
Early-Onset Familial Alzheimer Disease Variant PSEN2 N141I Heterozygosity is Associated with Altered Microglia Phenotype
Abstract
Background:
Early-onset familial Alzheimer disease (EOFAD) is caused by heterozygous variants in the presenilin 1 (PSEN1), presenilin 2 (PSEN2), and APP genes. Decades after their discovery, the mechanisms by which these genes cause Alzheimer’s disease (AD) or promote AD progression are not fully understood. While it is established that presenilin (PS) enzymatic activity produces amyloid-β (Aβ), PSs also regulate numerous other cellular functions, some of which intersect with known pathogenic drivers of neurodegeneration. Accumulating evidence suggests that microglia, resident innate immune cells in the central nervous system, play a key role in AD neurodegeneration.
Objective:
Previous work has identified a regulatory role for PS2 in microglia. We hypothesized that PSEN2 variants lead to dysregulated microglia, which could further contribute to disease acceleration. To mimic the genotype of EOFAD patients, we created a transgenic mouse expressing PSEN2 N141I on a mouse background expressing one wildtype PS2 and two PS1 alleles.
Results:
Microglial expression of PSEN2 N141I resulted in impaired γ-secretase activity as well as exaggerated inflammatory cytokine release, NFκB activity, and Aβ internalization. In vivo, PS2 N141I mice showed enhanced IL-6 and TREM2 expression in brain as well as reduced branch number and length, an indication of “activated” morphology, in the absence of inflammatory stimuli. LPS intraperitoneal injection resulted in higher inflammatory gene expression in PS2 N141I mouse brain relative to controls.
Conclusion:
Our findings demonstrate that PSEN2 N141I heterozygosity is associated with disrupted innate immune homeostasis, suggesting EOFAD variants may promote disease progression through non-neuronal cells beyond canonical dysregulated Aβ production.
INTRODUCTION
Heterozygous variants in presenilin 1 (PSEN1) and presenilin 2 (PSEN2) cause autosomal dominant, early-onset familial Alzheimer disease (EOFAD) through mechanisms that are not fully understood. Presenilins (PSs) are the enzymatic components of the membrane bound γ-secretase complex which cleaves amyloid-β protein precursor (AβPP) to yield amyloid-β (Aβ) peptides [1], the accumulation of which are a neuropathological hallmark of Alzheimer disease (AD). The association of nearly completely penetrant EOFAD variants and the production of Aβ led to the hypothesis that accumulation of amyloid peptide in the central nervous system (CNS) drives AD and has guided the therapeutic efforts for both EOFAD and sporadic AD [2]. While the altered neuronal production of Aβ species in monogenic AD is well established, the mechanisms that contribute to progressive neurodegeneration are not. This gap in understanding the pathogenesis of AD has hampered drug development efforts but may also represent opportunities for novel drug discovery.
The concept of neurodegeneration has developed from a previously “neuron centric” [3] model to one where glial cells play an early role in promoting disease and influencing phenotype [3]. Mechanistic drivers of AD are heterogeneous and attributable to multiple cell types across the temporal phases of disease [3, 4]. In particular, innate immunity has emerged as a pathogenic factor in the course of AD neurodegeneration [5, 6]. The glial-neuronal relationship in AD includes appropriate innate immune responses, such as forming protective barriers around Aβ to protect neurites [7], as well as injurious responses (release of neurotoxic molecules in the face of non-microbial stimuli) by non-neuronal cells [8, 9]. Glial cells can drive disease progression in monogenic forms of neurodegeneration. Studies of familial neurodegenerative diseases show that glial specific expression of disease associated genetic variants drives disease progression; in other words, a gene variant can alter normal function of multiple neural cell types which together contributes to disease phenotype [10–12]. Thus, the complex nature of immunity in AD means that developing effective therapies targeting novel candidate pathways such as inflammation, need to be tailored to restricting deleterious behavior while either boosting or at least not hampering appropriate immune activity.
Microglia, the resident innate immune cell in brain, are themselves multifaceted, maintaining homeostatic, developmental, and reparative roles in the CNS as well as clearing of debris and responding to pathogen-associated molecular pattern and damage-associated molecular pattern molecules [9, 13, 14]. We and others demonstrated that the familial AD gene, PSEN2 exerts a regulatory role on microglia that is relevant to AD progression. Presenilin 2 (PS2) protein modulates the phenotype of microglia, and the absence of PS2 promotes the microglial adoption of an exaggerated inflammatory response to lipopolysaccharide (LPS) and aggregated Aβ42 exposure [15–17]. Conditional PS knockout mice completely deficient in Psen2 and deficient in Psen1 only in forebrain neurons exhibit a widespread neuroinflammatory phenotype even in the absence of Aβ [18]. Taken together these studies suggest that PSEN2 may confer risk to AD not only through neuronal production of Aβ, but also through regulating the biology of CNS innate immune cells.
Presenilins are expressed ubiquitously and regulate a large array of cellular functions through proteolytic and non-enzymatic actions [19, 20]. As the enzymatic component of the γ-secretase complex, PSs cleave dozens of substrates beyond AβPP, including those relevant to neural and peripheral immune cells [21, 22]. Substrates of γ-secretase and PS2 activity that regulate immune cell function include TREM2, TNF Type Receptor 1, IL-1 receptor, IL-6 receptor, CX3CL1, and CSF1R [19, 23–25]. EOFAD associated PSEN variants compromise γ-secretase function and lead to a biochemical “loss-of-function”, causing dominant negative interference with the wildtype allele or reduced PS expression [26–30]. Consequently, PS dysfunction due to EOFAD PSEN2 variants could compromise CNS immune cell homeostasis similar to the absence of PS2 [15, 16], and thus contribute to AD pathogenesis.
We hypothesize that PSEN2 EOFAD variants are hypomorphic or impede normal PS2 function which leads to the dysregulation of innate immune physiology and AD progression. To better understand the immune state in human patients with PSEN2 FAD, we created a transgenic mouse model expressing the “Volga German” PSEN2 N141I variant on a background line expressing only one endogenous mouse PS2 [31].
The data presented here demonstrate a novel role for a PSEN2 disease associated variant in the disruption of innate immune homeostasis. Our findings support the hypothesis that PSEN EOFAD involves neuronal and glial dysfunction, broadening the scope of potential therapeutic targets.
METHODS
PSEN2 N141I transgenic mice
All breeding and experimental protocols were approved by the University of Washington IUCAC, Protocol # 3254-04. Mice were fed standard rodent chow and water ad libitum and maintained under specific pathogen–free conditions, according to standard animal husbandry procedures. The transgene, designated as Tg(CAG-loxP-EGFP-STOP-loxP-PSEN2(N141I) represents a PSEN2 cDNA (containing the N141I mutation) placed downstream of a floxed-stop cassette under the control of a chicken β-actin promoter. The GFP-STOP cassette prevents expression of the PSEN2 transgene prior to Cre excision. This PSEN2 expression vector was derived by releasing the 4E-BP1 transgene from vector 2383-4 by Nru, Sbfl (New England Biolabs, Ipswich, MA) restriction enzyme digestion [32]. The 1.4 kb PSEN2 N141I cDNA was PCR-amplified, cut with Nrul & Sbfl, and ligated into the corresponding sites of vector 2383, yielding a new PSEN2 expression vector designated 2740-B6. The 2740-B6 vector DNA was digested with ApaLI (New England BioLabs) and the 9.8 kb vector insert was gel purified away from the 1.2 kb vector backbone restriction fragment (Fig. 1A). This gel purified DNA was then run over a Detoxi-Gel Endotoxin Removal Column (Thermo Fisher Scientific, Waltham, MA) to further purify the DNA prior to microinjection into C57BL/6J6C3H/HeJ oocytes (purchased from Jackson Laboratory, Bar Harbor ME, and bred at UW). A Cre derived, germline excised, PSEN2(N141I) expressing sub-line of mice (designated Tg(CAG-loxP-PSEN2 (N141I)) was derived by crossing the floxed mice to a CMV-Cre deleter line that ubiquitously expresses Cre from the human cytomegalovirus promoter (purchased from Jackson Laboratory and bred at UW), yielding a second line of mice that stably and ubiquitously express PS2(N141I) from the chicken β-actin (CAG) promoter. These Cre derived mice were maintained and analyzed on a hemizygous Psen2 background and are referred to as “PS2 N141I”. The parental (non-excised) control transgenic mice, referred to as “TG Control”, express EGFP but not PSEN2 N141I due to the presence of the quadruplicated floxed-STOP cassette [33]. There were no other transgenes introduced or crossed into the PSEN2 N141I line.
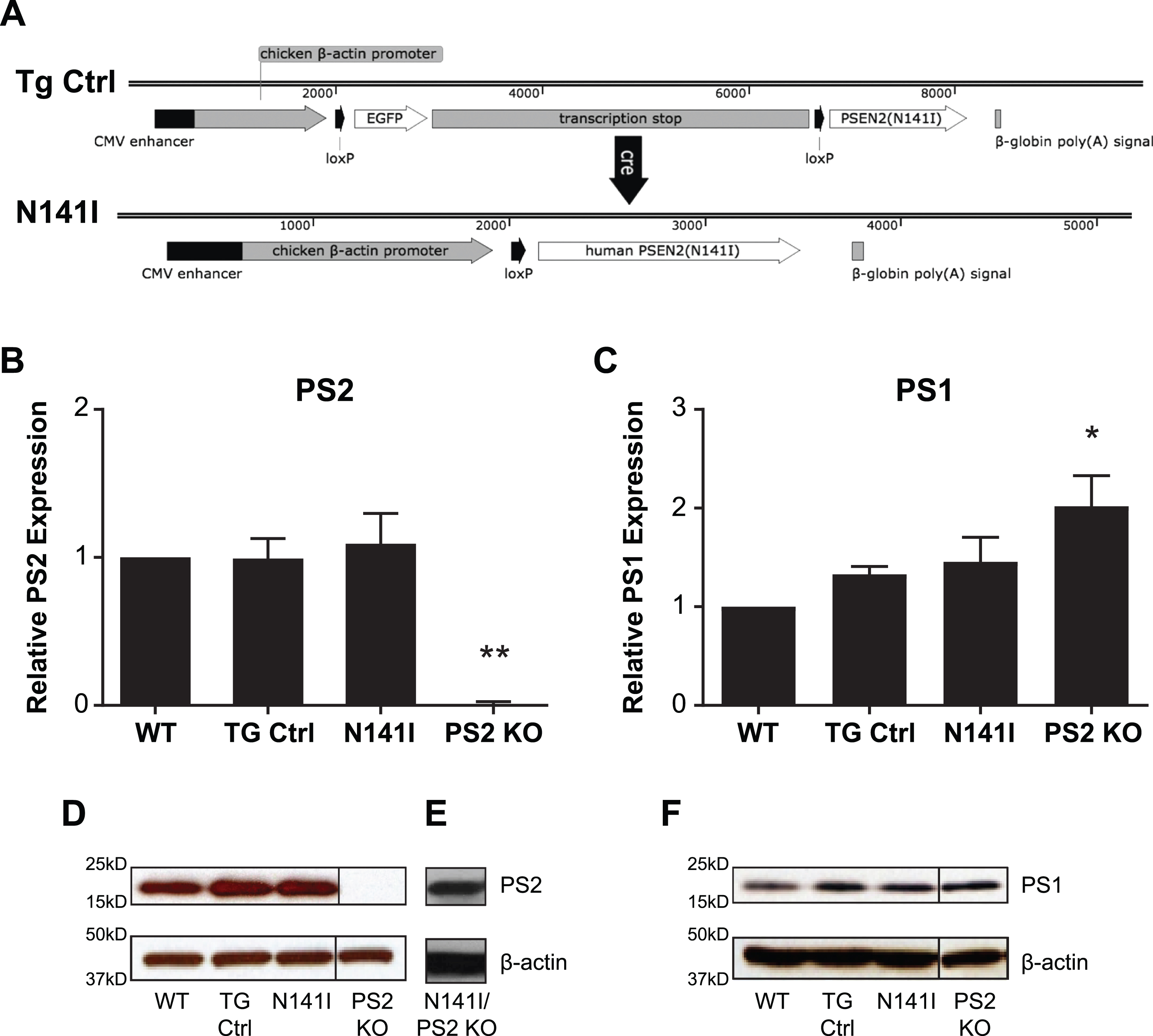
Fig. 1
PSEN2 N141I Transgene constructs and expression of PS2 N141I in microglia. A) Control mice (TG Ctrl) express a transgene containing the PSEN2 N141I cDNA that is downstream of a floxed stop sequence: Tg(CAG-loxP-EGFP-STOP-loxP-PSEN2(N141I). Upon Cre exposure, the stop cassette is excised resulting in the transgene: Tg(CAG-loxP-PSEN2(N141I) and expression of the PSEN2 N141I (N141I). B) Microglia with one copy of human PS2 N141I and one copy of wildtype mouse PS2 show similar PS2 protein expression compared to transgenic control (TG Ctrl) and wildtype microglia (WT). Data are mean±SEM, n = 3 and represent experiments from independent cultures, *p≤0.05; ***p≤0.001, t-test analysis. C) PS2 KO microglia express significantly increased PS1, while TG and N141I do not have significantly different levels from control. Data are mean±SEM, n = 3 and represent experiments from independent cultures, *p≤0.05; ***p≤0.001, t-test analysis D) Representative western blot of lysates analyzed in (B) prepared from primary microglia isolated from WT, TG Ctrl, and N141I on a hemizygous murine Psen2 background immunolabeled for PS2. PS2 KO lysate is used as negative control. E) Lysate prepared from microglia isolated from a PS2 N141I expressing animal crossed to PS2 KO background shows PS2 expression confirming that the transgene expresses mutant PS2. F) Representative western blot analysis of lysates analyzed in (C) illustrating PS1 expression. PS2 and PS1 expression is similar in PS2 N141I microglia relative to wildtype and TG Ctrl.
Microglia culture
Primary neonatal microglia were collected as previously described [16]. Briefly, mixed glial cell cultures were maintained in PDL-coated T75 or T175 flasks (Genesee Scientific, San Diego, CA) in D10C media: DMEM (Sigma-Aldrich, St. Louis, MO) supplemented with 10% heat-inactivated horse serum (Gemini Bio-Products, West Sacramento, CA), 10% F12 medium (Sigma), 2 mM L-glutamine (Gibco/Thermo Fisher Scientific), 5 mM HEPES (Gibco/Thermo Fisher Scientific), 100 IU/mL penicillin and 100 μg/mL streptomycin (Gibco/Thermo Fisher Scientific) for up to four weeks. Starting on week 2 and every week thereafter, floating and loosely adherent microglia were harvested by manual agitation.
Western blot analysis
Western blot analysis for PS2 and PS1 was performed on primary microglia as described previously [15, 16]. Briefly, plated cells were lysed and SDS–PAGE was performed with Invitrogen equipment and reagents using β-mercaptoethanol (Bio-Rad Laboratories, Hercules, CA), 4–12% Bis-Tris gradient gels (Invitrogen/Thermo Fisher Scientific), 3-(N-morpholino)-propanesuflonic acid - sodium dodecyl sulfate (MOPS-SDS) buffer (Invitrogen/Thermo Fisher Scientific), and polyvinylidene difluoride membrane (Bio-Rad Laboratories). Membranes were incubated in 5% milk/phosphate buffered saline (PBS) with 0.1% Tween-20 with primary rabbit-anti-PS2 antibody (ab51249; 1 : 1000 Abcam, Cambridge, MA), primary anti-PS1 (ab76083; 1 : 1000 Abcam), or primary mouse-anti-βactin antibody (A5441; 1 : 200,000 Sigma-Aldrich) followed by secondary antibody (either anti-rabbit IgG horseradish peroxidase (NA934; GE Healthcare Bio-Sciences, Pittsburg, PA) or anti-mouse IgG horseradish peroxidase [NA931V; GE Healthcare Bio-Sciences). Quantification of protein was performed employing Image J software (National Institutes of Health, Bethesda, MD).
NFκB and γ-secretase activity
Primary microglia were plated at 1×104 cells per well on PDL-coated 96-well plates in D10C. NFkB activity was assessed as previously described [15]. Cells were transduced at the time of plating with a lentivirus containing an expression vector with firefly luciferase expression driven by a minimal cytomegalovirus (CMV) promoter and tandem repeats of the NFκB transcriptional response element (Cignal Lenti E2F Reporter) (Qiagen, Valencia, CA). γ-secretase enzymatic activity was assessed as described previously [16] using lentivirus prepared through the Fred Hutchinson Cancer Research Center lentiviral core facility (Seattle, WA; NIH Grant DK 56465). For both assays, cells were also transduced with viral particles expressing Renilla luciferase driven by a CMV promoter as an internal control (Qiagen) at 5 viral particles/cell (MOI 5). Twenty-four hours after plating and infection, growth media were removed and replaced with Macrophage Serum Free Media (M-SFM) (Gibco). Following another 24 h, cells were treated with 100 ng/mL LPS in M-SFM for 4 h prior to NFkB measurement or 24 h prior to γ-secretase measurement. Media was removed and plates were subsequently stored at –80°C until luciferase activity assay was assessed using Promega Dual-Glo reagents according to manufacturer’s protocol (Promega, Madison, WI) and measured on a luminometer.
Cytokine release
Five×104 neonatal microglia per well were plated on PDL coated 96 well plates in D10C. After 24 h, cells were cultured in fresh D10C with or without 100 ng/mL LPS for an additional 24 h. Supernatant was transferred to a new 96 well plate and stored at –80°C until quantification. Analytes were assessed using a Mouse Cytokine/Chemokine Magnetic Bead panel –(Immunology Multiplex Assay, MCYTOMAG-70K; Millipore, Billerica, MA) according to manufacturer’s protocol.
Aβ phagocytosis
Fibrillar Aβ1–42 (Bachem, Torrance, CA) was prepared and conjugated to pHrodo™ Red (Thermo Fisher Scientific) as described previously [34]. Primary microglia were plated at 2×104 cells/well in 96-well Primaria plates in D10C media. The following day, cells were incubated with D10C (media only control) or 1 μM pHrodo-Aβ in D10C at 4°C or 37°C for 5 h. Cells were washed with cold PBS, trypsinized, and analyzed using standard flow cytometry techniques on an LSR II cytometer (BD Sciences, San Jose, CA). Quantification of phagocytosis was done by comparing the geometric mean fluorescence intensity (MFI) of phagocytized pHrodo-Aβ between genotypes relative to their respective controls using Flow Jo software version 10 (BD Biosciences). Data represents the mean of three separate experiments.
Apoptotic cell phagocytosis
Apoptosis was induced in SH-SY5Y neuroblastoma cells via camptothecin (CPT) as we have previously described [35]. SH-SY5Y5 cell membranes were fluorescently labeled with PKH Red Fluorescent Cell Linker Kit (Sigma-Aldrich) according to manufacturer’s instructions and incubated at 37°C in DMEM/F12 media with 10% FBS and 0.1% Pen/Strep for 24 h, followed by exposure to 500 nM camptothecin CPT (Sigma-Aldrich) for 18 h to induce cellular damage [35]. Primary microglia cell membranes were fluorescently labeled with PKH Green Fluorescent Cell Linker Kit (Sigma-Aldrich) according to manufacturer’s instructions, plated at 1×105 cells/well in 4-well plates in D10C media and incubated overnight at 37°C. The following day, labeled SH-SY5Y cells damaged with CPT treatment were harvested, resuspended in D10C media and added to wells containing labeled microglia at 1x105 SH-SY5Y cells/well. Cells were co-cultured at 4°C or 37°C for 5 h. Cells were washed with cold PBS, trypsinized, fixed, and analyzed using standard flow cytometry techniques on an LSR cytometer (BD Sciences). The population of microglia that internalized SH-SY5Y cells was determined by calculating the percent of events detected to be both PE and FITC positive relative to the number of FITC only positive for each genotype relative to their respective 4°C controls.
Gene expression
Snap frozen brain tissue was homogenized in Qiazol (Qiagen) with a TissueRuptor (Qiagen). Total RNA was extracted from the lysate using the RNeasy Micro Kit (Qiagen) according to manufacturer’s instructions. Total RNA was reverse transcribed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems/Thermo Fisher Scientific) according to the manufacturer’s instructions. Gene expression was assessed using pre-designed Assay on Demand Taqman Primers (Applied Biosystems/Thermo Fisher Scientific) and Taqman Universal Master Mix II (Applied Biosystems/Thermo Fisher Scientific) using the QuanStudio 6 Real-Time PCR Systems (Applied Biosystems/Thermo Fisher Scientific). Average raw cycle threshold data were normalized to the expression of GAPDH for each sample (ΔCt). Relative gene expression was normalized to the average ΔCt of the saline injected Tg Control animals by use of ΔΔCt calculations.
Microglia morphology
Immunohistochemistry was performed on PFA fixed, 30 μm sagittal sections. Sections were permeabilized with 0.4% triton X-100 and blocked with 5% donkey serum, 1% BSA, 0.1% triton X-100, 0.05% tween-20, and 0.05% sodium azide in PBS. Sections were then incubated with goat anti-Iba1 (1 : 300, Abcam, 5076) overnight at 4°C, washed three times in 1X PBS, and incubated with donkey anti-goat, Alexa Fluor 488 (1 : 500, Life technologies, A11055) at room temperature for 1 h, stained with DAPI and mounted using Fluoromount-G (Southern Biotech Birmingham, AL). Images were taken from the motor cortex and dorsal hippocampal cortex regions using a Zeiss Axiovert 200 M microscope (Leica Microsystems). Z-stack images were taken at 1.5 μm using a 20X objective. The z-stacks were deconvolved and projection images created using SlideBook 5 (Intelligent Imaging Innovations, Inc.). Cell counting and morphology analysis were performed using ImageJ [36]. Briefly, the images were converted to black and white, the brightness/contrast adjusted equally, and an unsharp mask applied to increase contrast. The images were converted to binary, pixels less than 2 pixels apart were joined, and despeckle was performed to remove noise. The images were skeletonized. Branches containing ≤2 endpoints and < 10 pixels in length were removed. Number of endpoints and average branch length were summed for each image and divided by the number of microglia somas. The number of somas in each image were manually counted by two unbiased raters blind to the condition of the animals. Interrater reliability was strong as assessed using intraclass correlation coefficient (ICC motor cortex = 0.94, dorsal hippocampus = 0.88). Five images per region were analyzed and averaged for each animal.
Systemic LPS administration
Ultrapure LPS from E. coli serotype 055:B5 (InvivoGen, San Diego, CA) at 1 mg/kg or saline control was administered to 22–26-week-old male TG Control or PS2 N141I mice by intraperitoneal injections for four consecutive days. Three and a half hours after the final (fourth) injection, anesthetized mice were perfused with PBS and brains were quickly removed and snap-frozen.
RESULTS
PS2 protein expression in the PS2N141I transgenic mouse
Previous studies described PSEN2 N141I expressing transgenic models, though these mice were either designed in the context of expressing mutant AβPP or both alleles of the endogenous murine Psen2 [37, 38]. Our goal was to create a mouse that more closely resembles the genetic phenotype of patients with EOFAD in order to survey the intrinsic immune dysfunction due to an EOFAD associated allele. We created a novel transgenic mouse expressing a human PSEN2 N141I allele on a hemizygous, murine Psen2+/– background (Fig. 1A). These mice demonstrated no significant difference with regard to fecundity or viability by age 15 months in normal breeding conditions.
To evaluate PS2 protein expression in the PSEN2 N141I transgenic, we performed western blot analysis on wildtype (WT), TG control, PS2 N141I, and Psen2 KO [16] microglia from three independent cultures. We found total PS2 protein level to be similar between WT, TG Control, and PS2 N141 (Fig. 1B). To confirm the transgene expresses human PS2 N141I protein, we crossed the PSEN2 N141I transgenic to the Psen2 KO line and detected PS2 protein by western blot confirming that the transgene is expressing PS2 N141I (Fig. 1E). We had previously demonstrated that PS2 protein levels influence PS1 levels in a compensatory manner such that decreased PS2 expression led to increased PS1 [16]. Because PS1 shares some functional overlap with PS2, we assayed for PS1 protein levels in the PS2 N141I microglia and found expression of the mutant PS2 N141I does not induce significantly higher levels of PS1 in contrast to Psen2 KO (Fig. 1F). This suggested that functional consequences of PS2 variant expression in microglia is unlikely to be secondary to enhanced PS1 expression.
γ-secretase activity is impaired in PS2 N141I expressing microglia
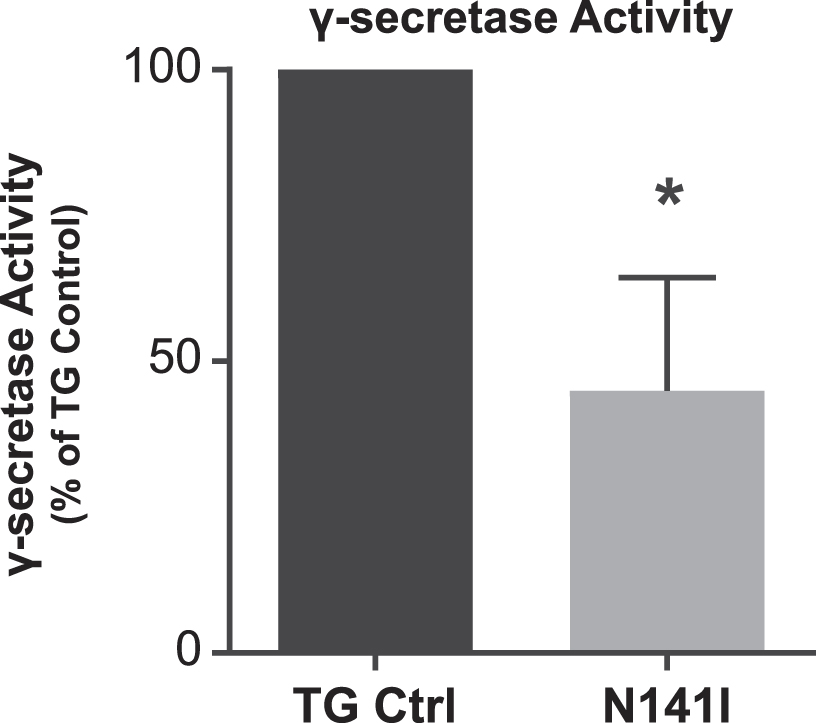
Presenilins regulate cellular function through enzymatic and non-enzymatic functions. To better understand the impact of EOFAD variants on PS2 enzymatic activity in microglia, we employed an in vitro luciferase based γ-secretase assay that measures production of the APP intracellular domain (AICD), the cleavage product of ɛ-site γ-secretase activity [39]. We have previously shown that PS2 is the predominant γ-secretase expressed by microglia and that the absence or decreased levels of PS2 results in decreased baseline γ-secretase activity [16]. We anticipated that PS2 N141I expressing microglia would demonstrate impaired γ-secretase activity despite having two wildtype Psen1 alleles and one wildtype Psen2 allele. Enzymatic activity from cultured neonatal microglia from PS2 N141I mice confirmed significantly decreased γ-secretase activity compared to transgene controls (Fig. 2) indicating that the PS2 variant impairs the biochemical activity of γ-secretase mediated AICD production previously demonstrated in Psen2 KO microglia [16].
Fig. 2
γ-secretase activity is decreased in PS2 N141I microglia. Transgenic control (TG Ctrl) and PS2 N141I (N141I) microglia were transduced with lentiviral γ-secretase assay constructs. Seventy-two hours post transduction, γ-secretase activity was measured by the induction of luciferase activity by an APP-Gal4 construct and normalized to TG Ctrl. PS2 N141I microglia show decreased γ-secretase activity compared TG Ctrl. Data are mean±SEM, n = 4 and represent experiments from independent cultures, *p≤0.05, t-test analysis.
Immune phenotype is altered in cultured PS2 N141I microglia
NFκB activation
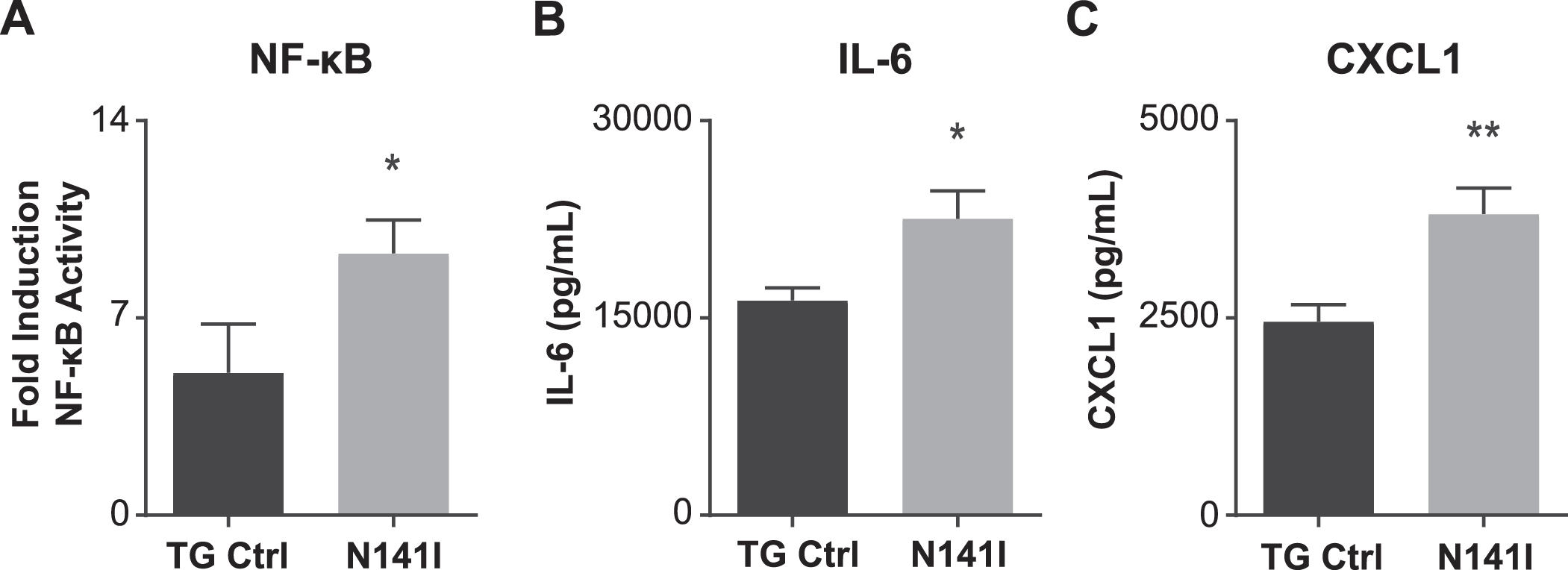
After determining that PS2 N141I expression impairs γ-secretase enzymatic function in microglia, we hypothesized that PS2 N141I influences the inflammatory microglial phenotype similar to Psen2 KO microglia [15]. We first assayed the activation of NFκB, a canonical innate immune effector molecule using a luciferase reporter assay [15]. After treatment with LPS for 4 h both PSEN2 N141I and TG Control neonatal microglia induce NFκB transcriptional activity. Similar to previous work demonstrating that microglia deficient in PS2 have exaggerated NFκB activity [15], microglia expressing PS2 N141I mount significantly greater NFκB activation relative to control (Fig. 3A). This observation is consistent with the hypothesis that the transcriptional activity of NFκB in PS2 FAD microglia is inherently dysregulated.
Fig. 3
NFκB transcriptional activity and inflammatory cytokine/chemokine secretion is enhanced in PS2 N141I microglia in response to LPS stimulation. A) Transgenic control (TG Ctrl) and PS2 N141I (N141I) microglia were transduced with lentiviral NFκB assay constructs. Forty-eight hours post transduction, microglia were exposed to 100 ng/mL LPS for 4 h and NFκB activity was measured by a luciferase reporter assay. PS2 N141I microglia show increased NFκB induction compared TG Ctrl. Data are mean±SEM, n = 4 and represent experiments from independent cultures, *p < 0.05, t-test analysis. Transgenic control (TG Ctrl) and PS2 N141I (N141I) primary microglia were stimulated with 100 ng/mL LPS for 24 h. B) IL-6 and C) KC (CXCL1) were significantly elevated in cell supernatants from PS2 N141I microglia relative to TG Ctrl as measured by multiplex cytokine assay. Data are mean±SEM, n = 4 and represent experiments from independent cultures, *p≤0.05, t-test analysis.
Cytokine release
To better understand how the PSEN2 N141I allele influences microglial immune-phenotype, a multi-plex cytokine analysis was performed on cell culture supernatant collected from TG control and PS2 N141I neonatal microglia with and without inflammatory stimulus. There was no measurable cytokine protein detectable in unstimulated cells. We observed that in response to LPS exposure, PS2 N141I microglia secrete increased levels of the inflammatory mediators IL-6 and CXCL1 (Fig. 3B, C), suggesting there is a phenotypic shift in the immune status of PS2 N141I microglia.
Phagocytosis
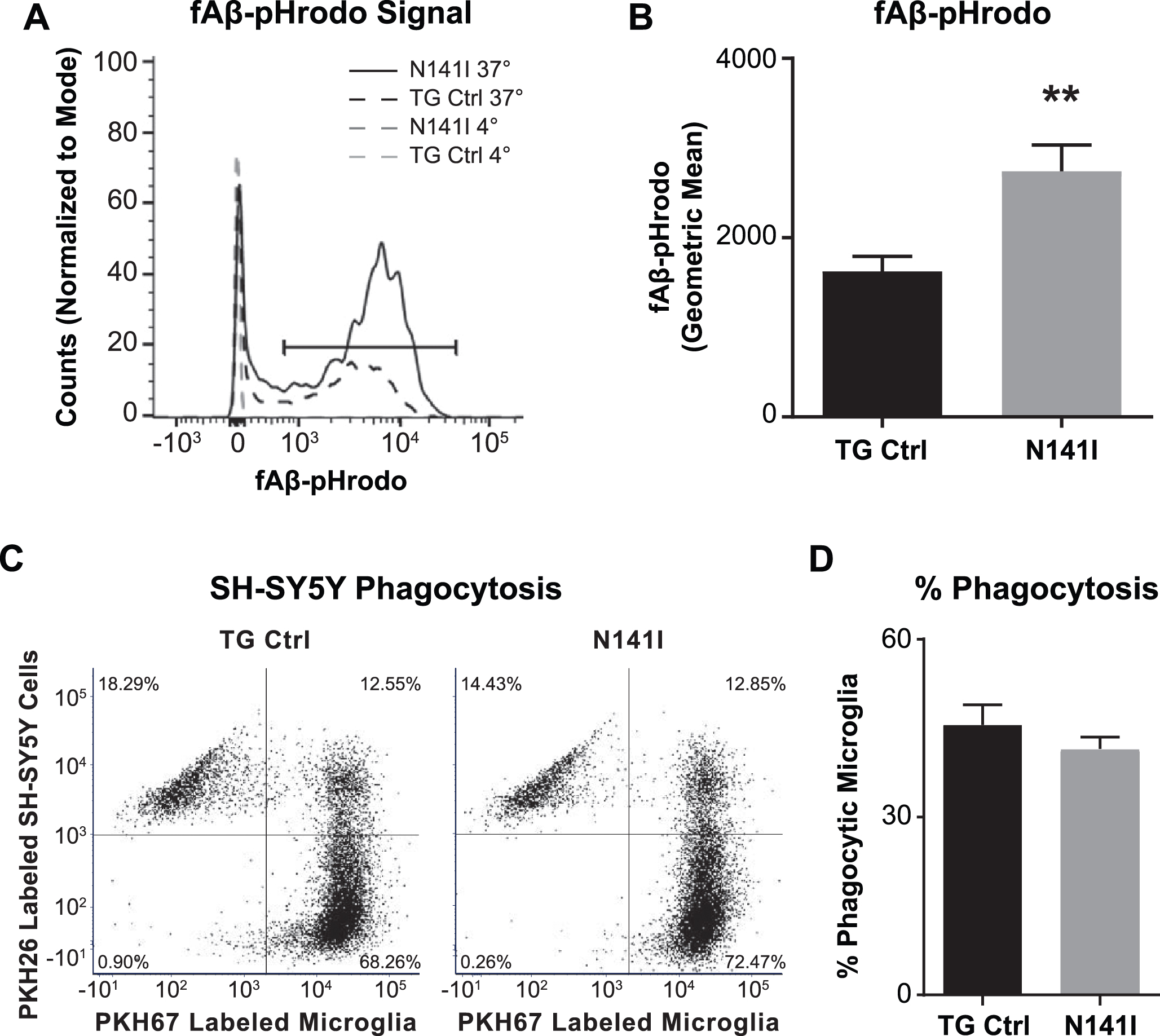
One of the many functions of microglia in the healthy and diseased CNS includes the clearing of debris through phagocytosis. In vivo, EOFAD patients display an aggressive deposition of Aβ plaques and previous studies demonstrate that microglia deficient in PS1 and PS2 have impaired Aβ phagocytosis in vitro [40]. This raised the possibility that PS2 N141I expression may similarly impair Aβ internalization by microglia thus contributing to neuropathology. Employing a standard Aβ fibrillation method [16], we assayed the capacity for PS2 N141I neonatal microglia to internalize Aβ. Contrary to our expectations, we found that expression of the PS2 N141I augmented the measure of internalized fluorescent Aβ following a 5 h incubation as compared to TG control indicating that the presence of the EOFAD variant enhances internalization of Aβ in vitro (Fig. 4A, B)
The regulation of microglia phagocytosis can be stimulus specific [41]. We queried if the observed enhanced internalization was common to additional substrates besides Aβ. Phagocytosis of chemically induced apoptotic neurons can be measured to assess the phagocytic phenotype of microglia in vitro [42]. We determined whether there is a similarly altered pattern of internalization of apoptotic neurons in PS2 N141I microglia. After incubating primary microglia from both genotypes with SH-SY5Y cells exposed to camptothecin to induce apoptosis, we found that while both microglia genotypes internalized labeled SH-SY5Y cell products, there was no difference in their relative capacity to internalize apoptotic cells at 5 h (Fig. 4C, D).
Fig. 4
PS2 N141I microglia show increased Aβ phagocytosis. Transgenic control (TG Ctrl) and PS2 N141I (N141I) microglia were exposed to 1 μM pHrodo-Aβ for 5 h and analyzed by flow cytometry. A) Phagocytosis was determined by a shift in Aβ-fluorophore cellular signal at 37°C versus signal at 4°C compared to media only controls for each genotype (as shown in representative histogram). B) PS2 N141I microglia demonstrate increased phagocytosis of Aβ-pHrodo relative to TG Ctrl measured by geometric mean. Data are mean±SEM, n = 3 and represent experiments from independent cultures, **p≤0.01, t-test analysis. C) Representative dot plot demonstrating that TG Ctrl and N141I microglia labeled with PKH67 internalize PKH26 labeled apoptotic SH5Y. D) Similar percentage of TG Ctrl and N141I microglia phagocytose apoptotic SH-SY5Y cells. Data are mean±SEM, n = 3 and represent experiments from independent cultures.
PS2 N141I expression alters microglia morphology and inflammatory gene expression in vivo
Microglia morphology
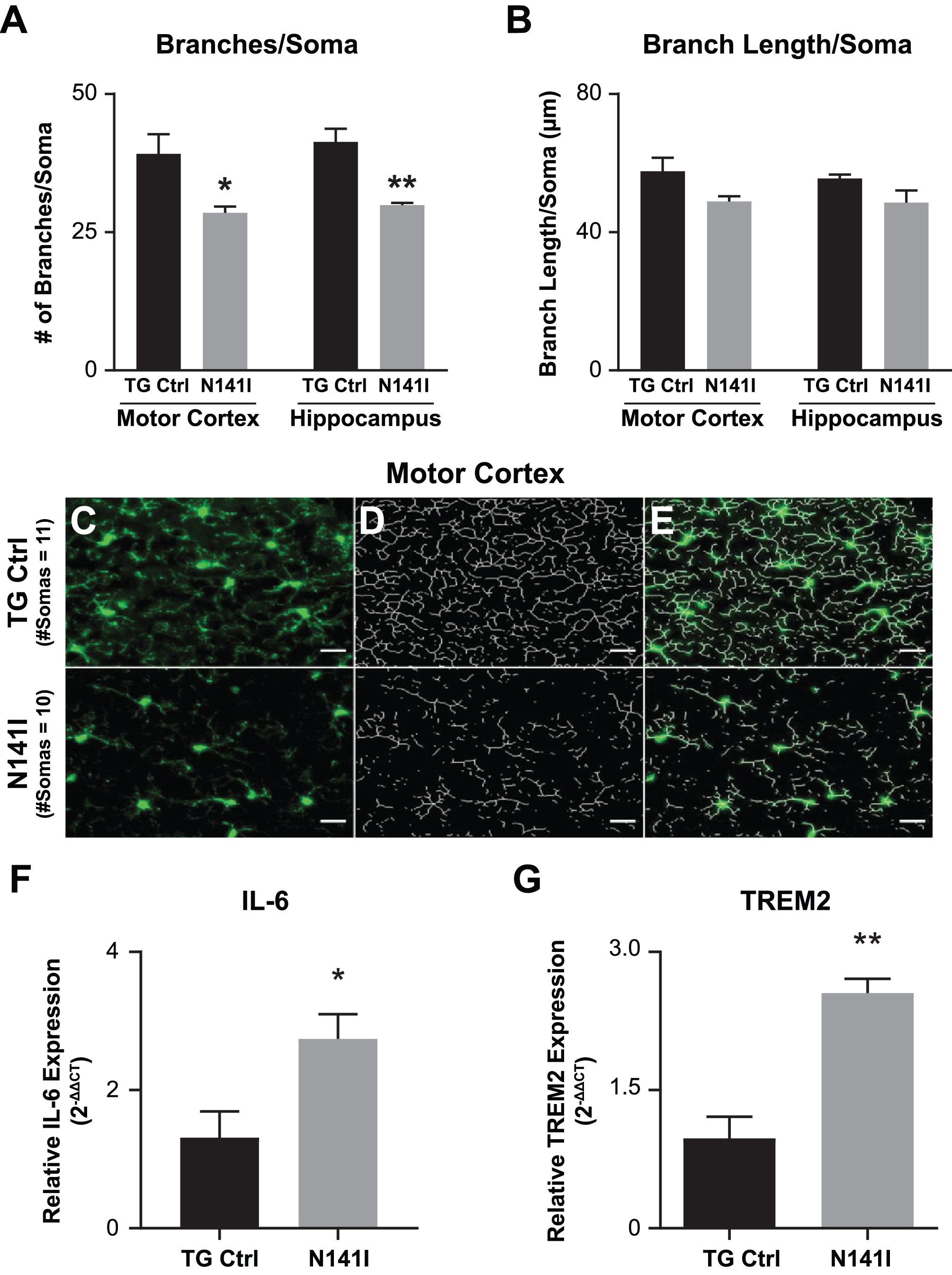
Microglia morphology is variable and correlated to state or function [43]. In homeostasis, microglia appear “ramified” with long complex branching processes. With activation microglia begin to assume a more amoeboid appearance in which branching complexity is decreased and processes are shorter [44]. To evaluate the impact of PS2 N141I expression on microglia in vivo without an inflammatory stimulus, we quantified the cell number, branch number, and average branch length of Iba1 labeled microglia in the motor cortex and dorsal hippocampus cortex from saline injected TG control and PS2 N141I mice (n = 3 each genotype). We found that there was no genotype difference in soma number in either motor or hippocampus sections (data not shown). However, we found in the saline control cohort that PS2 N141I was associated with an altered appearance of microglia which had significantly fewer branches (Fig. 5C) and no change in branch length (Fig. 5D). When combining quantification of branch number and branch length measured in both the PBS and the LPS injected mice (n = 6 LPS injected each genotype), we found significant differences between genotypes in both branch number and branch length suggesting that we may have been underpowered to detect differences in branch length from the saline injected cohort (data not shown). Decrease in branch number is a feature of the early stages of activation [45], therefore the results suggest that variant PS2 expression may prime microglia toward a more activated state even in the absence of a peripheral immune stimulus.
Fig. 5
Immune gene expression and microglial morphology are altered in brains of PS2 N141I mice. Male transgenic control (TG Ctrl) or PS2 N141I (N141I) mice were injected intraperitoneally (IP) with saline daily for four days. Whole brains were collected 3.5 h following the fourth injection as described in Materials and Methods. A) Soma number, branch number, and branch length quantification showed significant decrease in branch number/soma in N141I microglia from both motor cortex and hippocampal cortex (n = 3 mice each genotype, *p≤0.05, **p≤0.01, t-test analysis. B) Branch length was not statistically fewer in either region in PS2 N141I mice, n = 3 mice each genotype C) Iba-1 (green) immunostaining of TG Ctrl (upper panel) and PS2 N141I (lower panel) microglia (scale bar = 20 μm). D) Skeletonized images of microglia branches with Image J. E) Composite Iba1 and skeletonized images. RNA was isolated from brain homogenate and mRNA expression was determined by RT-qPCR analysis. mRNA transcript of F) IL-6 and G) TREM2 is elevated in the brains of PS2 N141I mice injected IP with saline relative to TG Ctrl animals (n = 4 for each genotype) *p≤0.05, **p≤0.01, t-test analysis.
In vivo gene expression
To test whether FAD PS2 influences neural immune homeostasis in vivo, we measured expression of inflammatory mediators after saline or LPS intraperitoneal injection. We found that in saline injected mice, IL-6 (Fig. 5A) and TREM2 (Fig. 5B), were elevated in whole brain homogenate of PS2 N141I animals suggesting a dysregulated immune environment at baseline or in the absence of inflammatory stimulation.
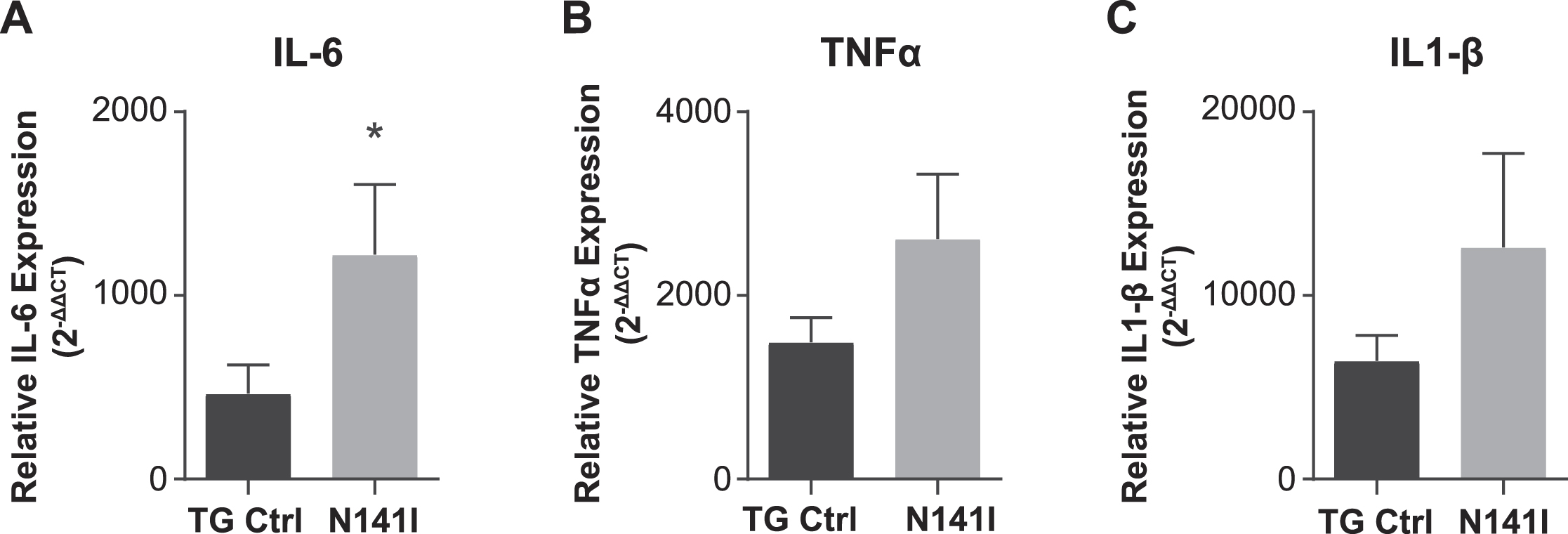
After LPS stimulation, both PS2 N141I and TG control animals mounted an immune response as reflected in increased inflammatory cytokine gene expression in whole brain homogenate compared to saline treated controls. The response in PS2 N141I animals was exaggerated compared to TG control mice, as suggested by significantly increased IL-6, demonstrating that the presence of PS2 N141I not only impacts the “resting” or baseline immune phenotype, but also the response to immune stimulus (Fig. 6).
Fig. 6
Inflammatory response to intraperitoneal LPS injection is exaggerated in PS2 N141I animals. Male transgenic control (TG Ctrl) or PS2 N141I (N141I) mice were injected intraperitoneally (IP) with ultra-pure LPS daily for four days. Whole brains were collected 3.5 h following the fourth injection as described in Materials and Methods. mRNA transcript of IL-6 is significantly elevated and TNFα and IL-1β are elevated in the brains of PS2 N141I mice measured after serial injection with 1 mg/kg LPS relative to TG Ctrl animals. Data are the mean±SEM and are representative of 4 (saline) or 10–14 (LPS injected) mice per group, *p≤0.05, t-test analysis.
DISCUSSION
Presenilins and AβPP were implicated in AD pathogenesis more than 25 years ago and significant progress has been made regarding the clinical, pathological, and molecular features of sporadic AD and EOFAD. The basic biochemical association between presenilins, AβPP, and neuropathologic amyloid has driven most therapeutic efforts to date. However, we remain in a period of open discovery regarding drug targeting for both familial and sporadic AD. While EOFAD variants lead to altered Aβ40/42 species, it is not fully established that the deposition/production of neuronal Aβ is sufficient to lead to the aggressive phenotype in EOFAD. The more “holistic” view, as suggested by De Strooper and Karran reconciles the initiation stages of AD related to proteopathy, followed by a complex multicellular stage mediated by neuronal and non-neuronal cells leading to disease progression and eventually clinically evident disease [3]. Our work explores the mechanisms by which an EOFAD variant could contribute to that cellular phase of AD during which biochemical abnormality converts to pathogenic glial responses.
Mouse models in other familial neurodegenerative disease have demonstrated that disease gene variants can exert pathogenic effects in multiple cell types resulting in both neuronal and glial dysfunction [10–12]. We hypothesized a similar pattern of multiple cell type dysfunction induced by disease associated EOFAD variants could be contributing to a deleterious CNS immune environment that further drives neurodegeneration. Our data shows that the PS2 N141I variant alters microglial phenotype leading to exaggerated inflammatory responses. This suggests that PSEN2 variants may contribute to AD through aggravation of injurious inflammatory pathways. Since these mice are not crossed to AβPP mutant or overexpressing mice to induce Aβ plaque, our findings underscore that the observed increased inflammatory phenotype is not a consequence of amyloid plaque.
Presenilins are essential for myriad cellular functions [46] and their roles as a γ-secretase have been implicated in the regulation of inflammation [18, 19]. We demonstrate here that the PSEN2 N141I variant results in decreased γ-secretase activity in microglia despite the presence of one wildtype Psen2 and two wild type Psen1 alleles. Given that the γ-secretase mediated endoproteolytic cleavage of several substrates including Notch1 and TREM2 regulate innate immune cell signaling [19, 47], we propose that the PS2 N141I mediated disruption of normal enzymatic activity leads to a loss of microglial homeostasis in EOFAD patients. Consequently, in addition to aberrant Aβ production, PSEN2 disease associated variants would impair the normal innate immune response to protein aggregates and promote an exaggerated, injurious inflammatory response. Our studies of PSEN2 N141I function are in a transgenic mouse model; however, we and others have demonstrated impaired Notch cleavage associated with PSEN variants in samples obtained from EOFAD cases suggesting that disrupted endoproteolytic activity is occurring in the patient setting as well [30, 48]. As PS2 also mediates cellular functions beyond its enzymatic role, future studies will explore how PS2 may function to direct innate immune cell activity though γ-secretase dependent or independent paths.
Absence of PS2 in microglia is associated with increased inflammatory markers in vivo and exaggerated inflammatory responses characterized by enhanced cytokine release, NFkB activation, and depressed anti-inflammatory microRNA levels in cultured microglia [15–17, 49]. Our study asked whether expression of one wildtype and one EOFAD variant PS2 allele similarly modulates the characteristics of microglia. We would predict that if the variant impairs normal PS2 activity in microglial function (i.e., through a hypomorphic allele or through dominant negative mechanisms [50]) microglia expressing PS2 N141I would show an exaggerated inflammatory state in our model.
Consistent with a disrupted homeostatic or hyper-reactive state, PS2 N141I induced aberrant cytokine expression. The enhanced expression of TREM2 and IL-6 in the brains of saline injected PS2 N141I mice is further evidence that the PSEN2 variant impacts the CNS immune environment even in the presence of a wildtype PS2 allele. IL-6 signaling is implicated in AD not only by biomarker assays, but also through studies demonstrating associations between genetic variants in IL-6 and the IL-6 receptor with AD [51, 52]. The role of TREM2 in AD pathogenesis is complex and an active focus of study. Recent studies have reported that TREM2 is an important modulator of innate immune physiology and elevated TREM2 is associated with a microglia disease state in AD murine models [53]. In this context, the finding that human PS2 N141I expressing mice show increased TREM2 supports the notion that PS2 N141I alters immune cell phenotype. Of note, the measures of cytokine expression in adult brain are not microglia specific and may represent the responses of multiple neural cell types.
We were able to assess microglia specific activation through evaluation of morphology which can reveal microglial phenotypes or states [9]. Homeostatic, surveilling microglia extend long ramified processes and as they proceed to activated, inflammatory states, branches of processes retract and decrease in number [36, 44, 54]. Saline injected PS2 N141I mice microglia displayed fewer branches compared to TG controls while branch length was not significantly reduced. The morphological changes observed in PS2 N141I microglia despite the absence of inflammatory stimulation (LPS injection) suggests a baseline skewing of PS2 N141I microglia toward activation. We did not find that there was a regional difference between motor cortex and hippocampus with regard to alteration of branch morphology by PSEN2 N141I mice indicating that this may be a brain-wide phenomenon.
The enhanced NFkB activity observed in PS2 N141 microglia co-occurred with increased phagocytosis of Aβ. What cannot be discerned in our in vitro studies is if the shift in inflammatory cytokine release or enhanced Aβ phagocytosis reflects a proximal phenotype change in all microglia or whether the EOFAD PS2 N141 variant influences the likelihood of a microglial cell within a population to adopt one phenotypic feature or another at baseline or after stimulus. There is considerable heterogeneity of microglia in vivo and subtyping of microglia populations manipulated in vitro may help to understand further the dynamics of microglial change at a cellular and population level [53, 55]. Also heterogeneous are multiple microglial phagocytosis pathways which are dependent on specific stimuli and environment [9]. In line with this, we found that PS2 variant function on internalization does not appear to be a generalized phenomenon. In contrast to augmented Aβ phagocytosis, we did not measure a significant difference of apoptotic cell internalization suggesting that PS2 may preferentially influence specific phagocytosis pathways. How the alteration of phagocytosis mechanics in the presence of PS2 N141I manifests in vivo and in a multicellular environment remains to be determined. Recent work has suggested that microglia internalization of Aβ itself may contribute to Aβ aggregation and is proposed to be an initial step in plaque formation [56]. In that context, we could speculate that PS2 N141I may potentially lead to increased pathology due to enhanced uptake of Aβ.
A limitation to the interpretation of our study is that the transgenic mouse is expressing the PS2 N141I constitutively and not under direct regulation of the endogenous promoter. A knock-in model to create a true heterozygous state would more accurately recapitulate the patient genotype. We also assessed immune gene expression relatively early in the adult life of these mice and therefore did not incorporate the impact of aging on the interaction of variant PS2 with immune function.
Conclusion
The pathophysiology of AD is complex and the biochemical connection between PSs, AβPP, and Aβ deposition support an important role for Aβ in AD. However, it is likely that factors which initiate neurodegeneration and progression are the consequence of dysfunction of multiple cell types. Our novel findings here reveal additional factors and cell types by which PSEN2 associated variants also influence the immune state. While the relationship between PSEN variants and Aβ is likely a relevant factor in EOFAD pathogenesis, our studies would suggest that EOFAD individuals may also benefit from immunomodulatory drugs relevant to the inflammatory biology driven by their disease variant. Therapeutic approaches targeting multiple pathways may be the future of AD therapy as immune targeted therapeutics develop [57, 58]. Understanding the innate immune dysfunction inherent in EOFAD and sporadic AD will be valuable to identify appropriate targets for future drug development.
ACKNOWLEDGMENTS
This work was supported by NIH R01NS072395 (S.J.) and 1RF1AG051437 (S.J., G.A.G.).
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/20-0492r1).
REFERENCES
[1] | Takami M , Nagashima Y , Sano Y , Ishihara S , Morishima-Kawashima M , Funamoto S , Ihara Y ((2009) ) gamma-Secretase: Successive tripeptide and tetrapeptide release from the transmembrane domain of beta-carboxyl terminal fragment. J Neurosci 29: , 13042–13052. |
[2] | Selkoe DJ , Hardy J ((2016) ) The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8: , 595–608. |
[3] | De Strooper B , Karran E ((2016) ) The cellular phase of Alzheimer’s disease. Cell 164: , 603–615. |
[4] | Herrup K ((2010) ) Reimagining Alzheimer’s disease–an age-based hypothesis. J Neurosci 30: , 16755–16762. |
[5] | Heneka MT , Kummer MP , Latz E ((2014) ) Innate immune activation in neurodegenerative disease. Nat Rev Immunol 14: , 463–477. |
[6] | Ransohoff RM , El Khoury J ((2015) ) Microglia in health and disease. Cold Spring Harb Perspect Biol 8: , a020560. |
[7] | Condello C , Yuan P , Schain A , Grutzendler J ((2015) ) Microglia constitute a barrier that prevents neurotoxic protofibrillar Abeta42 hotspots around plaques. Nat Commun 6: , 6176. |
[8] | Newcombe EA , Camats-Perna J , Silva ML , Valmas N , Huat TJ , Medeiros R ((2018) ) Inflammation: The link between comorbidities, genetics, and Alzheimer’s disease. J Neuroinflammation 15: , 276. |
[9] | Kettenmann H , Hanisch UK , Noda M , Verkhratsky A ((2011) ) Physiology of microglia. Physiol Rev 91: , 461–553. |
[10] | Boillee S , Yamanaka K , Lobsiger CS , Copeland NG , Jenkins NA , Kassiotis G , Kollias G , Cleveland DW ((2006) ) Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312: , 1389–1392. |
[11] | Ditsworth D , Maldonado M , McAlonis-Downes M , Sun S , Seelman A , Drenner K , Arnold E , Ling SC , Pizzo D , Ravits J , Cleveland DW , Da Cruz S ((2017) ) Mutant TDP-43 within motor neurons drives disease onset but not progression in amyotrophic lateral sclerosis. Acta Neuropathol 133: , 907–922. |
[12] | Louvi A , Chen L , Two AM , Zhang H , Min W , Gunel M ((2011) ) Loss of cerebral cavernous malformation 3 (Ccm3) in neuroglia leads to CCM and vascular pathology. Proc Natl Acad Sci U S A 108: , 3737–3742. |
[13] | Salter MW , Stevens B ((2017) ) Microglia emerge as central players in brain disease. Nat Med 23: , 1018–1027. |
[14] | Davalos D , Grutzendler J , Yang G , Kim JV , Zuo Y , Jung S , Littman DR , Dustin ML , Gan WB ((2005) ) ATP mediates rapid microglial response to local brain injury. Nat Neurosci 8: , 752–758. |
[15] | Jayadev S , Case A , Alajajian B , Eastman AJ , Moller T , Garden GA ((2013) ) Presenilin 2 influences miR146 level and activity in microglia. J Neurochem 127: , 592–599. |
[16] | Jayadev S , Case A , Eastman AJ , Nguyen H , Pollak J , Wiley JC , Moller T , Morrison RS , Garden GA ((2010) ) Presenilin 2 is the predominant gamma-secretase in microglia and modulates cytokine release. PLoS One 5: , e15743. |
[17] | Qin J , Zhang X , Wang Z , Li J , Zhang Z , Gao L , Ren H , Qian M , Du B ((2017) ) Presenilin 2 deficiency facilitates Abeta-induced neuroinflammation and injury by upregulating P2X7 expression. Sci China Life Sci 60: , 189–201. |
[18] | Saura CA ((2010) ) Presenilin/gamma-secretase and inflammation. Front Aging Neurosci 2: , 16. |
[19] | Walter J , Kemmerling N , Wunderlich P , Glebov K ((2017) ) gamma-secretase in microglia –implications for neurodegeneration and neuroinflammation. J Neurochem 143: , 445–454. |
[20] | Otto GP , Sharma D , Williams RS ((2016) ) Non-catalytic roles of presenilin throughout evolution. J Alzheimers Dis 52: , 1177–1187. |
[21] | Walter J ((2015) ) Twenty years of presenilins–important proteins in health and disease. Mol Med 21 Suppl 1: , S41–48. |
[22] | Haapasalo A , Kovacs DM ((2011) ) The many substrates of presenilin/gamma-secretase. J Alzheimers Dis 25: , 3–28. |
[23] | Chhibber-Goel J , Coleman-Vaughan C , Agrawal V , Sawhney N , Hickey E , Powell JC , McCarthy JV ((2016) ) gamma-secretase activity is required for regulated intramembrane proteolysis of tumor necrosis factor (TNF) receptor 1 and TNF-mediated pro-apoptotic signaling. J Biol Chem 291: , 5971–5985. |
[24] | Fan Q , Gayen M , Singh N , Gao F , He W , Hu X , Tsai LH , Yan R ((2019) ) The intracellular domain of CX3CL1 regulates adult neurogenesis and Alzheimer’s amyloid pathology. J Exp Med 216: , 1891–1903. |
[25] | Elzinga BM , Twomey C , Powell JC , Harte F , McCarthy JV ((2009) ) Interleukin-1 receptor type 1 is a substrate for gamma-secretase-dependent regulated intramembrane proteolysis. J Biol Chem 284: , 1394–1409. |
[26] | Chavez-Gutierrez L , Bammens L , Benilova I , Vandersteen A , Benurwar M , Borgers M , Lismont S , Zhou L , Van Cleynenbreugel S , Esselmann H , Wiltfang J , Serneels L , Karran E , Gijsen H , Schymkowitz J , Rousseau F , Broersen K , De Strooper B ((2012) ) The mechanism of gamma-Secretase dysfunction in familial Alzheimer disease. EMBO J 31: , 2261–2274. |
[27] | Shen J , Kelleher RJ 3rd ((2007) ) The presenilin hypothesis of Alzheimer’s disease: Evidence for a loss-of-function pathogenic mechanism. Proc Natl Acad Sci U S A 104: , 403–409. |
[28] | Jayne T , Newman M , Verdile G , Sutherland G , Munch G , Musgrave I , Moussavi Nik SH , Lardelli M ((2016) ) Evidence for and against a pathogenic role of reduced gamma-secretase activity in familial Alzheimer’s disease. J Alzheimers Dis 52: , 781–799. |
[29] | Ahmadi S , Wade-Martins R ((2014) ) Familial Alzheimer’s disease coding mutations reduce Presenilin-1 expression in a novel genomic locus reporter model. Neurobiol Aging 35: , 443 e445–443 e416. |
[30] | Braggin JE , Bucks SA , Course MM , Smith CL , Sopher B , Osnis L , Shuey KD , Domoto-Reilly K , Caso C , Kinoshita C , Scherpelz KP , Cross C , Grabowski T , Nik SHM , Newman M , Garden GA , Leverenz JB , Tsuang D , Latimer C , Gonzalez-Cuyar LF , Keene CD , Morrison RS , Rhoads K , Wijsman EM , Dorschner MO , Lardelli M , Young JE , Valdmanis PN , Bird TD , Jayadev S ((2019) ) Alternative splicing in a presenilin 2 variant associated with Alzheimer disease. Ann Clin Transl Neurol 6: , 762–777. |
[31] | Jayadev S , Leverenz JB , Steinbart E , Stahl J , Klunk W , Yu CE , Bird TD ((2010) ) Alzheimer’s disease phenotypes and genotypes associated with mutations in presenilin 2. Brain 133: , 1143–1154. |
[32] | Tsai SY , Rodriguez AA , Dastidar SG , Del Greco E , Carr KL , Sitzmann JM , Academia EC , Viray CM , Martinez LL , Kaplowitz BS , Ashe TD , La Spada AR , Kennedy BK ((2016) ) Increased 4E-BP1 expression protects against diet-induced obesity and insulin resistance in male mice. Cell Rep 16: , 1903–1914. |
[33] | Drago J , Padungchaichot P , Wong JY , Lawrence AJ , McManus JF , Sumarsono SH , Natoli AL , Lakso M , Wreford N , Westphal H , Kola I , Finkelstein DI ((1998) ) Targeted expression of a toxin gene to D1 dopamine receptor neurons by cre-mediated site-specific recombination. J Neurosci 18: , 9845–9857. |
[34] | Amos PJ , Fung S , Case A , Kifelew J , Osnis L , Smith CL , Green K , Naydenov A , Aloi M , Hubbard JJ , Ramakrishnan A , Garden GA , Jayadev S ((2017) ) Modulation of hematopoietic lineage specification impacts TREM2 expression in microglia-like cells derived from human stem cells. ASN Neuro 9: , 1759091417716610. |
[35] | Johnson MD , Kinoshita Y , Xiang H , Ghatan S , Morrison RS ((1999) ) Contribution of p53-dependent caspase activation to neuronal cell death declines with neuronal maturation. J Neurosci 19: , 2996–3006. |
[36] | Young K , Morrison H ((2018) ) Quantifying microglia morphology from photomicrographs of immunohistochemistry prepared tissue using ImageJ. J Vis Exp. doi: 10.3791/57648. |
[37] | Richards JG , Higgins GA , Ouagazzal AM , Ozmen L , Kew JN , Bohrmann B , Malherbe P , Brockhaus M , Loetscher H , Czech C , Huber G , Bluethmann H , Jacobsen H , Kemp JA ((2003) ) PS2APP transgenic mice, coexpressing hPS2mut and hAPPswe, show age-related cognitive deficits associated with discrete brain amyloid deposition and inflammation. J Neurosci 23: , 8989–9003. |
[38] | Oyama F , Sawamura N , Kobayashi K , Morishima-Kawashima M , Kuramochi T , Ito M , Tomita T , Maruyama K , Saido TC , Iwatsubo T , Capell A , Walter J , Grunberg J , Ueyama Y , Haass C , Ihara Y ((1998) ) Mutant presenilin 2 transgenic mouse: Effect on an age-dependent increase of amyloid beta-protein 42 in the brain. J Neurochem 71: , 313–322. |
[39] | Wiley JC , Hudson M , Kanning KC , Schecterson LC , Bothwell M ((2005) ) Familial Alzheimer’s disease mutations inhibit gamma-secretase-mediated liberation of beta-amyloid precursor protein carboxy-terminal fragment. J Neurochem 94: , 1189–1201. |
[40] | Farfara D , Trudler D , Segev-Amzaleg N , Galron R , Stein R , Frenkel D ((2011) ) gamma-Secretase component presenilin is important for microglia beta-amyloid clearance. Ann Neurol 69: , 170–180. |
[41] | Fu R , Shen Q , Xu P , Luo JJ , Tang Y ((2014) ) Phagocytosis of microglia in the central nervous system diseases. Mol Neurobiol 49: , 1422–1434. |
[42] | Witting A , Muller P , Herrmann A , Kettenmann H , Nolte C ((2000) ) Phagocytic clearance of apoptotic neurons by Microglia/Brain macrophages: Involvement of lectin-, integrin-, and phosphatidylserine-mediated recognition. J Neurochem 75: , 1060–1070. |
[43] | Kreutzberg GW ((1996) ) Microglia: A sensor for pathological events in the CNS. Trends Neurosci 19: , 312–318. |
[44] | Torres-Platas SG , Comeau S , Rachalski A , Bo GD , Cruceanu C , Turecki G , Giros B , Mechawar N ((2014) ) Morphometric characterization of microglial phenotypes in human cerebral cortex. J Neuroinflammation 11: , 12. |
[45] | Stence N , Waite M , Dailey ME ((2001) ) Dynamics of microglial activation: A confocal time-lapse analysis in hippocampal slices. Glia 33: , 256–266. |
[46] | Wolfe MS ((2020) ) Unraveling the complexity of gamma-secretase. Semin Cell Dev Biol–10.1016/j.semcdb.2020.01.005. |
[47] | Glebov K , Wunderlich P , Karaca I , Walter J ((2016) ) Functional involvement of gamma-secretase in signaling of the triggering receptor expressed on myeloid cells-2 (TREM2). J Neuroinflammation 13: , 17. |
[48] | Sannerud R , Esselens C , Ejsmont P , Mattera R , Rochin L , Tharkeshwar AK , De Baets G , De Wever V , Habets R , Baert V , Vermeire W , Michiels C , Groot AJ , Wouters R , Dillen K , Vints K , Baatsen P , Munck S , Derua R , Waelkens E , Basi GS , Mercken M , Vooijs M , Bollen M , Schymkowitz J , Rousseau F , Bonifacino JS , Van Niel G , De Strooper B , Annaert W ((2016) ) Restricted location of PSEN2/gamma-secretase determines substrate specificity and generates an intracellular Abeta pool. Cell 166: , 193–208. |
[49] | Beglopoulos V , Sun X , Saura CA , Lemere CA , Kim RD , Shen J ((2004) ) Reduced beta-amyloid production and increased inflammatory responses in presenilin conditional knock-out mice. J Biol Chem 279: , 46907–46914. |
[50] | Zhou R , Yang G , Shi Y ((2017) ) Dominant negative effect of the loss-of-function gamma-secretase mutants on the wild-type enzyme through heterooligomerization. Proc Natl Acad Sci U S A 114: , 12731–12736. |
[51] | Haddick PC , Larson JL , Rathore N , Bhangale TR , Phung QT , Srinivasan K , Hansen DV , Lill JR ; Alzheimer’s Disease Genetic Consortium (ADGC), Alzheimer’s Disease Neuroimaging Initiative (ADNI), Pericak-Vance MA , Haines J , Farrer LA , Kauwe JS , Schellenberg GD , Cruchaga C , Goate AM , Behrens TW , Watts RJ , Graham RR , Kaminker JS , van der Brug M ((2017) ) A common variant of IL-6R is associated with elevated IL-6 pathway activity in Alzheimer’s disease brains. J Alzheimers Dis 56: , 1037–1054. |
[52] | Papassotiropoulos A , Hock C , Nitsch RM ((2001) ) Genetics of interleukin 6: Implications for Alzheimer’s disease. Neurobiol Aging 22: , 863–871. |
[53] | Keren-Shaul H , Spinrad A , Weiner A , Matcovitch-Natan O , Dvir-Szternfeld R , Ulland TK , David E , Baruch K , Lara-Astaiso D , Toth B , Itzkovitz S , Colonna M , Schwartz M , Amit I ((2017) ) A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169: , 1276–1290 e1217. |
[54] | Karperien A , Ahammer H , Jelinek HF ((2013) ) Quantitating the subtleties of microglial morphology with fractal analysis. Front Cell Neurosci 7: , 3. |
[55] | Mathys H , Davila-Velderrain J , Peng Z , Gao F , Mohammadi S , Young JZ , Menon M , He L , Abdurrob F , Jiang X , Martorell AJ , Ransohoff RM , Hafler BP , Bennett DA , Kellis M , Tsai LH ((2019) ) Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570: , 332–337. |
[56] | Spangenberg E , Severson PL , Hohsfield LA , Crapser J , Zhang J , Burton EA , Zhang Y , Spevak W , Lin J , Phan NY , Habets G , Rymar A , Tsang G , Walters J , Nespi M , Singh P , Broome S , Ibrahim P , Zhang C , Bollag G , West BL , Green KN ((2019) ) Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat Commun 10: , 3758. |
[57] | Fani Maleki A , Rivest S ((2019) ) Innate immune cells: Monocytes, monocyte-derived macrophages and microglia as therapeutic targets for Alzheimer’s disease and multiple sclerosis. Front Cell Neurosci 13: , 355. |
[58] | Wes PD , Sayed FA , Bard F , Gan L ((2016) ) Targeting microglia for the treatment of Alzheimer’s disease. Glia 64: , 1710–1732. |


