
Shear-Induced Amyloid Aggregation in the Brain: V. Are Alzheimer’s and Other Amyloid Diseases Initiated in the Lower Brain and Brainstem by Cerebrospinal Fluid Flow Stresses?
Abstract
Amyloid-β (Aβ) and tau oligomers have been identified as neurotoxic agents responsible for causing Alzheimer’s disease (AD). Clinical trials using Aβ and tau as targets have failed, giving rise to calls for new research approaches to combat AD. This paper provides such an approach. Most basic AD research has involved quiescent Aβ and tau solutions. However, studies involving laminar and extensional flow of proteins have demonstrated that mechanical agitation of proteins induces or accelerates protein aggregation. Recent MRI brain studies have revealed high energy, chaotic motion of cerebrospinal fluid (CSF) in lower brain and brainstem regions. These and studies showing CSF flow within the brain have shown that there are two energetic hot spots. These are within the third and fourth brain ventricles and in the neighborhood of the circle of Willis blood vessel region. These two regions are also the same locations as those of the earliest Aβ and tau AD pathology. In this paper, it is proposed that cardiac systolic pulse waves that emanate from the major brain arteries in the lower brain and brainstem regions and whose pulse waves drive CSF flows within the brain are responsible for initiating AD and possibly other amyloid diseases. It is further proposed that the triggering of these diseases comes about because of the strengthening of systolic pulses due to major artery hardening that generates intense CSF extensional flow stress. Such stress provides the activation energy needed to induce conformational changes of both Aβ and tau within the lower brain and brainstem region, producing unique neurotoxic oligomer molecule conformations that induce AD.
INTRODUCTION
The tremendous current and potential future medical and social impacts on the world population of Alzheimer’s disease (AD) are daunting [1]. Amyloid-β (Aβ) and tau have been the primary AD clinical targets [2–8]. However, there has been no treatment yet based on these targets that stops or reverses the course of this amyloid brain disease. There is increasing pressure for a different research approach.
This paper offers such an approach. It integrates previous proposals by the author regarding flow stress-induced protein aggregation [9–15] within the glymphatic cerebrospinal fluid (CSF) flow system [16], a recent speculative synthesis of how Aβ and tau oligomers initiate and drive AD pathogenesis [17], and recent magnetic resonance imaging (MRI) experimental evidence regarding high intensity of chaotic CSF motion in the lower brain glymphatic region [18–21]. The proposed new approach both preserves Aβ and tau as primary clinical targets and provides answers to critical unanswered questions raised in the above synthesis.
In vivo CSF is far from quiescent. Flow through narrow channels has been shown to induce Aβ and other protein aggregation due to energetic shear and extensional flow forces [9, 14, 22–25]. The author has previously suggested that flow stress-induced aggregation of dissolved Aβ and tau may be responsible for the formation of neurotoxic oligomer seeds that initiate self-propagation of these seeds throughout the brain cortex, ultimately causing AD [15].
This paper suggests two possible locations in the lower brain and brainstem region as high-energy sites of formation of these neurotoxic oligomer seeds. These same sites are also those of the earliest Aβ and tau AD pathology. These are the cerebral aqueduct (CA) joining the third and fourth brain ventricles and the basal cistern containing the circle of Willis artery complex. It is proposed that hardening of major brain arteries produces enhanced CSF systolic pulse waves, especially in these lower brain regions closest to the heart. These pulse waves induce CSF flow stress pulses that induce otherwise unattainable conformation changes in dissolved Aβ and tau molecules. These excited, flow-stressed molecules then ultimately produce Aβ and tau oligomers or oligomer seeds, depending on their energy. This proposed mechanism explains the long-held but unexplained “heart-mind connection” between heart failure and AD [26]. It also explains possible locations and mechanisms for the origins of Aβ and tau AD pathology.
Flow stress-induced aggregation of tau and Aβ
Quiescent aqueous solutions of Aβ and other amyloid monomer molecules slowly aggregate naturally in the amyloid cascade [8]. These Aβ molecules aggregate first in low molecular weight oligomers, then protofibrils, fibrils, and plaques. Shear flow-induced aggregation to form Aβ amyloid fibrils was first reported by Dunstan et al. [27]. Cohen et al. [28] investigated the mechanism of this slow fibril formation in quiescent solutions, but also showed that increasing rates of solution agitation significantly increased the rate of Aβ fibril formation. Aβ oligomers have been identified as toxins responsible for AD neuron damage [29, 30].
The mechanisms for in vivo and in vitro Aβ and tau oligomer formation are still uncertain. A significant activation energy is required for conformation change from an Aβ random coil conformation into a cross-beta conformation that precedes Aβ self-aggregation to form oligomers [31]. Aβ oligomers extracted from AD patients and oligomers containing the same number of Aβ monomers but synthesized in the laboratory can have quite different chemical and biological characteristics [32, 33]. The author has proposed that the brain flow-stress and environment provide the activation energy needed for these conformation changes [15].
Surface- and flow-induced aggregation
Studies of many different non-amyloid proteins show that most resist aggregation even at very high shear rates [24]. However, in the presence of certain surfaces, solutions of these same proteins aggregate at much lower shear rates, some even without shear [25]. This property is a serious problem in the manufacture and shipment of proteins. Hydrophobic surfaces have been shown to be quite efficient in promoting agitated protein monomer aggregation [25]. Air-water surfaces, which behave as hydrophobic surfaces, are also very efficient in promoting protein aggregation. Surfaces containing electrostatic charges attract oppositely-charged protein surfaces resulting in protein aggregation. Thus, the combination of flow and surfaces would appear to be critical variables in amyloid monomer protein aggregation, which is, in turn, critical in initiating amyloid disease.
Because of its amino acid composition, Aβ is attracted to both hydrophobic and positive and negative surfaces [34]. Thus, in our work with small samples of Aβ injected into a mobile phase flowing through narrow bore stainless steel capillary tubing [14], nearly 100%of the Aβ was initially adsorbed on the tube’s inner walls. However, when flow was halted for a short period and flow was resumed, part of the adsorbed sample had been released back into the mobile phase, the amount depending on the flow rate. However, repeated sample injections demonstrated increasing retention of each subsequent injected sample, ultimately with no release of the Aβ wall aggregate upon flow cessation. The nature of the type of surface retention depended on both the shear rate and surface characteristics of the previously adsorbed Aβ.
Molecular dynamics studies of the interaction of Aβ with bilayer membranes without shear predict the formation of Aβ barrel structures with membrane pores that allow calcium ions to flow through the membrane [35]. This formation could inactivate a neuron or astrocyte of which that membrane is a part. These calculations did not take into account probable shear effects in CSF at the membrane wall that could alter the conformation of the Aβ just prior to adsorption, thus possibly affecting its subsequent fate on the surface.
EFFECTS OF CSF FLOW STRESS
Shear, shear rate, and strain
Any time that an aqueous liquid flows in a confined space, it will be subjected to shear flow. Since the liquid wets the wall, the velocity of the innermost water surface layer during flow is zero. However, with flow this generates a liquid velocity gradient as one moves away from the wall. A typical result is represented by the laminar flow profile shown in Fig. 1. As indicated in this figure, the action brought about by laminar flow interacting with a flexible molecule has two components. It is both rotated and distorted. The nature of the molecular distortion is determined by the relative velocities of the different water layers within this profile and the molecule’s resistance to distortion. In a complex system, such as an in vivo fluid like brain CSF, a particular velocity field can be very complex since the fluid flow path wall geometry may also be complex. Shear rates, generally given in sec–1, are related to the fractional distortion of the molecule’s dimension per unit time. The local shear rate depends on the flow system’s geometry. Changes in any part of this geometry can change the shear rates at relatively distant points. Input flow rates help determine both the flow and shear rates at any given point of an entire closed flow system. Thus, sometimes shear rate information about complex flow systems can only be determined by experimental measurements and computer calculations.
Fig. 1
a) Laminar shear flow: flexible molecule undergoing stretching, distortion, chemical excitation, and molecular rotation. b) Later time than (a) showing direction of rotation.
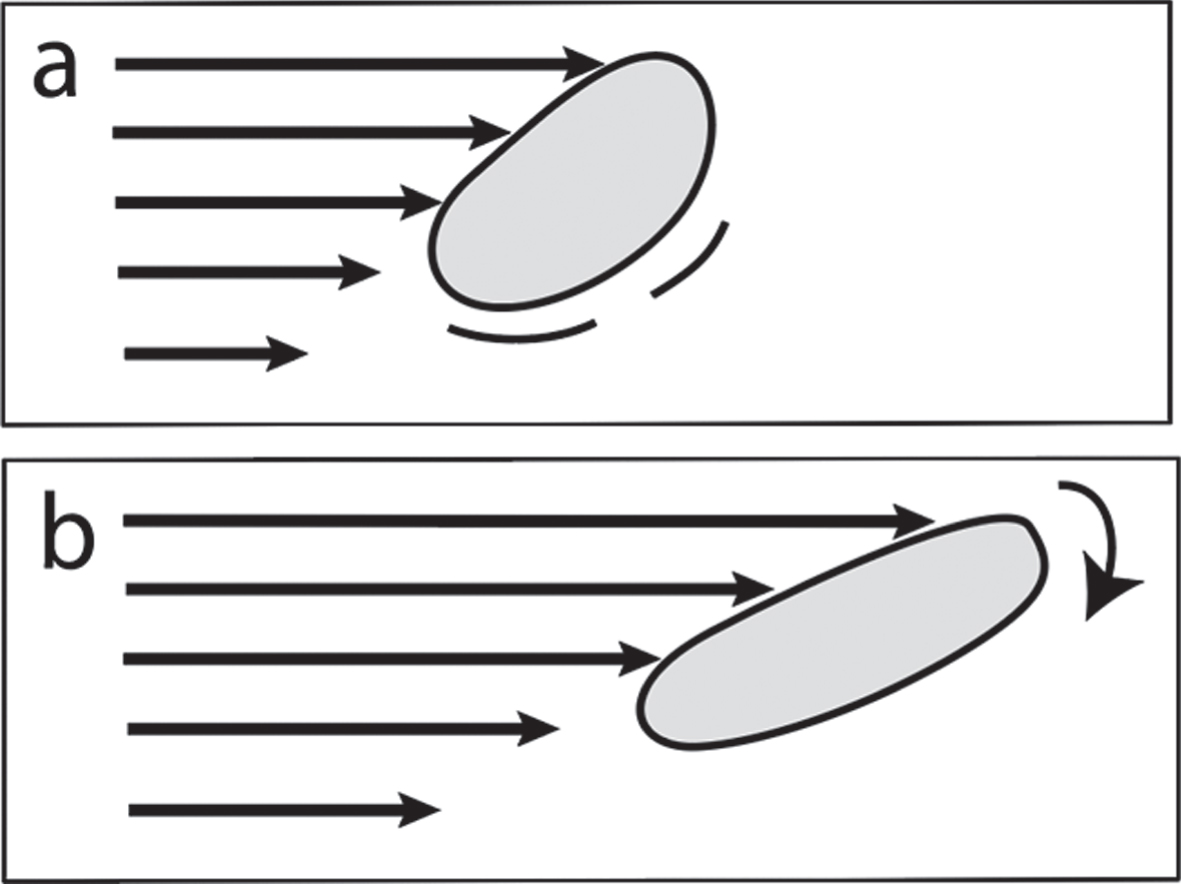
Strain is defined as the dimensionless magnitude of an object’s deformation, equal to the change in the dimension of the object such as a molecule divided by its original dimension. The strain rate is the rate of change of strain per unit time, generally given in units sec–1.
Extensional flow and protein aggregation
When fluid flow encounters a narrowing of the flow path or there is flow between flow-impeding objects, as indicated in Fig. 2, there is one particular region of interest. This is a region in which there is increasing acceleration as a dissolved solute molecule moves toward severe narrowing of the flow path. Such a flow situation produces a dominant extensional shear component and a molecule trapped in such a flow undergoes a non-uniform stretch designated as extensional flow of the type illustrated in Fig. 3. Such a flow-induced stretch can change the molecular conformation. This stretch is released after it passes through the AB “narrows” (Fig. 2). Both Aβ and tau are intrinsically disorganized proteins (IDPs) [15], which, when stretched and then relaxed, could well drastically change conformations under such dynamic stress conditions.
Fig. 2
In both cases, extensional shear is found on the left side. A lower extensional shear rate is found between B and C. Predominant laminar shear is found close to A, B, and C surfaces.
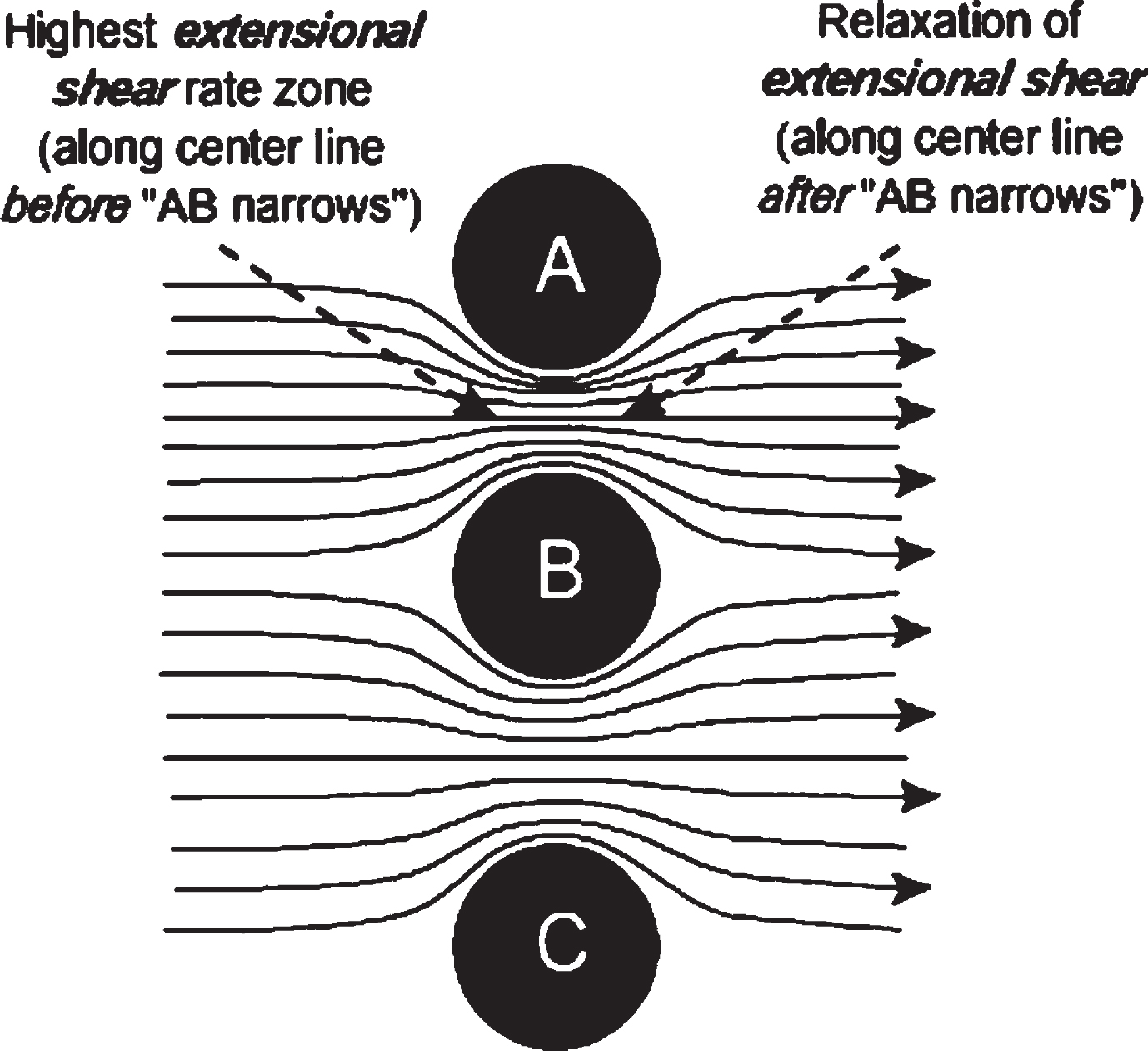
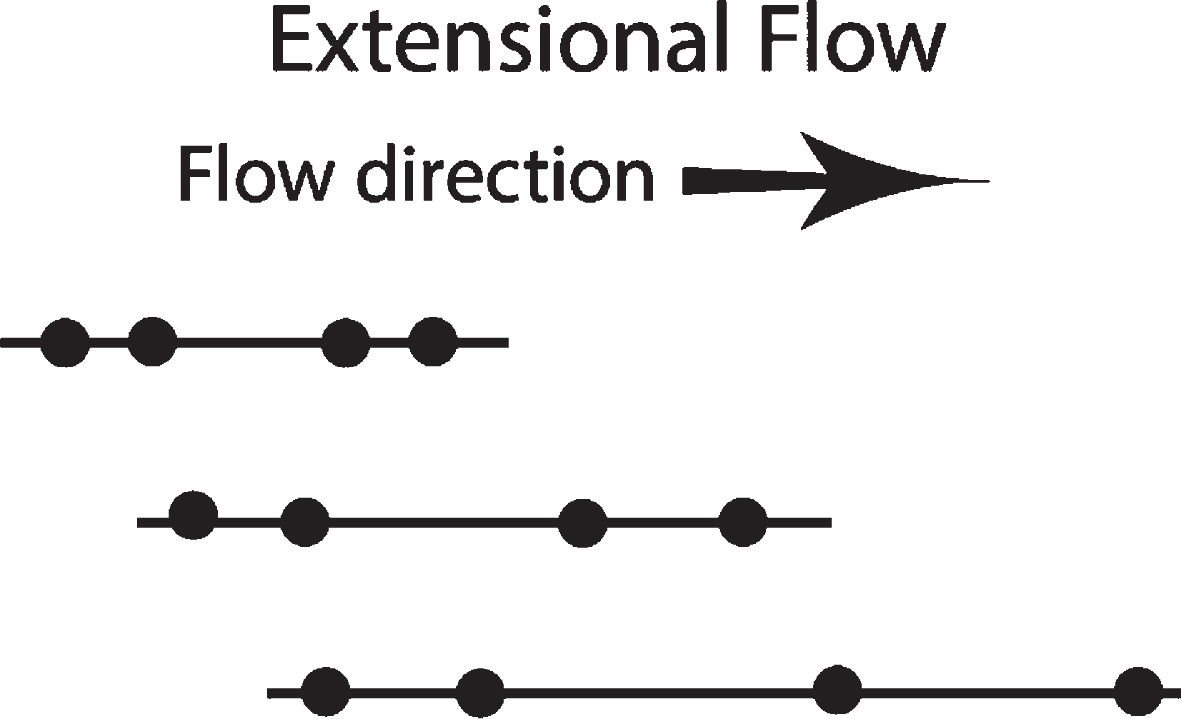
Fig. 3
Lines representing protein under extensional flow, with two sets of points, leading and lagging. The line is unevenly stretched, with the distance between the leading set of points accelerating faster than the distance between the lagging set of points. The molecule is moving in the direction of flow. This is the type of extensional strain found on the left side of Fig. 2 below between A and B objects, as well as between B and C.
Fluid flow regions with a dominant extensional stress component can induce molecular conformational transitions that are rarely, if ever, allowed under quiescent laboratory solution conditions. The type of transition depends on the amount and directions of kinetic energy transferred to the molecule from CSF kinetic flow energy. Increased chemical energy induced by flow stress is represented in this and previous papers of this author [10–15] by an asterisk (*), e.g., flow stress-excited amyloid beta is Aβ* in a low shear flow-stressed excited state.
Experiments reported in a paper by Dobson et al. [36] demonstrate that many dissolved proteins that do not have a tendency to self-aggregate under quiescent conditions aggregate to ultimately form fibrils when repeatedly exposed to extensional flow stresses. The experiment consisted of two syringes connected by a narrow bore capillary. The solution was passed through this capillary back and forth many times to induce this aggregation. Extensional flow is created when the fluid flow path is abruptly narrowed, thus increasingly accelerating the fluid flow as it approaches the entrance to the narrowed flow path. It is primarily in this small, suddenly accelerated flow region near the entry to the junction of the syringes and the capillary that extensional flow is generated. Thus, for every pass through the high extensional stress flow zone at these junctions, the times of exposure of protein molecules to extensional flow stresses were very short, on the order of tens of milliseconds. Calculations showed that the rate of energy transfer for extensional flow is orders of magnitude faster than for wall-generated laminar shear flows in the capillary in these experiments [36]. Shortening the length of the capillary did not change the results, indicating that the extensional flow-induced change was caused exclusively by extensional flow. The question arises, are there CSF flow situations within the brain that generate similar extensional flows?
THE GLYMPHATIC CSF FLOW SYSTEM
Arrows starting in the center of the brain cross section indicated in Fig. 4 illustrate the glymphatic CSF flow path origin within the human brain. CSF is mainly generated within the choroid plexus of each of the four brain ventricles and its net flow is from these ventricles into the various brain cisterns (see later Fig. 8 for locations of these), then to the subarachnoid space (SAS), and ultimately into the vein system. The emphasis on “net” is because, just as in the case of blood flow within the body, the CSF flow stream is not steady. Most of the cyclic glymphatic CSF flow appears to be generated by pulse waves emanating through various components of the vascular system arteries, arterioles, and under certain circumstances, capillaries. In much of the glymphatic system, this CSF flow is a cycling back-and-forth flow, with net flow from ventricles through cisterns to the subarachnoid space (Figs. 4, 5, 8). The driving forces involving the CSF flow pattern are complex, but include heartbeat rate, pulse strength and pattern, inspiration rate (mainly for CSF in the brainstem), blood vessel flexibility and dimensions, and the presence of flow-impeding objects, such as blood vessels, nerves and nerve bundles, and proteins projecting from membranes.
Fig. 4
Full glymphatic brain CSF flow system. Net flow starts in centrally-located lateral ventricles (right and left), through the interventricular foramen, to the third ventricle, then through the very narrow central aqueduct (CA) into the fourth ventricle. From there, the net CSF flow is into the brainstem and spinal cord, then into various cisterns (see Fig. 8), and finally into the subarachnoid space (SAS) covering the brain cortex. The CSF then either enters the brain in the perivascular space surrounding arteries entering the cortex or moves through the arachnoid granulations and exits from the glymphatic system. The CSF enters the cerebral cortex, but the mechanism and location of the CSF’s brain waste solutes exit from the cerebral cortex is under debate. Source: Wikipedia commons 1317_CFS_Circulation.jpg.
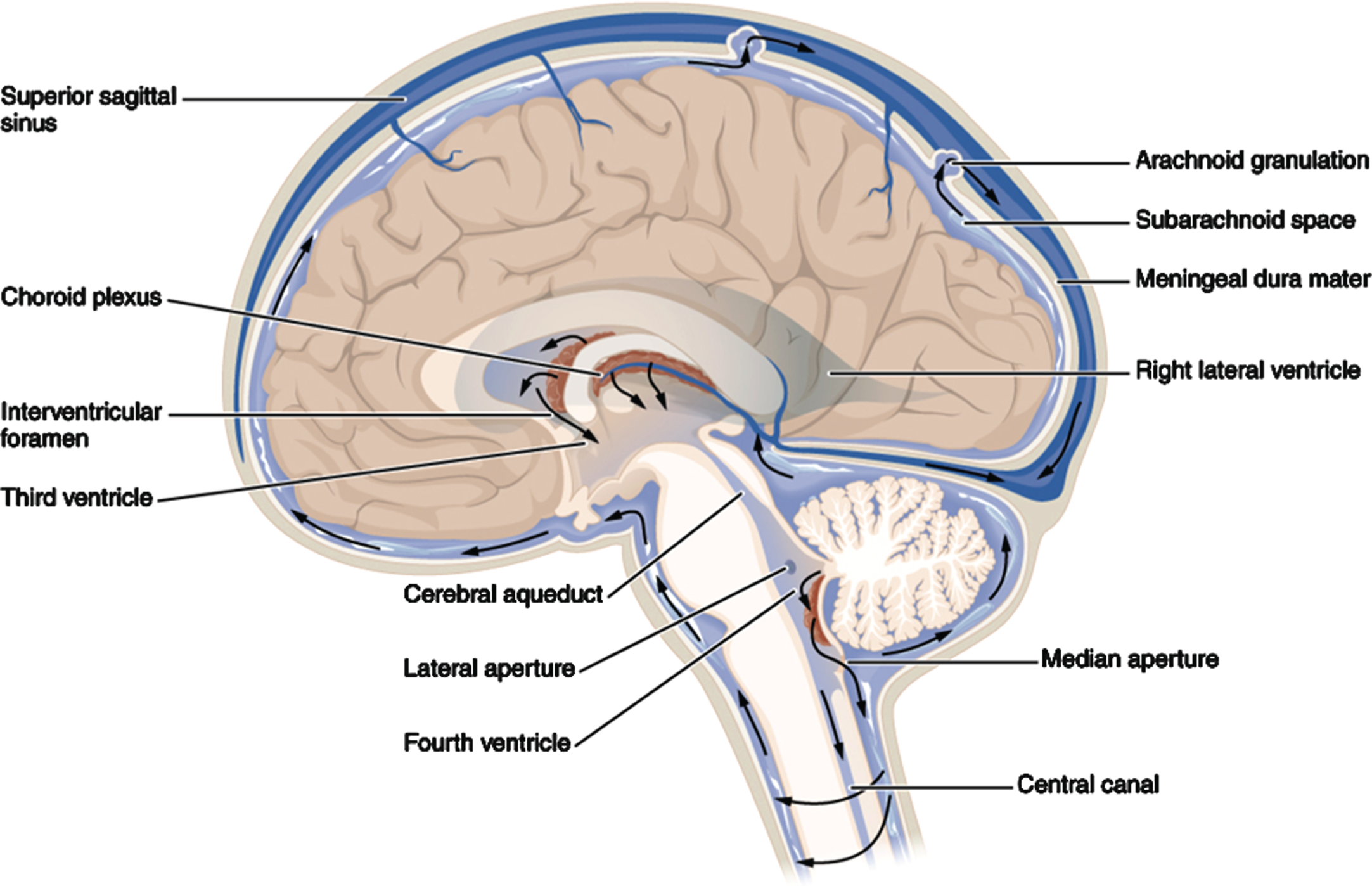
Fig. 8
Locations of the major brain CSF-filled cisterns, showing also all CSF motion paths (arrows) of the glymphatic system [16] with the exception of the part that includes the entries and exits to the cortex parenchyma. Particular attention is directed to the “basal cistern” which include the chiasmatic and interpeduncular cisterns (see text for discussion). These are upstream in the CSF motion path of the Aβ pathology in the orbitofrontal cortex, a lower portion of the frontal cortex, which also interfaces with the cistern of lamina terminalis. Source: https://ranzcrpart1.fandom.com/Fandom communities (known as “wikis”) is licensed under the Creative Commons License 3.0 (Unported) (CC-BY-SA).
![Locations of the major brain CSF-filled cisterns, showing also all CSF motion paths (arrows) of the glymphatic system [16] with the exception of the part that includes the entries and exits to the cortex parenchyma. Particular attention is directed to the “basal cistern” which include the chiasmatic and interpeduncular cisterns (see text for discussion). These are upstream in the CSF motion path of the Aβ pathology in the orbitofrontal cortex, a lower portion of the frontal cortex, which also interfaces with the cistern of lamina terminalis. Source: https://ranzcrpart1.fandom.com/Fandom communities (known as “wikis”) is licensed under the Creative Commons License 3.0 (Unported) (CC-BY-SA).](https://content.iospress.com:443/media/jad/2021/79-3/jad-79-3-jad201025/jad-79-jad201025-g008.jpg)
Fig. 5
Zones of chaotic/“turbulent” CSF motion in black (most intense) and shades of gray (less intense). These zones are found mainly around the brainstem, cisterns, third ventricle, spinal cord, and to a lesser extent, cortex SAS. This MRI study was on a healthy human subject. Source: [18] licensed under Creative Commons Attribution-Non Commercial-No Derivative International. Alteration permission granted by authors.
![Zones of chaotic/“turbulent” CSF motion in black (most intense) and shades of gray (less intense). These zones are found mainly around the brainstem, cisterns, third ventricle, spinal cord, and to a lesser extent, cortex SAS. This MRI study was on a healthy human subject. Source: [18] licensed under Creative Commons Attribution-Non Commercial-No Derivative International. Alteration permission granted by authors.](https://content.iospress.com:443/media/jad/2021/79-3/jad-79-3-jad201025/jad-79-jad201025-g005.jpg)
FLOW STRESSES WITHIN THE CEREBRAL CORTEX
Within the blood-brain barrier, inside the cortex parenchyma, the CSF counterpart of the glymphatic path is the interstitial fluid (ISF) path. ISF flow through the cerebral cortex parenchyma is still a subject of much debate [3, 4]. No bulk flow rates of ISF within regions of the cortex have been reported. There is little doubt that ISF bulk flow, if any, is much slower than the CSF flow in the rest of the glymphatic flow system outside the blood-brain barrier.
Cortex ISF flow path dimensions are very small, and the dimensional sensitivity to shear, e.g., in a capillary, is highly nonlinear. An added dimension in this consideration is that cortex flow rates are reported to increase during sleep [37, 38]. The case has been made by the author [10] for shear-induced aggregation producing Aβ-containing cerebral amyloid angioplasty (CAA) deposits in and around the arteries surrounding and entering the cortex. The cause of oligomer and plaque formation within the cortex parenchyma has yet to be established, although the author has suggested a potential shear-based mechanism if perivascular ISF flow is found within the parenchyma [15].
FLOW STRESSES WITHIN THE LOWER BRAIN AND BRAINSTEM
Recent MRI studies have been reported on CSF motion in the whole brain [18–21]. Dynamic improved motion-sensitized driven-equilibrium, steady-state free precession (Dynamic iMSDE SSFP) is reported to be able to detect energetic, chaotic, “turbulent” motion in aqueous fluids within the brain. As a result of these studies, the brain can be divided into high and low energy liquid flow regions, as illustrated in Fig. 5. The brain cortex areas in Fig. 5 clearly show minimal chaotic motion in that region. By comparison, the dark and shaded areas below the cortex detect the presence of energetic CSF chaotic flow in or around the third and fourth ventricles, brainstem, cisterns, spinal cord, and SAS between the skull and the cortex surface. It is especially useful to watch movies made from these MRI studies (movie links are in [20]). Strong, pulsing CSF flows correlated with the cardiac cycle in these high energy brain areas are quite prominent, especially in the third ventricle, as shown in these movies, Fig. 5, and later below in Fig. 7.
Fig. 7
Cyclic reciprocal CSF flow directions and rates in the third ventricle (top), cerebral aqueduct (CA) (middle tube), and top of third ventricle (bottom, widening) at two time points in the cardiac cycle, showing reversals in CSF flow direction (C1-flow toward fourth ventricle; B1-flow toward third ventricle). Flow rates are calculated from MRI data and hydrodynamic computations [39]. CSF flow rates are in mm/s–1 according to color coded lines and keys. Modified from [39] under Creative Commons CC-BY license.
![Cyclic reciprocal CSF flow directions and rates in the third ventricle (top), cerebral aqueduct (CA) (middle tube), and top of third ventricle (bottom, widening) at two time points in the cardiac cycle, showing reversals in CSF flow direction (C1-flow toward fourth ventricle; B1-flow toward third ventricle). Flow rates are calculated from MRI data and hydrodynamic computations [39]. CSF flow rates are in mm/s–1 according to color coded lines and keys. Modified from [39] under Creative Commons CC-BY license.](https://content.iospress.com:443/media/jad/2021/79-3/jad-79-3-jad201025/jad-79-jad201025-g007.jpg)
A recent high field MRI paper [23] reports studies of brain tissue strain as a function of the cardiac cycle. Figure 6 illustrates brain tissue mechanical strain (up-and down stretching is shown in this figure), during the cardiac cycle. High strain regions in Fig. 6 correlate with the same intensely chaotic motion regions reported in Fig. 5. These high field MRI studies report significant brain tissue strain in Fig. 6 correlated with the heart systole in tissue around the same chaotic basal cisterns, third and fourth ventricle, and brainstem regions. Such mechanical strain apparently stimulates the chaotic CSF motion in these regions shown in Fig. 6. By contrast, there are much less intense, broadly distributed tissue disturbances spread throughout the cerebral cortex. These studies confirm earlier 4D-PC MRI studies [19] in which there was an elevated pressure gradient observed in the basal cistern. Velocity imaging studies [19] also demonstrate significant CSF motion in the third ventricle, CA, fourth ventricle, and cisterna magna and in the pontomedullary and prepontine cisterns, and anterior parts of the interpeduncular and chiasmatic cisterns, which are together designated as the basal cistern (Fig. 8). Small MRI-sampled segments of water contained in CSF can reach instantaneous flow velocities on the order of several centimeters per second, but also have acceleration rates over one hundred cm/s2 on the brainstem surface [21]. The latter can cause significant strain within molecules dissolved in this CSF.
Fig. 6
Whole brain tissue mechanical strain at the peak systole maximum in comparison with that at early systole. Yellow is maximum strain in the direction of the head and blue is strain at the same time in the direction of the feet. CSO and BG are two different sampling regions. Strain intensity change regions in this figure correlate spatially with turbulence region data in Fig. 5. Source [23] licensed under Creative Commons 4.0.
![Whole brain tissue mechanical strain at the peak systole maximum in comparison with that at early systole. Yellow is maximum strain in the direction of the head and blue is strain at the same time in the direction of the feet. CSO and BG are two different sampling regions. Strain intensity change regions in this figure correlate spatially with turbulence region data in Fig. 5. Source [23] licensed under Creative Commons 4.0.](https://content.iospress.com:443/media/jad/2021/79-3/jad-79-3-jad201025/jad-79-jad201025-g006.jpg)
Figure 7 shows selected data obtained from MRI measurements and hydrodynamic calculations of the CSF flow rates in and between the third and fourth brain ventricles [39]. Reciprocal back-and-forth CSF flows in this region were correlated with the cardiac cycle. These flows show complex flow patterns in the third ventricle, including a persistent vortex effect seen in Fig. 7 in both CSF flow directions. Highly variable laminar and extensional flows are implied, but not reported. Extensional shear is found in those regions represented in Fig. 7 wherever there are abrupt changes in color along flow lines. Extensional shear rate intensity increases as the rate of change of color increases as one moves along any single flow line in the direction of flow. Thus, the highest extensional shear rates will be found, according to the data reported in Fig. 7, in two regions. One is in the CA itself, as anticipated, in the downward C1 flow region at the entrance to the CA from the third ventricle. The second, quite unanticipated, region is in the upward B1 flow in the region of the persistent vortex, where it would appear there is the highest extensional flow in the region of what may be the junction of a double vortex as the fluid exits from the CA into the third ventricle. It must be remembered that these two figures are merely for two time points in the cardiac cycle. Point B is taken in the fill period R-peak where the flow through the aqueduct is oriented in cranial direction, whereas point C corresponds to the flush period where CSF flow is in caudal direction.
Within the CA, laminar shear rates are estimated to be on the order of hundreds of reciprocal seconds, the same order of magnitude as seen in protein studies in the author’s laboratory [9]. In those early studies, samples of proteins that normally do not aggregate were shown to spontaneously plate out as solids on the inside walls of a stainless-steel capillary with inner diameter and flow rates similar to those detected in the CA in Fig. 7. Calculations of extensional shear rates are needed for the entire cardiac cycle at different systolic pulse intensities, as will be discussed below.
ORIGINS OF Aβ AND TAU AD PATHOLOGY
The earliest locations of a number of amyloid disease pathologies have been identified [40, 41]. Aβ plaque and tau tangle AD pathologies are both initiated in one location and spread widely to neighboring regions. Such progression has been attributed to seeding (propagation of templated conformational conversion), sometimes designated as “prion-like” seeding [42]. Figure 9 illustrates successive stages and phases for AD and Parkinson’s disease pathology [40].
Fig. 9
Propagation of inclusion pathologies of Aβ and tau in AD through different disease stages. source [40].
![Propagation of inclusion pathologies of Aβ and tau in AD through different disease stages. source [40].](https://content.iospress.com:443/media/jad/2021/79-3/jad-79-3-jad201025/jad-79-jad201025-g009.jpg)
Figure 9 demonstrates the distinctly different spatial origins of the two pathologies revealed by postmortem appearance of brain Aβ plaque and tau tangle aggregates.
Aβ plaque pathology is initiated in the basal temporal and orbitofrontal neocortex. Tau pathology is initiated in a much more limited space in a part of the midbrain near the pons wall of the fourth brain ventricle. This paper deals with the origins of this pathology, so that in Fig. 9, one should note carefully the pathology locations of the earliest times, either in stage I or phase 1. The origins of Aβ and tau pathology are clearly in different locations. Also note that both are either in the lower frontal cerebral cortex or the brainstem region.
Chen and Mobley have published [17] a comprehensive synthesis of AD pathology and amyloid literature focusing on studies involving Aβ and tau neurotoxic oligomers. This synthesis incorporates recent discoveries about the pathways that Aβ and tau oligomers take in migrating between the upper brain cortex region and the lower brain regions containing the entorhinal cortex (EC), the locus coeruleus (LC), and hippocampus. It concludes by offering a speculative synthesis for how oligomers of Aβ and tau are initiated and drive AD pathogenesis.
However, this synthesis is accompanied by three unanswered questions:
(1) What are the mechanisms for the formation of Aβ and tau oligomers and their seeds?
(2) What are the reasons for the earliest location of the tau pathology being in the region of the EC and LC?
(3) What is the reason for the earliest location of the Aβ pathology being in the basal temporal and orbitofrontal neocortex?
The author of this paper has suggested [10, 12, 15] partial answers to question (1). That is, briefly, the formation of Aβ oligomers can be initiated by a CSF or parenchymal ISF shear- or extensional flow-induced Aβ monomer conformational change. Rapid neurotoxic oligomer formation follows with collisions between two Aβ molecules, at least one of which is shear- or extensional flow-activated. The discussion below will suggest more detailed proposed answers to question (1) and propose answers to questions (2) and (3). The author suggests the answers lie in the highly energetic CSF motion regions (Fig. 5) accompanying intense tissue agitation (Fig. 6) generated by intense major artery pulse pressure waves in the circle of Willis arteries shown below in Fig. 10, resulting in the energetic and chaotic CSF flows that distort and change the conformations of dissolved or surface-adsorbed Aβ and tau molecules.
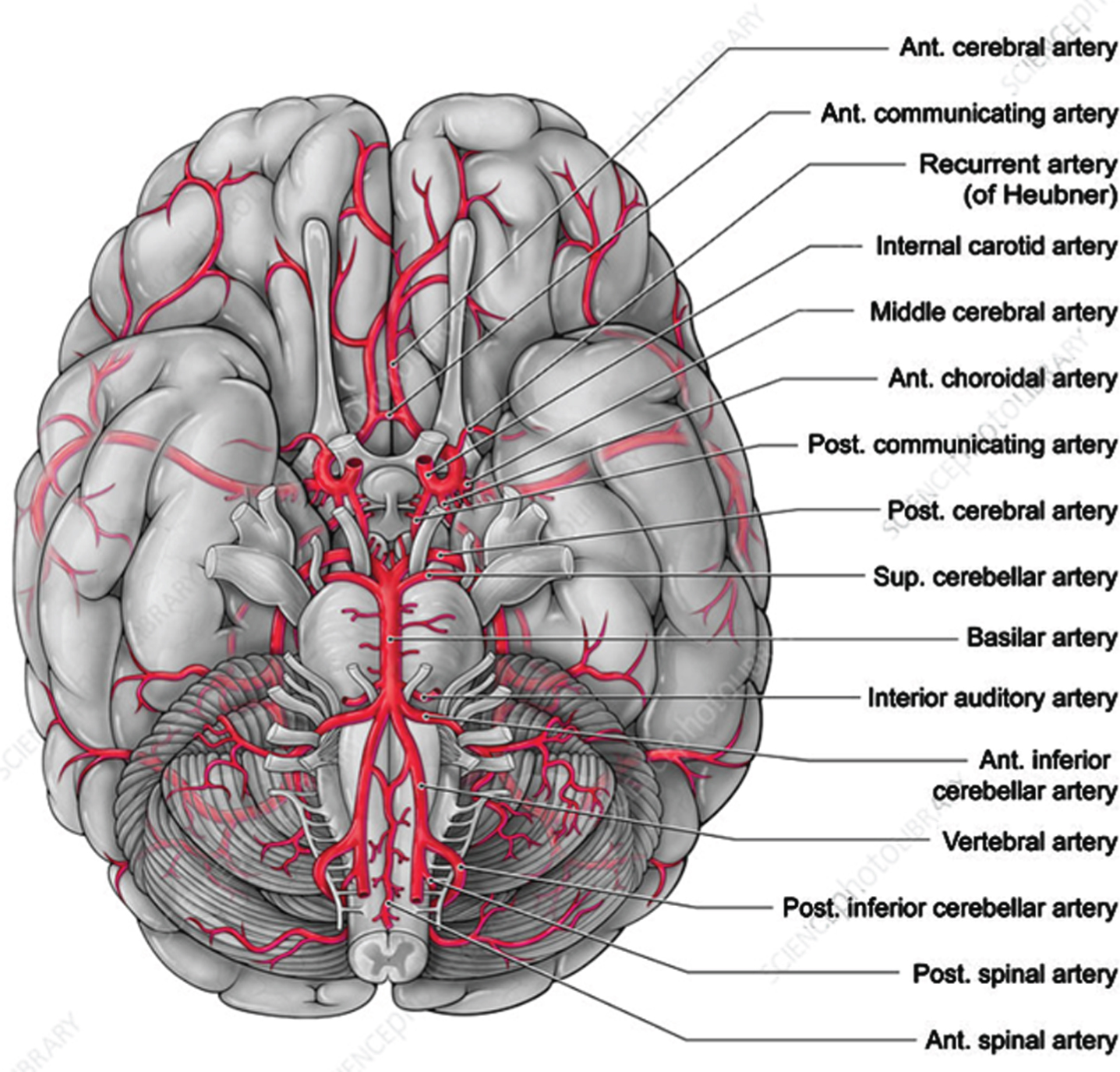
Fig. 10
Major arteries feeding the brain. This is a 3D view of the bottom of the brain. A major part of the glymphatic path travels up the center of this figure. In this view, the spine and brainstem are projecting out and down from the center of the brain. The arterial circle of Willis, which is enclosed on two sides by the temporal lobes, is located in the center of this figure. It indicates how the temporal lobes enclose the basal cistern walls just above the basilar artery. The region of the brain surrounding the circle of Willis in this figure is the general region of the cortex that is the origin of the Aβ AD pathology. Source: Alamy.com.
HYPOTHESES REGARDING THE ORIGIN OF Aβ AND TAU AD PATHOLOGY
The following are the author’s tentative hypotheses based on the above facts and discussion. These hypotheses are based on the existing literature, including previous speculative papers by the author [9–15]. The author looks forward to discussions generated by these suggestions as they are significantly different from existing theories.
The energy source for Aβ and tau conformation changes is CSF flow stress
Shear rates on the order of several hundred sec–1 are needed to change the Aβ conformation from the highly flexible ensemble IDP state [15] (the “random coil”) to the much more rigid cross-beta non-IDP conformation. Shear experiments using Raman spectra to explore protein backbone alterations have found that similar order of magnitude shear rates reversibly alter quite different protein backbone conformations [43]. These low shear rates contrast with some reported experiments where many orders of magnitude higher shear rates could not denature proteins. However, similar shear experiments in the presence of surfaces have demonstrated that, depending on the nature of the surface, certain surfaces can readily promote protein aggregation at very low shear rates [24, 44]. Hydrophobic and polar surfaces were found to be quite effective in this regard.
Biological membranes have distinct hydrophobic as well as polar character. Cellular membranes line CSF flow channels. CSF flow through confining surfaces generates laminar as well as extensional flow depending on the complex geometries of these channels. The highest shear rate in pure laminar flow is at the confining wall [10]. Shear-stressed CSF solute molecules such as Aβ will likely bond, at least temporarily, to the wall if their surface intermolecular bonding potential is high. Flowing Aβ will be increasingly shear-stressed as it approaches the confining wall and may stay stressed during and after its wall adsorption. When flow rates slow or reverse direction, there may be a tendency for the stressed molecule to return to at least some intramolecular Aβ structure, unless there is very strong intermolecular surface bonding. The latter could be the case where there is very high flow shear exposing long lengths of the Aβ protein molecule to the membrane surface. The adsorbed Aβ can migrate on a protein island membrane surface and be stabilized and form barrel-shaped complexes [35]. These complexes can form channels that leak calcium ions and thus inactivate neuron and astrocyte cells.
However, if there is weak intramolecular bonding, the molecule could regain partial structure during periods of diminished shear and return to the flowing CSF stream. Thus, the membrane adsorption process would be quite sensitive to flow rate.
Aβ and tau AD pathology and major flow stress are in the same lower brain regions
The earliest Aβ pathology is located near a highly complex region of the glymphatic flow pathway surrounding the basal cistern through which Aβ- and tau-containing CSF travels (Fig. 8). The critical pathological regions are the basal temporal and orbitofrontal neocortex. This is in the same location as one of the two major regions of strong CSF chaotic flow rates shown in Fig. 5. This path encompasses two relatively large CSF basilar cisterns (interpeduncular and chiasmatic), a major brain vascular complex (the circle of Willis), and neuron axon bundles that connect the brainstem with many parts of the brain cortex. It is also CSF downstream from the intensely chaotic CSF motion shown in Fig. 5 in the pontomedullary cistern. The basal frontal temporal brain region laterally contains the EC. The EC region is the epicenter and origin of tau tangle pathology.
The major vascular components of the circle of Willis are found in the center of Fig. 10, just above the basilar artery in this figure. These are contained within each of the cisterns making up the basal cistern (Fig. 8). They generate strong heart pulse waves [19] especially because of the presence of the major carotid arteries. These waves are major generators of the CSF back-and-forth flow in this CSF-filled cistern. There also may be CSF flow in the narrow space between the two brain hemispheres in the interhemispheric cistern (Fig. 8) that could produce Aβ shear-induced products in the lower frontal and temporal lobes because of the presence of the surface cerebral arteries. This results from the tissue strain (Fig. 6), giving rise to the additional chaotic flow (Fig. 5) in the third ventricle. Such chaotic flows could also give rise to shear-induced activation of Aβ or tau molecules within that ventricle.
Arteries such as the basilar, carotid, and cerebral arteries and their branches (Fig. 10) are covered by the arachnoid membrane within the SAS in the basal part of the brain. The pulse waves from these arteries result in reciprocal CSF pumping action within these glymphatic CSF paths in a number of cisterns. Thus, this cistern region and that surrounding the pons surface containing the major basilar artery show maximum chaotic flow in the MRI studies (Fig. 5) [18]. The chaotic CSF motion observed in this glymphatic region of interest results from intense pressure waves. These waves emanating from major arteries have been reported in MRI studies [19]. Thus, the same type of chaotic CSF motion observed in the pontomedullary cistern (Fig. 8) from the pulsation of the major basilar artery is apparently generated by pulsations of the circle of Willis arteries. The pontomedullary cistern, which is upstream from the basilar cisterns, delivers a heavy dose of chaotic energy to cistern amyloid monomer molecules. Could there already possibly be flow-stress altered Aβ and tau conformations formed even before their next, perhaps even greater, dose of chaotic motion in the basil cisterns?
Aβ aggregation is initiated by cardiac pulse-driven, CSF laminar and extensional stress
One possible molecular consequence of all this chaotic energy exposure is that many small, random conformational changes are induced by chaotic CSF flow surroundings. Since Aβ and tau are IDP molecules, each of these changes could cause small changes in the composition of their IDP conformation ensembles [15]. In each newly altered ensemble, one or more of the IDP conformations may exceed the activation energy and entropy requirements for the ultimate formation of a cross-beta conformation that is necessary for Aβ or tau monomer aggregation. Once the transition to a cross-beta conformation is completed, the Aβ* or tau* protein is now quite prone to aggregate with other monomers. If it is a seed molecule, it will convert other Aβ molecules to another cross-beta conformation and likely bond with the seed molecule, initiating a self-propagating aggregation reaction.
Thus, it is proposed that the initiation of Aβ pathology in the basil cistern is due to the molecular distortions of Aβ caused by the chaotic motions revealed in Fig. 5. The chemical reactions that form oligomers may take place on the frontal lobe wall membranes on which the distorted Aβ* molecule is adsorbed. If so, the wall acts as a catalyst for the formation of these seeds because of the lower activation energy for dimer and possibly multimer formation, initiating further aggregation to form Aβ oligomers. If these are seed Aβ oligomers, they can then propagate through the frontal lobe membrane and then through the rest of the cortex and follow the numerous neural paths to critical locations involved in memory formation and storage as suggested by Chen and Mobley [17].
Laboratory flow experiments have reported that Aβ aggregation is both initiated [27] and accelerated [28] by shear. However, the membrane surfaces that generate shear stresses are likely to play a critical role in Aβ protein aggregation [10]. Because back-and-forth CSF flow is observed both around cortex pial arteries [45, 46] and through brain ventricles [39], it is assumed that the same kind of directional oscillation of CSF motion is taking place in other regions of the glymphatic path, e.g., in the cisterns. Because of this CSF flow reciprocation, solutes such as Aβ and tau have ample time to diffuse to and interact with the wall under these flow conditions. Our experiments [14] have shown that when Aβ interacts under steady laminar flow conditions with a stainless steel wall, it both reversibly and irreversibly aggregates, depending on both flow rates and repeated exposure of solutes to the walls already containing previously adsorbed Aβ. Experiments with proteins other than Aβ have shown that, under shear stress, many adsorb to surfaces in several layers, one of these being a surface layer, but that flow stress can remove outer layers as microparticles [24]. Could this be a possible mechanism for the initial formation of Aβ plaque?
Extensional flow-stressed molecules are especially vulnerable to conformational change
As indicated in Fig. 1, although one component of shear experienced in laminar flows is extensional (elongational), leading to stretching of CSF solute molecules, there is also a rotational tumbling component. When there is free rotation in bulk CSF undergoing laminar flow, this causes a cyclic reversal of the stretching motion. At or near the wall, such rotation can force more intimate contact of the rotating, stretching molecule with the wall, providing increased opportunity for adsorption. However, if CSF is in a part of the fluid flow field resulting in primarily extensional flow with a minimal rotational component, there is primarily molecular stretching. That is, until there is a reversal in reciprocating bulk CSF flow direction, at which point the stretching is released. Thus, there is a time window in which the stretched molecule can reorganize into a different conformation. This could represent one possible mechanism for the conversion of the IDP random coil to cross-beta Aβ or tau conformation.
In free shear flow, during the initial stretch and rotation part of the cycle of a solute, a stretched molecule may not have an opportunity to react with a nearby stretched molecule to form a stretched dimer because its stretch has been relaxed by the rotation. However, at higher solute concentrations, if there is mainly extensional stress for a long enough period with little shear rotational contribution, there is a greater chance for two elongated molecules to meet and form a stretched dimer or a conformationally altered monomer (Fig. 11).
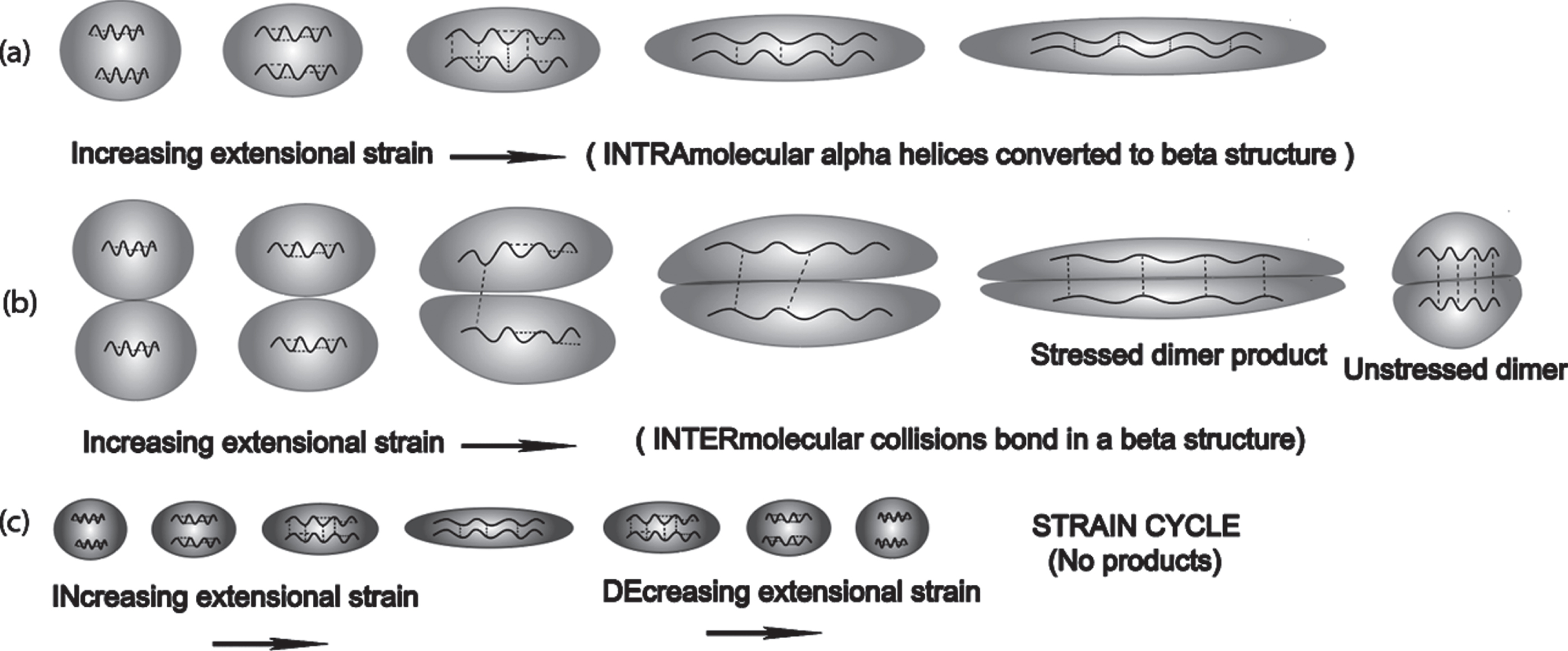
Fig. 11
Extensional flow-induced transformation from an alpha to beta conformation via intramolecular and intermolecular mechanisms: (a) Two nearby alpha helical segments within the same molecule are exposed to extensional flow to such a point where their protein backbones are exposed to each other resulting in an alpha to beta conformation change within a single molecule; (b) When the molecular concentration is high enough, a concentration dependent-bimolecular collision between extensional flow-stretched molecules can produce a dimer that is much more stable than the monomer; (c) However, if the strain-induced stretch is not sufficient to allow close proximity of each alpha helix in a single molecule, then there is a lack of intramolecular product formation, resulting in heat release.
If high amounts of elongational energy are very rapidly delivered, extensively stretching the molecule quickly, there would be an opportunity to rapidly form a conformationally high energy transformed monomer (Fig. 11a or Fig. 12). The latter could be a monomer precursor that ultimately produces a seed oligomer.
Fig. 12
Proposed mechanism for the rapid strain-induced transformation of Aβ from an IDP alpha conformation to a beta hairpin conformation through extensional stretching distortion. This particular alpha state was selected from favored IDP states in [60]. This beta sheet structured molecule may then react quickly with another and possibly form a dimer seed. This aggregation reaction could take place over many stretch-and-release cycles.
![Proposed mechanism for the rapid strain-induced transformation of Aβ from an IDP alpha conformation to a beta hairpin conformation through extensional stretching distortion. This particular alpha state was selected from favored IDP states in [60]. This beta sheet structured molecule may then react quickly with another and possibly form a dimer seed. This aggregation reaction could take place over many stretch-and-release cycles.](https://content.iospress.com:443/media/jad/2021/79-3/jad-79-3-jad201025/jad-79-jad201025-g012.jpg)
Experiments with pulsating and reciprocating flows with varying energies are needed to identify possible fluid flow-induced Aβ and tau chemical products. Such experiments may require different flow geometries that mimic those found within the glymphatic system. Since Aβ, tau, and phosphorylated tau are all found in CSF, it will be important to also test the effects of shear- and extensional flow-rates on mixtures of these molecules.
With Aβ-tau mixtures, there is also a possibility for the formation of a complex between shear- or extensional flow-induced tau and one or more flow-activated Aβ molecules that is a catalytic intermediate prior to the formation of a cross-beta tau conformer, a precursor to a tau oligomer or tau seed. Figures 11, 12 demonstrate possible extensional flow effects on molecules containing alpha helical segments that are flow stress-induced to produce potential Aβ seeds, seed precursors, or Aβ dimers.
It is claimed by Dunstan et al. [27] that: “The change in time scales for amyloid (Aβ) formation from sheared to un-sheared may be used to interpret the magnitude of energy change that is produced by shear . . . Assuming a single activated process, the ratio of these times gives us the difference in energy between the un-sheared and sheared samples, which is∼4.3 kT, or 1 weak hydrogen bond per entity in solution. Thus, we are studying a very weak perturbation on the protein structure.” The author has suggested [12] that there is a concerted shear-induced conformational change, similar to that shown in Figs. 11a or 12, involving the whole molecule that involves a relatively low energy, shear-induced transition state. This low energy event could possibly involve quantum tunneling in hydrogen bond exchanges [47]. That is, H atoms may tunnel through potential energy barriers at much lower energies than would be needed to actually break current H bonds and make new ones. The further suggestion is made here that it is not necessary to unfold the amyloid monomer in a reverse path of the protein folding process in order to convert it into a cross-beta structure. Instead, shear or extensional forces spread over the entire molecule such as that shown in Fig. 1 may require less energy because there are weak bonds being simultaneously broken at the same time as new ones are being formed as depicted in the transition states suggested in Figs. 11, 12.
Flow stress events in and around CA initiate tau pathology in the locus coeruleus (LC)
As mentioned previously by the author in a discussion of flow stress in the glymphatic path [15], the CA (Fig. 13) is the narrowest, most flow-restricting parts of the ventricular CSF path. The LC is a large bundle of neuron axon projections located within the CA, near its lower exit into the fourth ventricle, and which continues in two locations on or near the CA wall surface into the fourth ventricle. The LC is the principal site for brain synthesis of norepinephrine, which is a hormone and neurotransmitter that increases arousal and alertness, promotes vigilance, enhances formation and retrieval of memory and focuses attention. The LC nuclei send this hormone signal throughout the entire cerebral cortex as well as to a variety of other structures including the amygdala, hippocampus, cerebellum, and spinal cord. Thus, these LC neuron projections are connected to many different regions of the brain that deal with memory, arousal, sleep-wake cycle, attention, behavioral flexibility, behavioral inhibition, psychological stress, cognitive control, and emotions. These are all reflected in AD symptoms.
Fig. 13
Views of the brain ventricles, showing the narrowing of CSF motion channels in flowing from the third ventricle (“3V”) to the fourth ventricle (“4V”), with increased CSF extensional stress rates as the flow channel narrows in the CA (cerebral aqueduct; “Aq” in this illustration) and creating zones of dominant extensional stress. Source: [39] Licensed under Creative Commons CC-BY license.
![Views of the brain ventricles, showing the narrowing of CSF motion channels in flowing from the third ventricle (“3V”) to the fourth ventricle (“4V”), with increased CSF extensional stress rates as the flow channel narrows in the CA (cerebral aqueduct; “Aq” in this illustration) and creating zones of dominant extensional stress. Source: [39] Licensed under Creative Commons CC-BY license.](https://content.iospress.com:443/media/jad/2021/79-3/jad-79-3-jad201025/jad-79-jad201025-g013.jpg)
The LC is also reported to reveal the earliest pre-tangle [48, 49] phosphorylated tau (p-tau) AD pathology. This p-tau was found in the proximal axons of the LC, leading to speculation that p-tau was an even earlier pathological marker than tau tangle AD pathology in the EC. Aggregated tau tangles were observed first in the EC, but the locus of the earliest tau pathology is closer to that of Aβ.
MRI observations [19] demonstrate that the reciprocal back-and-forth CSF motion in the CA is not symmetrical. A well-regulated laminar flow was observed in this aqueduct, and a high-velocity downward motion was noted after a slow upward motion. This bidirectional CSF motion was seen in all healthy individuals.
Extensional fluid stress is felt by Aβ and tau molecules when CSF in the third and fourth brain ventricles approaches its top and bottom CA entrances. CSF is cycling back-and-forth with a net flow direction through the CA from third to fourth ventricle. However, a persistent vortex is reported near the entrance to the CA within the third ventricle (see Fig. 7 and [39] for details). One must remember that molecules dissolved in this fluid have already been exposed to highly chaotic flow within the third ventricle (Fig. 5). Thus, Aβ and tau molecules and possibly their oligomers dissolved in the CSF that are transiting through the CA are subjected multiple times to both chaotic flow and extensional flow at junctions of the CA within the third and fourth ventricles, and laminar flow within the CA. It is suggested here that, under these energy rich conditions, such flow-stressed molecules may have an opportunity to collide with another similarly stressed molecule to form a more stable dimer, possibly leading to higher neurotoxic oligomers. However, it is also possible that neurotoxic oligomers can form on the LC-containing walls that have adsorbed flow-stressed amyloid monomers of Aβ and tau. These molecules can form oligomers on this surface that can interact with the LC causing neural damage.
In flow experiments reported by Dobson et al. [36], it was suggested that the passage of protein solutions back and forth through the abrupt flow path dimensional changes was responsible for incremental changes in protein conformations that finally produced one that was responsible for initiating rapid protein aggregation. The aggregation process was protein concentration dependent, but it was also protein-type dependent. For example, how flexible is the protein when it is flow-stressed?
The biological surroundings of the many different types of glymphatic environments will also affect the conformational makeup of IDP ensembles. For example, high extensional flow regions will probably favor stretched ensemble conformations that rarely exist in quiescent solution.
Oligomers extracted from AD patients have different characteristics than oligomers of the same composition synthesized in the laboratory. It is suggested here that the reason for this is that human brain-derived oligomers were formed under unusual brain fluid flow conditions described above and thus have different fluid flow-stressed conformations during and after their formation. Therefore, it is suggested that laboratory experiments be conducted under these same flow stress conditions and oligomers be compared with those synthesized under quiescent conditions.
Pulse energy is a critical pathological variable
Low bulk ISF flow rates in the upper cortex, if bulk flow indeed exists [50], are the result of the very high density of parenchymal neurons, neuroglia, basal lamina, and the very small extracellular spaces that all serve to constrict ISF fluid flow channels. Blood flow intensity as well as CSF and ISF intensities generally decrease with distance along the vascular tree connections from the heart to the brain [23]. However, low flow rates do not necessarily guarantee low flow stress rates, since there is a nonlinear inverse relationship between flow path width dimensions and shear rate. However, there is still little evidence of high energy chaotic flow in the cerebral cortex (Fig. 5).
It would appear that there are only low energy flow stress events in the cortex parenchyma that may first produce a mechanically distorted Aβ* molecule that initiates the formation of a relatively mild neurotoxic oligomers [10]. This low energy is generated by slower cortex parenchymal fluid flow processes than those found in the lower brain and brainstem region. Higher energy CSF motion in the lower brain region causes more energetic fluid stress-induced molecular distortion of Aβ and tau, producing higher energy dissolved Aβ** and possibly tau** molecules. Such energetic distortion could possibly break or at least weaken hydrogen bonds that stabilize whatever characteristic protein structure that these IDP molecules might temporarily contain. It is expected that this chaotic higher energy can create flow stress-induced products containing longer, comparatively unstructured stretches of amino acid chains (Fig. 14). During flow reversal periods, such long, highly activated molecules could refold into different prion-like oligomer seeds containing cross-beta structure. Such potential seed structures may only be available initially in the lower brain regions where higher flow stress energies are available. These seeds could then produce the types of oligomers that are the keys to seed formation suggested in the Chen-Mobley synthesis for producing tau pathology [17].
Fig. 14
Extensional flow-induced intramolecular processes during CSF passage through the cerebral aqueduct. Column A: low-energy cycle with no conformation-altered monomer. Column B: High-energy toxic monomer seed formed that can react with locus coeruleus. Modified from [39]. Licensed under Creative Commons CC-BY.
![Extensional flow-induced intramolecular processes during CSF passage through the cerebral aqueduct. Column A: low-energy cycle with no conformation-altered monomer. Column B: High-energy toxic monomer seed formed that can react with locus coeruleus. Modified from [39]. Licensed under Creative Commons CC-BY.](https://content.iospress.com:443/media/jad/2021/79-3/jad-79-3-jad201025/jad-79-jad201025-g014.jpg)
Cerebral aqueduct MRIs might possibly be amyloid biomarkers
As indicated in Fig. 15, MRI is a means of determining in vivo CA dimensions and LC locations under full CSF motion. Because of the extensive flow-induced Aβ wall deposits on the walls of laboratory capillaries with roughly similar dimensions and flow rates as those measured in CA structures [14], the contrast in CA cross sections noted in Fig. 16 were of great interest. If we describe the CA cross section as a crown with a flat floor, the CA floor of the Parkinson’s disease patient in Fig. 16 looks as though it has been filled in. This considerably narrows the flow dimensions for CSF, increasing the CSF flow resistance in the CA in addition to also greatly increasing the shear rates experienced by any Aβ and tau molecules dissolved in the oscillating CSF within the CA of the Parkinson’s disease patient. The reason for this narrowing of the CA dimensions could well be the result of flow stress-induced aggregation processes (Fig. 17). Further investigations are needed involving longitudinal MRI studies to see whether this effect is correlated with disease progression. If so, laboratory flow experiments should be conducted to explore a possible mechanism for the buildup and explore whether MRI measurements in the CA and other ventricle and cistern flow stress hot spots can act as amyloid disease biomarkers.
Fig. 15
Visualization of human LC as an MRI hyperintense signal in the brainstem using T1-weighted MRI optimized to enhance neuromelanin LC content (white arrows), seen in axial (B) and coronal (C) planes acquired as shown in (A). Source [51] with permission.
![Visualization of human LC as an MRI hyperintense signal in the brainstem using T1-weighted MRI optimized to enhance neuromelanin LC content (white arrows), seen in axial (B) and coronal (C) planes acquired as shown in (A). Source [51] with permission.](https://content.iospress.com:443/media/jad/2021/79-3/jad-79-3-jad201025/jad-79-jad201025-g015.jpg)
Fig. 16
Contrast in LC (white arrow) environment in cerebral aqueduct in (A) a healthy patient; (B) Parkinson’s disease patient with early symptoms.Source [52] with permission.
![Contrast in LC (white arrow) environment in cerebral aqueduct in (A) a healthy patient; (B) Parkinson’s disease patient with early symptoms.Source [52] with permission.](https://content.iospress.com:443/media/jad/2021/79-3/jad-79-3-jad201025/jad-79-jad201025-g016.jpg)
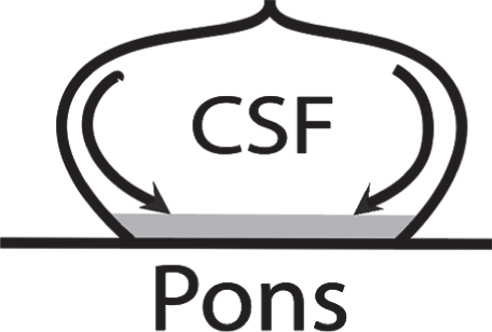
Fig. 17
Cross section of the cerebral aqueduct, which is surrounded above by the pons and that has a floor of the cerebellum. In addition to laminar shear flow, there may be secondary flow, indicated by the arrows causing flow toward the floor of flow-stressed molecules that aggregate on the floor.
A flow-stress energy continuum is responsible for a preclinical progression continuum to MCI
It is now known that decades before the first clinical AD symptoms appear, there are other preclinical and mild cognitive impairment (MCI) AD symptoms that can be observed, measured, and classified [53]. Among these are accumulation within CSF and ISF of Aβ in the form of soluble Aβ and tau monomers and oligomers, synaptic disfunction, tau mediated neuronal injury, and the initiation of the reduction of brain volume. MCI symptoms are codified indicators of mild cognitive dysfunction. It is within these first two periods in Fig. 18 that the author wishes to focus in an attempt to identify flow-stress events, locations, and mechanisms of initiating AD.
Fig. 18
Deterioration of cognitive function in AD with time correlated with postulated corresponding increases in flow-stress energized Aβ and tau molecules contained in brain CSF. Both of these are postulated to be smooth transitions. The dashed line is that for AD patients and the solid line for healthy individuals. Asterisks indicate the degree of flow-stress energy and aggregation in the amyloid monomers. Note that this diagram represents a hypothetical model for the pathological-clinical continuum of AD but does not imply that all individuals with biomarker evidence of AD-pathophysiological process will progress to the clinical phases of the illness. Aβ42* represents cortex plaque formation. Aβ40* represents low energy oligomers and CAA formation. Aβ** represents higher energy oligomer seed precursors. Tau** represents higher energy tau oligomer seed precursors. Figure modified (in red) from [53] with permission.
![Deterioration of cognitive function in AD with time correlated with postulated corresponding increases in flow-stress energized Aβ and tau molecules contained in brain CSF. Both of these are postulated to be smooth transitions. The dashed line is that for AD patients and the solid line for healthy individuals. Asterisks indicate the degree of flow-stress energy and aggregation in the amyloid monomers. Note that this diagram represents a hypothetical model for the pathological-clinical continuum of AD but does not imply that all individuals with biomarker evidence of AD-pathophysiological process will progress to the clinical phases of the illness. Aβ42* represents cortex plaque formation. Aβ40* represents low energy oligomers and CAA formation. Aβ** represents higher energy oligomer seed precursors. Tau** represents higher energy tau oligomer seed precursors. Figure modified (in red) from [53] with permission.](https://content.iospress.com:443/media/jad/2021/79-3/jad-79-3-jad201025/jad-79-jad201025-g018.jpg)
There are smooth, continuous transitions between three stages: preclinical, MCI, and AD dementia, as shown in Fig. 18 [53]. The symbols in red (Aβ*, Aβ**, and Tau**) represent the steadily increasing degree of flow-stress energy within the cortex and lower brain region, with resulting aggregation of Aβ and tau monomers, at various points in the AD timeline. At lower energies, Aβ42 is readily aggregated in the form of senile plaque in the cortex. At higher energies Aβ40, which escapes the cortex, is aggregated as perivascular CAA [10]. Note that proposed high amounts of stress energy are needed to induce tau to aggregate, whereas Aβ aggregates much more readily at lower energies and at earlier times. There would appear to be a higher energy Aβ** oligomer seed that spreads prion-like throughout the cortex, possibly formed within or on the walls of the basal cistern. Tau** apparently requires a more complex flow environment.
It is reported [54] that Aβ induces phosphorylation of tau (p-tau) that is released into ISF and CSF. p-tau is both water soluble and an IDP molecule. As such, it should be a relatively flexible molecule but probably with more alpha helical and possibly beta structure in the various members of its IDP p-tau conformational ensemble than that found in the Aβ ensemble.
Tau is a much larger IDP than Aβ and thus is a more complex molecule from a conformational change point of view. Flow-stressed conformational changes involving tau have more intricate energy and entropy demands. Increasing energy extensional flow should stretch tau into increasingly longer, relatively-linear ensemble conformations. This energy is probably available from chaotic processes in the lower brain regions. However, the conformational changes required to convert tau into a cross-beta seed conformation may require a number of sequential and focused molecular perturbation processes. These may only arise from cyclic extensional strain events initiated within the unique geometry of the combination of the third ventricle, CA, and fourth ventricle to accomplish the conversion of p-tau into a cross-beta conformation capable of ultimately forming a tau oligomer seed.
The smooth and continuous nature of these increasing energy transfer processes, according to this hypothesis, is made possible by the smooth and continuous hardening of the carotid and aorta arteries that produce smooth and continuous increases in systolic CSF pulse energies in the lower brain regions. It is acknowledged that some of these curves and data in Fig. 18 are hypothetical and oversimplify a much more complex situation, but nevertheless may offer a useful overall framework that is able to support the more complex theory outlined above.
Cystolic CSF spikes arising from hardened major arteries are the missing link to the origin of AD
Chaotic flow regions in the lower brain, cisterns, and around the brainstem are present in healthy adults, as indicated in Fig. 5. Thus, chaotic lower brain CSF flow per se does not cause AD. What, then, are the conditions that trigger AD and possibly other amyloid diseases? This paper suggests that the reason is, as the AD patient ages, the aorta and carotid arteries harden and lose their wall flexibility that helps even out sharp blood pressure pulses emanating from the heart [55–57]. As these arteries harden, the heart must work harder to make up for resulting lower total oxygen delivered to the brain. Thus, with increasing age, these hardened arteries pass along a sharper pulse intensity of the heart systole pressure pulse further down the vascular tree, shown symbolically in Fig. 19. Thus, the pulse becomes increasingly destructive, ultimately causing brain silent microbleeds in brain capillaries [55].
Fig. 19
Vascular pulsing that is primarily responsible for CSF flow –the effects of aging and artery hardening. The aorta and carotid harden and lose their flexibility that is responsible for steady capillary flow (upper figure). But artery hardening causes intense heart pulses to pass through these two major arteries and increases the pulse intensity motion in the circle of Willis, the minor arteries, and capillaries, causing microbleeds [55]. Thus, the increased pulse intensity in the circle of Willis with age should be responsible for increased chaotic CSF flow in the neighborhood of the circle of Willis, namely in the basal cistern and its direct CSF connections. This increased motion could lead to higher energy Aβ and tau molecular states that are precursors to neurotoxic seeds, thus possibly initiating Aβ seeds leading to AD.
![Vascular pulsing that is primarily responsible for CSF flow –the effects of aging and artery hardening. The aorta and carotid harden and lose their flexibility that is responsible for steady capillary flow (upper figure). But artery hardening causes intense heart pulses to pass through these two major arteries and increases the pulse intensity motion in the circle of Willis, the minor arteries, and capillaries, causing microbleeds [55]. Thus, the increased pulse intensity in the circle of Willis with age should be responsible for increased chaotic CSF flow in the neighborhood of the circle of Willis, namely in the basal cistern and its direct CSF connections. This increased motion could lead to higher energy Aβ and tau molecular states that are precursors to neurotoxic seeds, thus possibly initiating Aβ seeds leading to AD.](https://content.iospress.com:443/media/jad/2021/79-3/jad-79-3-jad201025/jad-79-jad201025-g019.jpg)
It is assumed that perivascular CSF motion is generated in the cisterns and CA by these same heart pressure pulse waves. If so, as one ages, there should also be increasing energy pulsed CSF waves and therefore increasing chaotic flow in the lower brain region. In turn, these waves should cause increased CSF flow-induced Aβ and tau molecule distortion. Such an increase in CSF pulse energy could reach a critical energy level such that it exceeds the activation energy for conversion of the comparatively large, complex tau molecule into a conformation that subsequently converts other tau molecules into tau tangles in the EC.
Could age-elevated CSF pulse intensity initiate both Aβ and tau AD pathology elsewhere?
During the history of the search for the AD’s origins, many have noted parallels between cognitive decline and vascular dysfunction [26, 56]. “In humans, a chronic increase in pulse pressure is associated with cognitive decline and dementia. More specifically, increased PWV (pulse wave velocity) is associated with memory loss, MCI, cognitive decline, and dementia” [56]. MCI patients have been shown to have the sharpest, most intense pulse blood flow when compared with AD patients and healthy controls [58]. Could age-related excess blood and CSF pulse energy be the critical AD-vascular connection? That is, if vascular degeneration of the aorta and carotid artery flexibility lead to increased systolic pulse pressure and PWV, does this also cause increased CSF chaotic flow? These highly directional kinetic energy CSF motions could overcome activation energy barriers, allowing conformational transformations of each monomer that lead to the formation of a catalytic Aβ-tau** transient, possibly on a CA surface. This could be followed by the formation and release of either a tau** seed or an amyloid-beta tau** oligomer seed from this surface that can spread in a prion-like fashion throughout the brain to initiate AD symptoms.
The locations of CSF chaotic motion hot spots are the same as Aβ and tau pathology
Aβ oligomer seeds can spread from the medial temporal and orbitofrontal neocortex throughout the anterior cortex. The Chen-Mobley synthesis [17] suggests that these oligomers migrate through neuronal circuits from the cortex to regions near the EC, LC, and the hippocampus, all of which are critical to functioning memory. However, Aβ as well as tau oligomers could also be formed within the CA and third and fourth ventricle. It is here in a highly localized pons location that tau seeds are apparently initially formed, not in the upper cortex.
Proposed mechanism for the formation of fluid stress-induced tau seeds
The above mechanism still neglects one critical prerequisite, the need for both Aβ and tau to somehow be involved in the critical step or steps leading to initiation of AD tau pathology. Why are the origins of Aβ and tau seeds located in different parts of the brain? It is suggested that Aβ seed is formed by the lower-energy chaotic motions within the Circle of Willis region because of the pulse-induced CSF motions in the MCI phase toward the beginning of the major artery hardening process. The larger tau molecule is harder to convert into a seed oligomer. It takes some combination of a more-focused, more highly energetic third ventricle and, in particular, a CA extensional flow pattern to convert tau into the necessary cross-beta conformation needed to form a neurotoxic tau seed. The energy needed for this tau conversion could be achieved at a later time in the artery hardening process when systolic pulse energy reaches a high critical energy level. El Sankeri et al. show [58] that a maximum systolic pulse flow is achieved in MCI patients when compared with healthy controls and AD patients.
DRUG STUDIES ON DRUG-AMYLOID MONOMER MIXTURES IN A FLOW STRESS ENVIRONMENT ARE NEEDED
Laboratory flow stress studies have shown that proteins aggregate much faster when agitated [28] and under high extensional flow conditions [36]. However, protein oligomer products generated under these flow-stress conditions have not been identified. But in vivo studies indicate that the brain environment somehow produces aggregated amyloids and amyloid species that are conformationally different from those that are formed under quiescent laboratory conditions [32]. The author has suggested [12, 15] that this is because shear- or extensional flow-induced conformation changes resulting from flow stress within the brain are different from those in quiescent conditions.
Most drugs operate on the idea that structure predicts function. But the structures (conformations) of both proteins [36] and antibodies [36, 59] have been shown to change with stresses associated with shear and extensional flows. If the effectiveness of a drug depends on its structure and the toxicity of the toxic amyloid depends on its structure, then what are the effects of chaotic flow stresses on the drug-amyloid combination? What would be the effects during their initial collisions under flow stress conditions? Fig. 20 illustrates in a purely geometric manner that the same type of fit may be still possible in the extensionally stressed pair but would involve a different number of chemical bonds. Thus, it is vitally important to expose to extensional flow both the amyloid monomer and the proposed AD drug in the same flow stress environment at the same time. The effects of both extensional flow rates and shear rates on proposed drugs need to be taken into consideration.
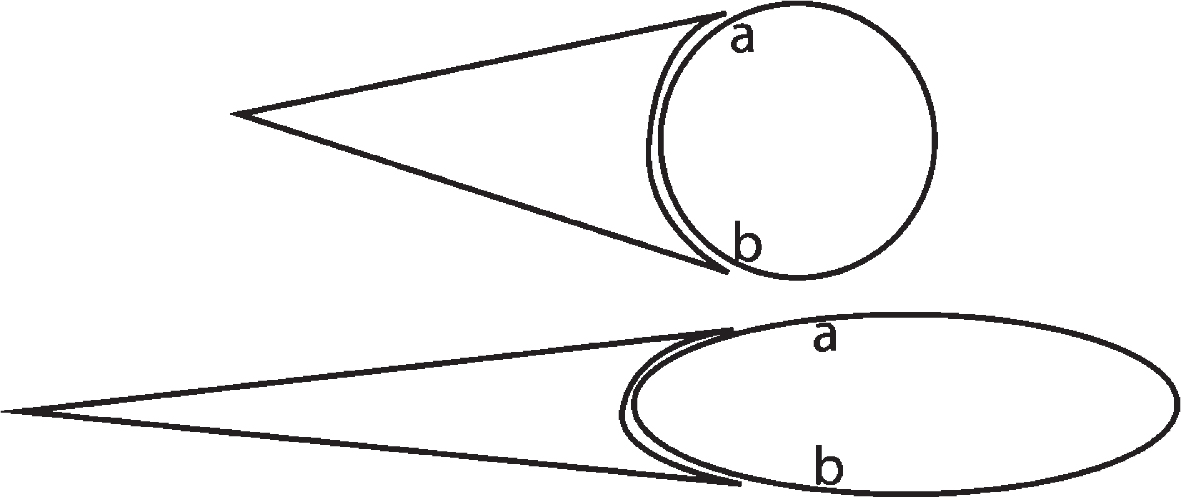
Fig. 20
Effect of extensional flow conditions on an antigen (circle)-antibody (cone) fit. Upper figures are unstrained and lower are both strained. Although there is a possibility of a strained fit, that fit would not cover the same percentage of the chemical surface at the intersection as shown by the fixed a and b points on the antigen. Although this is a purely geometric model, the author suggests that the same reasoning would probably apply at the molecular level. This would depend on the comparative geometric responses of the amyloid and antibody molecules to strain. In this figure, it is assumed that the strain responses are equal for antigen and antibody.
Thus, the experimental question raised by this author is, what are the in vivo effects of shear and extensional flow conditions that CSF solutions of Aβ and tau experience in the continuous, reciprocal back-and-forth CSF, heavily pulsed, motions in the vicinity of the CA? The experimental variables that need to be studied are those listed by Dobson et al. [36]: the applied extensional flow stress rate (proportional to flow rate), the concentration, and the protein structure. Both Aβ and tau are highly flexible IDPs. Thus, they both should be readily affected by extensional strain.
Increased temperature has been shown through molecular dynamics calculations to increase the fraction of the Aβ IDP conformations that are partially structured in alpha helical or beta sheet segments [60]. This is surprising because increased temperatures would be expected to unfold any protein and reduce the amount of structure in the molecule. If increased thermal free energy promotes additional structure in this IDP molecule, does increased extensional strain do the same thing? If so, what kind of structures are produced? Could such forces catalyze a random coil in converting from an alpha to a beta conformation in these amyloid monomers? These questions should be answered with appropriate experiments or molecular dynamics calculations if possible.
RECOMMENDED EXPERIMENTS AND CLINICAL STUDIES TO TEST THE ABOVE HYPOTHESES
The above hypotheses are just that. The author believes that they are based on a solid foundation of supporting literature. However, they admittedly lack supporting sufficient experimental support, which the author is unable to undertake. The needed experiments are many. A large number of them have been detailed in the author’s previous papers [11 (supplementary material), 15]. More based on this paper are listed below.
The Dobson et al. paper [36] demonstrates that the rate of extensional strain energy delivered to dissolved proteins is a very important variable with respect to the rate of protein aggregation. An increased extensional flow component also increased the energy transfer rate but decreased the amount of time a molecule would be exposed to such stresses while transiting this small extensional flow region. On the other hand, at lower flow rates, the transiting molecule is exposed to lower extensional strain rates over longer periods of time. Which is more effective in promoting molecular conformation change in amyloids? Are in vivo clues available to help answer this question?
The flow rates calculated by Siyahhan et al. [39] are important and provide interesting and useful flow rate data. However, corresponding calculations of extensional shear rates are badly needed so that experimental models can be constructed of those regions that have elevated extensional flow rates. Laboratory experiments need to be done in such regions to study the variables that affect the formation of Aβ and tau oligomers and the structures of the chemical products produced.
Previous papers by the author have suggested many other urgent experiments [11 (supplementary material), 15]. Perhaps the simplest, yet most basic experiment is a plug injection of a short cylindrical sample into a flowing mobile phase in an empty capillary at a flow rate where the Aβ, tau, or other CSF protein-containing molecules do not significantly diffuse during transit through the capillary. This should create a laminar flow profile of sample that exits the capillary in some respects in a series of short, thin cylindrical layer-by-short, thin cylindrical layer, with each subsequent layer being exposed to different shear rates and total amounts of shear. If the concentration of the exiting effluent is monitored as a function of time, it would show a sudden spike at the time of first sample exiting the capillary, followed by an exponential concentration decay. The first aliquot of sample-containing effluent from the center of the capillary will contain solution in the sudden high solute concentration spike zone that has been exposed to near zero shear rate along the centerline of the capillary (the tip of the bullet in a bullet-shaped sample profile). Each succeeding sample aliquot layer will have been exposed to longer time periods of shear, but more importantly, have been exposed to increasing shear rates. Such aliquots need to be tested for the presence of oligomers, especially when the solute Aβ or tau concentrations are increased, as well as flow rates and flow types, e.g., pulsed and cycling back-and-forth flow through the capillary.
Most important are experiments in which chemical products are identified in mixtures of Aβ and tau that are subjected to reciprocal flows in experimental models with realistic CA region geometry. Concentration ratios of these two amyloid monomer solutes and resulting products should also be studied. Among experimental variables would be flow rate, cyclic back-and-forth flow frequency, increasing energy pulsed flows, ratio of Aβ/tau concentrations, temperature, and flow stop and flow wall release experiments [15]. Stopped flow spectroscopy experiments (ultraviolet, circular dichroism, Raman, infrared) should also be undertaken to look for shear-excited transient molecular decay or aggregation reactions. Experiments contrasting the sensitivity to the different Aβ isoforms, Aβ40 and Aβ42, should be included. ELISA analyses could also be used to detect oligomeric Aβ.
Very important would also be longitudinal human in vivo MRI clinical studies following up on the CA narrowing results shown in Fig. 16. These studies are recommended as a possible CA biomarker at least for Parkinson’s disease as well as AD and possibly other amyloid diseases. Floor and/or wall deposits similar to those found in Fig. 16 could be measured as a function of disease progression. Further MRI investigations of other ventricle and cistern regions as a function of disease progression could possibly be revealing and provide possible biomarkers.
SUGGESTED FLOW STRESS ENERGY LEVELS AND THEIR CONSEQUENCES
Figure 21 illustrates some of the main hypotheses of this paper. Although the focus of AD research has been on chemical and biological processes primarily within the upper cortex, both AD pathology and the high energy, chaotic nature of CSF motion are in the same lower regions of the brain glymphatic system. MRI studies in this region have revealed chaotic CSF motion in the third ventricle and in a number of cisterns and brainstem regions shown as black regions in Fig. 21. Aβ and tau pathologies are located either in high chaotic regions or are in regions that not only have high chaotic energy, but also receive CSF containing Aβ and tau that has been just exposed to high chaotic flow stress. This highly energetic CSF motion has the energy necessary to promote severe Aβ and tau molecular distortion, possibly leading to the formation of templated Aβ and tau seeds that can spread throughout the cortex and cause amyloid diseases.
Fig. 21
Origins of Aβ and tau AD pathology correlates with black areas revealing regions in which CSF motion is either chaotic and/or flowing from a chaotic flow region. Parkinson’s and ALS pathology also originate in the lower brainstem region where motor nerves jut into chaotic CSF. Source: [18] Licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives International.
![Origins of Aβ and tau AD pathology correlates with black areas revealing regions in which CSF motion is either chaotic and/or flowing from a chaotic flow region. Parkinson’s and ALS pathology also originate in the lower brainstem region where motor nerves jut into chaotic CSF. Source: [18] Licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives International.](https://content.iospress.com:443/media/jad/2021/79-3/jad-79-3-jad201025/jad-79-jad201025-g021.jpg)
There are three different energetic types of flow field regions of interest encountered in amyloid flow stress studies: shear, extensional flow, and chaotic flow. Chaotic flow fields have not been fully characterized except for their being highly energetic. Shear causes molecular distortion and rotation, whereas predominantly extensional flow causes mostly molecular stretching of dissolved solutes. Extensional flow is caused by relatively sudden reductions in flow channel cross sectional dimensions, such as that observed in the CA in Fig. 21 found just above the location labeled “Tau pathology origin here”. Since net CSF flow is from the third to the fourth ventricle in this figure, any shear-induced neurotoxic oligomer Aβ and tau products formed in the third and fourth ventricles and the CA will be exposed to the critical LC located in and around the point of the “Tau pathology origin here” marker.
The Tau pathology origin is in the same exact location as the LC. This LC is exposed to CSF that has just recently passed through both a highly energetic chaotic CSF third ventricle zone and multiple high-energy extensional CA shear zones! In these regions, there should be sufficient accumulated flow-stress energy to overcome the activation energy to convert tau into neurotoxic seeds—if CSF pulse energy is sufficiently high. Neurotoxic damage to the LC affects the entire cortex and other critical parts of the lower brain and is among the earliest tau AD pathological events. Thus, it is suggested that high-energy flow stress events in the third ventricle/CA/fourth ventricle regions that produce neurotoxic oligomers could be the trigger that initiates AD.
Table 1 summarizes hypothetical comparative energy levels of flow-stress excited amyloid molecules in various brain locations, with suggested hypothetical outcomes. Aβ** monomer seeds have not been characterized, but oligomer seeds for both Aβ and tau have been isolated from AD brains that have different characteristics than similar oligomers synthesized in the laboratory. The suggestion in Table 1 is that for a particular amyloid monomer, there is a critical flow stress energy input, indicated by the dashed line, that alters the molecule’s conformation in such a way as to form either a templated seed or its precursor. This line is drawn below those products formed from flow stress generated by high energy vascular pulse waves that generate very high CSF motion energy, resulting in the ultimate formation of amyloid seeds.
Table 1
Relative flow stress energy levels and consequences
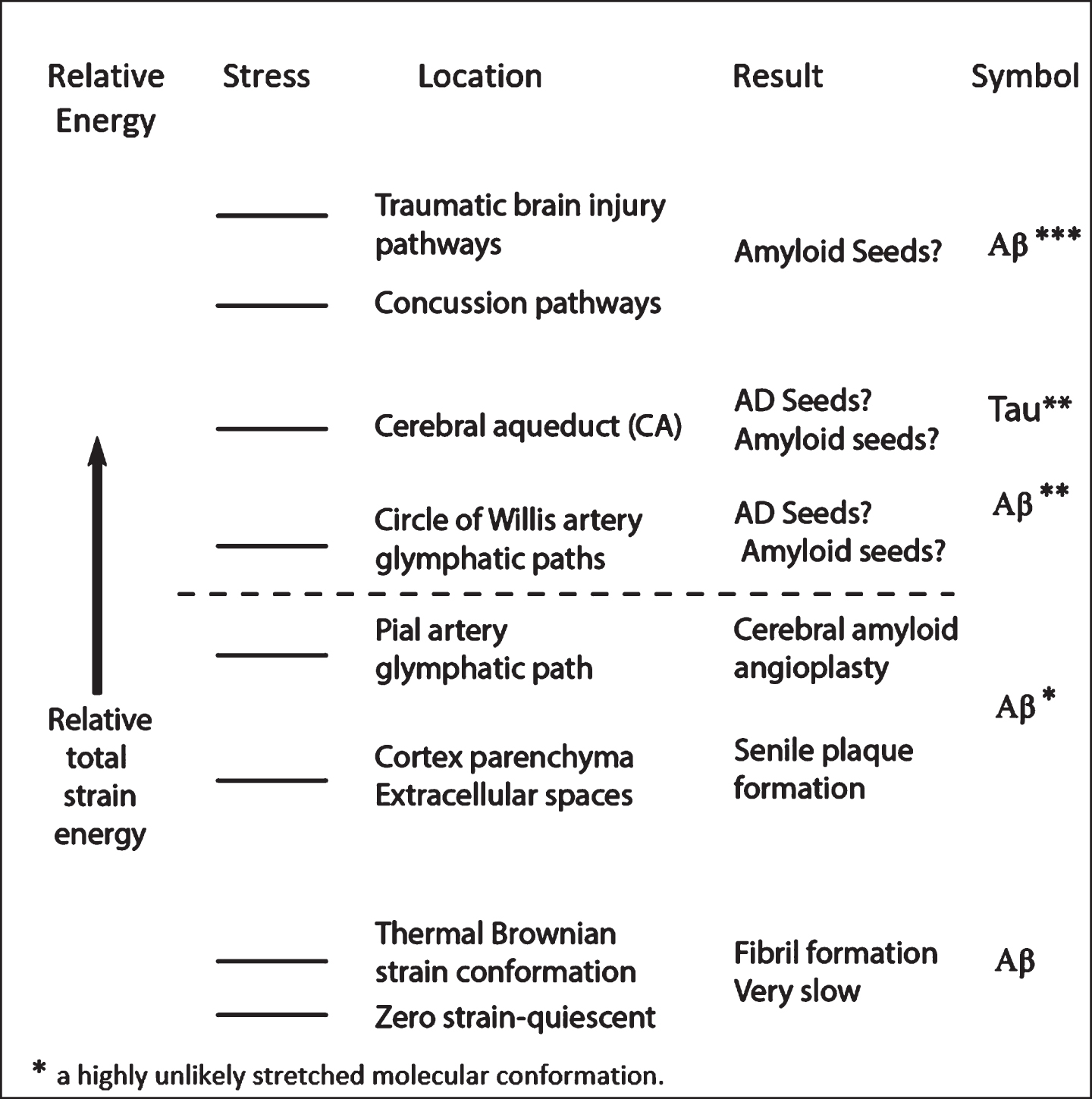 |
ARE THESE HYPOTHESES APPLICABLE TO AMYLOID DISEASES OTHER THAN AD?
A number of amyloid disease monomers other than Aβ and tau are also IDPs. Among these, there are many parallels with Aβ and tau, especially since there is a class of approximately twenty amyloid diseases, tauopathies, that are reported to involve tau. Some of these diseases have strains that are attributed to different molecular conformations of the same monomer. These different strains could be formed by either shear or extensional flow in different parts of the brain, particularly in the brainstem region because of the proximity of major blood vessels that generate significant mechanical pulse energy.
Nerves act as CSF flow-blocking obstacles and may initiate other amyloid diseases. CSF motion within the brain’s glymphatic flow path is not only often blocked by embedded arteries and veins but also by nerves and nerve bundles emanating from the brainstem and within cisterns. Figure 22 illustrates nerves that probably block CSF flow generated by pulse waves emanating from the major basilar and vertebral arteries.
Fig. 22
Brain stem pons and oblongata nerves (yellow) that project into the surrounding CSF and arteries, probably providing CSF flow-restricting objects that could, under the right pulse energy conditions, result in the formation of CSF flow stress-induced neurotoxic oligomers. These molecules could inactivate these nerves. The lower oblongata region is the pathological origin of Parkinson’s disease and ALS. Source [61] with permission.
![Brain stem pons and oblongata nerves (yellow) that project into the surrounding CSF and arteries, probably providing CSF flow-restricting objects that could, under the right pulse energy conditions, result in the formation of CSF flow stress-induced neurotoxic oligomers. These molecules could inactivate these nerves. The lower oblongata region is the pathological origin of Parkinson’s disease and ALS. Source [61] with permission.](https://content.iospress.com:443/media/jad/2021/79-3/jad-79-3-jad201025/jad-79-jad201025-g022.jpg)
Early-stage Parkinson’s disease and ALS (amyotrophic lateral sclerosis) patients show very early pathology in the lower medulla region in Fig. 22 that might arise from toxic amyloid products produced in this region by stresses generated from CSF flow around these nerves that are mostly associated with motor function. Is it possible that the strongly chaotic CSF flow generated by the heavily pulsing vertebral arteries around these projecting nerves could produce flow-induced neurotoxic oligomer products leading to Aβ*-induced motor neuron damage in these areas, thus leading to Parkinson’s and other motor nerve-involved amyloid diseases?
SUMMARY
• Regions of highly variable laminar, extensional, and chaotic flow stresses have been identified in the human brain. Low ISF stress energy is found in the cerebral cortex. Very high energy flow stress has been identified in CSF flow around the lower brain frontal lobe cortex cisterns, within and between certain brain ventricles, and around the brainstem.
• Therefore, dissolved Aβ and tau molecules within brain CSF are exposed to high energy mechanical forces that distort these molecules, altering their molecular conformations in ways that critically depend on the CSF flow path geometry and intensity of flow stress processes.
• It is known that the mechanical energy input from flow stresses accelerates the aggregation of Aβ and other protein molecules, especially in the presence of hydrophobic and polar surfaces.
• Aβ and tau oligomers have been identified as the neurotoxic agents in amyloid diseases. It has been suggested that these aggregates arise from “misfolded” protein molecules, but no energetic cause of this misfolding has thus far been accepted by the scientific community.
• Neurotoxic templated prion-like oligomer seeds extracted from the AD brain differ from those synthesized in the laboratory.
• The author proposes that fluid flow stresses, probably in conjunction with brain surfaces, provide the energy for a catalytic mechanism to form these conformation-altered oligomers.
• It is proposed that variable flow stress energy yields different oligomer amyloid strains, initiating different neurological malfunctions or destruction mechanisms. CSF stress with predominant extensional flow contribution is proposed to be most effective flow stress in production of neurotoxic Aβ and tau oligomers.
• However, the ultimate trigger that initiates AD symptoms is suggested to be the age-related increasing energy systolic cardiac pulse waves that drive increasingly chaotic CSF flow stress in lower brain regions. This increasing pulse energy arises from aortic and carotid artery hardening with increasing age and justifies the “heart-mind AD connection” theory.
• The highest chaotic energy levels that induce Aβ and tau pathology are two locations in the lower brain: 1) the basil cistern containing the circle of Willis arteries, and 2) the region including the third and fourth ventricles and the narrow CA connecting them. These two regions coincide, respectively, with the temporal origins of Aβ and tau pathology.
• Therefore, it would appear that Aβ and tau oligomers generated in the cerebral aqueduct and basil cistern are one key to initiating AD pathology. The other key is the age-related high energy systolic cardiac pulse waves arising from artery hardening and driving the CSF chaotic flow pulse that alters molecular conformations.
• Experiments are urgently needed to test the above hypotheses, especially longitudinal MRI studies in the lower brain regions of AD and other amyloid disease patients. Also quite critical are chemical analyses of oligomer flow-induced products in lower brain regions and models of these regions.
ACKNOWLEDGMENTS
The author gratefully acknowledges the editorial help of David Teplow, Aditya Raghunandan, and Elinor Thomforde.
The author’s disclosure is available online (https://www.j-alz.com/manuscript-disclosures/20-1025r2).
REFERENCES
[1] | Alzheimer’s Association ((2020) ) Alzheimer’s disease facts and figures. Alzheimers Dement 16: , 391–460. |
[2] | Cremades N , Dobson CM ((2018) ) The contribution of biophysical and structural studies of protein self-assembly to the design of therapeutic strategies for amyloid diseases. Neurobiol Dis 109: , 178–190. |
[3] | Chiti F , Dobson CM ((2017) ) Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Annu Rev Biochem 86: , 27–68. |
[4] | Cline EN , Bicca MA , Viola KL , Klein WL ((2018) ) The amyloid-beta oligomer hypothesis: Beginning of the third decade. J Alzheimers Dis 64: (s1), S567–S610. |
[5] | De Strooper B , Karran E ((2018) ) The cellular phase of Alzheimer’s disease. Cell 164: , 603–615. |
[6] | Mannini B , Chiti F ((2017) ) Chaperones as suppressors of protein misfolded oligomer toxicity. Front Mol Neurosci 10: , 98. |
[7] | Hane FT , Lee BY , Leonenko Z ((2017) ) Recent progress in Alzheimer’s disease research, part 1: Pathology. J Alzheimers Dis 57: , 1–28. |
[8] | Dobson CM ((2017) ) The amyloid phenomenon and its links with human disease. Cold Spring Harb Perspect Biol 9: , a023648. |
[9] | Trumbore CN , Tremblay R , Penrose J , Mercer M , Kelleher F ((1983) ) Unusual flow behavior in high-performance liquid chromatography capillary tubing. J Chromatogr 280: , 43–57. |
[10] | Trumbore CN ((2016) ) Shear-induced amyloid formation in the brain. I. Potential vascular and parenchymal processes. J Alzheimers Dis 54: , 457–470. |
[11] | Trumbore CN ((2017) ) Shear-induced amyloid formation in the brain: II. An experimental system for monitoring amyloid shear processes and investigating potential spinal tap problems. J Alzheimers Dis 59: , 543–557. |
[12] | Trumbore CN ((2018) ) Shear-induced amyloid formation in the brain: III. The roles of shear energy and seeding in a proposed shear model. J Alzheimers Dis 65: , 47–70. |
[13] | Trumbore CN ((2018) ) Shear-induced amyloid formation in the brain: IV. Liquid shear in synapse regions near senile plaques. J Alzheimers Dis 66: , 57–73. |
[14] | Trumbore CN , Paik J , Fay D , Vachet RW ((2019) ) Preliminary capillary flow experiments with amyloid-beta, possible needle and capillary Abeta adsorption, and a proposal for drug evaluation under shear conditions. J Alzheimers Dis 72: , 751–760. |
[15] | Trumbore CN ((2019) ) Shear-induced amyloid formation of IDPs in the brain. In Dancing protein clouds: Intrinsically disordered proteins in health and disease, Part A, Progress in Molecular Biology and Translational Science, Volume 166, Uversky VN, ed. Elsevier Inc., pp. 225–309. |
[16] | Plog BA , Nedergaard M ((2018) ) The glymphatic system in central nervous system health and disease: Past, present, and future. Annu Rev Pathol 13: , 379–394. |
[17] | Chen XQ , Mobley WC ((2019) ) Alzheimer disease pathogenesis: Insights from molecular and cellular biology studies of oligomeric Abeta and tau species. Front Neurosci 13: , 659. |
[18] | Atsumi H , Horie T , Kajihara N , Sunaga A , Sakakibara Y , Matsumae M ((2020) ) Simple identification of cerebrospinal fluid turbulent motion using a dynamic improved motion-sensitized driven-equilibrium steady-state free precession method applied to various types of cerebrospinal fluid motion disturbance. Neurol Med Chir (Tokyo) 60: , 30–36. |
[19] | Matsumae M , Hirayama A , Atsumi H , Yatsushiro S , Kuroda K ((2014) ) Velocity and pressure gradients of cerebrospinal fluid assessed with magnetic resonance imaging. J Neurosurg 120: , 218–227. |
[20] | Horie T , Kajihara N , Matsumae M , Obara M , Hayashi N , Hirayama A , Takizawa K , Takahara T , Yatsushiro S , Kuroda K ((2017) ) Magnetic resonance imaging technique for visualization of irregular cerebrospinal fluid motion in the ventricular system and subarachnoid space. World Neurosurg 97: , 523–531. |
[21] | Matsumae M , Kuroda K , Yatsushiro S , Hirayama A , Hayashi N , Takizawa K , Atsumi H , Sorimachi T ((2019) ) Changing the currently held concept of cerebrospinal fluid dynamics based on shared findings of cerebrospinal fluid motion in the cranial cavity using various types of magnetic resonance imaging techniques. Neurol Med Chir (Tokyo) 59: , 133–146. |
[22] | Duerkop M , Berger E , Durauer A , Jungbauer A ((2018) ) Impact of cavitation, high shear stress and air/liquid interfaces on protein aggregation. Biotechnol J 13: , e1800062. |
[23] | Sloots JJ , Biessel GJ , Zwanenburg JM ((2020) ) Cardiac and respiration-induced brain deformations in humans quantified with high-field MRI. Neuroimage 210: , 116581. |
[24] | Grigolato F , Arosio P ((2020) ) Synergistic effects of flow and interfaces on antibody aggregation. Biotechnol Bioeng 117: , 417–428. |
[25] | Banerjee S , Hashemi M , Lv Z , Maity S , Rochet JC , Lyubchenko YL ((2017) ) A novel pathway for amyloids self-assembly in aggregates at nanomolar concentration mediated by the interaction with surfaces. Sci Rep 7: , 45592. |
[26] | Sweeney MD , Montagne A , Sagare AP , Nation DA , Schneider LS , Chui HC , Harrington MG , Pa J , Law M , Wang DJJ , Jacobs RE , Doubal FN , Ramirez J , Black SE , Nedergaard M , Benveniste H , Dichgans M , Iadecola C , Love S , Bath PM , Markus HS , Salman RA , Allan SM , Quinn TJ , Kalaria RN , Werring DJ , Carare RO , Touyz RM , Williams SCR , Moskowitz MA , Katusic ZS , Lutz SE , Lazarov O , Minshall RD , Rehman J , Davis TP , Wellington CL , González HM , Yuan C , Lockhart SN , Hughes TM , Chen CLH , Sachdev P , O’Brien JT , Skoog I , Pantoni L , Gustafson DR , Biessels GJ , Wallin A , Smith EE , Mok V , Wong A , Passmore P , Barkof F , Muller M , Breteler MMB , Román GC , Hamel E , Seshadri S , Gottesman RF , van Buchem MA , Arvanitakis Z , Schneider JA , Drewes LR , Hachinski V , Finch CE , Toga AW , Wardlaw JM , Zlokovic BV ((2019) ) Vascular dysfunction-The disregarded partner of Alzheimer’s disease. Alzheimers Dement 15: , 158–167. |
[27] | Dunstan DE , Hamilton-Brown P , Asimakis P , Ducker W , Bertolini J ((2009) ) Shear flow promotes amyloid-beta fibrilization. Protein Eng Des Sel 22: , 741–746. |
[28] | Cohen SI , Linse S , Luheshi LM , Hellstrand E , White DA , Rajah L , Otzen DE , Vendruscolo M , Dobson CM , Knowles TP ((2013) ) Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc Natl Acad Sci U S A 110: , 9758–9763. |
[29] | Huang YR , Liu RT ((2020) ) The toxicity and polymorphism of beta-amyloid oligomers. Int J Mol Sci 21: , 4477. |
[30] | Hayden EY , Teplow DB ((2013) ) Amyloid beta-protein oligomers and Alzheimer’s disease. Alzheimers Res Ther 5: , 60. |
[31] | Kusumoto Y , Lomakin A , Teplow DB , George B , Benedek GB ((1998) ) Temperature dependence of amyloid β-protein fibrillization. Proc Natl Acad Sci U S A 95: , 12277–12282. |
[32] | Jucker M , Walker LC ((2013) ) Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501: , 45–51. |
[33] | Walker LC ((2016) ) Proteopathic strains and the heterogeneity of neurodegenerative diseases. Annu Rev Genet 50: , 329–346. |
[34] | Moores B , Drolle E , Attwood SJ , Simons J , Leonenko Z ((2011) ) Effect of surfaces on amyloid fibril formation. PLoS One 6: , e25954. |
[35] | Shafrir Y , Durell S , Arispe N , Guy HR ((2010) ) Models of membrane-bound Alzheimer’s Abeta peptide assemblies. Proteins 78: , 3473–3487. |
[36] | Dobson J , Kumar A , Willis LF , Tuma R , Higazi DR , Turner R , Lowe DC , Ashcroft AE , Radford SE , Kapura N , David J , Brockwell DJ ((2017) ) Inducing protein aggregation by extensional flow. Proc Natl Acad Sci U S A 114: , 4673–4678. |
[37] | Mander BA ((2020) ) Local sleep and Alzheimer’s disease pathophysiology. Front Neurosci 14: , 525970. |
[38] | Xie L , Kang H , Kang H , Xu Q , Chen MJ , Liao Y , Thiyagarajan M , O’Donnell J , Christensen DJ , Nicholson C , Iliff JJ , Takano T , Deane R , Nedergaard M ((2013) ) Sleep drives metabolite clearance from the adult brain. Science 342: , 373–377. |
[39] | Siyahhan B , Knobloch V , de Zélicourt D , Asgari M , Schmid Daners M , Poulikakos D , Kurtcuoglu V ((2014) ) . Flow induced by ependymal cilia dominates near-wall cerebrospinal fluid dynamics in the lateral ventricles. J R Soc Interface 11: , 20131189. |
[40] | Goedert M , Masuda-Suzukake M , Falcon B ((2017) ) Like prions: The propagation of aggregated tau and a-synuclein in neurodegeneration. Brain 140: , 266–278. |
[41] | Jucker M , Walker LC ((2018) ) Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat Neurosci 21: , 1341–1349. |
[42] | Colby D , Prusiner S ((2011) ) De novo generation of prion strains. Nat Rev Microbiol 9: , 771–777. |
[43] | Ashton L , Dusting J , Imomoh E , Balabani S , Blanch EW ((2010) ) Susceptibility of different proteins to flow-induced conformational changes monitored with Raman spectroscopy. Biophys J 98: , 707–714. |
[44] | Grigolato F , Colombo C , Ferrari R , Rezabkova L , Arosio P ((2017) ) Mechanistic origin of the combined effect of surfaces and mechanical agitation on amyloid formation. ACS Nano 11: , 11358–11367. |
[45] | Mestre H , Tithof J , Du T , Song W , Peng W , Sweeney AM , Olveda G , Thomas JH , Nedergaard M , Kelley DH ((2018) ) Flow of cerebrospinal fluid is driven by arterial pulsations and is reduced in hypertension. Nat Commun 9: , 4878. |
[46] | Thomas JH ((2019) ) Fluid dynamics of cerebrospinal fluid flow in perivascular spaces. J R Soc Interface 16: , 20190572. |
[47] | Iyengar SS , Sumner I , Jakowski J ((2008) ) Hydrogen tunneling in an enzyme active site: A quantum wavepacket dynamical perspective. J Phys Chem B 112: , 7601–7613. |
[48] | Betts MJ , Ehrenberg AJ , Hammerer Duzel E ((2018) ) Commentary: Locus coeruleus ablation exacerbates cognitive deficits, neuropathology, and lethality in P301S tau transgenic mice. Front Neurosci 12: , 401. |
[49] | Giorgi FS , Ryskalin L , Ruffoli R , Biagioni F , Limanaqi F , Ferrucci M , Busceti CL , Bonuccelli U , Fornai F ((2017) ) The neuroanatomy of the reticular nucleus locus coeruleus in Alzheimer’s disease. Front Neuroanat 11: , 80. |
[50] | Benveniste H , Liu X , Koundal S , Sanggaard S , Lee H , Wardlaw J ((2019) ) The glymphatic system and waste clearance with brain aging. Gerontology 65: , 106–119. |
[51] | Liu KY , Marijatta F , Hämmerer D , Acosta-Cabronero J , Düzel E , Howard RJ ((2017) ) Magnetic resonance imaging of the human locus coeruleus: A systematic review. Neurosci Biobehav Rev 83: , 325–355. |
[52] | Garcia-Lorenzo D , Santos CL , Ewenczyk C , Leu-Semenescu S , Gallea C , Quattrocchi G , Lobo PP , Poupon C , Benali. H , Arnulf I , Vidailhet M , Lehericy S ((2013) ) The coeruleus/subcoeruleus complex in rapid eye movement sleep behaviour disorders in Parkinson’s disease. Brain 136: , 2120–2129. |
[53] | Sperling RA , Aisen PS , Beckett LA , Bennett DA , Craft S , Fagan AM , Iwatsubo T , Jack CR Jr , Kaye J , Montine TJ , Park DC , Reiman EM , Rowe CC , Siemers E , Stern Y , Yaffe K , Carrillo MC , Thies B , Morrison-Bogorad M , Wagster MV , Phelps CH ((2011) ) Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7: , 280–292. |
[54] | Hernandez F , Gomez de Barreda E , Fuster-Matanzo A , Lucas JJ , Avila J ((2010) ) GSK3: A possible link between beta amyloid peptide and tau protein. Exp Neurol 223: , 322–325. |
[55] | Stone J , Johnstone DM , Mitrofanis J , O’Rourke M ((2015) ) The mechanical cause of age-relate dementia (Alzheimer’s disease): The brain is destroyed by the pulse. J Alzheimers Dis 44: , 355–373. |
[56] | Thorin-Trescases N , de Montgolfier O , Pincon A , Raignault A , Caland L , Labbe P , Thorin E ((2018) ) Impact of pulse pressure on cerebrovascular events leading to age-related cognitive decline. Am J Physiol Heart Circ Physiol 314: , H1214–H1224. |
[57] | Tublin JM , Adelstein JM , Del Monte F , Combs CK , Wold LE ((2019) ) Getting to the heart of Alzheimer disease. Circ Res 124: , 142–149. |
[58] | El Sankari S , Gondry-Jouet C , Fichten A , Godefroy O , Serot JM , Deramond H , Meyer ME , Balédent O ((2011) ) Cerebrospinal fluid and blood flow in mild cognitive impairment and Alzheimer’s disease: A differential diagnosis from idiopathic normal pressure hydrocephalus. Fluids Barriers CNS 8: , 12. |
[59] | Willis LF , Kumar A , Dobson J , Bond NJ , Lowe D , Turner R , Radford SE , Kapur N , Brockwell DJ ((2018) ) Using extensional flow to reveal diverse aggregation landscapes for three IgG1 molecules. Biotechnol Bioeng 115: , 1216–1225. |
[60] | Granata D , Baftizadeh F , Habchi J , Galvagnion C , De Simone A , Camilloni C , Laio A , Vendruscolo M ((2015) ) The inverted free energy landscape of an intrinsically disordered peptide by simulations and experiments. Sci Rep 5: , 15449. |
[61] | Hendrix P , Griessenauer CJ , Foreman P , Loukas M , Fisher WS , Rizk E , Shoja MH , Tubbs RS ((2014) ) Arterial supply of the lower cranial nerves: A comprehensive review. Clin Anat 27: , 108–117. |

