
Potential Novel Role of COVID-19 in Alzheimer’s Disease and Preventative Mitigation Strategies
Abstract
There are a number of potential implications for the field of Alzheimer’s disease (AD) stemming from the global spread of SARS-CoV-2. Neuroinflammation is known to be a prominent feature of neurodegeneration and plays a major role in AD pathology. Immune response and excessive inflammation in COVID-19 may also accelerate the progression of brain inflammatory neurodegeneration, and elderly individuals are more susceptible to severe outcomes after SARS-CoV-2 infection. Individuals with type 2 diabetes (T2D) are at an increased risk for AD as well as severe outcomes after SARS-CoV-2 infection. Genetic and socioeconomic factors influencing the rates of T2D, AD, and COVID-19 severity may create an exceptionally high-risk profile for certain demographics such as African Americans and Hispanic Americans. Type I interferon response plays an important role in both host response to viral infection, as well as AD pathology and may be a sensible therapeutic target in both AD and COVID-19.
The novel coronavirus SARS-CoV-2 emerged in China in late 2019 and its origin has been traced to a market in Wuhan, China [1]. The virus responsible for COVID-19 subsequently spread throughout the world and was declared to be a global pandemic by the World Health Organization on March 11, 2020 [2]. As of May 3, 2020, there were a total of 3,349,786 COVID-19 cases and 238,628 fatalities globally [3]. As our knowledge of the COVID-19 has progressed, advanced age has become recognized as one of the predominant risk factors associated with severe outcomes after infection with those over 59 years of age being 5 times more likely to die from COVID-19 than younger people [4]. The predominance of additional risk factors such as cardiovascular disease and metabolic disorders indicates that biological age (as opposed to chronological age) may be the single biggest risk factors toward severe or fatal outcomes [5]. Diabetes is considered a major comorbidity factor for COVID-19 related fatalities [6]. Type 2 diabetes (T2D), the most common form of diabetes, has also been linked with an increased predisposition to Alzheimer’s disease (AD) through shared genetic etiologies [7]. Genetic and socioeconomic factors have compounded the risk of AD, T2D, and COVID-19 in certain populations. African Americans and Hispanics had 1.5–2 times higher incidence of both T2D and AD compared to non-Hispanic whites [8, 9]. These racial and ethnic disparities are further exacerbated in COVID-19. For example, in Wisconsin and Louisiana, African Americans account for 70% of COVID-19 deaths despite accounting for only 26% and 32% of those populations, respectively [10]. In New York City, African Americans account for 24.3% of the population but accounted for 35.4% of COVID-19 related hospitalizations and 30% of COVID-19 related deaths. Similarly, Hispanics account for 29.1% of the population but accounted for 31.3% of COVID-19 related hospitalizations and 30.4% of COVID-19 related deaths [11]. Given the parallels between higher rates of T2D, AD, and COVID-19 along with the fact that diabetes has been identified as a major comorbidity factor for COVID-19, these demographics are at an increased risk of severe outcomes related to COVID-19.
Severe outcomes after SARS-CoV-2 infection are often associated with a “cytokine storm” of pronounced inflammation [12]. One of the reasons that elderly individuals are at a higher risk after infection may be due to a higher existing baseline of inflammation. Inflammation steadily increases with age in a process referred to as “inflamm-aging” [13]. The “cytokine storm” associated with COVID-19 has been associated with an increase of pro-inflammatory cytokines such as tumor necrosis factor-alpha, interleukin-1 (IL-1), and IL-6 all of which have been identified to be increased in the elderly [13, 14]. Thus, rendering the elderly particularly vulnerable to severe outcomes resulting from COVID-19. Interferon regulatory factor 5 (IRF5) plays a key role in facilitating exaggerated immune response (cytokine storm) after Influenza A virus infection, and is regulated by the glucose sensitive upstream factor O-GlcNAc transferase [15]. The potential for elevations in blood glucose to increase inflammation through IRF5 activity, along with the overlapping pathology of T2D and AD may create a “perfect storm” of excessive immune response after infection with SARS-CoV-2 in AD patients (Fig. 1). Increased IRF5 activity resulting from T2D comorbidity may also synergize with amyloid-stimulated Type I interferon (IFN) response (described below).
Fig.1
Potential Interactions Between SARS-CoV-2 and Alzheimer’s Disease. Type 2 diabetes (T2D) acts as predisposing factor for Alzheimer’s disease (AD). Elevations in blood glucose resulting from T2D may exacerbate pathology in both AD and COVID-19 or comorbidity of the two, through increased activity of Interferon regulatory factor 5 (IRF5). Type I interferons (IFN) mediate inflammation after viral infection, and in response to nucleic acid containing amyloid fibrils, eventually leading to synaptic loss. Amyloid fibrils may entrap viral particles, leading to further enhancement of IFN response. Solid line arrows indicate proven mechanisms, dotted line arrows indicate theoretical mechanisms.
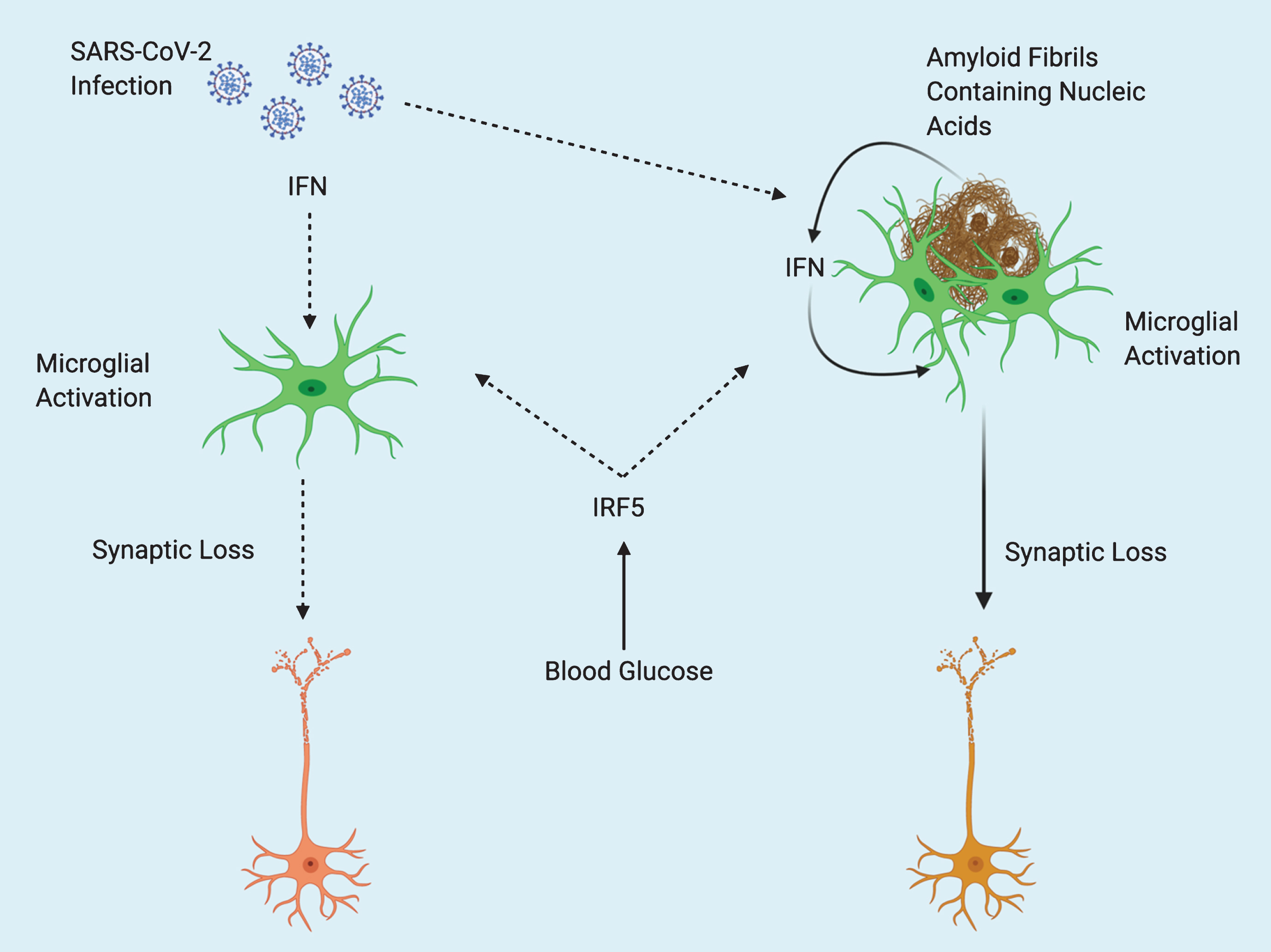
IFNs are a group of cytokines that play an important role in mediating host response to viral infection [16]. Roy and colleagues recently demonstrated the role of IFN in mediating AD pathology. Hippocampal tissues from multiple different transgenic mouse models of AD were all shown to have increased expression of genes known to be induced by IFN. Furthermore, nucleic acid containing amyloid fibrils were shown to stimulate the expression of genes known to be involved with IFN response both in vitro and in vivo. The subset of microglia known as the “microglial neurodegenerative phenotype” associated with nucleic acid containing amyloid plaques and displayed increased expression of IFN pathway related genes. IFN was found to directly activate microglia and stimulate a proinflammatory response. Furthermore, IFN was found to stimulate complement cascade activation and promote synapse elimination. Finally, interferon stimulated genes were also found to be upregulated in AD patient brains and correlated with increasing Braak scores [17]. The findings of Roy et al. are particularly interesting, because the amyloid fibrils which were found to induce interferon response contained nucleic acids (DNA and RNA). Previously, Eimer and colleagues showed that 5XFAD mice infected with herpes simplex virus 1 (a double stranded DNA virus) displayed accelerated amyloid fibrillization. Additionally, amyloid was found to entrap viral particles in a process that was suggested to be part of the CNS innate immune system [18]. Readhead and colleagues found increased levels of human herpesvirus 6 and human herpesvirus 7 across several regions in postmortem brains of AD patients [19]. Thus, it would appear that amyloid-β acts as a part of host response to infection. Conceivably, amyloid fibrils entrap microbial pathogens and subsequently induce microglial activation and IFN response (Fig. 1). It is not currently clear as to whether AD pathology is primarily driven by microbial infection or is a matter of this immune response gone awry. These ideas shed an interesting light on what we might expect in terms of neurological damage in survivors of COVID-19. SARS-CoV-2 entry into the brain has already been documented; this is interesting to consider in light of the fact that 43.2% of COVID-19 patients are asymptomatic [20, 21]. It is possible that there may be an existing population who have become unknowingly predisposed to neurodegeneration through silent viral entry into the brain. Additionally, pre-symptomatic individuals with undiagnosed AD may see an acceleration of symptoms due to a bout of systemic inflammation resulting from infection. The lasting implications of SARS-CoV-2 and its effects on the brain are not yet known, but its potential as a priming factor for neurodegeneration may be of great importance to the field of ADresearch.
While the potential for IFN response in AD and COVID19 to synergize and exacerbate pathology has yet to be explored, IFN response may also play a role in the immediate effects of COVID-19 on the brain. The acute neurological complications resulting from SARS-CoV-2 infection include meningitis and acute necrotizing encephalopathy, as well as gustatory and olfactory impairments [20, 22, 23]. IFN has previously been associated with viral encephalitis as well as the inherited encephalopathy Aicardi–Goutières syndrome [24, 25].
Suppression of IFN response in both AD and COVID-19 (or comorbidity of the two) may be an advantageous strategy for controlling excessive immune response. Additionally, it is interesting to speculate on how AD therapeutics might interact with SARS-CoV-2 infection. Currently, the approved treatments donepezil, rivastigmine, and galantamine all act by increasing acetylcholine levels by inhibiting its degrading enzyme acetylcholinesterase [26]. While not yet explored, the cholinergic anti-inflammatory pathway may be a viable target for controlling inflammation and preventing a “cytokine storm” after SARS-CoV-2 infection, and acetylcholinesterase (AChE) inhibitors could potentially be repurposed as an adjunctive therapy. Interestingly, the AChE inhibitor galantamine has been shown to decrease serum cytokines and chemokines such as TNF-α, IL-10, and MCP-1 in a rat model of arthritis [27]. Stimulation of the vagus nerve through α7 nicotinic receptors can have anti-inflammatory effects [28], leading some to even speculate that nicotine administration may be effective in preventing excessive inflammation in COVID-19 patient [29]. If stimulation of the cholinergic anti-inflammatory pathway were to be a beneficial strategy, the abundance of existing nicotinic compounds that have previously been developed to treat AD and other neurological/psychiatric disorders could provide a number of additional treatment options [30].
It is also important to recognize the indirect effects that the COVID-19 pandemic will likely have on AD patients. Because elderly individuals are at a higher risk for mortality after SARS-CoV-2 infection, it is necessary to isolate these individuals and limit their contact with other people during this pandemic. However, time spent with caretakers and family, as well as social interaction and daily activity are generally considered to help prevent cognitive decline in the elderly. Thus isolation, while necessary may unfortunately carry increased risk of cognitive decline in elderly individuals. While the sum total effects of the COVID19 pandemic are not yet known it will undoubtedly affect many AD patients either directly or indirectly. Only large-scale retrospective analysis in addition to preclinical research will reveal the full range of implications for aging and neurodegeneration.
ACKNOWLEDGMENTS
This study was supported by Grant Number P50 AT008661-01 from the NCCIH and the ODS. Dr. Pasinetti holds a Senior VA Career Scientist Award. We acknowledge that the contents of this study do not represent the views of the NCCIH, the ODS, the NIH, the U.S. Department of Veterans Affairs, or the United States Government. Figure 1 was made using Biorender.com.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/20-0537r1).
REFERENCES
[1] | Li Q , Guan X , Wu P , Wang X , Zhou L , Tong Y , Ren R , Leung KSM , Lau EHY , Wong JY , Xing X , Xiang N , Wu Y , Li C , Chen Q , Li D , Liu T , Zhao J , Liu M , Tu W , Chen C , Jin L , Yang R , Wang Q , Zhou S , Wang R , Liu H , Luo Y , Liu Y , Shao G , Li H , Tao Z , Yang Y , Deng Z , Liu B , Ma Z , Zhang Y , Shi G , Lam TTY , Wu JT , Gao GF , Cowling BJ , Yang B , Leung GM , Feng Z ((2020) ) Early transmission dynamics in Wuhan, China, of novel coronavirus–infected pneumonia. N Engl J Med 382: , 1199–1207. |
[2] | WHO ((2020) ) WHO Director-General’s opening remarks at the media briefing on COVID-19 – 11 March 2020, World Health Organization. |
[3] | WHO ((2020) ) Coronavirus disease (COVID-19) Situation Report – 104, World Health Organization. |
[4] | Wu JT , Leung K , Bushman M , Kishore N , Niehus R , de Salazar PM , Cowling BJ , Lipsitch M , Leung GM ((2020) ) Estimating clinical severity of COVID-19 from the transmission dynamics in Wuhan, China. Nat Med 26: , 506–510. |
[5] | Lauc G , Sinclair D ((2020) ) Biomarkers of biological age as predictors of COVID-19 disease severity. Aging (Albany NY) 12: , 6490–6491. |
[6] | Fang L , Karakiulakis G , Roth M ((2020) ) Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection? Lancet Respir Med 8: , e21. |
[7] | Hao K , Di Narzo AF , Ho L , Luo W , Li S , Chen R , Li T , Dubner L , Pasinetti GM ((2015) ) Shared genetic etiology underlying Alzheimer’s disease and type 2 diabetes. Mol Aspects Med 43-44: , 66–76. |
[8] | CDC ((2020) ) National Diabetes Statistics Report 2020 – Estimates of Diabetes and Its Burden in the United States. |
[9] | U.S. Department of Health & Human Services ((2014) ) Racial and ethnic disparities in Alzheimer’s disease: a literature review. |
[10] | Dyer O ((2020) ) Covid-19: Black people and other minorities are hardest hit in US. BMJ 369: , m1483. |
[11] | NYC Health Department ((2020) ) Age-adjusted rates of lab-confirmed COVID-19 non hospitalized cases, estimated non-fatal hospitalized cases, and total persons known to have died (lab-confirmed and probable) per 100,000 by race/ethnicity group. |
[12] | Ye Q , Wang B , Mao J ((2020) ) The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID-19. J Infect 80: , 607–613. |
[13] | Rea IM , Gibson DS , McGilligan V , McNerlan SE , Alexander HD , Ross OA ((2018) ) Age and age-related diseases: role of inflammation triggers and cytokines. Front Immunol 9: , 586. |
[14] | Scarpa R , Costa L , Del Puente A , Caso F ((2020) ) Role of thymopoiesis and inflamm-aging in COVID-19 phenotype. Pediatr Neonatol 61: , 364–365. |
[15] | Wang Q , Fang P , He R , Li M , Yu H , Zhou L , Yi Y , Wang F , Rong Y , Zhang Y , Chen A , Peng N , Lin Y , Lu M , Zhu Y , Peng G , Rao L , Liu S ((2020) ) -GlcNAc transferase promotes influenza A virus–induced cytokine storm by targeting interferon regulatory factor–5. Sci Adv 6: , eaaz7086. |
[16] | McNab F , Mayer-Barber K , Sher A , Wack A , O’Garra A ((2015) ) Type I interferons in infectious disease. Nat Rev Immunol 15: , 87–103. |
[17] | Roy ER , Wang B , Wan Y , Chiu G , Cole A , Yin Z , Propson NE , Xu Y , Jankowsky JL , Liu Z , Lee VM-Y , Trojanowski JQ , Ginsberg SD , Butovsky O , Zheng H , Cao W ((2020) ) Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J Clin Invest 130: , 1912–1930. |
[18] | Eimer WA , Vijaya Kumar DK , Navalpur Shanmugam NK , Rodriguez AS , Mitchell T , Washicosky KJ , György B , Breakefield XO , Tanzi RE , Moir RD ((2018) ) Alzheimer’s disease-associated β-amyloid is rapidly seeded by herpesviridae to protect against brain infection. Neuron 99: , 56–63.e3. |
[19] | Readhead B , Haure-Mirande J-V , Funk CC , Richards MA , Shannon P , Haroutunian V , Sano M , Liang WS , Beckmann ND , Price ND , Reiman EM , Schadt EE , Ehrlich ME , Gandy S , Dudley JT ((2018) ) Multiscale analysis of independent Alzheimer’s cohorts finds disruption of molecular, genetic, and clinical networks by human herpesvirus. Neuron 99: , 64–82.e7. |
[20] | Poyiadji N , Shahin G , Noujaim D , Stone M , Patel S , Griffith B ((2020) ) COVID-19–associated acute hemorrhagic necrotizing encephalopathy: CT and MRI features. Radiology, doi: 10.1148/radiol.2020201187. |
[21] | Lavezzo E , Franchin E , Ciavarella C , Cuomo-Dannenburg G , Barzon L , Del Vecchio C , Rossi L , Manganelli R , Loregian A , Navarin N , Abate D , Sciro M , Merigliano S , Decanale E , Vanuzzo MC , Saluzzo F , Onelia F , Pacenti M , Parisi S , Carretta G , Donato D , Flor L , Cocchio S , Masi G , Sperduti A , Cattarino L , Salvador R , Gaythorpe KAM , Imperial College London COVID-19 Response Team, Brazzale AR , Toppo S , Trevisan M , Baldo V , Donnelly CA , Ferguson NM , Dorigatti I , Crisanti A ((2020) ) Suppression of COVID-19 outbreak in the municipality of Vo, Italy. medRxiv 2020.04.17.20053157; doi: 10.1101/2020.04.17.20053157. |
[22] | Moriguchi T , Harii N , Goto J , Harada D , Sugawara H , Takamino J , Ueno M , Sakata H , Kondo K , Myose N , Nakao A , Takeda M , Haro H , Inoue O , Suzuki-Inoue K , Kubokawa K , Ogihara S , Sasaki T , Kinouchi H , Kojin H , Ito M , Onishi H , Shimizu T , Sasaki Y , Enomoto N , Ishihara H , Furuya S , Yamamoto T , Shimada S ((2020) ) A first case of meningitis/encephalitis associated with SARS-Coronavirus-2. Int J Infect Dis 94: , 55–58. |
[23] | Vavougios GD ((2020) ) Potentially irreversible olfactory and gustatory impairments in COVID-19: Indolent vs. fulminant SARS-CoV-2 neuroinfection. Brain Behav Immun, doi: 10.1016/j.bbi.2020.04.071. |
[24] | Delhaye S , Paul S , Blakqori G , Minet M , Weber F , Staeheli P , Michiels T ((2006) ) Neurons produce type I interferon during viral encephalitis. Proc Natl Acad Sci U S A 103: , 7835–7840. |
[25] | Hofer MJ , Campbell IL ((2013) ) Type I interferon in neurological disease—The devil from within. Cytokine Growth Factor Rev 24: , 257–267. |
[26] | Ellis JM ((2005) ) Cholinesterase inhibitors in the treatment of dementia. J Am Osteopath Assoc 105: , 145–158. |
[27] | Gowayed MA , Refaat R , Ahmed WM , El-Abhar HS ((2015) ) Effect of galantamine on adjuvant-induced arthritis in rats. Eur J Pharmacol 764: , 547–553. |
[28] | Ulloa L ((2005) ) The vagus nerve and the nicotinic anti-inflammatory pathway. Nat Rev Drug Discov 4: , 673–684. |
[29] | Farsalinos K , Niaura R , Le Houezec J , Barbouni A , Tsatsakis A , Kouretas D , Vantarakis A , Poulas K ((2020) ) Editorial: Nicotine and SARS-CoV-2: COVID-19 may be a disease of the nicotinic cholinergic system. Toxicol Rep 7: , 658–663. |
[30] | Bertrand D , Terry AV ((2018) ) The wonderland of neuronal nicotinic acetylcholine receptors. Biochem Pharmacol 151: , 214–225. |


