
Aluminum and Neurofibrillary Tangle Co-Localization in Familial Alzheimer’s Disease and Related Neurological Disorders
Abstract
Background:
Protein misfolding disorders are frequently implicated in neurodegenerative conditions. Familial Alzheimer’s disease (fAD) is an early-onset and aggressive form of Alzheimer’s disease (AD), driven through autosomal dominant mutations in genes encoding the amyloid precursor protein and presenilins 1 and 2. The incidence of epilepsy is higher in AD patients with shared neuropathological hallmarks in both disease states, including the formation of neurofibrillary tangles. Similarly, in Parkinson’s disease, dementia onset is known to follow neurofibrillary tangle deposition.
Objective:
Human exposure to aluminum has been linked to the etiology of neurodegenerative conditions and recent studies have demonstrated a high level of co-localization between amyloid-β and aluminum in fAD. In contrast, in a donor exposed to high levels of aluminum later developing late-onset epilepsy, aluminum and neurofibrillary tangles were found to deposit independently. Herein, we sought to identify aluminum and neurofibrillary tangles in fAD, Parkinson’s disease, and epilepsy donors.
Methods:
Aluminum-specific fluorescence microscopy was used to identify aluminum in neurofibrillary tangles in human brain tissue.
Results:
We observed aluminum and neurofibrillary-like tangles in identical cells in all respective disease states. Co-deposition varied across brain regions, with aluminum and neurofibrillary tangles depositing in different cellular locations of the same cell.
Conclusion:
Neurofibrillary tangle deposition closely follows cognitive-decline, and in epilepsy, tau phosphorylation associates with increased mossy fiber sprouting and seizure onset. Therefore, the presence of aluminum in these cells may exacerbate the accumulation and misfolding of amyloidogenic proteins including hyperphosphorylated tau in fAD, epilepsy, and Parkinson’s disease.
INTRODUCTION
Familial Alzheimer’s disease (fAD) is differentiated from the sporadic form of the disease by its early age of onset, typically occurring before the age of 65. This rare hereditary condition represents less than 5% of individuals, who go on to develop Alzheimer’s disease (AD) [1]. Mutations in genes encoding the amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2), mark the autosomal dominant pattern of fAD inheritance [2, 3]. Subsequently, the enhanced proteolytic processing of amyloid-β protein precursor (AβPP) through sequential cleavage by β-secretase (B-site APP-cleaving enzyme 1, BACE1) and γ-secretase, drives the formation of the pathogenic amyloid-β (Aβ) peptides [4]. Histological analysis of postmortem fAD brain tissue is principally characterized by the extracellular deposition of Aβ in senile plaques, intracellular neurofibrillary tangles (NFTs) of hyperphosphorylated tau protein, and cerebral amyloid angiopathy (CAA) [5].
As the second most common form of neurodegenerative disorder after AD, Parkinson’s disease (PD) shares protein misfolding abnormalities including tau and Aβ proteinopathies [6, 7]. The deposition of NFTs and senile plaques have been found in the brains of donors with PD, in comparable quantities and regions to those observed in AD [6]. Autosomal dominant mutations of the SNCA gene in PD are known to trigger disease-causing missense mutations of the α-synuclein protein, normally responsible for pre-synaptic signaling and membrane trafficking [8]. Subsequent molecular changes in α-synuclein cause the protein to misfold and deposit as Lewy bodies in neuronal somata and Lewy neurites in neuronal cell processes. Consequently, the concomitant loss of dopaminergic neurons in the substantia nigra pars compacta in the midbrain directly follows PD pathogenesis, as characterized by Braak staging of Lewy pathology [6, 7, 9]. Neuropathological forms of α-synuclein and tau are rarely found in isolation and their co-deposition with other amyloidogenic proteins including Aβ, have been associated with AD-like co-morbidities in vivo [10]. Those pathological conformations adopted, promote the cross-seeding of α-synuclein and tau. Further templating and protein aggregation may then occur through a prion-like transmission mechanism, promoting disease spread between adjacent neurons. Taken collectively, such has highlighted a synergistic role for tauopathies in accelerating aberrant α-synuclein inclusions and vice versa in PD brain tissues [6, 7, 10].
Aluminum is the third most abundant element and the most abundant metal in the Earth’s crust. Despite its ubiquity, aluminum is non-essential to life, participates in biochemical reactions, and accumulates over time in the central nervous system (CNS) [11, 12]. Aluminum is known to accumulate in human brain tissue of donors diagnosed with both neurodegenerative and neurodevelopmental disorders including AD [13, 14], PD [15, 16], and epilepsy [17]. Investigations into the distribution of aluminum in human brain tissue of donors diagnosed with fAD have revealed co-deposition of the metal ion in senile plaques [13, 14]. In a Colombian cohort of fAD donors presenting with a PSEN1-E280A mutation, aluminum was also identified in CAA-laden blood vessels, in which its co-localization with fibrillar Aβ was observed [14]. Donors with this mutation exhibit increased levels of cortical Aβ and early-onset and aggressive AD etiology [18]. Owing to the unique association of aluminum with Aβ and the high levels of aluminum found within these brain tissues relative to controls [19], such implicated a role for the metal in the neuropathology of fAD [14]. Elevated levels of aluminum have been reported in neuromelanin-containing neurons and in Lewy bodies of the substantia nigra region of PD donors [15, 16]. In addition, densely packed and phosphorylated neurofilaments of alpha-synuclein in Lewy bodies and neurites would be expected to bind aluminum with high affinity, thereby promoting its intracellular accumulation [7, 10, 15]. Furthermore, in the presence of aluminum, the rate of α-synuclein fibrillation has been shown to increase in vitro, inducing conformational changes of the protein to an aggregated insoluble form [20].
While the co-deposition of aluminum with Aβ has been suggested in fAD, such an association has yet to be confirmed with intraneuronal NFTs of hyperphosphorylated tau protein [13, 14]. In renal dialysis patients, elevated aluminum concentrations and its subsequent accumulation in brain tissue was associated with increased insoluble hyperphosphorylated tau and depleted normal tau protein in the cerebral cortex [21]. Owing to the high affinity of aluminum for phosphate groups, both adenosine triphosphate (ATP) and DNA are known to act as ligands for chelation of aluminum in intraneuronal pools [22, 23]. Therefore, the high number of inorganic phosphate ligands on intraneuronal tau protein in NFTs would be predicted in the cerebral cortex of both fAD and PD patients [10].
To assess such an association of aluminum with NFTs in vivo, we have made use of aluminum-specific fluorescence microscopy, utilizing the fluorophore lumogallion (4-chloro-3-(2,4-dihydroxyphenylazo)-2-hydroxybenzene-1-sulphonic acid). As a selective fluorescent molecular probe for the detection of intracellular aluminum in vivo, we have optimized its use for the detection of potential intraneuronal aluminum in Colombian PSEN1-E280A fAD, PD, and epilepsy donors [13, 14]. Herein, we have made use of thioflavin S as a fluorophore for the detection of NFTs of hyperphosphorylated tau protein. ThS is frequently used for the identification of NFTs in human brain tissue [7, 14, 17, 24, 25]. When stained with ThS, NFTs most notably produce characteristic flame-like morphologies in intraneuronal occlusions, distinctive from larger extracellular senile plaques that typically span tens of microns in diameter [26].
Previously, we have demonstrated the intracellular accumulation of aluminum in glial cells and neuronal debris in a case of epilepsy, brought on by aluminum poisoning in the individual’s potable water supply [17]. While extensive NFT deposition was noted in the frontal, parietal, occipital, and temporal lobes, no direct association with aluminum was identified [17]. A recent study of temporal lobe epilepsy (TLE) patients who underwent temporal lobe resection, demonstrated striking similarities with post-mortem temporal lobe specimens from AD patients [27]. Therein, increased phosphorylation of pathological tau was noted in NFTs in both TLE and AD tissues [27]. Therefore, we have additionally investigated potential similarities in the intraneuronal accumulation of aluminum and NFTs in a case study of an individual exposed to high levels of aluminum, that later died of asphyxiation through an epileptic fit [17]. Finally, we show preliminary data for aluminum and NFT-like deposition in PD, allowing for comparisons of their distribution in human brain tissue to be drawn across complex neurological disease states.
METHODS
Human brain tissue
Formalin-fixed paraffin-embedded (FFPE) brain tissue blocks tissue from Colombian fAD donors carrying a PSEN1-E280A mutation were obtained from the Universidad de Antioquia, Medellin, Colombia brain tissue bank, following ethical approval by Keele University, UK (ERP 2391) [14]. FFPE brain tissue blocks from a 60-year-old male donor who died as a consequence of asphyxiation following an epileptic fit were provided by University Hospitals Plymouth, NHS Trust, UK and sent to Keele University upon request of the coroner to investigate the content and distribution of aluminum. The deceased as described by the coroner, was a victim of the Lowermoor Treatment Works, Camelford, who in 1988 was exposed to high levels of aluminum in his potable water supply. Full details of the pathology of this case are described elsewhere [17]. PD brain tissue from an 87-year-old male donor was received as 5μm adjacent serial sections on electrostatically-charged glass slides from Parkinson’s UK Brain Bank at Imperial College London, funded by Parkinson’s UK (NREC approval no. 18/WA/0238).
Microtomy
All chemicals were from Sigma Aldrich, UK unless otherwise stated. Brain tissue received as FFPE tissue blocks were cooled on wet ice for 10 min and adjacent serial sections prepared at 5μm using a rotary RM2025 microtome, equipped with Surgipath DB80 LX low-profile stainless-steel microtome blades (both from Leica Microsystems, UK). Sections were floated onto ultrapure water (conductivity <0.067μS/cm) at 40°C and transferred onto SuperFrost® Plus adhesion slides (Thermo Scientific, UK). Excess wax was removed from dried sections by heating at 62°C for 20 min, before dewaxing and rehydration procedures.
Dewaxing and rehydration of tissue sections
All brain tissue sections were dewaxed with Histo-Clear (National Diagnostics, US) for 3 min, fresh Histo-Clear for 1 min and transferred into 100% v/v ethanol (HPLC grade used throughout) for 2 min to remove the clearing agent. Sections were subsequently rehydrated using an ethanol gradient from 95, 70, 50, and 30% v/v for 1 min in each solvent, before rehydration in ultrapure water for 35 s.
Lumogallion staining
All staining procedures were performed at ambient temperature, away from light. Rehydrated fAD tissue sections were fully immersed in Coplin jars containing 1 mM lumogallion (4-chloro-3-(2,4-dihydroxyphenylazo)-2-hydroxybenzene-1-sulphonic acid, TCI Europe N. V., Belgium) in 50mM PIPES pH 7.4 for 6 h. Autofluorescence controls were prepared by incubating sections in the buffer only. Epilepsy brain tissue sections were stained for 24 h in Coplin jars and PD sections for 45 min in humidity chambers, in the presence of the fluorophore or the buffer only for autofluorescence controls. Following staining, all sections were rinsed in the same PIPES buffer and washed for 30 s in ultrapure water, before mounting with Fluoromount™ under glass coverslips.
Thioflavin s staining
Following analysis of lumogallion stained sections via fluorescence microscopy, mounted sections on glass slides were placed in ultrapure water with gentle agitation provided by a stirrer bar, overnight. Once coverslips had lifted, lumogallion stained sections were outlined with a hydrophobic PAP pen, allowing for re-staining with thioflavin S (ThS) in humidity chambers. Sections were re-stained with ca 0.075% w/v ThS in 50% v/v ethanol for 8 min. Following ThS staining, slides were twice rinsed for 10 s in fresh 80% v/v ethanol and washed for 30 s in ultrapure water. Sections were subsequently mounted with Fluromount™ using new glass coverslips.
Microscopy
Sections were analyzed by use of an Olympus BX50 fluorescence microscope (mercury source) equipped with a BX-FLA reflected light attachment and vertical illuminator. Lumogallion fluorescence was acquired using a U-MNIB3 filter cube (bandpass λex: 470–495 nm, dichromatic mirror: 505 nm, longpass λem: 510 nm) and ThS fluorescence by use of a U-MWBV2 filter cube (bandpass λex: 400–440 nm, dichromatic mirror: 455 nm, longpass λem: 475 nm, both from Olympus, UK). Images were captured using a ColorView III CCD camera using the CellD software suite (Olympus, SiS Imaging Solutions, GmbH). Light transmission values and exposure settings were fixed across respective treatments. Merging of fluorescence channels was performed using Photoshop (Adobe Systems Inc., US).
RESULTS
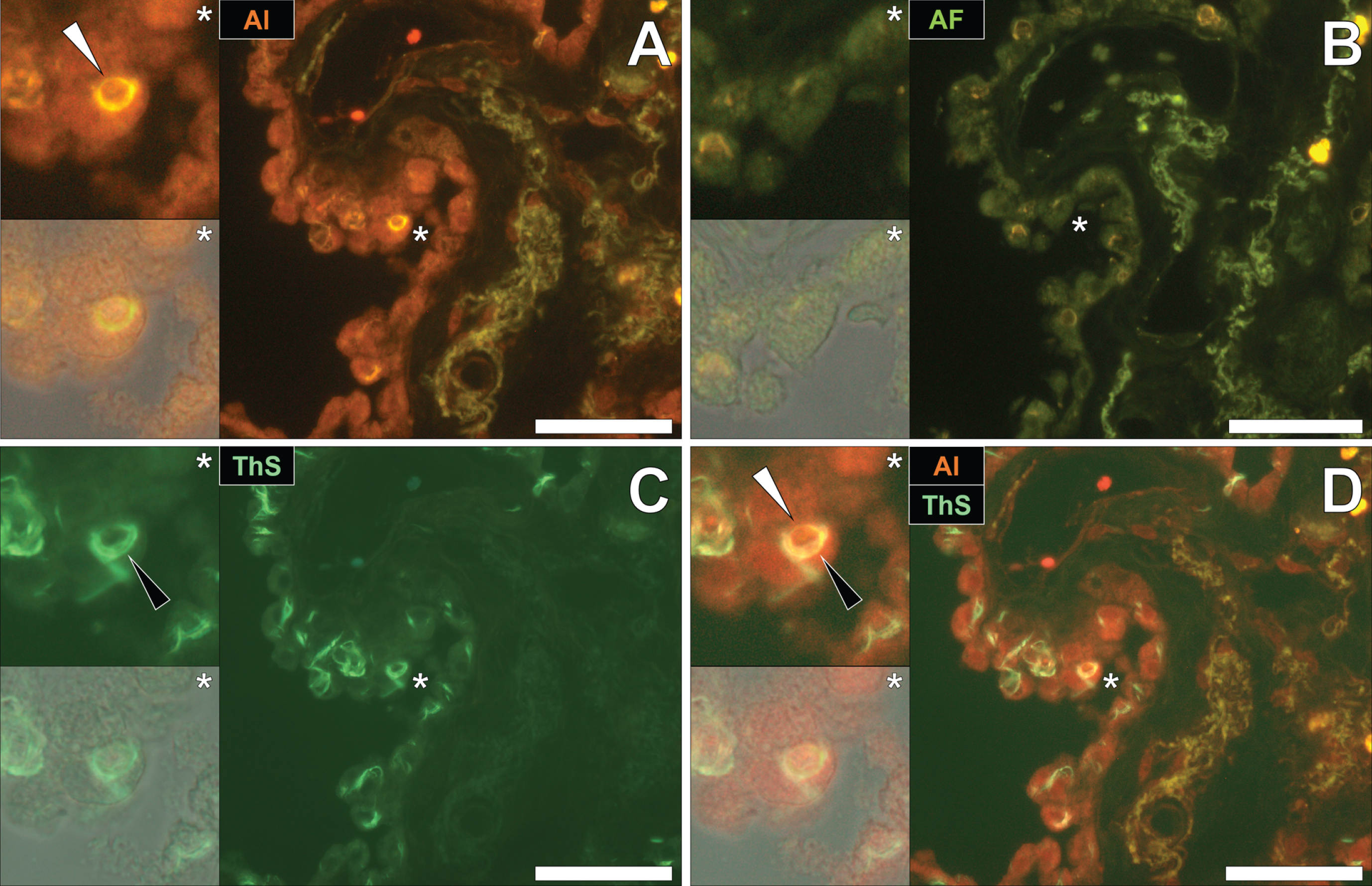
Aluminum and neurofibrillary tangle deposition in familial Alzheimer’s disease
To identify the potential deposition of aluminum and NFTs in fAD, sections were first stained with lumogallion and deposits of aluminum identified. Aluminum-specific fluorescence microscopy identified extracellular aluminum deposition in the temporal cortex of a 45-year-old female Colombian fAD donor (Fig. 1A). Higher magnifications revealed the presence of nearby neuronal cells loaded with punctate cytosolic deposits of the metal ion, via an intense orange fluorescence emission.
Numerous and frequently intraneuronal lipofuscin deposits were readily differentiated from lumogallion-reactive aluminum, by a weaker green/yellow fluorescence emission (see Supplementary Fig. 1). Upon re-staining of the section with ThS, the identical lumogallion-reactive neuron stained positively for intraneuronal NFTs at its periphery, via a green fluorescence emission (Fig. 1B). Merging of the fluorescence channels revealed that aluminum and NFTs were located in the same cell (Fig. 1C), with the brightfield overlay confirming their intracellular deposition (Fig. 1D).
Fig. 1
Intracellular aluminum co-located with ThS-reactive NFTs in the temporal cortex of a Colombian donor (Case 90: Female, aged 45) with fAD (PSEN1-E280A mutation). A) Punctate intracellular aluminum (orange, white arrows) in neuronal cells exhibiting positive (green) fluorescence for (B) intraneuronal NFTs (black arrows) with (C) merging of fluorescence channels and the brightfield overlay (D) depicting their co-deposition. Magnified inserts are denoted by asterisks in the respective fluorescence micrographs. Al, aluminum; ThS, thioflavin S. Magnification: X 400, scale bars: 50μm.
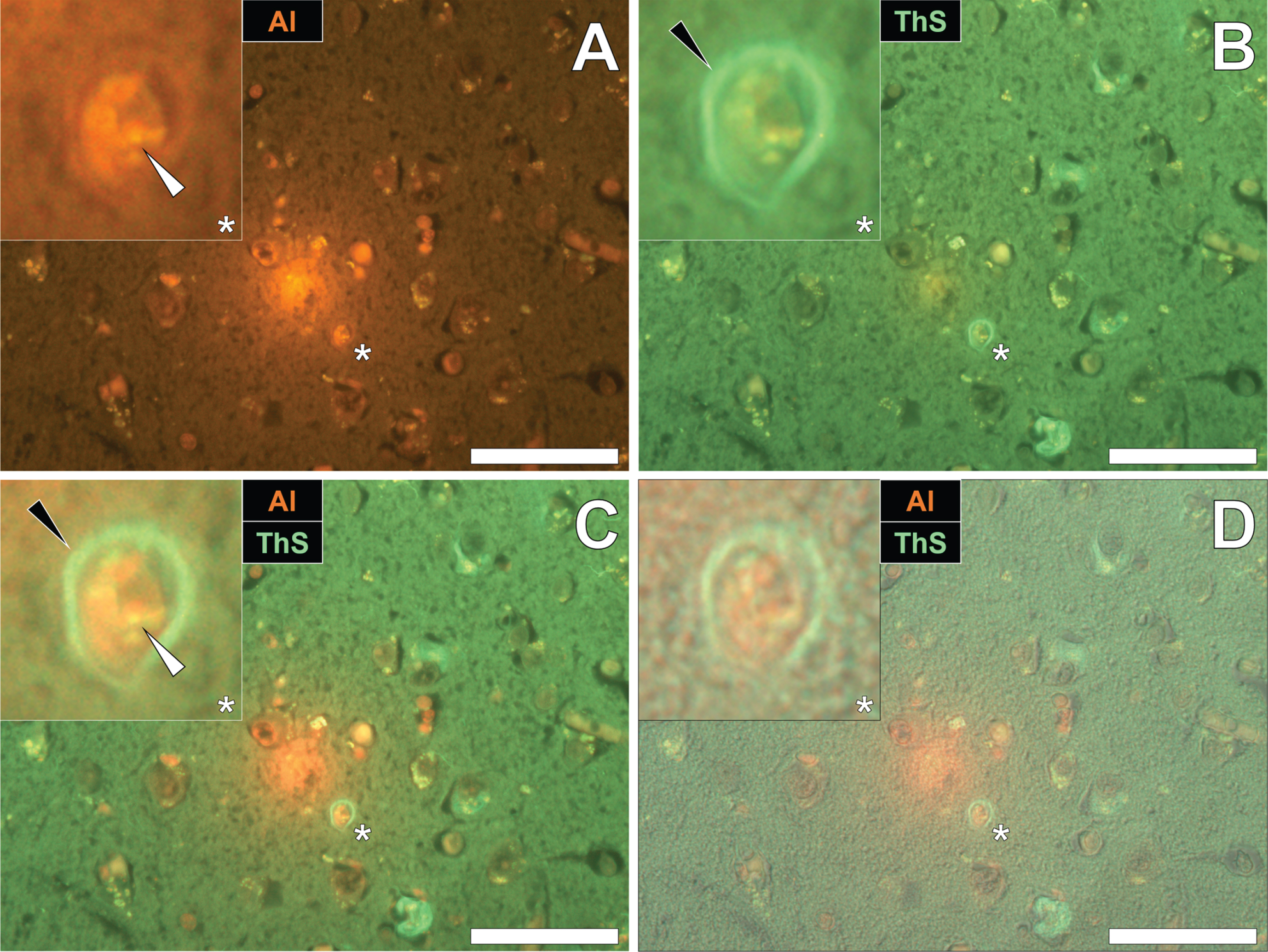
Similarly, intraneuronal aluminum appearing as punctate orange deposits were found in the parietal cortex of a 60-year-old male Colombian fAD donor (Fig. 2A). Such was differentiated from a green autofluorescence emission of the non-stained serial section (see Supplementary Fig. 2). The identical neuron revealed intracellular ThS-reactive NFTs, as highlighted by a green fluorescence emission (Fig. 2B). Interestingly, merging of the fluorescence channels identified the co-localization of lumogallion-reactive aluminum and ThS-reactive NFTs at the periphery of the cell (Fig. 2C). Overlaying of the brightfield channel revealed both deposits to be enclosed by a clear cell membrane, confirming their intracellular co-deposition (Fig. 2D).
Fig. 2
Intracellular aluminum co-located with ThS-reactive NFTs in the parietal cortex of a Colombian donor (Case 218: Male, aged 60) with fAD (PSEN1-E280A mutation). A) Punctate intracellular aluminum (orange, white arrows) in neuronal cells exhibiting positive (green) fluorescence for (B) intraneuronal NFTs (black arrows) with (C) merging of fluorescence channels and the brightfield overlay (D) depicting their co-localization. Magnified inserts are denoted by asterisks in the respective fluorescence micrographs. Al, aluminum; ThS, thioflavin S. Magnification: X 400, scale bars: 50μm.
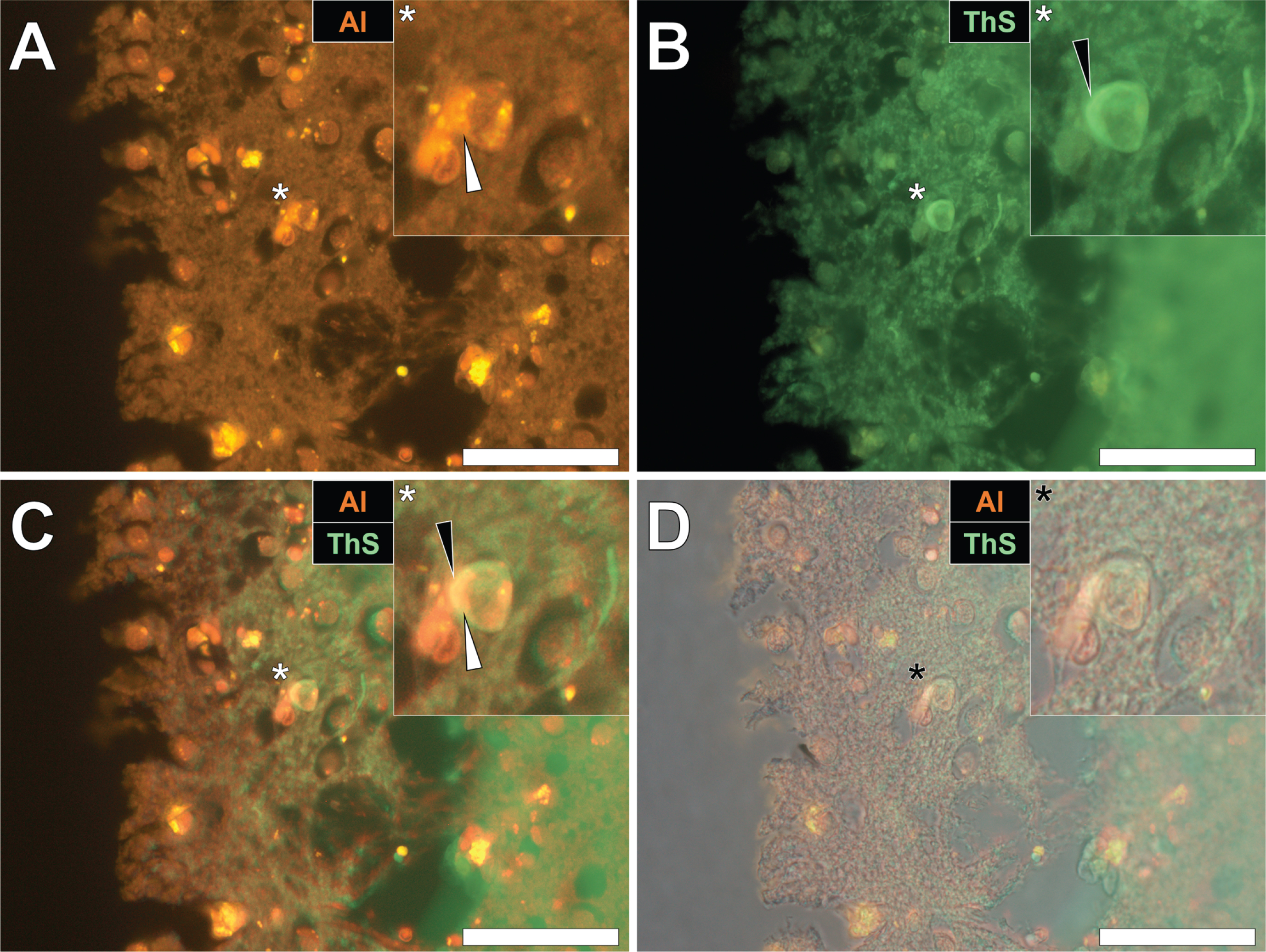
Herein, prolonged staining with lumogallion identified the presence of intracellular aluminum in the parietal cortex of a 57-year-old female Colombian fAD donor (Fig. 3A), versus a weak green autofluorescence emission of the non-stained serial section (see Supplementary Fig. 3). Microglial cells and neurons near cellular debris stained positively for aluminum (Fig. 3A) and ThS-reactive senile plaques (Fig. 3B) were observed. While aluminum appeared to be distributed in cell soma, ThS-reactive neuropil-like threads were highlighted via a green fluorescence emission in dendritic/axonal-like cell projections, upon merging of fluorescence (Fig. 3C) and brightfield (Fig. 3D) channels.
Fig. 3
Intracellular aluminum in glia and neurons co-located with ThS-reactive NFTs and amyloid-β in the parietal cortex of a Colombian donor (Case 260: Female, aged 57) with fAD (PSEN1-E280A mutation). A) Punctate intracellular aluminum (orange) in a microglial cell (white arrow). B) Intraneuronal NFTs (green, black arrows) with (C) merging of fluorescence channels depicting their co-deposition. D) The brightfield overlay depicts cell membranes. Magnified inserts are denoted by asterisks in the respective fluorescence micrographs. Al, aluminum; ThS, thioflavin S. Magnification: X 400, scale bars: 50μm.
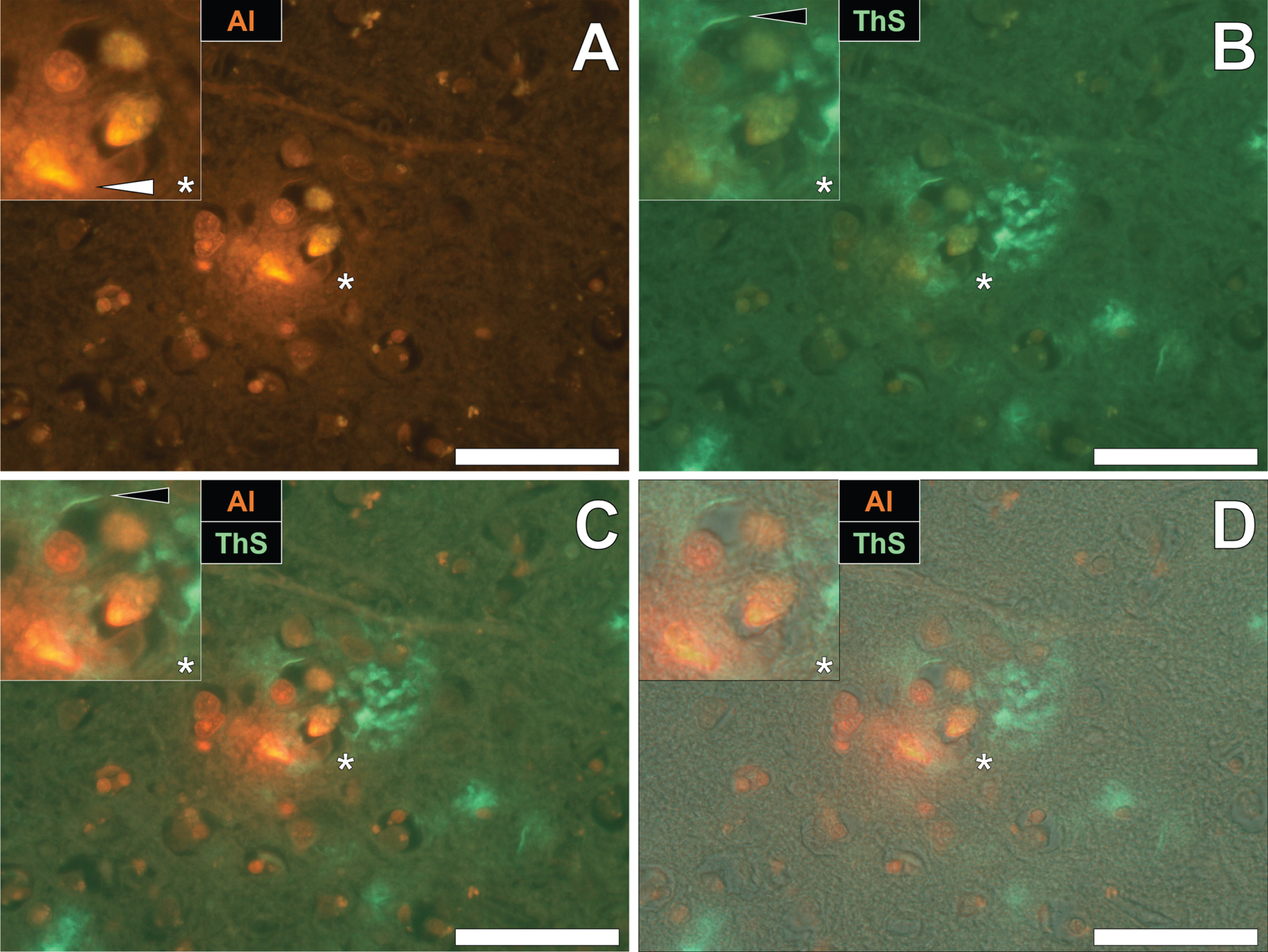
Aluminum and neurofibrillary tangle deposition in epilepsy
To draw comparisons between aluminum and NFT distribution in fAD and epilepsy, brain tissue from a 60-year old male donor who died as a consequence of an epileptic fit was sequentially stained with lumogallion and ThS. Prolonged staining with lumogallion revealed the presence of intracellular aluminum in neuronal cells in the temporal cortex, via an orange fluorescence emission (Fig. 4A). Analysis of the non-stained serial section revealed a green autofluorescence emission of brain parenchyma with punctate yellow intraneuronal deposits being confirmed in the same cells (Fig. 4B). ThS counter-staining of the identical lumogallion-stained section demonstrated the presence of NFT-like deposits in axons and dendrites of the same neuron, via a green fluorescence emission (Fig. 4C). Merging of fluorescence and brightfield channels confirmed the intraneuronal distribution of aluminum and axonal-like deposition of NFTs, in the same cell (Fig. 4D).
Fig. 4
Intracellular aluminum co-located with ThS-reactive NFTs in neurons in the temporal cortex of a 60-year-old male donor with epilepsy. A) Intranuclear aluminum (orange, white arrows) and (B) autofluorescence of the non-stained section. C) The identical neuronal cell exhibiting positive (green) fluorescence for NFTs (black arrows) with (D) merging of fluorescence channels depicting their co-localization. Magnified inserts are denoted by asterisks in the respective fluorescence micrographs of which merging of the brightfield overlay is depicted in the lower panels. Al, aluminum; ThS, thioflavin S. Magnification: X 400, scale bars: 50μm.
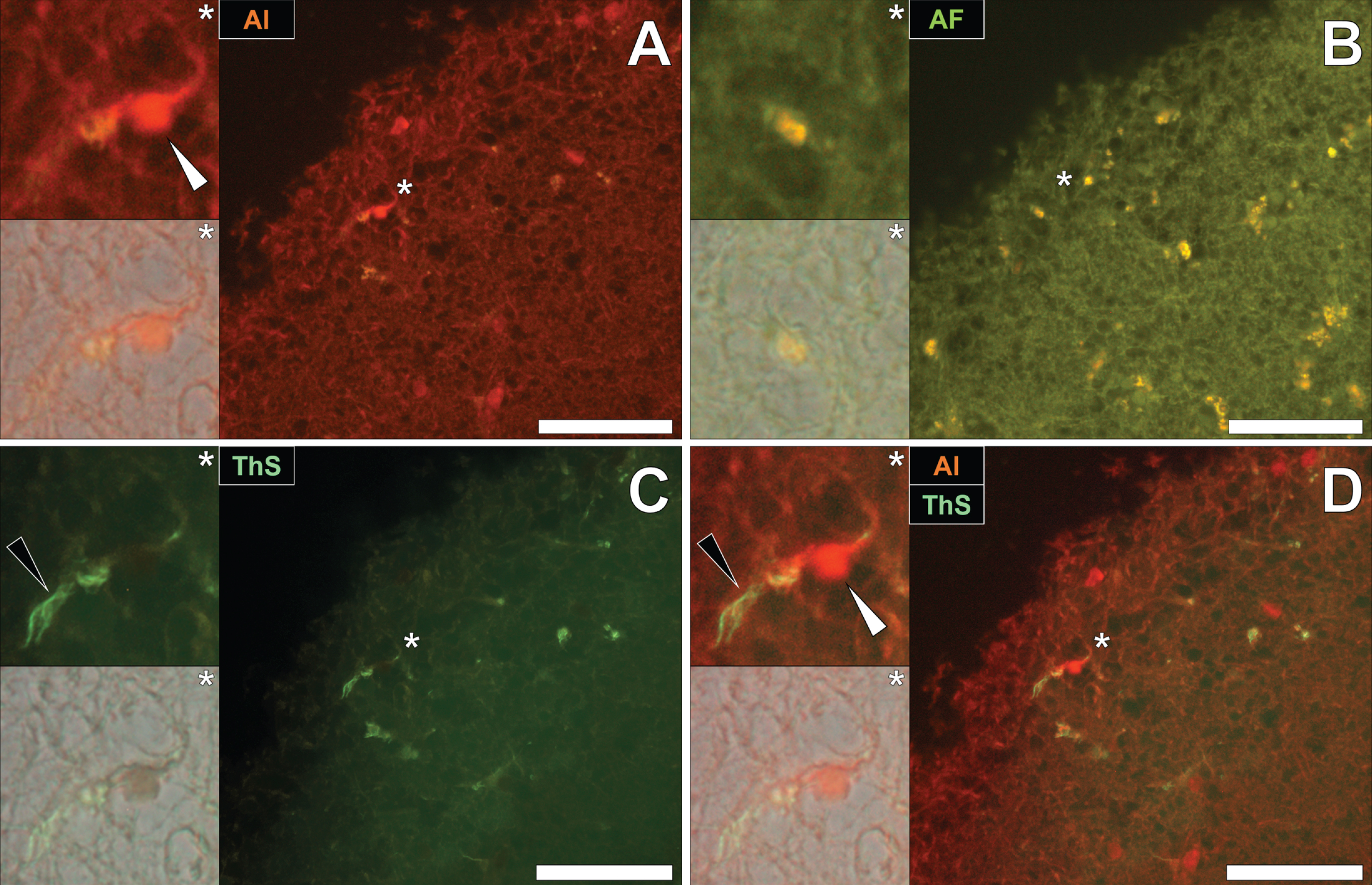
Aluminum and neurofibrillary tangle deposition in Parkinson’s disease
To draw comparisons to NFT and aluminum deposition in fAD and epilepsy, conventional lumogallion and ThS counter-staining were performed on donor tissues obtained from an 87-year-old male with PD [13, 17, 28]. Lumogallion staining revealed positive orange fluorescence of aluminum in tangle-like formations, in epithelial cells lining the choroid plexus of the hippocampus (Fig. 5A). Green autofluorescence and occasional lipofuscin deposition were noted in the same cells, in the non-stained adjacent serial section (Fig. 5B). ThS counter-staining identified Biondi ring-like tangles via an intensive green fluorescence emission, reminiscent of NFTs in the identical epithelial cells (Fig. 5C). Merging of the fluorescence and brightfield channels identified prominent aluminum deposition, co-located with Biondi ring-like tangles in the same epithelial cell (Fig. 5D).
Fig. 5
Intracellular aluminum co-located with ThS-reactive Biondi ring-like tangles in the choroid plexus (hippocampus) of an 87-year-old male donor with Parkinson’s disease. A) Intracellular aluminum (orange, white arrows) in epithelial cells lining the choroid plexus and (B) autofluorescence of the non-stained section. C) The identical epithelial cell exhibiting positive (green) fluorescence for Biondi ring tangles (black arrows) with (D) merging of fluorescence channels depicting their co-localization. Magnified inserts are denoted by asterisks in the respective fluorescence micrographs of which merging of the brightfield overlay is depicted in the lower panels. Al, aluminum; ThS, thioflavin S. Magnification: X 400, scale bars: 50μm.
DISCUSSION
We have demonstrated the presence of intracellular aluminum and NFTs in neurons in the cerebral cortex of both fAD and epilepsy donors and co-located with Biondi ring-like tangles in epithelial cells lining the choroid plexus in PD. In fAD, intracellular punctate deposits of aluminum were observed in neurons in the parietal and temporal cortex of three individual fAD donors, all carrying the PSEN1-E280A mutation. ThS-reactive NFTs were found in the identical neurons, upon counter-staining. Interestingly, the pattern of aluminum and NFT co-deposition was seen to vary in cortical neurons, with each depositing in different cellular locations. Typically, flame-like NFTs were observed at the periphery of neurons, with aluminum appearing to be deposited in cell nuclei. In fAD, only a single incidence of diffuse aluminum staining and potentially co-located intraneuronal NFTs were observed in the parietal cortex. Intracellular aluminum was also observed in microglial-like cells near ThS-reactive senile plaques, in the same donor.
Senile plaque morphologies and neuritic dystrophy have been shown to revert upon the activation of microglial cells in the 5xFAD murine model of AD [29]. Microglia are known to play a pivotal role in their ability to block aberrant tangle formation, though their high loading with aluminum observed in our study may prevent their ability to do so in vivo [14, 30, 31]. It is important to stress that while aluminum and NFTs were observed in the same cortical neurons, aluminum was predominantly observed to be co-located with ThS-reactive senile plaques, as has been previously reported in the same Colombian donor cohort [14]. Furthermore, several NFTs stained positively with ThS without producing any signal for aluminum upon prolonged lumogallion staining.
In the brain tissue of a donor with late-onset adult epilepsy, aluminum and NFTs were both found deposited in cortical neurons of the temporal lobe. We observed that while aluminum was generally found deposited in the nucleus, ThS-reactive NFTs were observed in axonal-like projections in the same neuron. Our previous report of aluminum distribution in the brain tissue of this donor, only identified aluminum in glial cell populations, thereby depositing at sites away from intraneuronal NFTs of hyperphosphorylated tau protein [17].
In a donor with PD, Biondi ring-like tangles were found in epithelial cells, lining the choroid plexus of the hippocampus. Those tangles identified were both lumogallion and ThS-reactive and thereby demonstrated the presence of aluminum within these fibrillar inclusions. We have previously made the observations of aluminum in epithelial cells of the choroid plexus in a donor with CAA and ThS-reactive Biondi ring-like tangles in the same donor with epilepsy; revisited in this study [17, 32]. Interestingly, both were victims of the now infamous Camelford aluminum poisoning incident and herein we report the first co-localization of these neuropathological hallmarks, within the same epithelial cells in PD.
Biondi ring tangles were originally thought to be artefacts described as “off-target” binding of the flortaucipir-based PET radioligand, [F-18]AV-1451 [33]. However, in a follow-up study by Ikonomovic and colleagues, immunolabelling of the choroid plexus of aged AD brains revealed the presence of phosphorylated tau with minimal immunoreactivity for Aβ [34]. Those tangles identified, were described as Biondi ring tangles that have been previously reported in aged healthy and AD brain tissues [34–36]. A continuing research effort is currently underway to better characterize PET tracers for tau imaging and the reasons underlying off-target labelling in living patients. However, these studies have continued to report the presence of tau in Biondi ring tangles, supporting our preliminary observations of these neuropathological hallmarks in a donor with PD [37, 38].
The blood-cerebrospinal fluid barrier has been suggested to act as a potential entry route of α-synuclein into the brain through its passage across choroid plexus epithelia via energy-dependent active transport [39]. Therefore, the presence of aluminum in these cells may exacerbate the accumulation and misfolding of amyloidogenic proteins including hyperphosphorylated tau and α-synuclein. Indeed, tau and α-synuclein can interact in cells and their aberrant cross-seeding has been suggested to synergistically enhance protein misfolding and fibrillar inclusions in vivo [7, 10]. In addition, PD-specific mutations including those in leucine-rich repeat kinase 2 (LRRK2), has been implicated in the hyperphosphorylation of tau and the subsequent deposition of NFTs [6, 40].
The intranuclear deposition of aluminum has been highlighted in the past, likely owing to the high affinity of aluminum binding to the phosphate backbone of DNA [41, 42]. We have previously reported the unequivocal presence of intranuclear aluminum in vitro in human spermatozoa and frequently in vivo, in the brain of donors with autism spectrum disorder, multiple sclerosis, epilepsy, and fAD [14, 17, 28, 43, 44]. While previous studies of fAD brain tissues revealed only occasional aluminum deposits in cortical neurons, pro-longed staining with the lumogallion fluorophore, herein, demonstrated an intense positive signal for the metal ion above background fluorescence. As aluminum readily binds to negatively charged phosphate moieties and also forms strong 1:1 complexes with lumogallion, such competitive binding equilibria may have shifted in favor of forming a fluorescent complex [23]. In this manner, Al3 + (aq) ions removed from nuclear DNA, may have allowed sufficient complexation with lumogallion to produce a positive intranuclear metal signal [45–47].
Aluminum is known to bind to the microtubule-associated protein tau and especially upon its hyperphosphorylation forming aberrant insoluble NFTs [48]. Intraneuronal NFTs are frequently observed in the cerebral cortex of fAD, PD, and epilepsy patients, collectively prompting our study to probe their intracellular presence [7, 14, 17, 27]. Epilepsy occurrence is more frequent in AD patients [49]. Concomitant with increased tau phosphorylation, increased mossy fiber sprouting has also been demonstrated in pentylenetetrazole-kindled rat models of epilepsy [50]. Furthermore, detailed histological analyses of human brain tissue excised from drug-resistant temporal lobe epilepsy (TLE) donors, have revealed intraneuronal tau phosphorylation, bearing striking similarities to those found in AD temporal lobe tissues [27]. Increased brain Aβ deposition has been suggested to be enhanced in the presence of aluminum, through its acceleration of proteolytic processing of AβPP via the amyloidogenic pathway [51]. Subsequently, the formation of Aβ fibrils is known to induce phosphorylation of tau in both in vivo and in vitro models of AD [52, 53].
While further research is needed to establish a role for aluminum in the catalysis of Aβ and subsequent NFT deposition in fAD, PD and epilepsy, our results now demonstrate the co-existence of aluminum in these neuropathological hallmarks [14]. We have used ThS for the detection of NFTs in fAD and epilepsy brain tissues and similarly unveiled the presence of Biondi ring tangles in PD. A limitation of our study is the sole use of ThS for this purpose, which is also known to bind to and visualize senile plaques of Aβ [54]. Kinetic-based studies monitoring fibrillation of the α-synuclein protein, have also shown reactivity to benzothiazole-based dyes, upon the formation of β-pleated sheet structures [7, 20]. Owing to the large size of extracellularly deposited senile plaques, these could be differentiated from smaller intracellular fibrillar morphologies and flame-like NFTs, herein observed in fAD and epilepsy brain tissues. Likewise, we could also identify characteristic Biondi ring tangles in the choroid plexus of a donor with PD that have frequently produced positive immunoreactivity against phosphorylated tau in previous studies [33–38]. Future research identifying specific phosphorylated tau residues and immunoreactivity against Aβ and α-synuclein is a logical next step to delineate aluminum accumulation in specific fibrillar assemblies. We now aim to perform immunolabelling against these specific neuropathological targets to shed light upon the role of aluminum in their mechanistic processes of assembly, in vivo.
Donors from the Colombian PSEN1-E280A fAD cohort are known to develop tauopathies later in life, as has been demonstrated by positron emission tomography (PET), in living patients [55]. Therein and similarly in PD, hyperphosphorylation of tau depositing as NFTs follows senile plaque deposition, before symptom onset and concurrent cognitive decline [7, 10, 56]. Future investigations of aluminum, Aβ, and NFT deposition in neurodegenerative and neurodevelopmental disorders would help to shed light upon the potentially shared pathological mechanisms underlying these complex disease states.
ACKNOWLEDGMENTS
MM is a Children’s Medical Safety Research Institute (CMSRI: a charity based in Washington DC, USA) Research Fellow. We are thankful to the families of all donors who donated tissues to the brain bank of the Universidad de Antioquia, Medellin, Colombia. Dr. Johana Gómez-Ramírez and Dr. Andrés Villegas-Lanau are thanked for tissue acquisition and processing for the delivery of FFPE tissue blocks to Keele University. Philp Edwards, University Hospitals Plymouth NHS Trust, is thanked for initial preparation of brain tissues of the epilepsy donor and we are thankful to the next of kin for their support and to the Taunton Coroner, Michael Rose, for his help in bringing about this research. Parkinson’s disease brain tissue samples (NREC no. 18/WA/0238) and associated clinical and neuropathological data were supplied by Parkinson’s UK Brain Bank at Imperial, funded by Parkinson’s UK, a charity registered in England and Wales (258197) and in Scotland (SC037554).
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/20-0838r1).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-200838.
REFERENCES
[1] | Zhu XC , Tan L , Wang HF , Jiang T , Cao L , Wang C , Wang J , Tan CC , Meng XF , Yu JT ((2015) ) Rate of early onset Alzheimer’s disease: A systematic review and meta-analysis. Ann Transl Med 3: , 38. |
[2] | Goate A , Chartierharlin MC , Mullan M , Brown J , Crawford F ((1991) ) Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s-disease. Nature 349: , 704–706. |
[3] | Sherrington R , Rogaev EI , Liang Y , Rogaeva EA , Levesque G ((1995) ) Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375: , 754–760. |
[4] | LaFerla FM , Green KN , Oddo S ((2007) ) Intracellular amyloid-β in Alzheimer’s disease. Nat Rev Neurosci 8: , 499–509. |
[5] | DeTure MA , Dickson DW ((2019) ) The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener 14: , 32. |
[6] | Kalia LV , Lang AE ((2015) ) Parkinson’s disease. Lancet 386: , 896–912. |
[7] | Irwin DJ , Lee VMY , Trojanowski JQ ((2013) ) Parkinson’s disease dementia: Convergence of α-synuclein, tau and amyloid-β pathologies. Nat Rev Neurosci 14: , 626–636. |
[8] | Stefanis L ((2012) ) α-synuclein in Parkinson’s disease. Cold Spring Harb Perspect Med 4: , a009399. |
[9] | Braak H , Tredici KD , Rüb U , de Vos RAI , Steur ENHJ , Braak E ((2003) ) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24: , 197–211. |
[10] | Yan X , Uronen RL , Huttunen HJ ((2020) ) The interaction of α-synuclein and tau: A molecular conspiracy in neurodegeneration? Semin Cell Dev Biol 99: , 55–64. |
[11] | Exley C , Mold M ((2019) ) Aluminium in human brain tissue: How much is too much? J Biol Inorg Chem 24: , 1279–1282. |
[12] | Exley C , Mold M ((2020) ) Imaging of aluminium and amyloid β in neurodegenerative disease. Heliyon 6: , e03839. |
[13] | Mirza A , King A , Troakes C , Exley C ((2017) ) Aluminium in brain tissue in familial Alzheimer’s disease. J Trace Elem Med Biol 40: , 30–36. |
[14] | Mold M , Linhart C , Gómez-Ramírez J , Villegas-Lanau A , Exley C ((2020) ) Aluminum and amyloid-β in familial Alzheimer’s disease. J Alzheimers Dis 73: , 1627–1635. |
[15] | Hirsch EC , Brandel JP , Galle P , Javoy-Agid F , Agid Y ((1991) ) Iron and aluminum increase in the substantia nigra of patients with Parkinson’s disease: An X-ray microanalysis. J Neurochem 56: , 446–451. |
[16] | Good PF , Olanow CW , Perl DP ((1992) ) Neuromelanin-containing neurons of the substantia nigra accumulate iron and aluminum in Parkinson’s disease: A LAMMA study. Brain Res 593: , 343–346. |
[17] | Mold M , Cottle J , Exley C ((2019) ) Aluminium in brain tissue in epilepsy: A case report from Camelford. Int J Environ Res Public Health 16: , 2129. |
[18] | Lopera F , Ardilla A , Martínez A , Madrigal L , Arango-Viana JC , Lemere CA , Arango-Lasprilla JC , Hincapíe L , Arcos-Burgos M , Ossa JE , Behrens IM , Norton J , Lendon C , Goate AM , Ruiz-Linares A , Rosselli M , Kosik KS ((1997) ) Clinical features of early-onset Alzheimer disease in a large kindred with an E280A presenilin-1 mutation. J Amer Med Assoc 277: , 793–799. |
[19] | Exley C , Clarkson E ((2020) ) Aluminium in human brain tissue from donors without neurodegenerative disease: A comparison with Alzheimer’s disease, multiple sclerosis and autism. Sci Rep 10: , 7770. |
[20] | Uversky VN , Li J , Fink AL ((2001) ) Metal-triggered structural transformations, aggregation and fibrillation of human α-synuclein. A possible molecular link between Parkinson’s disease and heavy metal exposure. J Biol Chem 276: , 44284–44296. |
[21] | Harrington CR , Wischik CM , McArthur FK , Taylor GA , Edwardson JA , Candy JM ((1994) ) Alzheimer’s-disease-like changes in tau protein processing: Association with aluminium accumulation in brains of renal dialysis patients. Lancet 343: , 993–997. |
[22] | Exley C , Birchall JD ((1996) ) Biological availability of aluminium in commercial ATP. J Inorg Biochem 63: , 241–252. |
[23] | Luque NB , Mujika JI , Rezabal E , Ugalde JM , Lopez X ((2014) ) Mapping the affinity of aluminum(III) for biophosphates: Interaction mode and binding affinity in 1:1 complexes. Phys Chem Chem Phys 16: , 20107–20119. |
[24] | Al-Shaikh FSH , Duara R , Crook JE , Lesser ER , Schaeverbeke J , Hinkle KM , Ross OA , Ertekin-Taner N , Pedraza O , Dickson DW , Graff-Radford NR , Murray ME ((2020) ) Selective vulnerability of the nucleus basalis of Meynert among neuropathologic subtypes of Alzheimer disease. JAMA Neurol 77: , 225–233. |
[25] | Sun A , Nguyen XV , Bing G ((2002) ) Comparative analysis of an improved thioflavin-S stain, Gallyas silver stain, and immunohistochemistry for neurofibrillary tangle demonstration on the same sections. J Histochem Cytochem 50: , 463–472. |
[26] | Cras P , van Harskamp F , Hendriks L , Ceuterick C , van Dujin CM , Stefanko SZ , Hofman A , Kros JM , Broeckhoven CV , Martin JJ , van Harskamp F ((1998) ) Presenile Alzheimer dementia characterized by amyloid angiopathy and large amyloid core type senile plaques in the APP 692Ala Gly mutation. Acta Neuropathol 96: , 253–260. |
[27] | Gourmaud S , Shou H , Irwin DJ , Sansalone K , Jacobs LM , Lucas TH , Marsh ED , Davis KA , Jensen FE , Talos DM ((2020) ) Alzheimer-like amyloid and tau alterations associated with cognitive deficit in temporal lobe epilepsy. Brain 143: , 191–209. |
[28] | Mold M , Umar D , King A , Exley C ((2018) ) Aluminium in brain tissue in autism. J Trace Elem Med Biol 46: , 76–82. |
[29] | Casali BT , MacPherson KP , Reed-Geaghan EG , Landreth GE ((2020) ) Microglia depletion rapidly and reversibly alters amyloid pathology by modification of plaque compaction and morphologies. Neurobiol Dis 142: , 104956. |
[30] | Condello C , Yuan P , Schain A , Grutzendler J ((2015) ) Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nat Commun 6: , 6176. |
[31] | Song WM , Colonna M ((2018) ) The identity and function of microglia in neurodegeneration. Nat Immunol 19: , 1048–1058. |
[32] | Mold M , Cottle J , King A , Exley C ((2019) ) Intracellular aluminium in inflammatory and glial cells in cerebral amyloid angiopathy: A case report. Int J Environ Res Public Health 16: , 1459. |
[33] | Johnson KA , Schultz A , Betensky RA , Becker JA , Sepulcre J , Rentz D , Mormino E , Chhatwal J , Amariglio R , Papp K , Marshall G , Albers M , Mauro S , Pepin L , Alverio J , Judge K , Philiossaint M , Shoup T , Yokell D , Dickerson B , Gomez-Isla T , Hyman B , Vasdev N , Sperling R ((2016) ) Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann Neurol 79: , 110–119. |
[34] | Ikonomovic MD , Abrahamson EE , Price JC , Mathis CA , Klunk WE ((2016) ) [F-18]AV-1451 Positron emission tomography retention in choroid plexus: More than “off-target” binding. Ann Neurol 80: , 307–308. |
[35] | Miklossy J , Kraftsik R , Pillevuit O , Lepori D , Genton C , Bosman FT ((1998) ) Curly fiber and tangle-like inclusions in the ependyma and choroid plexus –a pathogenetic relationship with cortical Alzheimer-type changes? J Neuropathol Exp Neurol 57: , 1202–1212. |
[36] | Wen GY , Wisniewski HM , Kascsak RJ ((1999) ) Biondi ring tangles in the choroid plexus of Alzheimer’s disease and normal aging brains: A quantitative study. Brain Res 832: , 40–46. |
[37] | Saint-Aubert L , Lemoine L , Chiotis K , Leuzy A , Rodriguez-Vieitez E , Nordberg A ((2017) ) Tau PET imaging: Present and future directions. Mol Neurodegener 12: , 19. |
[38] | Lemoine L , Leuzy A , Chiotis K , Rodriguez-Vieitez E , Nordberg A ((2018) ) Tau positron emission tomography imaging in tauopathies: The added hurdle of off-target binding. Alzheimers Dement 10: , 232–236. |
[39] | Bates CA , Zheng W ((2014) ) Brain disposition of α-Synuclein: Roles of brain barrier systems and implications for Parkinson’s disease. Fluids Barriers CNS 11: , 17. |
[40] | Shanley MR , Hawley D , Leung S , Zaidi NF , Dave R , Schlosser KA , Bandopadhyay R , Gerber SA , Liu M ((2015) ) LRRK2 facilitates tau phosphorylation through strong interaction with tau and cdk5. Biochemistry 54: , 5198–5208. |
[41] | Sheet SK , Sen B , Thounaojam R , Aguan K , Khatua S ((2017) ) Highly selective light-up Al3 + sensing by a coumarin based Schiff base probe: Subsequent phosphate sensing DNA binding and live cell imaging. J Photochem Photobiol A Chem 332: , 101–111. |
[42] | Exley C , House E ((2011) ) Aluminium in the human brain. Monatsh Chem 142: , 357–363. |
[43] | Klein JP , Mold M , Mery L , Cottier M , Exley C ((2014) ) Aluminum content of human semen: Implications for semen quality. Reprod Toxicol 50: , 43–48. |
[44] | Mold M , Chmielecka A , Rodriguez MRR , Thom F , Linhart C , King A , Exley C ((2018) ) Aluminium in brain tissue in multiple sclerosis. Int J Env Res Pub Health 15: , 1777. |
[45] | Wu J , Zhou CY , Chi H , Wong MK , Lee HK , Ong HY , Ong CN ((1995) ) Determination of serum aluminium using an ion-pair reversed phase high-performance liquid chromatographic-fluorimetric system with lumogallion. J Chromatogr B Biomed Appl 663: , 247–253. |
[46] | Hydes DJ , Liss PS ((1976) ) Fluorimetric method for the determination of low concentrations of dissolved aluminium in natural waters. Analyst 101: , 922–931. |
[47] | Ren JL , Zhang J , Luo JQ , Pei XK , Jiang ZX ((2001) ) Improved fluorimetric determination of dissolved aluminium by micelle-enhanced lumogallion complex in natural waters. Analyst 126: , 698–702. |
[48] | Mujika JI , Torre GD , Formoso E , Grande-Aztatzi R , Grabowski SJ , Exley C , Lopez X ((2018) ) Aluminum’s preferential binding site in proteins: Sidechain of amino acids versus backbone interactions. J Inorg Biochem 181: , 111–116. |
[49] | Giorgi FS , Saccaro LF , Busceti CL , Biagioni F , Fornai F ((2020) ) Epilepsy and Alzheimer’s disease: Potential mechanisms for an association. Brain Res Bull 160: , 107–120. |
[50] | Liu X , Chen L , Chen Y ((2017) ) N-methyl-D-aspartate receptors mediate epilepsy-induced axonal impairment and tau phosphorylation via activating glycogen synthase kinase-3β and cyclin-dependent kinase 5. Discov Med 23: , 221–234. |
[51] | Clauberg M , Joshi JG ((1993) ) Regulation of serine protease activity by aluminum: Implications for Alzheimer disease. Proc Natl Acad Sci U S A 90: , 1009–1012. |
[52] | Prema A , Thenmozhi AJ , Manivasagam T , Essa MM , Guillemin GJ ((2017) ) Fenugreek seed powder attenuated aluminum chloride-induced tau pathology, oxidative stress, and inflammation in a rat model of Alzheimer’s disease. J Alzheimers Dis 60: , S209–S220. |
[53] | Stoothoff WH , Johnson GV ((2005) ) Tau phosphorylation: Physiological and pathological consequences. Biochim Biophys Acta 1739: , 280–297. |
[54] | Guntern R , Bouras C , Hof PR , Vallet PG ((1992) ) An improved thioflavine S method for staining neurofibrillary tangles and senile plaques in Alzheimer’s disease. Experientia 48: , 8–10. |
[55] | Quiroz YT , Sperling RA , Norton DJ , Baena A , Arboleda-Velasquez JF , Cosio D , Schultz A , Lapoint M , Guzman-Velez E , Miller JB , Kim LA , Chen K , Tariot PN , Lopera F , Reiman EM , Johnson KA ((2018) ) Association between amyloid and tau accumulation in young adults with autosomal dominant Alzheimer disease. JAMA Neurol 75: , 548–556. |
[56] | McDade E , Bateman RJ ((2018) ) Tau positron emission tomography in autosomal dominant Alzheimer disease: Small windows, big picture. JAMA Neurol 75: , 536–538. |


