
Molecular Processing of Tau Protein in Progressive Supranuclear Palsy: Neuronal and Glial Degeneration
Abstract
Background:
Alzheimer’s disease (AD) and progressive supranuclear palsy (PSP) are examples of neurodegenerative diseases, characterized by abnormal tau inclusions, that are called tauopathies. AD is characterized by highly insoluble paired helical filaments (PHFs) composed of tau with abnormal post-translational modifications. PSP is a neurodegenerative disease with pathological and clinical heterogeneity. There are six tau isoforms expressed in the adult human brain, with repeated microtubule-binding domains of three (3R) or four (4R) repeats. In AD, the 4R:3R ratio is 1:1. In PSP, the 4R isoform predominates. The lesions in PSP brains contain phosphorylated tau aggregates in both neurons and glial cells.
Objective:
Our objective was to evaluate and compare the processing of pathological tau in PSP and AD.
Methods:
Double and triple immunofluorescent labeling with antibodies to specific post-translational tau modifications (phosphorylation, truncation, and conformational changes) and thiazin red (TR) staining were carried out and analyzed by confocal microscopy.
Results:
Our results showed that PSP was characterized by phosphorylated tau in neurofibrillary tangles (NFTs) and glial cells. Tau truncated at either Glu391 or Asp421 was not observed. Extracellular NFTs (eNFTs) and glial cells in PSP exhibited a strong affinity for TR in the absence of intact or phosphorylated tau.
Conclusion:
Phosphorylated tau was as abundant in PSP as in AD. The development of eNFTs from both glial cells and neuronal bodies suggests that truncated tau species, different from those observed in AD, could be present in PSP. Additional studies on truncated tau within PSP lesions could improve our understanding of the pathological processing of tau and help identify a discriminatory biomarker for AD and PSP.
INTRODUCTION
Progressive supranuclear palsy (PSP) is a neurodegenerative disease [1] first described by Steele and colleagues in 1964 [2]. PSP is characterized by onset over 40 years of age, with a mean survival of patients from symptom onset of six years, and culmination in the death of patients. It belongs to a group of over 20 disorders that are termed tauopathies, of which Alzheimer’s disease (AD) is the most common [3]. PSP presents a wide variety of signs and symptoms that can overlap with other neurodegenerative disorders [4]. PSP affects movement, control of walking (gait) and balance, speech, swallowing, and vision (blurring of vision and problems controlling eye movement). People with PSP may also exhibit parkinsonism, photophobia, disturbances of sleep and emotions, depression, anxiety, and dementia [1, 5–7]. PSP has a prevalence of 5–8/100,000 individuals [8, 9], but the clinical diagnosis can only be confirmed by postmortem pathological analysis. PSP brain exhibits significant atrophy in the brain convolutions, brain stem, basal ganglia (substantia nigra, subthalamic nucleus, and globus pallidus), cerebral cortex (frontal lobe), dentate nucleus of the cerebellum, and spinal cord. Tau protein is pathologically modified and forms aggregates in neuronal and glial bodies in PSP [10]. Under normal conditions, tau stabilizes the microtubules favoring the movement of vesicles and organelles along the axon.
Tau undergoes cycles of phosphorylation/dephosphorylation to carry out its function. Six isoforms of tau are expressed in the adult human brain, which results from alternative splicing of pre-mRNA generated from a single, MAPT gene. These isoforms (ranging from 352 to 441 amino acids in length) differ in the presence of N-terminal inserts (0, 1, or 2) and the number of C-terminal repeats (3R or 4R). The microtubule-binding region (MTBR) is composed of either 3 or 4 repeats. Whereas the ratio of 3R and 4R isoforms is 1:1 in the healthy brain, there are many 4R isoforms in PSP disease [11]. PSP is a sporadic onset disease with a few cases of familial origin associated with mutations in the MAPT gene [12]. These mutations are not exclusive to PSP and can occur in other neurodegenerative disorders. The cause of the onset of PSP pathology is still unknown. It has been suggested that altered mitochondrial function involved in neurodegeneration could occur at the beginning of PSP. Most of the current studies are focused on the relationship of the tau protein with neuronal and glial cell death. Tau post-translational modifications, including acylation, O-GlcNAcylation, O-glycosylation, and truncation, have been related to neurodegeneration. These modifications lead to tau accumulation in the neuronal soma (pre-neurofibrillary tangles (NFT) and NFT), oligodendrocytes (coiled body), glial cells (tuft-shaped astrocytes, TAs), and neuronal processes (neuropil threads) [13]. In PSP, the neuropathological changes do not correlate with the patient deterioration. However, the presence of the lesions in different brain areas exemplifies the phenotypic variability [14]. Soluble and insoluble pathological tau is found in the cell soma in a toxic form, affecting neuronal and glial cells.
The particular sequence of the pathological tau processing has been derived from studies using specific immunological markers in AD. In this work, we have compared post-translational changes in tau protein in NFTs and glial cells in PSP and AD brains. We have used a panel of antibodies that recognize different aspects of tau protein in order to understand the differing states of tau protein during aggregation and NFT development in the two disorders.
MATERIALS AND METHODS
PSP brains
The striatum, amygdala, and hippocampus from PSP brains were obtained from the Brain Bank of the Institute of Neuropathology of Bellvitge University Hospital, Barcelona, Spain. The age range of brain donors was from 57 to 86 years old (Table 3). The diagnosis was confirmed by histopathological analysis. Formalin-fixed paraffin-embedded tissue blocks were cut into 6-μm thick sections and mounted on glass slides for further processing.
AD brains
Human brain tissue from six AD patients (age range 47 to 90 years, mean of 67.5 years, and with 2–6 h postmortem delay) was used (Table 3). Tissue was obtained from the National Dementia BioBank, Mexico, following the institutional bioethics guidelines. The diagnosis of AD followed the NIA-NINCDS group criteria [15]. Blocks of the hippocampus and adjacent entorhinal cortex were fixed by immersion in a solution of 4% paraformaldehyde in phosphate-buffered saline (PBS), pH 7.4, at 4°C, for seven days. The pathological severity of AD was determined by the density of NFTs and neuritic plaques, using the immunoperoxidase technique and thiazine red (TR) staining.
Immunofluorescent labeling
Tissue slices were deparaffinized in xylene and then rehydrated in serial solutions of ethanol and distilled water. Antigen retrieval was achieved by treatment at 100°C for 20 min in citrate buffer (0.1 M citric acid, 0.1 M sodium citrate, pH 6.0). Lipofuscin autofluorescence was removed with Sudan B Black (SBB) solution (0.1% SBB in 70% ethanol) for 15 min. Non-specific sites were blocked with 10% fetal bovine serum in PBS for 20 min at room temperature. Tissues were then incubated with the primary antibody cocktail (Table 1) overnight at 4°C, and then with the secondary antibodies (Table 2). Sections were counterstained with TR to differentiate non-fibrillar and fibrillar tau aggregates [16, 17] and mounted in Immuno-Flour mounting medium (ICN Biomedicals, Aurora, OH).
Table 1
List of primary antibodies used in this study. The antibodies recognize different tau epitopes including phosphorylation sites, conformational changes, and truncated species
| Primary antibody | Epitope | Isotype | Reference or source |
| 499 | Intact tau at 14–26 | Mo IgG | [52] |
| Tau7 | Intact tau at 426–441 | Mo IgG | [38] |
| pT231 | Phosphorylation at Thr231 | Rb IgG | Invitrogen |
| AT8 | Phosphorylation at Ser202, Thr205 | Mo IgG | Invitrogen |
| TG3 | Regional conformational change, dependent on phosphorylation at Thr231, Ser235 | Mo IgM | [53] |
| AT100 | Regional conformational change, dependent on phosphorylation at Thr212, Ser214 | Mo IgG | Invitrogen |
| Alz50 | Amino acids: 5–15, 312–322 | Mo IgM | [44, 54] |
| Structural conformational change | |||
| pS396 | Phosphorylation at Ser396 | Rb IgG | Invitrogen |
| PHF1 | Phosphorylation at Ser396, Ser404 | Mo IgG | [55] |
| TauC3 | Truncation at Asp421 | Mo IgG | [38] |
| 423 | Truncation at Glu391 | Mo IgG | [34] |
Table 2
List of secondary antibodies used in this study. Antibody-specific fractions were used to avoid cross-reaction between antibodies. Mo, mouse; Rb, rabbit
| Fluorescent Conjugate | Isotype |
| Alexa488 | Goat anti-Mo IgG (γ) |
| Goat anti-Mo IgM (μ) | |
| Goat anti-Rb IgG (H + L) | |
| Alexa546 | Goat anti-Mo IgG (γ) |
| Goat anti-Mo IgM (μ) | |
| Goat anti-Rb IgG (H + L) | |
| CY5 | Goat anti-Mo IgG (γ) |
| Goat anti-Mo IgM (μ) | |
| Goat anti-Rb IgG (H + L) |
Table 3
Cases used in this study
| Diagnosis | Sex | Age | Brain bank |
| Progressive supranuclear palsy | M | 75 | Brain Bank of the Institute of Neuropathology of Bellvitge University Hospital, Barcelona, Spain |
| M | 85 | ||
| M | 70 | ||
| M | 77 | ||
| F | 80 | ||
| F | 86 | ||
| F | 79 | ||
| F | 75 | ||
| Alzheimer’s disease | F | 77 | National Dementia BioBank, FESc, UNAM |
| F | 83 | ||
| F | 70 | ||
| M | 78 | ||
| M | 80 | ||
| F | 75 | ||
| F | 70 | ||
| F | 80 |
Confocal microscopy
Double and triple-immunolabeled sections were examined using a confocal laser scanning microscope (TCS-SP8, Leica, Heidelberg), and a 100x oil-immersion plan Apochromatic objective (NA 1.4). Ten to fifteen consecutive individual sections were sequentially scanned at 0.5–0.8μm intervals for two or three channels throughout the z-axis of the sample. The collected images were projected and analyzed onto the two-dimensional plane using a pseudocolor display of green (FITC), red (TRITC), and blue (CY5). Fluorochromes were excited at 488 nm (for FITC), 540 nm (for TRITC), and 650 nm (for CY5).
RESULTS
The pathological processing of the tau protein occurs in a wide variety of diseases called tauopathies, including AD and PSP. Identifying similarities and differences in the pathological tau processing can favor the understanding of tauopathies and, hopefully, the finding of an appropriate biomarker and treatment.
Neuropathology of AD
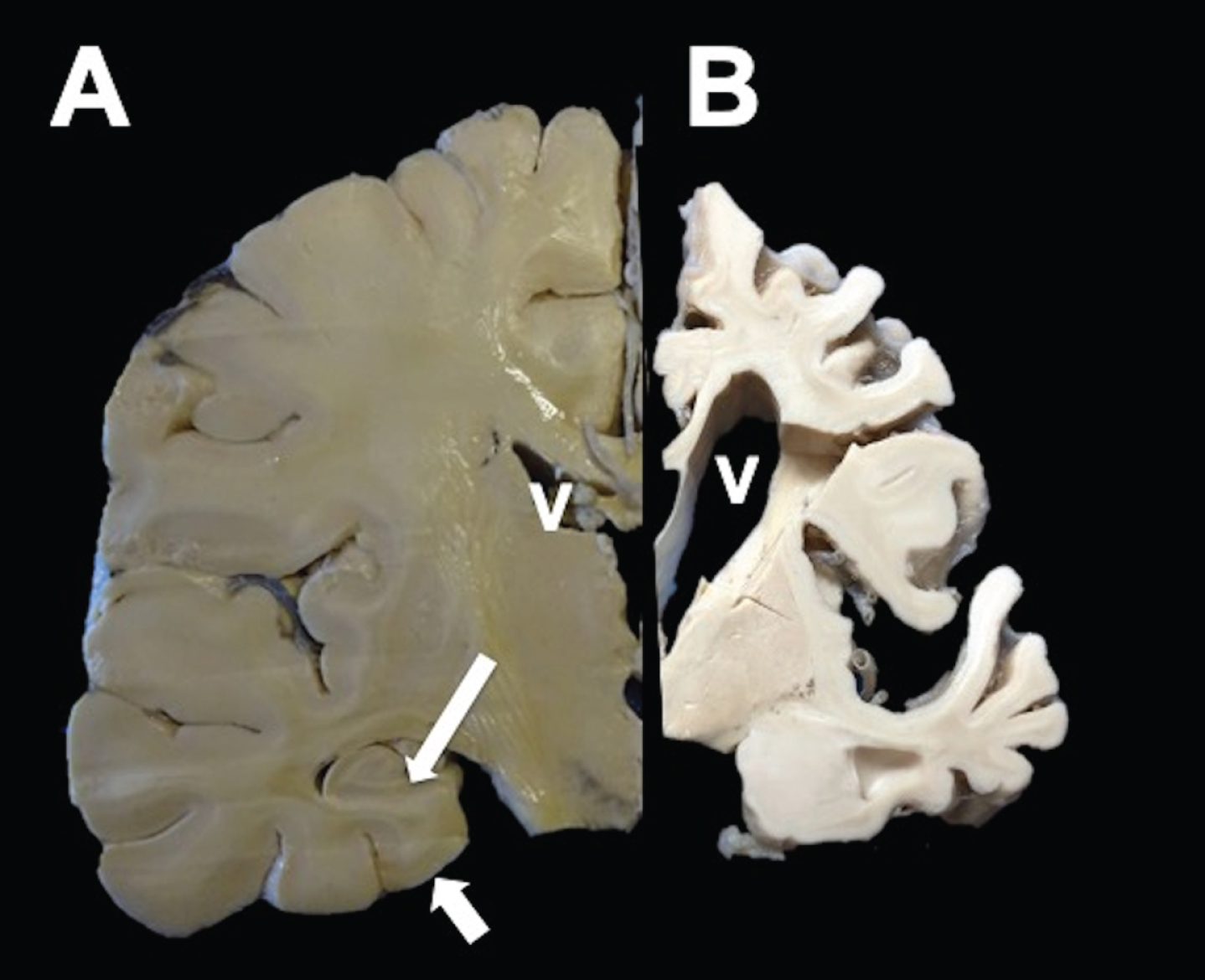
Macroscopically, AD brain showed a considerable reduction in the brain volume (Fig. 1B) compared to a control case (Fig. 1A). Aside from enlargement of the sulci and ventricles, hippocampal atrophy is prominent in AD (Fig. 1B).
Fig.1
Coronal section of a healthy (left) and AD hemisphere (right). In AD, a considerable reduction in the brain volume with enlargement of the grooves between folds, prominent ventricles (V), and hippocampal atrophy (arrows) was observed.
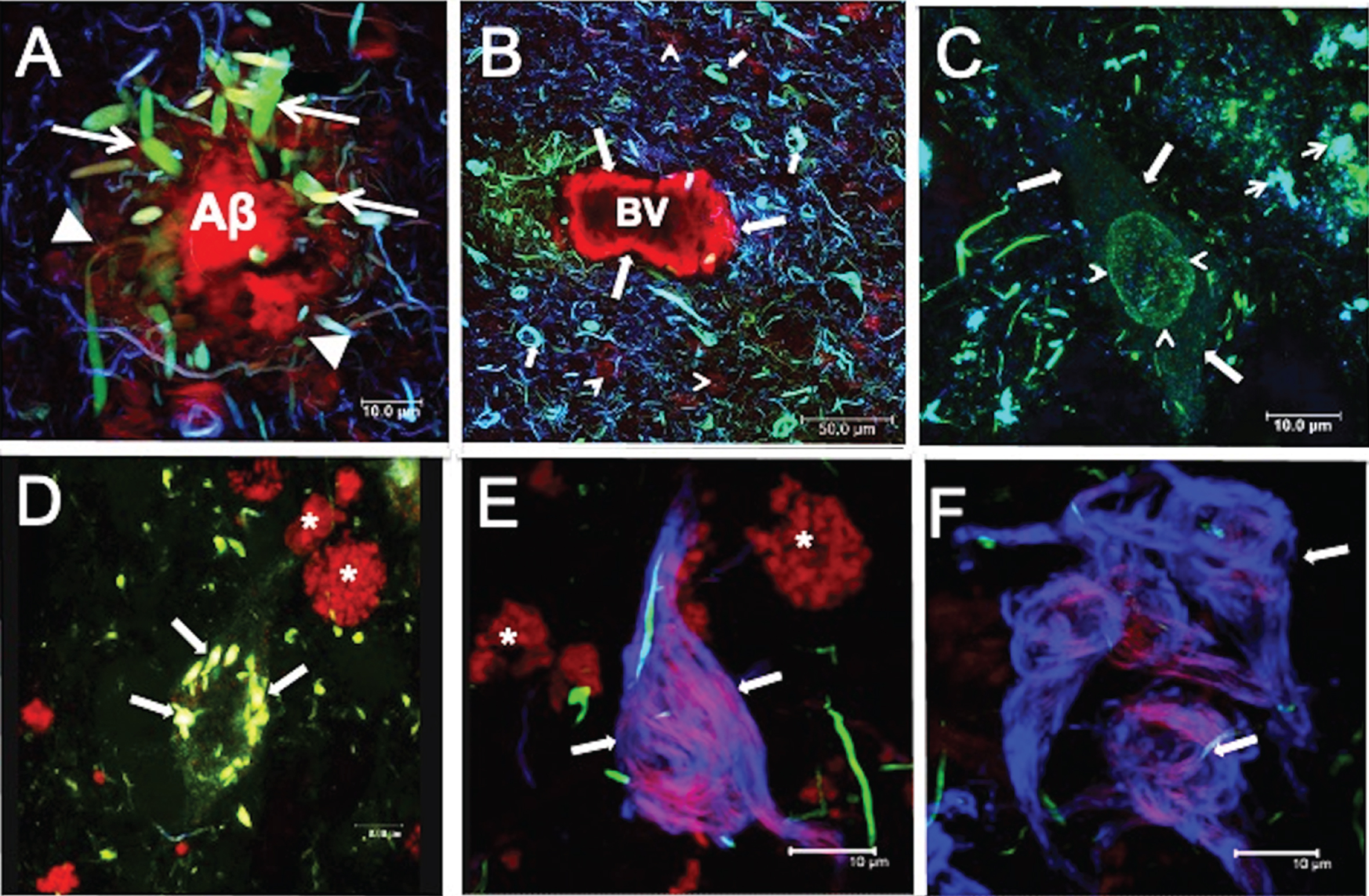
Histopathological lesions in AD
In AD, the characteristic histopathological lesions are the neuritic plaques and NFTs (Fig. 2). Neuritic plaques comprise a dense core surrounded by a diffuse corona of amyloid-β peptide (Aβ) (Fig. 2A; arrowheads), and numerous dystrophic neurites (DNs) (Fig. 2A; arrows). Neuritic plaques showed colocalization of the antibodies pT231 (green channel) and TG-3 (blue channel), with TR (red channel). TR is a fluorescent marker with an affinity for the insoluble filaments of Aβ and tau, in neuritic plaques and NFTs, respectively [16, 17]. Fibrillar TR-positive Aβ aggregates were commonly observed in blood vessels (Fig. 2B; large arrows). NFTs positive for all three markers (pT231/TG-3/TR) were found in the periphery (Fig. 2B; small arrows). Neuropil threads and DNs were observed with a variable extent of colocalization for these markers. Aggregation of tau protein in the neuronal soma was also seen as diffuse granular staining called pre-NFTs (Fig. 2C; arrows). Pre-NFTs frequently showed dense granular perinuclear staining (Fig. 2C; arrowhead), immunoreactive with pT231 antibody (green channel), and colocalizing with the Alz50 antibody (blue channel). Pre-NFTs did not show an affinity for the TR dye. In a more advanced stage, pre-NFTs were distinguished in the periphery as small aggregates immunoreactive with these markers (Fig. 2C; small arrows). These first dense aggregates are referred to as small NFTs. They are observed in Fig. 2D, where colocalization of TG-3 and TR was observed (Fig. 2D; arrows). DNs with varying degrees of colocalization were found in the neighborhood. Intracellular NFTs (iNFTs) were flame-shaped (Fig. 2E; arrows) and recognized by TR and the antibody 423 (blue channel). Autofluorescent lipofuscin granules were observed in the rhodamine channel (*). The S199 antibody strongly recognized some DNs in the vicinity. Extracellular NFTs (eNFTs; Fig. 2F; arrows) were reactive with only antibody 423 (blue channel), that was colocalized with TR to varying extents. S199 antibody-positive DNs were detected in the periphery.
Fig.2
Characteristic lesions in AD. A) Neuritic plaques (NPs) bound thiazine red (TR). Associated with NPs, dystrophic neurites (DNs) (arrows) were immunoreactive with both pT231 (green channel) and TG3 (blue channel) antibodies, and these demonstrated varying extents of colocalization. B) Amyloid angiopathy. A blood vessel (BV) was labeled with TR (arrows). Many DNs and neurofibrillary tangles (NFTs) were observed at the proximity of the BV, and these showed colocalization with pT231/TG3/TR to varying extents. C-F) Aggregation pattern of tau in the neuronal soma. C) The pre-NFTs were characterized by diffuse granular staining with by RZ3 and Alz50 (arrow). Perinuclear staining (arrowhead) with no affinity for TR was also observed. Small dense tau aggregates (arrowheads) were observed in the vicinity of pre-NFTs. D) Small NFTs were recognized by pT231 and strongly colocalized with TR (arrows). E) Intracellular NFTs (iNFTs) recognized only by TR and colocalizing with the antibody 423; fibers tended to be compact (arrows). S199-reactive DNs were observed. F) Extracellular NFTs (eNFTs) showed immunoreactivity mainly with antibody 423 but with partial colocalization with TR. The eNFTs are a loose structure (arrows). Images obtained with the Leica SP8 Confocal Microscope. *Lipofuscin: autofluorescent granular structures. Scale bars, 10 μm, except for (B), 50 μm.
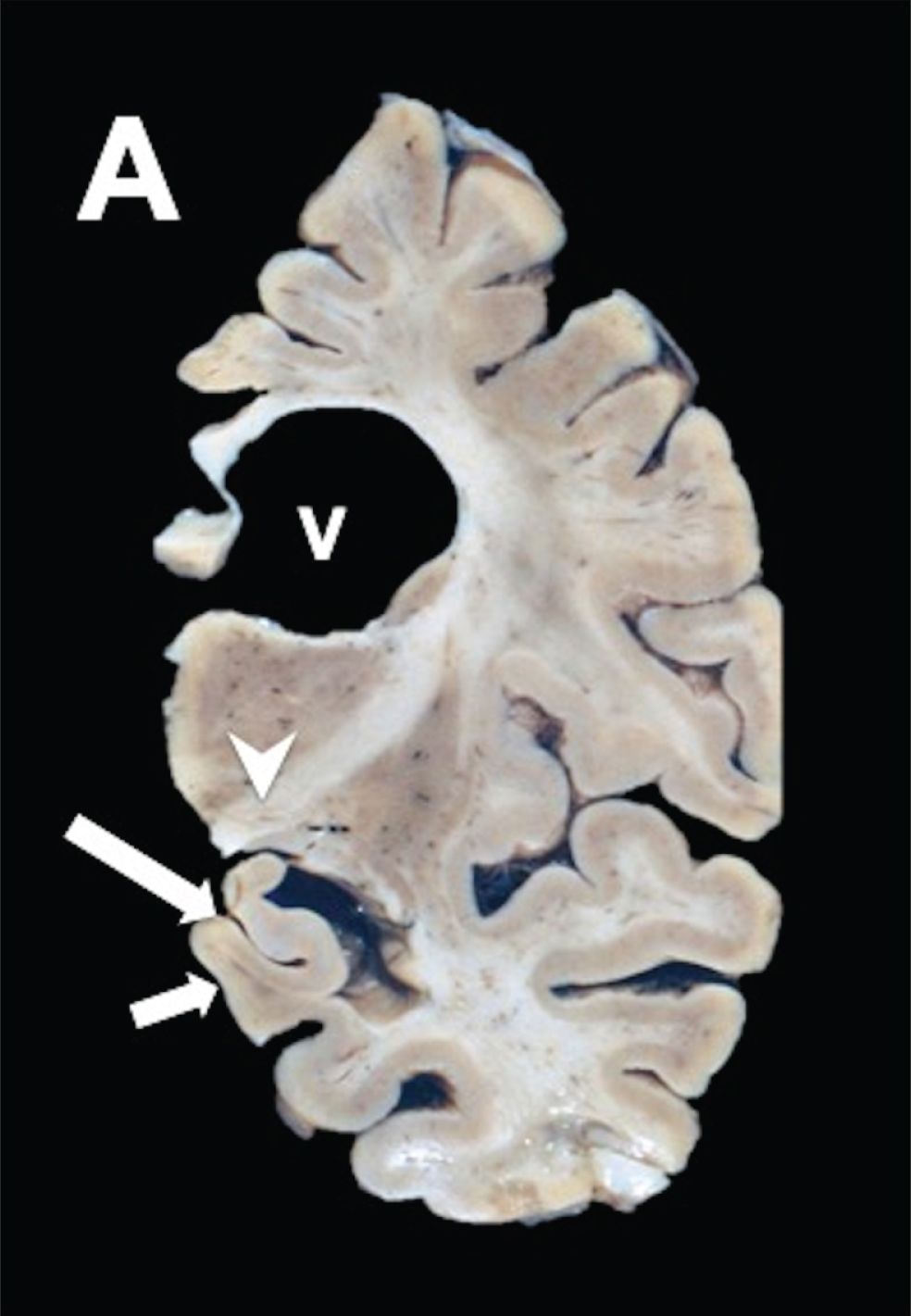
Neuropathological anatomy in PSP
Macroscopically, coronal sections at the level of the subthalamic nucleus showed that there was atrophy of the globus pallidus and subthalamic nucleus (Fig. 3A; arrowhead) in PSP brain. The hippocampus showed considerable atrophy in advanced stages (Fig. 3A; small arrow) and ventricles appeared enlarged (Fig 3A; V). The midbrain showed tegmental atrophy and loss of pigmentation in the substantia nigra (not shown). The locus coeruleus showed a relatively preserved pigmentation in the pontine section. The cerebellum presented with atrophy in the dentate nucleus (not shown).
Fig.3
PSP coronal brain section of the brain stem shows discrete frontotemporal atrophy, quadrigeminal cistern atrophy, and sieve state in frontal periventricular white matter, and thalamus.
Tau pathology in PSP
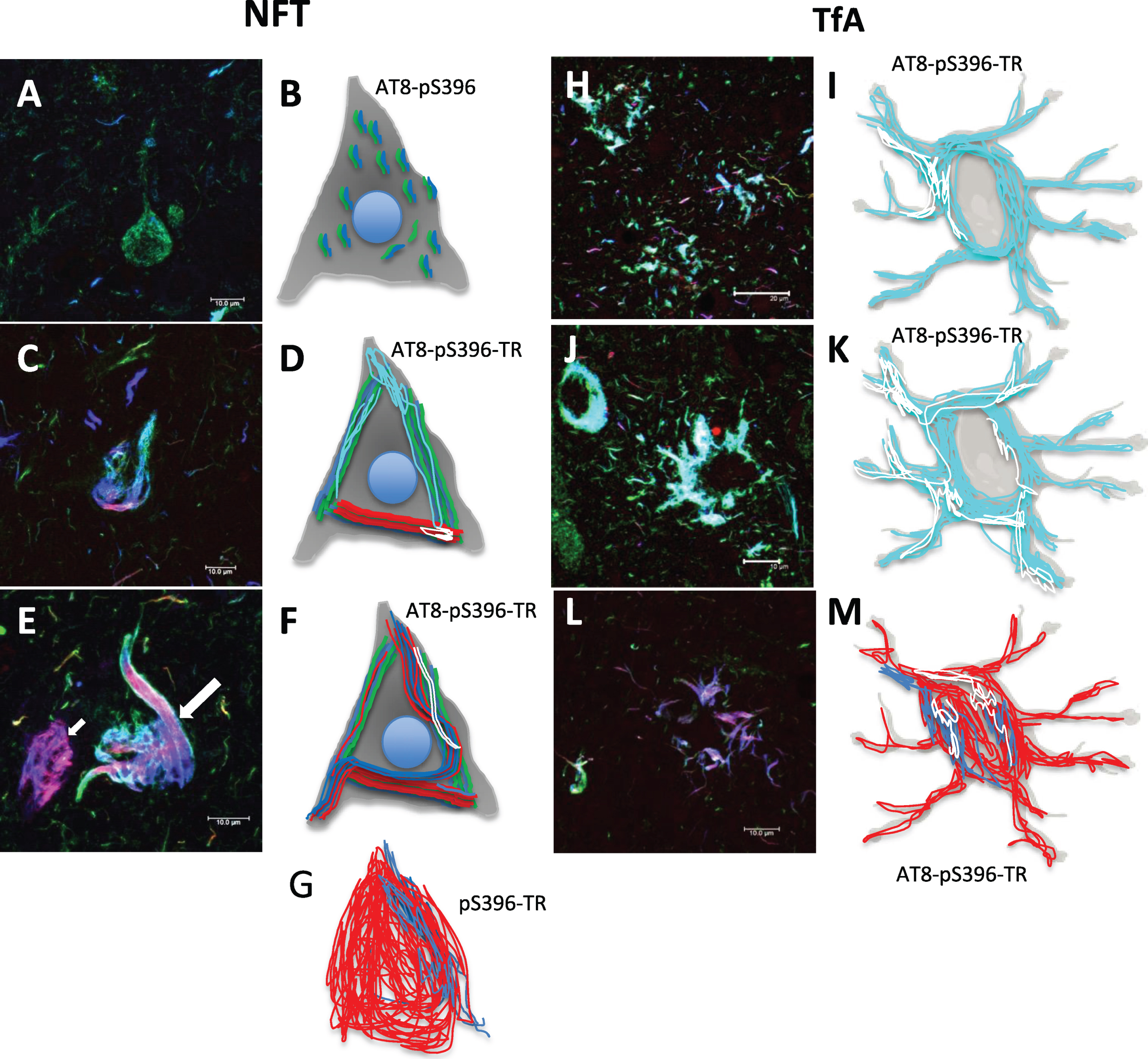
In PSP, the histopathological lesions are characterized by hyperphosphorylated tau aggregates predominantly of the 4R isoform. These aggregates are found in neurons, glial cells, and oligodendrocytes [18]. Fibrillar tau aggregates in TAs bound TR dye and were phosphorylated at both AT8 (green channel) and pS396 sites (Fig. 4A; arrows). This staining of small fibrils is called pre-NFT in PSP (Fig. 4; A*). Phosphorylated tau was expressed as fibrillar aggregates in globose NFTs (gNFTs), where there was colocalization of AT8/pS396 (Fig. 4B; large arrow). A gNFT population also showed that some aggregates were TR-positive (Fig. 4B; small arrow); others showed a complete absence of AT8-immunoreactivity. In Fig. 4C, pS396-positive NFTs characterized by a loose fibrillar structure, showed colocalizion with TR (Fig. 4C; arrow). Additionally, a dense spherical structure was found continuously within the neuronal bodies (Fig. 4D. Large arrows). This structure reacted with all three markers (AT8/TR/pS396). However, some small fibrils, positive only for the AT8 antibody (Fig. 4D; small arrows), were observed in the apical neurite.
Fig.4
Characteristic lesions in PSP immunostained with AT8 and pS396, counterstained with TR. A) Tufted astrocytes. B) Intracellular globose NFTs. C) Extracellular NFT. D) Pre-NFT positive for antibody pT231 (green channel) in the apical soma and dendrite. A dense spherical body was commonly observed in the neuronal soma, reactive with both antibodies and TR. Scale bars, 10 μm.
Tau aggregates in the neuronal soma in PSP brain
NFTs, DNs, and neuropil threads are present in both AD and PSP [19]. In PSP brains, several patterns of aggregation and polymerization of the tau protein in neuronal bodies were discernable. The first tau aggregation pattern was seen using the AT8 antibody, which labeled a large number of small fibrils throughout the neuronal soma (Fig. 5A; arrows). These fibrils were not recognized by either TR or pS396 antibody. These fibrils were also labeled by antibodies that recognize tau protein with intact N- (499) and C-terminal (Tau7) region. In the second aggregation pattern, small dense packets of tau were immunoreactive for AT8, pS396, and TR (Fig. 5B; arrows). Some fibrils, with the same characteristics described in Fig. 5A, were observed in the neuronal cytoplasm. DNs with similar staining characteristics were found in this vicinity. The small NFTs tended to elongate and fuse, invading the neuronal soma as a trabecula (Fig. 5C; arrow), positive for all three markers (AT8/TR/pS396). The tau aggregation process for the formation of iNFTs is shown in Fig. 5D-F. An immunoreactivity for markers AT8 and pS396, partially colocalized with TR, was observed. In the iNFTs, the TR staining was predominant and compact (Fig. 5F; arrows). The eNFTs were characterized by a loose fibrillar structure with scattered bundles of filaments (Fig. 5G; arrows). This fibrillar structure was labeled largely with TR and showed colocalization with pS396 only in discrete areas. AT8-positive DNs with varying degrees of TR colocalization were noticed in the periphery.
Fig.5
Aggregation pattern of tau in the neuronal soma in PSP. Double immunostaining with AT8 and pS396 antibodies, counterstained with TR. A) Pre-NFT labeled with AT8 antibody (arrows). Tau was aggregated as small fibrils that invade the neuronal soma completely. A large number of dystrophic neurites and neuropil threads were noticed, which colocalize with TR and are recognized by the pS396 antibody. B) Small NFTs. Both antibodies recognized small dense bundles of tau that invade the neuronal soma and apical dendrite. C) The small NFTs showed a tendency to fuse, forming an intracellular trabecula and invading the neuronal soma. In these, the three markers colocalized (arrows). D) Another common aggregation pattern observed was bundles of filaments recognized by TR colocalizing with both antibodies to various extents (arrows). E,F) Intracellular NFTs showed a tendency to fill the neuronal body with highly insoluble filaments. G) Finally, extracellular NFTs (eNFTs) were observed. The eNFTs were characterized by loosely packed fibers, recognized by the pS396 antibody and TR. In the periphery, dystrophic neurites immunoreactive only with AT8 (green channel) were found. Scale bars, 10 μm.
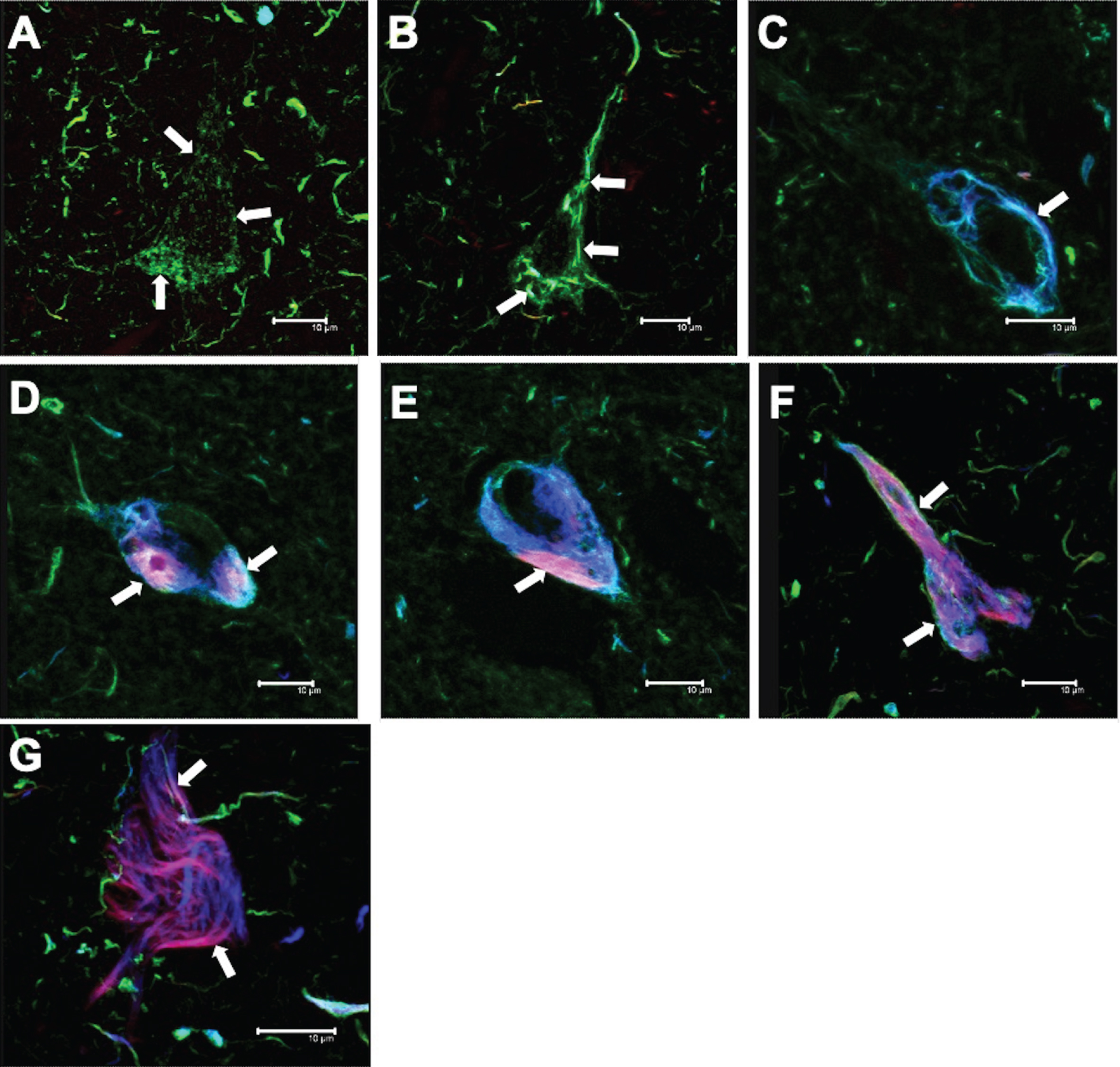
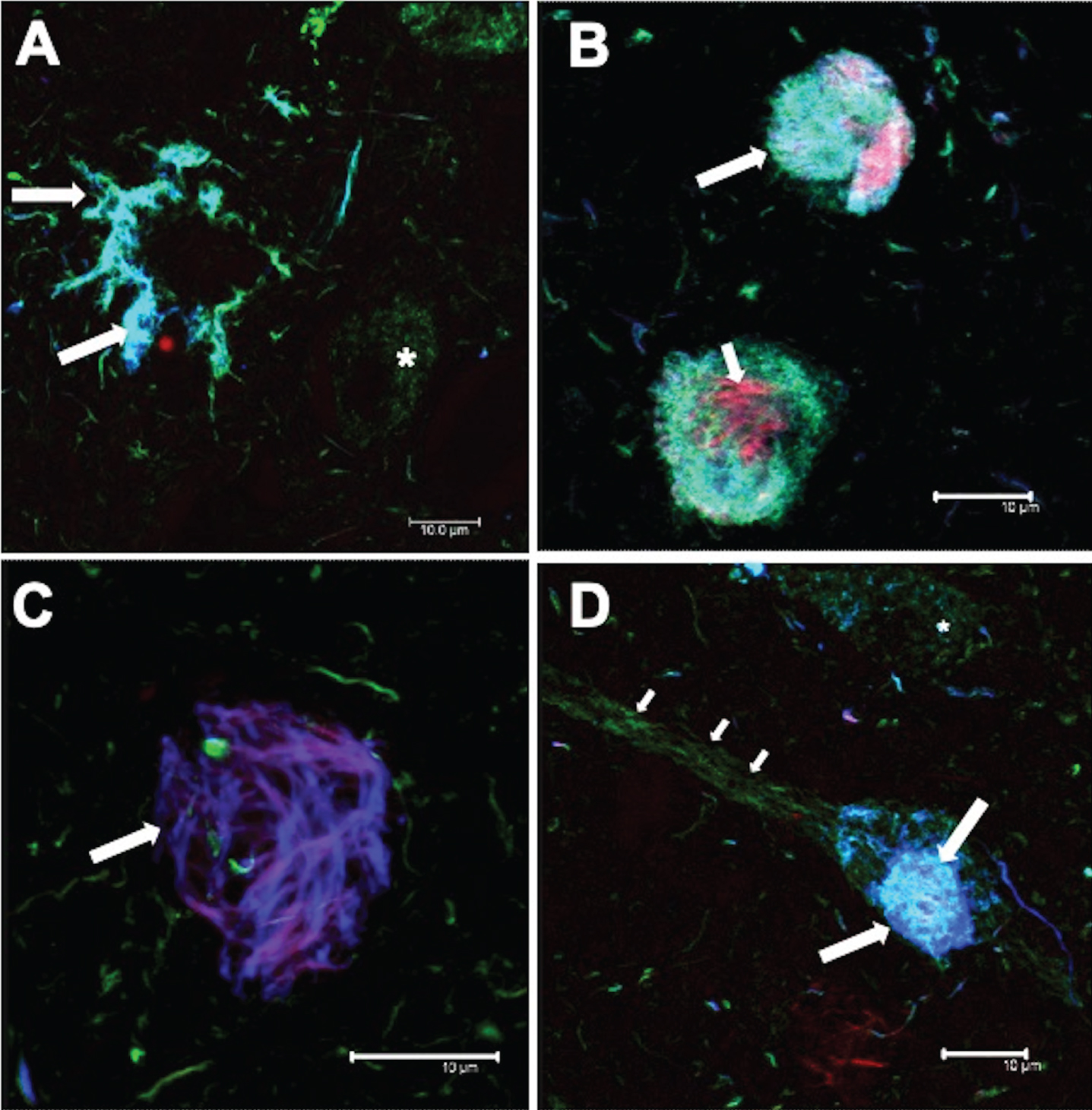
Tufted astrocytes
The following patterns of immunoreactivity were observed for TAs (Fig. 6). Individual glial cells and pairs of glial cells were observed (Fig. 6A; arrows). These cells were immunoreactive for both AT8 and pS396 antibodies, and showed a high affinity for TR. As the number of glial cells increased, a central space appeared (Fig. 6B; arrows). Glial cell aggregates of different sizes (Fig. 6C; arrows) formed the pathological TAs (Fig. 6D; arrows). These glial cells were TR-positive (Fig. 6D; small arrow).
Fig.6
Different tau expression patterns in tufted astrocytes (TAs) in PSP. TAs were strongly recognized by AT8 and pS396 and colocalized with TR.
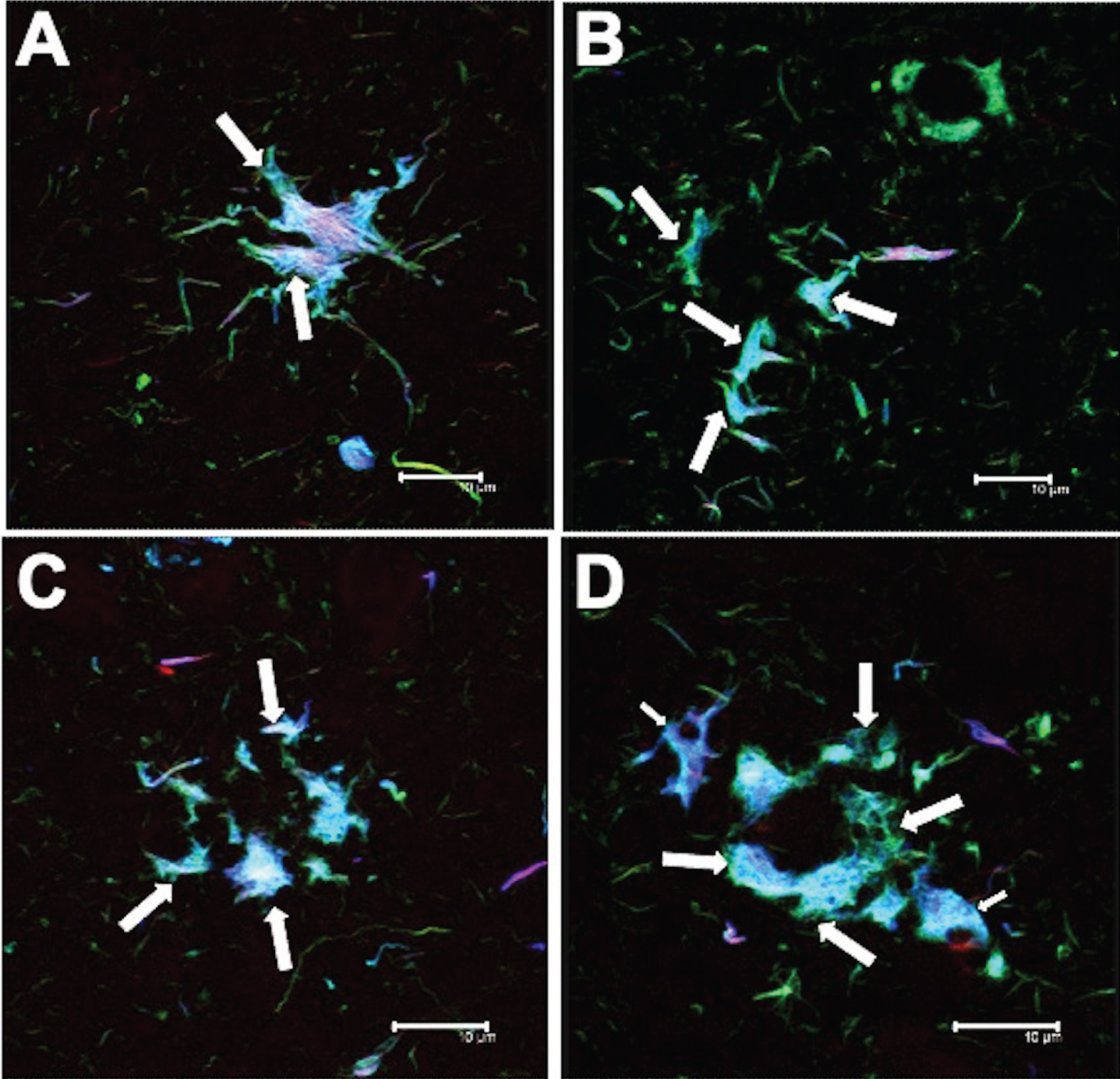
Molecular processing of tau in PSP
Intact tau protein
The presence of N-terminal tau in pre-NFTs, iNFTs, and TAs was demonstrated by an intermediate level of reactivity with the antibody 499 (Table 4). However, the C-terminal region of tau protein, recognized by the Tau7 antibody, was present only infrequently in the pre-NFTs and iNFTs (Table 4). Tau7-negative and TR-positive TAs were observed.
Table 4
Qualitative expression of post-translational modifications of pathological tau recognized by antibodies recognizing different epitopes. This analysis is exclusive for PSP cases. C-terminal tau protein (Tau-7 antibody) was detected weakly in the neurofibrillary tangles (NFTs). However, the tufted astrocytes (TAs) showed no positivity for this antibody. Phosphorylation at Thr231 not associated with conformational change was abundant in NFTs and TAs. There was no evidence of C-terminal truncation based on absence of immunoreactivity with TauC–3 and 423. Thiazine red (TR) recognized a population of NFTs and TAs
| Antibody | PreNFT | iNFT | Tufted astrocytes |
| 499 | ++ | ++ | ++ |
| TAU7 | +/– | +/– | – |
| pT231 | ++ | +++ | +++ |
| AT8 | ++ | ++ | +++ |
| TG3 | + | + | + |
| AT100 | + | + | + |
| ALZ50 | ++ | ++ | ++++ |
| pS396 | ++ | ++ | +++ |
| PHF1 | ++ | ++ | +++ |
| 499 | ++ | ++ | – |
| 423 | – | – | – |
| TAU–C3 | – | + | – |
| TR | – | ++ | +++ |
Phosphorylation of the N-terminal region of tau
The antibodies that recognize the phosphorylation sites at pT231 (Fig. 7A-C), pS202 and pT205 (AT8; Fig. 7D-F) in the tau N-terminal region, showed strong reactivity for pre-NFTs (Fig. 7; A++, D++) and iNFTs (Fig. 7B+++, E++). In Fig. 7A and D, preNFT with small fibers distributed in the neuronal soma (arrows) were observed. Both antibodies pT231 and AT8 showed immunoreactivity in various NFT populations at different stages of neurodegeneration (Fig. 7B, E). An NFT population appeared with a compact and dense structure (Fig. 7E); another NFT population showed a more flexible pattern. DNs were observed in the vicinity (small arrow). TAs showed a strong reactivity for both antibodies (Fig. 7C, F; arrows).
Fig.7
Expression of different tau epitopes in pre-NFT, iNFT, and tufted astrocytes in PSP. Left) A-C) pT231, D-F) AT8, G-I) TG3, J-L) AT100, M-O) Alz50, P-R) pS396, and S-U) PHF1. Right) Schematic with the location of the tau epitopes. Although all antibodies have affinity for the structures shown, the expression pattern was qualitatively different (see Table 4). Scale bars, 10 μm.
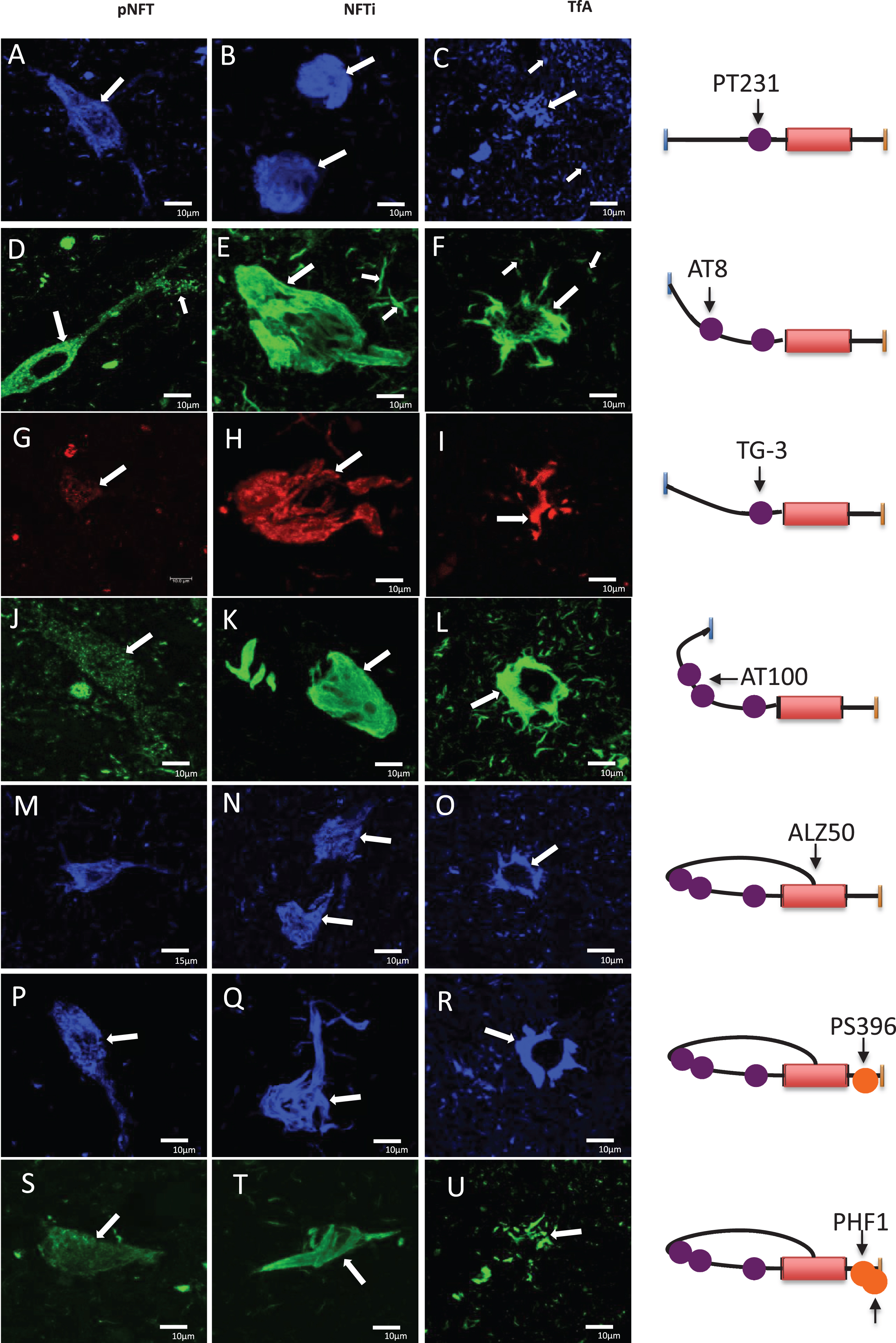
Regional conformational changes
The conformational changes were investigated using by TG-3 (Fig. 7G-I) and AT100 (Fig. 7J-L). Qualitatively, the abundance of immunoreactive structures with both antibodies was limited. Moderate reactivity for TG-3 and AT100 was observed in preNFTs (Fig. 7G+, J+), iNFTs (Fig. 7H+, K+), and TAs (Fig. 7I+, K+), and with the same characteristics previously described.
Structural conformational changes
The structural conformational changes recognized by the antibody Alz50 recognizes tau with its N-terminal portion intact. The Alz50-immunoreactivity in preNFTs showed small fibrils (Fig 7M, O; arrows). The reactivity in preNFTs and iNFTs was moderate (++). This staining did not recognize the entire structure of the iNFTs (Fig. 7N; arrows). TAs were strongly reactive with Alz50 (+++, Fig. 7R; arrows).
Phosphorylation of the C-terminal region of tau
The antibodies pS396 (Fig. 7P-R) and PHF1 (Fig. 7S-U) labeled preNFTs (Fig. 7P, S) and iNFTs with moderate abundance (++, Fig. 7Q, T; arrows). These antibodies reacted abundantly with the TAs (+++). Additionally, the pS396 antibody evidenced transitional NFTs, from iNFTs to eNFTs (Fig. 7Q; arrow).
Extracellular neurofibrillary tangles
In AD, eNFTs are composed primarily of the protease-resistant PHF-core comprised of a 92–95 amino acid fragment culminating at Glu391 and recognized by the 423 antibody. Truncation at Asp421, generated by caspase-3, has been associated with the early molecular stages of neurodegeneration in AD [20]. Truncation at Asp421 and Glu391 favor the polymerization of tau protein into highly insoluble filaments.
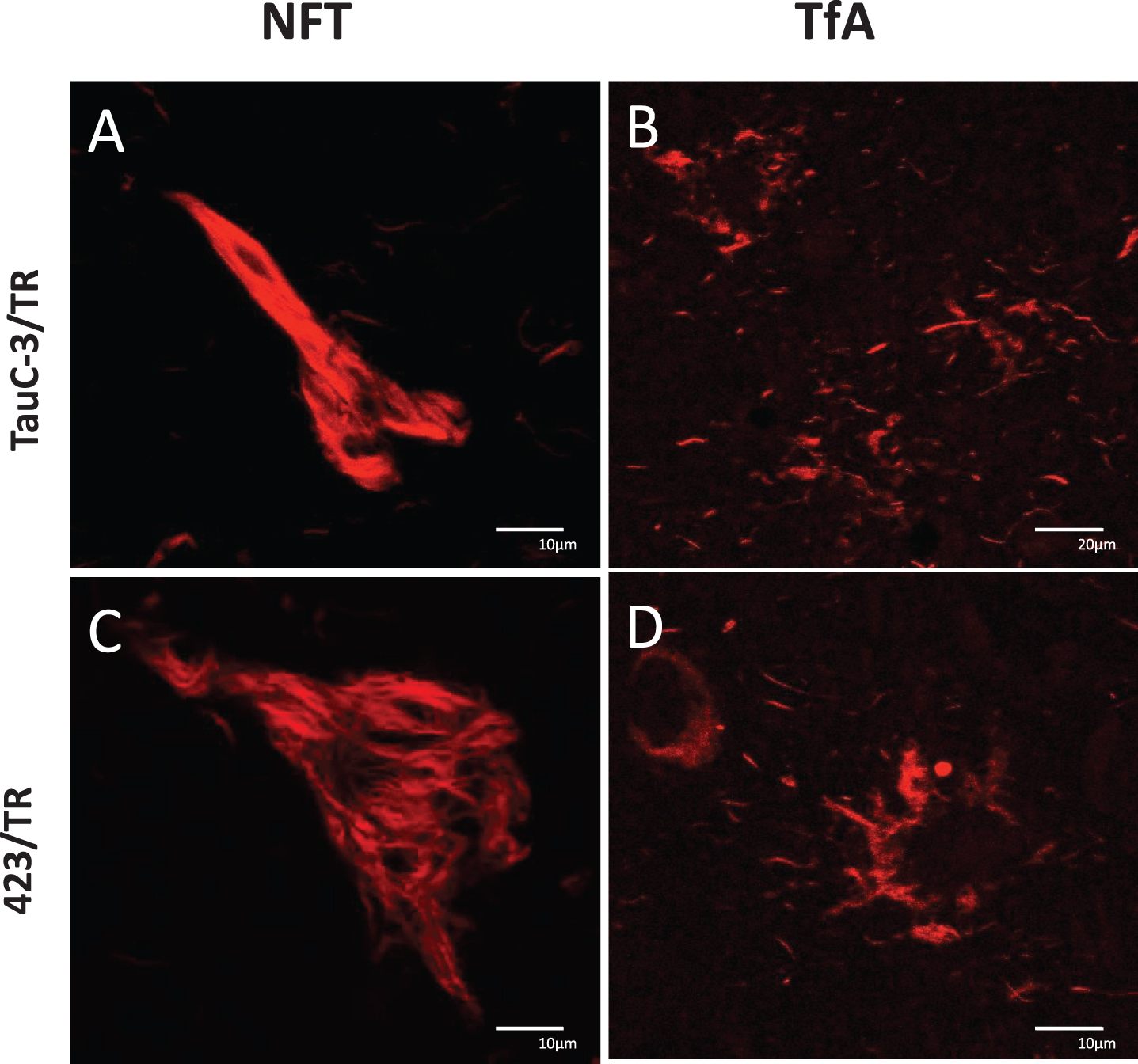
Truncated tau
Tau truncated at Asp421 and Glu391 is recognized by TauC-3 and 423, respectively. The iNFTs and eNFTs in PSP were not detected with either of these antibodies (Fig. 8A, C, respectively). These tangles bound TR. In contrast, the TAs in PSP were labeled by TR but not by Tau-C3 or 423.
Fig.8
Absence of tau truncation in PSP: A, B) TauC-3 and C, D) 423. Lesions labeled by TR: A) iNFT, C) eNFT, and in B, D) tufted astrocytes. Scale bars, 10 μm, except for (B), 20 μm.
DISCUSSION
PSP is a disorder with a very low prevalence and that has been little studied to date. The histopathological lesions in PSP are NFTs and TAs [21]; with the 4R-tau isoforms predominant in PSP [22]. Few studies have focused on describing the morphological development of tau pathology in this disorder. A more in-depth analysis may favor a better understanding of the disease and the discovery of specific markers for this and other tauopathies.
Aggregation of tau protein in NFT
In AD, the tau protein aggregation in the neuronal body is characterized by the presence of diffuse granular staining and various stages of NFT development [23]. Tau protein referred to as pre-NFT is characterized by a diffuse granular distribution in the cytoplasm and perinuclear region. Antibodies directed against phosphorylated tau in these granular structures did not colocalize with TR [24, 25]. However, tau showed a strong immunoreactivity for phosphorylated tau markers in the neuronal soma. This pattern of immunoreactivity in AD differs from that found in PSP. In PSP, the first stages of aggregation showed abundant individual fibrils throughout the neuronal cytoplasm. TR did not bind to this structure, indicating that such aggregates are soluble [26, 27]. Later, dense aggregates were observed in the neuronal soma in both AD and PSP cases in our study. In the latter aggregates, a considerable accumulation of tau was observed, that was colocalized with TR, suggesting that these small bundles contain fibrillar structures with a β-folded conformation. These aggregates are similar in AD and PSP, with small tangles that tend to elongate and fuse, forming a trabecula of bundles of fibrillar structures, eventually invading the neuronal soma (iNFT), until tangles become extracellular [16, 23–25]. The eNFTs are characterized by having a loose morphology and being exposed to the extracellular space [16, 17, 24, 26, 28, 29].
Molecular processing of the tau protein
The pathological processing of tau is initiated by the presence of a truncated tau fragment of 92–95 amino acids in AD [25]. It is the minimal paired helical filament core fragment (PHF-core) [30–32] with a truncation at Glu391 [33], and which is recognized by the antibody 423 [34]. Recent studies have suggested that the PHF-core is an early initiating event of AD pathology [20]. It has been shown that the C-terminal region of tau is very susceptible to proteolysis [29, 35]. Caspase-3 can proteolyze tau at Asp421 and Binder developed an antibody, TauC-3, that recognizes this truncation [36]. In vitro studies have shown that the presence of truncated tau favors its polymerization [37–41].In PSP, study of the expression of tau with its intact C-terminus has not been studied. It is the opposite of the situation found in AD. Whereas the N- and C-terminal domains of full-length tau are abundant in iNFTs in AD, we did not see the C-terminus in PSP. This suggests that proteolytic processing of the C-terminus is more accelerated in PSP. Phospho-tau epitopes are abundant in both AD and PSP [42]. In contrast, tau phosphorylated at Ser396, that is abundantly observed in the first aggregation stage in the neuronal soma in PSP, is not observed in the pre-NFTs in AD [35]. The specific sequence of tau phosphorylation (as detected using pT231, TG3, AT8, and AT100 antibodies) may favor both conformational and regional changes (TG3 and AT100) [43], and generate structural changes (Alz50) [24, 25, 44]. Phosphorylation of tau at Ser396 has been associated with late events in tau processing [25]. Previous biochemical studies, using different antibodies against phosphorylated tau (Thr181, Ser202, Ser214, Ser396, and Ser422), showed bands at 72, 50/55, and 37 kDa. Bands at 60, 42, 33, and 29 kDa were also observed. The different molecular weights and antibody affinities are associated with the proteolysis of tau [45]. In AD, bands at 75–60–65 kDa corresponded to full-length, phosphorylated tau. The 45 and 12 kDa bands were compatible with truncated forms at Asp421 and Glu391 (recognized by the TauC-3 and 423 antibodies, respectively). It has been described that the protein fragment corresponding to the PHF-core forms straight and helical filaments with a beta-folded conformation under physiological conditions. It shares a macromolecular structure similar to the purified filaments from the AD brains [46]. In our analysis, no immunoreactivity was observed with the antibody 423. Truncation at Asp421 was reported in occasional NFTs in PSP previously [45]. This suggests that truncation at Glu391 is not a feature of PSP, according to Binder [42]. Some eNFT and glial cell populations, labeled only by TR, were observed in PSP. TR has an affinity for fibrillar structures with beta-folded conformation. This suggests that one of the main differences in tau processing between tauopathies is the presence of additional truncation sites that favor the tau polymerization and insolubility. Further studies are required to verify this hypothesis. Our results suggest that other forms of truncated tau could act as differential markers between tauopathies.
Processing and polymerization of tau in neuronal and glial cells in PSP
Tau aggregation occurs with intact and variably phosphorylated tau protein molecules. Truncated tau expression favors polymerization. In Fig. 9, we summarize the observations described in this work. The pre-NFT stage (Fig. 9A, B) is characterized by the aggregation of tau into small fibrils, in which TR has no affinity. In AD, tau phosphorylated at Ser396 is not observed in pre-NFTs (results not shown). iNFTs are a massive aggregation of TR-related filaments (Fig. 9C, D). Sporadically, presence of moderate levels of tau truncated at Asp421 was observed in the iNFTs. An eNFT population with a high affinity for TR and partially immunoreactive to the antibody pS396 was found (Fig. 9E; arrow, Fig. 9G). Tau aggregation appears to be a much more accelerated process in TAs than in neuronal NFTs. Tau occurs as abundant dense aggregates related to TR in the glial population. Fibrillar TR-positive tau was observed (Fig. 9G, H, I, J). This could be due to faster processing of tau by glial cells; these cells are involved in degradation and their proteolytic enzymes could accelerate tau proteolysis. However, no immunoreactivity of the antibody 423 to Glu-391 truncation was observed in neuronal or glial NFTs. As previously suggested, this fragment is highly toxic and favors the tau polymerization. The glial cell population showed a strong affinity for TR. Therefore, this result suggests that there could be variability in the truncation sites that favor the polymerization of tau for different disorders and there may be truncations that are both specific and characteristic for PSP.
Fig.9
Schematic representation of the tau aggregation process in neuronal and glial degeneration in PSP. A-G) Neuronal degeneration, H-M) Glial degeneration. A) Neurofibrillary pre-tangle. B) Representation showing a staining of small fibrils positive to the AT8 and pS396 antibodies and distributed throughout the neuronal soma. C, D, F. Arrow) Representation of different tau aggregation events, positive for TR and colocalizing to varying degrees with AT8 and pS396 antibodies. E, G) NFT populations were recognized by TR and partially by the pS396 antibody. H, I, J, K) Tau aggregation pattern in glial cells, which colocalize to varying extents for AT8 and pS396 antibodies. L, M) Glial cells were mostly TR-positive. Scale bars, 100 μm.
Clinical implications of the tau pathological processing in AD and PSP
In AD, there is a clear clinicopathological correlation between cognitive deterioration and the presence of NFTs in the hippocampus. Braak and Braak described a staging sequence for the appearance of NFT in the entorhinal cortex and hippocampus [47]. Later studies showed the relationship between these lesions and cognitive impairment in AD [48]. An attempt has been made to correlate the presence of NFTs and tau aggregation in glial cells with the clinical evolution of PSP pathology. Zuzana Stejskalova analyzed the alteration of the pyramidal motor system for various types of PSP (probable, possible or suggestive, PSP-Richarson PSP-Parkinsonism). Although a greater burden of histopathological lesions was observed in PSP-Richarson, a significant correlation between clinical symptoms and pathology was not found [49]. On the other hand, areas of the pallido-nigro-Luysial axis have recently been analyzed in different clinical subtypes of PSP. Tau in NFTs and glial cells suggest that there are different processes regarding the clinical course of the patient and the distribution of these lesions in the specific regions. The vulnerability of cell populations in different areas needs to be determined as does the spread of tau. Pathological tau can be transmitted between neurons in a prion-like manner, as suggested for other neurodegenerative diseases [41].
In PSP-Richardson, NFT accumulation has been observed initially in subcortical and brainstem nuclei. Early oligodendroglial damage in the globus pallidus and astroglial involvement in the striatum has been found. Subsequently, tau in astroglial cells continues in cortical areas, which precede the accumulation of cortical and oligodendroglial NFTs. This continues together in a temporal sequence from frontoparietal-temporal to occipital cortex. In PSP, it has been proposed that NFTs can be found in early affected areas. This could be sought, through neuroimaging studies, in preclinical forms to understand the earliest pathogenic events [50].
The presence of tau in NFTs, glial and microglial cells are considered as pathological structures. In AD, NFTs are correlated with the clinical deterioration of patients. However, although a greater number of lesions have been evidenced in PSP compared to AD, no correlation with abnormal tau has been reported in PSP. In PSP, pathological structures are the products of much earlier events, for example, a sustained inflammatory activity of glial and microglial cells. This inflammation is produced by interleukins, cytokines, and abundant reactive oxygen species, which affect the neuronal metabolism and favor the pathological tau processing, including hyperphosphorylation and truncation [51].
CONCLUSION
The molecular processing of phosphorylated tau differs between PSP and AD. In this study, we observed the proteolytic processing of the C-terminal portion of tau in AD and PSP. The regional (TG3 and AT100) and conformational (Alz-50) changes, using specific markers, were described in PSP. Contrary to AD, phosphorylation at Ser396 was observed in pre-NFTs in PSP. The truncation event of tau may be pivotal in finding specific and differential markers for AD, PSP, and other tauopathies.
ACKNOWLEDGMENTS
This work was supported by Fondo Nacional de Ciencia y Tecnologia, FONDOCyT, from the Ministry of Higher Education, Science and Technology, Dominican Republic (2015-3A2-127 to MP-H) and (2018-2019-2A3-208 to JL-M and MP-H).
The authors want to express their gratitude to the following: Dr. P. Davies† (Albert Einstein College of Medicine, Bronx, NY, USA) and Lester I. Binder† (North Western, Chicago, IL, USA) for the generous gift of mAbs TG–3 and Alz–50, and Tau–1, Tau–5 and Tau–7, respectively; Tec. Amparo Viramontes Pintos for the handling of the brain tissue; support in the confocal microscopy unit of CIIDIR Durango, Instituto Politécnico Nacional; Union Medica University Clinic, Dominican Republic, for their support and collaboration in the development of this research project. We also want to express our gratitude to the Mexican families who have donated the brain of their loved ones affected with Alzheimer’s disease and made our research possible. This work is dedicated to the memory of Professor Dr. José Raúl Mena López†. †Deceased.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/20-1139r2).
REFERENCES
[1] | Boxer AL , Yu JT , Golbe LI , Litvan I , Lang AE , Hoglinger GU ((2017) ) Advances in progressive supranuclear palsy: New diagnostic criteria, biomarkers, and therapeutic approaches. Lancet Neurol 16: , 552–563. |
[2] | Steele JC , Richardson JC , Olszewski J ((1964) ) Progressive supranuclear palsy. A heterogeneous degeneration involving the brain stem, basal ganglia and cerebellum with vertical gaze and pseudobulbar palsy, nuchal dystonia and dementia. Arch Neurol 10: , 333–359. |
[3] | Iqbal K , Alonso Adel C , Chen S , Chohan MO , El-Akkad E , Gong CX , Khatoon S , Li B , Liu F , Rahman A , Tanimukai H , Grundke-Iqbal I ((2005) ) Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta 1739: , 198–210. |
[4] | Respondek G , Hoglinger GU ((2016) ) The phenotypic spectrum of progressive supranuclear palsy.. Parkinsonism Relat Disord 22: (Suppl 1), S34–36. |
[5] | Grimm MJ , Respondek G , Stamelou M , Arzberger T , Ferguson L , Gelpi E , Giese A , Grossman M , Irwin DJ , Pantelyat A , Rajput A , Roeber S , van Swieten JC , Troakes C , Meissner WG , Nilsson C , Piot I , Compta Y , Rowe JB , Hoglinger GU , Movement Disorder Society-Endorsed PSP Study Group ((2020) ) Clinical Conditions “suggestive of progressive supranuclear palsy”-diagnostic performance. Mov Disord 35: , 2301–2313. |
[6] | Hoglinger GU , Respondek G , Stamelou M , Kurz C , Josephs KA , Lang AE , Mollenhauer B , Muller U , Nilsson C , Whitwell JL , Arzberger T , Englund E , Gelpi E , Giese A , Irwin DJ , Meissner WG , Pantelyat A , Rajput A , van Swieten JC , Troakes C , Antonini A , Bhatia KP , Bordelon Y , Compta Y , Corvol JC , Colosimo C , Dickson DW , Dodel R , Ferguson L , Grossman M , Kassubek J , Krismer F , Levin J , Lorenzl S , Morris HR , Nestor P , Oertel WH , Poewe W , Rabinovici G , Rowe JB , Schellenberg GD , Seppi K , van Eimeren T , Wenning GK , Boxer AL , Golbe LI , Litvan I , Movement Disorder Society-endorsed PSP Study Group ((2017) ) Clinical diagnosis of progressive supranuclear palsy: The movement disorder society criteria. Mov Disord 32: , 853–864. |
[7] | Reich SG ((2020) ) The movement disorder society clinical criteria for progressive supranuclear palsy: The movie version. Parkinsonism Relat Disord 78: , 198–199. |
[8] | Fleury V , Brindel P , Nicastro N , Burkhard PR ((2018) ) Descriptive epidemiology of parkinsonism in the Canton of Geneva, Switzerland. Parkinsonism Relat Disord 54: , 30–39. |
[9] | Nath U , Ben-Shlomo Y , Thomson RG , Morris HR , Wood NW , Lees AJ , Burn DJ ((2001) ) The prevalence of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome) in the UK. Brain 124: , 1438–1449. |
[10] | Avila J ((2000) ) Tau aggregation into fibrillar polymers: Taupathies. FEBS Lett 476: , 89–92. |
[11] | Rosler TW , Tayaranian Marvian A , Brendel M , Nykanen NP , Hollerhage M , Schwarz SC , Hopfner F , Koeglsperger T , Respondek G , Schweyer K , Levin J , Villemagne VL , Barthel H , Sabri O , Muller U , Meissner WG , Kovacs GG , Hoglinger GU ((2019) ) Four-repeat tauopathies. Prog Neurobiol 180: , 101644. |
[12] | Rojo A , Pernaute RS , Fontan A , Ruiz PG , Honnorat J , Lynch T , Chin S , Gonzalo I , Rabano A , Martinez A , Daniel S , Pramstaller P , Morris H , Wood N , Lees A , Tabernero C , Nyggard T , Jackson AC , Hanson A , de Yebenes JG ((1999) ) Clinical genetics of familial progressive supranuclear palsy. Brain 122: (Pt 7), 1233–1245. |
[13] | Rossor MN , Brown J ((1995) ) Progressive supranuclear palsy: Neuropathologically based diagnostic clinical criteria. J Neurol Neurosurg Psychiatry 59: , 343. |
[14] | Baba Y , Putzke JD , Whaley NR , Wszolek ZK , Uitti RJ ((2006) ) Progressive supranuclear palsy: Phenotypic sex differences in a clinical cohort. Mov Disord 21: , 689–692. |
[15] | McKhann G , Drachman D , Folstein M , Katzman R , Price D , Stadlan EM ((1984) ) Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34: , 939–944. |
[16] | Luna-Munoz J , Peralta-Ramirez J , Chavez-Macias L , Harrington CR , Wischik CM , Mena R ((2008) ) Thiazin red as a neuropathological tool for the rapid diagnosis of Alzheimer’s disease in tissue imprints. Acta Neuropathol 116: , 507–515. |
[17] | Mena R , Edwards PC , Harrington CR , Mukaetova-Ladinska EB , Wischik CM ((1996) ) Staging the pathological assembly of truncated tau protein into paired helical filaments in Alzheimer’s disease. Acta Neuropathol 91: , 633–641. |
[18] | Dickson DW ((1999) ) Neuropathologic differentiation of progressive supranuclear palsy and corticobasal degeneration. J Neurol 246: (Suppl 2), II6–15. |
[19] | Bancher C , Lassmann H , Budka H , Grundke-Iqbal I , Iqbal K , Wiche G , Seitelberger F , Wisniewski HM ((1987) ) Neurofibrillary tangles in Alzheimer’s disease and progressive supranuclear palsy: Antigenic similarities and differences. Microtubule-associated protein tau antigenicity is prominent in all types of tangles. Acta Neuropathol 74: , 39–46. |
[20] | Luna-Viramontes NI , Campa-Córdoba BB , Ontiveros-Torres MÁ , Harrington CR , Villanueva-Fierro I , Guadarrama-Ortíz P , Garcés-Ramírez L , de la Cruz F , Hernandes-Alejandro M , Martínez-Robles S , González-Ballesteros E , Pacheco-Herrero M , Luna-Muñoz J ((2020) ) PHF-core tau as the potential initiating event for tau pathology in Alzheimer’s disease. Front Cell Neurosci 14: , 1–11. |
[21] | Murakami A , Nakamura M , Kaneko S , Lin WL , Dickson DW , Kusaka H ((2018) ) Aberrant accumulation of ErbB4 in progressive supranuclear palsy. Neuropathol Appl Neurobiol 44: , 563–573. |
[22] | Sergeant N , Wattez A , Delacourte A ((1999) ) Neurofibrillary degeneration in progressive supranuclear palsy and corticobasal degeneration: Tau pathologies with exclusively “exon 10” isoforms. J Neurochem 72: , 1243–1249. |
[23] | Mena R , Wischik CM , Novak M , Milstein C , Cuello AC ((1991) ) A progressive deposition of paired helical filaments (PHF) in the brain characterizes the evolution of dementia in Alzheimer’s disease. An immunocytochemical study with a monoclonal antibody against the PHF core. J Neuropathol Exp Neurol 50: , 474–490. |
[24] | Luna-Munoz J , Chavez-Macias L , Garcia-Sierra F , Mena R ((2007) ) Earliest stages of tau conformational changes are related to the appearance of a sequence of specific phospho-dependent tau epitopes in Alzheimer’s disease. J Alzheimers Dis 12: , 365–375. |
[25] | Luna-Munoz J , Garcia-Sierra F , Falcon V , Menendez I , Chavez-Macias L , Mena R ((2005) ) Regional conformational change involving phosphorylation of tau protein at the Thr231, precedes the structural change detected by Alz-50 antibody in Alzheimer’s disease. J Alzheimers Dis 8: , 29–41. |
[26] | Mena R , Edwards P , Perez-Olvera O , Wischik CM ((1995) ) Monitoring pathological assembly of tau and beta-amyloid proteins in Alzheimer’s disease. Acta Neuropathol 89: , 50–56. |
[27] | ReschJF, LehrGS, Wischik CM ((1991) ) Design and synthesis of a potential affinity/cleaving reagent for beta-pleated sheet protein structures. Bioorg Med Chem Lett 1: , 519–522. |
[28] | Galvan M , David JP , Delacourte A , Luna J , Mena R ((2001) ) Sequence of neurofibrillary changes in aging and Alzheimer’s disease: A confocal study with phospho-tau antibody, AD2. J Alzheimers Dis 3: , 417–425. |
[29] | Garcia-Sierra F , Ghoshal N , Quinn B , Berry RW , Binder LI ((2003) ) Conformational changes and truncation of tau protein during tangle evolution in Alzheimer’s disease. J Alzheimers Dis 5: , 65–77. |
[30] | Wischik CM , Crowther RA , Stewart M , Roth M ((1985) ) Subunit structure of paired helical filaments in Alzheimer’s disease. J Cell Biol 100: , 1905–1912. |
[31] | Novak M , Jakes R , Edwards PC , Milstein C , Wischik CM ((1991) ) Difference between the tau protein of Alzheimer paired helical filament core and normal tau revealed by epitope analysis of monoclonal antibodies 423 and 7.51. Proc Natl Acad Sci U S A 88: , 5837–5841. |
[32] | Novak M , Kabat J , Wischik CM ((1993) ) Molecular characterization of the minimal protease resistant tau unit of the Alzheimer’s disease paired helical filament. EMBO J 12: , 365–370. |
[33] | Wischik CM , Novak M , Edwards PC , Klug A , Tichelaar W , Crowther RA ((1988) ) Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci U S A 85: , 4884–4888. |
[34] | Novak M , Wischik CM , Edwards P , Pannell R , Milstein C ((1989) ) Characterisation of the first monoclonal antibody against the pronase resistant core of the Alzheimer PHF. Prog Clin Biol Res 317: , 755–761. |
[35] | Flores-Rodriguez P , Ontiveros-Torres MA , Cardenas-Aguayo MC , Luna-Arias JP , Meraz-Rios MA , Viramontes-Pintos A , Harrington CR , Wischik CM , Mena R , Floran-Garduno B , Luna-Munoz J ((2015) ) The relationship between truncation and phosphorylation at the C-terminus of tau protein in the paired helical filaments of Alzheimer’s disease. Front Neurosci 9: , 33. |
[36] | Gamblin TC , Berry RW , Binder LI ((2003) ) Tau polymerization: Role of the amino terminus. Biochemistry 42: , 2252–2257. |
[37] | Gamblin TC , Berry RW , Binder LI ((2003) ) Modeling tau polymerization in vitro: A review and synthesis. Biochemistry 42: , 15009–15017. |
[38] | Gamblin TC , Chen F , Zambrano A , Abraha A , Lagalwar S , Guillozet AL , Lu M , Fu Y , Garcia-Sierra F , LaPointe N , Miller R , Berry RW , Binder LI , Cryns VL ((2003) ) Caspase cleavage of tau: Linking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proc Natl Acad Sci U S A 100: , 10032–10037. |
[39] | Wegmann S , Medalsy ID , Mandelkow E , Muller DJ ((2013) ) The fuzzy coat of pathological human Tau fibrils is a two-layered polyelectrolyte brush.E. Proc Natl Acad Sci U S A 110: , 313–321. |
[40] | Wischik CM ((1989) ) Cell biology of the Alzheimer tangle. Curr Opin Cell Biol 1: , 115–122. |
[41] | Wischik CM , Schelter BO , Wischik DJ , Storey JMD , Harrington CR ((2018) ) Modeling prion-like processing of tau protein in Alzheimer’s disease for pharmaceutical development. J Alzheimers Dis 62: , 1287–1303. |
[42] | Berry RW , Sweet AP , Clark FA , Lagalwar S , Lapin BR , Wang T , Topgi S , Guillozet-Bongaarts AL , Cochran EJ , Bigio EH , Binder LI ((2004) ) Tau epitope display in progressive supranuclear palsy and corticobasal degeneration. J Neurocytol 33: , 287–295. |
[43] | Zheng-Fischhofer Q , Biernat J , Mandelkow EM , Illenberger S , Godemann R , Mandelkow E ((1998) ) Sequential phosphorylation of Tau by glycogen synthase kinase-3beta and protein kinase A at Thr212 and Ser214 generates the Alzheimer-specific epitope of antibody AT100 and requires a paired-helical-filament-like conformation. Eur J Biochem 252: , 542–552. |
[44] | Carmel G , Mager EM , Binder LI , Kuret J ((1996) ) The structural basis of monoclonal antibody Alz50’s selectivity for Alzheimer’s disease pathology. J Biol Chem 271: , 32789–32795. |
[45] | Puig B , Rey MJ , Ferrer I ((2005) ) Individual and regional variations of phospho-tau species in progressive supranuclear palsy. Acta Neuropathol 110: , 261–268. |
[46] | Al-Hilaly YK , Foster BE , Biasetti L , Lutter L , Pollack SJ , Rickard JE , Storey JMD , Harrington CR , Xue WF , Wischik CM , Serpell LC ((2020) ) Tau (297-391) forms filaments that structurally mimic the core of paired helical filaments in Alzheimer’s disease brain. FEBS Lett 594: , 944–950. |
[47] | Braak H , Braak E ((1991) ) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82: , 239–259. |
[48] | Garcia-Sierra F , Hauw JJ , Duyckaerts C , Wischik CM , Luna-Munoz J , Mena R ((2000) ) The extent of neurofibrillary pathology in perforant pathway neurons is the key determinant of dementia in the very old. Acta Neuropathol 100: , 29–35. |
[49] | Stejskalova Z , Rohan Z , Rusina R , Tesar A , Kukal J , Kovacs GG , Bartos A , Matej R ((2019) ) Pyramidal system involvement in progressive supranuclear palsy - a clinicopathological correlation. BMC Neurol 19: , 42. |
[50] | Kovacs GG , Lukic MJ , Irwin DJ , Arzberger T , Respondek G , Lee EB , Coughlin D , Giese A , Grossman M , Kurz C , McMillan CT , Gelpi E , Compta Y , van Swieten JC , Laat LD , Troakes C , Al-Sarraj S , Robinson JL , Roeber S , Xie SX , Lee VM , Trojanowski JQ , Hoglinger GU ((2020) ) Distribution patterns of tau pathology in progressive supranuclear palsy. Acta Neuropathol 140: , 99–119. |
[51] | Fernandez JA , Rojo L , Kuljis RO , Maccioni RB ((2008) ) The damage signals hypothesis of Alzheimer’s disease pathogenesis. J Alzheimers Dis 14: , 329–333. |
[52] | Wischik CM , Edwards PC , Lai RY , Roth M , Harrington CR ((1996) ) Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc Natl Acad Sci U S A 93: , 11213–11218. |
[53] | Jicha GA , Lane E , Vincent I , Otvos L Jr. , Hoffmann R , Davies P ((1997) ) A conformation- and phosphorylation-dependent antibody recognizing the paired helical filaments of Alzheimer’s disease. J Neurochem 69: , 2087–2095. |
[54] | Jicha GA , Bowser R , Kazam IG , Davies P ((1997) ) Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res 48: , 128–132. |
[55] | Bhaskar K , Konerth M , Kokiko-Cochran ON , Cardona A , Ransohoff RM , Lamb BT ((2010) ) Regulation of tau pathology by the microglial fractalkine receptor. Neuron 68: , 19–31. |


