the endogenous cerebral anti-TNF agent, is available. In contrast, in the severe abrupt-onset trio, such as in TBI patients who neither regain consciousness nor survive for long, the LC can be severely damaged, presumably by hypoxia and/or damage-induced TNF released soon after the acute event [141]. In milder cases, in contrast, NE turnover, implying release, occurs [142]. Healthy LCs, implying successful endogenous anti-TNF activity, have also been documented in those who lived out their normal lifespans with mild cases of chronic traumatic encephalopathy [143]. In short, survivors of this acute-onset trio, for whom the remorseless clinical progression seen in AD and PD is absent, usually retain or even improve their LC activity, which generates NE, with its endogenous cerebral anti-TNF activity. The growth of a pathogen being a much slower event than trauma or acute hypoxia, PAMP involvement is clearly precluded from contributing to the abrupt-onset syndromes.
Fig.1
A neuroinflammatory perspective of the mechanism of onset and survival in abrupt-onset compared to gradual-onset non-infectious neurodegenerative states. Contrast in rate and persistence of TLR ligands, and the health of LC, including its release of NA, the endogenous inhibitor of cerebral TNF production, are taken into account.
At AD onset, arguably pre-clinically, the isodendritic core—a phylogenetically conserved subcortical brain region that includes the LC in the pons, the dorsal raphe nucleus, and the substantia nigra (SN) in the midbrain—is prone to accumulate neurofibrillary lesions comprising p-tau [144, 145]. Most work focusses on the LC, the main NE-generating region of the brain, the loss of which has been studied in detail in the context of preclinical, and early clinical, AD [146, 147]. The functional complexity of the LC is currently under close examination and revision [148]. A decade earlier it had been demonstrated that activated microglia and the microglial-derived proinflammatory cytokine TNF can induce accumulation of the aggregation-prone tau molecules at this site [71]. Moreover minocyline, which reduces proinflammatory cytokines, also reduces p-tau [149]. The same has been reported of infliximab, the first of the anti-TNF biologicals [18]. Since, as outlined in the next paragraph, LC neurons generate most of the cerebral NE, one of the early harmful effects of chronically increased cerebral TNF is likely to be to deprive the brain of its endogenous anti-TNF agent, to the further detriment of the host. Should, on the other hand, the initial jolt of TNF in the sudden-onset neurodegenerative states, such as stroke, be severe enough, a fatal outcome is more likely the more the LC is severely damaged from the onset, partially removing the endogenous brake on cerebral TNF generation [141]. As noted above, the opposite effect has been noted in milder TBI, with compensatory NE release being increased [142].
THE CEREBRAL ANTI-TNF ROLE OF NE FROM THE LC
Why do abruptly-acquired neurodegenerative states not exhibit the remorseless clinical progression seen in those with gradual onsets? Now that Aβ has been relegated from dominating the mechanistic aspect of AD, instead being appreciated simply as one of a team of similarly acting DAMPs [99, 150], the harmfulness of excess cerebral levels of proinflammatory cytokines, in particular TNF, is rapidly gaining traction as an important mechanism of neurodegenerative disease. Five years before Aβ had its debut in the AD literature, it had already been shown that the same basal areas of the brain of AD patients contained subnormal levels of NE [151], and that degeneration of LC neurons, from whence cerebral NE largely arises, was responsible [152]. It subsequently became appreciated, initially in vitro [153, 154], as well as in vivo in the brain [155, 156], that this catecholamine is an endogenous anti-TNF agent. Subsequently it was reported that this inhibition of TNF production by NE operates, in vivo, by enhancing levels of the anti-TNF cytokine, IL-10 [157]. It follows, therefore, that modifying the sensitivity of the β-adrenergic receptor would modify TNF production, as indeed was demonstrated in 1995 [158]. Since then this has been shown for a number of synthetic agonists, isoproterenol [159], salmeterol [160], and salbutamol [161, 162]. The implications of these observations are expanded upon in the section of this text dealing with therapy. It suffices here to note that it took some years for the physiopathological implications of these observations to be understood, since awareness of the effects of TNF on brain function, barely in its infancy [163], was little-appreciated. Seven years on, a well-constructed physiological interpretation on the implications of this interaction between NE and TNF on phenomena such as homeostatic synaptic scaling had been generated [164]. So too had a series of publications on the implication of chronically reduced noradrenergic signaling through LC damage on the consequences of worsening neuroinflammatory change for AD [165–167].
The same broad principle evidently applies to PD, TNF having been reported for over 15 years to have four times the level in cerebrospinal fluid and the dopaminergic region of the brain than found in control patients [168]. As discussed earlier, a model of this condition is ameliorated by a novel double negative anti-TNF agent [19] and a large population-based study reports a much lower PD incidence in a clinical population treated with a specific anti-TNF biological for another purpose [26]. A cerebral deficiency of NE is also implicated in PD [169]. Moreover, the LC is regarded, as well being as a major NE source, as one of the main orchestrators of the other major monoaminergic nuclei, such as the SN, from whence dopamine neurons are lost in PD [170]. Notably, the degree of disease-related neuronal loss in this region has been reported to be more severe in the LC than the SN in both AD and PD, with the SN being less damaged in AD [171]. The implications of early inflammatory change in this brain region for understanding the clinical similarities (executive function loss, sleep disorders, neurogenic pain, fatigue) and differences between AD and the non-motor aspects of PD will clearly encourage further consideration of this aspect of its neuropathophysiology.
This avenue of enquiry is consistent with both of these slow onset states originating from excess chronic cerebral TNF, as we have referred to earlier in this text from fields from the TREM2, APOE4, specific anti-TNF agent, and Pb toxicity literatures. Targeting neuronal deficiency in the LC is under active consideration in both AD and PD [170, 172]. For instance, reduced cerebrospinal fluid levels of 3-methoxy-4-hydroxyphenylethyleneglycol (MHPG), a metabolite of NA, have recently been utilized to link certain clinical aspects of AD with loss of NA-generating neurons in the LC [173]. The practicality of LC imaging across the range of neurodegenerative diseases has recently been discussed in detail [174]. This comprehensive recent text covers this aspect of a number of the slow-onset neurodegenerative states, including PD. Clearly, through the reduced LC activity early in AD and PD, and thus loss of cerebral NE, the endogenous cerebral anti-TNF activity of this catecholamine is now well recognized. As is predictable from this reasoning, LC integrity has recently been associated with better memory performance in older adults [175]. Conceptually, this is consistent with the uncontrolled and self-amplifying inflammatory processes that Gao and Hong proposed in their prescient 2003 and 2008 texts [30, 176], and the outcome of Tobinick’s open trial of perispinal etanercept for AD in 2006 [23].
LC’S ROLE IN TNF BALANCE AT CLINICAL ONSET OF THE GRADUAL NEURODEGENERATIVE STATES
Typically, LC loss, and thus endogenous anti-TNF activity depletion, is already underway when clinical onset of the gradual neurodegenerative states discussed in this text is evident. There is, for example, evidence for a 30% neuronal loss before AD patients manifest even mild cognitive impairment [146]. These authors significantly associated the degree of LC neuronal loss with the ebbing of a number of cognitive skills. By this time others had quantified the local accretion of p-tau [177] and documented increases in expression of IL-1β, IL-6, and TNF genes [178] in the LC in early stage AD patients. Clearly, this implies the consequences of PAMPs and DAMPs acting on TLRs, generating excessive TNF and related cytokines in the brain, including the LC. Recently, the complexities of this chronic activation for the subtleties of brain function, resulting in enduring shifts in the homeostatic baseline, with long-lasting consequences for cerebral function and behavior, have been admirably discussed [179]. So too has the concept of the LC now being appreciated to serve a surprisingly wide array of cerebral functions [148]. This is consistent with the now well-appreciated pleiotropic cerebral functions of TNF being significantly controlled by the LC. Notably, TNF appears to be incriminated in damage within the LC in early stage AD [178]. In addition, the role of the gut microbiome in the development of AD and PD is nowadays expressed in terms of overstimulation of innate immunity by PAMPs of bacterial origin [180, 181].
Given the central roles of TNF in these events, it seems logical that a full complement of fit LC neurons, which generate and distribute NE, an inhibitor of this cytokine [153, 154, 156], is a key determinant of whether certain neurogenerative diseases remain static or worsen. Nevertheless, the literature on survivors of the abrupt-onset trio (stroke, TBI and cardiac arrest) entails essentially the same range of cells, soluble mediators, and other biomarkers, and therefore the same basic mechanism of pathogenesis, as do publications on the gradual onset syndromes such as AD and PD. Specific anti-TNF therapies are therefore logical for each group and are indeed developing.
SYNTHESIS
The patterns we describe in this article are consistent with the proposal that all the neurodegenerative syndromes discussed here are offspring of the loss of cerebral homeostasis, brought about by the brain’s basic response to an insult, whether acute or chronic. This varies much in degree, but little in principle. We discuss two distinct categories. One group has a dramatically sudden onset of high fluxes of DAMPs, released acutely and non-cumulatively into a brain with an intact LC, complete with its NE-mediated anti-TNF activity [153, 154, 156]. This, we suggest, explains the abrupt-onset syndrome that typically does not chronically worsen, yet, as observed post-stroke, post-TBI, and post cardiac arrest, does persist. Self-perpetuating autocrine microglial activation then ensues, and this can be expected to be removed through anti-TNF therapy, administered such that it gets to where it is needed [21]. The second group, syndromes with an imperceptibly slow onset but remorselessly increasing severity, as characterized by AD, are essentially a response to long term, slow onset but persistent, fluxes of DAMPs and/or PAMPs that can act in concert, although conceivably at times alone. This is in contrast to all except the most severe of the acute onset conditions, in which the LC quickly collapses, and decline ensues through abrupt loss of endogenous anti-TNF capacity. Importantly, the net capacity of these DAMPs and PAMPs to generate TNF in this second group, which includes AD, becomes increasing effective though early and worsening LC damage—plausibly initiated by the TNF that these DAMPs and PAMPs induce—rendering the endogenous anti-TNF activity of cerebral NE progressively powerless. Accordingly, functional and structural damage worsens, with an associated worsening clinical state. This said, there is always the possibility that predispositions for these two classes of neurological diseases are yet to be unearthed. Presently unsuspected classes of change, in, for example, the affinity of adrenergic receptors, is one possibility.
In summary, we propose that the three typically non-worsening neurodegenerative syndromes under discussion, post-stroke, post-TBI, and post cardiac arrest, eventually become and remain static because of excess cerebral TNF induced by the initial dramatic peak keeping microglia chronically activated through the autocrine loop discussed earlier [140]. Nevertheless, possession of an intact LC, and therefore an intact NE-mediated endogenous anti-cerebral TNF system, combined with the DAMP input having essentially stopped, allows these patients a strong long-term survival advantage over AD and PD sufferers. In contrast, AD and PD patients remorselessly worsen, being handicapped by sustained, albeit gradual, DAMP and PAMP inputs continuing to generate TNF, as well as loss of the LC-based endogenous anti-cerebral TNF system.
A SEA CHANGE IN VIEWS ON REVERSIBILITY OF LOSS OF BRAIN FUNCTION?
Clearly, in extreme stroke, TBI, or cardiac arrest the degree and duration of hypoxia can be fatal to cells as well as the patient, or cause permanent loss of cerebral function in survivors. Unfortunately, this concept seems to have been extended to become the logic behind a concept that remains a standard tenet of neurology: that if functional loss from acquired brain injury, be it stroke, TBI, or cardiac arrest, is still present some six months after the event, it is permanent because of cerebrocellular death from hypoxia, the histological details of the particular post-stroke brain reflecting its pattern of irreversible damage. Thus treatment would be fruitless. However, perispinal etanercept reversing functional loss of post-stroke syndromes years after the event [20, 21] shows every sign of rendering this traditional stance untenable. Even so, the American Academy of Neurology still officially advises clinicians against this approach on what could be called specious grounds [182]. This prediction of a sea change was reinforced by improved outcomes in a Phase I/IIa trial in SB623 cell transfers into brains of 16 patients six months after their stroke [183]. This technique did not pass an initial random trial, but individual continued improvements were apparently undeniable (https://scopeblog.stanford.edu/2016/06/02/stroke-of-luck-stem-cell-transplants-show-strong-signs-of-efficacy-in-clinical-safety-trial-for-stroke/). The author noted, as Tobinick’s 2011 and 2012 case studies had implied [20, 21], that, contrary to dogma, many injured brains are not permanently damaged, but can recover function. The outcome of an independent random controlled trial of perispinal etanercept [33] early this year is surely on the way to settling the matter. In short, the novel argument is that excess hypoxia-induced TNF [184, 185] often merely makes neural circuits dormant by, for example, excessively altering synaptic scaling. Neutralizing this excess with anti-TNF evidently brings these circuits back online.
IMPLICATIONS FOR TREATMENT OF NEURODEGENERATIVE DISEASES
As we have recently reviewed [1], there is now much interest in ensuring that anti-TNF biologicals, limited as they are by their molecular size, are administered so they enter the brain, where they are needed. This route limitation does not, of course, apply with small anti-TNF molecules, such as the 3,61-dithiothalidomides [186]. Should outcomes of impending random controlled trials open interest in basic research in this area, the patterns of interactions between DAMPs, PAMPs, LC damage, and NE production described above will be useful to rationalize the dose and frequency of treatment with this class of agent across the spectrum of these conditions. It could also minimize LC damage. Moreover, the effects of compensating for any diminution of NE through administering a synthetic beta2 adrenergic receptor agonist, such as isoproterenol, salmeterol, and salbutamol, as discussed earlier, could be usefully explored here. More research is needed, but it is encouraging that two profoundly large scale surveys, focused respectively on analyzing for the effect on PD incidence of repeated subcutaneous anti-TNF biologicals [26] and inhaled salbutamol [187], found a significant reduction. Each agent had been administered, on a grand scale, for another purpose. One implication is that beta2 adrenergic receptor agonist and anti-TNF biologicals warrant joint investigation.
More treatment vistas open as we gain more understanding of how TNF and associated cytokines act in the normal and damaged brain. Indeed, a novel perspective has been brought about in post-stroke treatment possibilities through incorporating this understanding of brain TNF. In particular this applies to the capacity of cerebral DAMPs and PAMPs to cause self-perpetuating autocrine microglial activation. Importantly, and in contrast to the behavior of macrophages outside the brain [188], once a TLR agonist induces microglia to generate TNF, its production can continue through generating an autocrine activation of the TNF/TNFR signaling pathway [140] until the excess TNF is neutralized by introducing an anti-TNF agent. This self-perpetuating microglial loop has been confirmed by others, who incriminate C1q, a component of the complement pathway [189], brain-derived neurotropic factor [190], and N-glycosylation of TFNR1 [191]. These events combine to cause an enduring downwards shift in the homeostatic baseline state [179], manifesting, through lowered synaptic scaling under the influence of TNF [192], as aspects of well-recognized clinical entities.
IMPLICATIONS FOR LONG-TERM NEUROLOGICAL ASPECTS OF COVID-19
Fatigue, delirium, “brain fog”, poor cognition, and memory failure, all with a tendency to persist, are well-described in post-stroke syndromes. For some months now all have been appreciated to occur, and persist, in COVID-19, even without severe respiratory symptoms having occurred [193, 194]. Above all, finding an effective way to treat these aspects of COVID-19 requires establishing whether the causative virus itself, or the PAMP-initiated cytokines generated in response to its presence, actually brings them about [195, 196]. Regarding the examples of fatigue and delirium, one obvious approach to resolving this question is to consider whether these symptoms occur in the absence of cerebral pathogens such as SARS-Cov-2 but in the presence of DAMPs that chronically raise levels of these same cytokines. Post-stroke [197], post-TBI [198, 199], and post-surgery states [200] fit this pattern. If, in contrast, the pathogen itself is a direct cause of these neurological changes, we are required to accommodate how bacteria [201], and protozoa [202] and viruses [195], all very different organisms, generate these same changes to cerebral function in any way other than through the common array of increased inflammatory cytokines their PAMP activity generates. Most likely, therefore, perispinal delivery of etanercept, as used to treat post-stroke syndromes [4, 20, 21, 33] is presently the most plausible treatment to consider. This is surely an uncontroversial proposal, since other specific anti-TNF biologicals are, or are about to be, trialed for the systemic aspects of COVID-19 [203], (http://www.chictr.org.cn/showprojen.aspx?proj=49889). Moreover, the widespread availability of these agents as biosimilars is a distinct advantage in these straightened times.
ACKNOWLEDGMENTS
The authors have no conflicts of interest that are relevant to the content of this article.
Funding from the Boyarski family is gratefully acknowledged.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/20-1186r1).
REFERENCES
[1] | Clark IA , Vissel B ((2019) ) Neurodegenerative disease treatments by direct TNF reduction, SB623 cells, maraviroc and irisin and MCC950, from an inflammatory perspective - a Commentary. Expert Rev Neurother 19: , 535–543. |
[2] | Tobinick EL ((2016) ) Perispinal delivery of CNS drugs. CNS Drugs 30: , 469–480. |
[3] | LaMacchia ZM , Spengler RN , Jaffari M , Abidi AH , Ahmed T , Singh N , Tobinick EL , Ignatowski TA ((2019) ) Perispinal injection of a TNF blocker directed to the brain of rats alleviates the sensory and affective components of chronic constriction injury-induced neuropathic pain. Brain Behav Immun 82: , 93–105. |
[4] | Clark IA ((2020) ) Randomized controlled trial validating the use of perispinal etanercept to reduce post-stroke disability has wide-ranging implications. Expert Rev Neurother 20: , 203–205. |
[5] | Dams-O’Connor K , Gibbons LE , Bowen JD , McCurry SM , Larson EB , Crane PK ((2013) ) Risk for late-life re-injury, dementia and death among individuals with traumatic brain injury: A population-based study. J Neurol Neurosurg Psychiatry 84: , 177–182. |
[6] | Djordjevic J , Sabbir MG , Albensi BC ((2016) ) Traumatic brain injury as a risk factor for Alzheimer’s disease: Is inflammatory signaling a key player? Curr Alzheimer Res 13: , 730–738. |
[7] | Perry DC , Sturm VE , Peterson MJ , Pieper CF , Bullock T , Boeve BF , Miller BL , Guskiewicz KM , Berger MS , Kramer JH , Welsh-Bohmer KA ((2016) ) Association of traumatic brain injury with subsequent neurological and psychiatric disease: A meta-analysis. J Neurosurg 124: , 511–526. |
[8] | Crane PK , Gibbons LE , Dams-O’Connor K , Trittschuh E , Leverenz JB , Keene CD , Sonnen J , Montine TJ , Bennett DA , Leurgans S , Schneider JA , Larson EB ((2016) ) Association of traumatic brain injury with late-life neurodegenerative conditions and neuropathologic findings. JAMA Neurol 73: , 1062–1069. |
[9] | Gardner RC , Byers AL , Barnes DE , Li Y , Boscardin J , Yaffe K ((2018) ) Mild TBI and risk of Parkinson disease: A chronic effects of neurotrauma consortium study. Neurology 90: , e1771–e1779. |
[10] | Hachinski V ((2018) ) The convergence of stroke and dementia. Arq Neuropsiquiatr 76: , 849–852. |
[11] | Huang YF , Yeh CC , Chou YC , Hu CJ , Cherng YG , Shih CC , Chen TL , Liao CC ((2019) ) Stroke in Parkinson’s disease. QJM 112: , 269–274. |
[12] | Iverson GL , Keene CD , Perry G , Castellani RJ ((2018) ) The need to separate chronic traumatic encephalopathy neuropathology from clinical features. J Alzheimers Dis 61: , 17–28. |
[13] | LoBue C , Munro C , Schaffert J , Didehbani N , Hart J , Batjer H , Cullum CM ((2019) ) Traumatic brain injury and risk of long-term brain changes, accumulation of pathological markers, and developing dementia: A review. J Alzheimers Dis 70: , 629–654. |
[14] | Pluta R , Ulamek-Koziol M , Januszewski S , Czuczwar SJ ((2017) ) Dysregulation of Alzheimer’s disease-related genes and proteins following cardiac arrest. Folia Neuropathol 55: , 283–288. |
[15] | Clausen B , Degn M , Martin N , Couch Y , Karimi L , Ormhoj M , Mortensen ML , Gredal H , Gardiner C , Sargent II , Szymkowski DE , Petit GH , Deierborg T , Finsen B , Anthony D , Lambertsen K ((2014) ) Systemically administered anti-TNF therapy ameliorates functional outcomes after focal cerebral ischemia. J Neuroinflammation 11: , 203. |
[16] | Bonetti NR , Diaz-Canestro C , Liberale L , Crucet M , Akhmedov A , Merlini M , Reiner MF , Gobbato S , Stivala S , Kollias G , Ruschitzka F , Luscher TF , Beer JH , Camici GG ((2019) ) Tumour necrosis factor-alpha inhibition improves stroke outcome in a mouse model of rheumatoid arthritis. Sci Rep 9: , 2173. |
[17] | Chio CC , Chang CH , Wang CC , Cheong CU , Chao CM , Cheng BC , Yang CZ , Chang CP ((2013) ) Etanercept attenuates traumatic brain injury in rats by reducing early microglial expression of tumor necrosis factor-alpha. BMC Neurosci 14: , 33. |
[18] | Shi JQ , Shen W , Chen J , Wang BR , Zhong LL , Zhu YW , Zhu HQ , Zhang QQ , Zhang YD , Xu J ((2011) ) Anti-TNF-alpha reduces amyloid plaques and tau phosphorylation and induces CD11c-positive dendritic-like cell in the APP/PS1 transgenic mouse brains. Brain Res 1368: , 239–247. |
[19] | Barnum CJ , Chen X , Chung J , Chang J , Williams M , Grigoryan N , Tesi RJ , Tansey MG ((2014) ) Peripheral administration of the selective inhibitor of soluble tumor necrosis factor (TNF) XPro(R)1595 attenuates nigral cell loss and glial activation in 6-OHDA hemiparkinsonian rats. J Parkinsons Dis 4: , 349–360. |
[20] | Tobinick E ((2011) ) Rapid improvement of chronic stroke deficits after perispinal etanercept: Three consecutive cases. CNS Drugs 25: , 145–155. |
[21] | Tobinick E , Kim NM , Reyzin G , Rodriguez-Romanacce H , DePuy V ((2012) ) Selective TNF inhibition for chronic stroke and traumatic brain injury: An observational study involving 629 consecutive patients treated with perispinal etanercept. CNS Drugs 26: , 1051–1070. |
[22] | Tobinick E , Rodriguez-Romanacce H , Levine A , Ignatowski TA , Spengler RN ((2014) ) Immediate neurological recovery following perispinal etanercept years after brain injury. Clin Drug Investig 34: , 361–366. |
[23] | Tobinick EL , Gross H , Weinberger A , Cohen H ((2006) ) TNF-alpha modulation for treatment of Alzheimer’s disease: A 6- month pilot study. MedGenMed 8: , 25. |
[24] | Chen YM , Chen HH , Lan JL , Chen DY ((2010) ) Improvement of cognition, a potential benefit of anti-TNF therapy in elderly patients with rheumatoid arthritis. Joint Bone Spine 77: , 366–367. |
[25] | Chou RC , Kane M , Ghimire S , Gautam S , Gui J ((2016) ) Treatment for rheumatoid arthritis and risk of Alzheimer’s disease: A nested case-control analysis. CNS Drugs 30: , 1111–1120. |
[26] | Peter I , Dubinsky M , Bressman S , Park A , Changyue L , Chen N , Wang A ((2018) ) Anti–tumor necrosis factor therapy and incidence of Parkinson disease among patients with inflammatory bowel disease. JAMA Neurol 75: , 939–946. |
[27] | Simpkins JW , Gatson JW , Wigginton JG ((2009) ) Commentary on “a roadmap for the prevention of dementia II. Leon Thal Symposium 2008.” Rationale and recommendations for first evaluating anti-Alzheimer’s disease medications in acute brain injury patients. Alzheimers Dement 5: , 143–146. |
[28] | Becker RE , Kapogiannis D , Greig NH ((2018) ) Does traumatic brain injury hold the key to the Alzheimer’s disease puzzle? Alzheimers Dement 14: , 431–443. |
[29] | Minniti CJ ((2015) ) Sandoz v. Amgen: Why current interpretation of the Biologic Price Competition and Innovation Act of 2009 is flawed and jeopardizes future competition. J Pat Trademark Off Soc 97: , 172–190. |
[30] | Gao HM , Hong JS ((2008) ) Why neurodegenerative diseases are progressive: Uncontrolled inflammation drives disease progression. Trends Immunol 29: , 357–365. |
[31] | Clark IA , Alleva LM , Vissel B ((2010) ) The roles of TNF in brain dysfunction and disease. Pharmacol Ther 128: , 519–548. |
[32] | Schett G , Elewaut D , McInnes IB , Dayer JM , Neurath MF ((2013) ) How cytokine networks fuel inflammation: Toward a cytokine-based disease taxonomy. Nat Med 19: , 822–824. |
[33] | Ralph SJ , Weissenberger A , Bonev V , King LD , Bonham MD , Ferguson S , Smith AD , Goodman-Jones AA , Espinet AJ ((2020) ) Phase I/II parallel double-blind randomized controlled clinical trial of perispinal etanercept for chronic stroke: Improved mobility and pain alleviation. Expert Opin Investig Drugs 29: , 311–326. |
[34] | Gordon R , Albornoz EA , Christie DC , Langley MR , Kumar V , Mantovani S , Robertson AAB , Butler MS , Rowe DB , O’Neill LA , Kanthasamy AG , Schroder K , Cooper MA , Woodruff TM ((2018) ) Inflammasome inhibition prevents alpha-synuclein pathology and dopaminergic neurodegeneration in mice. Sci Transl Med 10: , 1–12. |
[35] | Joy MT , Ben Assayag E , Shabashov-Stone D , Liraz-Zaltsman S , Mazzitelli J , Arenas M , Abduljawad N , Kliper E , Korczyn AD , Thareja NS , Kesner EL , Zhou M , Huang S , Silva TK , Katz N , Bornstein NM , Silva AJ , Shohami E , Carmichael ST ((2019) ) CCR5 is a therapeutic target for recovery after stroke and traumatic brain injury. Cell 176: , 1143–1157.e1113. |
[36] | Low PC , Manzanero S , Mohannak N , Narayana VK , Nguyen TH , Kvaskoff D , Brennan FH , Ruitenberg MJ , Gelderblom M , Magnus T , Kim HA , Broughton BR , Sobey CG , Vanhaesebroeck B , Stow JL , Arumugam TV , Meunier FA ((2014) ) PI3Kdelta inhibition reduces TNF secretion and neuroinflammation in a mouse cerebral stroke model. Nat Commun 5: , 3450. |
[37] | Martinez-Marmol R , Mohannak N , Qian L , Wang T , Gormal RS , Ruitenberg MJ , Vanhaesebroeck B , Coulson EJ , Meunier FA ((2019) ) p110delta PI3-Kinase inhibition perturbs APP and TNFalpha trafficking, reduces plaque burden, dampens neuroinflammation, and prevents cognitive decline in an Alzheimer’s disease mouse model. J Neurosci 39: , 7976–7991. |
[38] | Low PC , Misaki R , Schroder K , Stanley AC , Sweet MJ , Teasdale RD , Vanhaesebroeck B , Meunier FA , Taguchi T , Stow JL ((2010) ) Phosphoinositide 3-kinase delta regulates membrane fission of Golgi carriers for selective cytokine secretion. J Cell Biol 190: , 1053–1065. |
[39] | Lannutti BJ , Meadows SA , Herman SE , Kashishian A , Steiner B , Johnson AJ , Byrd JC , Tyner JW , Loriaux MM , Deininger M , Druker BJ , Puri KD , Ulrich RG , Giese NA ((2011) ) CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood 117: , 591–594. |
[40] | Singh A , Jones OD , Mockett BG , Ohline SM , Abraham WC ((2019) ) Tumor necrosis factor-alpha-mediated metaplastic inhibition of LTP Is constitutively engaged in an Alzheimer’s disease model. J Neurosci 39: , 9083–9097. |
[41] | Griciuc A , Patel S , Federico AN , Choi SH , Innes BJ , Oram MK , Cereghetti G , McGinty D , Anselmo A , Sadreyev RI , Hickman SE , El Khoury J , Colonna M , Tanzi RE ((2019) ) TREM2 acts downstream of CD33 in modulating microglial pathology in Alzheimer’s disease. Neuron 103: , 820–835.e7. |
[42] | Wu R , Li X , Xu P , Huang L , Cheng J , Huang X , Jiang J , Wu LJ , Tang Y ((2017) ) TREM2 protects against cerebral ischemia/reperfusion injury. Mol Brain 10: , 20. |
[43] | Rajan KB , Aggarwal NT , Schneider JA , Wilson RS , Everson-Rose SA , Evans DA ((2016) ) Role of APOE epsilon4 allele and incident stroke on cognitive decline and mortality. Alzheimer Dis Assoc Disord 30: , 318–323. |
[44] | Merritt VC , Clark AL , Sorg SF , Evangelista ND , Werhane ML , Bondi MW , Schiehser DM , Delano-Wood L ((2018) ) Apolipoprotein E (APOE) epsilon4 genotype is associated with reduced neuropsychological performance in military veterans with a history of mild traumatic brain injury. J Clin Exp Neuropsychol 40: , 1050–1061. |
[45] | Mahley RW , Weisgraber KH , Huang Y ((2006) ) Apolipoprotein E4: A causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc Natl Acad Sci U S A 103: , 5644–5651. |
[46] | Pankratz N , Byder L , Halter C , Rudolph A , Shults CW , Conneally PM , Foroud T , Nichols WC ((2006) ) Presence of an APOE4 allele results in significantly earlier onset of Parkinson’s disease and a higher risk with dementia. Mov Disord 21: , 45–49. |
[47] | Gale SC , Gao L , Mikacenic C , Coyle SM , Rafaels N , Murray Dudenkov T , Madenspacher JH , Draper DW , Ge W , Aloor JJ , Azzam KM , Lai L , Blackshear PJ , Calvano SE , Barnes KC , Lowry SF , Corbett S , Wurfel MM , Fessler MB ((2014) ) APOepsilon4 is associated with enhanced in vivo innate immune responses in human subjects. J Allergy Clin Immunol 134: , 127–134. |
[48] | Franchi L , Eigenbrod T , Nunez G ((2009) ) Cutting edge: TNF-alpha mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J Immunol 183: , 792–796. |
[49] | Oppenheim JJ , Tewary P , de la Rosa G , Yang D ((2007) ) Alarmins initiate host defense. Adv Exp Med Biol 601: , 185–194. |
[50] | Janeway CA Jr. ((1989) ) Pillars article: Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol 54: , 1–13. |
[51] | Clark IA , Virelizier J-L , Carswell EA , Wood PR ((1981) ) Possible importance of macrophage-derived mediators in acute malaria. Infect Immun 32: , 1058–1066. |
[52] | Clark IA ((2007) ) How TNF was recognized to be a key mechanism of disease. Cytokine Growth Factor Rev 18: , 335–343. |
[53] | Matzinger P ((1994) ) Tolerance, danger, and the extended family. Annu Rev Immunol 12: , 991–1045. |
[54] | Gallucci S , Matzinger P ((2001) ) Danger signals: SOS to the immune system. Curr Opin Immunol 13: , 114–119. |
[55] | Matzinger P ((2002) ) The danger model: A renewed sense of self. Science 296: , 301–305. |
[56] | Poltorak A , He X , Smirnova I , Liu MY , Van Huffel C , Du X , Birdwell D , Alejos E , Silva M , Galanos C , Freudenberg M , Ricciardi Castagnoli P , Layton B , Beutler B ((1998) ) Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in Tlr4 gene. Science 282: , 2085–2088. |
[57] | Zhang Q , Raoof M , Chen Y , Sumi Y , Sursal T , Junger W , Brohi K , Itagaki K , Hauser CJ ((2010) ) Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464: , 41–42. |
[58] | Rrapaj E , Trisolini E , Bertero L , Salvo M , Indellicato R , Andorno S , Garcia-Manteiga JM , Rena O , Boldorini RL ((2018) ) Expression analysis of HMGB1 in histological samples of malignant pleural mesothelioma. Histopathology 72: , 1039–1050. |
[59] | Schulze J , Zierath D , Tanzi P , Cain K , Shibata D , Dressel A , Becker K ((2013) ) Severe stroke induces long-lasting alterations of high-mobility group box 1. Stroke 44: , 246–248. |
[60] | Parker TM , Nguyen AH , Rabang JR , Patil AA , Agrawal DK ((2017) ) The danger zone: Systematic review of the role of HMGB1 danger signalling in traumatic brain injury. Brain Inj 31: , 2–8. |
[61] | Sugita A , Kinoshita K , Sakurai A , Chiba N , Yamaguchi J , Kuwana T , Sawada N , Hori S ((2017) ) Systemic impact on secondary brain aggravation due to ischemia/reperfusion injury in post-cardiac arrest syndrome: A prospective observational study using high-mobility group box 1 protein. Crit Care 21: , 247. |
[62] | Ge YW , Lahiri DK ((2002) ) Regulation of promoter activity of the APP gene by cytokines and growth factors: Implications in Alzheimer’s disease. Ann N Y Acad Sci 973: , 463–467. |
[63] | Zhao A , Li Y , Deng Y ((2020) ) TNF receptors are associated with tau pathology and conversion to Alzheimer’s dementia in subjects with mild cognitive impairment. Neurosci Lett 738: , 135392. |
[64] | Whiten DR , Brownjohn PW , Moore S , De S , Strano A , Zuo Y , Haneklaus M , Klenerman D , Livesey FJ ((2020) ) Tumour necrosis factor induces increased production of extracellular amyloid-β- and α-synuclein-containing aggregates by human Alzheimer’s disease neurons. Brain Commun 2: , fcaa146. |
[65] | Cui G , Wang H , Li R , Zhang L , Li Z , Wang Y , Hui R , Ding H , Wang DW ((2012) ) Polymorphism of tumor necrosis factor alpha (TNF-alpha) gene promoter, circulating TNF-alpha level, and cardiovascular risk factor for ischemic stroke. J Neuroinflammation 9: , 235. |
[66] | Taupin V , Toulmond S , Serrano A , Benavides J , Zavala F ((1993) ) Increase in IL-6, IL-1 and TNF levels in rat brain following traumatic lesion. Influence of pre- and post-traumatic treatment with Ro5 4864, a peripheral-type (p site) benzodiazepine ligand. J Neuroimmunol 42: , 177–185. |
[67] | Salminen A , Ojala J , Kauppinen A , Kaarniranta K , Suuronen T ((2009) ) Inflammation in Alzheimer’s disease: Amyloid-beta oligomers trigger innate immunity defence via pattern recognition receptors. Prog Neurobiol 87: , 181–194. |
[68] | Cook C , Kang SS , Carlomagno Y , Lin WL , Yue M , Kurti A , Shinohara M , Jansen-West K , Perkerson E , Castanedes-Casey M , Rousseau L , Phillips V , Bu G , Dickson DW , Petrucelli L , Fryer JD ((2015) ) Tau deposition drives neuropathological, inflammatory and behavioral abnormalities independently of neuronal loss in a novel mouse model. Hum Mol Genet 24: , 6198–6212. |
[69] | Yang W , Yu S ((2017) ) Synucleinopathies: Common features and hippocampal manifestations. Cell Mol Life Sci 74: , 1485–1501. |
[70] | Sznejder-Pacholek A , Joniec-Maciejak I , Wawer A , Ciesielska A , Mirowska-Guzel D ((2017) ) The effect of alpha-synuclein on gliosis and IL-1alpha, TNFalpha, IFNgamma, TGFbeta expression in murine brain. Pharmacol Rep 69: , 242–251. |
[71] | Gorlovoy P , Larionov S , Pham TT , Neumann H ((2009) ) Accumulation of tau induced in neurites by microglial proinflammatory mediators. FASEB J 23: , 2502–2513. |
[72] | Ihle-Hansen H , Hagberg G , Fure B , Thommessen B , Fagerland MW , Oksengard AR , Engedal K , Selnes P ((2017) ) Association between total-Tau and brain atrophy one year after first-ever stroke. BMC Neurol 17: , 107. |
[73] | Kulbe JR , Hall ED ((2017) ) Chronic traumatic encephalopathy-integration of canonical traumatic brain injury secondary injury mechanisms with tau pathology. Prog Neurobiol 158: , 15–44. |
[74] | Kim T , Mehta SL , Kaimal B , Lyons K , Dempsey RJ , Vemuganti R ((2016) ) Poststroke induction of alpha-synuclein mediates ischemic brain damage. J Neurosci 36: , 7055–7065. |
[75] | Impellizzeri D , Campolo M , Bruschetta G , Crupi R , Cordaro M , Paterniti I , Cuzzocrea S , Esposito E ((2016) ) Traumatic brain injury leads to development of Parkinson’s disease related pathology in mice. Front Neurosci 10: , 458. |
[76] | Acosta SA , Tajiri N , Sanberg PR , Kaneko Y , Borlongan CV ((2017) ) Increased amyloid precursor protein and tau expression manifests as key secondary cell death in chronic traumatic brain injury. J Cell Physiol 232: , 665–677. |
[77] | Lee PH , Bang OY , Hwang EM , Lee JS , Joo US , Mook-Jung I , Huh K ((2005) ) Circulating beta amyloid protein is elevated in patients with acute ischemic stroke. J Neural Transm (Vienna) 112: , 1371–1379. |
[78] | Starzl R , Wolfram D , Zamora R , Jefferson B , Barclay D , Ho C , Gorantla V , Brandacher G , Schneeberger S , Andrew Lee WP , Carbonell J , Vodovotz Y ((2015) ) Cardiac arrest disrupts caspase-1 and patterns of inflammatory mediators differently in skin and muscle following localized tissue injury in rats. Front Immunol 6: , 587. |
[79] | Clausen BH , Lambertsen KL , Dagnaes-Hansen F , Babcock AA , von Linstow CU , Meldgaard M , Kristensen BW , Deierborg T , Finsen B ((2016) ) Cell therapy centered on IL-1Ra is neuroprotective in experimental stroke. Acta Neuropathol 131: , 775–791. |
[80] | Yue Y , Shang C , Dong H , Meng K ((2019) ) Interleukin-1 in cerebrospinal fluid for evaluating the neurological outcome in traumatic brain injury. Biosci Rep 39: , BSR20181966. |
[81] | Pluta R , Kida E , Lossinsky AS , Golabek AA , Mossakowski MJ , Wisniewski HM ((1994) ) Complete cerebral ischemia with short-term survival in rats induced by cardiac arrest. I. Extracellular accumulation of Alzheimer’s beta-amyloid protein precursor in the brain. Brain Res 649: , 323–328. |
[82] | Mailliot C , Podevin-Dimster V , Rosenthal RE , Sergeant N , Delacourte A , Fiskum G , Buee L ((2000) ) Rapid tau protein dephosphorylation and differential rephosphorylation during cardiac arrest-induced cerebral ischemia and reperfusion. J Cereb Blood Flow Metab 20: , 543–549. |
[83] | Randall J , Mortberg E , Provuncher GK , Fournier DR , Duffy DC , Rubertsson S , Blennow K , Zetterberg H , Wilson DH ((2013) ) Tau proteins in serum predict neurological outcome after hypoxic brain injury from cardiac arrest: Results of a pilot study. Resuscitation 84: , 351–356. |
[84] | Tae HJ , Kang IJ , Lee TK , Cho JH , Lee JC , Shin MC , Kim YS , Cho JH , Kim JD , Ahn JH , Park JH , Kim IS , Lee HA , Kim YH , Won MH , Lee YJ ((2017) ) Neuronal injury and tumor necrosis factor-alpha immunoreactivity in the rat hippocampus in the early period of asphyxia-induced cardiac arrest under normothermia. Neural Regen Res 12: , 2007–2013. |
[85] | Li Q , Yu B , Yang P ((2015) ) Hypoxia-induced HMGB1 in wound tissues promotes the osteoblast cell proliferation via activating ERK/JNK signaling. Int J Clin Exp Med 8: , 15087–15097. |
[86] | Lee S , Kim S , Kang HY , Lim HR , Kwon Y , Jo M , Jeon YM , Kim SR , Kim K , Ha CM , Lee S , Kim HJ ((2020) ) The overexpression of TDP-43 in astrocytes causes neurodegeneration via a PTP1B-mediated inflammatory response. J Neuroinflammation 17: , 299. |
[87] | Tribett T , Erskine B , Bailey K , Brown T , Castellani RJ ((2019) ) Chronic traumatic encephalopathy pathology after shotgun injury to the brain. J Forensic Sci 64: , 1248–1252. |
[88] | Fang P , Schachner M , Shen YQ ((2012) ) HMGB1 in development and diseases of the central nervous system. Mol Neurobiol 45: , 499–506. |
[89] | Petzold A , Jenkins R , Watt HC , Green AJ , Thompson EJ , Keir G , Fox NC , Rossor MN ((2003) ) Cerebrospinal fluid S100B correlates with brain atrophy in Alzheimer’s disease. Neurosci Lett 336: , 167–170. |
[90] | Xu M , Zhou GM , Wang LH , Zhu L , Liu JM , Wang XD , Li HT , Chen L ((2016) ) Inhibiting High-Mobility Group Box 1 (HMGB1) attenuates inflammatory cytokine expression and neurological deficit in ischemic brain injury following cardiac arrest in rats. Inflammation 39: , 1594–1602. |
[91] | Shi X , Li M , Huang K , Zhou S , Hu Y , Pan S , Gu Y ((2017) ) HMGB1 binding heptamer peptide improves survival and ameliorates brain injury in rats after cardiac arrest and cardiopulmonary resuscitation. Neuroscience 360: , 128–138. |
[92] | Pokela M , Anttila V , Rimpilainen J , Hirvonen J , Vainionpaa V , Kiviluoma K , Romsi P , Mennander A , Juvonen T ((2000) ) Serum S-100beta protein predicts brain injury after hypothermic circulatory arrest in pigs. Scand Cardiovasc J 34: , 570–574. |
[93] | Faraco G , Fossati S , Bianchi ME , Patrone M , Pedrazzi M , Sparatore B , Moroni F , Chiarugi A ((2007) ) High mobility group box 1 protein is released by neural cells upon different stresses and worsens ischemic neurodegeneration in vitro and in vivo. J Neurochem 103: , 590–603. |
[94] | Shi K , Tian DC , Li ZG , Ducruet AF , Lawton MT , Shi FD ((2019) ) Global brain inflammation in stroke. Lancet Neurol 18: , 1058–1066. |
[95] | Shi E , Shi K , Qiu S , Sheth KN , Lawton MT , Ducruet AF ((2019) ) Chronic inflammation, cognitive impairment, and distal brain region alteration following intracerebral hemorrhage. FASEB J 33: , 9616–9626. |
[96] | Shi K , Zhang J , Dong JF , Shi FD ((2019) ) Dissemination of brain inflammation in traumatic brain injury. Cell Mol Immunol 16: , 523–530. |
[97] | Youngquist ST , Shah AP , Rosborough JP , Niemann JT ((2016) ) High serum tumor necrosis factor levels in the early post-cardiac arrest period are associated with poor short-term survival in a swine model of ventricular fibrillation. J Interferon Cytokine Res 36: , 575–579. |
[98] | Bro-Jeppesen J , Johansson PI , Kjaergaard J , Wanscher M , Ostrowski SR , Bjerre M , Hassager C ((2017) ) Level of systemic inflammation and endothelial injury is associated with cardiovascular dysfunction and vasopressor support in post-cardiac arrest patients. Resuscitation 121: , 179–186. |
[99] | Clark IA , Vissel B ((2015) ) Amyloid beta: One of three danger-associated molecules that are secondary inducers of the proinflammatory cytokines that mediate Alzheimer’s disease. Br J Pharmacol 172: , 3714–3727. |
[100] | Niklowitz WJ , Mandybur TI ((1975) ) Neurofibrillary changes following childhood lead encephalopathy. J Neuropathol Exp Neurol 34: , 445–455. |
[101] | Basha MR , Wei W , Bakheet SA , Benitez N , Siddiqi HK , Ge YW , Lahiri DK , Zawia NH ((2005) ) The fetal basis of amyloidogenesis: Exposure to lead and latent overexpression of amyloid precursor protein and beta-amyloid in the aging brain. J Neurosci 25: , 823–829. |
[102] | White LD , Cory-Slechta DA , Gilbert ME , Tiffany-Castiglioni E , Zawia NH , Virgolini M , Rossi-George A , Lasley SM , Qian YC , Basha MR ((2007) ) New and evolving concepts in the neurotoxicology of lead. Toxicol Appl Pharmacol 225: , 1–27. |
[103] | Mason LH , Harp JP , Han DY ((2014) ) Pb neurotoxicity: Neuropsychological effects of lead toxicity. Biomed Res Int 2014: , 840547. |
[104] | Zhang J , Cai T , Zhao F , Yao T , Chen Y , Liu X , Luo W , Chen J ((2012) ) The role of alpha-synuclein and tau hyperphosphorylation-mediated autophagy and apoptosis in lead-induced learning and memory injury. Int J Biol Sci 8: , 935–944. |
[105] | Li N , Yu ZL , Wang L , Zheng YT , Jia JX , Wang Q , Zhu MJ , Liu XL , Xia X , Li WJ ((2010) ) Increased tau phosphorylation and beta amyloid in the hippocampus of mouse pups by early life lead exposure. Acta Biol Hung 61: , 123–134. |
[106] | Gassowska M , Baranowska-Bosiacka I , Moczydlowska J , Tarnowski M , Pilutin A , Gutowska I , Struzynska L , Chlubek D , Adamczyk A ((2016) ) Perinatal exposure to lead (Pb) promotes Tau phosphorylation in the rat brain in a GSK-3beta and CDK5 dependent manner: Relevance to neurological disorders. Toxicology 347-349: , 17–28. |
[107] | Li N , Yu ZL , Wang L , Zheng YT , Jia JX , Wang Q , Zhu MJ , Liu XH , Xia X , Li WJ ((2009) ) Early-life lead exposure affects the activity of TNF-alpha and expression of SNARE complex in hippocampus of mouse pups. Biol Trace Elem Res 132: , 227–238. |
[108] | Kumawat KL , Kaushik DK , Goswami P , Basu A ((2014) ) Acute exposure to lead acetate activates microglia and induces subsequent bystander neuronal death via caspase-3 activation. Neurotoxicology 41: , 143–153. |
[109] | Li N , Liu F , Song L , Zhang P , Qiao M , Zhao Q , Li W ((2014) ) The effects of early life Pb exposure on the expression of IL1-beta, TNF-alpha and Abeta in cerebral cortex of mouse pups. J Trace Elem Med Biol 28: , 100–104. |
[110] | Clark IA , Vissel B ((2013) ) Treatment implications of the altered cytokine-insulin axis in neurodegenerative disease. Biochem Pharmacol 86: , 862–871. |
[111] | Dosunmu R , Alashwal H , Zawia NH ((2012) ) Genome-wide expression and methylation profiling in the aged rodent brain due to early-life Pb exposure and its relevance to aging. Mech Ageing Dev 133: , 435–443. |
[112] | Schneider JS , Kidd SK , Anderson DW ((2013) ) Influence of developmental lead exposure on expression of DNA methyltransferases and methyl cytosine-binding proteins in hippocampus. Toxicol Lett 217: , 75–81. |
[113] | Bihaqi SW , Huang H , Wu J , Zawia NH ((2011) ) Infant exposure to lead (Pb) and epigenetic modifications in the aging primate brain: Implications for Alzheimer’s disease. J Alzheimers Dis 27: , 819–833. |
[114] | Deng GM , Liu ZQ , Tarkowski A ((2001) ) Intracisternally localized bacterial DNA containing CpG motifs induces meningitis. J Immunol 167: , 4616–4626. |
[115] | Walko TD , Bola RA , Hong JD , Au AK , Bell MJ , Kochanek PM , Clark RS , Aneja RK ((2014) ) Cerebrospinal fluid mitochondrial DNA: A novel DAMP in pediatric traumatic brain injury. Shock 41: , 499–503. |
[116] | Sanchez OF , Lee J , Yu King Hing N , Kim SE , Freeman JL , Yuan C ((2017) ) Lead (Pb) exposure reduces global DNA methylation level by non-competitive inhibition and alteration of dnmt expression. Metallomics 9: , 149–160. |
[117] | Coon S , Stark A , Peterson E , Gloi A , Kortsha G , Pounds J , Chettle D , Gorell J ((2006) ) Whole-body lifetime occupational lead exposure and risk of Parkinson’s disease. Environ Health Perspect 114: , 1872–1876. |
[118] | Weisskopf MG , Weuve J , Nie H , Saint-Hilaire MH , Sudarsky L , Simon DK , Hersh B , Schwartz J , Wright RO , Hu H ((2010) ) Association of cumulative lead exposure with Parkinson’s disease. Environ Health Perspect 118: , 1609–1613. |
[119] | Weuve J , Press DZ , Grodstein F , Wright RO , Hu H , Weisskopf MG ((2013) ) Cumulative exposure to lead and cognition in persons with Parkinson’s disease. Mov Disord 28: , 176–182. |
[120] | Ingram JH , Stone M , Fisher J , Ingham E ((2004) ) The influence of molecular weight, crosslinking and counterface roughness on TNF-alpha production by macrophages in response to ultra high molecular weight polyethylene particles. Biomaterials 25: , 3511–3522. |
[121] | Calderon-Garciduenas L , Azzarelli B , Acuna H , Garcia R , Gambling TM , Osnaya N , Monroy S , MR DELT , Carson JL , Villarreal-Calderon A , Rewcastle B ((2002) ) Air pollution and brain damage. Toxicol Pathol 30: , 373–389. |
[122] | Calderon-Garciduenas L , Solt AC , Henriquez-Roldan C , Torres-Jardon R , Nuse B , Herritt L , Villarreal-Calderon R , Osnaya N , Stone I , Garcia R , Brooks DM , Gonzalez-Maciel A , Reynoso-Robles R , Delgado-Chavez R , Reed W ((2008) ) Long-term air pollution exposure is associated with neuroinflammation, an altered innate immune response, disruption of the blood-brain barrier, ultrafine particulate deposition, and accumulation of amyloid beta-42 and alpha-synuclein in children and young adults. Toxicol Pathol 36: , 289–310. |
[123] | Calderon-Garciduenas L , de la Monte SM ((2017) ) Apolipoprotein E4, gender, body mass Index, inflammation, insulin resistance, and air pollution interactions: Recipe for Alzheimer’s disease development in Mexico City young females. J Alzheimers Dis 58: , 613–630. |
[124] | Win-Shwe TT , Yamamoto S , Fujitani Y , Hirano S , Fujimaki H ((2008) ) Spatial learning and memory function-related gene expression in the hippocampus of mouse exposed to nanoparticle-rich diesel exhaust. Neurotoxicology 29: , 940–947. |
[125] | Zhang X , Chen X , Zhang X ((2018) ) The impact of exposure to air pollution on cognitive performance. Proc Natl Acad Sci U S A 115: , 9193–9197. |
[126] | Chen H , Kwong JC , Copes R , Hystad P , van Donkelaar A , Tu K , Brook JR , Goldberg MS , Martin RV , Murray BJ , Wilton AS , Kopp A , Burnett RT ((2017) ) Exposure to ambient air pollution and the incidence of dementia: A population-based cohort study. Environ Int 108: , 271–277. |
[127] | Carswell EA , Old LJ , Kassel RL , Green S , Fiore N , Williamson B ((1975) ) An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci U S A 72: , 3666–3670. |
[128] | Clark IA , Cowden WB ((2003) ) The pathophysiology of falciparum malaria. Pharmacol Ther 99: , 221–260. |
[129] | Wu X , Gowda NM , Kumar S , Gowda DC ((2010) ) Protein-DNA complex is the exclusive malaria parasite component that activates dendritic cells and triggers innate immune responses. J Immunol 184: , 4338–4348. |
[130] | Lillie PJ , Duncan CJ , Sheehy SH , Meyer J , O’Hara GA , Gilbert SC , Hill AV ((2012) ) Distinguishing malaria and influenza: Early clinical features in controlled human experimental infection studies. Travel Med Infect Dis 10: , 192–196. |
[131] | Pisa D , Alonso R , Fernandez-Fernandez AM , Rabano A , Carrasco L ((2017) ) Polymicrobial infections In brain tissue from Alzheimer’s disease patients. Sci Rep 7: , 5559. |
[132] | Monastero R , Caruso C , Vasto S ((2014) ) Alzheimer’s disease and infections, where we stand and where we go. Immun Ageing 11: , 26. |
[133] | Itzhaki RF , Lathe R , Balin BJ , Ball MJ , Bearer EL , Braak H , Bullido MJ , Carter C , Clerici M , Cosby SL , Del Tredici K , Field H , Fulop T , Grassi C , Griffin WS , Haas J , Hudson AP , Kamer AR , Kell DB , Licastro F , Letenneur L , Lovheim H , Mancuso R , Miklossy J , Otth C , Palamara AT , Perry G , Preston C , Pretorius E , Strandberg T , Tabet N , Taylor-Robinson SD , Whittum-Hudson JA ((2016) ) Microbes and Alzheimer’s disease. J Alzheimers Dis 51: , 979–984. |
[134] | Brown GC ((2019) ) The endotoxin hypothesis of neurodegeneration. J Neuroinflammation 16: , 180. |
[135] | Shin SH , Kim EK , Lee KY , Kim HS ((2019) ) TNF-alpha antagonist attenuates systemic lipopolysaccharide-induced brain white matter injury in neonatal rats. BMC Neurosci 20: , 45. |
[136] | Itzhaki RF , Lathe R ((2018) ) Herpes viruses and senile dementia: First population evidence for a causal link. J Alzheimers Dis 64: , 363–366. |
[137] | Tzeng NS , Chung CH , Lin FH , Chiang CP , Yeh CB , Huang SY , Lu RB , Chang HA , Kao YC , Yeh HW , Chiang WS , Chou YC , Tsao CH , Wu YF , Chien WC ((2018) ) Anti-herpetic medications and reduced risk of dementia in patients with herpes simplex virus infections-a nationwide, population-based cohort study in Taiwan. Neurotherapeutics 15: , 417–429. |
[138] | Alvarez-Arellano L , Maldonado-Bernal C ((2014) ) Helicobacter pylori and neurological diseases: Married by the laws of inflammation. World J Gastrointest Pathophysiol 5: , 400–404. |
[139] | Kountouras J , Boziki M , Gavalas E , Zavos C , Deretzi G , Chatzigeorgiou S , Katsinelos P , Grigoriadis N , Giartza-Taxidou E , Venizelos I ((2010) ) Five-year survival after Helicobacter pylori eradication in Alzheimer disease patients. Cogn Behav Neurol 23: , 199–204. |
[140] | Kuno R , Wang J , Kawanokuchi J , Takeuchi H , Mizuno T , Suzumura A ((2005) ) Autocrine activation of microglia by tumor necrosis factor-alpha. J Neuroimmunol 162: , 89–96. |
[141] | Valko PO , Gavrilov YV , Yamamoto M , Noain D , Reddy H , Haybaeck J , Weis S , Baumann CR , Scammell TE ((2016) ) Damage to arousal-promoting brainstem neurons with traumatic brain injury. Sleep 39: , 1249–1252. |
[142] | Levin BE , Brown KL , Pawar G , Dunn-Meynell A ((1995) ) Widespread and lateralization effects of acute traumatic brain injury on norepinephrine turnover in the rat brain. Brain Res 674: , 307–313. |
[143] | McKee AC , Stern RA , Nowinski CJ , Stein TD , Alvarez VE , Daneshvar DH , Lee HS , Wojtowicz SM , Hall G , Baugh CM , Riley DO , Kubilus CA , Cormier KA , Jacobs MA , Martin BR , Abraham CR , Ikezu T , Reichard RR , Wolozin BL , Budson AE , Goldstein LE , Kowall NW , Cantu RC ((2013) ) The spectrum of disease in chronic traumatic encephalopathy. Brain 136: , 43–64. |
[144] | Theofilas P , Dunlop S , Heinsen H , Grinberg LT ((2015) ) Turning on the light within: Subcortical nuclei of the isodentritic core and their role in Alzheimer’s disease pathogenesis. J Alzheimers Dis 46: , 17–34. |
[145] | Stratmann K , Heinsen H , Korf HW , Del Turco D , Ghebremedhin E , Seidel K , Bouzrou M , Grinberg LT , Bohl J , Wharton SB , den Dunnen W , Rub U ((2016) ) Precortical phase of Alzheimer’s disease (AD)-related tau cytoskeletal pathology. Brain Pathol 26: , 371–386. |
[146] | Kelly SC , He B , Perez SE , Ginsberg SD , Mufson EJ , Counts SE ((2017) ) Locus coeruleus cellular and molecular pathology during the progression of Alzheimer’s disease. Acta Neuropathol Commun 5: , 8. |
[147] | Theofilas P , Ehrenberg AJ , Nguy A , Thackrey JM , Dunlop S , Mejia MB , Alho AT , Paraizo Leite RE , Rodriguez RD , Suemoto CK , Nascimento CF , Chin M , Medina-Cleghorn D , Cuervo AM , Arkin M , Seeley WW , Miller BL , Nitrini R , Pasqualucci CA , Filho WJ , Rueb U , Neuhaus J , Heinsen H , Grinberg LT ((2018) ) Probing the correlation of neuronal loss, neurofibrillary tangles, and cell death markers across the Alzheimer’s disease Braak stages: A quantitative study in humans. Neurobiol Aging 61: , 1–12. |
[148] | Chandler DJ , Jensen P , McCall JG , Pickering AE , Schwarz LA , Totah NK ((2019) ) Redefining noradrenergic neuromodulation of behavior: Impacts of a modular locus coeruleus architecture. J Neurosci 39: , 8239–8249. |
[149] | Garwood CJ , Cooper JD , Hanger DP , Noble W ((2010) ) Anti-inflammatory impact of minocycline in a mouse model of tauopathy. Front Psychiatry 1: , 136. |
[150] | Morris GP , Clark IA , Vissel B ((2014) ) Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol Commun 2: , 135. |
[151] | Adolfsson R , Gottfries CG , Roos BE , Winblad B ((1979) ) Changes in the brain catecholamines in patients with dementia of Alzheimer type. Br J Psychiatry 135: , 216–223. |
[152] | Mann DM , Lincoln J , Yates PO , Stamp JE , Toper S ((1980) ) Changes in the monoamine containing neurones of the human CNS in senile dementia. Br J Psychiatry 136: , 533–541. |
[153] | Hu XX , Goldmuntz EA , Brosnan CF ((1991) ) The effect of norepinephrine on endotoxin-mediated macrophage activation. J Neuroimmunol 31: , 35–42. |
[154] | Feinstein DL , Heneka MT , Gavrilyuk V , Dello Russo C , Weinberg G , Galea E ((2002) ) Noradrenergic regulation of inflammatory gene expression in brain. Neurochem Int 41: , 357–365. |
[155] | Heneka MT , Galea E , Gavriluyk V , Dumitrescu-Ozimek L , Daeschner J , O’Banion MK , Weinberg G , Klockgether T , Feinstein DL ((2002) ) Noradrenergic depletion potentiates beta -amyloid-induced cortical inflammation: Implications for Alzheimer’s disease. J Neurosci 22: , 2434–2442. |
[156] | Kaneko YS , Mori K , Nakashima A , Sawada M , Nagatsu I , Ota A ((2005) ) Peripheral injection of lipopolysaccharide enhances expression of inflammatory cytokines in murine locus coeruleus: Possible role of increased norepinephrine turnover. J Neurochem 94: , 393–404. |
[157] | Ağaç D , Estrada LD , Maples R , Hooper LV , Farrar JD ((2018) ) The β2-adrenergic receptor controls inflammation by driving rapid IL-10 secretion. Brain Behav Immun 74: , 176–185. |
[158] | Ignatowski TA , Spengler RN ((1995) ) Regulation of macrophage-derived tumor necrosis factor production by modification of adrenergic receptor sensitivity. J Neuroimmunol 61: , 61–70. |
[159] | Chou RC , Stinson MW , Noble BK , Spengler RN ((1996) ) Beta-adrenergic receptor regulation of macrophage-derived tumor necrosis factor-alpha production from rats with experimental arthritis. J Neuroimmunol 67: , 7–16. |
[160] | Qian L , Wu HM , Chen SH , Zhang D , Ali SF , Peterson L , Wilson B , Lu RB , Hong JS , Flood PM ((2011) ) beta2-adrenergic receptor activation prevents rodent dopaminergic neurotoxicity by inhibiting microglia via a novel signaling pathway. J Immunol 186: , 4443–4454. |
[161] | Romberger DJ , Heires AJ , Nordgren TM , Poole JA , Toews ML , West WW , Wyatt TA ((2016) ) beta2-Adrenergic agonists attenuate organic dust-induced lung inflammation. Am J Physiol Lung Cell Mol Physiol 311: , L101–110. |
[162] | Keranen T , Hommo T , Moilanen E , Korhonen R ((2017) ) beta2-receptor agonists salbutamol and terbutaline attenuated cytokine production by suppressing ERK pathway through cAMP in macrophages. Cytokine 94: , 1–7. |
[163] | Pickering M , Cumiskey D , O’Connor JJ ((2005) ) Actions of TNF-alpha on glutamatergic synaptic transmission in the central nervous system. Exp Physiol 90: , 663–670. |
[164] | O’Donnell J , Zeppenfeld D , McConnell E , Pena S , Nedergaard M ((2012) ) Norepinephrine: A neuromodulator that boosts the function of multiple cell types to optimize CNS performance. Neurochem Res 37: , 2496–2512. |
[165] | Heneka MT , Nadrigny F , Regen T , Martinez-Hernandez A , Dumitrescu-Ozimek L , Terwel D , Jardanhazi-Kurutz D , Walter J , Kirchhoff F , Hanisch UK , Kummer MP ((2010) ) Locus ceruleus controls Alzheimer’s disease pathology by modulating microglial functions through norepinephrine. Proc Natl Acad Sci U S A 107: , 6058–6063. |
[166] | Feinstein DL , Kalinin S , Braun D ((2016) ) Causes, consequences, and cures for neuroinflammation mediated via the locus coeruleus: Noradrenergic signaling system. J Neurochem 139: (Suppl 2), 154–178. |
[167] | Leanza G , Gulino R , Zorec R ((2018) ) Noradrenergic hypothesis linking neurodegeneration-based cognitive decline and astroglia. Front Mol Neurosci 11: , 254. |
[168] | Mogi M , Harada M , Riederer P , Narabayashi H , Fujita K , Nagatsu T ((1994) ) Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from Parkinsonian patients. Neurosci Lett 165: , 208–210. |
[169] | Gesi M , Soldani P , Giorgi FS , Santinami A , Bonaccorsi I , Fornai F ((2000) ) The role of the locus coeruleus in the development of Parkinson’s disease. Neurosci Biobehav Rev 24: , 655–668. |
[170] | Vermeiren Y , De Deyn PP ((2017) ) Targeting the norepinephrinergic system in Parkinson’s disease and related disorders: The locus coeruleus story. Neurochem Int 102: , 22–32. |
[171] | Zarow C , Lyness SA , Mortimer JA , Chui HC ((2003) ) Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch Neurol 60: , 337–341. |
[172] | O’Neill E , Harkin A ((2018) ) Targeting the noradrenergic system for anti-inflammatory and neuroprotective effects: Implications for Parkinson’s disease. Neural Regen Res 13: , 1332–1337. |
[173] | Jacobs HIL , Riphagen JM , Ramakers I , Verhey FRJ ((2019) ) Alzheimer’s disease pathology: Pathways between central norepinephrine activity, memory, and neuropsychiatric symptoms. Mol Psychiatry, doi: 10.1038/s41380-019-0437-x. |
[174] | Betts MJ , Kirilina E , Otaduy MCG , Ivanov D , Acosta-Cabronero J , Callaghan MF , Lambert C , Cardenas-Blanco A , Pine K , Passamonti L , Loane C , Keuken MC , Trujillo P , Lusebrink F , Mattern H , Liu KY , Priovoulos N , Fliessbach K , Dahl MJ , Maass A , Madelung CF , Meder D , Ehrenberg AJ , Speck O , Weiskopf N , Dolan R , Inglis B , Tosun D , Morawski M , Zucca FA , Siebner HR , Mather M , Uludag K , Heinsen H , Poser BA , Howard R , Zecca L , Rowe JB , Grinberg LT , Jacobs HIL , Duzel E , Hammerer D ((2019) ) Locus coeruleus imaging as a biomarker for noradrenergic dysfunction in neurodegenerative diseases. Brain 142: , 2558–2571. |
[175] | Dahl MJ , Mather M , Duzel S , Bodammer NC , Lindenberger U , Kuhn S , Werkle-Bergner M ((2019) ) Rostral locus coeruleus integrity is associated with better memory performance in older adults. Nat Hum Behav 3: , 1203–1214. |
[176] | Liu B , Hong JS ((2003) ) Role of microglia in inflammation-mediated neurodegenerative diseases: Mechanisms and strategies for therapeutic intervention. J Pharmacol Exp Ther 304: , 1–7. |
[177] | Ehrenberg AJ , Nguy AK , Theofilas P , Dunlop S , Suemoto CK , Di Lorenzo Alho AT , Leite RP , Diehl Rodriguez R , Mejia MB , Rub U , Farfel JM , de Lucena Ferretti-Rebustini RE , Nascimento CF , Nitrini R , Pasquallucci CA , Jacob-Filho W , Miller B , Seeley WW , Heinsen H , Grinberg LT ((2017) ) Quantifying the accretion of hyperphosphorylated tau in the locus coeruleus and dorsal raphe nucleus: The pathological building blocks of early Alzheimer’s disease. Neuropathol Appl Neurobiol 43: , 393–408. |
[178] | Andres-Benito P , Fernandez-Duenas V , Carmona M , Escobar LA , Torrejon-Escribano B , Aso E , Ciruela F , Ferrer I ((2017) ) Locus coeruleus at asymptomatic early and middle Braak stages of neurofibrillary tangle pathology. Neuropathol Appl Neurobiol 43: , 373–392. |
[179] | Tchessalova D , Posillico CK , Tronson NC ((2018) ) Neuroimmune activation drives multiple brain states. Front Syst Neurosci 12: , 39. |
[180] | Osorio C , Kanukuntla T , Diaz E , Jafri N , Cummings M , Sfera A ((2019) ) The post-amyloid era in Alzheimer’s disease: Trust your gut feeling. Front Aging Neurosci 11: , 143. |
[181] | Caputi V , Giron MC ((2018) ) Microbiome gut-brain axis and Toll-like receptors in Parkinson’s isease. Int J Mol Sci 19: , 1689. |
[182] | Clark IA ((2017) ) Editorial: An unsound AAN Practice Advisory on poststroke etanercept. Expert Rev Neurother 17: , 215–217. |
[183] | Steinberg GK , Kondziolka D , Wechsler LR , Lunsford LD , Coburn ML , Billigen JB , Kim AS , Johnson JN , Bates D , King B , Case C , McGrogan M , Yankee EW , Schwartz NE ((2016) ) Clinical outcomes of transplanted modified bone marrow-derived mesenchymal stem cells in stroke: A Phase 1/2a study. Stroke 47: , 1817–1824. |
[184] | Ghezzi P , Dinarello CA , Bianchi M , Rosandich ME , Repine JE , White CW ((1991) ) Hypoxia increases production of interleukin-1 and tumor necrosis factor by human mononuclear cells. Cytokine 3: , 189–194. |
[185] | Mukandala G , Tynan R , Lanigan S , O’Connor JJ ((2016) ) The effects of hypoxia and inflammation on synaptic signaling in the CNS. Brain Sci 6: , 6. |
[186] | Tweedie D , Sambamurti K , Greig NH ((2007) ) TNF-alpha inhibition as a treatment strategy for neurodegenerative disorders: New drug candidates and targets. Curr Alzheimer Res 4: , 378–385. |
[187] | Mittal S , Bjornevik K , Im DS , Flierl A , Dong X , Locascio JJ , Abo KM , Long E , Jin M , Xu B , Xiang YK , Rochet JC , Engeland A , Rizzu P , Heutink P , Bartels T , Selkoe DJ , Caldarone BJ , Glicksman MA , Khurana V , Schule B , Park DS , Riise T , Scherzer CR ((2017) ) beta2-Adrenoreceptor is a regulator of the alpha-synuclein gene driving risk of Parkinson’s disease. Science 357: , 891–898. |
[188] | Qin LY , Wu XF , Block ML , Liu YX , Bresse GR , Hong JS , Knapp DJ , Crews FT ((2007) ) Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 55: , 453–462. |
[189] | Farber K , Cheung G , Mitchell D , Wallis R , Weihe E , Schwaeble W , Kettenmann H ((2009) ) C1q, the recognition subcomponent of the classical pathway of complement, drives microglial activation. J Neurosci Res 87: , 644–652. |
[190] | Zhang X , Zeng L , Yu T , Xu Y , Pu S , Du D , Jiang W ((2014) ) Positive feedback loop of autocrine BDNF from microglia causes prolonged microglia activation. Cell Physiol Biochem 34: , 715–723. |
[191] | Han L , Zhang D , Tao T , Sun X , Liu X , Zhu G , Xu Z , Zhu L , Zhang Y , Liu W , Ke K , Shen A ((2015) ) The role of N-glycan modification of TNFR1 in inflammatory microglia activation. Glycoconj J 32: , 685–693. |
[192] | Stellwagen D , Malenka RC ((2006) ) Synaptic scaling mediated by glial TNF-alpha. Nature 440: , 1054–1059. |
[193] | Fotuhi M , Mian A , Meysami S , Raji CA ((2020) ) Neurobiology of COVID-19. J Alzheimers Dis 76: , 3–19. |
[194] | Kotfis K , Williams Roberson S , Wilson JE , Dabrowski W , Pun BT , Ely EW ((2020) ) COVID-19: ICU delirium management during SARS-CoV-2 pandemic. Crit Care 24: , 176. |
[195] | Azizi SA , Azizi SA ((2020) ) Neurological injuries in COVID-19 patients: Direct viral invasion or a bystander injury after infection of epithelial/endothelial cells. J Neurovirol 26: , 631–641. |
[196] | Marshall M ((2020) ) How COVID-19 can damage the brain. Nature 585: , 342–343. |
[197] | Barbour VL , Mead GE ((2012) ) Fatigue after stroke: The patient’s perspective. Stroke Res Treat 2012: , 863031. |
[198] | Beaulieu-Bonneau S , Morin CM ((2012) ) Sleepiness and fatigue following traumatic brain injury. Sleep Med 13: , 598–605. |
[199] | Clark IA , Vissel B ((2018) ) The inflammatory nature of post-surgical delirium predicts benefit of agents with anti-TNF effects, such as Dexmedetomidine. Front Neurosci 12: , 257. |
[200] | Inouye SK , Westendorp RG , Saczynski JS ((2014) ) Delirium in elderly people. Lancet 383: , 911–922. |
[201] | Tauber SC , Djukic M , Gossner J , Eiffert H , Brück W , Nau R ((2020) ) Sepsis-associated encephalopathy and septic encephalitis: An update. Expert Rev Anti Infect Ther, doi: 10.1080/14787210.2020.1812384. |
[202] | Bruneel F ((2019) ) Human cerebral malaria: 2019 mini review. Rev Neurol (Paris) 175: , 445–450. |
[203] | Feldmann M , Maini RN , Woody JN , Holgate ST , Winter G , Rowland M , Richards D , Hussell T ((2020) ) Trials of anti-tumour necrosis factor therapy for COVID-19 are urgently needed. Lancet 395: , 1407–1409. |

