
Working Towards a Blood-Derived Gene Expression Biomarker Specific for Alzheimer’s Disease
Abstract
Background:
The typical approach to identify blood-derived gene expression signatures as a biomarker for Alzheimer’s disease (AD) have relied on training classification models using AD and healthy controls only. This may inadvertently result in the identification of markers for general illness rather than being disease-specific.
Objective:
Investigate whether incorporating additional related disorders in the classification model development process can lead to the discovery of an AD-specific gene expression signature.
Methods:
Two types of XGBoost classification models were developed. The first used 160 AD and 127 healthy controls and the second used the same 160 AD with 6,318 upsampled mixed controls consisting of Parkinson’s disease, multiple sclerosis, amyotrophic lateral sclerosis, bipolar disorder, schizophrenia, coronary artery disease, rheumatoid arthritis, chronic obstructive pulmonary disease, and cognitively healthy subjects. Both classification models were evaluated in an independent cohort consisting of 127 AD and 687 mixed controls.
Results:
The AD versus healthy control models resulted in an average 48.7% sensitivity (95% CI = 34.7–64.6), 41.9% specificity (95% CI = 26.8–54.3), 13.6% PPV (95% CI = 9.9–18.5), and 81.1% NPV (95% CI = 73.3–87.7). In contrast, the mixed control models resulted in an average of 40.8% sensitivity (95% CI = 27.5–52.0), 95.3% specificity (95% CI = 93.3–97.1), 61.4% PPV (95% CI = 53.8–69.6), and 89.7% NPV (95% CI = 87.8–91.4).
Conclusions:
This early work demonstrates the value of incorporating additional related disorders into the classification model developmental process, which can result in models with improved ability to distinguish AD from a heterogeneous aging population. However, further improvement to the sensitivity of the test is still required.
INTRODUCTION
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder affecting an estimated one in nine people over the age of 65 years of age, making it the most common form of dementia worldwide [1]. Current clinical diagnosis of the disease is primarily based on a time-consuming combination of physical, mental, and neuropsychological examinations. With the rapid increase in the prevalence of the disease, there is a growing need for a more accessible, cost-effective, and time-effective approach for diagnosing and monitoring AD.
For research purposes, brain positron emission tomography (PET) scans and cerebrospinal fluid can be used to suggest AD. In particular, decreased amyloid-β (Aβ) and increased tau levels in cerebrospinal fluid have been successfully used to distinguishing between AD, mild cognitive impairment, and cognitive healthy individuals with high accuracy. However, as a relatively invasive and costly procedure, it may not appeal to the majority of patients or be practical on a large-scale trial basis for screening the population [2–4]. A peripheral blood-derived biomarker for AD would be advantageous.
Blood is a complex mixture of fluid and multiple cellular compartments that are consistently changing in protein, lipid, RNA, and other biochemical entity concentrations [5], which may be useful for AD diagnosis. Recently, a study successfully used APP669–711/Aβ1–42 and Aβ1–40/Aβ1–42 ratios and their composites, to predict individual brain Aβ load when compared to Aβ-PET imaging [6]. However, the test predicts Aβ deposition, which is also found in other brain disorders such as frontotemporal dementia, and therefore, the test requires AD specificity evaluation. Another study reviewed 163 candidate blood-derived proteins from 21 separate studies as a potential biomarker for AD [7]. The overlap of biomarkers between studies was limited, with only four biomarkers, α-1-antitrypsin, α-2-macroglobulin, apolipoprotein E, and complement C3, found to replicate in five independent cohorts. However, a follow-on study discovered these biomarkers were not specific to AD, and were also discovered to be associated with other brain disorders including Parkinson’s disease (PD) and schizophrenia (SCZ) [8], once again, suggesting the need to consider other neurological and related disorders in study designs to enable the discovery of biomarkers specific to AD.
Several studies have also attempted to exploit blood transcriptomic measurements for AD biomarker discovery. Initial research was limited to the analysis of single differentially expressed genes (DEG) as a means to distinguish AD from cognitively healthy individuals [2, 9]. However, the limited overlap and reproducibility of DEG from independent cohorts suggests this method alone is not reliable enough [2]. A solution to this problem would be to use machine learning algorithms to identify combinations of gene expression changes that may represent a biomarker for AD. This technique has been applied in multiple studies, which have demonstrated to some extent, the ability to differentiate AD from non-AD subjects [3, 10–13]. However, small sample size and lack of independent validation datasets may have led to overfitting. The decrease in costs associated with microarray technologies led a study developing an AD classification model based on a larger training set of 110 AD and 107 controls and validating in an independent cohort of 118 AD and 118 controls. The model achieved 56% sensitivity, 74.6% specificity, and an accuracy of 66%, which equated to 69.1% positive predictive power (PPV) and 63% negative predictive power (NPV) [11]. This was one of the first studies to demonstrate some validation in an independent cohort; however, the classification model still lacked the 90% predictive power desired from a clinical diagnostic test [14].
Previous studies have demonstrated the potential use of blood transcriptomic levels to differentiate between AD and cognitively healthy individuals; however, they are yet to be precise enough for clinical utility and are yet to be extensively evaluated on specificity by assessing model performance in a heterogeneous aging population of multiple diseases. This validation process is critical to determine whether the classification model is indeed disease-specific, a general indication of ill health, or an overfit.
This study developed a microarray gene expression processing pipeline with reproducibility and clinical utility in mind. New subjects could be independently processed and predicted through the same classification models without using any prior knowledge on gene expression variation of the data used to develop the classification model and without making any alteration to the classification models itself. XGBoost classification models were developed using the typical approach of training in blood transcriptomic profiling from AD and cognitively healthy controls. The models were evaluated in an independent testing set mimicking a heterogeneous aging population consisting of AD, related mental disorders (PD, multiple sclerosis [MS], bipolar disorder [BD], SCZ), common elderly health disorders and other related diseases (coronary artery disease [CD], rheumatoid arthritis [RA], chronic obstructive pulmonary disease [COPD]), and cognitively healthy subjects to assess the models ability to distinguish AD from related diseases and otherwise healthy subjects. In addition, a second approach was used where XGBoost classification models were developed using AD, mental health disorders, common elderly health disorders, and cognitively healthy subjects. The second approach used independent non-AD samples, and was evaluated on the same independent testing set as the first approach to investigate the effects on model performance when incorporating additional related disorders into the AD classification development process.
METHODS
Data acquisition
Microarray gene expression studies were sourced from publicly available repositories Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/) and ArrayExpress (https://www.ebi.ac.uk/arrayexpress/) in May 2018. Study inclusion criteria were: 1) microarray gene expression profiling must be performed on a related, common elderly health, or mental health disorder; 2) RNA was extracted from whole blood or a component of blood; 3) study must contain at least ten human subjects; and 4) data was generated on either the Illumina or Affymetrix microarray platform using an expression BeadArray containing at least 20,000 probes. The microarray platform was restricted to Affymetrix and Illumina only, as replication between the two platforms is generally very high [15–18], and expression BeadArrays restricted to a minimum of 20,000 probes to maximize the overlap of genes across studies, while also optimizing the number studies available for inclusion.
Data processing
The data processing pipeline was designed with reproducibility and clinical utility in mind. New subjects could be independently processed and predicted through the same classification models without using any prior knowledge on gene expression variation of the data used to develop the classification model and without making any alteration to the classification models itself. All data processing was undertaken in RStudio (version 1.1.447) using R (version 3.4.4). Microarray gene expression studies were acquired from public repositories using the R packages “GEOquery” (version 2.46.15) and “ArrayExpress” (version 1.38.0). For longitudinal studies involving treatment effects, placebo subjects or initial gene expression profiling from baseline subjects before treatment were used. Studies consisting of multiple disorders were separated by disease into datasets consisting of diseased subjects and corresponding healthy controls if available.
Raw gene expression data generated on the Affymetrix platform were “mas5” background corrected using the R package “affy” (version 1.42.3), log2 transformed and then Robust Spline Normalized (RSN) using the R package “lumi” (version 2.16.0). Datasets generated on the Illumina platform were available in either a “raw format” containing summary probes and control intensities with corresponding p-values or a “processed format” where data had already been processed and consisted of a subset of probes and samples deemed suitable by corresponding study authors. When acquiring studies, preference was given to “raw format” data where possible, and when available, was “normexp” background corrected, log2 transformed, and quantile normalized using the “limma” R package (version 3.20.9).
Sex was then predicted using the R package “massiR” (version 1.0.1) and subjects with discrepancies between predicted and recorded sex removed from further analysis. Then, within each gender and disease diagnosis group of a dataset, probes above the “X” percentile of the log2 expression scale in over 80% of the samples were deemed “reliably detected”. To account for the variation of redundant probes across different BeadArrays, the “X” percentile threshold value was manually adjusted until a variety of robust literature defined house-keeping genes were correctly defined as expressed or unexpressed in their corresponding gender groups [19]. Any probe labelled as “reliably detected” in any group (based on gender and diagnosis) was taken forward for further analysis from all samples within that dataset. This process substantially eliminates noise [20] and ensures disease and gender-specific signatures are captured within each dataset.
Next, to ensure homogeneity within biological groups, outlying samples were iteratively identified and removed using the fundamental network concepts described in [21]. Finally, to enable cross-platform probes to be comparable, platform-specific probe identifiers were annotated to their corresponding universal Entrez gene identifiers using the appropriate BeadArray R annotation files; “hgu133plus2.db”, “hgu133a.db”, “hugene10sttranscriptcluster.db”, “illuminaHumanv4.db”, and “illuminaHumanv3.db”.
Cross-platform normalization and sample correlation analysis
A rescaling technique, the YuGene transform, was applied to each dataset independently to enable transcriptomic information between datasets to be directly comparable. YuGene assigns modified cumulative proportion value to each measurement, without losing essential underlying information on data distributions, allowing the transformation of independent studies and individual samples [22]. This enables new data to be added without global renormalization and allows the training and testing set to be independently rescaled. Common “reliably detected” probes across all processed datasets that contained both female and male subjects were extracted from each dataset and independently rescaled using the R package YuGene (version 1.1.5). YuGene transformation assigns a value between 0 and 1 to each gene, where 1 is highly expressed. As samples originated from publicly available datasets, potential duplicate samples may exist in this study. Therefore, correlation analysis was performed on all samples using the common probes to investigate duplicate samples across different studies.
Training set and testing set assignment
Multiple datasets from the same disease were available, allowing entire datasets to be assigned to either the “Training Set” for classification model development or the “Testing Set” for independent external validation. Larger datasets from the same disease were prioritized to the training set, allowing the machine learning algorithm to learn in a larger discovery set.
Individual subjects within the training and testing set were assigned a “0” class if the subject was AD or “1” if the subject was non-AD (includes healthy controls and non-AD diseased subjects). Grouping the non-AD subjects into a single class effectively mimics a large heterogeneous aging population where subjects may have a related mental disorder, neurodegenerative disease, common elderly health disorder, or are considered relatively healthy.
Classification model development
Two types of classification models were created. The first was developed using the typical approach, training in AD subjects and their associated cognitively healthy control samples only. This model is referred to as the “AD vs Healthy Control” classification model. The second classification model was developed using the same AD and healthy control samples used for the “AD vs Healthy Control” classification; however, additional related disorders and their associated healthy controls were introduced as additional controls. This model is referred to as the “AD vs Mixed Control” classification model.
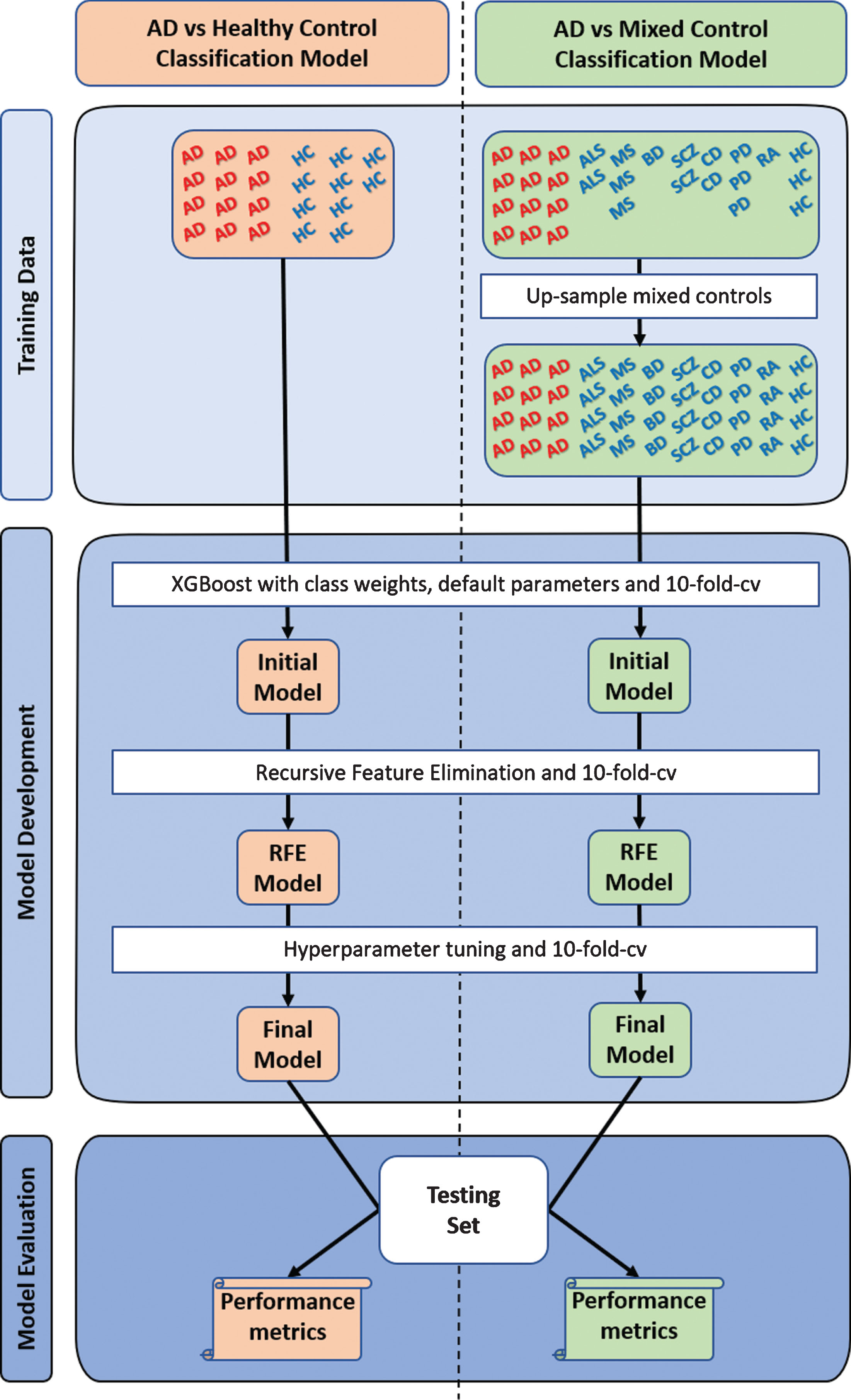
The control group of the “AD vs Mixed Control” classification model consisted of multiple diseases and their complementary healthy controls; however, the number of samples across the individual diseases in this mixed control group were unbalanced. As all non-AD samples would be assigned a “1”, the disorder with the largest number of samples would influence the classification model development process more. Therefore, to address this issue, all the complementary healthy subjects from all diseased dataset were assumed to be disease-free and were pooled to create a “pooled controls” set. Then, samples within each disorder were upsampled with replacement to match the total number of samples in the “pooled controls” group (excludes AD). This process balances the number of samples across disorders in the mixed control group, which essentially balances the probability of a sample being selected from any one of the non-AD diseases or “pooled controls” during the classification model development process. This process is further illustrated in Fig. 1.
Fig.1
Overview of study design. Two types of XGBoost classification models were developed, optimized, and evaluated. The first (“AD vs Healthy Control”) used the typical approach, training in Alzheimer’s disease (AD) and cognitively healthy controls (HC), while the second (“AD vs Mixed Control”) was trained in AD and a mixed controls group. The mixed control group consisted of Parkinson’s disease (PD), multiple sclerosis (MS), amyotrophic lateral sclerosis (ALS), bipolar disorder (BD), schizophrenia (SCZ), coronary artery disease (CD), rheumatoid arthritis (RA), chronic obstructive pulmonary disease (not represented in the figure), and cognitively healthy subjects. The individual groups within the mixed controls were upsampled with replacement to avoid sampling biases during model development. To account for the randomness, a thousand “AD vs Healthy Control” and a thousand “AD vs Mixed Control” classification models were developed and evaluated. cv, cross-validation; RFE, recursive feature elimination.
Classification models were built using the tree boosting algorithm, XGBoost, as implemented in the R package “xgboost” (version 0.6.4.1) [23]. The tree learning algorithm uses parallel and distributed computing, is approximately 10 times faster than existing methods, and allows several hyperparameters to be tuned to reduce the possibility of overfitting [24]. Default tuning parameters were set to eta = 0.3, max_depth = 6, gamma = 0, min_child_weight = 1, subsample = 1, colsample_bytree = 1, objective = “binary:logistic”, nrounds = 10000, early_stopping_rounds parameters = 20 and eval_metric = ”logloss”. Due to the unbalanced classes between AD and non-AD samples, the scale_pos_weight function was incorporated to assign weights to the smallest class, ensuring the machine learning algorithm did not bias towards the largest class during the classification model development. The initial model was built and internally evaluated using 10-fold cross-validation with stratification which calculates a test logloss mean at each nrounds iteration, stopping if an improvement to the test logloss means is not achieved in the last 20 iterations. The nrounds iteration that achieved the optimal test logloss mean was used to build the initial classification model, reducing the chance for an “overfit” model.
During the internal cross-validation process, each feature (gene) was assigned an importance value (“variable importance feature”), which is based on how well the gene contributed to the correct prediction of individuals in the training set. The higher the variable importance value for a gene, the more useful that gene was in distinguishing AD subjects from non-AD individuals. The genes contributing to the initial XGBoost model were each assigned a variable importance value. The least two variable important features were then iteratively removed, classification models re-built, and logloss performance measures re-evaluated. This process was repeated through all available baseline features, with the minimum logloss from all iterations used to determine the most predictive genes. This process is referred to as “recursive feature elimination” and has been shown to improve classification model performance and reduce model complexity by removing weak and non-predictive features [25].
Following the identification of the most predictive genes, the classification model was further refined by iteratively tuning through the following hyperparameter values: max_depth (2 : 20, 1), min_child_weight (1 : 10, 1), gamma (0 : 10, 1), subsample (0.5 : 1, 0.1), colsample_bytree (0.5 : 1, 0.1), alpha (0 : 1, 0.1),), lambda (0 : 1, 0.1), and eta (0.01 : 0.2, 0.01), while performing a 10-fold cross-validation with stratification and evaluating the test logloss mean to select the optimum hyperparameters. Finally, for reproducibility purposes, the same seed number was consistently used throughout the upsampling and model development process. However, to account for the randomness introduced during the bootstrap upsampling and model development processes, and to provide an insight into the stability of the results, a thousand “AD vs Healthy Control” and a thousand “AD vs Mixed Control” classification models were developed, refined, and evaluated. Upsampling and model development was performed using a different seed number ranging from 1 : 1000. This would ensure the subjects that were upsampled were randomized across the 1,000 different “AD vs Mixed Control” classification models, and as each classification model was initially developed using a different randomized number, this would result in 1,000 different classification models that attempt to solve the same problem.
Classification model evaluation
Each classification model was validated on the independent unseen testing set, predicting the diagnosis of all subjects as a probability ranging from 0 to 1, where AD≤0.5 > non-AD. The prediction accuracy, sensitivity, specificity, PPV, and NPV were calculated to evaluate the overall classification model’s performance. To aid in the interpretation of the sensitivity and specificity of the classifiers, AUC scores were generated using the R package “ROCR” (version 1.07) with the following recommended diagnostic interpretations used: “excellent” (AUC = 0.9–1.0), “very good” (AUC = 0.8–0.9), “good” (AUC = 0.7–0.8), “sufficient“ (AUC = 0.6–0.7), “bad” (AUC = 0.5–0.6), and “test not useful” when AUC value is < 0.5 [26].
Furthermore, the clinical utility metrics were calculated to evaluate the clinical utility of the classification models. The positive Clinical Utility Index (CUI+) was calculated as PPV * (sensitivity/100) and the negative Clinical Utility Index (CUI –) calculated as NPV * (sensitivity/100). The Clinical Utility Index (CUI) essentially corrects the PPV and NPV values for occurrence of that test in each respective population and scores can be converted into qualitative grades as recommended: “excellent utility” (CUI≥0.81), “good utility” (CUI≥0.64) and “satisfactory utility” (CUI≥0.49) and “poor utility” (CUI < 0.49) [27]. As a thousand “AD vs Healthy Control” and a thousand “AD vs Mixed Control” classification models were evaluated, the average performance for each metric is calculated along with the 95% confidence interval (CI). An overview of the classification model development and evaluation process is provided in Fig. 1.
The biological importance of predictive features
The “AD vs Mixed Control” classification models contain a list of ranked genes derived from analyzing multiple disorders, which collectively attempt to differentiate AD from non-AD subjects. The predictive genes were analyzed using an Over-Representation Analysis (ORA) implemented through the ConsensusPathDB (http://cpdb.molgen.mpg.de) web-based platform (version 33) [28] in November 2018 to assess their collective biological significance. For pathway enrichment analysis, a background gene list was included, and a minimum overlap of the query signature and database was set as 2.
Data availability
The data used in this study were all publicly available with accession details provided in Table 1. All analysis scripts used in this study are available at https://doi.org/10.5281/zenodo.3371459.
Table 1
Dataset demographics
| Disorder | Study ID (associated publication) | Platform | BeadArray | Tissue source | Demographics before QC | Samples removed during QC | Demographics after QC | Training and testing set assignment | |||||||
| No. probes | Case sex (M/F) | Control sex (M/F) | No. samples | No. gender mismatches | No. outlying sample | No. probes | Case sex (M/F) | Control sex (M/F) | No. samples | ||||||
| Alzheimer’s Disease | GSE63060 ([31]) | I | HT-12 v3.0 | WB | 38323 | 46/99 | 42/62 | 249 | 2 | 10 | 5364 | 45/93 | 40/59 | 237 | Training |
| GSE63061 ([31]) | I | HT-12 v4.0 | WB | 32049 | 51/81 | 55/87 | 274 | 5 | 4 | 5241 | 48/79 | 54/84 | 265 | Testing | |
| E-GEOD-6613 ([32]) | A | HG U133A | WB | 22283 | 8/15 | 11/11 | 45 | 0 | 1 | 4184 | 8/14 | 11/11 | 44 | Training | |
| Parkinson’s Disease | E-GEOD-6613 ([32]) | A | HG U133A | WB | 22283 | 38/12 | 0/0 | 50 | 0 | 0 | 3674 | 38/12 | 0/0 | 50 | Training |
| E-GEOD-72267 ([33]) | A | HG U133A 2.0 | PBMC | 22277 | 23/17 | 8/11 | 59 | 0 | 0 | 8742 | 23/17 | 8/11 | 59 | Testing | |
| Multiple Sclerosis | GSE24427 ([34]) | A | HG U133A | WB | 22283 | 9/16 | 0/0 | 25 | 0 | 0 | 6633 | 9/16 | 0/0 | 25 | Testing |
| E-GEOD-16214 ([35]) | A | HG U133 plus 2.0 | PBMC | 54675 | 11/71 | 0/0 | 82 | 0 | 3 | 8098 | 11/68 | 0/0 | 79 | Training | |
| E-GEOD-41890 ([36]) | A | Exon 1.0 ST | PBMC | 33297 | 20/24 | 12/12 | 68 | 0 | 1 | 8157 | 19/24 | 12/12 | 67 | Training | |
| Schizophrenia | GSE38484 ([37]) | I | HT-12 v3.0 | WB | 48743 | 76/30 | 42/54 | 202 | 9 | 5 | 6700 | 69/28 | 39/52 | 188 | Training |
| E-GEOD-27383 ([38]) | A | HG U133 plus 2.0 | WB | 54675 | 43/0 | 29/0 | 72 | 0 | 1 | 11297 | 42/0 | 29/0 | 71 | Testing | |
| GSE38481 ([37]) | I | Human-6 v3 | WB | 24526 | 4/11 | 16/6 | 37 | 2 | 1 | 8106 | 11/3 | 15/5 | 34 | Testing | |
| Bipolar Disorder | E-GEOD-46449 ([39]) | A | HG U133 plus 2.0 | L | 54675 | 28/0 | 25/0 | 53 | 0 | 0 | 9882 | 28/0 | 25/0 | 53 | Training |
| GSE23848 ([40]) | I | Human-6 v2 | WB | 48701 | 6/14 | 5/10 | 35 | 0 | 0 | 7211 | 6/14 | 5/10 | 35 | Testing | |
| Cardiovascular Disease | E-GEOD-46097 ([41]) | A | HG U133A 2.0 | PBMC | 22277 | 102/36 | 60/180 | 378 | 0 | 24 | 7676 | 94/36 | 57/167 | 354 | Training |
| GSE59867 ([42]) | A | Exon 1.0 ST | WB | 33297 | 85/26 | 0/0 | 111 | 0 | 3 | 7936 | 82/26 | 0/0 | 108 | Testing | |
| E-GEOD-12288 ([43]) | A | HG U113A | WB | 22283 | 88/22 | 84/28 | 222 | 0 | 8 | 4815 | 83/22 | 82/27 | 214 | Training | |
| Rheumatoid Arthritis | E-GEOD-74143 ([44]) | A | HT HG U113 plus | WB | 54715 | 81/296 | 0/0 | 377 | 1 | 23 | 8112 | 80/273 | 0/0 | 353 | Training |
| E-GEOD-54629 ([45]) | A | Exon 1.0 ST | WB | 33297 | 11/58 | 0/0 | 69 | 0 | 0 | 11931 | 11/58 | 0/0 | 69 | Testing | |
| E-GEOD-42296 ([46]) | A | Exon 1.0 ST | PBMC | 33297 | 4/15 | 0/0 | 19 | 0 | 0 | 10417 | 4/15 | 0/0 | 19 | Testing | |
| Chronic Obstructive Pulmonary Disease | E-GEOD-54837 ([47]) | A | HG U133 plus 2.0 | WB | 54675 | 91/45 | 57/33 | 226 | 0 | 16 | 5531 | 83/44 | 52/31 | 210 | Training |
| E-GEOD-42057 ([48]) | A | HG U133 plus 2.0 | WB | 54675 | 52/42 | 22/20 | 136 | 3 | 4 | 6445 | 49/39 | 21/20 | 129 | Testing | |
| ALS | E-TABM-940 | A | HG U133 plus 2.0 | WB | 54675 | 27/26 | 18/19 | 90 | 3 | 10 | 10442 | 27/25 | 15/10 | 77 | Training |
| Total | 904/956 | 486/533 | 2879 | 25 | 114 | 870/906 | 465/49 | 2740 | |||||||
Each study is accompanied by its corresponding publication (if available), where individual study design can be obtained. When possible, datasets were obtained in their raw format, except for GSE63060, GSE63061, E-GEOD-41890, GSE23848, E-GEOD74143, E-GEOD-54629, and E-GEOD-42296 which were only available in a processed form where the dataset had already been background corrected, log2 transformed, and normalized by techniques stated in corresponding publications. Multiple datasets from the same disease existed in this study. The dataset with the largest number of diseased subjects was prioritized into the training set for better discovery. Study IDs initiating with “GSE” and “E-GEOD” were obtained from GEO and ArrayExpress, respectively. I, Illumina; A, Affymetrix; WB, whole blood; PBMC, peripheral blood mononuclear cell; L, lymphocytes.
RESULTS
Summary of data processing
Twenty-one publicly available studies were identified, acquired, and processed. Separating studies by disease status resulted in 22 datasets, which consisted of 3 AD, 3 MS, 3 SCZ, 3 CD, 3 RA, 2 COPD, 2 BD, 2 PD, and 1 ALS orientated dataset. Fifteen datasets contained both diseased and complementary healthy subjects, and the remaining 7 contained only diseased subjects. An overview of the demographics of each dataset is provided in Table 1.
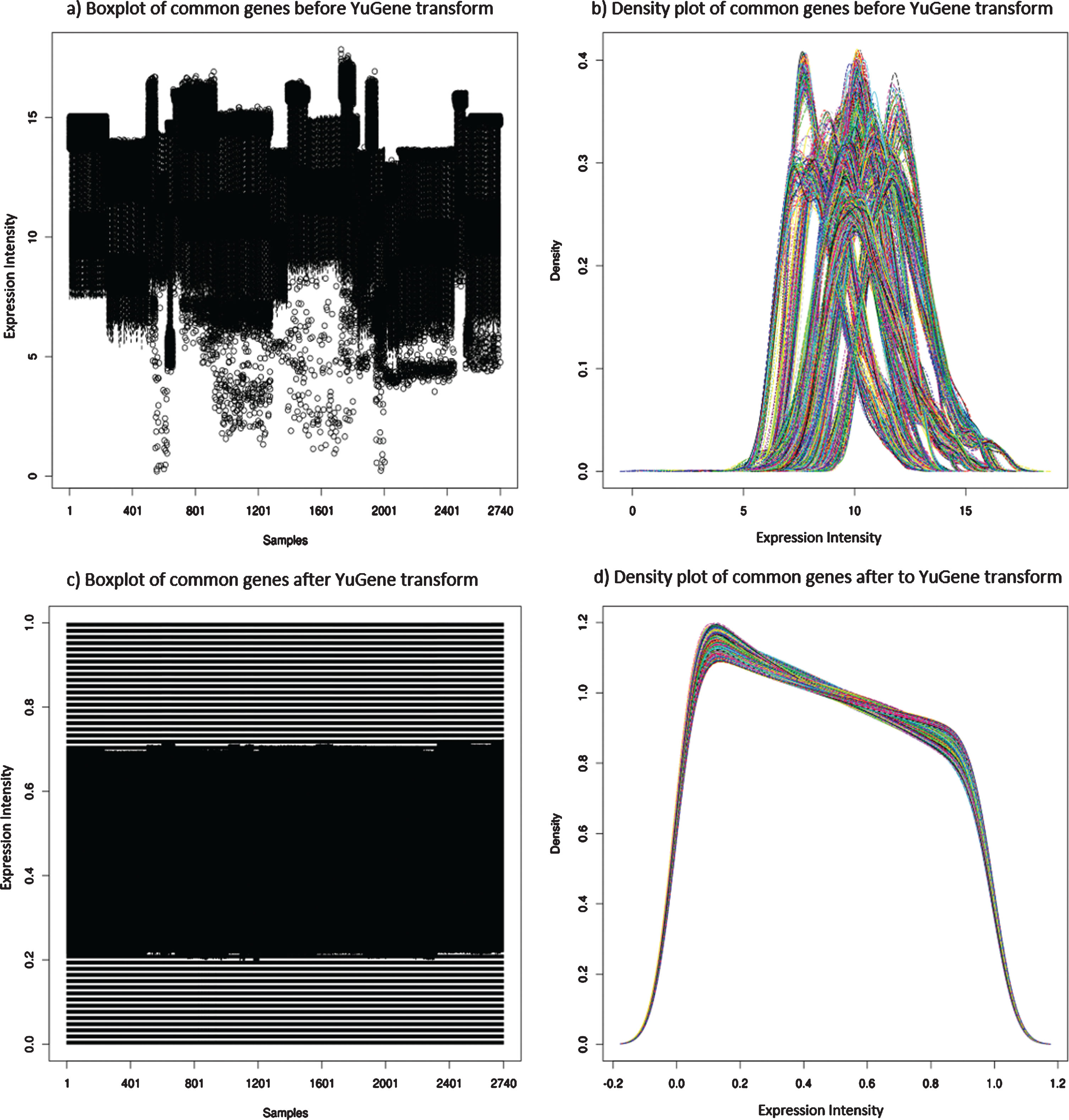
Independently processing the 22 datasets resulted in a total of 2,740 samples after quality control (QC), of which 287 samples were AD. Since 11 different BeadArrays had been used to expression profile the 9 different diseases, and as 7 datasets were only available in a “processed format” (GSE63060, GSE63061, E-GEOD-41890, GSE23848, E-GEOD74143, E-GEOD-54629, and E-GEOD-42296), each dataset varied in the number of “reliably detected” genes after QC (detailed in Table 1). Initially, any probe deemed “reliably detected” in any one of the 22 datasets was compiled, resulting in 7,452 genes. In theory, this would ensure all measurable sex and disease-specific genes were potentially captured within the data. However, following the independent transformation of each dataset, platform and BeadArray-specific batch effects were observed. This can be primarily explained by different platforms having different probe designs to target different transcripts of the same gene, leading to significant discrepancies and even absence in the measurement of the same gene by different platforms [15]. Therefore, to address this platform and BeadArray-specific batch effect, 1,681 common “reliably detected” genes across all datasets that contained both male and female subjects (20 datasets) were extracted from each dataset and independently YuGene transformed. Essentially, these 1,681 genes are expressed at a level deemed “reliably detected” in all 11 different BeadArrays and across both male and female subjects. The expression distribution of the,1681 genes in each subject is shown in Figure 2. The variation across the 1,681 “reliably detected” genes prior to YuGene transform is significantly different across samples and datasets (Fig. 2a,b), making the data from different datasets and microarray platforms incomparable. However, this was addressed by independently normalizing each sample using only the 1,681 “reliably detected” common genes, which resulted in a more evenly distributed gene expression profile across all samples (Fig. 2c,d), a characteristic desired by machine learning algorithms.
Fig.2
Distribution of gene expression across all 2,740 subjects in this study. Plots a) and c) are boxplots, where each vertical line represents an individual, while plots b) and d) represents the expression density of the same 2,740 subjects where each line represents a different individual. Plots a) and b) shows the variation of the gene expression across subjects prior to YuGene transformation, providing evidence of batch effects between samples and datasets. In contrast, plots c) and d) reveals a more evenly distributed gene expression profile across all 2,740 subjects when extracting the 1,681 common “reliably detected” genes, and independently YuGene transforming each sample.
Correlation analysis was then performed on all samples, which suggested all samples were highly correlated, with the maximum per sample correlation coefficients ranging from 0.86–0.99. No sample was deemed to be a duplicate, and therefore, no additional sample was removed following QC.
Training set and testing set demographics
Multiple datasets from the same disease were obtained in this study, with the largest dataset from each disease assigned to the training set to improve discovery. However, three AD datasets were available, and the two largest datasets were generated on the Illumina platform with the third originating from the Affymetrix platform. To address any subtle differences in gene expression, which may still exist in the data due to platform differences, the largest Illumina AD and the Affymetrix AD datasets were both assigned to the training set.
Following dataset assignment, the training set consisted of 160 AD subjects and 1,766 non-AD subjects, while the testing set consisted of 127 AD subjects and 687 Non-AD subjects. The Non-AD group in both the training and testing set consisted of subjects with either PD, MS, SCZ, BD, CD, RA, COPD, or were relatively healthy. Only one ALS dataset suitable for this study was identified and was deemed too small to split into the training and testing set. Therefore, the ALS dataset was assigned to the training set, allowing the machine learning algorithm to learn multiple disease expression signatures, which could further aid in differentiating AD from Non-AD subjects.
Upsampling was performed on the mixed control group to balance the number of samples across the individual diseases, preventing bias toward the majority classes during model development. The “pooled controls” contained 702 samples, and was the largest group in the training set; therefore, the remaining diseases were upsampled to the same number. This resulted in the “AD vs Mixed Controls” being trained on 160 AD samples and 6,318 non-AD samples. An overview of subjects in the training and testing set is provided in Table 2.
Table 2
Overview Training and Testing set subjects
| Dataset | Training set | Testing set | Class assignment for XGBoost | |
| AD vs Healthy Control | AD vs Mixed Control | |||
| Alzheimer’s Disease | 160* | 160* | 127 | 0 |
| Parkinson’s Disease | 0 | 702 (50) | 40 | 1 |
| Multiple Sclerosis | 0 | 702 (122*) | 25 | 1 |
| Schizophrenia | 0 | 702 (97*) | 56* | 1 |
| Bipolar Disorder | 0 | 702 (28) | 20 | 1 |
| Cardiovascular Disease | 0 | 702 (235*) | 108 | 1 |
| Rheumatoid Arthritis | 0 | 702 (353) | 88* | 1 |
| Chronic Obstructive Pulmonary Disease | 0 | 702 (127) | 88 | 1 |
| ALS | 0 | 702 (52) | 0 | 1 |
| Pooled Controls | 127* | 702* | 262 | 1 |
Entire datasets from each disease were assigned to either the “Training Set” for classification model development or the “Testing Set” for validation purposes. Datasets with the larger number of diseased subjects were prioritized into the training set to increase discovery. Two types of classification models were developed, the first (“AD vs Healthy Control”) was developed using only the 160 AD and associated 127 healthy control samples, and the second (“AD vs Mixed Controls”) was developed using the same 160 AD samples, and 6,318 upsampled mixed controls. The pooled controls in the “AD vs Healthy Control” training set originates only from AD datasets. Sample numbers provided in brackets are before upsampling. Sample numbers with an asterisk (*) indicates multiple datasets were available, and subject numbers shown are a sum across these datasets.
The “AD vs Healthy Control” classification model development and performance
The “AD vs Healthy Control” classification models were developed using the only two AD datasets (GSE63060 and E-GEOD-6613) available in the training set, which consisted of 160 AD and 127 cognitively healthy controls. A thousand models were developed, refined and evaluated, each using a different seed number. The models were initially built using default parameters, however, after model refinement, an average of 57 predictive genes (95% CI = 18–101) were selected with optimum hyperparameters identified as eta = 0.13 (95% CI = 0.02–0.2), max_depth = 6.3 (95% CI = 5–10), gamma = 0.2 (95% CI = 0–1.5), min_child_weight = 1.01 (95% CI = 1–1), subsample = 0.99 (95% CI = 0.95–1), colsample_bytree = 0.99 (95% CI = 0.8–1), alpha = 0.1 (95% CI = 0–0.8), lambda = 0.9 (95% CI = 0.2–1), and nrounds = 54.4 (95% CI = 18–211).
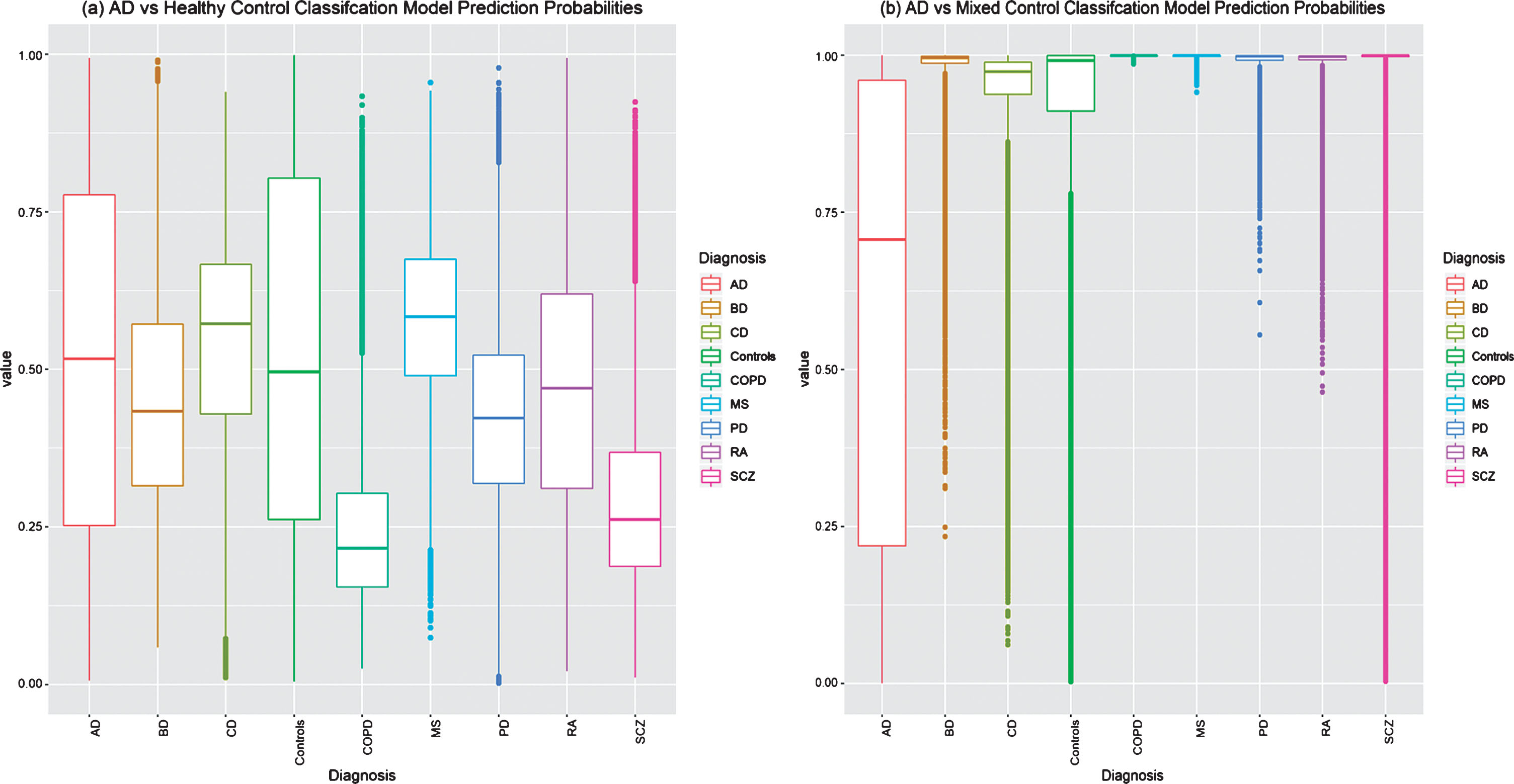
The “AD vs Healthy Control” classification models were evaluated in the independent testing set and achieved an average sensitivity of 48.7% (95% CI = 34.7–64.6), a specificity of 41.9% (95% CI = 26.8–54.3), and a balanced accuracy of 45.3% (95% CI = 36.0–56.0). Additional classification performance metrics are provided in Table 3. As this model was developed and evaluated a thousand times, each sample in the testing set was predicted a thousand times, each by a different classification model. The raw probability predictions of all the samples in the testing set by each of the thousand “AD vs Healthy Control” classification models are shown in Figure 3a, where high misclassification can be observed in all disease groups and controls, demonstrating an increased false-positive rate and the inability of the classification models to confidently assign a positive (0) or negative (1) class to each subject.
Table 3
Classification model performance
| AD vs Healthy Control | AD vs Mixed Control | |
| Sensitivity | 48.7% (34.7–64.6) | 40.8% (27.5–52.0) |
| Specificity | 41.9% (26.8–54.3) | 95.22% (93.3–97.1) |
| PPV | 13.6% (9.9–18.5) | 61.35% (53.8–69.6) |
| NPV | 81.1% (73.3–87.7) | 89.7% (87.8–91.4) |
| Balanced Accuracy | 45.3% (36.0–56.0) | 67.99% (61.9–72.9) |
| AUC | 0.45 (0.34–0.60) | 0.86 (0.82–0.90) |
| AUC Rating | Test not useful | Very Good |
| CUI+ve | 0.07 (0.04–0.12) | 0.25 (0.16–0.32) |
| CUI+ve Rating | Poor | Poor |
| CUI –ve | 0.34 (0.2–0.46) | 0.85 (0.84–0.87) |
| CUI –ve Rating | Poor | Excellent |
The table provides the average performance measurements form validating a thousand “AD vs Healthy Control” and a thousand “AD vs Mixed Control” classification models on the same testing set. A students T-test between the “AD vs Healthy Control” and “AD vs Mixed Control” classification performances reveals a significant difference for all metrics (p < 2.20e–16). The values provided in brackets () are the 95% confidence interval.
Fig.3
Testing set raw prediction comparison by (a) the thousand “AD vs Healthy Control” classification models and (b) the thousand “AD vs Mixed Control” Classification models. Samples with a probability of≤0.5 are predicted to be AD. Controls represent pooled non-diseased subjects from all datasets. AD, Alzheimer’s disease; BD, bipolar disease; CD, coronary artery disease; COPD, chronic obstructive pulmonary disease; MS, multiple sclerosis; PD, Parkinson’s disease; RA, rheumatoid arthritis; SCZ, schizophrenia.
The average AUC was calculated as 0.45 (95% CI = 0.34–0.60), which translates to “test is not useful” as a diagnostic test [26]. The average positive (CUI+ve) and negative (CUI –ve) clinical utility values are calculated as 0.07 (95% CI = 0.04–012) and 0.34 (95% CI = 0.2–0.46), respectively. These clinical utility scores suggest the classification model is “poor” at detecting the presence and absence of AD, and based on current validation results, has no real clinical utility [27].
The “AD vs Mixed Control” classification model development and performance
The thousand “AD vs Mixed Control” classification models were developed using the entire training set, which, after bootstrap upsampling, consisted of 160 AD and 6,318 non-AD subjects. The models were initially built using default parameters; however, after model refinement, an average of 89.4 predictive genes (95% CI = 66.0–116.0) were selected with the optimum hyperparameters identified as eta = 0.12 (95% CI = 0.01–0.20), max_depth = 4.1 (95% CI = 2–5), gamma = 0 (95% CI = 0–0), min_child_weight = 1 (95% CI = 1–1), subsample = 1 (95% CI = 0.95–1), colsample_bytree = 0.77 (95% CI = 0.5–1), alpha = 0.02 (95% CI = 0–0.1), lambda = 0.9 (95% CI = 0.1–1), and nrounds = 1173.1 (95% CI = 297.9–6956.3).
The “AD vs Mixed Control” classification models were evaluated in the testing set and achieved an average 40.8% (95% CI = 27.5–52.0) sensitivity, 95.2% (95% CI = 93.3–97.1) specificity, and a balanced accuracy of 68.0% (95% CI = 61.9–72.9). Additional classification performance metrics are provided in Table 3. A students T-test detects a significant difference (p < 2.20e–16) between all of the “AD vs Healthy Control” and “AD vs Mixed Control” performance metrics. The “AD vs Mixed Control” classification performance outperforms the typical “AD vs Healthy Control” classification models in all performance metrics, except for sensitivity, where a decrease in performance is observed from 48.7% to 40.8%. Nevertheless, due to the “AD vs Mixed Control” classification model predicting less false positives, an increase in the average PPV (61.4%, 95% CI = 53.8–69.6) is observed when compared to the “AD vs Healthy Control” classification models average PPV (13.6%, 95% CI = 9.9–18.5). This is further emphasized in Fig. 3b, where the raw probability predictions for all individuals in the testing set are more correctly and confidently predicted by the “AD vs Mixed Control” Classification models when compared to the typical “AD vs Healthy Control” classification models.
The “AD vs Mixed Control” classification model average AUC score is 0.86 (95% CI = 0.82–0.9) which translates to a “very good” diagnostic test [26]; however, the average clinical utility values (CUI+ve = 0.25 [95% CI = 01.6–032] and CUI –ve = 0.85 [95% CI = 0.84–0.87]) suggests this classification model is “poor” in detecting AD but “excellent” to rule out “AD” [27].
The “AD vs Mixed Control” classification model’s predictive features
The thousand “AD vs Mixed Control” classification models identified, on average, 89 predictive features (genes) to discriminate between AD and non-AD subjects with an average balanced accuracy of 68% (95% CI = 61.9–72.9). Only 800 of the 1,681 available genes were selected by anyone of the thousand models as a predictive feature, with 11 being consistently selected by all one thousand models. These 11 genes are KDM3B, TH1L, RARA, SPEN, NDUFA1, THYN1, UBR4, BSDC1, LDHB, LPP, and BAG5. Gene set enrichment on these genes identified “The citric acid (TCA) cycle and respiratory electron transport” (q-value = 0.03) and HIV Infection (q-value = 0.03) as the only biological pathways significantly enriched; however, when incorporating a background gene list (the 1,681 genes available for selection by the classification model algorithm), no pathway was significantly enriched.
DISCUSSION
Previous attempts to identify a blood-derived gene expression signature for AD diagnosis have relied on the typical approach of training machine learning algorithms on AD and cognitively healthy subjects only. This may inadvertently lead to classification models learning expression signatures that may be of general illness rather than being disease-specific. Validating such a classification model in a heterogeneous aging population may fail to distinguish AD from similar mental health disorders, neurodegenerative diseases, and common elderly health disorders. To explore this potential issue, two AD classification models were developed and evaluated. The first model (“AD vs Healthy Control”) was developed in 160 AD and 127 complementary cognitive healthy subjects, and the second (“AD vs Mixed Control”) was developed in 160 AD and 6,318 upsampled non-AD subjects comprising of PD, MS, BD, SCZ, CD, RA, COPD, ALS, and healthy subjects.
Both types of classification models were evaluated in the same external independent cohort comprising of AD, PD, MS, BD, SCZ, CD, RA, COPD, and healthy subjects totaling 814 subjects. A thousand “AD vs Healthy Control” and a thousand “AD vs Mixed Control” classification models were developed, refined, and evaluated to account for the randomness introduced during the bootstrap upsampling and the model development process.
The “AD vs Healthy Control” classification models perform poorly in a heterogeneous aging population
The typical approach of developing a classification model trained on AD and complementary cognitive healthy control subjects produced models with an average sensitivity of 48.7% (95% CI = 34.7–64.6) in an independent cohort of 127 AD subjects. On average, these models perform worse than a previous attempt which attained a sensitivity of 56.8% when validated in an independent testing set of 118 AD subjects [11]. However, the study in question only built and evaluated a single model and in this study, 97/1000 models attained a higher sensitivity. Nevertheless, on average, the “AD vs Healthy Control” models in this study are very much similar to identifying AD samples based on complete randomness alone (assumed to be 50%). Furthermore, when evaluating these models in a heterogeneous aging population, a process often neglected by previous studies, low average specificity of 41.9% (95% CI = 26.8–54.3) was attained, which equates to a very low average PPV of only 13.6% (26.8–54.3). This is reiterated in the high misclassification of PD, MS, BD, SCZ, CD, RA, COPD, and healthy subjects as AD in the testing set. Since misclassification was observed in all groups, including large portions of the healthy controls, the “AD vs Healthy Control” classification models are most likely not capturing signals of AD, dementia, or general illness, but is most likely a result of technical noise, individual study batch effects, and overfitting. This is mirrored in the model’s performance metrics, which translates to a “poor” clinical utility in detecting the presence and absence of AD. Overall, the typical approach of AD classification model development failed to accurately distinguish AD subjects in a heterogeneous aging population consisting of PD, MS, BD, SCZ, CD, RA, COPD, ALS, and relatively healthy controls.
The “AD vs Mixed Control” classification models outperforms the typical “AD vs Healthy Control” classification models
The “AD vs Mixed Control” classification models attained a validation PPV average of 61.4% (95% CI = 53.8–69.6) and an NPV average of 89.7% (95% CI = 87.8–91.4), which outperforms the validation PPV average of 13.6% (26.8–54.3) and NPV average of 81.1% (73.3–87.7) achieved by the “AD vs Healthy Control” classification models. However, this improvement was at the cost of sensitivity, which was reduced from an average of 48.7% (“AD vs Healthy Control”) to an average of 40.8% (“AD vs Mixed Control”). Nevertheless, an overall increase in the clinical utility of the “AD vs Mixed Control” classification model was measured and according to the recommended CUI interpretations in [27], the model is “poor” in “ruling in” AD but “excellent” in “ruling out” AD.
The increase performance of the “AD vs Mixed Control” classification model is most likely the result of incorporating additional related mental health and common elderly health disorders into the classification model development process, which allowed the machine learning algorithm to learn more complex relationships between genes to differentiate between AD and non-AD subjects. This is reflected in the average 57 (95% CI = 18–101) genes and 54 (95% CI = 18–211) nrounds (trees) being used for prediction in the “AD vs Healthy Control” classification models, which is increased to an average 89 (95% CI = 66–116) genes and 1173 (95% CI = 298–6956) nrounds for the “AD vs Mixed Control” classification models. Together with the CUI interpretations, the classification model seems to have learned expression signatures that are typically not AD, rather than identifying AD. Although this has improved the ability to distinguish AD from other related diseases and cognitively healthy controls, the sensitivity of the model was reduced and needs to be further improved for this type of research to be beneficial in the clinical setting.
Predictive features consist of age-related markers
Age is one of the most significant risk factors for AD, and the prevalence of the disease is known to increase with age. A meta-analysis study investigating blood transcriptional changes associated with age in 14,983 humans, identified 1,496 differentially expressed genes with chronical age [29], of which two genes (LDHB and LPP) are consistently used as a predictive feature in all one thousand “AD vs Mixed Control” classification models. The datasets used in this study were publicly available, and as such, were accompanied with limited phenotypic information, including age. Therefore, age was not accounted for during the classification model developmental process. However, as this study uses a variety of common elderly health disorders, in addition to the 3 AD datasets, and study designs generally incorporate complementary age-matched controls, it is highly unlikely the classification model is predicting age alone but is more likely using a combination of signals including age to distinguish AD. Without age information for all subjects, this study is unable to conclude how age is influencing the model prediction process.
Limitations
All data used in this study were publicly available, and as such, many were accompanied by limited phenotypic information, including sex, which was predicted based on gene expression when missing. Therefore, this study was unable to incorporate additional phenotypic information during the classification model building process, which has been shown to improve model performance [11]. Information such as comorbidities, age, and medications are unknowns, which could be affecting model performances in this study. For instance, control subjects in this study that originated from non-AD datasets were screened negative for their corresponding disease of interest but were not screened for cognitive function, i.e., control subjects from the CD datasets were included in their retrospective dataset if they did not have CD, they were not necessarily checked for cognitive impairment. Therefore, some misclassified control subjects may indeed be on the AD spectrum, and it is important to note subjects from the pooled control group were most misclassified as AD by the “AD vs Mixed Control” classification models. However, it is also important to note the training set used to develop the “AD vs Mixed Control” classification model also contains these controls which have not been screened for AD. If these controls or age-related disease subjects are comorbid with AD, the classification model may have inadvertently learned to be biased toward a subgroup of AD subjects with no comorbid with any other disease, hence the low sensitivity validation performance when introducing additional datasets into the classification model developmental process.
This study involved a number of subjects clinically diagnosed with a health issue, and therefore were most likely on some sort of therapeutic treatment to manage or treat the underlying disease, another piece of vital information generally missing from publicly available datasets and from this study. As therapeutic drugs have been well-known to affect gene expression profiling, including memantine, a common drug used to treat AD symptoms [30], the “AD vs Mixed Control” classification models may have inadvertently learned gene expression perturbations due to therapeutic treatment rather than disease biology, and would, therefore, fail in the clinical setting to diagnose AD subjects who are not already on medication. To address this issue along with co-morbidity, clear and detailed phenotypic information would be needed for all subjects, which is encouraged for future studies planning to submit genetic data to the public domain.
Finally, this study used datasets generated on 11 different microarray BeadArrays, resulting in datasets ranging from 22277–54715 probes prior to any QC. Coupled with differences in BeadArrays designs across platforms, the overlap of genes was drastically reduced to 1,681 common “reliable detected” genes across all datasets, and most likely may have also inadvertently lost some disease-specific changes. To address this issue, these subjects need to be expression profiled on the same microarray platform and ideally the same expression BeadArray, which currently does not exist in the public domain. However, the advances in sequencing technologies, which can capture expression changes across the whole transcriptome, can potentially solve this issue and future studies are encouraged to replicate this study design with RNA-Seq data with detailed phenotypic information when/if available, albeit, this may bring new challenges.
Conclusion
This study relied on publicly available microarray gene expression data, which too often lacks detailed phenotypic information for appropriate data analysis and needs to be addressed by future studies. Nevertheless, with the available phenotypic information and limited common “reliably detected” genes across the different microarray platforms and BeadArrays, this study demonstrated the typical approach of developing an AD blood-based gene expression classification model using only AD and complementary healthy controls fails to accurately distinguish AD from a heterogeneous aging population. However, by incorporating additional related mental health and common elderly health disorders from different microarray platforms and expression chips into the classification model development process can result in a model with improved “predictive power” in distinguishing AD from a heterogeneous aging population. Nevertheless, further improvement is still required in order to identify a robust blood transcriptomic signature more specific to AD.
ACKNOWLEDGMENTS
This study presents independent research supported by the NIHR BioResource Centre Maudsley at South London and Maudsley NHS Foundation Trust (SLaM) & Institute of Psychiatry, Psychology and Neuroscience (IoPPN), King’s College London. The views expressed are those of the author(s) and not necessarily those of the NHS, NIHR, Department of Health or King’s College London.
RJBD and SJN are supported by: 1) Health Data Research UK, which is funded by the UK Medical Research Council, Engineering and Physical Sciences Research Council, Economic and Social Research Council, Department of Health and Social Care (England), Chief Scientist Office of the Scottish Government Health and Social Care Directorates, Health and Social Care Research and Development Division (Welsh Government), Public Health Agency (Northern Ireland), British Heart Foundation and Wellcome Trust; and 2) The National Institute for Health Research University College London Hospitals Biomedical Research Centre.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/19-1163r1).
REFERENCES
[1] | ((2018) ) 2018 Alzheimer’s disease facts and figures includes a special report on the financial and personal benefits of early diagnosis. Alzheimers Dement 14: , 367–429. |
[2] | Han G , Wang J , Zeng F , Feng X , Yu J , Cao H-YY , Yi X , Zhou H , Jin L-WW , Duan Y , Wang Y-JJ , Lei H ((2013) ) Characteristic transformation of blood transcriptome in Alzheimer’s disease. J Alzheimers Dis 35: , 373–86. |
[3] | Lunnon K , Sattlecker M , Furney SJ , Coppola G , Simmons A , Proitsi P , Lupton MK , Lourdusamy A , Johnston C , Soininen H , Kłoszewska I , Mecocci P , Tsolaki M , Vellas B , Geschwind D , Lovestone S , Dobson R , Hodges A ((2013) ) A blood gene expression marker of early Alzheimer’s disease. J Alzheimers Dis 33: , 737–753. |
[4] | Henriksen K , O’Bryant SE , Hampel H , Trojanowski JQ , Montine TJ , Jeromin A , Blennow K , Lönneborg A , Wyss-Coray T , Soares H , Bazenet C , Sjögren M , Hu W , Lovestone S , Karsdal MA , Weiner MW ((2014) ) The future of blood-based biomarkers for Alzheimer’s disease. Alzheimers Dement 10: , 115–131. |
[5] | Thambisetty M , Lovestone S ((2010) ) Blood-based biomarkers of Alzheimer’s disease: Challenging but feasible. Biomark Med 4: , 65–79. |
[6] | Nakamura A , Kaneko N , Villemagne VL , Kato T , Doecke J , Doré V , Fowler C , Li Q-X , Martins R , Rowe C , Tomita T , Matsuzaki K , Ishii K , Ishii K , Arahata Y , Iwamoto S , Ito K , Tanaka K , Masters CL , Yanagisawa K ((2018) ) High performance plasma amyloid-β biomarkers for Alzheimer’s disease. Nature 554: , 249–254. |
[7] | Kiddle SJ , Sattlecker M , Proitsi P , Simmons A , Westman E , Bazenet C , Nelson SK , Williams S , Hodges A , Johnston C , Soininen H , Kłoszewska I , Mecocci P , Tsolaki M , Vellas B , Newhouse S , Lovestone S , Dobson RJB ((2014) ) Candidate blood proteome markers of Alzheimer’s disease onset and progression: A systematic review and replication study. J Alzheimers Dis 38: , 515–531. |
[8] | Chiam JTW , Dobson RJB , Kiddle SJ , Sattlecker M ((2014) ) Are blood-based protein biomarkers for Alzheimer’s disease also involved in other brain disorders? A systematic review. J Alzheimers Dis 43: , 303–314. |
[9] | Rye PD , Booij BB , Grave G , Lindahl T , Kristiansen L , Andersen HM , Horndalsveen PO , Nygaard H a. , Naik M , Hoprekstad D , Wetterberg P , Nilsson C , Aarsland D , Sharma P , Lönneborg A ((2011) ) A novel blood test for the early detection of Alzheimer’s disease. J Alzheimers Dis 23: , 121–129. |
[10] | Booij BB , Lindahl T , Wetterberg P , Skaane NV , Sæbø S , Feten G , Rye PD , Kristiansen LI , Hagen N , Jensen M , Bårdsen K , Winblad B , Sharma P , Lönneborg A ((2011) ) A gene expression pattern in blood for the early detection of Alzheimer’s disease. J Alzheimers Dis 23: , 109–119. |
[11] | Voyle N , Keohane A , Newhouse S , Lunnon K , Johnston C , Soininen H , Kloszewska I , Mecocci P , Tsolaki M , Vellas B , Lovestonea S , Hodges A , Kiddle S , Dobson RJB ((2016) ) A pathway based classification method for analyzing gene expression for Alzheimer’s disease diagnosis. J Alzheimers Dis 49: , 659–669. |
[12] | Roed L , Grave G , Lindahl T , Rian E , Horndalsveen PO , Lannfelt L , Nilsson C , Swenson F , Lönneborg A , Sharma P , Sjögren M ((2013) ) Prediction of mild cognitive impairment that evolves into alzheimer’s disease dementia within two years using a gene expression signature in blood: A pilot study. J Alzheimers Dis 35: , 611–621. |
[13] | Fehlbaum-Beurdeley P , Jarrige-Le Prado AC , Pallares D , Carrière J , Guihal C , Soucaille C , Rouet F , Drouin D , Sol O , Jordan H , Wu D , Lei L , Einstein R , Schweighoffer F , Bracco L ((2010) ) Toward an Alzheimer’s disease diagnosis via high-resolution blood gene expression. Alzheimers Dement 6: , 25–38. |
[14] | Huynh RA , Mohan C ((2017) ) Alzheimer’s disease: Biomarkers in the genome, blood, and cerebrospinal fluid. Front Neurol 8: , 102. |
[15] | Barnes M , Freudenberg J , Thompson S , Aronow B , Pavlidis P ((2005) ) Experimental comparison and cross-validation of the Affymetrix and Illumina gene expression analysis platforms. Nucleic Acids Res 33: , 5914–5923. |
[16] | Maouche S , Poirier O , Godefroy T , Olaso R , Gut I , Collet J-P , Montalescot G , Cambien F ((2008) ) Performance comparison of two microarray platforms to assess differential gene expression in human monocyte and macrophage cells. BMC Genomics 9: , 302. |
[17] | MAQC Consortium, Shi L , Reid LH , Jones WD , Shippy R , Warrington JA , Baker SC , Collins PJ , de Longueville F , Kawasaki ES , Lee KY , Luo Y , Sun YA , Willey JC , Setterquist RA , Fischer GM , Tong W , Dragan YP , Dix DJ , Frueh FW , Goodsaid FM , Herman D , Jensen RV , Johnson CD , Lobenhofer EK , Puri RK , Schrf U , Thierry-Mieg J , Wang C , Wilson M , Wolber PK , Zhang L , Amur S , Bao W , Barbacioru CC , Lucas AB , Bertholet V , Boysen C , Bromley B , Brown D , Brunner A , Canales R , Cao XM , Cebula TA , Chen JJ , Cheng J , Chu TM , Chudin E , Corson J , Corton JC , Croner LJ , Davies C , Davison TS , Delenstarr G , Deng X , Dorris D , Eklund AC , Fan XH , Fang H , Fulmer-Smentek S , Fuscoe JC , Gallagher K , Ge W , Guo L , Guo X , Hager J , Haje PK , Han J , Han T , Harbottle HC , Harris SC , Hatchwell E , Hauser CA , Hester S , Hong H , Hurban P , Jackson SA , Ji H , Knight CR , Kuo WP , LeClerc JE , Levy S , Li QZ , Liu C , Liu Y , Lombardi MJ , Ma Y , Magnuson SR , Maqsodi B , McDaniel T , Mei N , Myklebost O , Ning B , Novoradovskaya N , Orr MS , Osborn TW , Papallo A , Patterson TA , Perkins RG , Peters EH , Peterson R , Philips KL , Pine PS , Pusztai L , Qian F , Ren H , Rosen M , Rosenzweig BA , Samaha RR , Schena M , Schroth GP , Shchegrova S , Smith DD , Staedtler F , Su Z , Sun H , Szallasi Z , Tezak Z , Thierry-Mieg D , Thompson KL , Tikhonova I , Turpaz Y , Vallanat B , Van C , Walker SJ , Wang SJ , Wang Y , Wolfinger R , Wong A , Wu J , Xiao C , Xie Q , Xu J , Yang W , Zhang L , Zhong S , Zong Y , Slikker W Jr. ((2006) ) The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat Biotechnol 24: , 1151–1161. |
[18] | Chen JJ , Hsueh H-M , Delongchamp RR , Lin C-J , Tsai C-A ((2007) ) Reproducibility of microarray data: A further analysis of microarray quality control (MAQC) data. BMC Bioinformatics 8: , 412. |
[19] | Chang CW , Cheng WC , Chen CR , Shu WY , Tsai ML , Hsu IC , Huang CL , Hsu IC ((2011) ) Identification of human housekeeping genes and tissue-selective genes by microarray meta-analysis. PLoS One 6: , e22859. |
[20] | Lazar C , Meganck S , Taminau J , Steenhoff D , Coletta A , Molter C , Weiss-Solís DY , Duque R , Bersini H , Nowé A ((2013) ) Batch effect removal methods for microarray gene expression data integration: A survey. Brief Bioinform 14: , 469–490. |
[21] | Oldham MC , Langfelder P , Horvath S ((2012) ) Network methods for describing sample relationships in genomic datasets: Application to Huntington’s disease. BMC Syst Biol 6: , 63. |
[22] | LêCao K-A , Rohart F , McHugh L , Korn O , Wells CA ((2014) ) YuGene: A simple approach to scale gene expression data derived from different platforms for integrated analyses. Genomics 103: , 239–251. |
[23] | Chen T , Guestrin C ((2016) ) XGBoost: A Scalable Tree Boosting System. |
[24] | Dhaliwal S , Nahid A-A , Abbas R , Dhaliwal SS , Nahid A-A , Abbas R ((2018) ) Effective intrusion detection system using XGBoost. Information 9: , 149. |
[25] | Guyon I , Weston J , Barnhill S , Labs T , Bank R ((2013) ) Tracking cellulase behaviors. Biotechnol Bioeng 110: , fmvi. |
[26] | Šimundić AM ((2009) ) Measures of diagnostic accuracy: Basic definitions. EJIFCC 19: , 203–211. |
[27] | Mitchell AJ ((2011) ) Sensitivity×PPV is a recognized test called the clinical utility index (CUI+). Eur J Epidemiol 26: , 251–252; author reply 252. |
[28] | Kamburov A , Wierling C , Lehrach H , Herwig R ((2009) ) ConsensusPathDB - A database for integrating human functional interaction networks. Nucleic Acids Res 37: , 623–628. |
[29] | Peters MJ , Joehanes R , Pilling LC , Schurmann C , Conneely KN , Powell J , Reinmaa E , Sutphin GL , Zhernakova A , Schramm K , Wilson YA , Kobes S , Tukiainen T , NABEC/UKBEC Consortium, Ramos YF , Göring HHH , Fornage M , Liu Y , Gharib SA , Stranger BE , De Jager PL , Aviv A , Levy D , Murabito JM , Munson PJ , Huan T , Hofman A , Uitterlinden AG , Rivadeneira F , van Rooij J , Stolk L , Broer L , Verbiest MMPJ , Jhamai M , Arp P , Metspalu A , Tserel L , Milani L , Samani NJ , Peterson P , Kasela S , Codd V , Peters A , Ward-Caviness CK , Herder C , Waldenberger M , Roden M , Singmann P , Zeilinger S , Illig T , Homuth G , Grabe H-J , Völzke H , Steil L , Kocher T , Murray A , Melzer D , Yaghootkar H , Bandinelli S , Moses EK , Kent JW , Curran JE , Johnson MP , Williams-Blangero S , Westra H-J , McRae AF , Smith JA , Kardia SLR , Hovatta I , Perola M , Ripatti S , Salomaa V , Henders AK , Martin NG , Smith AK , Mehta D , Binder EB , Nylocks KM , Kennedy EM , Klengel T , Ding J , Suchy-Dicey AM , Enquobahrie DA , Brody J , Rotter JI , Chen Y-DI , Houwing-Duistermaat J , Kloppenburg M , Slagboom PE , Helmer Q , den Hollander W , Bean S , Raj T , Bakhshi N , Wang QP , Oyston LJ , Psaty BM , Tracy RP , Montgomery GW , Turner ST , Blangero J , Meulenbelt I , Ressler KJ , Yang J , Franke L , Kettunen J , Visscher PM , Neely GG , Korstanje R , Hanson RL , Prokisch H , Ferrucci L , Esko T , Teumer A , van Meurs JBJ , Johnson AD ((2015) ) The transcriptional landscape of age in human peripheral blood. Nat Commun 6: , 8570. |
[30] | Huang H , Nguyen T , Ibrahim S , Shantharam S , Yue Z , Chen JY ((2015) ) DMAP: A connectivity map database to enable identification of novel drug repositioning candidates. BMC Bioinformatics 16: , S4. |
[31] | Sood S , Gallagher IJ , Lunnon K , Rullman E , Keohane A , Crossland H , Phillips BE , Cederholm T , Jensen T , van Loon LJC , Lannfelt L , Kraus WE , Atherton PJ , Howard R , Gustafsson T , Hodges A , Timmons JA ((2015) ) A novel multi-tissue RNA diagnostic of healthy ageing relates to cognitive health status. Genome Biol 16: , 185. |
[32] | Scherzer CR , Eklund AC , Morse LJ , Liao Z , Locascio JJ , Fefer D , Schwarzschild MA , Schlossmacher MG , Hauser MA , Vance JM , Sudarsky LR , Standaert DG , Growdon JH , Jensen RV , Gullans SR ((2007) ) Molecular markers of early Parkinson’s disease based on gene expression in blood. Proc Natl Acad Sci U S A 104: , 955–960. |
[33] | Calligaris R , Banica M , Roncaglia P , Robotti E , Finaurini S , Vlachouli C , Antonutti L , Iorio F , Carissimo A , Cattaruzza T , Ceiner A , Lazarevic D , Cucca A , Pangher N , Marengo E , di Bernardo D , Pizzolato G , Gustincich S ((2015) ) Blood transcriptomics of drug-naïve sporadic Parkinson’s disease patients. BMC Genomics 16: , 876. |
[34] | Goertsches RH , Hecker M , Koczan D , Serrano-Fernandez P , Moeller S , Thiesen HJ , Zettl UK ((2010) ) Long-term genome-wide blood RNA expression profiles yield novel molecular response candidates for IFN-beta-1b treatment in relapsing remitting MS. Pharmacogenomics 11: , 147–161. |
[35] | De Jager PL , Jia X , Wang J , De Bakker PIW , Ottoboni L , Aggarwal NT , Piccio L , Raychaudhuri S , Tran D , Aubin C , Briskin R , Romano S , Baranzini SE , McCauley JL , Pericak-Vance MA , Haines JL , Gibson RA , Naeglin Y , Uitdehaag B , Matthews PM , Kappos L , Polman C , McArdle WL , Strachan DP , Evans D , Cross AH , Daly MJ , Compston A , Sawcer SJ , Weiner HL , Hauser SL , Hafler DA , Oksenberg JR ((2009) ) Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat Genet 41: , 776–782. |
[36] | Irizar H , Muñoz-Culla M , Sepú Lveda L , Sáenz-Cuesta M , Prada A , Castillo-Triviño T , Zamora-Ló Pez G , De Munain AL , Olascoaga J , Otaegui D ((2014) ) Transcriptomic profile reveals gender-specific molecular mechanisms driving multiple sclerosis progression. PLoS One 9: , e90482. |
[37] | de Jong S , Boks MPM , Fuller TF , Strengman E , Janson E , de Kovel CGF , Ori APS , Vi N , Mulder F , Blom JD , Glenthøj B , Schubart CD , Cahn W , Kahn RS , Horvath S , Ophoff RA ((2012) ) A gene co-expression network in whole blood of schizophrenia patients is independent of antipsychotic-use and enriched for brain-expressed genes. PLoS One 7: , e39498. |
[38] | van Beveren NJM , Buitendijk GHS , Swagemakers S , Krab LC , Röder C , de Haan L , van der Spek P , Elgersma Y ((2012) ) Marked reduction of AKT1 expression and deregulation of AKT1-associated pathways in peripheral blood mononuclear cells of schizophrenia patients. PLoS One 7: , e32618. |
[39] | Clelland CL , Read LL , Panek LJ , Nadrich RH , Bancroft C , Clelland JD ((2013) ) Utilization of never-medicated bipolar disorder patients towards development and validation of a peripheral biomarker profile. PLoS One 8: , e69082. |
[40] | Beech RD , Lowthert L , Leffert JJ , Mason PN , Taylor MM , Umlauf S , Lin A , Lee JY , Maloney K , Muralidharan A , Lorberg B , Zhao H , Newton SS , Mane S , Epperson CN , Sinha R , Blumberg H , Bhagwagar Z ((2010) ) Increased peripheral blood expression of electron transport chain genes in bipolar depression. Bipolar Disord 12: , 813–824. |
[41] | Ellsworth DL , Croft DT , Weyandt J , Sturtz LA , Blackburn HL , Burke A , Haberkorn MJ , McDyer FA , Jellema GL , Van Laar R , Mamula KA , Chen Y , Vernalis MN ((2014) ) Intensive cardiovascular risk reduction induces sustainable changes in expression of genes and pathways important to vascular function. Circ Cardiovasc Genet 7: , 151–160. |
[42] | Maciejak A , Kiliszek M , Michalak M , Tulacz D , Opolski G , Matlak K , Dobrzycki S , Segiet A , Gora M , Burzynska B ((2015) ) Gene expression profiling reveals potential prognostic biomarkers associated with the progression of heart failure. Genome Med 7: , 26. |
[43] | Sinnaeve PR , Donahue MP , Grass P , Seo D , Vonderscher J , Chibout S-D , Kraus WE , Sketch M , Nelson C , Ginsburg GS , Goldschmidt-Clermont PJ , Granger CB ((2009) ) Gene expression patterns in peripheral blood correlate with the extent of coronary artery disease. PLoS One 4: , e7037. |
[44] | Walsh AM , Whitaker JW , Huang CC , Cherkas Y , Lamberth SL , Brodmerkel C , Curran ME , Dobrin R ((2016) ) Integrative genomic deconvolution of rheumatoid arthritis GWAS loci into gene and cell type associations. Genome Biol 17: , 79. |
[45] | Sellam J , Marion-Thore S , Dumont F , Jacques S , Garchon HJ , Rouanet S , Taoufik Y , Hendel-Chavez H , Sibilia J , Tebib J , Le Loët X , Combe B , Dougados M , Mariette X , Chiocchia G ((2014) ) Use of whole-blood transcriptomic profiling to highlight several pathophysiologic pathways associated with response to rituximab in patients with rheumatoid arthritis: Data from a randomized, controlled, open-label trial. Arthritis Rheumatol 66: , 2015–2025. |
[46] | Mesko B , Poliska S , Váncsa A , Szekanecz Z , Palatka K , Hollo Z , Horvath A , Steiner L , Zahuczky G , Podani J , Nagy L ((2013) ) Peripheral blood derived gene panels predict response to infliximab in rheumatoid arthritis and Crohn’s disease. Genome Med 5: , 59. |
[47] | Singh D , Fox SM , Tal-Singer R , Bates S , Riley JH , Celli B ((2014) ) Altered gene expression in blood and sputum in copd frequent exacerbators in the eclipse cohort. PLoS One 9: , e107381. |
[48] | Bahr TM , Hughes GJ , Armstrong M , Reisdorph R , Coldren CD , Edwards MG , Schnell C , Kedl R , LaFlamme DJ , Reisdorph N , Kechris KJ , Bowler RP ((2013) ) Peripheral blood mononuclear cell gene expression in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 49: , 316–323. |


