
Dual Bioorthogonal Labeling of the Amyloid-β Protein Precursor Facilitates Simultaneous Visualization of the Protein and Its Cleavage Products
Abstract
The amyloid-β protein precursor (AβPP) is critical in the pathophysiology of Alzheimer’s disease (AD), since two-step proteolytic processing of AβPP generates the neurotoxic amyloid-β peptide (Aβ). We developed a dual fluorescence labeling system to study the exact subcellular location of γ-secretase cleavage of AβPP. The C-terminal tail of AβPP was fluorescently labeled using a SNAP-tag, while the Aβ region of AβPP was fluorescently tagged with a dye at a genetically-encoded noncanonical amino acid (ncAA). The ncAA was introduced at specific positions in AβPP using a genetic code expansion strategy and afterwards, the reactive side-chain of the ncAA was coupled to the dye using a bioorthogonal labeling chemistry. In proof-of-concept experiments, HEK293T cells were transfected with plasmids containing engineered AβPP harboring an amber mutation and an amber codon suppression system with an evolved tRNA synthetase/tRNA pair and grown in the presence of a lysine-derived ncAA. Processing of the AβPP variants was validated with ELISA and immunoblotting, and seven AβPP mutants that showed similar cleavage pattern as wild-type AβPP were identified. The AβPP mutant was fluorescently labeled with 6-methyl-tetrazine-BDP-FL and TMR-Star at the ncAA and SNAP-tag, respectively. Using this approach, AβPP was fluorescently labeled at two sites in living cells with minimal background to allow monitoring of Aβ and C-terminal cleavage products simultaneously. The method described provides a powerful tool to label Aβ with minimal perturbations of its processing, thus enabling studies of the trafficking of the cleavage products of AβPP.
INTRODUCTION
Alzheimer’s disease (AD) is the most common form of dementia. The primary risk factor for AD is age and the prevalence of AD is expected to double every 20 years [1]. The amyloid-β peptides (Aβ), which are 39- to 43-amino acid residue peptides derived from the amyloid-β protein precursor (AβPP) are involved in the molecular pathogenesis of AD. Three membrane-bound enzymes are largely responsible for AβPP processing: α-, β-, and γ-secretase. In cases where AβPP is first cleaved by β-secretase and then by γ-secretase, Aβ is produced [2]. The longer forms of Aβ—Aβ42 and Aβ43—are toxic to neurons and trigger further manifestations of AD such as tangle formation, neuroinflammation, and apoptosis [3]. Besides AβPP, γ-secretase cleaves more than 90 substrates, including Notch and the receptor tyrosine-protein kinase ErbB. Therefore, inhibition of γ-secretases also interferes with other essential physiological processes and is associated with adverse side effects [4, 5]. A better understanding of AβPP processing, including the spatial and temporal parameters of AβPP proteolysis, is crucial for developing novel therapies for targeting γ-secretase.
One approach to study AβPP processing in live cell systems is to use protein labels or chemical probes. Tracking the production of AβPP and subsequent processing both with respect to the timing and location of the cleavages would provide novel information. However, commonly used direct or indirect labeling techniques, such as fluorescent protein fusion or immunofluorescence staining, have limitations. For example, due to their size, fluorescent proteins cannot be introduced at or near the Aβ sequence within AβPP without perturbing its processing or subcellular localization. Therefore, we aimed to develop a minimally disruptive dual-labeling strategy to study relevant AβPP fragments in live cells.
To introduce a fluorescent label into the Aβ region of AβPP, genetic code expansion for site-specific incorporation of a noncanonical amino acid (ncAA) at defined positions can be used. The genetic code encodes 20 canonical amino acids, in addition to selenocysteine, in mammals. The expansion of this code enables site specific and genetically directed incorporation of analogues of natural AAs, which can be used to label proteins with minimal changes of the size, structure or properties of the proteins, and visualize proteins using microscopy [6]. Expansion of the genetic code requires 1) a tRNA-synthetase/tRNA pair, 2) a ncAA, and 3) a blank codon. The ncAA is incorporated into AβPP by the pyrrolysyl-synthetase (PylRS)/tRNA pair from Methanosarcina mazei at an amber codon. PylRSAF, an engineered variant of PylRS, has a broad specificity for bulky amino acids, which allows it to incorporate trans-cyclooctene lysine (TCO*K) [7, 8]. The aminoacyl-tRNA-synthetase/tRNA pairs must be orthogonal in the organism in which they are used, meaning that the synthetases aminoacylate their matching tRNA, but not tRNAs of the host organism. The orthogonal tRNA is then only aminoacylated by the orthogonal synthetase, but not by any endogenous synthetase [9]. Moreover, the ncAA should only be recognized by the orthogonal tRNA-synthetase/tRNA pair and not by any endogenous tRNA-synthetase/tRNA pair. The incorporation process of the ncAA is a competition with translation release factors, which recognize the stop codons and trigger the termination of the translation.
The incorporation of a ncAA into AβPP can be used for labeling the Aβ sequence with various fluorescent dyes. The click chemistry reaction between 1,2,4,5-tetrazines and strained alkene dienophiles has several advantages, including a fast reaction time, high selectivity and minimal perturbation of the protein moiety [10]. The reaction rate between TCO*K and tetrazine is indeed up to 105 M–1 s–1 [6]. Another orthogonal labeling strategy is the SNAP-tag. It is an O6-alkyl guanine-DNA alkyltransferase (AGT) and can be used to label proteins with small molecules such as fluorophores in living cells [11]. Compared with fluorescent proteins like GFP, the use of a SNAP-tag labeling accommodates a range of organic dyes, which can be brighter and more stable fluorophores [12]. In this study we combined two fluorescence labels reacting with a ncAA in the Aβ sequence and a SNAP-tag in the C-terminus. Thus, we have designed a method that enables real-time monitoring of γ-secretase cleavage of AβPP as well as trafficking of the substrate and both products.
MATERIALS AND METHODS
Reagents and plasmids
The pSNAP-tag(m) plasmid, which was used as backbone, is available at Addgene (#101135). The transfection reagent TransIT-LT-1 was purchased from Mirus (Lot81023060) and the ncAA TCO*K (axial trans-cyclooct-2-ene–L–lysine; CAS 1801936-26-4) was purchased from SiChem. We used four fluorescent dyes for labeling. The SNAP-Cell® 647-SiR (S9102 S) and the SNAP-Cell® TMR-Star (S9105 S) were purchased from New England BioLabs and the 6-methyl-tetrazine-BDP-FL is produced by Jena Bioscience (RK011-001). The SiR-DNA kit (SC007) was purchased by Spirochrome. For detecting Aβ, we used the High sensitive IBL-ELISA: Amyloid-beta (1–42) (LOT. 1D-818) and Amplex® UltraRed from Molecular Probes, Thermo Fisher Scientific. The 4–20% polyacrylamide Bis-Tris gels were purchased by BioRad. The cOmplete™, EDTA-free Protease Inhibitor Cocktail was purchased from Sigma (000000005056489001). The primary antibodies we used are the Anti-SNAP-tag® antibody (Polyclonal), (1 : 200) from New England BioLabs (P9310 S) and the Anti-β-Amyloid, 1–16, Clone: 6E10 (1 : 1000) from BioLegend (803001). The secondary antibodies are the IRDye 680RD Donkey anti-Rabbit (926-68073) and IRDye 800 CW Donkey anti-mouse (92632212) from LI-COR Biosciences.
Constructs, cloning, and mutagenesis
The Plasmid pSNAP-tag(m) was shortened by XbaI and NotI. Afterwards the tRNA cassette (previously described by Schmied et al. [13]) was inserted by an In-fusion method. Briefly, the fragment was amplified using the primer Forward (5′-TTGATTATTGACTAGTAAGGCCTCCAAGGCC-3′) and Reverse (5′-ACTATTAATAACTAGTGGAGCGATCGCAGATCCACTAG -3′). The gene encoding hAβPP695 was amplified using the primer AβPP-F (5′-CGAGCTCGGATCGATATCCACCATGCTGCCCGGTTTGGC-3′) and AβPP-R (5′-TGTCCATGGTGGGGCAGGCACTTCCAAATTCGTTCTGCATCTGCTCAAAGAAC-3′). Both genes were cloned into the vector constructed as described above via EcoRV and EcoRI. The mutations in AβPP were inserted by Site-directed mutagenesis by primer extension. The gene encoding the PylRSAF was amplified from a plasmid carrying PylRSAF using the primer PylRS-F (5′-CTCACTCGAGGGATCCTTATTACAGGTTCGTAGAGATC-3′) and PylRS-R (5′-CAGGTTGGTGCTGATGCCGTTATAATACGATTCAGAACGG–3′) and cloned into the vector described above via EcoRV and BamHI.
Transient transfection and labeling of HEK293T-cells
HEK293T cells were seeded in ViewPlate-96 PDL coated, black, glass bottom plates. After 24 h the HEK293T cells were transfected with 0.02 μg PylRSAF-vector and 0.08 μg gene-of-interest vector. As a transfection reagent, TransIT-LT-1 was used according to the manufacturer’s protocol. 0.1 mM TCO*K (axial trans-cyclooct-2-ene–L–lysine) from a 100 mM stock solution was added directly after transfection, and cells were incubated for 40 h before labeling. The SNAP-Cell® TMR-Star stock solution was diluted 1 : 200 in Dulbecco’s Modified Eagle’s Medium (DMEM)/10% Fetal Bovine Serum (FBS) medium to yield a labeling medium of 3 μM dye substrate. After replacing the cell culture medium with the SNAP-Cell® TMR-Star labeling medium, the cells were incubated at 37°C and 5% CO2 for 30 min. Next, the cells were washed three times with cell culture medium and incubated in fresh medium for 120 min. The medium was replaced one more time. 5 μM 6-methyl-tetrazine-BDP-FL in medium was added and the cells were incubated at 37°C, 5% CO2 for 30 min. After washing three times with cell culture medium, the cells were incubated at 37°C, 5% CO2 for 120 min before a last rinsing. Afterwards, the cells were labeled with 1 μM SiR-DNA for 1 h.
Determination of levels of Aβ42 via ELISA and AβPP fragments via western blot
5×104 HEK293T cells were seeded in 24-well plates. After 24 h, the HEK293T cells were transfected with the PylRSAF-vector and the gene-of-interest vector in a ratio of 1 : 4 (0.4 μg DNA in total per well). As a transfection reagent, TransIT-LT-1 was used according to the manufacturer’s protocol. 0.5 mM TCO*K from a 100 mM stock solution was added directly after transfection, and cells were incubated for 40 h. Aβ42 levels in the conditioned medium were determined by a commercial sandwich ELISA modified with the fluorescent substrate Amplex® UltraRed to increase sensitivity as described previously [14]. Instead of adding stop solution, samples were transferred to a new plate. The fluorescence was measured by a Fluostar Galaxy plate reader (Emission Filter 590-12).
Endogenous AβPP and AβPP-SNAP levels in the cell lysates were determined by semi-quantitative western blotting. 20 μg protein were loaded in each well. Samples were loaded on 4–20% polyacrylamide Bis-Tris gels. The protein was transferred to nitrocellulose membranes, probed with 6E10 and anti-SNAP antibodies and imaged with the Odyssey® CLx Imaging System.
Determination of labeled AβPP-SNAP and fragments via SDS-PAGE
5×105 HEK293T cells were seeded in 12-well plates. After 24 h, the cells were transfected with the PylRSAF-vector and the gene-of-interest vector in a ratio of 1 : 4. TransIT-LT-1 was used as a transfection reagent, according to the manufacturer’s protocol. 0.1 mM TCO*K was added from a 100 mM stock solution directly after transfection, followed by incubation of the cells for 40 h before labeling. To remove the excessive TCO*K, the cells were washed with DMEM+10% FBS for 30 min. The cells were subsequently washed three times with cell culture medium and incubated in fresh medium for 120 min. The medium was replaced one more time. 5 μM 6-methyl-tetrazine-BDP-FL was added and the cells were incubated at 37°C, 5% CO2 for 30 min. After washing three times with cell culture medium, the cells were incubated at 37°C, 5% CO2 for 120 min before a last washing step. The cells were lysed in lysis buffer (dH2O+0.1% SDS with completeTM protease inhibitor) and sonicated. The samples were centrifuged to pellet cell debris and concentrated to dryness by speed vac. The remaining solids were taken up in 1X dye-less Laemmli-buffer and transferred on a Biorad TGX gel. Images of the gels were taken with an Amersham Imager 600, using the 525BP20 emission filter to detect 6-methyl-tetrazine-BDP-FL.
Imaging and image analysis
Cells were imaged using a Zeiss LSM780 laser scanning confocal microscope. Lasers and filters were chosen to illuminate at fluorophore excitation maximum. Cy5 channel was used for nuclei staining, 488 nm channel was used for mT-BDPFL, 543 nm channel was used for TMR-Star. Images were processed using the software ImageJ. Experimental conditions were kept constant for all experiments. For the cell analysis, the software CellProfiler was used to quantify the mean fluorescence intensity in each image, corrected for the number of cells.
Time series of dual labeled AβPP-SNAP in HEK293T-cells
HEK293T cells were seeded in ViewPlate-96 PDL coated, black, Glass Bottom plates. After 24 h the HEK293T cells were transfected with 0.02 μg PylRSAF-vector and 0.08 μg gene-of-interest vector. As a transfection reagent, TransIT-LT-1 was used according to the manufacturer’s protocol. 0.1 mM TCO*K (axial trans-cyclooct-2-ene–L–lysine from a 100 mM stock solution was added directly after transfection, and cells were incubated for 40 h before labeling. The SNAP-Cell® SiR stock solution was diluted 1 : 200 in DMEM/10% FBS medium to yield a labeling medium of 3 μM dye substrate. After replacing the cell culture medium with the SNAP-Cell® TMR-Star labeling medium, the cells were incubated at 37°C and 5% CO2 for 30 min. Next, the cells were washed three times with cell culture medium and incubated in fresh medium for 120 min. The medium was replaced one more time. 5 μM 6-methyl-tetrazine-BDP-FL in medium was added and the cells were incubated at 37°C, 5% CO2 for 30 min. After washing three times with cell culture medium, the cells were incubated at 37°C, 5% CO2 for 120 min before a last rinsing. Cells were imaged using a Zeiss LSM780 laser scanning confocal microscope. Lasers and filters were chosen to illuminate at fluorophore excitation maximum. Cy5 channel was used for SNAP-Cell SiR, 488 nm channel was used for mT-BDPFL.
Deglycosylation of AβPP
The same lysates from HEK293T cells, which were used for the Western immunoblot, were used and mixed with 10X Glycoprotein Denaturing Buffer (NEB) and heated at 100°C for 10 min. The lysate was stored on ice and centrifuged for 10 seconds. Afterwards 10x G7 Reaction Buffer (NEB) and 1% NP40 (NEB) was added. This sample was divided into two parts. The first part serves as a control, whereas 1 μl Neuraminidase (NEB), 1 μl O-glycosidase (NEB), and 1 μl PNGaseF (NEB) was added to the second part. The protein mix was incubated overnight at 37°C and thereafter separated by SDS-PAGE and transferred to a PVDF membrane.
Changes of AβPP processing through γ-secretase inhibition
5×104 HEK293T cells were seeded in 24-well plates. After 24 h, the HEK293T cells were transfected with the PylRSAF-vector and the gene-of-interest vector in a ratio of 1 : 4 (0.4 μg DNA in total per well). As a transfection reagent, TransIT-LT-1 was used according to the manufacturer’s protocol. 0.5 mM TCO*K from a 100 mM stock solution was added directly after transfection, and cells were incubated for 48 h. Furthermore, samples were treated for the whole 48 h or just the last 24 h with the γ-secretase inhibitor L685,458. As a control transfected cells, which were not treated with inhibitor were used. Endogenous AβPP and AβPP-SNAP levels in the cell lysates were determined by semi-quantitative western blotting. 20 μg protein were loaded in each well. Samples were loaded on 4–20% polyacrylamide Bis-Tris gels. The protein was transferred to nitrocellulose membranes, probed with 6E10 and SNAP antibodies and imaged with the Odyssey® CLx Imaging System.
RESULTS
Incorporation of ncAAs at ten positions of hAβPP695-SNAP
A bioorthogonal labeling reaction between TCO*K and 6-methyl-tetrazine-BODIPY-FL (mT-BDP-FL) (Supplementary Figure 1) enabled us to label the Aβ region in living cells. Ten different AβPP variants were constructed, each of them with an amber codon in, or close to, the Aβ region of hAβPP695. Five of the mutations were located in the extracellular/luminal region of Aβ, three in the transmembrane region of Aβ and two close to the γ-secretase cleavage site in the transmembrane region outside the Aβ sequence (Fig. 1B, C). Since the ncAA used is a lysine-derivative, amino acid sites that either feature basic (R, H) or bulky (Y, H, F, I, L) side chains were chosen as replacement sites. We hypothesized that ncAAs near to the membrane border are more accessible for labeling, because of a lower steric inhibition than in the transmembrane region and being in a hydrophobic environment. Therefore, M51 > amber and L52 > amber were exchanged because of their position at the border of the transmembrane region. However, the M51 > amber and L52 > amber could have an influence on the γ-secretase cleavage site. Moreover, it was taken into consideration that no amino acids affecting the α- or β-secretase cleavage sites were exchanged [15, 16].
Fig.1
Vectors used for amber codon suppression in AβPP-SNAP and the cleavage sites of the resulting protein products. A) Schematic diagrams of the vectors for amber codon suppression in AβPP-SNAP. The vectors contain a bidirectional cytomegalovirus promotor (CMV), which initiates the transcription of the gene for the orthogonal synthetase PylRSAF or the gene of interest (AβPP-SNAP) and a U6 promotor expression cassette with pyrrolysyl-tRNAs (PylT). B) AβPP-SNAP processing in the non-amyloidogenic pathway. C) AβPP-SNAP processing in the amyloidogenic pathway. The amino acid positions in green show the selected sites for incorporation of ncAAs by amber codon suppression. The grey vertical lines between the amino acid sequences indicate the membrane borders.
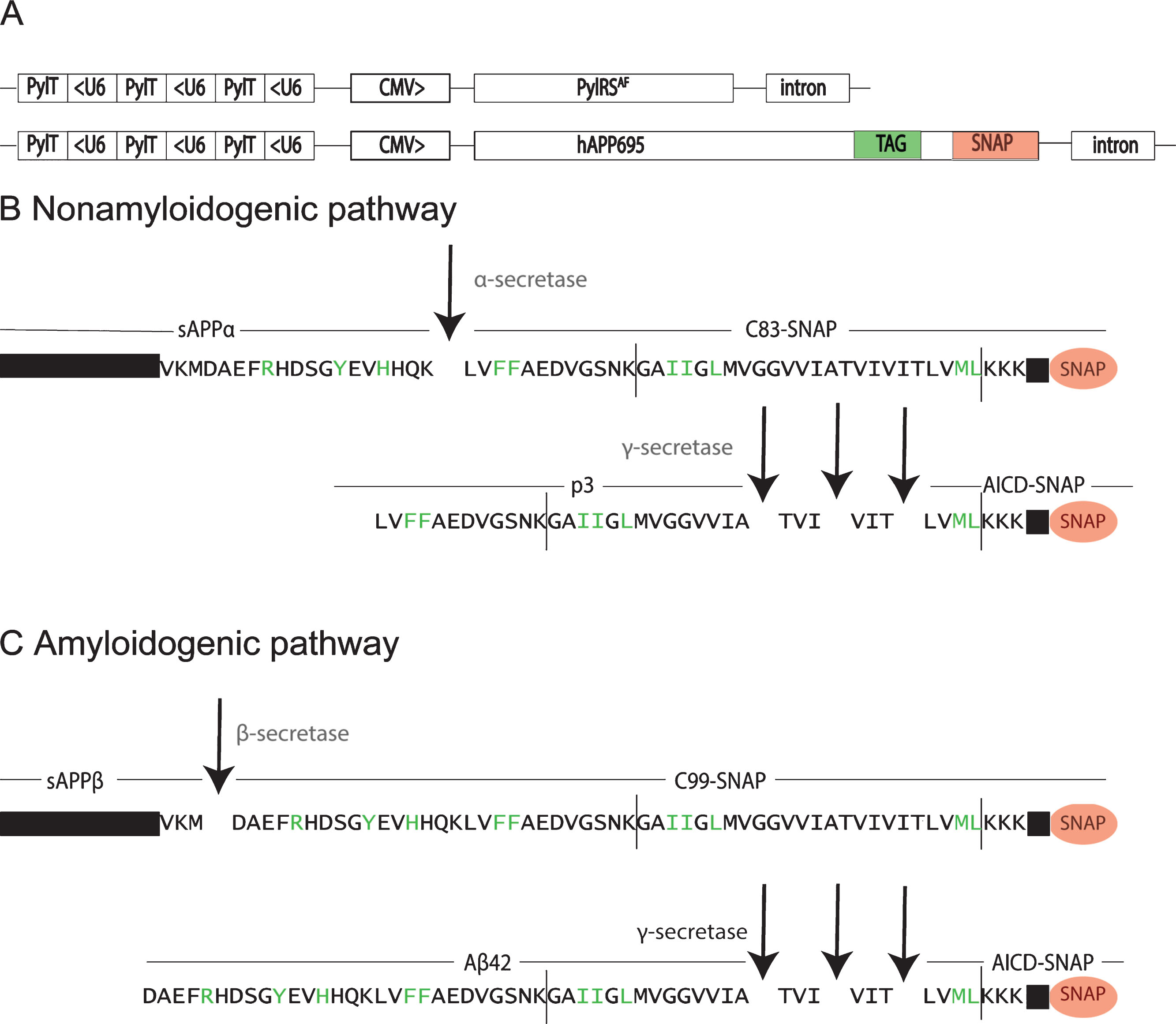
In eight of the ten mutations, we detected full length AβPP by western blotting (Supplementary Figures 2 and 3). Four of these, H13 > amber, F20 > amber, L34 > amber and M51 > amber, were chosen for further validation. This enables us to label different cleavage products of AβPP (Fig. 1). In case we use the mutation H13 > amber, we label full-length AβPP, sAβPPα, C99, and Aβ. With incorporation of the ncAA at position F20 > amber or L34 > amber, we can label full-length AβPP, C83, C99, p3, and Aβ. With a mutation at position M51 > amber, full-length AβPP, C83, C99, and AICD can be labeled.
Validation of mutated hAβPP695-SNAP processing by ELISA and western blotting
Since it is important that the processing and trafficking of the mutated AβPP-SNAP are not altered in comparison to wt AβPP-SNAP, the cleavage pattern of AβPP-SNAP after introduction of a ncAA into AβPP-SNAP was evaluated. To this end, Aβ42 levels in the medium of transfected HEK293T cells were measured using an Aβ42 sandwich ELISA. Aβ42 production of cells transfected with mutated AβPP-SNAP was compared with wt AβPP-SNAP. The H13 > amber mutation resulted in similar amount of Aβ42 as wt. In contrast, L34 > amber resulted in an increased cleavage, whereas F20 > amber resulted in a reduced cleavage. In controls lacking ncAA, only endogenous Aβ42 levels were detected. For the M51 > amber mutation without ncAA the Aβ42 production was higher in comparison with non-transfected cells and the other constructs without ncAA. The M51 > amber mutation is located downstream of the Aβ42 sequence so that the whole Aβ42 sequence is expressed also without ncAA (i.e., Aβ42 can be produced also in the absence of ncAA) (Fig. 2A).
Fig.2
ELISA and western blotting experiments confirming the expression and processing of full-length amber-suppressed AβPP-SNAP variants. A) HEK293T cells were transfected with hAβPP695-SNAP vectors with different mutations and the PylRSAF. The mutations are labeled with the amino acids number in Aβ. TCO*K was used as ncAA. AβPP-SNAP without mutation (wt) was used as a standard to compare the Aβ42 production of the different mutants. Untransfected cells were included as a second control to measure the endogenous Aβ42 levels (M). Aβ42 levels in the conditioned medium were determined by a commercial sandwich ELISA modified with the fluorescent substrate Amplex® UltraRed to increase sensitivity. n.s., not significant, **p-value < 0.01 ***p-value < 0.001. B, C) Antibody 6E10 (green) and antibody Anti-SNAP (red) were used to visualize AβPP-SNAP and its cleavage products. In the table, the estimated sizes of all detectable cleavage products in the Western blot are listed.
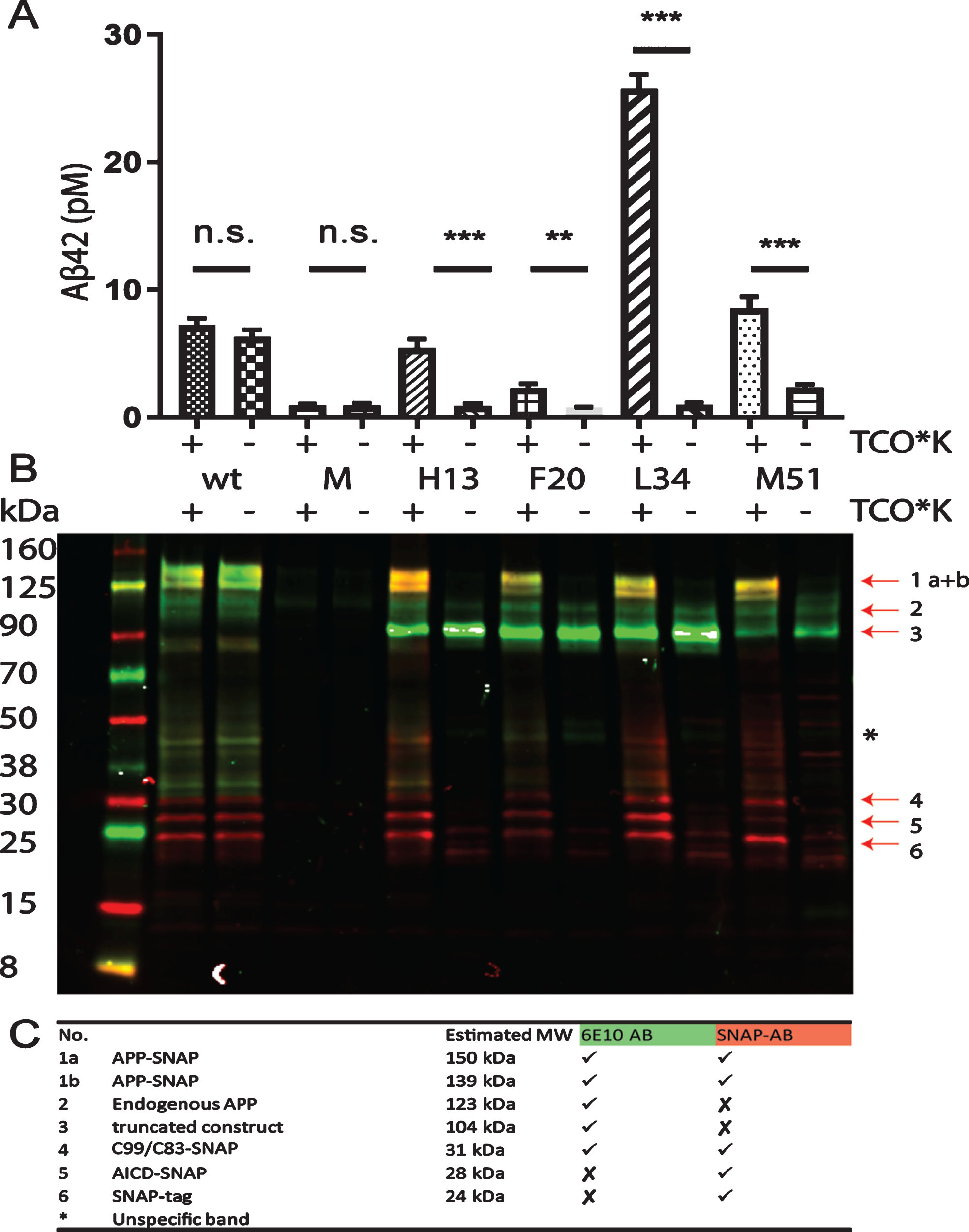
Western blotting provided further evidence for successful AβPP processing. The antibody 6E10 (green) recognizes AβPP, sAβPPα, Aβ, and C99, while the SNAP-tag antibody (red) detects the SNAP-tag at the C-terminus (Fig. 2B). The double band at 139–150 kDa represents non-glycosylated and glycosylated AβPP-SNAP, since AβPP has several glycosylation sites (Supplementary Figure 4) [17, 18]. Between 31 kDa and 24 kDa, C99/C83-SNAP, AICD-SNAP and SNAP-tag can be detected (Fig. 2 C). For unequivocal identification of the bands, the effect of treatment with the γ-secretase inhibitor L685,458 was evaluated. As expected, the AICD-SNAP band disappeared while C83/C99-SNAP was the most prominent cleavage product after treatment with the inhibitor (Supplementary Figure 5). Since H13 > amber was observed to be processed in a similar manner as wildtype AβPP-SNAP, we selected this mutation for further studies.
Validation of the labeling specificity between TCO*K and mt-BDP-FL
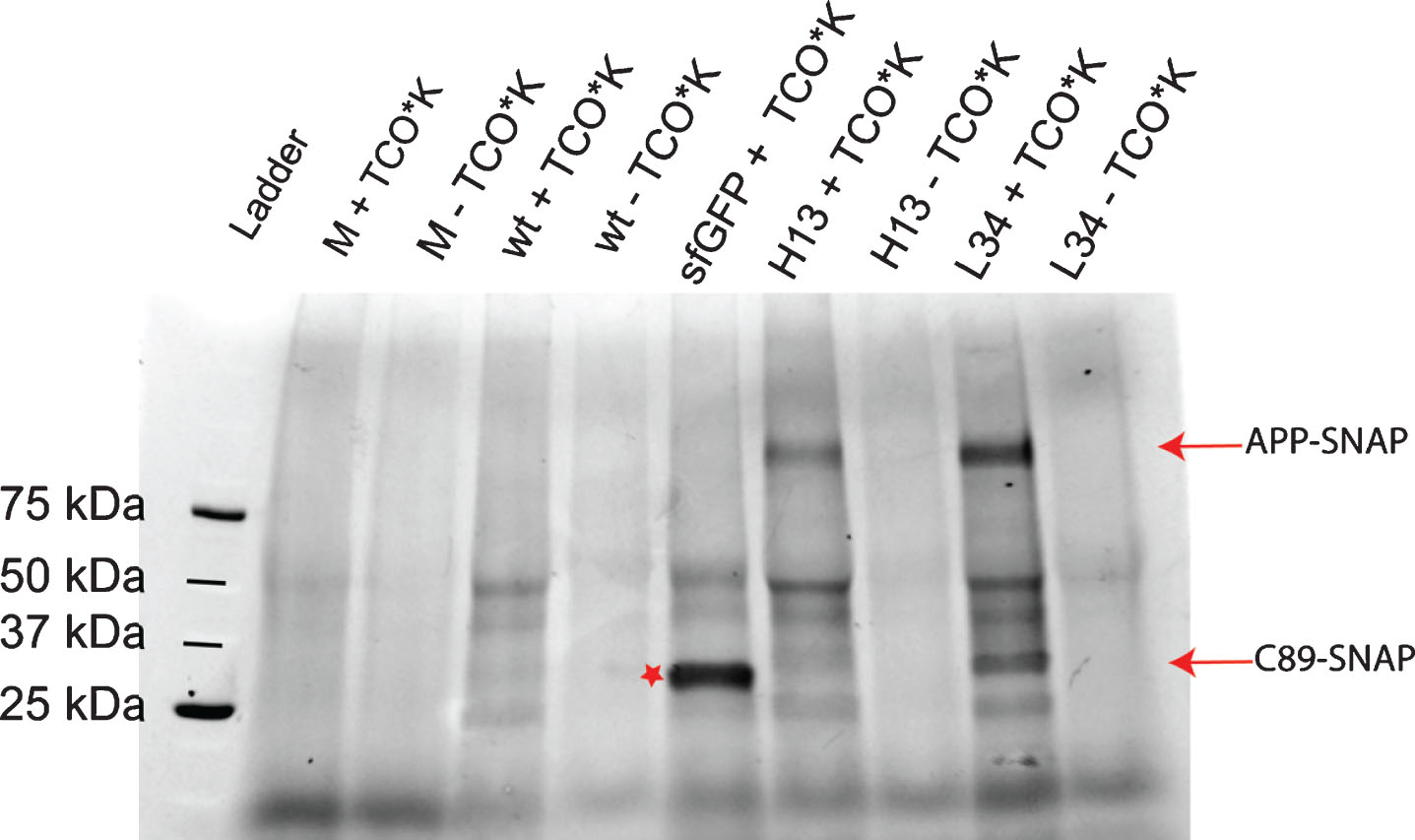
After having shown with epitope-specific antibodies that AβPP-SNAP is expressed and processed, we sought to leverage the ncAA introduced into the Aβ region to validate the labeling specificity between TCO*K and mt-BDP-FL. To this end, excessive ncAA was washed out from the HEK293T cell expression system and mT-BDP-FL added for 30 min. First, labeling and its specificity in the living cells were evaluated using SDS-PAGE (Fig. 3). Full-length AβPP-SNAP (L34 and H13 + TCO*K) and C89-SNAP (L34 + TCO*K) could be detected, suggesting that the ncAA reacted with mT-BDP-FL. The C89-SNAP was not detected with the H13 > amber mutation since the ncAA is located N-terminal to the α-secretase cleavage site (see Fig. 1). sfGFP with an amber codon at position 150 was used as a positive control [19]. A band around 50 kDa and a few other faint bands were detected in all conditions where amber suppression was active, suggesting that ncAA is also incorporated at endogenous amber codons (Fig. 3). However, these could be distinguished from Aβ labelled AβPP due to the dual labeling as well as different subcellular location (see below).
Fig.3
Evaluation of the specificity of mT-BDP-FL incorporation into amber-suppressed AβPP-SNAP variants by SDS-PAGE. HEK293T cells were transfected with AβPP-SNAP vectors with different mutations and the PylRSAF. The mutations are labeled with the amino acid residue number in Aβ. TCO*K was used as ncAA. Live cells were labeled with mT-BDP-FL (Excitation maximum at 488 nm), and lysates prepared subsequently. Images of the gels were taken by an Amersham Imager 600, filter 525BP20. Full-length AβPP-SNAP (L34 and H13 + TCO*K) and C89-SNAP (L34 + TCO) could be detected. As controls we labeled cells, which were transfected with AβPP-SNAP without amber codon (wt) and untransfected cells (M). As a further control, sfGFP (Excitation maximum of 503 nm) with an amber codon at position 150 was used (marked with a star).
Live cell imaging of dual labeled mutated hAβPP-SNAP
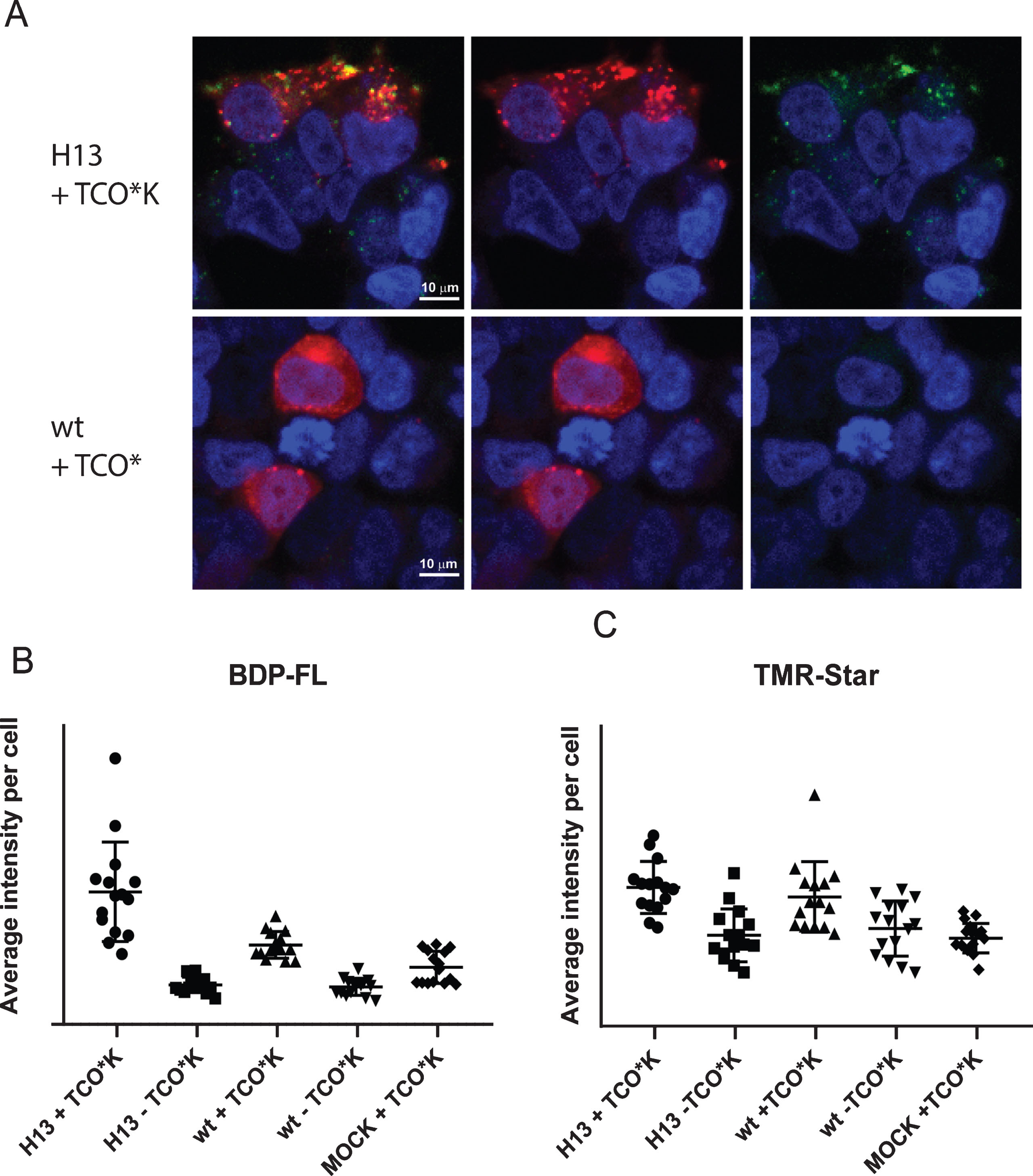
Next, we evaluated fluorescence images of transfected cells. Still images and time series were done on live cells, since the fluorescence signal of mt-BDP-FL was stronger without fixation. In this paper we use two different dyes, which react with the SNAP-tag. For the still images, SNAP-Cell® TMR-Star was used, which has an excitation maximum at 554 nm and an emission maximum at 580 nm. To avoid cross-talk between the channels, we imaged the channels sequentially. Dual-labeled cells were observed after transfection of H13 > amber and addition of TCO*K (Fig. 4A and Supplementary Figure 6). As expected, the average intensity of mT-BDP-FL fluorescence was slightly increased in the wt with TCO*K sample in comparison to the other controls, because cells transfected with the PylRSAF/tRNA pair incorporate the ncAA into endogenous amber codons found naturally in other genes (Fig. 4B, C). In support of this explanation, the labeling pattern of the cells differed. While the fluorescence was present in vesicular structures in the cells that were transfected with the H13 > amber mutation, the fluorescence pattern observed in the wt+TCO*K samples was weaker and more diffuse, in line with cytosolic labeling. Thus the unspecific staining doesn’t interfere with the specific labeling of Aβ. Colocalization of the peak signals from both dyes at vesicular structures suggest that indeed we were able to label AβPP at the two intended sites.
Fig.4
A) Confocal microscopy of HEK293T cells verifies dual labeling of the expressed AβPP-SNAP protein. HEK293T cells were transfected with the amber codon machinery and AβPP H13 > amber or wt. Live cells were labeled with TMR-Star (red) and mT-BDP-FL, which reacts with the ncAA (green). Afterwards, the nuclei were labeled with SiR-HOECHST. Labeled cells were imaged sequentially by confocal microscopy (40× magnification). B, C) Mean intensity of mT-BDP-FL and TMR per cell in each image. Number of experiments = 3. Five pictures were taken in each well.
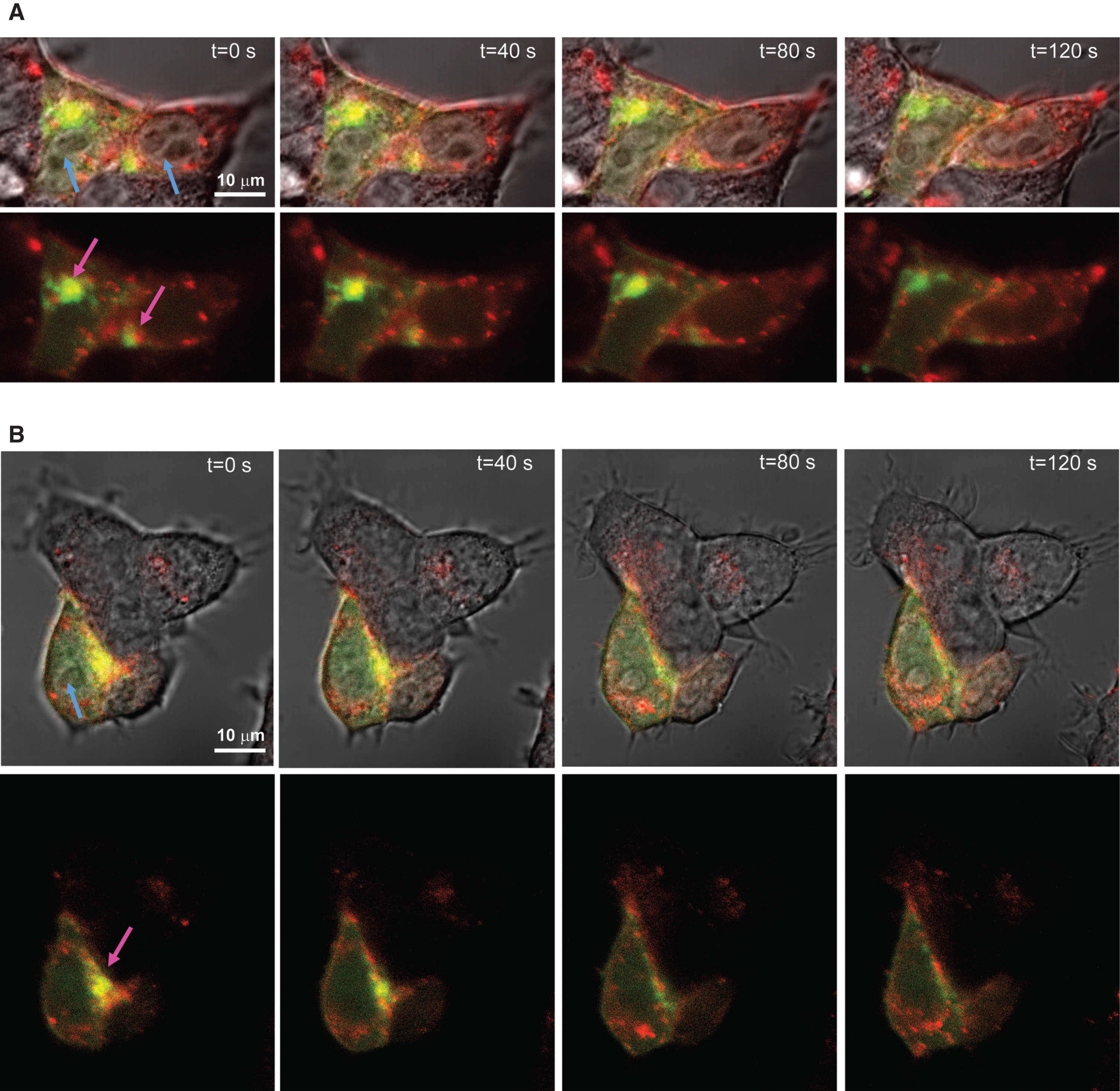
Time series of dual labeled mutated hAβPP-SNAP
We could also specifically visualize vesicle movement in live cell imaging of HEK293T cells (Fig. 5 and Supplementary Videos 1 and 2), further suggesting that the double-labeling method did not interfere with trafficking of AβPP cleavage products. Therefore, we imaged the SNAP-dye and mt-BDP-FL simultaneously to detect vesicular movement. We used SNAP-Cell® 647-SiR, which has an excitation maximum of 645 nm and an emission maximum at 661 nm to avoid cross-talk between the dyes.
Fig.5
A, B) Time series of dual labelled AβPP-SNAP protein over a time frame of two minutes shows vesicular movement. HEK293T cells were transfected with the amber codon machinery and AβPP H13 > amber. Live cells were labeled with SNAP-Cell® 647-SiR (red) and mT-BDP-FL, which reacts with the ncAA (green). Labeled cells were imaged simultaneous by confocal microscopy (63× magnification). Blue arrows indicate nuclei, pink arrows indicate subcellular locations, where most AβPP is located.
DISCUSSION
Revealing the trafficking and processing of AβPP is necessary for understanding the molecular and cellular pathophysiology of AD. Most studies about AβPP processing using cell lysates or measure changes in Aβ production via ELISA. However, detailed information about the subcellular locations of the processing is missing. Furthermore, the exact localization of γ-secretase cleavage is necessary to find out where Aβ is produced in the cells and if there is a difference between the localization of the Aβ42 and Aβ40 production. If so, this can be addressed in future studies to see if the localization of γ-secretase cleavage is changing with age. Therefore, we have established a dual labeling system using amber codon suppression and SNAP-tag labeling of AβPP with minimal changes in the size and structure of the protein and its processing. Our approach using genetic code expansion is the first approach described for direct labelling of Aβ in full length AβPP in living cells till today.
Genetic code expansion and the labeling of the ncAAs has two main limitations. Since the incorporation of TCO*K is always in competition with the translation release factors, truncated AβPP is produced. This limitation can be mitigated by codon engineering or by using genetic code expansion in engineered organisms, where the release factors do not recognize amber codons [13, 20, 21]. The second limitation is the background labeling, which can be minimized by using fluorophores whose fluorescence increase upon the labeling reaction in living cells. Thus, the “turn-on” property of mT-BDP-FL leads to an increase of the signal-to-background ratio and makes the mT-BDP-FL dye suitable for live cell imaging of intracellular targets. Fluorescence of the free dye is significantly quenched by the tetrazine moiety, and the quenching is relieved after undergoing cycloaddition with TCO*K. The fluorescence intensity of the uncharged and lipophilic dye is increased 15 times upon reacting with the dienophile of the ncAA [22]. As Serfling et al. show, high intracellular labeling background can be a problem for TAMRA and SiR-tetrazines because of excessive TCO*K and tRNA–TCO*K inside the cells [23]. However, we detect background labeling only when we express the PylRSAF/tRNA pair. In the SDS-PAGE in Figure 3, we show that the background labeling comes from incorporation of the ncAA into endogenous proteins with amber codon. However, amber codon is the least used codon in mammalian cells and since AβPP is a membrane protein we can distinguish between other cytosolic proteins and AβPP, since the latter are found in vesicular structures.
A number of recent studies monitored fluorescently labeled AβPP in living cells, but in none of these was directly labeled Aβ used. Tam et al. studied AβPP processing by adding paGFP at the C-terminus of AβPP and measured the decrease of fluorescence signal at the lysosomes. Also in other studies AβPP was labeled either in the C- or the N-terminus, often in combination with labeling of other proteins to either study protein-protein interaction or in assays for measuring BACE activity [24–26]. However, those studies cannot distinguish between the different secretase cleavage reactions or follow Aβ trafficking. Therefore, the use of genetic code expansion technology will pave the way for detailed AβPP processing studies, because it offers the great advantage that the fluorescent tag is minimally perturbing and spatially controlled. Thus, the method could also be well suited to make use of the full potential of the new generation of super-resolution microscopes, because of the close distance of the fluorophore to the protein in comparison with fluorescently labeled antibodies [27]. Furthermore, this powerful technique could be used for other types of biochemical studies, such as cross-linking studies in which the exact cross-linking position is known, by using photoactivatable ncAAs.
In conclusion, we have developed a method, which allows real-time monitoring of γ-secretase activity by dual labeling of AβPP with fluorophores on both sides of the γ-secretase cleavage site. This was achieved by linking one fluorophore directly to the Aβ sequence and another fluorophore to a SNAP-tag in the C-terminus of AβPP. We provided proof-of concept by using both ELISA, SDS-PAGE, western blotting, and image analysis that the labeling system worked and that AβPP processing is not altered. Finally, using confocal microscopy, we were able to visualize AβPP and its cleavage products, as well as monitor movement of the cleavage products, in live cells.
ACKNOWLEDGMENTS
We acknowledge the support from Thomas Olausson (L. S. van Husen), Swedish Alzheimer Foundation (Alzheimerfonden) (L. O. Tjernberg), Swedish Research Council (L. O. Tjernberg), Hjärnfonden (L. O. Tjernberg), Stiftelsen för Gamla Tjänarinnor (S. Schedin-Weiss), Gun and Bertil Stohnes Foundation (S. Schedin-Weiss), Karolinska Institutet, SFO Molecular Biosciences and Vetenskapsrådet (S. J. Elsässer), and Margaretha af Ugglas‘ Stiftelse (B. Winblad).
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/19-0898).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JAD-190898.
REFERENCES
[1] | Winblad B , Amouyel P , Andrieu S , Ballard C , Brayne C , Brodaty H , Cedazo-Minguez A , Dubois B , Edvardsson D , Feldman H , Fratiglioni L , Frisoni GB , Gauthier S , Georges J , Graff C , Iqbal K , Jessen F , Johansson G , Jönsson L , Kivipelto M , Knapp M , Mangialasche F , Melis R , Nordberg A , Rikkert MO , Qiu C , Sakmar TP , Scheltens P , Schneider LS , Sperling R , Tjernberg LO , Waldemar G , Wimo A , Zetterberg H ((2016) ) Defeating Alzheimer’s disease and other dementias: a priority for European science and society. Lancet Neurol 15: , 455–532. |
[2] | De Strooper B , Saftig P , Craessaerts K , Vanderstichele H , Guhde G , Annaert W , Von Figura K , Van Leuven F ((1998) ) Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 391: , 387–90. |
[3] | Hardy JA , Higgins GA , Hardy JA , Higgins GA ((1992) ) Alzheimer’s disease: the amyloid cascade hypothesis. Science 256: , 184–185. |
[4] | De Strooper B , Annaert W , Cupers P , Saftig P , Craessaerts K , Mumm JS , Schroeter EH , Schrijvers V , Wolfe MS , Ray WJ , Goate A , Kopan R ((1999) ) A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracelluar domain. Nature 398: , 518–522. |
[5] | Haaspasalo A , Kovacs DM ((2012) ) The many substrates of presenilin/gamma-secretase. J Alzheimers Dis 25: , 3–28. |
[6] | Lang K , Davis L , Wallace S , Mahesh M , Cox DJ , Blackman ML , Fox JM , Chin JW ((2012) ) Genetic encoding of bicyclononynes and trans-Cyclooctenes for site- specific protein labeling in vitro and in live mammalian cells via rapid fluorogenic Diels–Alder Reactions. J Am Chem Soc 134: , 10317–10320. |
[7] | Yanagisawa T , Ishii R , Fukunaga R , Kobayashi T , Sakamoto K , Yokoyama S ((2008) ) Multistep engineering of Pyrrolysyl-tRNA synthetase to genetically encode Nɛ-(o-Azidobenzyloxycarbonyl) lysine for site-specific protein modification. Chem Biol 15: , 1187–1197. |
[8] | Nikić I , Plass T , Schraidt O , Szymański J , Briggs JAG , Schultz C , Lemke EA ((2014) ) Minimal tags for rapid dual-color live-cell labeling and super-resolution microscopy. Angew Chem Int Ed 53: , 2245–2249. |
[9] | Liu DR , Schultz PG ((1999) ) Progress toward the evolution of an organism with an expanded genetic code. Chem Biochem 96: , 4780–4785. |
[10] | Blackman ML , Royzen M , Fox JM ((2008) ) Tetrazine ligation: fast bioconjugation based on inverse-electron-demand Diels–Alder reactivity. J Am Chem Soc 130: , 13518–13519. |
[11] | Keppler A , Gendreizig S , Gronemeyer T , Pick H , Vogel H , Johnsson K ((2003) ) A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotechnol 21: , 86–89. |
[12] | van de Linde S , Heilemann M , Sauer M ((2012) ) Live-cell super-resolution imaging with synthetic fluorophores. Annu Rev Phys Chem 63: , 519–540. |
[13] | Schmied WH , Elsässer SJ , Uttamapinant C , Chin JW ((2014) ) Efficient multisite unnatural amino acid incorporation in mammalian cells via optimized pyrrolysyl tRNA synthetase/tRNA expression and engineered eRF1. J Am Chem Soc 136: , 15577–15583. |
[14] | Hashimoto M , Bogdanovic N , Volkmann I , Aoki M , Winblad B , Tjernberg LO ((2010) ) Analysis of microdissected human neurons by a sensitive ELISA reveals a correlation between elevated intracellular concentrations of Aβ42 and Alzheimer’s disease neuropathology. Acta Neuropathol 119: , 543–554. |
[15] | Smith DG , Cappai R , Barnham KJ ((2007) ) The redox chemistry of the Alzheimer’s disease amyloid β peptide. Biochim Biophys Acta 1768: , 1976–1990. |
[16] | Kummer MP , Heneka MT ((2014) ) Truncated and modified amyloid-beta species. Alzheimers Res Ther 6: , 28. |
[17] | Frykman S ((2017) ) Maturation and processing of the amyloid precursor protein is regulated by the potassium/sodium hyperpolarization-activated cyclic nucleotide-gated ion channel 2 (HCN2). Biochem Biophys Res Commun 483: , 352–358. |
[18] | Schedin-Weiss S , Winblad B , Tjernberg LO ((2014) ) The role of protein glycosylation in Alzheimer disease. FEBS J 281: , 46–62. |
[19] | Elsässer SJ , Ernst RJ , Walker OS , Chin JW ((2016) ) Genetic code expansion in stable cell lines enables encoded chromatin modification. Nat Methods 13: , 158–64. |
[20] | Zhang B , Yang Q , Chen J , Wu L , Yao T , Wu Y , Xu H , Zhang L , Xia Q , Zhou D ((2016) ) CRISPRi-manipulation of genetic code expansion via RF1 for reassignment of amber codon in bacteria. Sci Rep 6: , 20000. |
[21] | Schwark D , Schmitt M , Fisk J ((2018) ) Dissecting the contribution of release factor interactions to amber stop codon reassignment efficiencies of the Methanocaldococcus jannaschii orthogonal pair. Genes 9: , 546. |
[22] | Devaraj NK , Hilderbrand S , Upadhyay R , Mazitschek R , Weissleder R ((2010) ) Bioorthogonal turn-on probes for imaging small molecules inside living cells. Angew Chem Int Ed Engl 49: , 2869–2872. |
[23] | Serfling R , Seidel L , Bock A , Lohse MJ , Annibale P , Coin I ((2019) ) Quantitative single-residue bioorthogonal labeling of G protein-coupled receptors in live cells. ACS Chem Biol 14: , 1141–1149. |
[24] | Das U , Wang L , Ganguly A , Saikia JM , Wagner SL , Koo EH , Roy S ((2015) ) Visualizing APP and BACE-1 approximation in neurons yields insight into the amyloidogenic pathway. Nat Neurosci 19: , 55–64. |
[25] | Tam JH , Seah C , Pasternak SH ((2014) ) The amyloid precursor protein is rapidly transported from the Golgi apparatus to the lysosome and where it is processed into beta-amyloid. Mol Brain 7: , 54. |
[26] | Coughlan K , Huang X , He X , Chung CHY , Li G , Tang J ((2013) ) Expression and processing of fluorescent fusion proteins of amyloid precursor protein (APP). Biochim Biophys Acta 1833: , 1562–1571. |
[27] | Balzarotti F , Eilers Y , Gwosch KC , Gynnå AH , Westphal V , Stefani FD , Elf J , Hell SW ((2016) ) Nanometer resolution imaging and tracking of fluorescent molecules with minimal photon fluxes. Science 355: , 606–612. |


