
Generation of Transgenic Cynomolgus Monkeys Overexpressing the Gene for Amyloid-β Precursor Protein
Abstract
Alzheimer’s disease (AD) is the most common cause of dementia and understanding its pathogenesis should lead to improved therapeutic and diagnostic methods. Although several groups have developed transgenic mouse models overexpressing the human amyloid-β precursor protein (APP) gene with AD mutations, with and without presenilin mutations, as well as APP gene knock-in mouse models, these animals display amyloid pathology but do not show neurofibrillary tangles or neuronal loss. This presumably is due to differences between the etiology of the aged-related human disease and the mouse models. Here we report the generation of two transgenic cynomolgus monkeys overexpressing the human gene for APP with Swedish, Artic, and Iberian mutations, and demonstrated expression of gene tagged green fluorescent protein marker in the placenta, amnion, hair follicles, and peripheral blood. We believe that these nonhuman primate models will be very useful to study the pathogenesis of dementia and AD. However, generated Tg monkeys still have some limitations. We employed the CAG promoter, which will promote gene expression in a non-tissue specific manner. Moreover, we used transgenic models but not knock-in models. Thus, the inserted transgene destroys endogenous gene(s) and may affect the phenotype(s). Nevertheless, it will be of great interest to determine whether these Tg monkeys will develop tauopathy and neurodegeneration similar to human AD.
INTRODUCTION
Dementia has become an important disease that all aging societies are facing. It is caused by multiple diseases, but among them, Alzheimer’s disease (AD) is the most common cause [1, 2]. To develop effective treatments for AD and related dementias, it is first necessary to understand the pathogenesis of AD, and then it will be possible to develop diagnostic methods and therapeutics for this disease. To accomplish this, animal models that accurately reflect the clinical and pathological features of the human disease are needed.
The neuropathological hallmarks of AD are senile plaques and neurofibrillary tangles [3]. The main components of senile plaques and neurofibrillary tangles are amyloid-β peptide (Aβ) and hyperphosphorylated tau protein [3]. The first genetic mutations causing hereditary early-onset (familial) AD were discovered in the amyloid-β precursor protein (APP) gene [4]. In addition to this gene, mutations in presenilin 1 (PS-1) [5] and presenilin 2 (PS-2) genes [6, 7] were identified as causes of familial AD. Mutations in any of these three genes resulted in increased production of Aβ [8]. The “amyloid hypothesis” resulting from these genetic findings proposes that the deposition of toxic Aβ must be the primary event in AD pathology [8, 9].
Experimental animal models of AD are critical to clarify its pathogenesis and to assess the potential of novel therapeutic and diagnostic agents. Several groups have developed Aβ plaque-developing transgenic (Tg) mouse models by overexpressing the human APP gene with different mutations, including some with presenilin mutations (for reviews, see [10, 11]). A knock-in mouse model has now been generated [12] in which expression of humanized mutated APP resulted in mice that overproduce pathogenic Aβ without overexpressing AβPP or its subfragments [12]. These AD model mice have contributed to understand AD pathology and develop novel diagnostic and therapeutic methods for AD [11]. Interestingly, however, these models display amyloid pathology but not neurofibrillary tangles or neuronal loss [10, 11]. It remains unknown why mouse models of AD show only amyloid pathology but fail to exhibit tau pathology or neuronal loss. There are several explanations for the discrepancy between human AD and mouse models. First, the lifespan of mice is too short to generate tau pathology [11]. The other possible reason is species differences between rodents and humans [11]. For example, there are several differences in amino acid sequences between the human and mouse Aβ. Primate models of AD should help resolve these discrepancies.
The cynomolgus monkey (Macaca fascicularis), a nonhuman primate, belongs to the Old World group of monkeys and is closer to humans in terms of its anatomy and genomic conservation than New World monkeys, including the common marmoset (Callithrix jacchus). Cynomolgus monkeys have an advantage for research purposes over other macaque species because they breed year-round rather than seasonally, a feature of rhesus monkeys (Macaca mulatta). Therefore, they have been used widely for modeling human disorders, including Parkinson’s disease [13].
Here we report that Tg cynomolgus monkey blastocysts produced by intracytoplasmic sperm injection (ICSI) were vitrified and thawed at high efficiency and resulted in live births. Using these reproductive techniques, we have succeeded in generating two Tg cynomolgus monkeys overexpressing the APP gene containing Swedish mutations (K595 N/M596 L), the Artic mutation (E618 G) and the Iberian mutation (I641F).
MATERIALS AND METHODS
Animals
All experimental procedures were approved by the Animal Care and Use Committee of Shiga University of Medical Science and were carried out in accordance with approved guidelines (Approval number: 2016-10-1, 2019-10-1). Oocytes were collected from 14 sexually mature female cynomolgus monkeys, aged 4–13 years and weighing 2.5–3.9 kg. Eighty-one sexually mature females aged 4 years old and weighing 2.0–3.8 kg, were used as recipients. Semen was collected from three sexually mature male monkeys, aged 9–18 years and weighing 4.5–7.0 kg, by penile electroejaculation as described [14]. Temperature and humidity in the animal rooms were maintained at 25±2°C and 50±5%, respectively. The light cycle was regulated at 12 h light and 12 h dark. In the morning, each monkey was fed 20 g/kg of body weight of commercial pellet monkey chow (CMK-1; CLEA Japan), supplemented with 20–50 g of sweet potatoes or bananas in the afternoon. Water was available ad libitum.
Lentiviral vector construction
Three pathogenic mutations for the gene causing familial AD, which result in amino acid substitutions of K595 N/M596 L (known as the Swedish mutation), E618 G (the Arctic mutation), and I641F (the Iberian mutation), were introduced in cDNA for the neuronal splicing isoform of human APP (NM_201414.2) using site-directed mutagenesis. Additionally, DNA fragments for FLAG epitope tag (DYKDDDDK) and P2A peptide were inserted downstream of the signal sequence and the stop codon of APP, respectively, by polymerase chain reaction (PCR)-based mutagenesis. To construct pCSII-CAG-FLAG-APP-P2A-GFP, fragments of the CAG promoter derived from pCAGGS (provided by Dr. Hitoshi Niwa, Kumamoto University), a FLAG-APP-P2A fragment and green fluorescent protein (GFP) cDNA were inserted in the multiple cloning sites of pCSII-EF-MCS-IRES2-Venus plasmids.
Lentiviral vector package and transduction
Lentiviral transduction was performed as described [14]. In brief, viral particles were obtained through cationic dPEI (PEI Max MW 25,000 kDa; Polysciences Inc., Warrington, PA, USA) transfection in 293FT cells, and with packaging plasmids VSVG, RSV-Rev, and HIVgp plasmids. Viral supernatants were harvested after 48 h of transfection. The supernatant then was passed through a PVDF filter (pore size 0.22 μm) and concentrated by ultracentrifugation (50,000 g for 2 h at 4°C). The pellet was suspended in Connaught Medical Research Laboratories (CMRL) Medium-1066 (Thermo Fisher Scientific, Waltham, MA, USA) and centrifuged on a 20% (w/v) sucrose cushion. After the viral pellet had been resuspended in CMRL medium, the infectious unit (IU) value was determined using Lenti-Xtrademark p24 Rapid Titer kits (Takara Bio, Shiga, Japan).
Lentiviral infection of 293FT cells
The 293FT cells were plated at 5 × 105 cells on 30 mm dishes, and then infected with lentiviral particles at concentrations of 1, 10 or 100 IU; 48 h later the cells were collected.
Production of transgenic (Tg) cynomolgus monkeys
Oocyte collection, virus injection into embryos, ICSI, embryo transfer, pregnancy detection and observation of EGFP fluorescence in Tg offspring were carried out as described [15]. The oocytes were collected by laparoscopy. The ten oocyte donors underwent superovulation for the first time and four oocyte donors underwent superovulation for the second time [16]. Each received subcutaneous infusions of human follicle-stimulating hormone (hFSH; 15 IU/kg, Asuka Pharmaceutical, Tokyo, Japan) via a micro-infusion pump (iPRECIO SMP-200; Primetech Corp, Tokyo, Japan) at 7 μl/h for 10 days. On day 10, the animals received an intramuscular injection of human chorionic gonadotropin (Puberogen; Nippon Zenyaku Kogyo, Fukushima, Japan), and oocytes were aspirated laparoscopically after 40 h with the monkeys under general anesthesia. The collected oocytes were immediately assessed for nuclear maturity under an inverted microscope. Those in which the first polar body was extruded were selected and matured in m-TALP medium, a modified Tyrode’s solution containing HEPES, and injected with lentiviruses: ICSI was performed 3–4 h after virus injection. The fertilized oocytes were cultured in CMRL Medium-1066 containing 20% (v/v) fetal bovine serum (FBS) at 38°C in 5% CO2 and 5% O2. When embryos had developed to blastocysts, one or two were transferred into each female recipient.
Vitrification and thawing of blastocysts
Vitrification and thawing of blastocysts were performed according to the instructions of the Vitrification and Thawing kits (VT601-TOP/602-KIT; Kitazato, Shizuoka, Japan). Briefly, one to three blastocysts were transferred to equilibration solution for 15 min. The blastocysts were then transferred into vitrification solution for 1 min and subsequently placed on a Cryotop freezing device (Kitazato). The blastocysts were stored under liquid nitrogen (–196°C).
For thawing, the Cryotop was immersed in thawing solution for 1 min. Then blastocysts were then transferred to diluent solution for 3 min. Each was washed twice in washing solution for 6 min and cultured in CMRL medium-1066 containing 20% (v/v) FBS for 12 h. A blastocyst was regarded as having survived if the blastocoele re-expanded.
Immunocytochemistry
Blastocysts were fixed in 4% paraformaldehyde (PFA) at room temperature (RT) for 30 min and treated with blocking solution (Blocking One; Nacalai Tesque, Kyoto, Japan) including 0.1% Triton X-100 for 30 min at RT, and then incubated with a mouse anti-CDX2 antibody (MU392A-UC; BioGenex, Fremont, CA, USA, 1 : 500) overnight at 4°C in the blocking solution. After three washes in Blocking One solution in phosphate-buffered saline (PBS), the blastocysts were incubated with anti-mouse AlexaFluor 647-conjugated antibodies for 1 h at RT. After three washes in blocking solution in PBS, the blastocysts were incubated with rabbit anti-GFP Alexa Fluor 488-conjugated antibodies in blocking solution with Hoechst or DAPI for 1 h at RT. Hair roots were fixed in 4% PFA at RT for 30 min, and treated with Blocking One solution containing 0.1% TritonX-100 for 30 min at RT, and then incubated with rabbit anti-GFP AlexaFluor 488-conjugated antibodies in blocking solution with Hoechst for 1 h at RT. Images were taken using a Leica TCS SP8 confocal microscope (Leica Microsystems, Wetzlar, Germany).
For detecting cell surface APP, HeLa cells were transiently transfected with pCSII-CAG-GFP or pCAII-CAG-FLAG-APP-P2A-GFP using Lipofectamine 2000 (Thermo Fisher, Waltham, MA, USA) according to the manufacturer’s instructions. On the next day, the cells were fixed with 4% PFA for 10 min at RT and treated with 3% normal goat serum in PBS for 30 min at RT to minimize nonspecific reactions. Then, the cells were stained with a mouse anti-FLAG antibody (M2, Sigma-Aldrich, St. Louis, MO, USA) at 1 : 10000 for 60 min without permeabilization with Triton X-100, followed by visualization with an Alexa 594 conjugated goat anti-mouse secondary antibody (Thermo Fisher Scientific, 1 : 400). After staining, the cells were imaged using a Leica TCS SP8 confocal microscope.
Immunohistochemistry
Organs from aborted fetuses were fixed using 4% PFA in 100 mM phosphate buffer saline (PBS; pH 7.4) for 7 days, and then stored in 15% sucrose in PBS at 4°C. They were embedded in OCT compound (Sakura Finetek, Tokyo Japan) and frozen in the liquid nitrogen. Frozen sections were cut at a thickness of 10 μm at –20°C. The sections were reacted with rat anti-GFP (GF090 R; Nacalai Tesque, 1 : 500), rabbit anti-GFP (MBL, 1 : 500) or mouse anti-NeuN (A60; Merck Millipore, Burlington, MA, USA, 1 : 1000) antibodies overnight at 4°C. Then, the sections were further visualized with Alexa 488-conjugated anti-rat IgG, anti-rabbit-IgG or Alexa 594-conjugated anti-mouse IgG secondary antibodies (Thermo Fisher Scientific) for 1 h at RT, and observed using confocal microscopy as above.
Genomic PCR
Genomic DNA was extracted from umbilical cords, placentas and cerebral cortexes using lysis buffer (10 mM Tris-HCl (pH 8.0), 100 mM NaCl, 50 mM EDTA, 0.5% SDS, and 0.5 mg/ml proteinase K). The lysate was treated with phenol and phenol–chloroform and precipitated with ethanol. PCR was carried out using BioTaqtrademark DNA polymerase (Bioline, London, UK). PCR amplification was performed with the following profile: one cycle at 94°C for 4 min, 35 cycles at 94°C for 30 s, 60°C for 30 s, and 72°C for 30 s and one cycle at 72°C for 7 min.
Reverse transcription (RT)–PCR
Total RNA was extracted from cells or tissues using RNeasy Mini kits (Qiagen, Hilden, Germany) or NucleoSpin RNA Plus (Macherey-Nagel, Duren, Germany). For reverse transcription, ReverTra Ace (Toyobo, Osaka, Japan), Superscript III (Thermo Fisher Scientific) and oligo (dT) 20 primers were used. PCR was carried out using BioTaqtrademark DNA polymerase (Bioline) and the following primers: GFP forward 5′-AAGTCGTGCTGCTTCATGTG-3′ and reverse 5′-ACGTAAACGGCCACAAGTTC-3′; glyceraldehyde 3-phosphate dehydrogenase (GAPDH) forward 5′-ATTCCACCCATGGCAAGTTC-3′ and reverse 5′-ATCGCCCCACTTGATTTTGG-3′.
Western blot analysis
Western blotting was carried out as described [15], with some modifications. Briefly, 1 mm3 blocks of tissues were incubated in RIPA buffer (50 mM Tris-HCl, 150 mM NaCl, 0.5% sodium deoxycholate, 1% NP40, and 0.1% SDS) with protease and phosphatase inhibitors for 10 min at 4°C. Samples were diluted in sample buffer (Sample Buffer Solution, 198-13282, Wako, Osaka, Japan) at a final concentration of 10 μg/μl and stored at –20°C before assessment. After denaturing by boiling at 95°C for 5 min, 2.5 μl aliquots of samples separated by SDS–PAGE on 10% polyacrylamide gel at 250 V for 80 min and then transferred onto a PVDF membrane (Merck Millipore). The membrane was blocked using Blocking One solution, and then incubated with the following antibodies: rabbit anti-GFP (1 : 4,000), mouse anti-actin (C4; Santa Cruz Biotechnology, Dallas, TX, USA; 1 : 1000), rabbit anti-C-terminal APP (A8714; Merck Millipore 1 : 10,000), mouse anti-N-terminal APP (MAB348; Merck Millipore, 1 : 1,000), mouse anti-FLAG (M2; Sigma-Aldrich, 1 : 4,000), rabbit anti-FLAG (D6W5B, CST, Danvers, MA, USA) or rabbit horseradish peroxidase (HRP)-conjugated anti-β-actin (PM053-7: MBL, Nagoya, Japan, 1 : 4,000) overnight at 4°C in Blocking One solution. After three washes in Tween20-PBS (T-PBS), the membrane were incubated with appropriate secondary HRP-labeled anti-rabbit immunoglobulin G (Invitrogen, 1 : 2,000) or HRP-labeled anti-mouse immunoglobulin G (Abcam, Cambridge, UK, 1 : 2,000), in Blocking One solution for 1 h at RT. After one wash of 15 min and five washes of 5 min each with T-PBS, peroxidase activity was visualized using the Chemi-Lumi One system (Nacalai Tesque) according to the manufacturer’s instructions.
Flow cytometry analysis
Samples of 0.5 ml blood were collected from the femoral vein using a 27-gauge needle and centrifuged at 1,730 g for 5 min to remove whole blood cells. Hemolysis was performed with Lysing buffer (BD Biosciences, Franklin Lakes, NJ, USA) to collect mononuclear cells. These were washed with PBS and suspended in PBS+2% (v/v) FBS. The pellet was incubated with mouse Alexa Fluor 647-conjugated anti-human CD20 (302318: BioLegend, San Diego, CA, USA; 1 : 10), mouse phycoerythrin-conjugated anti-human CD3 (552127: BD Biosciences,1 : 10), and allophycocyanin-conjugated anti-mouse/human CD11b (101212: BioLegend, 1 : 100) antibodies for 1 h on ice. Samples were washed with PBS and resuspended in 300 μl PBS containing 0.1 mg/ml propidium iodide. Fluorescence-activated cell sorting (FACS) analysis was then performed using a FACSCalibur instrument (BD Biosciences).
Brain tissue preparation of aborted monkeys
Frozen brain samples were first homogenized on ice in Tris-buffered saline (TBS; 25 mM Tris-HCl, 150 mM NaCl, pH 7.5) (1 mL/150 mg wet weight) containing protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany), sonicated, and then centrifuged in a TLA110 rotor (Beckman Coulter, Brea, CA, USA) at 104,300 x g for 60 min at 4°C. The supernatant from each sample was collected as a TBS-soluble fraction, and each pellet was then re-suspended in 70% formic acid (FA) in water, sonicated, and centrifuged at 104,300 x g for 60 minutes at 4°C. The supernatant was recovered and neutralized with a 20-fold dilution in 1 M Tris base. Protein concentration of each sample was determined by the Protein Assay Bicinchoninate kit (Nacalai tesque, Kyoto, Japan).
Enzyme-linked immunosorbent assay (ELISA)
The levels of Aβ40 and Aβ42 were measured using commercially available ELISA kits for human Aβ40 (FUJIFILM Wako Pure Chemical, Osaka, Japan; code no. 298-64601) and human Aβ42 (FUJIFILM Wako Pure Chemical; code no. 298-64401) according to the manufacturer’s instructions
Immunohistochemical examinations of brains from aborted monkeys
Paraffin embedded brain tissues from the aborted monkeys and an AD patient were sliced at a thickness of 6 μm. For detection of Aβ plaques and phosphorylated tau, the sections were treated with 100% formic acid for 1 min at RT and 0.3% H2O2 for 10 min at RT, respectively, after deparaffinization. The sections were reacted with anti-Aβ (4G10, Biolegend, 1 : 1000) or phosphorylated tau (AT8, Invitrogen, 1 : 1000) antibodies overnight at 4 C followed by detection of bound antibodies with HRP-conjugated anti-mouse IgG. Immunoreactivity signals were visualized by incubation in diaminobenzidine substrate. To visualize nuclei, sections were counterstained with 0.1% cresyl violet.
Statistical analysis
Statistical comparisons of all data were carried out using unpaired Student’s t tests and multiple one-way analysis of variance (ANOVA) using GraphPad Prism 8 software (https://www.graphpad.com/scientific-software/prism/); p < 0.05 was considered statistically significant.
RESULTS
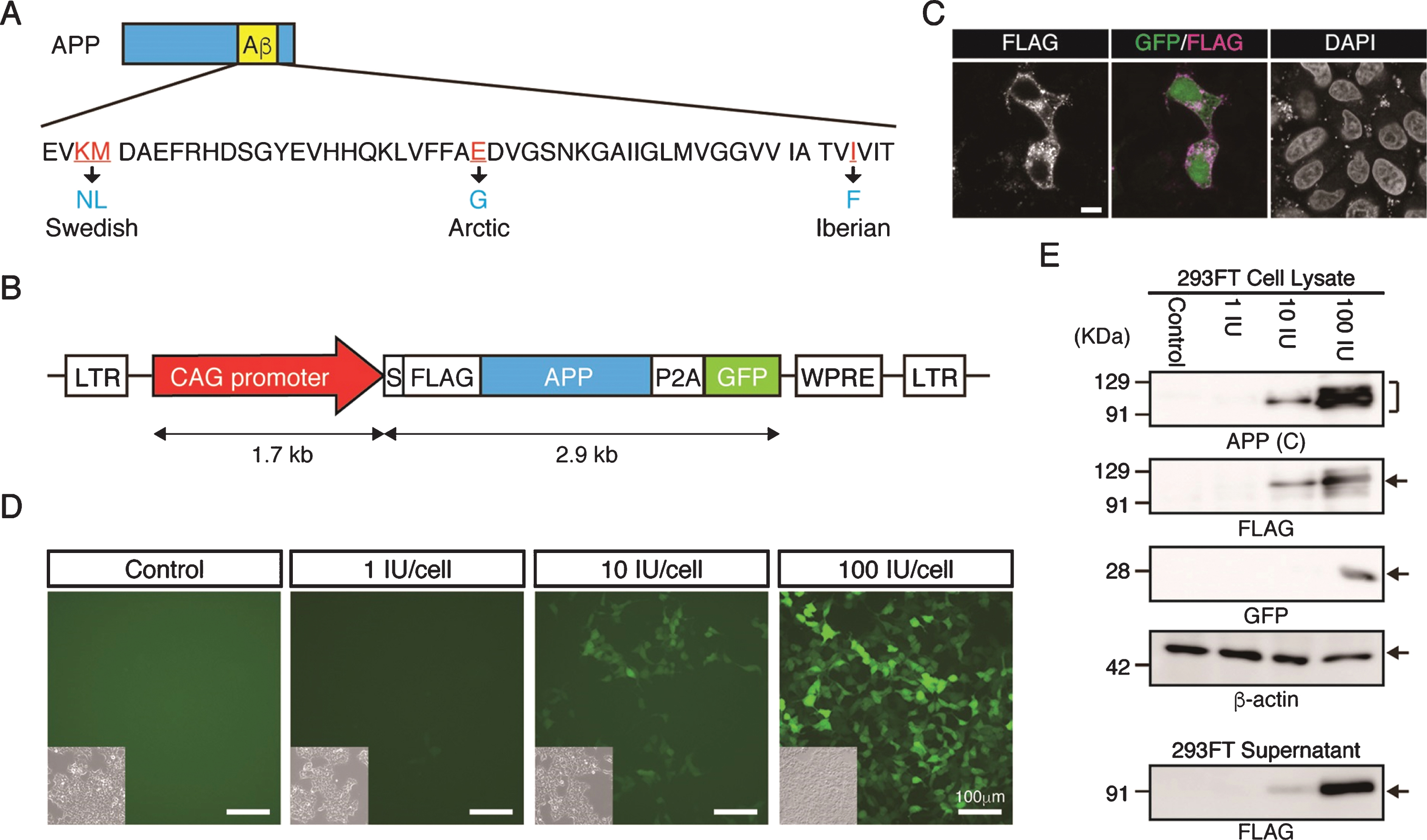
To generate Tg cynomolgus monkeys carrying the gene for APP with familial mutations, the CAG promoter was used because our previous work indicated it is the most effective for overexpressing a transgene in neural cells of cynomolgus monkeys or neural cells derived from cynomolgus monkey embryonic stem cells, with minimal silencing [14, 15]. We designed the APP lentiviral construct carrying familial mutations to facilitate amyloid formation (Fig. 1A), linked to P2A-GFP to provide a surrogate marker for APP expression (Fig. 1B). When we introduced the construct into HeLa cells transiently, anti-FLAG antibody clearly detected the signal in plasma membrane and inside the cell (Fig. 1 C), indicating that the FLAG-tagged APP protein had been processed successfully. GFP fluorescence was observed successfully in lentivirus infected cells (Fig. 1D). Western blot analysis indicted that APP protein was detected by antibodies against the C-terminal region of APP or its FLAG-epitope (Fig. 1E). FLAG-APP was also detected in the supernatant of the cells, indicating that APP-P2A-GFP was processed as a secreted protein (Fig. 1E).
Fig.1
Generation of the APP-P2A-GFP lentiviral vector. A) Design of the APP mutations. The Swedish, Arctic and Iberian mutations were introduced into the human APP gene. B) Schematic representation of the lentiviral vector used for generation of APP Tg monkeys. LTR, long terminal repeat; S, signal peptide sequence; FLAG, FLAG epitope tag sequence; P2A, self-cleaving 2A peptide sequence; GFP, green fluorescent protein; WPRE, woodchuck hepatitis posttranscriptional regulatory element. C) Localization of FLAG-tagged APP protein. Scale bars = 10 μm. D) Fluorescent images of 293FT cells 48 h after transduction with an APP-P2A-GFP lentivirus. Insets in each panel show brightfield images. Scale bars = 100 μm. E) APP protein expression in the lentivirus-infected 293FT cells and culture supernatants. APP (C), APP-C terminal.
To generate Tg cynomolgus monkeys, we injected APP-P2A-GFP-expressing lentiviruses into the perivitelline space of oocytes to facilitate infection followed by ICSI. At 7 days after the injection, GFP expression was observed throughout the blastocyst stage (Fig. 2A). Confocal microscopy showed GFP in most—if not all—blastomeres (Fig. 2B). We injected 447 oocytes and transferred 63 blastocysts to recipient female monkeys, but because of the limited number of recipients, we needed to vitrify the remainder. Previously, no cynomolgus monkeys have been produced from cryopreserved blastocysts, while the use of cryopreserved embryos at earlier stages has succeeded [18, 19]. When cryopreserved blastocysts were thawed, 26 out of 54 vitrified APP-lentivirus injected blastocysts expanded successfully (48.1%), while some of them showed retarded growth (Fig. 2 C), suggesting that vitrification and thawing caused some damage (Fig. 2D). When the normal looking expanded blastocysts were transferred into recipient monkeys, two out of 18 gave birth successfully, indicating that the vitrified–thawed cynomolgus monkey blastocyst can survive to term (Fig. 2B, Table 1). We also noticed that the implantation rate of APP-lentivirus injected blastocysts tended to be lower than that of wild-type (WT) blastocysts, although this difference was not statistically significant (Fig. 2E). To improve the pregnancy rate of APP-lentivirus injected blastocysts, we transferred pairs of blastocysts to recipient females and found that the rate of implantation increased to 30%, though this change was not statistically significant (Fig. 2E).
Fig.2
Generation of APP-GFP Tg cynomolgus monkeys. A) Images of cynomolgus monkey blastocysts 7 days after infection with the lentivirus. Scale bars = 100 μm. The left panel shows a brightfield image and the right panel shows a fluorescence image. B) Immunohistochemistry of the blastocysts 7 days after infection with anti-CDX2 and GFP antibodies. Scale bars = 100 μm. C) Images of vitrified and thawed cynomolgus monkey blastocysts. Upper panels show expanded blastocysts at 12 h after thawing. Lower panels show unexpanded blastocysts at the same time. D) Efficiency of recovery rate after vitrification and thawing. E) Implantation rate of WT blastocysts, APP lentivirus-infected blastocyst (one or two embryos per recipient). **p < 0.01 between treatments.
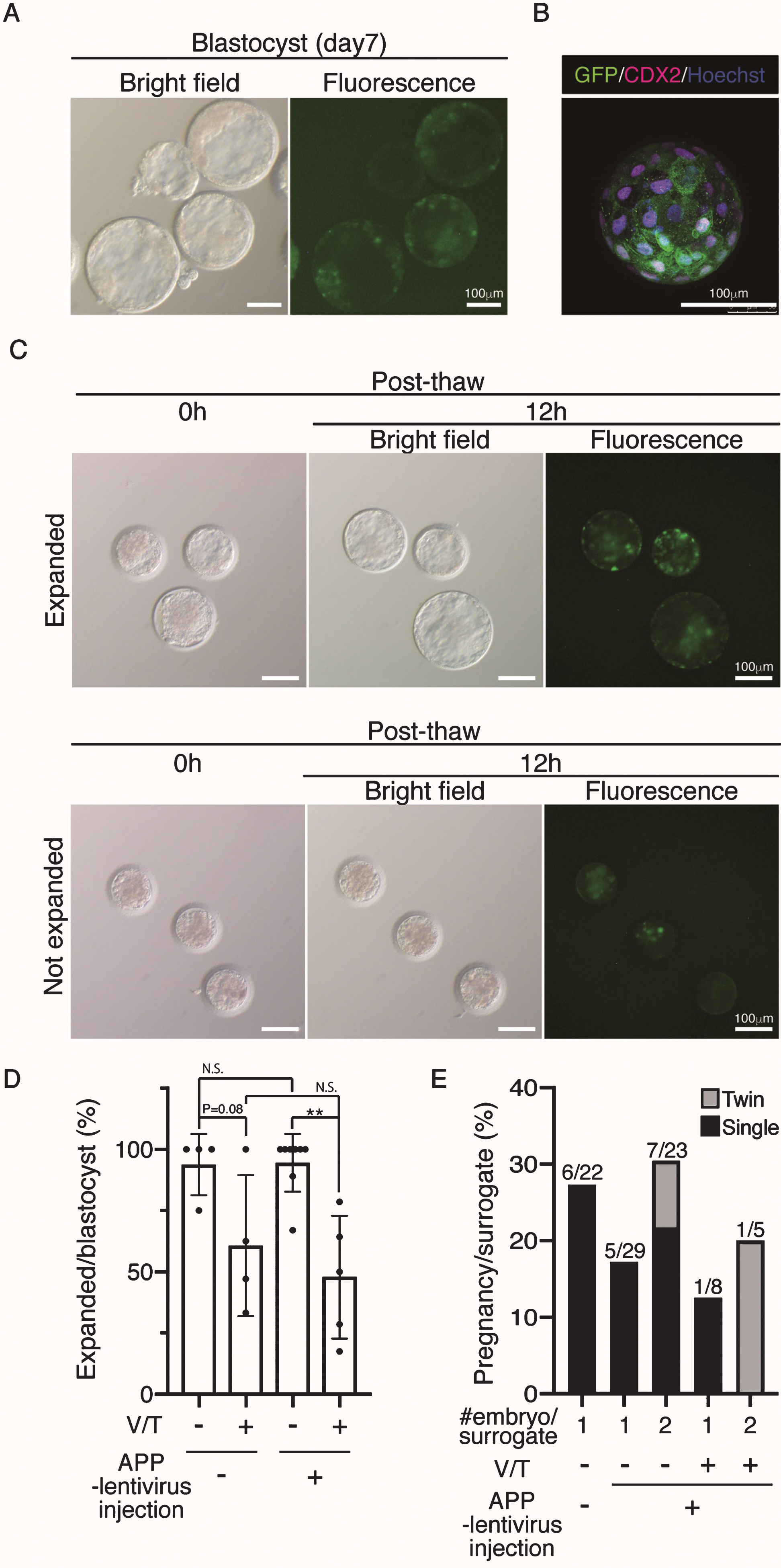
Table 1
In vitro and in vivo development of embryos after lentivirus injection
| Group | APP-lentivirus injected (Fresh) | APP-lentivirus injected (Vitrified/thawed) | WT (Fresh) | ||
| 1 embryo/surrogate | 2 embryos/surrogate | 1 embryo/surrogate | 2 embryos/surrogate | 1 embryo/surrogate | |
| No. of MII oocyte used for ICSI | 518 | 134 | |||
| No. of 2 cell stage | 323 (62.4) | 104 (77.6) | |||
| No. of blastocyst (% per 2 cell) | 177 (54.8) | 65 (62.5) | |||
| No. of blastocyst vitrified | 82 | – | |||
| No. of blastocyst thawed | – | – | 54 | – | |
| No. of post-thawed blastocyst expanded | – | – | 26 (48.1) | – | |
| ETs | 29 | 46 | 8 | 10 | 22 |
| No. of surrogates | 29 | 23 | 8 | 5 | 22 |
| No. of pregnancies at 30 days after ICSI | 5 (17.2) | 7 (30.4) | 1 (12.5) | 1 (20) | 6 (27.3) |
| Twin | – | 2 | – | 1 | – |
| Spontaneous miscarriage | 2 | 6 | 0 | 1 | 3 |
| Live birth | 3 | 3 | 1 | 1 | 3 |
| GFP+offspring | 2 | 0 | 0 | 1 | – |
ICSI, intracytoplasmic sperm injection; ET, embryo transfer; GFP, green fluorescent protein.
In humans and marmosets, oocytes can be collected repeatedly through ovarian stimulation by hFSH, and the collected oocytes contribute to in vivo development after in vitro fertilization or ICSI [20–22]. In rhesus monkeys, oocytes were obtained successfully from a second cycle of superovulation by hFSH [23]. Previously, we reported that oocytes were collected successfully after a second round of hFSH stimulation, but it was not known whether these oocytes could contribute to in vivo development after ICSI. Here, we found that 122 mature oocytes recovered by second ovarian stimulation developed into 43 blastocysts after ICSI; subsequently these gave rise to two live offspring (Table 2).
Table 2
In vitro and in vivo development of 2nd Ovum pick up (OPU) embryos after lentivirus injection
| Group | APP-lentivirus injected | ||
| 1st OPU | 2nd OPU | 3rd OPU | |
| No. of MII oocyte used for ICSI | 326 | 122 | 70 |
| No. of 2 cell stage | 194 (59.5) | 73 (59.8) | 56 (80) |
| No. of blastocyst (% per 2cell) | 99 (51.0) | 43 (58.9) | 35 (62.5) |
| ETs | 51 | 12 | 12 |
| No. of surrogates | 40 | 6 | 6 |
| No. of pregnancies at 30 days after ICSI | 9 (22.5) | 2 (33.3) | 1 (16.7) |
| Twin | 1 | 0 | 1 |
| Spontaneous miscarriage | 6 | 0 | 2 |
| Live birth | 4 | 2 | 0 |
MII, Metaphase II; ICSI, intracytoplasmic sperm injection; ETs, embryo transfers.
The twins were aborted at embryo day (E)101 and E102 (Fig. 3A) and GFP fluorescence was evident in the skin and skeletal muscle of both Tg offspring (Fig. 3A, Supplementary Figure 2). Integration of GFP was confirmed by genomic and RT–PCR (Fig. 3B, C), and GFP was seen in NeuN-positive neural cells in both Tg offspring (#101 and #102; Fig. 3D and Supplementary Figure 2). In total, eight monkeys were born and two showed clear GFP fluorescence in the face, placenta and amnion, compared with the WT neonates (Fig. 4, Table 3). One additional baby monkey showed weak GFP fluorescence, but we could not take a clear picture because of the monkey’s quick movements (Table 3). We stopped taking pictures because the handling was causing distress. Because two of the baby monkeys showed clear green fluorescence, we investigated whether this was derived from GFP. To investigate this, the placenta was analyzed for GFP expression at genomic, mRNA and protein levels (Fig. 5). Genomic PCR indicated both monkeys were GFP-positive (Fig. 5A). Semi-quantitative RT–PCR analysis indicated that GFP mRNA expression was detectable in the umbilical cord, placenta and amnion of monkey #1 and in the placenta of monkey #2 (Fig. 5B). Western blot analysis showed that FLAG-tagged APP and GFP expressions were abundant in the placenta of monkey #1 and at low levels in the placenta of #2 (Fig. 5 C). To analyze the two Tg monkeys, we investigated GFP expression in hair follicles and leukocytes and observed it in the hair follicles of both (Fig. 6A, B). FACS analysis of the tissues from these monkeys showed that 15.2% of T cells, 22.5% of B cells, 87.4% of granulocytes and 31.75% of monocytes were GFP-positive (Fig. 6 C, D, E). Together, these data confirm that we had successfully generated two APP Tg cynomolgus monkeys.
Table 3
List of live offsprings derived from APP-lentivirus injected oocytes
| Offsprings | ID# | Bith date | V/T | ET | Gender | GFP | ||
| PCR | skin | FACS of bloods | ||||||
| #1 | CE2159F | 4/17/2017 | – | Single | F | + | + | + |
| #2 | CE2165F | 7/17/2017 | – | Single | F | + | + | + |
| #3 | CE2195F | 10/18/2017 | – | Single | F | – | – | – |
| #4 | CE2343M | 3/26/2018 | + | Single | M | + | – | + |
| #5 | CE2349F | 8/3/2018 | – | Double | F | – | – | – |
| #6 | CE2354M | 10/10/2018 | – | Double | M | + | – | – |
| #7 | CE2355F | 10/10/2018 | – | Double | F | + | – | + |
| #8 | CE2385F | 12/24/2018 | + | Double | F | + | +/– | + |
ET, embryo transfer; GFP, green fluorescent protein; FACS, fluorescence-activated cell sorting; V/T, Vitrification/thawing.
Fig.3
GFP expression of aborted twin APP Tg monkeys. A) Bright-field and fluorescence microscopy images of the fetal Tg monkeys. B) Detection of the integrated transgene by genomic PCR. DNA from a normal placenta was used as a control. C) Detection of the transcript from the transgene by RT–PCR. RNA from a normal placenta was used as a control. D) Immunohistochemistry of the cerebral cortex of the aborted Tg and age-matched non-transgenic fetuses. Sections of the cerebral cortex were subjected to immunohistochemistry with anti-GFP and anti-NeuN antibodies, together with DAPI for nuclear staining, and they were observed using a confocal microscope (SP-8, Leica). Arrows indicate NeuN- and GFP-positive cells. Dashed lines in the low magnification images of the WT fetuses indicate the top of the cerebral wall. Inserts are enlarged images of square areas on the lower magnification micrographs. Scale bars = 250 μm (low magnification) or 50 μm (high magnification).
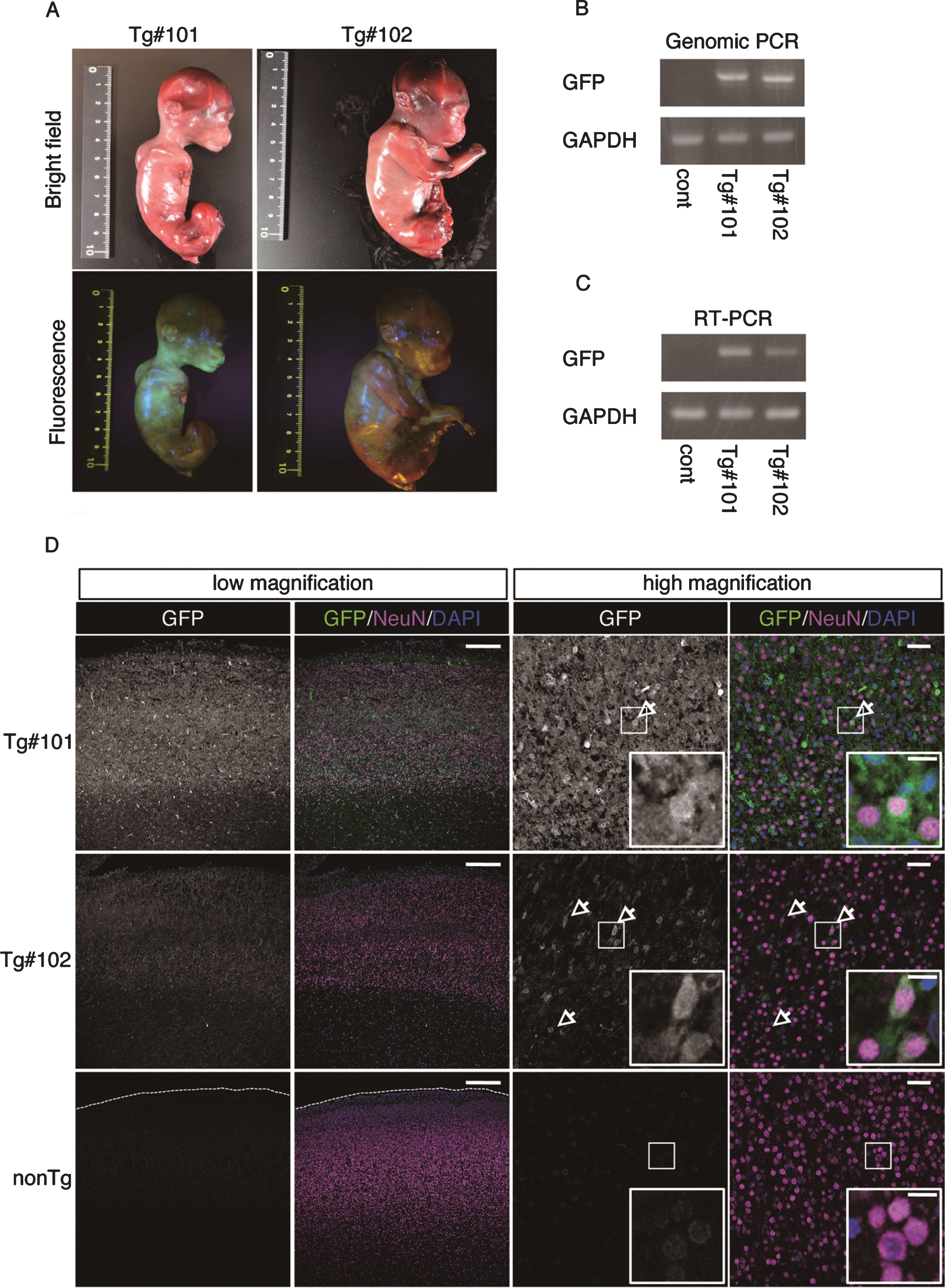
Fig.4
Fluorescence images of the Tg cynomolgus monkeys. Upper panels showing fluorescence images of the face, placenta and amnion of APP-GFP Tg offspring #1. Lower panels showing fluorescence images of the face and placenta of APP-GFP offspring #2. Insets in each panel show brightfield images.
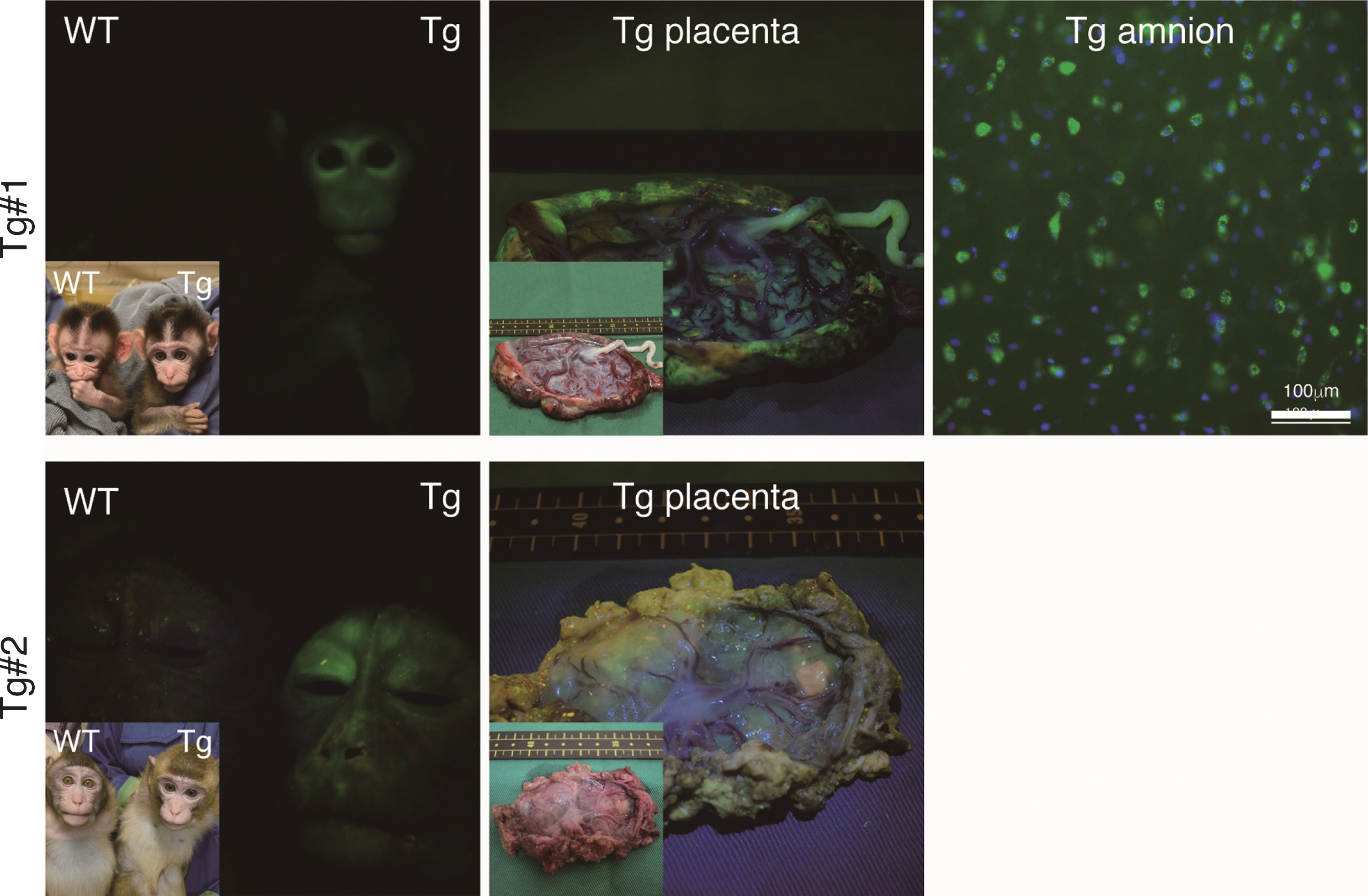
Fig.5
APP expression in the placenta. A) Genomic PCR analysis with DNAs from Tg #1, Tg #2 and WT offspring. PC, positive control. B) RT–PCR with cDNAs from Tg #1, Tg #2 and WT offspring. C) Western blot analysis with proteins extracted from the placentas of Tg #1, Tg #2 and WT offspring. APP (C), APP-C terminal; APP (N), APP-N terminal; N.S., nonspecific.
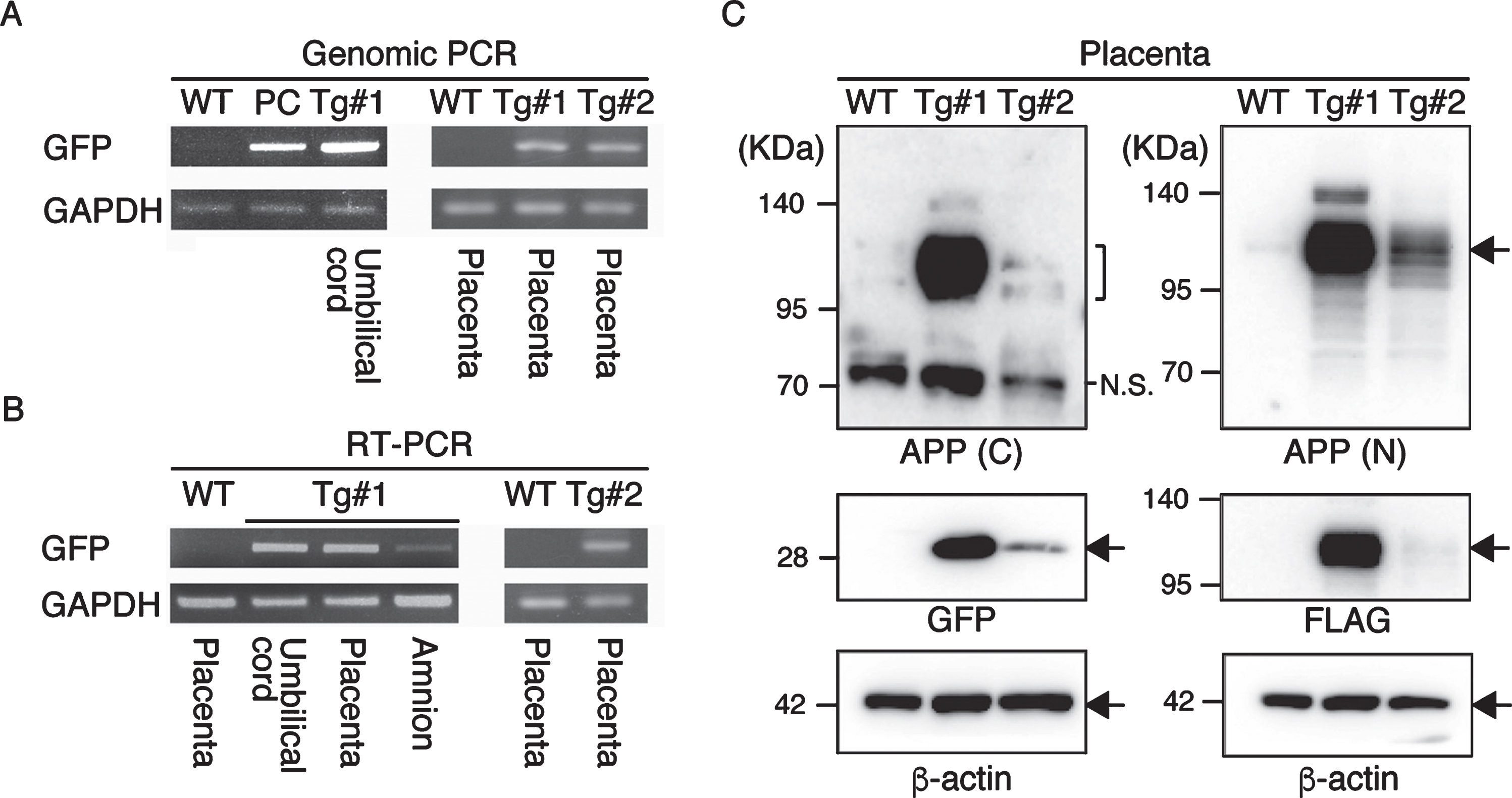
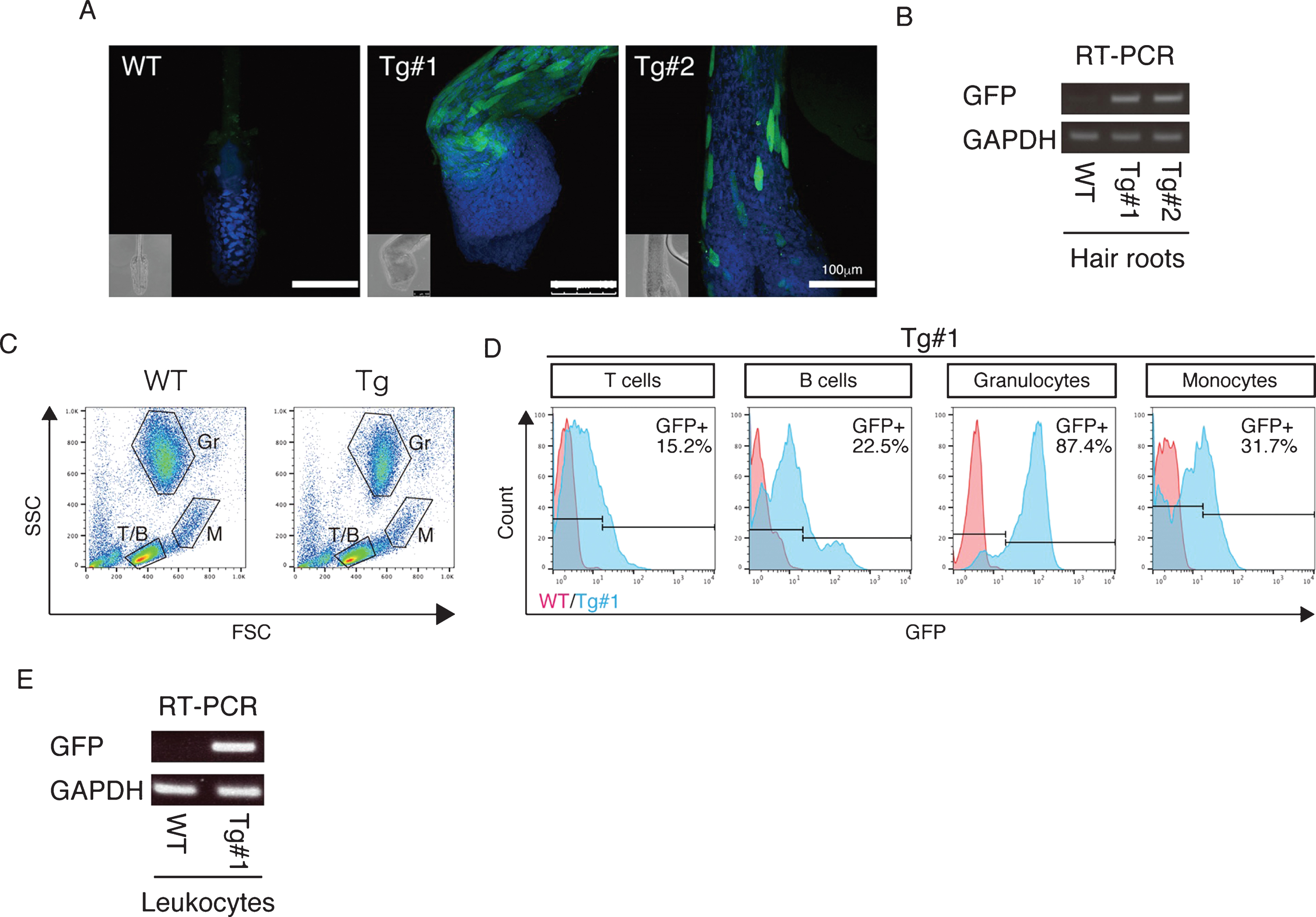
Fig.6
APP expression in the Tg cynomolgus monkeys. A) Immunohistochemistry of hair roots from WT, Tg #1 and Tg #2 offspring with anti-GFP antibodies detected by confocal microscopy. Images were taken under the same instrument settings (same laser intensity). Scale bars = 100 μm. B) RT–PCR with cDNAs of hair roots from WT, Tg #1 and Tg #2 offspring. C) FSC/SSC plots for T cells (T/B), B cells (T/B), granulocytes (Gr) and monocytes (M). D) GFP expression in blood cells from WT (red) and Tg #1 (blue) offspring. E) RT–PCR with cDNAs of leukocytes from WT and Tg #1 offspring.
Plasma Aβ levels were measured in a WT monkey and the two transgenic monkeys (Table 4). In the transgenic monkeys, the plasma Aβ40 levels were approximately double, while the plasma Aβ42 levels were approximately 50-fold, compared with those of the WT monkey resulting in approximately 20-fold increase in ratio of Aβ42 to Aβ40 in the transgenic monkeys compared with the WT monkey.
Table 4
Plasma Aβ levels in transgenic cynomolgus monkeys
| Aβ40 (pM) | Aβ42 (pM) | Aβ42/Aβ40 | |
| WT | 25.2 | 1.4 | 0.06 |
| Tg#1 | 55.5 | 61.5 | 1.11 |
| Tg#2 | 42.6 | 64.7 | 1.52 |
Soluble and insoluble (formic acid soluble) Aβ levels were measured in the brain of aborted WT and transgenic monkeys (Table 5). The levels of insoluble Aβ42 were much higher in the transgenic monkeys (147.5 and 1,016.9 pmol/g protein compared to the WT monkey (19.3 pmol/g). The ratios of Aβ42 to Aβ40 in the transgenic monkeys showed a moderate increase in the soluble fraction and a marked increase in the insoluble fraction compared with the WT monkey.
Table 5
Brain Aβ levels in aborted transgenic cynomolgus monkeys. The Aβ levels are expressed as pmol/g protein
| Soluble fraction | Insoluble fraction | |||||
| Aβ40 | Aβ42 | Aβ42/Aβ40 | Aβ40 | Aβ42 | Aβ42/Aβ40 | |
| WT | 4.6 | 0.6 | 0.13 | 150 | 19.3 | 0.13 |
| Tg#101 | 4.5 | 1.8 | 0.39 | 310.6 | 1,016.90 | 3.27 |
| Tg#102 | 1.7 | 0.8 | 0.44 | 81.5 | 147.5 | 1.81 |
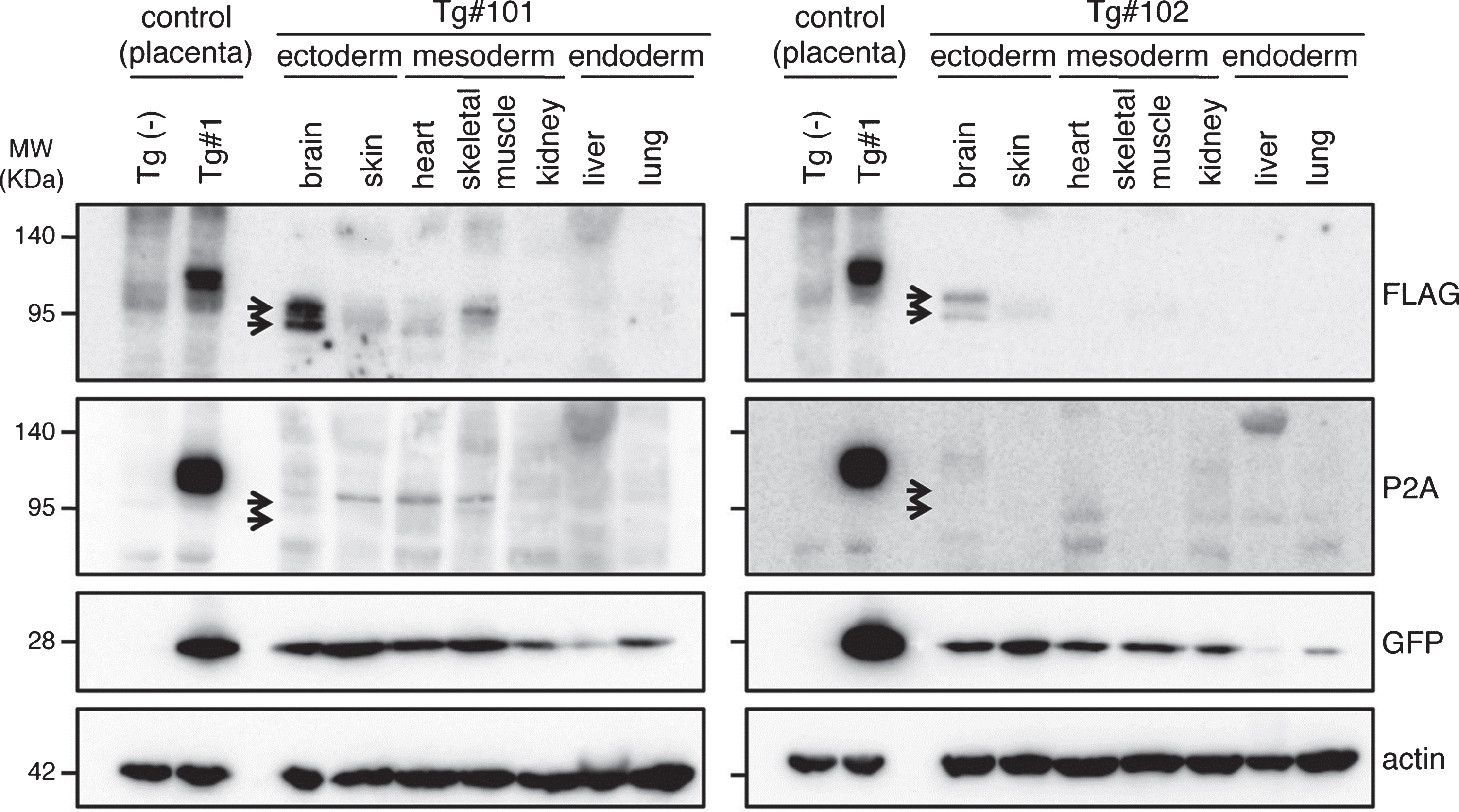
To confirm the expression of the transgene products in the aborted fetuses, protein samples from the fetuses were analyzed by western blotting (Fig. 7 and Supplementary Figure 3). GFP was detected in all tissue samples (indicated by GFP in Fig. 7). However, the anti-FLAG antibody, which recognizes the N-terminal of the exogenous APP, only reacted strongly with the brain-derived lysates identifying two bands; faint traces of GFP were detectable in some of the other tissues (indicated by FLAG in Fig. 7). Since the molecular sizes of the APP bands in the brain were smaller than that observed in the placenta from Tg#1, the same proteins samples were further blotted with an antibody specific for the C-terminal of APP. This antibody could detect APP in the lysate from the Tg placenta at the same size recognized by the N-terminal antibody, but it never reacted with the brain protein samples from the tissues of the aborted animals (indicated by P2A in Fig. 7).
Fig.7
Western blotting analysis of APP expression in placenta of wild and Tg#1 cynomolgus monkeys and several tissues in aborted Tg monkeys. Protein samples extracted from the indicated tissues of Tg #101 (left) and Tg #102 (right) were separated by 5–20% of SDS-PAGE, and subjected to western blotting analysis probed with antibodies against FLAG, P2A, GFP, and actin. As controls, the lysates from the placentas of Tg #1 and wild-type animal were also analyzed. Arrows indicate the positions of bands recognized by the anti-FLAG antibody.
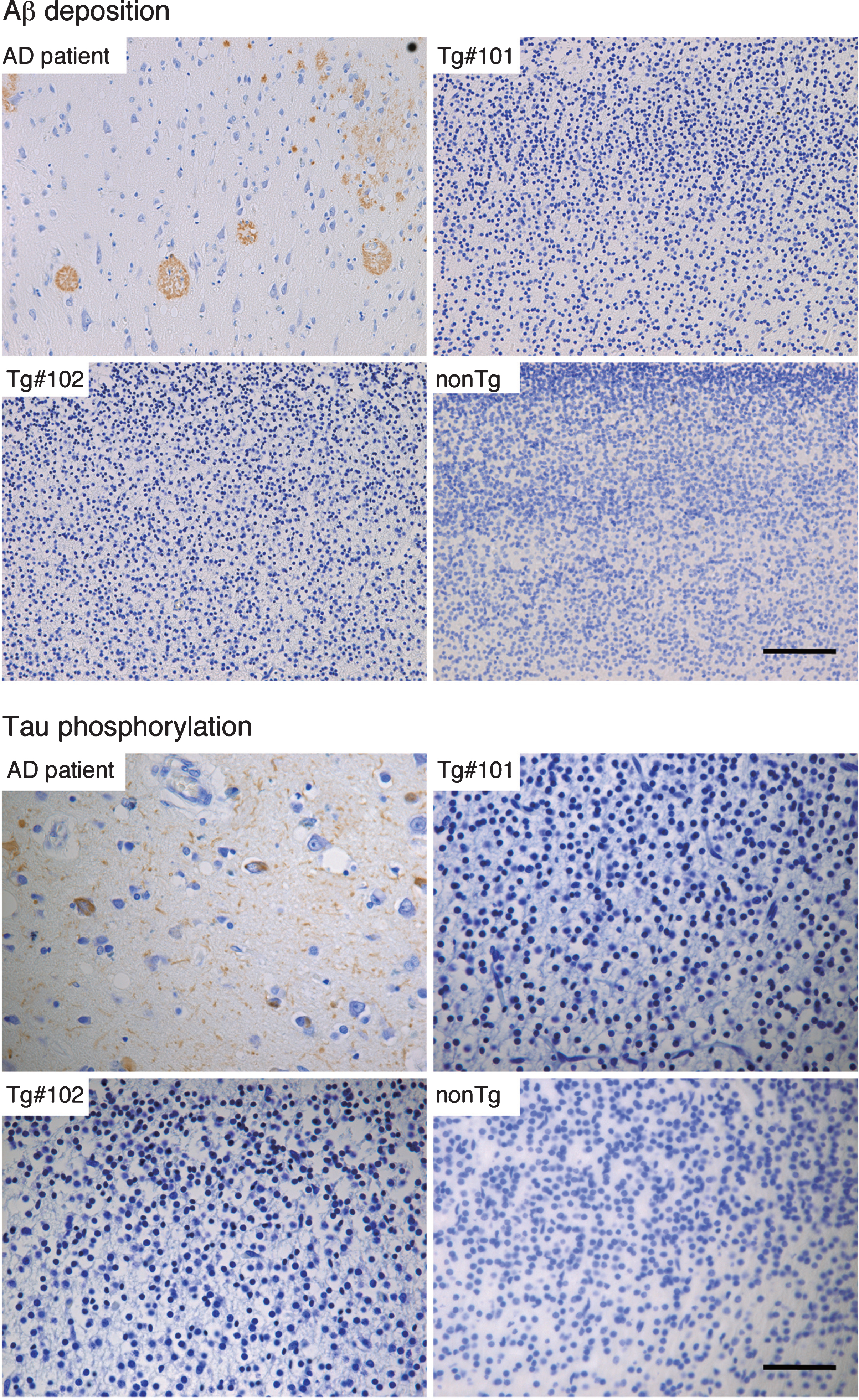
By immunohistochemistry, Aβ deposition and phosphorylated tau could be detected in brain tissues from an AD patient (Fig. 8), but not in the Tg or nonTg monkeys (Fig. 8).
Fig.8
Immunohistochemistry for Aβ and phosphorylated tau in the brains of wild and the Tg#1 and Tg #2 cynomolgus monkeys compared with an Alzheimer’s disease case. Detection of AD-related pathologies in the cerebral cortex of aborted Tg and age-matched non-transgenic fetuses. The sections of the brain tissues from the monkey embryos and an AD patient were stained with anti-Aβ (upper) and anti-phosphorylated tau antibodies (lower). Scale bar = 50 μm.
DISCUSSION
In this report, we describe the initial production of a new primate Tg models of AD using the expression construct pCSII-CAG-FLAG-APP-P2A-GFP. This is the first report describing the generation of Tg cynomolgus monkeys as potential experimental models for AD. The Tg monkeys were detectable at birth by GFP fluorescence. Although several Tg monkeys were aborted, two Tg monkeys were born alive.
In the transgenic monkeys, plasma Aβ40 levels were approximately double, while the plasma Aβ42 levels were approximately 50-fold compared with those of the WT monkey. Consequently, there was an approximately 20-fold increase in ratio of Aβ42 to Aβ40 in the transgenic monkeys, compared with the WT monkey. In the aborted Tg monkeys, however, there were no differences in the brain levels of soluble Aβ levels between aborted WT and transgenic monkeys, but there were noticeable differences in levels of insoluble Aβ42. Ratios of Aβ42 to Aβ40 in the transgenic monkeys showed a small increase in the soluble fraction and a marked increase in the insoluble fraction.
There appeared to be a discrepancy between plasma Aβ levels in living Tg monkeys and brain Aβ levels in the aborted animals. One possibility is that the expression level of mutant APP may be low in the aborted Tg monkeys. This was shown by the low expression levels of GFP and APP in the brain of aborted monkey compared with placenta from living Tg monkeys. The other possibility is that the expression level of APP increase after birth. By immunohistochemistry, no amyloid deposition and phosphorylated tau were observed in the brains of the aborted monkeys. In wild-type cynomolgus monkeys, mature (classical and primitive) plaques appeared in monkeys at age of more than 20 years old [24]. We used a transgene carrying the Swedish, Arctic, and Beyreuther/Iberian mutations that have been reported to accelerate amyloid deposition [12]. Saito et al. reported that knock-in-mouse carrying the same mutations showed amyloid deposition that began by 2 months and plateaued by 7 months [12]. From this, we assume that amyloid plaques will develop at about 5 years of age in our model monkey. According to Bateman et al. [25], amyloid deposition occurs 20–30 years before the onset of AD, and tau pathology as well as neurodegeneration develop later. Therefore, it can be anticipated that it will take more time for tau pathology and neurodegeneration in the brain of Tg monkey after amyloid deposits. As tau pathology does not occur in any mouse model carrying APP mutations [10], it will be of great interest to clarify whether these Tg monkeys will develop tauopathy and neurodegeneration similar to human AD.
Rodent models have been very useful for research on AD and for the development of diagnostic and therapeutic agents. However, we should consider the limitations of such models. For example, the first vaccine-based therapy for AD showed dramatic effects in a mouse model [26], but, when the vaccine was administered to human patients, severe side-effects occurred such as neuroinflammation [27–29]. The failure of that first vaccine trial indicates that we should test potential therapeutic agents for adverse effects using animal models that closely model humans, such as monkeys [30].
It must be appreciated that even Tg AD monkeys will have some limitations. We employed the CAG promoter, which will promote gene expression in a non-tissue specific manner. Although western blotting analysis indicated that expression of APP in Tg monkeys was higher in the brain than other tissues, the other tissues also expressed APP, including blood cells. Thus, these monkeys may develop a systemic amyloidosis-like disease. Refinements to these models will be possible once the pathological features are more fully investigated. Inducible knock-in of gene expression at later ages, and more specific cellular promoters can be considered but to be a valid human model, all features of this primate model will require a number of years to see pathological effects.
In summary, we generated Tg monkeys carrying mutations identified to cause familial AD. To confirm whether such monkeys will be suitable as a model for AD, we will need to examine their phenotypes. However, we will have to wait for several years until these monkeys develop AD-like pathology.
ACKNOWLEDGMENTS
This study was supported in part by JSPS KAKENHI Grant Numbers JP17K14977 to Y.S., 17K17812 to N.W., 17K08077 to T.M., 17H03560 to I.T., and 19H03546 to M.N., by the Strategic Research Program for Brain Sciences from Japan Agency for Medical Research and Development (17dm0107141h0002) to M.N., and by an in-house grant from Shiga University of Medical Science to M.E. and Y.S. We thank James Cummins, PhD, from Edanz Group (http://www.edanzediting.com/ac) for editing a draft of this manuscript. We thank Dr. Douglas G Walker and Dr. Jean-Pierre Bellier for the technical comments.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/19-1081r2).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-191081.
REFERENCES
[1] | Ikejima C , Hisanaga A , Meguro K , Yamada T , Ouma S , Kawamuro Y , Hyouki K , Nakashima K , Wada K , Yamada S , Watanabe I , Kakuma T , Aoyama Y , Mizukami K , Asada T ((2012) ) Multicentre population-based dementia prevalence survey in Japan: A preliminary report. Psychogeriatrics 12: , 120–123. |
[2] | Winblad B , Amouyel P , Andrieu S , Ballard C , Brayne C , Brodaty H , Cedazo-Minguez A , Dubois B , Edvardsson D , Feldman H , Fratiglioni L , Frisoni GB , Gauthier S , Georges J , Graff C , Iqbal K , Jessen F , Johansson G , Jönsson L , Kivipelto M , Knapp M , Mangialasche F , Melis R , Nordberg A , Rikkert MO , Qiu C , Sakmar TP , Scheltens P , Schneider LS , Sperling R , Tjernberg LO , Waldemar G , Wimo A , Zetterberg H ((2016) ) Defeating Alzheimer’s disease and other dementias: A priority for European science and society. Lancet Neurol 15: , 455–532. |
[3] | Braak H , Braak E ((1996) ) Evolution of the neuropathology of Alzheimer’s disease. Acta Neurol Scand Suppl 165: , 3–12. |
[4] | Goate A , Chartier-Harlin M-C , Mullan M , Brown J , Crawford F , Fidani L , Giuffra L , Haynes A , Irving N , James L , Mant R , Newton P , Rooke K , Roques P , Talbot C , Pericak-Vance M , Roses A , Williamson R , Rossor M , Owen M , Hardy J ((1991) ) Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349: , 704–706. |
[5] | Sherrington R , Rogaev EI , Liang Y , Rogaeva EA , Levesque G , Ikeda M , Chi H , Lin C , Li G , Holman K , Tsuda T , Mar L , Foncin J-F , Bruni AC , Montesi MP , Sorbi S , Rainero I , Pinessi L , Nee L , Chumakov I , Pollen D , Brookes A , Sanseau P , Polinsky RJ , Wasco W , Da Silva HAR , Haines JL , Pericak-Vance MA , Tanzi RE , Roses AD , Fraser PE , Rommens JM , St George-Hyslop PH ((1995) ) Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375: , 754–760. |
[6] | Rogaev EI , Sherrington R , Rogaeva EA , Levesque G , Ikeda M , Liang Y , Chi H , Lin C , Holman K , Tsuda T , Mar L , Sorbi S , Nacmias B , Piacentini S , Amaducci L , Chumakov I , Cohen D , Lannfelt L , Fraser PE , Rommens JM , George-Hyslop PHS ((1995) ) Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 376: , 775–778. |
[7] | Levy-Lahad E , Wasco W , Poorkaj P , Romano DM , Oshima J , Pettingell WH , Yu CE , Jondro PD , Schmidt SD , Wang K ((1995) ) Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 269: , 973–977. |
[8] | Hardy J , Selkoe DJ ((2002) ) The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 297: , 353–356. |
[9] | Hardy JA , Higgins GA ((1992) ) Alzheimer’s disease: The amyloid cascade hypothesis. Science 256: , 184–185. |
[10] | Sasaguri H , Nilsson P , Hashimoto S , Nagata K , Saito T , De Strooper B , Hardy J , Vassar R , Winblad B , Saido TC ((2017) ) APP mouse models for Alzheimer’s disease preclinical studies. EMBO J 36: , 2473–2487. |
[11] | Drummond E , Wisniewski T ((2017) ) Alzheimer’s disease: Experimental models and reality. Acta Neuropathol 133: , 155–175. |
[12] | Saito T , Matsuba Y , Mihira N , Takano J , Nilsson P , Itohara S , Iwata N , Saido TC ((2014) ) Single App knock-in mouse models of Alzheimer’s disease. Nat Neurosci 17: , 661–663. |
[13] | KikuchiT, MorizaneA, DoiD, MagotaniH, OnoeH, HayashiT, MizumaH, TakaraS, TakahashiR, InoueH, MoritaS, YamamotoM, OkitaK, NakagawaM, ParmarM, TakahashiJ (2017) Human iPS cell-derived dopaminergic neurons function in a primate Parkinson’s disease model. Nature 548: , 592–596. |
[14] | Seita Y , Tsukiyama T , Azami T , Kobayashi K , Iwatani C , Tsuchiya H , Nakaya M , Tanabe H , Hitoshi S , Miyoshi H , Nakamura S , Kawauchi A , Ema M ((2019) ) Comprehensive evaluation of ubiquitous promoters suitable for the generation of transgenic cynomolgus monkeys. Biol Reprod 100: , 1440–1452. |
[15] | Seita Y , Tsukiyama T , Iwatani C , Tsuchiya H , Matsushita J , Azami T , Okahara J , Nakamura S , Hayashi Y , Hitoshi S , Itoh Y , Imamura T , Nishimura M , Tooyama I , Miyoshi H , Saitou M , Ogasawara K , Sasaki E , Ema M ((2016) ) Generation of transgenic cynomolgus monkeys that express green fluorescent protein throughout the whole body. Sci Rep 6: , 24868. |
[16] | Seita Y , Iwatani C , Tsuchiya H , Nakamura S , Kimura F , Murakami T , Ema M ((2019) ) Poor second ovarian stimulation in cynomolgus monkeys (Macaca fascicularis) is associated with the production of antibodies against human follicle-stimulating hormone. J Reprod Dev 65: , 267–273. |
[17] | Podlisny MB , Tolan DR , Selkoe DJ ((1991) ) Homology of the amyloid beta protein precursor in monkey and human supports a primate model for beta amyloidosis in Alzheimer’s disease. Am J Pathol 138: , 1423–1435. |
[18] | Balmaceda JP , Heitman TO , Garcia MR , Pauerstein CJ , Pool TB ((1986) ) Embryo cryopreservation in cynomolgus monkeys. Fertil Steril 45: , 403–406. |
[19] | Yamasaki J , Iwatani C , Tsuchiya H , Okahara J , Sankai T , Torii R ((2011) ) Vitrification and transfer of cynomolgus monkey (Macaca fascicularis) embryos fertilized by intracytoplasmic sperm injection. Theriogenology 76: , 33–38. |
[20] | Steptoe PC , Edwards RG ((1978) ) Birth after the reimplantation of a human embryo. Lancet 2: , 366. |
[21] | Palermo G , Joris H , Devroey P , Van Steirteghem AC ((1992) ) Pregnancies after intracytoplasmic injection of single spermatozoon into an oocyte. Lancet 340: , 17–18. |
[22] | Marshall VS , Browne MA , Knowles L , Golos TG , Thomson JA ((2003) ) Ovarian stimulation of marmoset monkeys (Callithrix jacchus) using recombinant human follicle stimulating hormone. J Med Primatol 32: , 57–66. |
[23] | Iliff SA , Molskness TA , Stouffer RL ((1995) ) Anti-human gonadotropin antibodies generated during in vitro fertilization (IVF)-related cycles: Effect on fertility of rhesus macaques. J Med Primatol 24: , 7–11. |
[24] | Nakamura S , Nakayama H , Goto N , Ono F , Sakakibara I , Yoshikawa Y ((1998) ) Histopathological studies of senile plaques and cerebral amyloidosis in cynomolgus monkeys. J Med Primatol 27: , 244–252. |
[25] | Bateman RJ , Xiong C , Benzinger TL , Fagan AM , Goate A , Fox NC , Marcus DS , Cairns NJ , Xie X , Blazey TM , Holtzman DM , Santacruz A , Buckles V , Oliver A , Moulder K , Aisen PS , Ghetti B , Klunk WE , McDade E , Martins RN , Masters CL , Mayeux R , Ringman JM , Rossor MN , Schofield PR , Sperling RA , Salloway S , Morris JC ; Dominantly Inherited Alzheimer Network ((2012) ) Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med 367: , 795–804. |
[26] | Schenk D , Barbour R , Dunn W , Gordon G , Grajeda H , Guido T , Hu K , Huang J , Johnson-Wood K , Khan K , Kholodenko D , Lee M , Liao Z , Lieberburg I , Motter R , Mutter L , Soriano F , Shopp G , Vasquez N , Vandevert C , Walker S , Wogulis M , Yednock T , Games D , Seubert P ((1999) ) Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400: , 173–177. |
[27] | Bishop GM , Robinson SR , Smith MA , Perry G , Atwood CS ((2002) ) Call for Elan to publish Alzheimer’s trial details. Nature 416: , 677. |
[28] | Check E ((2002) ) Nerve inflammation halts trial for Alzheimer’s drug. Nature 415: , 462–462. |
[29] | Nicoll JAR , Wilkinson D , Holmes C , Steart P , Markham H , Weller RO ((2003) ) Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: A case report. Nat Med 9: , 448–452. |
[30] | Robinson SR , Bishop GM , Lee H-G , Münch G ((2004) ) Lessons from the AN 1792 Alzheimer vaccine: Lest we forget. Neurobiol Aging 25: , 609–615. |


