
Alzheimer’s Disease-Like Neurodegeneration in Porphyromonas gingivalis Infected Neurons with Persistent Expression of Active Gingipains
Abstract
Background:
Porphyromonas gingivalis (P. gingivalis) and its gingipain virulence factors have been identified as pathogenic effectors in Alzheimer’s disease (AD). In a recent study we demonstrated the presence of gingipains in over 90% of postmortem AD brains, with gingipains localizing to the cytoplasm of neurons. However, infection of neurons by P. gingivalis has not been previously reported.
Objective:
To demonstrate intraneuronal P. gingivalis and gingipain expression in vitro after infecting neurons derived from human inducible pluripotent stem cells (iPSC) with P. gingivalis for 24, 48, and 72 h.
Methods:
Infection was characterized by transmission electron microscopy, confocal microscopy, and bacterial colony forming unit assays. Gingipain expression was monitored by immunofluorescence and RT-qPCR, and protease activity monitored with activity-based probes. Neurodegenerative endpoints were assessed by immunofluorescence, western blot, and ELISA.
Results:
Neurons survived the initial infection and showed time dependent, infection induced cell death. P. gingivalis was found free in the cytoplasm or in lysosomes. Infected neurons displayed an accumulation of autophagic vacuoles and multivesicular bodies. Tau protein was strongly degraded, and phosphorylation increased at T231. Over time, the density of presynaptic boutons was decreased.
Conclusion:
P. gingivalis can invade and persist in mature neurons. Infected neurons display signs of AD-like neuropathology including the accumulation of autophagic vacuoles and multivesicular bodies, cytoskeleton disruption, an increase in phospho-tau/tau ratio, and synapse loss. Infection of iPSC-derived mature neurons by P. gingivalis provides a novel model system to study the cellular mechanisms leading to AD and to investigate the potential of new therapeutic approaches.
INTRODUCTION
Mounting evidence has identified Porphyromonas gingivalis (P. gingivalis) as a driving factor in the progression of Alzheimer’s disease (AD) [1, 2]. P. gingivalis specific cell free DNA can be detected in the cerebrospinal fluid and its protease virulence factors, arginine-gingipain (Rgp) and lysine-gingipain (Kgp), are present in the brains of over 90% of AD patients and correlate with tau and ubiquitin pathology [1]. P. gingivalis has been identified as a keystone pathogen for periodontitis [3, 4], which has been shown to be a risk factor for AD and a more rapid cognitive decline [5–10]. After oral infection in mice, P. gingivalis specific DNA and gingipains can be detected in the brain [1, 2, 11]. In one study using wild type mice, the major hallmarks of AD pathology, i.e., amyloid plaques, inflammation, microglial activation, hyperphosphorylated tau, and neuron loss, were reported after 22 weeks of infection [2]. To date, the mechanism of P. gingivalis-induced AD has not been elucidated, and it is imperative to develop a relevant in vitro model to advance our understanding of the underlying processes and to support the development of new therapies.
In our previous work, we demonstrated the susceptibility of tau protein to gingipain fragmentation [1], underlining the importance to study P. gingivalis-induced neurodegeneration in the context of functional tau protein. Tau is expressed in mature neurons only, and specific mechanisms that involve tau phosphorylation and aggregation have often been studied in transformed cell lines transfected with full-length tau protein [12]. Here, we use human inducible pluripotent stem cells (iPSC)-derived neuron cultures that express tau and display extensive neurite networks with functional synapses that allow the investigation of neuron-specific degenerative endpoints, such as hyperphosphorylation of tau or synapse loss [13].
In vitro cell infection studies with P. gingivalis have been conducted in a variety of cell types: gingival epithelial cells and fibroblasts, oral keratinocytes, endothelial cells from coronary arteries or umbilical cords, smooth muscle cells, hepatic cells, and dendritic cells [14–24]. The differences in attachment, invasion, persistence, and survival of P. gingivalis in the different culture systems are significant and highlight the need to better understand specific P. gingivalis-host interactions in highly specialized cells such as neurons.
P. gingivalis is an anaerobic, asaccharolytic, Gram-negative bacterium, and its survival in tissue depends heavily on its ability to invade and persist in host cells. The gingipains, Kgp and Rgp, are cysteine proteases attached on the bacterial surface or secreted into the environment. The gingipains are the major virulence factors that enable evasion of the cellular defenses from the host cell [25, 26]. For example, in coronary endothelial cells, gingipains have been shown to prevent the fusion of autophagosomes containing P. gingivalis with lysosomes, enabling a persistent residence of P. gingivalis in autophagosomes [24].
In this study, we describe an in vitro culture system to study P. gingivalis-host interactions of mature neurons and provide an initial characterization of AD-related neurodegeneration. We found that P. gingivalis can invade and survive in neurons and produce intraneuronal gingipains that are proteolytically active. After infection, 25% of neurons are lost in a time dependent manner. In surviving cells, full length tau was reduced with an increase in phosphorylation ratio over time, and this finding was associated with loss of neuronal synapses. At the ultrastructural level, intraneuronal P. gingivalis appeared free in cytoplasm or resided in lysosome-like structures, and infected neurons displayed accumulation of autophagic vacuoles and multivesicular bodies, a hallmark of dystrophic neurites in AD brains. Our results reveal that P. gingivalis can invade and survive inside neurons which leads to neuronal damage associated with AD.
MATERIALS AND METHODS
Aim and design
The aim of this study was to demonstrate that P. gingivalis can invade and persist in mature neurons. The result is an in vitro model system to study the neurodegenerative effects of P. gingivalis virulence on neurons. The study was designed to define the experimental parameters of neuron cultures and infection, to demonstrate viability of host and pathogen, and to provide an initial characterization of the time dependent effects on host and pathogen.
NPC-derived neuronal cultures
Two neural precursor cell lines (NPCs XCL1 [#70901], XCL4[#70902]) were purchased from StemCell Technology, Inc. (Vancouver, Canada) and differentiated into neurons according to the manufacturer’s protocol (catalog numbers 08500 and 08510). Briefly, NPCs were plated onto Poly-l-Ornithine (PLO) and Laminin (MilliporeSigma, Burlington, MA) coated plates and cultured in STEMdiff™ Neuron Differentiation Media for 2 to 4 passages. Neuron maturation was achieved by changing from Neuron Differentiation to STEMdiff™ Neuron Maturation Media or Brainphys media (BrainPhys™ hPSC Neuron Kit, StemCell Technology) for 3 to 4 weeks. Neuronal maturation was confirmed in each culture by immunohistochemical staining of neuronal markers such as Tuj1 and Map2a. Cells were maintained at 5% oxygen and 5% carbon dioxide, and media changes were done every other day. All experiments were done in triplicates with 6 to 8 replicates in each experiment.
Bacterial culture and inoculum preparation
P. gingivalis, ATCC 33277 (ATCC, Manassas, VA) was streaked onto Brucella Blood Agar with Hemin and Vitamin K plates (Hardy Diagnostics, Santa Maria, CA) and grown under anaerobic conditions at 37°C for 5–7 days. For infection, colonies were inoculated in tryptic soy broth (TSB; Hardy Diagnostics) with 0.5 mg/mL L-cysteine, 5μg/mL hemin, and 2μg/mL vitamin K and grown under anaerobic conditions at 37°C for 24–72 h. Cultures were then diluted 1 to 5 or 1 to 2.5 and grown under anaerobic conditions at 37°C for 16–24 h to OD600 0.5–0.8. Cultures were centrifuged at 3220 g room temperature (RT) for 20 min. Supernatant was discarded and bacterial pellet re-suspended and labeled with CellVue Claret Fluorescent Cell Linker Kit (MilliporeSigma). Dye was inactivated after a 30-s incubation with 5–10 mL 1 mg/mL bovine serum albumin (BSA; VWR, Radnor, PA). Bacteria was centrifuged at 3220 g RT for 20 min, supernatant discarded, and re-suspended in 1 mL 1X Dulbecco’s phosphate-buffered saline (PBS; ThermoFisher Scientific, Carlsbad, CA). Bacterial prep was mixed by pipette and an aliquot was taken for quantitative polymerase chain reaction (qPCR) analysis to determine the bacterial genome copy number per mL for the appropriate bacteria to cell ratio (BCR). The infectious bacterial titer was confirmed by colony forming unit (CFU) analysis.
qPCR
Bacterial DNA was released by incubating samples at 95°C for 10 min. Primers and detection probes (MilliporeSigma) specific for the arginine-gingipain A (Rgp), lysine-gingipain (Kgp), and 16 s rRNA genes were used to detect and quantify P. gingivalis DNA. Primer sequences used: Rgp A fwd: 5’-GCTCTTTCCATAAACGTAAG-3’; Rgp A rev: 5’-GACACTCGTGAGATGAAG-3’; Kgp fwd: 5’-CGAAGCTGAAGTAGGAAC-3’; Kgp rev 5’-CAACCAAAGCCAAGAAGA-3’; 16 s fwd: 5’-ACGAGTATTGCATTGAATG-3’; 16 s rev: 5’-ACCCTTTAAACCCAATAAATC-3’. Detection probes used: Rgp A: FAM-TTCGGATCTTCGTTACGCATAATCA-BHQ-1; Kgp: Cyanine5-CACTAGCTGCCAATCCATCATT-BHQ-3; 16 s Hex-CGCTCGCATCCTCCGTATTAC-BHQ-1. Final concentrations of primers and probes were 0.5μM and 0.15μM, respectively. Samples were assayed in a 20μL reaction using 2X Kapa Probe Fast qPCR Master Mix (Kapa Biosystems, USA). Cycling conditions were based on manufacturer instruction, enzyme activation for 3 min at 95°C followed by 40 PCR cycles (95°C for 3 s, 60°C for 10 s). The standard curve was prepared using 1:10 dilutions of a GBLOCK DNA sequence (IDT, Coralville, IA).
Cell lysis and pooling for CFU assay
For CFU assay, cells were washed once with 1X PBS, then lysed with DI water for 30 min at 37°C. Lysate was collected, pooled by group, and kept on ice for analysis. Bacteria in lysate were quantified by CFU and qPCR analysis.
RT-qPCR
Aliquots of cell lysate were added to two volumes of RNA Protect (Qiagen), RNA was extracted with a Lucigen MasterPure™ Complete DNA and RNA Purification Kit (Middleton, WI) according to manufacturer protocol. RNA was converted to cDNA by SuperScript™ IV First-Strand Synthesis System (ThermoFisher Scientific) according to manufacturer protocol. RT-qPCR was performed using the same conditions as described for qPCR. Primer and detection probes (IDT) specific for arginine gingipain B (RgpB), Kgp, and 16 s rRNA were used to quantify cDNA. Primer sequences used: RgpB fwd: 5’- CTTCCACTTTCACATCCTTTA-3’; RgpB rev: 5’- CTGACGATGGTGATATGG-3’; Kgp fwd: 5’- CAACCAAAGCCAAGAAGA-3’; Kgp rev: 5’- CGAAGCTGAAGTAGGAAC-3’; 16 s fwd: 5’- ACGAGTATTGCATTGAATG-3’; 16 s rev: 5’- ACCCTTTAAACCCAATAAATC-3’. Detection probes used RgpB: 5’-TEX615-CCTTATTGTATCCTACTACGGTGAGC-IAbRQSp-3’; Kgp: 5’-Cy5- CACTAGCTGCCAATCCATCATT-IAbRQSp-3’; 16s: 5’-Hex- AGAATAAGCATCGGCTAACTCCGT-IABkFQ-3’.
Kgp and Rgp enzyme activity assays
Rgp enzymatic activity was measured using a kinetic colorimetric assay as previously described in Dominy et al. 2019 [12]. Samples were assayed in 100 mM Tris, 200 mM glycyl-glycine, 5 mM CaCl2, 10 mM Cys-HCl, pH 7.6 buffer with 1 mM Nα-Benzoyl-L-arginine 4-nitroanilide hydrochloride (L-BAPNA, MilliporeSigma) as the substrate. Optical density (OD) 405 nm was read every 1.5 min for 1.5 h at 37°C. Purified Rgp (a kind gift from Barbara Potempa, University of Louisville) at a known concentration was used to generate the standard curve. Kgp enzymatic activity was measured using a kinetic fluorescent assay. Samples were assayed in 100 mM Tris, 75 mM NaCl, 2.5 mM CaCl2, 10 mM Cys-HCl buffer with 10μM Z-His-Glu-Lys-MCA (MyBioSource, Inc., San Diego, CA) as the substrate. Assay was read at a wavelength of 460 nm on a Synergy H1 microplate reader (BioTek, Winooski, VT) every 1.5 min for 30 min at 37°C. Purified Kgp (a kind gift from Barbara Potempa, University of Louisville) at a known concentration was used to generate the standard curve.
Kgp and Rgp activity probes
Fluorescent activity probes and methods associated with their use for irreversible binding of Kgp (COR553) or Rgp (COR619) were as described [1] with the below modifications. The detailed chemical synthesis and structure of compounds in the structural series for lysine gingipain inhibitors utilized to make these fluorescent probes is found in PCT/US2016/061197 and, referenced in application 62/459,456. Probe detection of active Kgp and Rgp was performed as described previously [1] Briefly, 1μM COR553 or COR619 Cy5 probe, respectively was added into the medium of infected neuron cultures for 1 h at 37°C. Cells were washed in PBS and then lysed with DI water and centrifuged at 10000×g for 10 min at 4°C and resuspended in B-PER (ThermoFisher) at 10-fold concentration. The sample was prepared for blotting with Nupage LDS sample buffer (ThermoFisher) at 95°C and loaded onto denaturing AnyKd gel (Bio-Rad, Hercules, CA). After protein separation, the gel was imaged on ChemiDoc MP Imaging System (Bio-Rad) with Image Lab 6.01 software on the Cy5 channel.
Western blots
Cells were lysed in RIPA buffer (MilliporeSigma) supplemented with proteinase (ThermoFisher) and phosphatase (PhosSTOP, Roche Diagnostics, Indianapolis, IN) inhibitors. The concentration of cell lysates was determined using bicinchoninic acid assay (BCA) (ThermoFisher). Samples were loaded onto 4–20% Mini-Protean TGX Precast Gels (Bio-Rad) and then transferred to PVDF membranes (Bio-Rad) using Trans-Blot Turbo System (Bio-Rad). Membranes were incubated with following primary antibodies: mouse anti-total tau A0024 (DAKO; 1:1000, # A0024), mouse anti-synapsin (ThermoFisher; 1:1000, MA531919), rabbit anti-Gapdh-HRP (Cell Signaling; 1:2000, #8884), rabbit anti-Rgp antibody CAB101 (1:1000) [1], and rabbit anti-Kgp antibody CAB102 (1:1000) [1]. All secondary antibodies were horseradish protein (HRP) conjugated (Vector; 1:5000 or 1:10000). Blots were imaged by chemiluminescent detection using Supersignal West Fento (ThermoFisher) and the ChemiDoc imaging system. Quantification was performed with Image Lab 6.0.1 software (Bio-Rad) by dividing the integrated OD of all bands of interest by the OD of GAPDH detected on the same blot.
ELISA
Levels of phosphorylated tau and total tau in cell lysates were measured with the phospho-tau (Thr231)/total tau Kit from Meso Scale Diagnostics (Rockville, MD, K15121D) according to the manufacturer’s instructions. The cell lysates described above were diluted 50-fold in Tris Wash buffer containing 1% Blocker A (provided by the Meso Scale Diagnostic Phospho-Tau kit, Rockville, MD, K15121D) and incubated overnight in the Multi-spot 96 well 4-spot Phospho (Thr231)/total Tau plate shaking at 4°C. The next day, after washing and incubation with detection antibody according to the manufacturer’s instructions, phosphorylated tau and total tau were quantified with the MESO QuickPlex SQ 120 instrument (Meso Scale Diagnostics).
Cell death assay
To determine cell death, the Image-iT® DEAD Green™ viability stain (ThermoFisher, I10291) was added to the neuronal cultures at a final concentration of 100 nM and incubated for 1 h at 37°C. Cells where then washed with PBS and fresh media with NucBlue nuclear dye (NucBlue™ Live ReadyProbes™ Reagent, ThermoFisher, R37605) added before image acquisition. Dead cells were identified by the colocalization of DEAD Green dye with NucBlue nuclear dye. The percent cell death was calculated by the number of DEAD Green cells divided by the total number of NucBlue nuclear dye -positive cells within a well.
Immunohistochemistry
Cells were fixed in 4% paraformaldehyde for 1 h, then permeabilized and blocked with 0.1% of Triton X-100 and 5 % donkey serum (Jackson ImmunoResearch Laboratories, West Grove, PA) to reduce nonspecific background. After an overnight incubation at 4°C with the primary antibodies [anti-β-tubulin III (StemCell Technology, 60100AD), anti-Map2a (Abcam, ab5392), and anti-Thy1 (Abcam, ab133350)], cells were washed with PBS and incubated with secondary AlexaFluor-conjugated antibodies appropriate for the species (Jackson ImmunoResearch Laboratories, 1:500).
BacMam infection for live imaging
For fluorescent labeling of early endosome, late endosome, lysosomes, and synaptophysin, the cells were infected with CellLight™ Early Endosomes-GFP (C10586), CellLight™ Lysosomes-GFP (C10507), CellLight™ Late Endosomes-RFP (C10589), or CellLight™ Synaptophysin-RFP (C10610) (BacMam Technology, ThermoFisher Scientific, Carlsbad, CA), respectively, according to the supplier’s instructions 48 h before bacterial infection. For live imaging NucBlue nuclear dye was added before image acquisition. For live imaging of P. gingivalis internalization, the NeuO neuron-specific tracer (NeuroFluor™ NeuO, StemCell Technology, 01801) and the Lysotracker (LysoTracker™ Red DND-99, ThermoFisher, L7528) was added to the cultures 1 h before acquiring the images and 24 h after P. gingivalis infection.
Image acquisition and analysis
Images were acquired with the ImageXpress Micro Confocal High-Content Imaging System (Molecular Devices, Sunnyvale, CA) with a 40x objective. Series of images were acquired at different planes along the focal axis (Z-stack). Stacks from 7 up to 75 planes separated by 2 or 0.2μm, respectively, were acquired, covering approximately 15μm in depth. For 3D rendering, all individual images were saved; for 2D analysis, 2D maximum projection images were saved. Automated, built-in MetaXpress Analysis Macros such as Count Nuclei, Live/dead, and Cell scoring were used to quantify NucBlue-positive cells, dead cell, and neuron numbers, respectively. For the quantification of synaptophysin positive boutons, P. gingivalis positive puncta, and counts of early endosome, late endosome, and lysosome, the MetaXpress custom module Top Hat followed by the Count Round Objects was utilized. Co-localization of P. gingivalis in the different cellular compartments was quantified with Logical Operation using the AND function. Percent P. gingivalis within a compartment was calculated by the number of co-localized P. gingivalis divided by the total number of P. gingivalis. The number of synaptophysin-positive boutons was standardized to the average counts of non-infected neurons at 24-h time point.
Transmission electron microscopy
Neurons were grown on PLO and Laminin coated Permanox culture dishes (19340-72, Electron Microscopy Sciences, EMS, Hatfield, PA) for 3 weeks. 48 h after P. gingivalis infections, the samples were rinsed in PBS and fixed in 2.5% glutaraldehyde in PBS (EMS, 15980) overnight at 4°C. The next day, cells were post-fixed in 1% osmium tetroxide for 1 h at RT, rinsed in phosphate buffer (EMS, 19340-72), dehydrated in a graded ethanol series, and infiltrated with Poly/Bed® epoxy resin (Poly/Bed® 812 Mini Kit, 21959-1) according to the manufacturer’s instructions (Polysciences, Warrington, PA) using propylene oxide as a transition fluid. The samples were embedded in resin and polymerized for 24 h at 60°C. Ultrathin sections (70–80 nm) were cut using a diamond knife, collected on copper grids (200 mesh), and stained with 3% alcoholic uranyl acetate (6 min) and Reynold’s lead citrate (2 min). Grids were examined using the JEOL JEM-1400 transmission electron microscope. Representative digital images were obtained using the Gatan Orius SC1000 CCD Camera and DigitalMicrograph digital imaging software.
Statistical analysis
All statistical analyses were done in GraphPad Prism. The analysis used for each data set is described in the accompanying figure legend. Data are expressed as mean±95% confidence interval or if presented in log scale as geometric mean±geometric standard deviation. Statistical analyses for significant differences were performed with Student’s t-test, two-way ANOVA (parametric data sets), or Kruskal-Wallis (non-parametric analysis) followed by Tukey’s posthoc test or Dunn’s multiple comparison where appropriate. The criterion for statistical significance was p < 0.05.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding authors on reasonable request.
RESULTS
Neurotoxicity of P. gingivalis infection in neuron cultures is concentration and time dependent
To determine the ability of P. gingivalis to invade and persist in neuron cultures, we cocultured P. gingivalis and neurons in a low oxygen incubator (5% O2, 5% CO2) for 24 h. The partial oxygen pressure in the brain has been reported as 20–25 mmHg [27] and culture conditions at 1–5% oxygen model the physiological level for neurons which is beneficial for long term cultures [28]. P. gingivalis, an obligate anaerobe, has been shown to tolerate up to 6% O2 in reduced hemin cultures [29] enabling a neuron infection model in oxygen conditions suitable for both pathogen and host cells. To optimize bacteria to cell ratio (BCR) for a prolonged neuron infection model, we titrated the BCR to determine the best infection rate with highest neuronal viability for up to 72 h. Neurons were infected with a range of 30 to 600 BCR. Bacteria that were not attached, tightly associated with, or internalized in the neurons were washed off and the number of P. gingivalis per nucleus after 24 h of co-culture were quantified using confocal high content screening (HCS) (Fig. 1A). A dose response to infection was seen with higher BCR producing higher load of P. gingivalis infection and the most significant increase between 100 and 300 BCR.
Fig.1
Viability of iPSC derived neurons and P. gingivalis for up to 72 h after infection. A) The number of P. gingivalis colocalized with neurons increases with increased numbers of P. gingivalis added to cultures. Kruskal-Wallis test with Dunn’s multiple comparison test: ****p < 0.001. B) Neuron cell death assay quantified by the ratio of dead cells over the total number of nuclei 24 h after infection with different BCRs. Kruskal-Wallis test with Dunn’s multiple comparison test: ****p < 0.0001. C) Neuron cell death assay 24 h, 48 h, and 72 h after infection with BCR 300. Two-way ANOVA with Tukey’s multiple comparison test: **p < 0.01, ***p < 0.001. D) Quantification of bacteria 24 h, 48 h, and 72 h after infection with BCR 300 by colony assay (CFU), qPCR, and high content screening (HCS). E) RT-qPCR analysis of neuron lysates for Kgp, Rgp mRNA, and P. gingivalis specific 16 S rRNA. A-C) Mean with 95% confidence intervals. D-E) Geometric mean with geometric standard deviation.
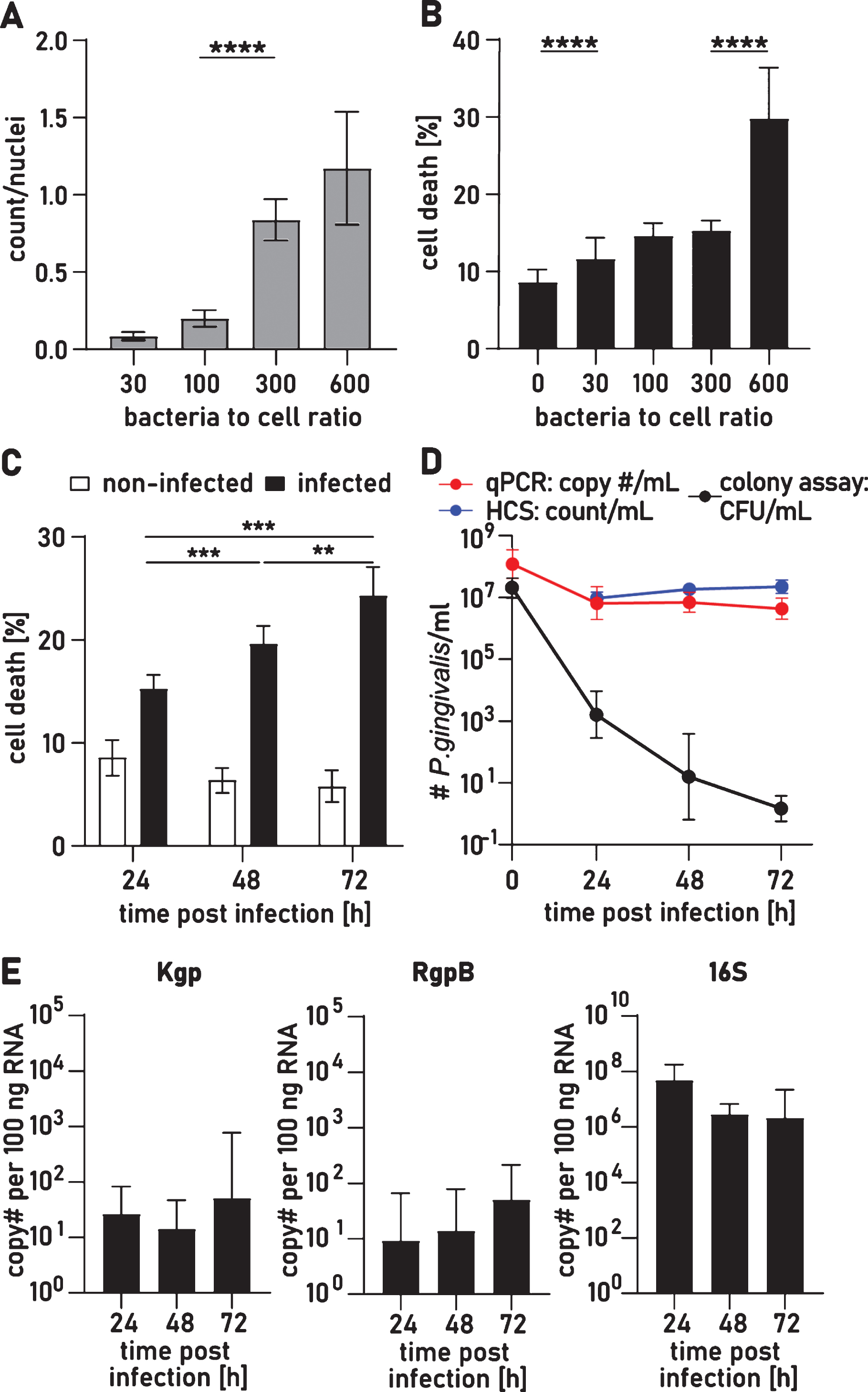
After 24 h, P. gingivalis induced cytotoxicity was significantly higher for BCR 30, 100, and 300 with averages of 11.7±5.4%, 14.6±3.4%, and 15.3±5.5% dead cells, respectively, compared to 8.6±6.0% in control cultures (Fig. 1B) but with no dose dependent increase. Infection with BCR 600, however, significantly increased the number of dead cells to 29.8±5.3%. Since BCR 300 resulted in the highest infection rate with low acute cytotoxicity, we quantified cell death at 48 h and 72 h after infection with BCR 300 (Fig. 1C). There was a significant time dependent increase in cell death to 19.7±4.7% at 48 h and to 24.35±7.6% at 72 h. 75% of neurons survived for at least 72 h of infection allowing these cultures to be utilized for further investigation of mechanistic effects of P. gingivalis and its proteolytic gingipains on human neurons.
Infection of neurons remains stable for up to 72 h and P. gingivalis enters a viable but non- culturable state
To determine the ongoing persistence of infection, we removed nonattached bacteria with PBS washes and, at multiple timepoints, quantified the remaining attached and intraneuronal bacteria by confocal HCS of the cultures followed by qPCR analysis of the neuron cell lysates. The HCS and qPCR analysis showed comparable neuron infection of P. gingivalis which remained stable from 24 h to 72 h (Fig. 1D). A colony forming unit assay (CFU assay) found that the infection rate at 24 h was 0.1%, and CFU numbers dropped another 100-fold from 24 h to 48 h (Fig. 1D). After 72 h of infection, very few cultures could be grown from cell lysates. Since neuron associated P. gingivalis persisted at such a high level, we surmised that after infecting neurons P. gingivalis might enter a viable but not culturable (VBNC) state. P. gingivalis displaying VBNC has been previously reported and VBNC is a common adaptation for intracellular pathogens to persist in a hostile environment [21, 30, 31]. To demonstrate viability of non-culturable P. gingivalis, we performed RT-qPCR analysis to detect active transcription of P. gingivalis specific genes to mRNA. RNA is quickly degraded after transcription and the presence of specific RNA has been described as a gold standard method to investigate VBNC [32, 33]. RNA was isolated from infected neuron cultures at 24 h, 48 h, and 72 h and mRNA was detected and quantified for Lysine gingipain (Kgp), arginine gingipain (RgpB) as well as P. gingivalis specific 16 S rRNA at all time points (Fig. 1E).
P. gingivalis aggregates are engulfed in neuronal membrane and express Rgp and Kgp
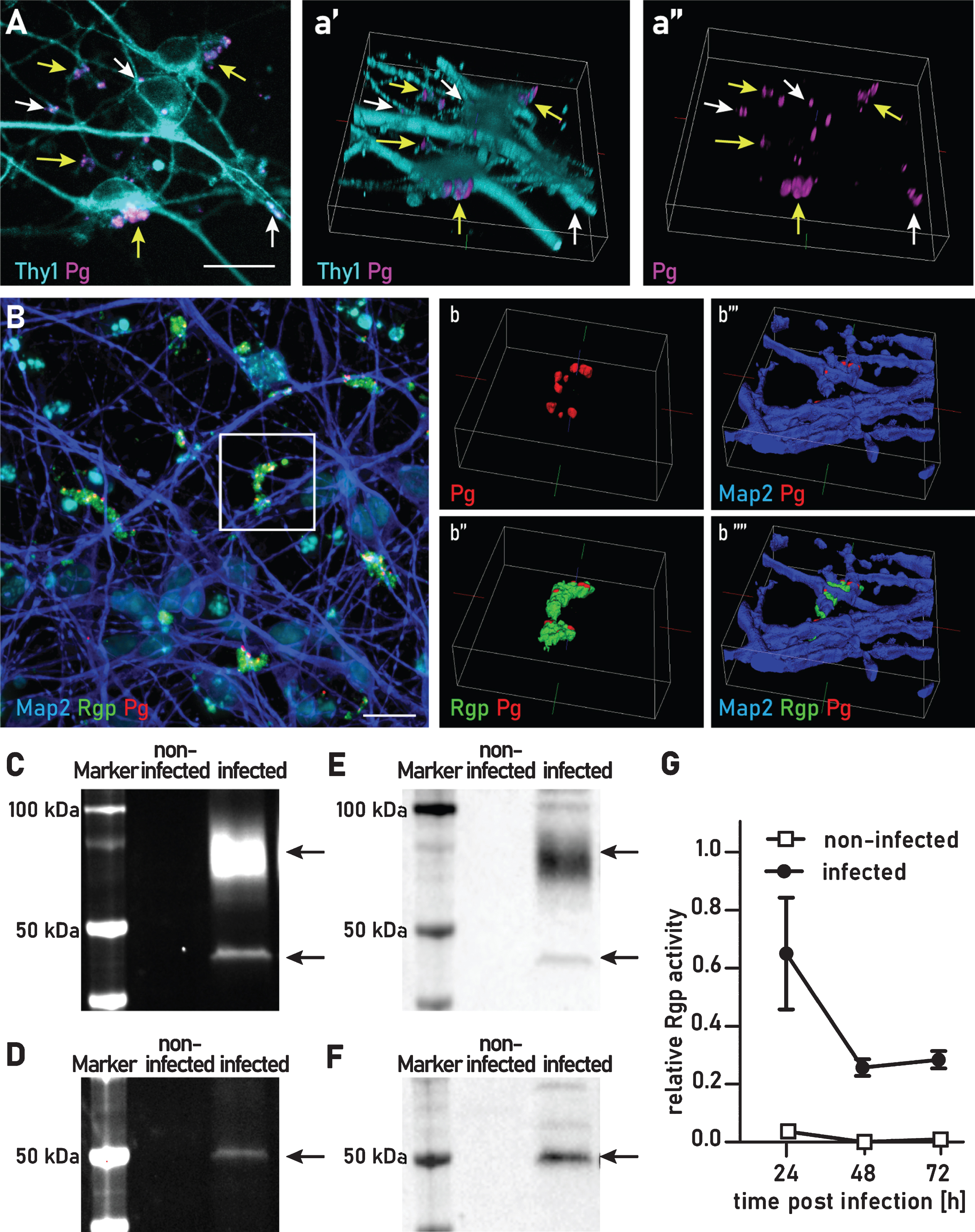
3D reconstructions from multichannel confocal image stacks were used to investigate if P. gingivalis was internalized into neurons in culture 24 and 48 h after infection, P. gingivalis was labeled with a fluorescence marker covalently bound to the bacterial membrane, and neurons were stained by immunofluorescence with the neuron-specific membrane protein marker Thy1/CD90. (Fig. 2A). Various sized aggregates of P. gingivalis appeared to reside in a membrane swelling on neurites and cell bodies. However, some of the aggregates appeared to be only partially covered by Thy1 positive membrane. Immunostaining with microtubule associate protein 2 (MAP2) showed MAP2 positive particles in and around the bacterial aggregate, supporting the finding that these aggregates are surrounded by neuronal cell products (Fig. 2B). P. gingivalis survival in the host cell has been shown to depend on the activity of Kgp and Rgp [1, 24–26]. Consequently, we investigated whether Kgp and Rgp were present in infected neuron cultures. Immunofluorescence staining with CAB101, an antibody raised against Rgp [5], formed a large diffuse field (green) around intraneuronal P. gingivalis (red) 48 h after infection suggesting consistent gingipain secretion of gingipains and confirming transcriptional activity of gingipain described above (Fig. 2B).
Fig.2
P. gingivalis is internalized and secretes active gingipains for up to 72 h. A) Maximal projection of a confocal image stack of P. gingivalis (magenta) infected neurons stained for the membrane specific protein Thy1 (cyan). a’) 3D reconstruction. a”) Same as a’ without the Thy1 channel. Yellow arrows point to aggregates of P. gingivalis partially engulfed in membrane. White arrows point at P. gingivalis completely internalized in the neuronal membrane. Scale bar 20μm. B) Maximal projection of confocal image stack immunofluorescence stained for Rgp (green), P. gingivalis (red), and MAP2 (blue): secreted Rgp creates a field around neuron associated bacterial clusters. b-b””) 3D iso-surface reconstruction of the area indicated in B. b) MAP2 and Rgp channels omitted. b”) MAP2 channel omitted. b”’) Rgp channel omitted. b””) All channels visible. Gel electrophoresis of cell lysates incubated with a Cy-5 conjugated activity probe for Rgp (C) and Kgp (D). Arrows indicate the expected protein sizes at 45 kDa and 70 kDa for Rgp (C) and 50 kDa for Kgp (D). E and F are the corresponding western blots to the gels depicted in C and D, respectively. Blots were stained with antibodies against Rgp (E) and Kgp (F). Full length gels and western blots are presented in Supplementary Figure 5. G) Colorimetric activity assay for Rgp activity from cell lysates at 24 h, 48 h, and 72 h after infection.
Gingipains in infected neuron cultures are proteolytically active for at least 72 h after infection
To determine if Kgp and Rgp retain active protease activity, we incubated the infected neuron cultures with Cy5-conjugated activity probes specific for Rgp and Kgp (COR619 and COR553, respectively). These probes are designed to be highly specific and to irreversibly bind to the active sites of Rgp and Kgp. Only active Rgp or Kgp is detected with this method. To probe intracellular and tightly cell-associated gingipains, cultures were washed with PBS before lysing. Active gingipains were then detected with the addition of the gingipain activity probes. At 24 h after infection, we were able to detect Rgp activity as measured by fluorescent probe detection of Rgp at the predicted molecular weights of 45 kDa and at 70 kDa (Fig. 2C). In the complementary experiment, we were able to detect active Kgp with a single band of 50 kDa (Fig. 2D). As a further control for the identity of Rgp and Kgp detected with the activity probes the identity of these bands was confirmed by immunoblotting with CAB101 and CAB102, polyclonal antibodies raised against Rgp and Kgp, respectively (Fig. 2E, F).
By using a highly sensitive substrate-based enzyme activity assay we were able to quantify Rgp activity of neuron internalized P. gingivalis for at least 72 h. We found a slight drop of activity when comparing 24 h of infection with 48 h, but this activity remained stable from 48 h to 72 h (Fig. 2G). We also assayed for Kgp activity, however Kgp activity was below the detection level of this Kgp substrate-based assay. Thus, in neuron cultures infected with P. gingivalis proteolytically active Kgp and Rgp are actively produced for at least 72 h after infection.
P. gingivalis interacts with the neuron membrane and is internalized
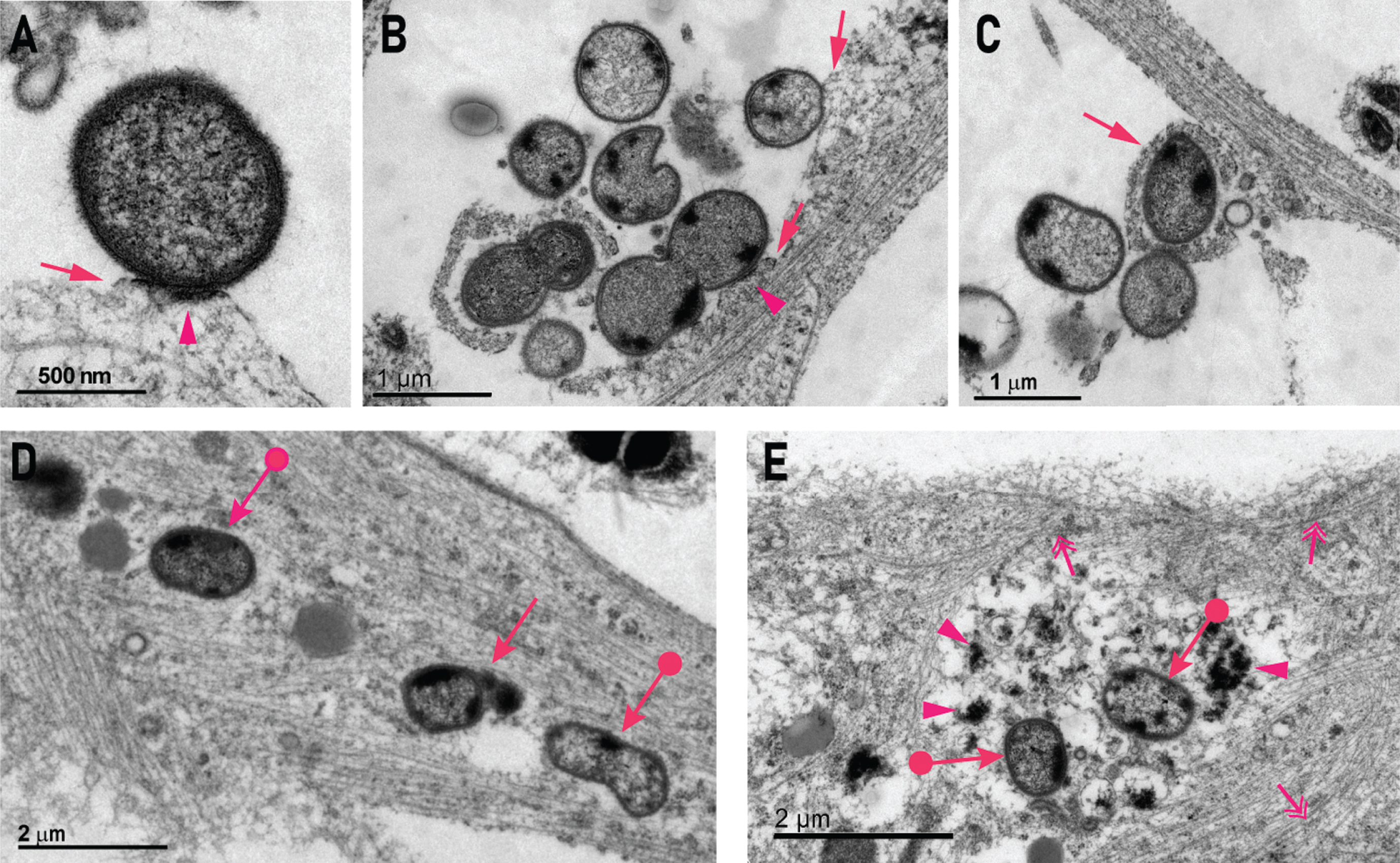
Transmission electron microscopy (TEM) was used to investigate internalized and attached P. gingivalis at the ultrastructural level. In the TEM images, healthy bacteria could be identified by a round structure, an electron dense double cell membrane and visible fimbriae (Fig. 3A-C). Extracellular bacteria could be seen contacting the neurite membrane and an electron dense band resembling a pedestal was occasionally visible. Such pedestals have also been described in scanning electron microscopy studies on P. gingivalis infected coronary artery endothelial cells and human umbilical vein endothelial cells [23, 34]. Pedestal formation is induced by pathogen-host interactions that are highly specific and not well understood for P. gingivalis, but commonly involve the rearrangement of the actin cytoskeleton and result in the pathogen entering the host cell [20, 35]. Some bacteria appeared to be in the process of being endocytosed into the cell (Fig. 3B, C). None of the bacteria observed during endocytosis were enclosed in an endocytic membrane (Fig. 3C, D), resulting in non-compartmentalized, free bacteria in the cytoplasm. Neurofilaments and microtubules around free bacteria were disrupted and the bacteria were surrounded by electron dense cell debris (Fig. 3E).
Fig.3
P. gingivalis attaches to neurons, is internalized, and is associated with disruption of the cytoskeleton. A,B) Examples of extracellular P. gingivalis contacting a neurite and inducing a membrane swelling resembling an actin pedestal (arrow). This was associated with an electron dense band along the contact surface to P. gingivalis (arrowhead). C) Some bacteria were internalized by membrane swellings on neurites (arrow). D) An example of two P. gingivalis organisms (circle arrows) residing within a neurite without being bound by a distinct surrounding membrane and one P. gingivalis inside a membrane (arrow). E) Two P. gingivalis within the cell body that are non-membrane bound (circle arrows) and are surrounded by heterogenous electron dense matrix (arrowheads) with a peripheral zone of neurofilaments and microtubules (double headed arrows).
Over time intraneuronal P. gingivalis increasingly reside in lysosomes
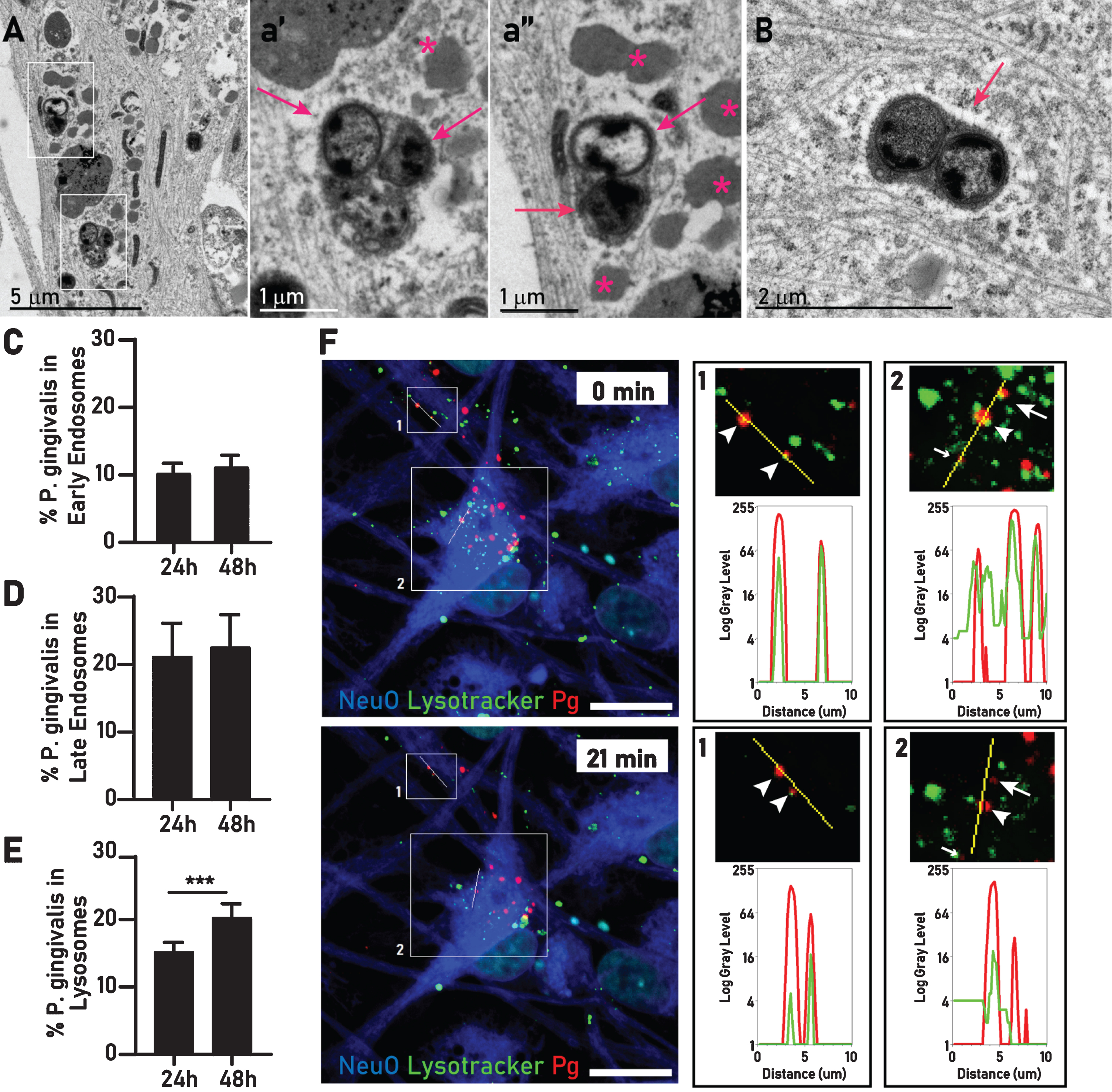
Much of the intracellular bacteria were located inside a single membrane compartment resembling a lysosome. These bacteria appeared to be healthy or in various stages of disintegration (Fig. 4A, B). Commonly, around compartmentalized bacteria we observed an electron light halo surrounding the membrane with the adjacent cytoplasm intact (Fig. 4B). To further characterize the lysosome-like structure observed in TEM, we expressed early endosome, late endosome, and lysosome specific markers (Rab5, Rab7, and Lamp1, respectively) in neurons using a baculovirus-based transduction system (BacMam) and quantified the colocalization with P. gingivalis by HCS at 24 h and 48 h after infection. The percentage of P. gingivalis in early endosomes and in late endosomes remained stable between 24 h and 48 h (Fig. 4C-E). However, the percentage of P. gingivalis colocalizing with lysosomes increased by 33% at 48 h compared to 24 h (Fig. 4E), suggesting that over time P. gingivalis may accumulate in these organelles. Using lysotracker staining in neurons infected with fluorescently labeled P. gingivalis we observed the colocalization of P. gingivalis and lysosomes over time and colocalization of P. gingivalis with lysotracker could be confirmed for bacteria moving in neurites and in cell bodies (Fig. 4F, Supplementary video).
Fig.4
Intraneuronal P. gingivalis is found in the endosomal/lysosomal pathway. A) Two groups of P. gingivalis (white rectangles) within membrane bound structures resembling lysosomes. a’) Enlarged inset of A (lower square) showing an aggregate of two intact bacteria (arrows) and a partially degraded bacterium a”) Enlarged inset of A (upper square) a pair of intact bacteria (arrows) surrounded by an accumulation of lipid droplets (stars). B) Two P. gingivalis inside a single membrane (arrow), the surrounding cytosol contains rough endoplasmic reticulum and neurofilament looks unperturbed except for a small electron lucent halo surrounding the bacteria containing compartment. Percentage of internalized P. gingivalis colocalizing with early endosomes (C), late endosomes (D), and lysosomes (E). Mean with 95% confidence interval. Student’s t-test, ***p < 0.005. F) Live imaging of P. gingivalis (red), lysotracker (green), and NeuO (blue). Line scans in a neurite (inset 1) and the cell body (inset 2) confirm colocalization of P. gingivalis with migrating lysosomes over time. In inset 1, both bacteria continue to colocalize with lysotracker (arrowheads). In inset 2, three bacteria colocalize at time 0. At 21 min, one remains in the lysosome (arrowhead), one has moved away from the line scan (small arrow), and one is not colocalizing with lysotracker anymore (arrow).
P. gingivalis infection induces multivesicular bodies and dystrophic neurites containing autophagic vacuoles and secondary lysosomes
Neuron cultures infected with P. gingivalis displayed cytopathological features in areas where no discernable bacteria were observed. In some neurons, multivesicular bodies and lipid droplets accumulated in the perikaryon (Fig. 5 A, B). In noninfected neuron cultures, only very few multivesicular bodies were present; however, we did find lipid droplets close to lysosomes (Supplementary Figures 2 and 3). Additional TEM images of noninfected neurons are presented in Supplementary Figures 1-4.
Fig.5
Infection with P. gingivalis causes an accumulation of autophagosomes and multivesicular bodies. A) Accumulation of multivesicular bodies (arrow heads) in the perikaryon of neuron infected with P. gingivalis. B) Magnified inset of A. C,D) Two examples of variably sized membrane bound vesicles containing heterogeneous amorphous electron dense material interpreted as secondary lysosomes (arrowheads) or occasional concentric lamellar bodies interpreted as autophagosomes (arrows). Quantification of mean neurite thickness (E) and mean neurite area (F) showed significant increases in infected neuron cultures. Mean with 95% confidence interval; **p < 0.01, ***p < 0.001, ****p < 0.0001.
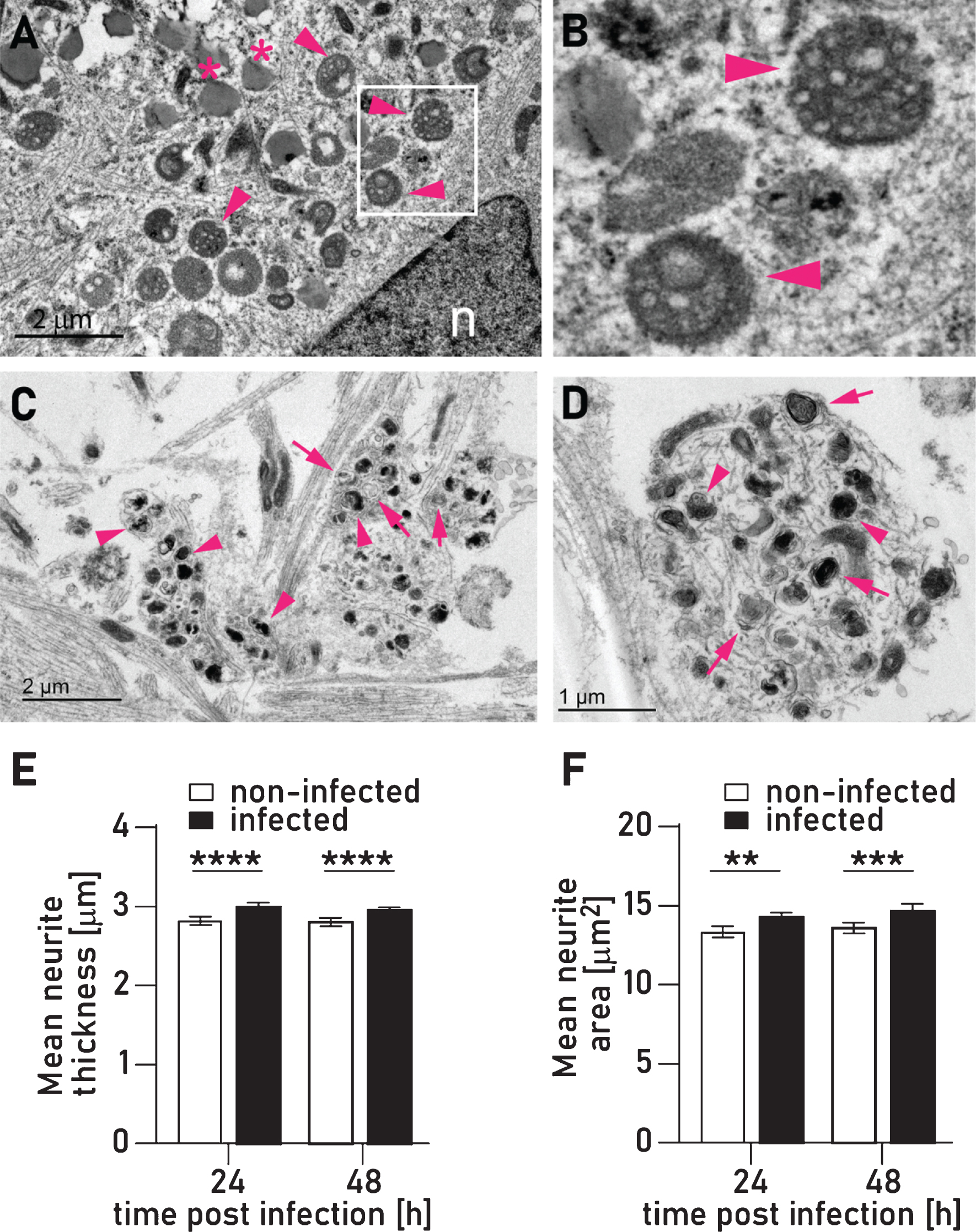
In addition to the pedestals induced by direct interaction with P. gingivalis described above, our TEM analysis revealed an additional type of membrane swelling where the membrane appeared to be intact and was filled with variably sized (less than 1μm diameter) membrane bound vesicles containing heterogeneous amorphous electron dense material we have interpreted as secondary lysosomes or concentric lamellar bodies interpreted as autophagosomes (Fig. 5C, D). These structures very closely resemble autophagic vacuoles that form in dystrophic neurites in the brains of AD patients [36, 37]. By using image analysis, we were able to quantify and confirm that mean neurite thickness and area is increased in P. gingivalis infected neurons (Fig. 5E, F).
Tau protein is degraded in P. gingivalis-infected neurons and phosphorylated over time
We have previously shown that recombinant tau-441 protein and high molecular weight tau in SH-SY5Y cells is highly susceptible to protease fragmentation by gingipains [1]. Western blot analysis of lysates from 24 h and 48 h infected neurons showed strong degradation of tau compared to non-infected controls (Fig. 6A, 6B) which confirms our previous findings. One of the hallmarks of the AD brain is an abnormal hyperphosphorylation of tau [38–40] and we investigated whether tau hyperphosphorylation might be a direct consequence of P. gingivalis infection. We performed western blot analysis for ptau(S396), a site that has been reported to be phosphorylated early in the onset of AD [38–40] and found that in infected cultures ptau was comparably degraded to the tau blot (Supplementary Figure 7). Because in the western blot analysis tau was fragmented to a degree that the quantitation of ptau/total tau was not feasible, we used a multiplex ELISA assay to quantify both total tau and ptau (T231), a phosphorylation site identified in AD brains [41, 42] to determine potential effects on hyperphosphorylation. Interestingly, ptau/tau ratio was increased by 23% after 48 h but not after 24 h (Fig. 6F). Both tau and ptau were equally reduced at 24 h but at 48 h tau was comparably more reduced than ptau (T231) (Fig. 6D, 6E).
Fig.6
P. gingivalis degrades tau, increases the ratio of ptau/tau and decreases synapse density. A) Western blots stained with total tau and synapsin antibodies for infected and noninfected lysates at 24 h and 48 h after infection. GAPDH was included as a loading control. Full length western blots are presented in Supplementary Figure 6. B,C) Densitometry of western blots depicted in A normalized to GAPDH. Mean with 95% confidence interval, 1-way ANOVA with Tukey’s multiple comparison test ****p < 0.0001, ***p < 0.001. Multiplex ELISA quantification of ptau(T231) (D), total tau (E), and the calculated ptau(T231)/total tau ratio (F). Mean with 95% confidence interval, 1-way ANOVA with Tukey’s multiple comparison test: *p < 0.05, **p < 0.01, ***p < 0.001. G) Multichannel maximum projection of a confocal stack with synaptophysin (green), P. gingivalis (red), and a universal membrane marker (blue) used as a cell mask for high content screening. The blue channel has been omitted in the right-side panel (g). Arrows point at P. gingivalis associated with neurons; arrow heads point at synaptophysin positive synaptic buttons. Scale bar 50μm. H) Quantification of synaptic buttons 24 h and 48 h after infection. Mean with 95% confidence interval, 2-way ANOVA with Dunnett’s multiple comparison test: **p < 0.01, ***p < 0.005.
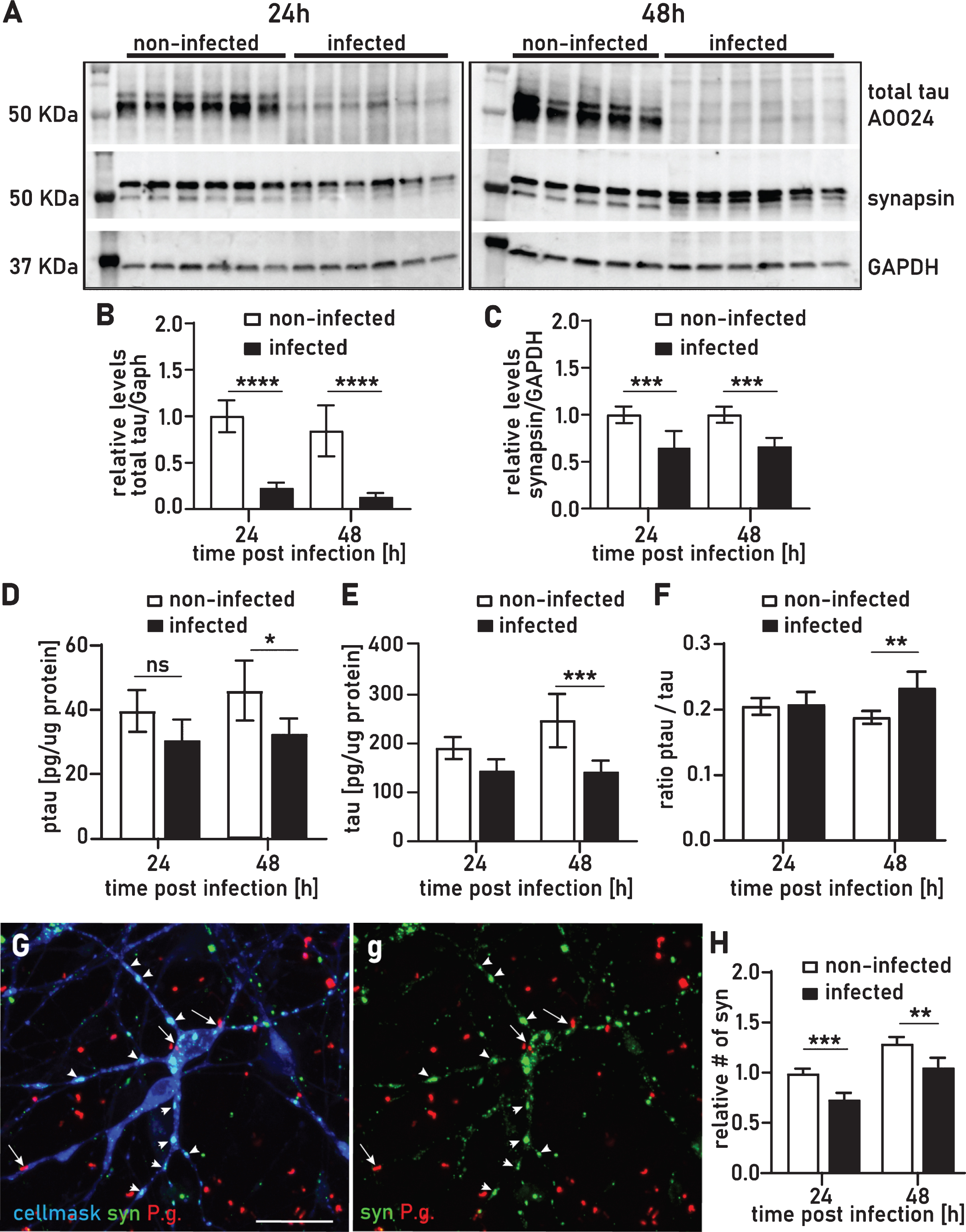
P. gingivalis neuronal infection results in synapse loss
High content screening analysis with immunofluorescence staining for synaptophysin revealed a 26% and 19% decrease in synapse density on neurons at 24 h and 48 h of infection, respectively (Fig. 6G, 6H). This finding was corroborated by a comparable decrease in synapsin detected by western blot (Fig. 6C). Synapse loss is one of the earliest pathological events in the onset of AD and both synaptophysin and synapsin have been reported to be decreased in vulnerable areas of AD affected brains [43].
DISCUSSION
This study demonstrates the infection of neurons with P. gingivalis in vitro. Intraneuronal P. gingivalis was detected within single membrane, lysosome-like structures or unbound in the cytoplasm. Confocal high content screening found colocalization with endosome markers Rab5 and Rab7 as well as the lysosome markers Lamp1 and lysotracker. In addition, we demonstrate that after 72 h of infection, P. gingivalis is in a viable but non-culturable state and expresses both active Rgp and Kgp. Larger aggregates of bacteria appeared partially engulfed by neuronal membrane and produced significant amounts of gingipain detectable by immunohistochemistry. Given the cluster formation and apparent coating with gingipains, we hypothesize these aggregates might represent biofilm covered repositories of P. gingivalis [44]. Consequently, comparable to periodontal P. gingivalis colonies, these aggregates appear to be producing gingipains and bacteria and continuously infect neurons over longer time periods.
In the ultrastructural analysis, the cytoskeleton surrounding cytosolic P. gingivalis appeared to be disintegrating and analysis of the cell lysates revealed significant degradation of tau protein. We have previously shown the proteolytic activity of gingipains on tau protein and that fragmentation of tau after P. gingivalis infection of SH-SY5Y cells was gingipain dependent [1] and active gingipains were demonstrated in infected neurons in the current study.
Intriguingly, the ratio of phosphorylated tau increased only 48 h after infection but was unaffected at 24 h, even though total tau was decreased at both times. This suggests that either hyperphosphorylated tau has greater stability and might be a protective mechanism against gingipain mediated protein digestion and/or phosphorylation of tau is increased after prolonged infection with P. gingivalis. It has previously been shown that phosphorylation of tau protects tau against proteolytic degradation [45]. This result is consistent with a recent study showing that oral infection with P. gingivalis in wild type mice resulted in gingipains being identified inside neurons along with hyperphosphorylated tau that was not seen in uninfected mice [2].
It is generally accepted that one of the main functions of tau is the stabilization of microtubules [46–48]. Degradation of tau by gingipains and resulting hyperphosphorylation may result in destabilization or dysfunction of microtubule structures. Microtubules are crucial for axonal transport mechanisms such as vesicle or lysosome transport [49], and the maintenance of synapses depends on microtubule-based transport of synapsin and synaptophysin [50]. In our neuron cultures, we observed a significant loss of synapse density after infection with P. gingivalis. This was normalized to the MAP2 positive area or total protein and therefore is independent of neurotoxicity induced by infection. Synapse loss and decreased synapsin is one of the first neuropathological events in the onset of AD related dementia [43]. Synapse loss in AD has been attributed to impaired axonal transport, and ultrastructural analysis in the AD brain found comparable neuronal abnormalities to iPSC neurons infected with P. gingivalis, including an accumulation of multivesicular bodies in the perikaryon and a buildup of autophagic vacuoles [36, 51, 52].
In our cultures, we found autophagic vacuoles only in infected cultures. This may represent a defense mechanism by infected cells to fight intracellular bacteria [53]; however, there is also experimental evidence that autophagic vacuoles accumulate when retrograde transport is inhibited [36, 51]. The resulting phenotype in the TEM analysis is remarkably comparable to autophagic vacuoles we observed in infected neurons.
Thus, the hypothesis that P. gingivalis infection results in impaired microtubule transport is supported by synaptic loss, decreased synapsin, multivesicular bodies accumulating in the perikaryon, and dystrophic neurites containing autophagic vacuoles. Importantly, these effects have all been described as early events in the development of AD pathology. Therefore, our initial characterization of P. gingivalis infected neurons suggests that this is a model to study early neuropathological events of AD-related dementia.
The primary aim of this study was to demonstrate the ability of P. gingivalis to invade and persist in neurons, and we optimized the BCR to our investigation time frame within the first 72 h of infection. Consequently, our study was performed with a BCR of 300, which is likely much higher than what a neuron in the brain might encounter and the resulting degenerative effects might be more pronounced and more toxic than what is encountered at physiological levels. However, if we extrapolate the neurodegenerative effects of this subacute infection to a progressive, repeated low level, chronic infection state, the resulting neuropathological effects appears very comparable to an AD brain.
In conclusion, P. gingivalis can infect neurons and persistently express gingipains. The neuropathological phenotype after 48 h of P. gingivalis infection includes autophagic vacuoles in neurites, an accumulation of multivesicular bodies in the perikaryon, degradation and phosphorylation of tau, and the loss of synapses. Comparable features of neuronal degeneration combined with presence of gingipains are detected in AD brains, and we propose that human iPSC-derived neurons infected with P. gingivalis are a novel in vitro model to investigate the neuropathological mechanisms and treatment of AD.
ACKNOWLEDGMENTS
This work was funded by Cortexyme Inc.
S.S.D. and C.C.L. are inventors on two issued patents both entitled “Inhibitors of lysine gingipain” (US/9,975,473 and US/9.988,375) as well as two pending patent applications entitled “Inhibitors of arginine gingipain” (PCT/US2016/061197, filed on 9 November 2016) and “Ketone inhibitors of lysine gingipain” (PCT/US2017/051912, filed on 15 September 2017). S.S.D. and C.C.L. are inventors on a patent application entitled “Methods of use for therapeutics targeting the pathogen P. gingivalis” (US2017/ 0014468, filed on 29 April 2015). L.J.H. is an inventor on a patent application entitled “Gingipain activity probes” (62/459,456; filed on 15 February 2017). Cortexyme is the assignee of these patents and applications.
Authors’ disclosures are available online (https://www.j-alz.com/manuscript-disclosures/20-0393).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-200393.
REFERENCES
[1] | Dominy SS , Lynch C , Ermini F , Benedyk M , Marczyk A , Konradi A , Nguyen M , Haditsch U , Raha D , Griffin C , Holsinger LJ , Arastu-Kapur S , Kaba S , Lee A , Ryder MI , Potempa B , Mydel P , Hellvard A , Adamowicz K , Hasturk H , Walker GD , Reynolds EC , Faull RLM , Curtis MA , Dragunow M , Potempa J ((2019) ) Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv 5: , eaau3333. |
[2] | Ilievski V , Zuchowska PK , Green SJ , Toth PT , Ragozzino ME , Le K , Aljewari HW , O’Brien-Simpson NM , Reynolds EC , Watanabe K ((2018) ) Chronic oral application of a periodontal pathogen results in brain inflammation, neurodegeneration and amyloid beta production in wild type mice. PLoS One 13: , e0204941. |
[3] | Hajishengallis G , Lamont RJ ((2014) ) Breaking bad: Manipulation of the host response by Porphyromonas gingivalis. Eur J Immunol 44: , 328–338. |
[4] | Darveau RP , Hajishengallis G , Curtis MA ((2012) ) Porphyromonas gingivalis as a potential community activist for disease. J Dent Res 91: , 816–820. |
[5] | Kaye EK , Valencia A , Baba N , Spiro A 3rd , Dietrich T , Garcia RI ((2010) ) Tooth loss and periodontal disease predict poor cognitive function in older men. J Am Geriatr Soc 58: , 713–718. |
[6] | Gatz M , Mortimer JA , Fratiglioni L , Johansson B , Berg S , Reynolds CA , Pedersen NL ((2006) ) Potentially modifiable risk factors for dementia in identical twins. Alzheimers Dement 2: , 110–117. |
[7] | Stein PS , Desrosiers M , Donegan SJ , Yepes JF , Kryscio RJ ((2007) ) Tooth loss, dementia and neuropathology in the Nun study. J Am Dent Assoc 138: , 1314–1322; quiz 1381-1312. |
[8] | Kamer AR , Pirraglia E , Tsui W , Rusinek H , Vallabhajosula S , Mosconi L , Yi L , McHugh P , Craig RG , Svetcov S , Linker R , Shi C , Glodzik L , Williams S , Corby P , Saxena D , de Leon MJ ((2015) ) Periodontal disease associates with higher brain amyloid load in normal elderly. Neurobiol Aging 36: , 627–633. |
[9] | Noble JM , Borrell LN , Papapanou PN , Elkind MS , Scarmeas N , Wright CB ((2009) ) Periodontitis is associated with cognitive impairment among older adults: Analysis of NHANES-III. J Neurol Neurosurg Psychiatry 80: , 1206–1211. |
[10] | Ide M , Harris M , Stevens A , Sussams R , Hopkins V , Culliford D , Fuller J , Ibbett P , Raybould R , Thomas R , Puenter U , Teeling J , Perry VH , Holmes C ((2016) ) Periodontitis and cognitive decline in Alzheimer’s disease. PLoS One 11: , e0151081. |
[11] | Poole S , Singhrao SK , Chukkapalli S , Rivera M , Velsko I , Kesavalu L , Crean S ((2015) ) Active invasion of Porphyromonas gingivalis and infection-induced complement activation in ApoE-/- mice brains. J Alzheimers Dis 43: , 67–80. |
[12] | Lim S , Haque MM , Kim D , Kim DJ , Kim YK ((2014) ) Cell-based models to investigate tau aggregation. Comput Struct Biotechnol J 12: , 7–13. |
[13] | Bardy C , van den Hurk M , Eames T , Marchand C , Hernandez RV , Kellogg M , Gorris M , Galet B , Palomares V , Brown J , Bang AG , Mertens J , Bohnke L , Boyer L , Simon S , Gage FH ((2015) ) Neuronal medium that supports basic synaptic functions and activity of human neurons in vitro. Proc Natl Acad Sci U S A 112: , E2725–2734. |
[14] | Al-Taweel FBH , Al-Magsoosi MJN , Douglas CWI , Whawell SA ((2018) ) Identification of key determinants in Porphyromonas gingivalis host-cell invasion assays. Eur J Oral Sci 126: , 367–372. |
[15] | Belton CM , Izutsu KT , Goodwin PC , Park Y , Lamont RJ ((1999) ) Fluorescence image analysis of the association between Porphyromonas gingivalis and gingival epithelial cells. Cell Microbiol 1: , 215–223. |
[16] | Dorn BR , Dunn WA Jr , Progulske-Fox A ((2001) ) Porphyromonas gingivalis traffics to autophagosomes in human coronary artery endothelial cells. Infect Immun 69: , 5698–5708. |
[17] | El-Awady AR , Miles B , Scisci E , Kurago ZB , Palani CD , Arce RM , Waller JL , Genco CA , Slocum C , Manning M , Schoenlein PV , Cutler CW ((2015) ) Porphyromonas gingivalis evasion of autophagy and intracellular killing by human myeloid dendritic cells involves DC-SIGN-TLR2 crosstalk. PLoS Pathog 10: , e1004647. |
[18] | Ho MH , Huang L , Goodwin JS , Dong X , Chen CH , Xie H ((2016) ) Two small molecules block oral epithelial cell invasion by Porphyromons gingivalis. PLoS One 11: , e0149618. |
[19] | Jang JY , Baek KJ , Choi Y , Ji S ((2017) ) Relatively low invasive capacity of Porphyromonas gingivalis strains into human gingival fibroblasts in vitro. Arch Oral Biol 83: , 265–271. |
[20] | Lamont RJ , Chan A , Belton CM , Izutsu KT , Vasel D , Weinberg A ((1995) ) Porphyromonas gingivalis invasion of gingival epithelial cells. Infect Immun 63: , 3878–3885. |
[21] | Li L , Michel R , Cohen J , Decarlo A , Kozarov E ((2008) ) Intracellular survival and vascular cell-to-cell transmission of Porphyromonas gingivalis. BMC Microbiol 8: , 26. |
[22] | Rodrigues PH , Reyes L , Chadda AS , Belanger M , Wallet SM , Akin D , Dunn W Jr. , Progulske-Fox A ((2012) ) Porphyromonas gingivalis strain specific interactions with human coronary artery endothelial cells: A comparative study. PLoS One 7: , e52606. |
[23] | Walter C , Zahlten J , Schmeck B , Schaudinn C , Hippenstiel S , Frisch E , Hocke AC , Pischon N , Kuramitsu HK , Bernimoulin JP , Suttorp N , Krull M ((2004) ) Porphyromonas gingivalis strain-dependent activation of human endothelial cells. Infect Immun 72: , 5910–5918. |
[24] | Yamatake K , Maeda M , Kadowaki T , Takii R , Tsukuba T , Ueno T , Kominami E , Yokota S , Yamamoto K ((2007) ) Role for gingipains in Porphyromonas gingivalis traffic to phagolysosomes and survival in human aortic endothelial cells. Infect Immun 75: , 2090–2100. |
[25] | Imamura T , Travis J , Potempa J ((2003) ) The biphasic virulence activities of gingipains: Activation and inactivation of host proteins. Curr Protein Pept Sci 4: , 443–450. |
[26] | Olsen I , Potempa J ((2014) ) Strategies for the inhibition of gingipains for the potential treatment of periodontitis and associated systemic diseases. J Oral Microbiol 6: , doi: 10.3402/jom.v6.24800 |
[27] | Ortiz-Prado E , Dunn JF , Vasconez J , Castillo D , Viscor G ((2019) ) Partial pressure of oxygen in the human body: A general review. Am J Blood Res 9: , 1–14. |
[28] | Zhu J , Aja S , Kim EK , Park MJ , Ramamurthy S , Jia J , Hu X , Geng P , Ronnett GV ((2012) ) Physiological oxygen level is critical for modeling neuronal metabolism in vitro. J Neurosci Res 90: , 422–434. |
[29] | Diaz PI , Rogers AH ((2004) ) The effect of oxygen on the growth and physiology of Porphyromonas gingivalis. Oral Microbiol Immunol 19: , 88–94. |
[30] | Li L , Mendis N , Trigui H , Oliver JD , Faucher SP ((2014) ) The importance of the viable but non-culturable state in human bacterial pathogens. Front Microbiol 5: , 258. |
[31] | Rafferty B , Jonsson D , Kalachikov S , Demmer RT , Nowygrod R , Elkind MS , Bush H Jr. , Kozarov E ((2011) ) Impact of monocytic cells on recovery of uncultivable bacteria from atherosclerotic lesions. J Intern Med 270: , 273–280. |
[32] | Lleo MM , Pierobon S , Tafi MC , Signoretto C , Canepari P ((2000) ) mRNA detection by reverse transcription-PCR for monitoring viability over time in an Enterococcus faecalis viable but nonculturable population maintained in a laboratory microcosm. Appl Environ Microbiol 66: , 4564–4567. |
[33] | Trevors JT ((2011) ) Viable but non-culturable (VBNC) bacteria: Gene expression in planktonic and biofilm cells. J Microbiol Methods 86: , 266–273. |
[34] | Progulske-Fox A , Kozarov E , Dorn B , Dunn W Jr. , Burks J , Wu Y ((1999) ) Porphyromonas gingivalis virulence factors and invasion of cells of the cardiovascular system. J Periodontal Res 34: , 393–399. |
[35] | Campellone KG , Siripala AD , Leong JM , Welch MD ((2012) ) Membrane-deforming proteins play distinct roles in actin pedestal biogenesis by enterohemorrhagic Escherichia coli. J Biol Chem 287: , 20613–20624. |
[36] | Nixon RA , Wegiel J , Kumar A , Yu WH , Peterhoff C , Cataldo A , Cuervo AM ((2005) ) Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study. J Neuropathol Exp Neurol 64: , 113–122. |
[37] | Nixon RA ((2007) ) Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci 120: , 4081–4091. |
[38] | Mondragon-Rodriguez S , Perry G , Luna-Munoz J , Acevedo-Aquino MC , Williams S ((2014) ) Phosphorylation of tau protein at sites Ser(396-404) is one of the earliest events in Alzheimer’s disease and Down syndrome. Neuropathol Appl Neurobiol 40: , 121–135. |
[39] | Simic G , Babic Leko M , Wray S , Harrington C , Delalle I , Jovanov-Milosevic N , Bazadona D , Buee L , de Silva R , Di Giovanni G , Wischik C , Hof PR ((2016) ) Tau protein hyperphosphorylation and aggregation in Alzheimer’s disease and other tauopathies, and possible neuroprotective strategies. Biomolecules 6: , 6. |
[40] | Wang JZ , Xia YY , Grundke-Iqbal I , Iqbal K ((2013) ) Abnormal hyperphosphorylation of tau: Sites, regulation, and molecular mechanism of neurofibrillary degeneration. J Alzheimers Dis 33: (Suppl 1), S123–139. |
[41] | Cavallini A , Brewerton S , Bell A , Sargent S , Glover S , Hardy C , Moore R , Calley J , Ramachandran D , Poidinger M , Karran E , Davies P , Hutton M , Szekeres P , Bose S ((2013) ) An unbiased approach to identifying tau kinases that phosphorylate tau at sites associated with Alzheimer disease. J Biol Chem 288: , 23331–23347. |
[42] | Schwalbe M , Kadavath H , Biernat J , Ozenne V , Blackledge M , Mandelkow E , Zweckstetter M ((2015) ) Structural impact of tau phosphorylation at threonine 231. Structure 23: , 1448–1458. |
[43] | Scheff SW , Price DA , Ansari MA , Roberts KN , Schmitt FA , Ikonomovic MD , Mufson EJ ((2015) ) Synaptic change in the posterior cingulate gyrus in the progression of Alzheimer’s disease. J Alzheimers Dis 43: , 1073–1090. |
[44] | Bao K , Belibasakis GN , Thurnheer T , Aduse-Opoku J , Curtis MA , Bostanci N ((2014) ) Role of Porphyromonas gingivalis gingipains in multi-species biofilm formation. BMC Microbiol 14: , 258. |
[45] | Litersky JM , Johnson GV ((1992) ) Phosphorylation by cAMP-dependent protein kinase inhibits the degradation of tau by calpain. J Biol Chem 267: , 1563–1568. |
[46] | Barbier P , Zejneli O , Martinho M , Lasorsa A , Belle V , Smet-Nocca C , Tsvetkov PO , Devred F , Landrieu I ((2019) ) Role of tau as a microtubule-associated protein: Structural and functional aspects. Front Aging Neurosci 11: , 204. |
[47] | Kadavath H , Hofele RV , Biernat J , Kumar S , Tepper K , Urlaub H , Mandelkow E , Zweckstetter M ((2015) ) Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc Natl Acad Sci U S A 112: , 7501–7506. |
[48] | Qiang L , Sun X , Austin TO , Muralidharan H , Jean DC , Liu M , Yu W , Baas PW ((2018) ) Tau does not stabilize axonal microtubules but rather enables them to have long labile domains. Curr Biol 28: , 2181–2189 e2184. |
[49] | Franker MA , Hoogenraad CC ((2013) ) Microtubule-based transport - basic mechanisms, traffic rules and role in neurological pathogenesis. J Cell Sci 126: , 2319–2329. |
[50] | Tang Y , Scott D , Das U , Gitler D , Ganguly A , Roy S ((2013) ) Fast vesicle transport is required for the slow axonal transport of synapsin. J Neurosci 33: , 15362–15375. |
[51] | Boland B , Kumar A , Lee S , Platt FM , Wegiel J , Yu WH , Nixon RA ((2008) ) Autophagy induction and autophagosome clearance in neurons: Relationship to autophagic pathology in Alzheimer’s disease. J Neurosci 28: , 6926–6937. |
[52] | Von Bartheld CS , Altick AL ((2011) ) Multivesicular bodies in neurons: Distribution, protein content, and trafficking functions. Prog Neurobiol 93: , 313–340. |
[53] | Cemma M , Brumell JH ((2012) ) Interactions of pathogenic bacteria with autophagy systems. Curr Biol 22: , R540–545. |


