
Molecular Mechanisms and Genetics of Oxidative Stress in Alzheimer’s Disease
Abstract
Alzheimer’s disease is the most common neurodegenerative disorder that can cause dementia in elderly over 60 years of age. One of the disease hallmarks is oxidative stress which interconnects with other processes such as amyloid-β deposition, tau hyperphosphorylation, and tangle formation. This review discusses current thoughts on molecular mechanisms that may relate oxidative stress to Alzheimer’s disease and identifies genetic factors observed from in vitro, in vivo, and clinical studies that may be associated with Alzheimer’s disease-related oxidative stress.
INTRODUCTION
Alzheimer’s disease (AD) is a neurodegenerative disorder with memory deficits and executive dysfunction as characteristic clinical features [1]. Investigations show that hallmarks of oxidative stress are observed early in the progress of AD [2–7]. Related to this, pre-symptomatic AD has been associated with mitochondrial deficiency resulting in disturbed bioenergetics [8]. Apart from reducing the generation of ATP, mitochondrial deficiency results in excessive production of reactive oxygen species (ROS). These ROS, in turn, have been related to membrane damage, cytoskeletal alterations, and cell death [9]. Other than features of oxidative stress, progress of AD is characterized by extracellular accumulation of aggregated amyloid-β (Aβ), and intracellular neurofibrillary tangles containing hyperphosphorylated tau. The precise nature of the association between oxidative stress and other hallmarks of AD pathology is unknown although some molecular mechanisms have been suggested which will be discussed in this review. Further, the excessive generation of ROS as well as the neutralization of their damaging effects in a neurodegenerative condition such as AD will be covered.
OXIDATIVE STRESS
Oxidative stress is a state in which either increased levels of cellular ROS are generated and/or cellular mechanisms to reduce the potentially damaging impact of ROS are of insufficient capacity [10, 11]. This definition inherently dictates that as long as the antioxidant defense system is sufficiently capable of scavenging the generated reactive species, increased ROS or reactive nitrogen species (RNS) production generally does not provide sufficient leverage to cause pathology. However, in some cases aging-related increased production of free radical species coincides with a decreased capacity of the endogenous antioxidant defense system [12, 13]. Even though normal aging coincides with increased levels of oxidative stress, a very diverse range of diseases demonstrates a more pronounced level of oxidative stress including attention deficit hyperactivity disorder (ADHD) [14], cancer (e.g., [15]), Parkinson’s disease [16], atherosclerosis [17], myocardial infarction [18], sickle cell disease [19], Down’s syndrome [20], depression [21], and diabetes mellitus [22].
OXIDATIVE DAMAGE TO BIOMACROMOLECULES IN ALZHEIMER’S DISEASE
Even though low levels of ROS are crucial for normal physiological functioning, increased ROS levels are associated with oxidative damage of various cellular compartments and molecules. For example, structural and functional impairments of membrane-associated macromolecules such as lipids and proteins in several regions of the brain have been observed in response to ROS associated damage [23–25]. Analysis of lipid rafts in AD brain tissue samples showed that increased levels of membrane-associated oxidative stress correlated with accumulation of cholesterol and ceramides into clustered microdomains which could be prevented by the antioxidant vitamin E and ceramide inhibitors [26]. The mechanisms by which such microdomains assemble have been elaborately studied but perhaps one of the key observations was that extracellular Aβ aggregation in close proximity of the cell membrane induces membrane-associated oxidative stress. Membrane-associated oxidative stress involves lipid peroxidation and generation of aldehyde 4-hydroxynonenal (HNE), a neurotoxic aldehyde that can be detected at early stages of disease progress in the AD brain [27]. Interestingly, HNE levels were observed to be proportional to the extent of neuronal lesions [28, 29]. Oxidative stress can also lead to activation of pathways involved in AD pathogenesis. For example, one member of the mitogen-activated protein kinases (MAPKs) family, namely p38, is activated during Aβ-mediated oxidative stress. Among the different roles of p38, it was observed to induce tau phosphorylation in a primary neuronal model, which could be prevented by pretreatment with an inhibitor of p38 or vitamin E [30]. These findings were confirmed in vivo using a transgenic APP/PS1 mouse model for AD [31]. AD-related oxidative stress is also reflected by extensive oxidative damage to nucleic acids leading to alterations in DNA structure [32, 33]. Apart from in AD, oxidation of mitochondrial DNA and RNA are observed in a number of other pathologies [34]. One feature of DNA/RNA oxidation is the oxidation of the base guanosine to produce 8-hydroxyguanosine (8-oxoG) [35]. High levels of 8-oxoG were observed in neurons within the hippocampus, subiculum, entorhinal cortex, and frontal, temporal, and occipital neocortex in autoptic brain tissues of patients affected by AD [36]. Moreover, RNA oxidation was found to be significantly increased in the hippocampus, cortical neurons, white matter and in the frontoparietal cortex of aged rats [37]. These findings imply a role of oxidative-stress induced damage of DNA and RNA in neurodegenerative disease and aging.
Also, Aβ and tau have been reported to undergo a number of modifications as a function of oxidative stress. Tau plays a role in microtubule organization by dynamically interacting with the formed microtubules [38]. Intracellular dynamics of microtubule organization were observed to be disrupted in AD patients [39]. Various cell lines, including ventricular myocytes, neuro-2A cells, rat pheochromocytoma PC12, and pancreatic epithelial cell line AR42J, when exposed to H2O2 or HNE, show a decreased growth of the microtubular network as a result of increased microtubular catastrophe rate [40–45] largely mediated by Michael addition reactions [45]. This paragraph discusses the types of modification that tau and Aβ are subject to under conditions of oxidative stress.
Copper-induced dityrosine cross-linking of Aβ
A specific type of Aβ assembly involves dityrosine cross-linking which has been associated with clinical markers of oxidative stress in AD but also other neurodegenerative diseases [46]. Increased levels of oxidative stress in the brain are reflected by increased brain content of copper (Cu) and zinc (Zn), specifically in the neuropil and in AD plaques [47, 48]. Copper was shown to catalyze hydroxyl radical, peroxynitrite, nitrosoperoxycarbonate, and lipid hydroperoxide-mediated dityrosine cross-linking [49, 50] in monomeric and, at a lower rate, fibrillar Aβ1-40 [51] in a concentration-dependent manner [51]. The precise mechanism of crosslinking has been subject of study [52], but it was shown that the picomolar affinity of Aβ for copper [53] drives the generation of H2O2, which, in turn, promotes the formation of SDS-resistant dityrosine cross-linked Aβ1-28, Aβ1-40, and Aβ1-42 [54, 55]. It has also been shown that Aβ1-42, the 42-residue more amyloidogenic version of Aβ, has higher affinity to bind Cu2+ than Aβ1-40, the 40-residue version of Aβ [55]. One of the hypotheses by which binding of Aβ to Cu2+ can induce the formation of H2O2 required for Aβ crosslinking is by its ability to undergo Fenton redox cycling [56]. Consistent with this thought, histidines 6, 13, and 14 in Aβ that were identified to be involved in the redox cycling of bound Cu2+ [43] are located in close proximity to tyrosine 10. Density functional theory calculations and tyrosine-to-alanine mutational studies experimentally demonstrated that indeed tyrosine residue 10 in Aβ critically determines the generation of H2O2 mediated by Aβ-Cu2+ interaction [57]. The resulting crosslinked species were shown to accumulate in the AD brain, and to exert high levels of toxicity to neuronal cells [54, 58, 59]. Using tandem mass spectrometry, it was observed that dityrosine cross-linked forms of Aβ can also be generated in vitro under conditions of oxidative stress induced by enzymatic peroxidation [60]. A recent paper showed that exposure of in vitro generated Aβ1-40 fibrils to Cu2+ significantly reduced fibril length as a result of fibril fragmentation [51]. Even though exposure of Aβ1-40 to Cu2+ was shown to induce thioflavin T (ThT) positive fibril assembly [51, 61, 62], the addition of H2O2 inhibited the further assembly process [51] possibly stabilizing potent neurotoxic Aβ species.
Methionine-35 oxidation of Aβ
A second commonly detected Cu2+-induced modification of Aβ in plaques is the reversible modification of oxidation-sensitive methionine 35 to its sulfoxide [48, 63] or its further irreversible oxidation product methionine sulfone. APP23 transgenic mice show methionine oxidized forms of Aβ1-40 [64] and methionine oxidized Aβ is also abundantly detected in AD patient brains [38, 63, 64]. The sulfoxide intermediate can be reduced by the action of peptide–methionine sulfoxide reductase [65], although levels of this enzyme in the AD brain were reportedly reduced [66]. In line with this observation, upon knock-out of methionine sulfoxide reductase A in a human amyloid-β protein precursor (AβPP) mouse model, levels of soluble methionine sulfoxide Aβ were increased and associated with defects in mitochondrial respiration and cytochrome c oxidase activity [67]. In turn, exposure of rat neuroblastoma cell line IMR-32 to methionine-oxidized Aβ1-42 induced an increase in levels of mRNA expression and activity of methionine sulfoxide reductase type A [68], suggesting that levels of methionine sulfoxide reductase A and methionine-oxidized Aβ1-42 may affect each other in a bidirectional manner. Somewhat conflicting results have been published on the effect of methionine-35 oxidation on Aβ aggregation. For example, it was shown that H2O2/Cu2+-induced methionine-35 oxidation slows down ThT-positive Aβ fibril formation of commercially derived Aβ1-40 and Aβ1-42 compared to wild type Aβ without affecting morphological features of the formed Aβ fibrils as observed by transmission electron microscopy (TEM) [69]. Marked differences in response to H2O2 induced methionine oxidation of Aβ1-40 and Aβ1-42 were observed in a different study showing that oxidation of Aβ1-40 increases fibril formation kinetics while slowing down fibril formation of Aβ1-42 [70] suggesting an isoform differential effect. Imaging of the resulting fibers using TEM showed that fibers generated by oxidized Aβ1-40 and Aβ1-42 were both highly fragmented compared to unoxidized peptide [70]. In another study, methionine 35 of synthesized Aβ1-42 was oxidized by exposure to H2O2 and oxidation was validated using mass spectrometry. Subsequent atomic force microscopy (AFM) and circular dichroism spectroscopy showed that methionine oxidation in this way hindered the typical random coil to β-sheet conversion and filamentous morphology characteristic for Aβ fibril formation [71]. Early aggregate formation of methionine oxidized Aβ1-40 was studied using electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry showing that trimer formation was inhibited without affecting dimer assembly [72]. One of the mechanisms suggested to affect the decreased aggregation propensity of Aβ upon oxidation of methionine 35 was that oxidation results in a reduced hydrophobicity of Aβ [71, 73], while hydrophobicity is one of the main driving forces for Aβ self-assembly. An oxidation-induced change in hydrophobicity was experimentally illustrated for apolipoprotein A-I, which, upon oxidation, affected the ability of this protein to interact with lipids [73]. It is difficult to delineate the precise origin of the diversity in results that have been obtained in aggregation studies of Aβ in response to methionine oxidation, but it is likely that variations in sample preparation, origin and incubation conditions may contribute as aggregation properties are sensitively affected by these parameters.
4-hydroxynonenal modification of Aβ
A third type of oxidative stress related feature in the AD brain is the accumulation of HNE [29, 74, 75]. HNE generation has been detected both in vitro and in vivo as a result of lipid peroxidation [76, 77]. A 1990 hypothesis paper proposed several multi-step iron-catalyzed chemical routes for the generation of HNE through the oxidation of n-6 polyunsaturated fatty acids, particularly linoleic, γ-linoleate, and arachidonic acid [78]. Further, the presence of Aβ was shown to induce Cu2+-mediated production of HNE from lipids [79], and that, in turn, the released HNE can conjugate with Aβ and induce assembly of Aβ into high molecular weight species and increase the generation of Aβ by modulating β-secretase (BACE) activity [80–82]. Collectively, these data suggest that a number of in-brain factors interrelate to generate a downward spiral that is possibly associated with the observed pathogenic progress of AD. Metals were shown to regulate HNE modification of Aβ. For example, it was observed that HNE modification of Aβ in vitro can be achieved by means of coincubation of Aβ with HNE upon overnight incubation only in PBS that is free from magnesium and calcium [83] consistent with the finding that physiological levels of calcium effectively inhibit HNE modification of Aβ [82]. Also HNE conjugation to a truncated form of Aβ, Aβ1-16, was shown by means of mass spectrometry to be prevented by calcium and copper [83]. Of interest then was the observation that HNE conjugation of Aβ is a ROS-induced modification often encountered in amyloid plaques [2, 27, 84, 85], while in plaque levels of calcium and copper are reportedly high. A study mimicking in vivo in plaque conditions, involving physiological levels of calcium and high levels of copper, demonstrated indeed that HNE-adducts and Aβ were both recognized, though not colocalized, in cerebral vessels [83]. These data perhaps demonstrate that the raised levels of metals in plaques locally inhibit HNE conjugation to Aβ in these plaques [82]. Lysine and histidine residues in Aβ seem to be the most reactive residues toward HNE adduct formation, and it was suggested that the microenvironment of a specific residue determines the actual reactivity to HNE [86]. Two chemical reactions were identified that dictate the HNE-Aβ adduct formation: via formation of a Schiff’s base or by Michael addition [86]. Consistent with this thought, the conjugation reaction can be quenched by azide, primary amines, ammonia, Tris, DTT [83] or trifluoroacetic acid [79]. Also the addition of antioxidants hydralazine [83] or 3,5-di-tert-butylhydroxytoluene (BHT), or copper gelator diethylenetriaminepentaacetic acid (DTPA) [79], were reported to inhibit HNE-modification of Aβ.
Heme-complex formation of Aβ
Heme-complexed Aβ adducts have been postulated to affect cytochrome c oxidase (COX) activity, a mitochondrial electron transport chain enzyme which is significantly decreased in the AD brain [87]. COX requires heme-a, of which regulatory heme is a precursor, to assemble and perform its function [88, 89]. In turn, heme-a levels were observed to be significantly decreased in the temporal lobes of AD patient brains compared to age-matched controls [90]. A potential Aβ-mediated role in the availability of heme in the AD brain came to light when it was shown that the presence of heme dose-dependently inhibits oligomer formation of both Aβ1-40 and Aβ1-42 in an immunoassay and prevented loss of cellular viability upon addition of the complex to human neuroblastoma cell line IMR32 [91]. It was thought that, by competitive binding to heme, Aβ could deplete the availability of regulatory heme leading to deprived COX functionality and energy deficiency in AD. Similarly, the presence of heme was found to inhibit activation of Aβ-induced inflammatory response in primary mouse astrocytes [92]. At the same time, Aβ and heme, in the presence of H2O2, were reported to increase tyrosyl radical formation in Aβ1-16 and mediate its dimerization through 3,3’-dityrosine cross-linking [93–95], a reaction that was observed to be competitively inhibited by NaNO2 [94]. A direct and rapid interaction between heme and Aβ was shown upon addition of heme-a or heme-b to Aβ1-40 or a mixture of Aβ1-40 and Aβ1-42 which resulted in an immediate spectral shift of heme [90]. Aβ histidine residues were speculated as potential binding site via involvement of the π-electrons of the histidine imidazole rings, as addition of copper and zinc ions competitively inhibited the interaction of Aβ with heme, but only when heme was added to the reaction mixture after copper and zinc [90, 91]. In a subsequent site-directed mutagenesis study, using voltammetry, histidines 13 and 14 were specifically identified as heme binding sites in Aβ [96]. In addition to this, an NMR-based spectroscopic study showed that heme-b binds to Aβ1-16 with higher affinity compared to free histidine or other histidine-containing peptides indicating that other parts of the Aβ peptide contribute to the interaction with heme-b [95]. Heme-Aβ conjugates have also been found in AD plaques and conjugation to heme was shown to inhibit Aβ aggregate formation in a cell-free system and to dissociate existing aggregates [97]. Spectroscopic studies shed more light on the structural implications for Aβ1-16 upon interaction with heme-b [95]. This study showed that two complexes can be formed that exist in equilibrium, a low spin six-coordinated 1:2 heme/Aβ1-16 stoichiometry and a high-spin heme-(Aβ1-16) species. Aβ-heme adducts were found to exercise peroxidase activity [98] although in vivo relevance of this catalytic activity was questioned as a result of the reported very low kcat value of the complex of 0.016 s-1 at 278 K compared with a reference value of 45.5 s-1 for horse radish peroxidase [95]. However, a substantial effort has since gone into understanding the structural basis of this peroxide activity. One study showed that mutation of either histidine 13 or 14, but not both, does not affect peroxidase activity of the Aβ1-16-heme complex [94]. Free histidine, similar to the unmutated Aβ1-16-heme complex, induced peroxidase activity as observed using an 2,2’-azinobis(3-ethylbenzothiazoline-6-sulphonic acid) diammonium salt (ABTS) oxidation assay [94]. Apart from a regulating role by histidines, peroxidase activity of the Aβ-heme complex was shown to involve arginine 5 as proton donating residue cleaving the O-O bond of the peroxide [94, 96, 98]. At the same time, the addition of free arginine to heme failed to induce peroxidase activity demonstrating that the structural incorporation of arginine 5 within a protein environment is somehow relevant for its action [94, 95]. Peroxidase activity of the complex was reported to depend on the heme-Aβ ratio and temperature, with increasing Aβ1-16 to heme and temperature inducing more potent peroxidase activity [95, 99].
Oxidative damage colocalizes with tau neurofibrillary tangles
Oxidative damage was found to colocalize with tau enriched neurofibrillary tangles [100]. In this study, hippocampal tissue from AD patients was subjected to postmortem analysis investigating the localization of the enzyme dimethylarginase. This enzyme regulates the activity of nitric oxide synthase [101]. In AD hippocampal tissue, neurons that contain neurofibrillary tangles also stain positive for dimethylargininase providing a first indication that nitric oxide is generated in close proximity to the tau that makes up the neurofibrillary tangles. In line with these observations, an antibody that recognizes an HNE-lysine adduct was found to colocalize with endogenously obtained paired helical tau filaments from AD brains [29]. Also, acrolein, which is an aldehyde product of lipid peroxidation, was observed to colocalize with neurofibrillary tangles in AD patient brains [102]. Further, the antibody Alz50 [103], which recognizes a conformational change in tau [104], coincides with heme oxygenase-1 (HO-1), which is an antioxidant enzyme [29], levels of which are strongly increased in the AD and mild cognitive impaired (MCI) brain [105]. Whether HO-1 activity is beneficial in terms of alleviating oxidative stress or can induce neurotoxicity in the MCI and AD brain has been subject of debate as increased HO-1 levels were also correlated with increased phosphorylation of tau serine residues [105].
Oxidation of tau affects filament assembly
Ascorbate/Fe(III)/O2-induced oxidation of bovine tau was shown to induce the assembly of tau into filaments in vitro [106]. The oxidation of one of the cysteine residues was found to be involved in the induction of the assembly of a recombinant fetal isoform of tau into such assemblies [107]. These data suggest that the generation of tau filaments is a disulfide bond mediated process while oxidation modulates the ability to self-assemble. Consistent with this thought it was recently shown that Zn2+ interacts with the cysteine residue of a truncated version of tau containing only the third repeat unit of the microtubule-binding domain accelerating its aggregation rate and toxicity in a Neuro-2A cell line [108]. A more direct role for oxidative stress in tau assembly was demonstrated upon administration of the anti-oxidants 2,4-disulfonyl α-phenyl tertiary butyl nitrone and N-acetylcysteine which reduced immunoreactivity against tau oligomers [109]. Of interest was the observation that peroxynitrite treatment of tau induced nitration, S-nitrosylation and oxidation of methionine, as observed by HPLC-electrospray ionization tandem mass spectrometry while markedly reducing aggregation, as analyzed by light scattering and electron microscopy [110]. Collectively, these observations suggest that oxidation of tau may modulate aggregation by either inducing or inhibiting the self-assembly process and that the specific outcome may depend on the type of oxidant and the specific amino acid residue involved. Phosphorylation was shown to importantly regulate HNE-induced assembly of tau as exposure of phosphorylated tau, as opposed to unmodified tau, induced misfolding of tau which was recognized by antibody Alz50, and the formation of tau aggregates [29]. In line with this, the self-assembly of a tau fragment including the first and third tubulin-binding domains showed that the presence of HNE mediated polymerization of phosphorylated tau [111]. At a molecular level, a link between phosphorylation and oxidative stress was revealed when a study showed that the activity of alkaline phosphatase was inhibited in the presence of HNE. Exposure of tau to HNE hence resulted in the generation of a tau species resistant against dephosphorylation [112].
AGE-conjugated tau is associated with oxidative stress markers
Advanced glycation end products (AGEs) are the oxidation product of sugars that interact with proteins and their accumulation has been related to amyloid deposition in AD [113]. Tau assembled into paired helical filaments has been shown to be immunoreactive against Nɛ-(carboxymethyl)lysine, one of the major AGEs [114]. Interaction of recombinantly produced tau with ribose-derived AGE products was shown to result in the generation of reactive oxygen intermediates, which, in turn, activate NFκb to induce amyloidogenic processing of AβPP to generate Aβ [115]. Uptake of AGE-glycated tau into SH-SY5Y neuroblastoma cells was associated with malondialdehyde and HO-1 detection which was prevented by the exposure of these cells to N-acetylcysteine and probucol, two antioxidant compounds [116]. Diffuse cytosolic immunoreactivity against AGE was shown in many neurons of post-mortem AD brains that also contain hyperphosphorylated tau [117]. Astrocytes residing in the temporal cortex of medium to severely affected AD subjects were found to be immunoreactive for inducible nitric oxide synthase (iNOS) as well as AGEs [118]. Thus far it is unclear whether AGE-glycation of tau has implications for the physiological role of this tubulin binding protein in the cytoskeletal organization or what the hierarchical correlation is between AGE formation and tau assembly into filaments.
Collectively, a clinical link between mitochondrial dysfunction and AD has been firmly established, with a central role for AD hallmark proteins Aβ and tau. While various types of ROS-mediated modifications of Aβ and tau have been investigated and play a potential role the precise implications of these species on disease progress have not been investigated.
EFFECT OF OXIDATIVE STRESS ON MITOCHONDRIA IN ALZHEIMER’S DISEASE
AD brain originating neurons containing defective mitochondria show loss of dendritic spines and abbreviation of dendritic arborization [119]. Differences in CA1 hippocampal mitochondria structure have been detected using 3-dimensional electron microscopy. Instead of the uniformly elongated mitochondrial morphology observed in wild type mice, human AD brain and hippocampal mitochondria in mice carrying mutations for presenilin-1 (psen1), AβPP, and tau, have an ovoid or teardrop profile [115]. Further, AD mouse models and AD patients show the presence of multiple small mitochondria and exaggerated mitochondrial division [120] suggesting that the mitochondrial fission process is altered in AD. The mitochondrial fission process relies on dynamin related protein 1 (Drp1) and mitochondrial fission protein 1 (Fis1) [121, 122]. Recent research observed the presence of elongated interconnected organelles where multiple teardrop shaped mitochondria were connected by thin double membranes. This structure, referred to as “Mitochondria-on-a-string (MOAS)”, has been identified in an AD mouse model together with increased Drp1 phosphorylation, causing incomplete fission. Even though altered mitochondrial fission processes in neurodegenerative diseases have been viewed primarily as a pathological feature, in cardiomyocytes Drp1 induced mitochondrial fission was shown to exert a protective effect against cellular apoptosis by enabling the cells to meet altered energetic demands [123]. An alternative role of Drp1 was suggested with the observation that reduced association of Drp1 with the mitochondrial membrane induced a lack of mitochondrial fusion, which, in turn, induces high levels of mitochondrial oxidative stress [124]. The fusion process should be in balance with mitochondrial fission to maintain mitochondrial homeostasis. Mitochondrial fusion is mediated by inner membrane fusion factor optic atrophy-1 (OPA1). Addition of H2O2 to an osteosarcoma and a cardiomyoblast cell line lead to inhibited mitochondrial fusion as a result of loss of OPA1 activity through cleavage mediated by metalloendopeptidase OMA1 [125, 126].
ENDOGENOUS ANTIOXIDANT ACTIVITY IS COMPROMISED IN ALZHEIMER’S DISEASE
The endogenous antioxidant capacity is a multi-component system targeted at neutralizing ROS and RNS to prevent damage of cellular compartments. Many of the factors involved in endogenous antioxidant capacity are affected in AD, and experimental evidence for this will be discussed in this section.
Glutathione
Glutathione (GSH) is one of the prime endogenous antioxidants in the brain. GSH is a tripeptide thiol-containing antioxidant that is synthesized by the conjugation of the amino acids glutamate, cysteine, and glycine mediated by the enzymes γ-glutamyl cysteine synthetase and glutathione synthetase [127]. GSH acts by scavenging ROS, and, in the process, becomes reversibly oxidized to form glutathione disulfate (GSSG) [127, 128]. Oxidative stress induces the expression of the NADPH-dependent enzyme glutathione reductase, which reverts oxidized GSSG to its reduced form GSH [129]. A study involving 74 human subjects demonstrated that GSH levels of autopsied brains did not significantly decrease with aging [128]. At the same time, whole-brain GSH levels were shown to be profoundly reduced in individuals suffering from AD compared to age-matched controls [130] although another study reports that GSH levels in AD brains are not significantly different from those found in age-matched control brains [131]. Region-specific differences were identified showing increased GSH levels in the hippocampus and midbrain of AD patients without significant difference in GSSG levels [132]. Moreover, a correlation between peripheral and brain levels of GSH exists as it was demonstrated that levels of erythrocytic GSH in elderly patients with MCI and AD were substantially decreased compared to a control group [130]. A study investigating human AD patient lymphocytes showed that decreased GSH levels correlated with increased GSSG levels [133]. Moreover, basal blood levels of GSSG/GSH ratios in control, mild, moderate or severe dementia patients showed a significant correlation with progression of disease [134]. Aging related reduction of brain GSH was shown to go hand in hand with decreased gene expression of γ-glutamyl cysteine synthetase in the brain [135]. While whole-brain levels of GSH transferase in AD brains were not significantly different from age-matched control brains [131], mRNA expression levels of γ-glutamyl cysteine synthetase vary per region in the brain with high expression levels in cortex, cerebellum and hippocampus and low expression in the neostriatum of mice [136, 137], and it was suggested that these regional differences in de novo GSH generation can explain regional differences in susceptibility to oxidative stress [137].
Melatonin
Melatonin, or N-acetyl-5-methoxytryptamine, is involved in various homeostatic functions to aid cellular protection. It is an electroreactive neurohormone with antioxidant activity that is synthesized and secreted in the brain from mitochondria of pinaelocytes, cells of the pineal gland [138–140]. Also the metabolites of melatonin, N1-acetyl-N2-formyl-5-methoxykynuramine (AFMK) and N1-acetyl-5-methoxykynuramine (AMK), demonstrate antioxidant activity, either directly by scavenging a variety of free radicals including hydroxyl, peroxyl, superoxide, peroxide and peroxynitrite (ONOO-) [141, 142], or indirectly by inducing antioxidant enzymes including superoxide dismutase (SOD), glutathione peroxidase (GPx), and GSH reductase [143], increasing GSH synthesis [144], and inhibiting prooxidant enzymes RNS, xanthine oxidase, and myeloperoxidase [145]. Even though aging is related to a decrease in CSF melatonin levels, presenile and senile AD patients demonstrated an even stronger reduction in melatonin levels that was shown to be dependent on apolipoprotein genotype [146], one of the strongest identified genetic correlates with AD. How these factors and processes are associated is currently unclear.
Transcriptional control of the endogenous antioxidant system by Nrf2
The neuron-glial unit, the main interaction site between neurons and cells of glial origin such as astrocytes, regulates oxidative stress levels through an intimately linked intercellular mechanism for maintaining redox homeostasis [147, 148]. Brain oxidative stress levels are maintained within strict limits as a result of the astrocytic nuclear factor erythroid 2 (NFE2)-related factor 2 (Nrf2) homeostatic pathway [149]. Upon translocation to the nucleus, Nrf2 binds to antioxidant response element (ARE), a promotor element present on antioxidant genes [150]. Nrf2 degradation is controlled by ubiquitin-mediated degradation, which, in turn, is regulated by cytoskeleton associated Kelch-like protein, Keap1 [151–154]. In the absence of oxidative stress, Nrf2 is transcriptionally inactive as its activity is repressed by Keap1 [154]. Under conditions of oxidative stress Keap1 is oxidized inhibiting the degradation of Nrf2. Transcriptional activity of Nrf2 was shown to decline upon aging [155, 156]. One study showed that AD progression was linked with haplotype allele variation in the NFE2L2 gene promotor which encodes for NRF2 [157] while therapeutic administration of a lentiviral vector encoding for human Nrf2 was shown to improve cognitive dysfunction in APP/PS1 [158], and APP/PS1DeltaE9 mice [159]. Furthermore, a recent transcriptomics study demonstrated that NRF2 knockout leads to early onset cognitive dysfunction, plaque deposition and tau tangle formation [160]. Other recent experimental evidence linking AD to the Nrf2 pathway showed that methysticin, a kavalactone activating the Nrf2 signaling pathway, reduced neuroinflammation, loss of memory and damage as a result of oxidative stress in the hippocampus of APP/Psen1 mice [161]. Even though the Nrf2 signaling pathway is highly active in astrocytic cells, this pathway is virtually absent in cells of neuronal origin [162, 163] while the capacity of neurons to degrade Nrf2 is high as a result of abundant neuronal expression of the protein cullin 3 which leads to destabilization of neuronal Nrf2 [162]. These observations argue for a high level of functional integration of astrocytes and neurons in the brain to regulate oxidative stress levels.
ALZHEIMER’S DISEASE RELATED OXIDATIVE STRESS
Disturbed metal ion homeostasis in Alzheimer’s disease
Metal ions such as Cu2+ and Zn2+ play an important role in regulating synaptic functioning by inhibiting the rat excitatory NMDA receptor [164], and rat GABA receptor [164, 165]. Iron ion (Fe2+) has been documented to regulate synaptic plasticity and synaptogenesis as well as myelination [166] as illustrated by the neuronal expression of iron transporter DMT1 [167–169]. The levels of these metal ions are normally strictly regulated to prevent oxidative stress resulting from interaction of Fe2+ or Cu2+ with oxygen to generate radicals such as superoxide ions or hydroxyl radicals. Disruption of metal ion homeostasis has been observed in various neurodegenerative disorders including AD [170]. A patient study using instrumental neutron activation analysis demonstrated that levels of Cu2+ were decreased while Zn2+ and Fe2+ levels were elevated in the hippocampus and amygdala of AD patients which correlated with observed histopathological changes in these regions [171]. On the other hand, serum levels of Cu2+ were shown to be increased in AD patients compared to control subjects [172]. Also in preclinical stages and MCI Fe2+ levels were increased in the cortex and cerebellum and correlated with generation of radicals [173]. Compared with the neuropil of the amygdala of AD patients, senile plaques were observed to contain increased levels of Zn2+, Fe2+, and selectively in the rim of the plaques, Cu2+ [47]. As Zn2+, Fe2+, and Cu2+ have been shown to interact with Aβ in vitro [174], metal ion dyshomeostasis has been postulated as potential mechanism by which AD pathology may be modulated.
Spatial link between amyloid plaques and cells exhibiting oxidative damage
A multiphoton microscopy-based study using the genetically encoded calcium indicator Yellow Cameleon 3.6 packaged into an adeno-associated virus (AAV2) and expressed in the brains of adult transgenic APP/PS1 mice showed that calcium overloaded neurites in living animals were more likely to be located in close proximity (<25μm) of a plaque [175]. This observation suggests a direct or spatial link between pathological alterations in neurons and the formation of senile plaques. A second marker that indicates that there is a spatial link between AD-related deposits in the brain and neuronal functioning was the receptor for advanced glycation end products (RAGE). Neuronal cells adjacent to senile plaques display increased RAGE expression while little change in expression was demonstrated in brain regions remote from plaques [176]. Two other markers that have been used to topologically differentiate subpopulations of cells affected by oxidative stress include p50, which is a DNA binding subunit of transcription factor NFκB [177, 178], and HO-1. Cellular structures containing accumulations of Aβ displayed increased levels of oxidative stress as demonstrated by elevated levels of HO-1, and p50 [176]. Inactive NFκB resides in the cytosol and is bound to inhibitory protein IκB which prevents nuclear translocation of NFκB. Phosphorylation, ubiquitination, and degradation of IκB drives the activation of NFκB [179]. The redox state regulates activation and nuclear translocation of NFκB [180], and, as such, ROS was found to induce phosphorylation of IκB via activation of responsible kinases [181, 182]. Using p50 and HO-1, it was observed that the spatial link found between Aβ deposits and induction of cellular ROS is not limited to CSF residing neurons, but this observation extends to endothelial and smooth muscle cells in cerebral blood vessels. The expression of HO-1 was found to be elevated in AD injured neuronal cells, a feature that was more pronounced in regions close to neurofibrillary tangles and Aβ plaque deposits [183].
Oxidative stress is an early stage pathological feature
A redox proteomics study of the brain of Down syndrome (DS) patients prior to onset of AD provided insight into the role of oxidative damage in the development of DS related early onset AD [184]. Male and female DS and control brains were analyzed postmortem for carbonylation levels of proteins as hallmark of oxidative stress. DS brains showed increased carbonylation of six proteins including cathepsin D, glial fibrillary acidic protein and succinyl-CoA:3-ketoacid-coenzyme A transferase 1 mitochondrial protein. Carbonylation affected protein functionality, while at the same time, proteasome activity and autophagy activity were decreased [184] potentially leading to loss of functional protein. Even though this study was conducted on a small number of subjects, it did provide important insight into the potential role of oxidative stress in early stages of disease. A larger scale study using human peripheral blood mononuclear cells (PBMCs) derived from 104 MCI subjects similarly showed increased oxidative stress markers as detected by the fluorescent probe DCFH2-DA [185]. Also, in MCI and mild AD patient PBMCs homeostasis of ER stress-mediated Ca2+ was disturbed with decreased SOD1 levels [185]. Analysis of lymphocytes obtained from MCI subjects and AD patients similarly showed increased ROS levels, detected by 8OHdG, compared to lymphocytes derived from an age-matched control population [186]. The validity of using 8OHdG brain levels as a biomarker to detect oxidative stress-related damage to DNA in AD patients has been questioned [187]. However, the detection of increased levels in the frontal cortex of other modified macromolecules such as F2-isoprostanes as well as 3-NT and oxidized glutathione detected in patients with probable AD further corroborates the thought that oxidative stress is an early stage pathological feature of AD [188]. The work by Ansari and Scheff also compared oxidative stress levels in age-matched groups with progressive forms of cognitive disorder, from non-cognitively impaired to AD, and showed that oxidative stress progressively worsened with cognitive decline. In addition to this, activities of SOD and catalase in post mitochondrial supernatant and in mitochondrial and synaptosomal fractions of the frontal cortex were significantly declined already in MCI subjects [188]. Consistent with this, an earlier longitudinal study on autopsied control and patient brains demonstrated that levels of isoprostane (F2) and F4-neuroprostane were increased in both amnestic MCI and late stage AD patients in various regions of the brain [189].
A vascular component
The microcerebrovascular structure showed age-dependent changes [190] which are more pronounced in cognitive disorders such as dementia [191, 192]. For example, the basement membranes of cortical capillaries of patients suffering from cognitive disorders were significantly thicker than those of age-matched controls [192]. Smooth muscle atrophy and general disorganization of these cells was consistently observed in AD subjects although these features seemed unrelated to the deposition of Aβ [193]. These structural changes translate into a decreased capillary flow in aged (16 months old) compared to young (2 months old) mice [194] as well as aggravated loss of blood flow rate in an aged APPswe/PS1ΔE9 transgenic mouse model [195]. The observed structural and functional changes in the microvascular organization thus lead to hypoperfusion and a general inability of the cerebral vasculature to meet the metabolic needs of the brain while this was partly compensated for by an increased ability to extract oxygen from the remaining blood flow [196]. However, the remaining metabolic deficiency is of sufficient magnitude to result in neural hypoxia [196]. Various conditions have been associated with increased brain oxidative stress and neuronal apoptosis in response to hypoxia, including sleep apnea [197, 198], exposure to carbon monoxide [199], and ischemia [200]. Sleep apnea co-occurs frequently with AD [201] while prevalence of sleep apnea positively correlates with aging [202, 203], and treatment of sleep apnea slows down the rate of cognitive decline in patients diagnosed with mild-to-moderate AD [201]. Further, hypoxia induced oxidative stress in the brain has been shown to induce cognitive deficits in rats [204]. The mechanisms by which the brain adapts to hypoperfusion-induced hypoxia have been explored and most proposed mechanisms are centered around the thought that activation of hypoxia-inducible factor 1α (HIF-1α) plays an important role. HIF-1α is a component of a heterodimeric complex with the aryl hydrocarbon nuclear translocator (ARNT or HIFβ) [205]. Under normoxic conditions, HIFα is dissociated from this complex and unstable as a result of its hydroxylation which targets it for ubiquitination and proteasomal degradation [206–208]. Hypoxia prevents hydroxylation of HIFα by inhibition of the two hydroxylating enzymes, factor inhibiting HIF-1 and prolyl hydroxylase enzymes [209, 210]. This stabilization induces its nuclear localization and heterodimeric complexation with ARNT. Subsequent co-recruitment of this complex with transcription coactivators p300 and CREB binding protein (CBP) initiates gene transcription. Hypoxic conditions are considered to raise cytosolic ROS levels and, in this way, induce the activation of HIF (reviewed in [211]) probably in a mitochondrial complex III-dependent manner [212]. The HIF-dependent hypoxia-inducible genes are generally involved in processes aimed at promotion cellular survival under hypoxic conditions. A study investigating mRNA expression in adult rat brains upon occlusion of the middle cerebral artery demonstrated that glucose transporter-1 (GLUT-1) and glycolytic enzymes (phosphofructokinase, aldolase, and pyruvate kinase) were upregulated to increase transport of glucose and glycolysis [213].
GENETIC AND OTHER FACTORS THAT CORRELATE OXIDATIVE STRESS TO ALZHEIMER’S DISEASE
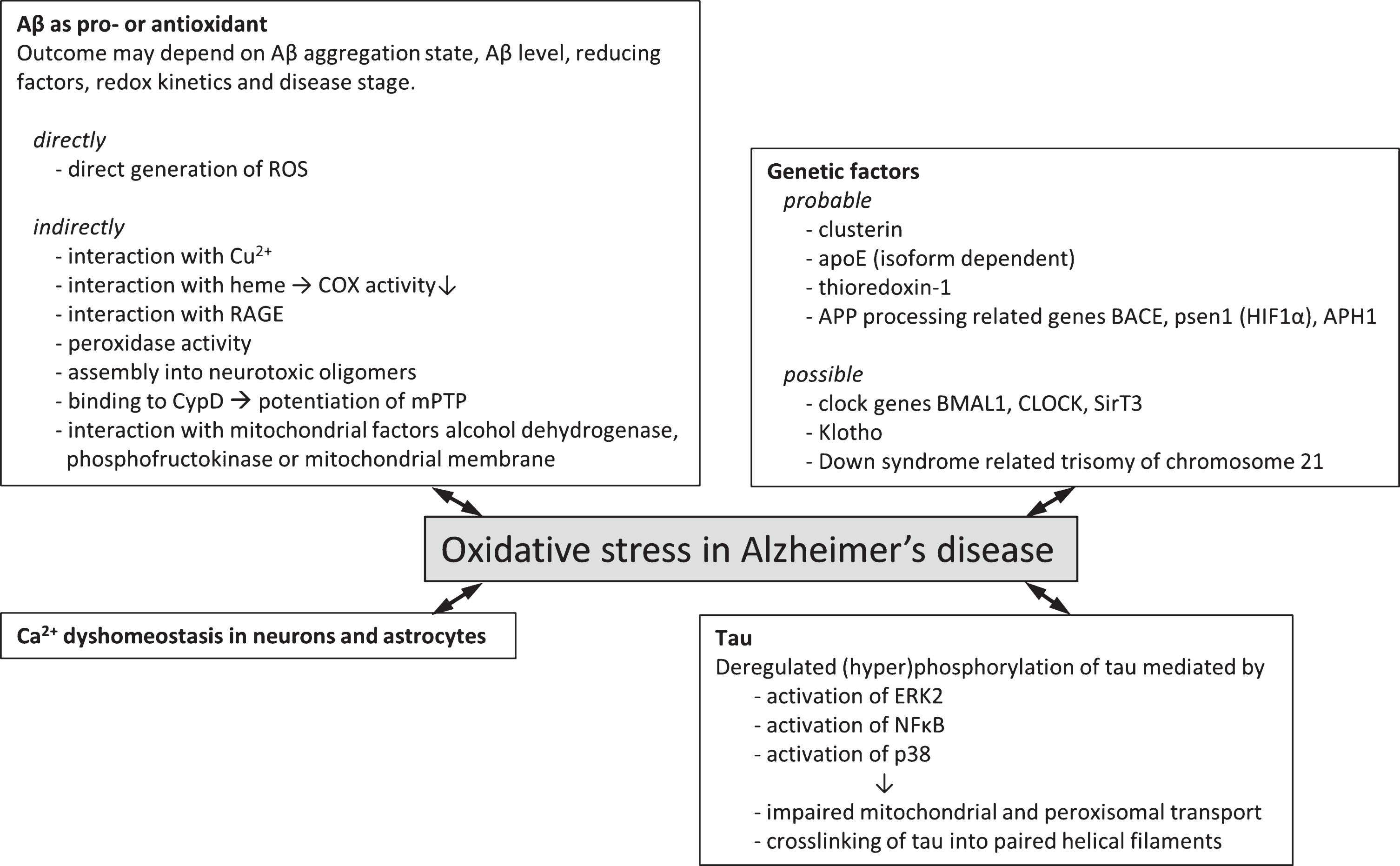
This paragraph will first review the established experimental evidence that has demonstrated a connection between oxidative stress and AD, such as clusterin, apolipoprotein E (ApoE), and genes related to AβPP processing machinery. Second, this paragraph will also highlight some potential interactions that have yet to be experimentally established but for which observations have shown to connect to both AD and oxidative stress. These factors include Klotho, and circadian clock genes and we envisage that future investigation into these factors and their relation to AD and oxidative stress levels may highlight alternative or additional mechanisms for the interaction of these clinical features. Figure 1 summarizes the genetic factors associating AD and oxidative stress to date.
Fig.1
Genetic factors and molecular mechanisms of oxidative stress in Alzheimer’s disease. Overview of probable (experimental evidence available in literature) and possible (no direct experimental evidence available) genetic factors that associate oxidative stress with Alzheimer’s disease. Various molecular mechanisms by which oxidative stress and Alzheimer’s disease may be associated have been described. These often involve the two hallmark proteins Aβ and tau and effects may be directly involving the generation of ROS or indirectly via interaction with various cellular factors giving rise to increased ROS generation or lowered endogenous antioxidant capacity.
Clusterin
Apolipoprotein J is a ubiquitously expressed secreted glycoprotein which is also known as clusterin (CLU). Aging induces elevated levels of CLU gene expression [214, 215] and plasma CLU [216]. In a genome-wide association study CLU has been identified as a genetic determinant of AD [217] and plasma CLU levels were associated with atrophy of the entorhinal cortex and clinical progression of the disease [218] as well as with longitudinal brain atrophy in MCI patients [219]. Apart from aging, expression of the CLU gene was demonstrated to be sensitive to heat-shock induced changes in the organism or the direct environment of the organism as a result of the presence of activating protein-1 (AP-1) and CLU-specific element regulatory elements in its promotor [220]. Consistent with the idea that CLU plays a role in stress-associated coping of cellular response a study in H9c2 cardiomyocytes revealed that the Akt/GSK-3β pathway may be involved in the anti-oxidant and anti-apoptotic effect of CLU in a megalin-dependent manner [221]. Multi-ligand receptor megalin has been identified to also act as receptor of clusterin [222]. Various cellular stress stimuli have been shown to regulate transcriptional activity of AP-1 [223]. Differential CLU expression was similarly observed in other oxidative stress-related pathologies including asthma [224], atopic dermatitis [225], diabetes type 2 [226], coronary heart disease [226], and cancer [227]. Oxidative stress increases CLU expression. This was demonstrated in a study in which human diploid fibroblasts were treated with H2O2 which resulted in increased mRNA levels of CLU [228]. In human neuroblastoma cell lines SH-SY5Y and IMR-32 both mRNA and protein levels of CLU were found to be upregulated in response to pro-oxidant pair iron-ascorbate [229]. In line with these observations, CLU was originally identified to function as a chaperone protein where its activity was reported to depend on cellular redox state [230]. CLU was shown to protect against oxidative stress in various cellular systems including fibroblasts and prostate cancer cells [221, 231] but also in vivo in a Drosophila melanogaster model [232]. In neuroblastoma N2a and SH-SY5Y cells knockdown of CLU by short hairpin RNA interference was found to down-regulate antioxidant capacity [233]. The precise anti-oxidant mechanism of CLU is not known although blockage of the sulfhydryl groups contained in the sequence of the protein resulted in abolishment of its oxidative stress preventive activity [232]. A review covering the involvement of CLU in oxidative stress detection and action has been published before [234]. Apart from an antioxidative effect of CLU, an indirect role of CLU actually promoting oxidative stress has been described showing that the presence of CLU induces the formation of slowly sedimenting complexes composed of SDS-resistant synthetic Aβ assemblies that, in turn, induced oxidative stress in PC12 cells [235].
Apolipoprotein E and Thioredoxin-1
Apolipoprotein E4 (ApoE4) was identified as one of the major genetic risk factors for AD [236–239]. Apolipoprotein E exists in three isoforms, ɛ2, ɛ3, and ɛ4, which vary in their amino acid composition. Carriers of the ɛ4-allele have an increased risk of developing AD [236] as well as a decreased age of AD onset [237] compared to non-ɛ4 carriers. The pathogenic origins of ApoE4 have been studied to great length and indicate that ApoE4 is involved in processes such as aggregation and clearance of Aβ [240, 241], mitochondrial dysfunction, and impairment of calcium [242, 243] or cellular iron homeostasis [244], and ApoE4 affects synaptic architecture and functioning [245, 246]. A potential connection between the ApoE allele, AD, and oxidative stress was first deduced from the observation that the extent of oxidative stress and anti-oxidant defense is related to ApoE genotype in mice and in patients [13, 244, 247–249]. ApoE was demonstrated to act, directly or indirectly, as an antioxidant against hydrogen peroxide-induced cytotoxicity in a B12 ApoE expressing cell line [250]. Elevated levels of peroxidized plasma low-density lipoproteins were observed in ApoE-deficient mice [251]. Levels of lipid oxidation were significantly increased in the frontal cortex of AD patients that were homozygous or heterozygous for the ɛ4-allele of ApoE compared to homozygous ɛ3 carriers and controls [13]. Upregulation of catalase activity was exclusively observed in frontal cortex tissue of homozygous ApoE4 carriers while SOD activity and concentrations of glutathione were not different from that of controls [13]. Additionally, levels of HNE were increased in ɛ4-carriers [84]. Further, mouse brain synaptosomes expressing human ApoE4 were more susceptible to Aβ42-associated oxidative stress than synaptosomes from mice expressing human ApoE2 or ApoE3 [252]. Various experimental findings shed light on the potential molecular mechanism underlying these observations. They show involvement of thioredoxin-1 (Trx1), an endogenous antioxidant with a downregulating role in apoptosis signal-regulating kinase-1 (Ask-1) [253]. Thioredoxin reductases are reducers of Trx1 [254]. Levels of Trx1 were reduced in AD brains [255, 256] depending on ApoE genotype, but also in ApoE4 expressing mouse hippocampi, and human primary cortical neurons and neuroblastoma cells to which ApoE4 was supplemented with the culture medium, compared to ApoE3 [257]. At the same time, Trx1 mRNA levels in ApoE4 TR mouse hippocampi were elevated consistent with findings reporting increased Trx1 expression in conditions of oxidative stress [258]. Persson and colleagues suggested that increased mRNA levels of Trx1 potentially act as a compensatory mechanism for the increased cathepsin D-induced Trx1 turnover as observed in SH-SY5Y neuroblastoma cells [257]. Moreover, Aβ1-42 was demonstrated to cause transient oxidation of Trx1 [255] as well as ApoE4-induced downregulation of this protein which resulted in activation of an apoptotic pathway involving the translocation of Death-Domain Associated Protein-6 [255, 257, 259] without affecting catalase and GSH activities [257].
Down syndrome, a trisomy of AβPP encoding chromosome 21
Individuals with DS are prone to develop early-age AD with pronounced oxidative stress. DS is characterized by trisomy of chromosome 21 (HSA21), which encodes AβPP as well as some proteins of relevance to redox homeostasis providing an interesting group of patients to study early stage aspects of oxidative stress in AD pathogenesis in response to a defined genetic condition. Comparable to observations in AD patients, mouse models of DS demonstrated deficits in hippocampal learning and memory as well as neurodegeneration of cholinergic basal forebrain neurons [260, 261]. DS patients display features of cellular energy impairment [4]. Recent transcriptomic profiling of the skeletal muscle of a DS mouse model showed that among the identified differentially expressed protein-coding genes in this tissue, two, Sod1 and Runx1, were implicated in oxidative stress [262]. Chromosome HSA21 also codes for SOD explaining why expression levels of SOD are increased in DS [263]. Transgenic mice overexpressing SOD1 demonstrate excessive levels of oxidative stress [264] because concentrations of CAT and GPx, two enzymes that act to neutralize hydrogen peroxide, the product of SOD1 activity, do not rise accordingly. Besides SOD1 fifteen other genes on HSA21 were predicted to play a role in mitochondrial energy generation and the metabolism of ROS [265]. Levels of various ROS, RNS and aldehyde products of lipid peroxidation were found to be increased in brain [20, 266] and urine [267] of DS humans and animals [266] indicating that oxidative stress may play a role in the pathogenesis of DS associated AD. Levels of oxidative stress, i.e., high ratio between SOD1 and GPx, correlated with cognitive phenotype [268]. However, a recent study showed that administration of melatonin at the pre- and post-natal stages partially alleviated oxidative stress but did not improve cognitive function in a mouse model [269]. The process of programmed cell death was found to coincide with increased levels of oxidative stress, compared to control cells. Programmed cell death could be rescued by administration of free radical scavengers including vitamin E, and N-tert-butyl-2-sulphophenylnitrone [263] but dietary parameters did not alleviate oxidative stress biomarkers in young adult DS patients [20]. Similar to AD, the DS brain shows features of oxidative stress at very early stage. For example, the DS fetal brain cortex was observed to show increased levels of thiobarbituric acid reactive substances (TBARs), HNE, and protein carbonyl groups compared to controls [270]. Also end-products of non-enzymatic glycation, pyrraline and pentosidine, were increased in DS fetal tissue [270] and in amniotic fluid of DS pregnancies [271].
AβPP processing machinery
Cells of both neuronal and non-neuronal origin exposed to H2O2 or HNE generate increased levels of intracellular and secreted Aβ [272–275]. The role of oxidative stress in Aβ generation was further demonstrated in Tg19959 mice, which overexpress a double mutated form of AβPP. Upon crossing this mouse line with a mouse in which one allele of MnSOD was knocked out, brain Aβ levels and Aβ plaque load were significantly increased [276]. Similarly, hypoxia treated transgenic APP23 mice that were subjected to hypoxia conditions demonstrated increased memory deficits and deposition of Aβ into plaques [277]. Repeated exposure to hypoxia facilitated progress of AD-like pathology in aged APPSwe/PS1A246E transgenic mice [278]. Vascular deposition of Aβ on the surface of cerebral endothelial cells results in vascular degeneration which has been observed in AD and leads to a condition termed cerebral amyloid angiopathy [279]. Exposure of primary cerebral endothelial cells derived from 2-month-old Tg2576 mouse brains to H2O2 resulted in upregulation of AβPP expression and altered AβPP processing to favor the amyloidogenic pathway [280]. Also in humans it was found that oxidative stress induced by hypoxia due to cardiac arrest increased serum Aβ levels [281] suggesting that the machinery that generates this peptide is upregulated under pro-oxidative stress conditions in a wide range of disease models affecting various regions of the brain.
Oxidative stress has an Aβ species specific effect
Aβ is generated as a heterogeneous pool of peptides which vary in the number of C-terminal amino acids. The two most prevalent types of Aβ are the 40-amino acid (Aβ1-40) and the 42-amino acid (Aβ1-42) isoforms. It has been demonstrated that the longer Aβ1-42 peptide is inherently more amyloidogenic than Aβ1-40. Analysis of the specific species of Aβ that were generated under HNE-induced oxidative stress conditions using a TN2 cell culture revealed a 70-80% and 60-140% increase of intracellular Aβx-40 and Aβx-42, respectively. Secreted levels of Aβx-40 were not affected by oxidative stress while secreted Aβx-42 was increased by approximately 50% compared to control cells [274]. These findings are potentially pathologically relevant as it was reported previously by our group and others that a marginally increased Aβ1-42:Aβ1-40 ratio has severe implications for synaptotoxic response [282–284]. Aβ is generated by sequential cleavage of AβPP by two enzymes, γ-secretase and BACE, by a process termed amyloidogenic pathway. Alternatively, AβPP can be cleaved into an N-terminally truncated fragment of the Aβ peptide, called the p3 peptide, by γ- and α-secretase-mediated processing. Details of AβPP processing have been extensively covered in a number of reviews [81, 285].
Both presenilin and anterior pharynx-defective-1, components of γ-secretase, are upregulated in conditions of oxidative stress
Psen1 constitutes the catalytic site of the AβPP cleaving enzyme γ-secretase. In concerted action with BACE, psen1 is responsible for the generation of Aβ (reviewed in [286]). Clinical mutations in psen1 cause familial forms of early onset AD (reviewed in [287]) and can affect γ-secretase mediated processing of AβPP in various ways [288]. Generally, mutations in psen1 comprise the composition of the heterogeneous Aβ mixture by shifting the ratio between the various Aβ peptides generated [289–291]. In human SK-N-BE neuroblastoma cells, which were exposed to HNE- or H2O2-mediated oxidative stress, an increase in Aβ level was found that could be attributed indirectly to γ-secretase in a c-jun N-terminal kinase (JNK)/c-jun pathway/BACE1 dependent manner [75]. However, a direct relation between hypoxia-induced oxidative stress and γ-secretase functionality exists. This was later demonstrated in zebrafish by showing that HIF-1α induces increased mRNA expression levels of zebrafish related PSEN1 [292]. In line with these observations, PSEN1 -/- fibroblasts demonstrated impaired induction of HIF-1α [293], suggesting an apparent bi-directional interaction between psen1 and HIF-1α. Importantly, this factor plays a crucial role in the regulation of oxygen homeostasis and the expression and stability of one of the HIF-1α domains is regulated by oxygen levels [205, 209, 210]. Anterior pharynx-defective-1 (APH1) is another component of γ-secretase and it was shown that Hela cells express increased levels of APH1α in response to chemical hypoxia induced activation of HIF-1α resulting in increased AβPP and Notch processing [294]. NF-kB has been identified as important regulator of HIF-1α expression [295]. NF-κB was shown to become activated and translocated to the nucleus in response to oxidative stress by addition of metformin, a pro-oxidative biguanide, to LAN5 neuroblastoma cells. This directly induced transcriptional activation of AβPP and psen1 and ultimately into increased AβPP cleavage, and intracellular accumulation of Aβ which promoted Aβ aggregation [296].
β-secretase is upregulated at a transcriptional level by oxidative stress
BACE1 is an integral part of the amyloidogenic processing pathway of AβPP and the functionality of this enzyme was, similar to γ-secretase, reported to be affected by oxidative stress. Low micromolar concentrations of HNE produced by pro-oxidant stimuli ascorbic acid/FeSO4 or H2O2/FeSO4 or by direct addition to cell medium of NT2 cells in culture were reported to induce BACE1 activity. This induction leads to an increased production of APP C-terminal fragments without affecting AβPP synthesis. Pretreatment of these NT2 cells with α-tocopherol prevented BACE induction and CTF generation demonstrating direct involvement of oxidative stress in inducing BACE activity [81, 297]. Also in APP/PS1 mice, it was shown that administration of an antioxidant compound, tricyclodecan-9-yl-xanthogenate (D609), which is a GSH-mimetic compound, leads to decreased levels of oxidative stress as well as a reduction in BACE1 levels resulting in decreased AβPP processing into Aβ [298]. Various other publications similarly reported that BACE1 protein expression levels were increased in response to oxidative stress [80, 274, 299–305]. A similar observation has been reproduced in various model systems. For example, a developing (48-h post fertilization) zebrafish animal model exposed to hypoxia showed that the mRNA levels and activity of zebrafish bace1, the zebrafish orthologue to human BACE1, were affected by oxidative stress. At the same time, the level of CAT was found to be increased upon exposure of zebrafish to hypoxia [292]. Also, in murine primary cortical neuronal cultures, severe and cytotoxic levels of oxidative stress lead to an increased BACE1 expression. Mild oxidative stress conditions were found to result in subcellular redistribution of BACE1 that promoted amyloidogenic processing of AβPP [306]. Apart from in various animal and cellular models, increased BACE1 levels and activity were also found in brains of sporadic AD patients [307–309]. To understand the cellular signaling pathways involved in oxidative stress-regulated expression of BACE1, an NT2 cell-based assay was used. In this system, pharmacological based inhibition of C-Jun N-terminal Kinase (JNK) and p38MAPK, involved in the stress activated protein kinase (SAPKs)/JNK signaling pathway, but not Akt signaling, was found to inhibit transcriptional regulation of BACE1 [274]. P38MAPK was also reported to be active and identified in Aβ deposits of AβPP tg mice [310]. These observations are consistent with an earlier report that showed that Sp1 regulates transcription of BACE1, where expression levels of Sp1 were positively correlated with the generation of BACE1 and AβPP processing [311]. In line with this, using a lipofuscinfluorphore A2E-mediated photo-oxidation model to investigate the role of BACE1 in age-related macular degeneration, it was shown that BACE1 expression is competitively regulated by Sp1 and DNA methyltransferase 1 (DNMT1) after photo-oxidation [313]. DNMT1 levels were reportedly decreased resulting in demethylation of specific loci within the BACE1 gene promotor [312]. Moreover, members of the SAPK family were also found to be upregulated in AD patient brains [30, 313] and are activated by various stress signals including oxidative stress [314, 315]. The oxidative-stress regulated involvement of JNK has been further demonstrated in studies using transgenic mouse models. For example, JNK was found to be significantly activated in mutant AβPP tg mice with extensive oxidative damage but not in mutant AβPP tg mice with little oxidative damage [100].
Downregulation of non-amyloidogenic AβPP processing
While it was consistently reported that the amyloidogenic processing pathway of AβPP is upregulated by various direct and indirect mechanisms, the non-amyloidogenic pathway, involving sequential cleavage of AβPP by α-secretase and γ-secretase, was found to be downregulated under conditions of oxidative stress [280]. Multiple lines of evidence have shown that γ-secretase is upregulated under conditions of oxidative stress. As such, it was anticipated that the net lowering of the non-amyloidogenic processing of AβPP should be accommodated for by a decrease in α-secretase activity. It was indeed shown that human neuroblastoma cell line SH-SY5Y exposed to hypoxic conditions decreases the expression of disintegrin and metalloproteinase domain-containing protein 10, or ADAM-10, also called α-secretase, in an O2-dose dependent manner [316]. Similarly, human MSN cells exposed to oxidative stress induced by H2O2 or FeCl2 were shown to downregulate the active form of ADAM10 [303]. Mechanisms that explain the downregulation of α-secretase under conditions of oxidative stress have not been explored in great detail. One of the hypotheses that has been postulated involves the JNK3-dependent phosphorylation of Thr668 of AβPP which is considered to be a direct target for BACE1 [303, 317].
Circadian clock genes
Even though the connection between circadian clock, oxidative stress, and AD has been little investigated and may not be directly related, the findings that have been reported on this topic show that a potential interaction between these features may well exist and warrants further investigation. This paragraph summarizes the experimental evidence in supports of such a connection. Circadian rhythm disturbances and associated disorders of the sleep-wake cycle, i.e., fragmentation of sleep and decreased duration of rapid eye movement sleep (REM), occur at various stages of AD-related neurodegeneration and symptoms are generally more severe with increasing Aβ burden and tau pathology [318–321]. One report shows that extensive loss of sleep reduced the activity of SOD and the production of ATP in rat hippocampi [322], features that are strikingly similar in the AD brain. In a follow-up report, apart from reduced SOD activity, it was found that the activity of glutathione peroxidase was also decreased while liver malondialdehyde levels were increased with the extent of sleep deprivation [323]. On the other hand, brain and peripheral tissues were shown to differ in their peroxiredoxin oxidation rhythms [324] as well as in other clock components [325]. This means that peripheral observations cannot be automatically extrapolated to brain processes. During periods of REM sleep firing rates of wake-active noradrenergic locus coeruleus neurons, cells that display high sensitivity to metabolic stress [326–328], are profoundly reduced [329]. Extensive wakefulness induces loss of locus coeruleus neurons and sirtuin type 3 (SirT3) was observed to be involved in this neurodegenerative process [330]. NAD+-dependent deacetylase SirT3 is an important regulator of energy production and redox response by reduction of GSH [331, 332], mediating the upregulation of SOD2 and catalase [333], and activation of SOD2 [334]. Extensive deprivation of sleep is related to reduced levels of SirT3 in young adult wild type mice locus coeruleus neurons while oxidative stress levels increase presumably as a result of a combined increase in metabolic activity and decline in antioxidant response [330]. Other circadian clock related factors associated with redox homeostasis of NAD cofactors include transcriptional activator complex BMAL1 and its binding partners CLOCK and NPAS2 [335]. These clock genes were observed to be involved in glucose metabolism and redox homeostasis in peripheral tissues [335–337]. Expression levels of the master circadian clock regulator genes Bmal1 and Clock are significantly decreased in the cerebral cortex of aged mice [338] although expression levels of these genes in AD brains have not been published. Mice generated with a deletion of Bmal1 demonstrated increased systemic [337, 339] and low levels of brain oxidative stress [340] mediated by a disturbance of its transcriptional targets, including Period Circadian Regulator 2 (Per2) and albumin D-element binding protein (Dbp) [341] as well as neuropathologies and synaptic degeneration [337, 341]. For example, the kinetic occipital region in the brain of these Bmal1 knock-out mice showed a three-fold increase in level of F4-neuroprostanes when measured indicative of increased lipid peroxidation levels in neuronal cells [341]. The deletion also resulted in increased neurodegeneration caused by mitochondrial 3-nitropropionic acid which was suggested to be a direct consequence of the decrease in BMAL1 transcription [341]. Both SirT3 and BMAL1 levels are tightly regulated by circadian rhythmicity of oxidoreductase factor nicotinamide adenine dinucleotide (NAD+) [342], while, in turn cellular redox status affects the activity of clock transcription factors [335]. Proteasome expression levels and activity were observed to follow circadian oscillations that correlated with the level of carbonylated proteins [343] demonstrating that clearance of oxidized proteins are also showing circadian rhythm. It would be of interest to investigate Bmal1 and Clock expression levels in AD patient brains to establish a possible connection between the observed symptoms of sleep deprivation in AD, oxidative stress status and cognitive dysfunction.
Klotho, the aging suppressor gene, regulated by oxidative stress
Klotho is a single-pass transmembrane protein hormone containing a long type I transmembrane domain and a short secreted domain [344]. The latter is released into the extracellular space upon insulin-mediated release by ADAM family members ADAM10 and ADAM17 [345]. Mutations in the KLOTHO gene were observed to induce a human aging resembling phenotype in a transgenic mouse model which could be genetically rescued by exogenous expression of klotho cDNA [346]. Consistent with this, aging was found to be suppressed in a klotho overexpressing mouse model [347]. Single nucleotide polymorphisms of KLOTHO affect trafficking and catalytic activity of klotho which was associated to onset of aging in a human population based study [348]. Expression of the protein declines with age as was demonstrated by microarray analysis of the aging brain of a rhesus monkey model [349]. Subsequently, the klotho gene has been dubbed an aging suppressor gene which acts by regulating oxidative stress [350] because it was shown to effectively reduce urinary excretion of 8-OHdG upon renal overexpression in mice [351]. This hypothesis was further supported by the finding that different cell types incubated with Klotho protein were protected from oxidative stress and apoptosis induced by paraquat [350], hydrogen peroxide [352], glutamate and oligomeric Aβ [353]. Apart from in the kidney, the protein is also expressed in the choroid plexus in the brain with low levels of expression in the hippocampus [346, 354]. In this brain region Klotho plays a role in hippocampus-dependent memory by regulating adult hippocampal neurogenesis [355]. Mutation of klotho in a transgenic mouse model demonstrated increased levels of 8-OHdG and malondialdehyde in the hippocampus in an age-dependent manner [356]. One of the molecular models by which secreted Klotho has been suggested to regulate oxidative stress includes the insulin/IGF-1 growth signaling pathway [357]. Klotho inhibits this pathway leading to activation of Forkhead box O (FOXO) transcription factor. This, in turn, enhances the expression of ROS scavenging enzyme mitochondrial manganese SOD2 [350] indicating that Klotho may perhaps lend its anti-aging capacity by indirect regulation of the generation of an antioxidant enzyme.
PUTATIVE MOLECULAR MECHANISMS OF ALZHEIMER’S DISEASE-RELATED OXIDATIVE STRESS
One of the striking observations is that microglia in close proximity to amyloid plaques are often found to be activated and release O2·– and H2O2 [358]. Clearly, the multicellular organization of the brain may be a relevant determinant in outcome of oxidative stress in AD, and potentially also other, neurodegenerative diseases. For both Aβ and tau a number of potential contributory as well as inhibitory pathways in the process of oxidative stress generation have been proposed. For example, Aβ can reduce Cu2+ to Cu+ in a catalytic cycle that uses O2 and biological reducing agents as substrates while generating neurotoxic H2O2 [359, 360] and a peptide radical inducing oxidation and dityrosine cross-linking of Aβ were observed [93, 361]. Figure 1 provides an overview of the various cellular factors and mechanisms that are thought to associate oxidative stress and AD to date.
A protective or advancing effect of Aβ on oxidative stress may be subtly defined
A potential anti-oxidant effect of Aβ has been attributed to the reported ability of Aβ to sequestrate redox-active metals [54, 361–367]. As a result of this binding, it has for example been proposed that Aβ may quench Cu2+ preventing Cu2+ from generating H2O2. In contrast, other reports have shown that the binding of Aβ to such transition metal ions actually results in enhanced formation of ROS [368] by the reported ability of Aβ to reduce Cu2+, upon binding, to Cu1 + [369]. A subsequent publication questioned the role of Cu2+ binding in generation of ROS as it was shown that rodent Aβ1-42, in which Cu2+ binding is ameliorated, results in similar levels of oxidative stress as human Aβ1-42 [369]. Heme-a, an essential component of mitochondrial complex IV, was shown to interact with Aβ1-40, resulting in decreased assembly of this complex into a functional electron transport chain complex [90]. These in vitro observations are supported by the finding that mitochondrial whole brain levels of cytochrome-a are decreased with 25% in the AD brain [370, 371]. Both these protective and toxic roles of Aβ-metal complexation in oxidative stress receive ample support in the scientific field and perhaps the actual outcome whether metal ion binding is toxic is more subtly defined by factors such as Aβ level, Aβ aggregation state, co-occurring factors such as the availability of a reducing agent [359], redox kinetics [372], or disease stage. Supportive of such a hypothesis is the observation that Aβ was found to act as a neurotrophic agent selectively at low nM concentrations while at higher peptide concentrations neurotrophic functionality was abolished [363, 373, 374]. Even though the functional role of Aβ has been heavily debated, it has been argued that antioxidant activity may be the primary role of this peptide in the brain [374]. However, a compelling finding arguing against an anti-oxidant function as primary role for Aβ was that oligomeric human Aβ, but not rodent Aβ, can bind two molecules of heme with sub μM affinity which results in the generation of a peroxidase [362]. Moreover, rat Aβ1-40 was reported to interact with zinc with lower affinity than human Aβ1-40. Such species specificity directly argues against a primary anti-oxidant role of Aβ although it does not rule out the potential of the peptide to also, next to its yet to identify primary role, demonstrate anti-oxidant activity via indirect routes.
Tau plays an indirect role in oxidative stress via organelle transport
Upon overexpression tau inhibits kinesin-dependent transport of mitochondria and peroxisomes into neuronal processes while the microtubular network remains intact [375]. Lack of transport of these two organelles was shown to deplete neurites from ATP and protection mechanisms against oxidative stress as was illustrated by an increased vulnerability of tau overexpressing differentiated N2a cells upon exposure to H2O2 [375]. Another parameter proposed to play a role in tau-induced oxidative stress includes tau aggregation into paired helical filaments [376]. Addition of synthesized HNE at micromolar concentrations to retinoic acid differentiated P19 embryonal carcinoma cells induced tau to crosslink into high molecular weight species [376]. Crosslinking of tau into paired helical filaments was shown to be driven by abnormal levels of phosphorylation, or hyperphosphorylation, of tau, which renders the protein insoluble and dysfunctional [377, 378]. The role of phosphorylation of tau in this process and the link to oxidative stress were highlighted by three subsequent publications showing that 1) phosphorylation of tau is partly regulated by the extracellular signal-regulated protein kinase (ERK2) which becomes activated upon exposure to H2O2 [379], and also, 2) by rapid and potent activation of transcription factor NFκB by reactive oxygen intermediate-mediated release of inhibitory factor IκB from NFκB [181], and 3) acrolein, a peroxidation product of arachidonic acid, induced p38 stress-kinase-mediated tau phosphorylation [380]. An in vitro follow up study demonstrated that co-incubation of a pseudo-phosphorylation mimicking form of tau with acrolein and methylglyoxal induced the formation of tau dimers and high molecular weight oligomers [381]. Other than phosphorylation, also the glycation of tau was observed to connect tau tangle formation with oxidative stress. Using SH-SY5Y cells, advanced glycation end product-recognizing antibodies, increased HO-1 and malondialdehyde reactivity were shown to colocalize with tau paired helical filaments [111].
Direct generation of ROS by Aβ fragments
A pioneering publication in 1994 used mass spectrometry and electron paramagnetic resonance spin trapping to demonstrate that Aβ in vitro under cell free conditions itself can fragment into free radical peptides in an oxygen-dependent but metal-independent manner [382]. The generated Aβ25-35 fragment was capable of inactivating the enzymes glutamine synthetase and creatine kinase. The authors suggested that methionine 35 may be capable of reacting with oxygen to produce sulfoxide, which, in turn, can result in radical generation [382, 383] although this hypothesis has not been experimentally verified.
Aβ aggregation state
Aβ was demonstrated to accumulate into various aggregation states ranging from monomeric to larger assemblies into amyloid plaques found upon postmortem analysis of AD patient brains. In-between these two states an apparent continuum of oligomeric aggregates with different aggregation numbers, e.g., the number of monomers per oligomer, exists, and many studies have attempted to identify and characterize the most toxic species within this range. Detailed reviews have been published on this specific topic highlighting the potential toxic role of these intermediates and their relation with the clinicopathological features of AD [384–387].
The dynamic nature of Aβ assembly hampers studies into oxidative stress
Generally, experimental studies indicate that particularly the intermediate soluble aggregated forms of Aβ are highly toxic and these species are commonly referred to as oligomers or pre- or protofibrils [388, 389]. More specifically, soluble SDS-stable dimers, extracted from AD brains [390], up to 56 kDa soluble Aβ assemblies that are capable of inducing cognitive impairment in Tg2576 mice [391] have been identified as potential toxic species acting upon AD progress. While most of these studies merely investigate the association between assembly state and loss of cellular viability, synaptic function, or cognition, consistent with these observations, a study investigating the effects of different Aβ aggregate species on oxidative stress showed that specifically prefibrillar and oligomeric Aβ1-42 potently increased levels of oxidative stress in NT2 cells as detected by HNE and H2O2 generation [392]. Using a combination of electron spin resonance spectroscopy coupled to spin trapping, a short burst of H2O2 generation was observed during early aggregation stages of Aβ1-40 [393]. Recently, incubation of Aβ with Cu2+ and H2O2 were shown to modulate Aβ self-assembly. Depending on the Cu2+ to H2O2 ratio, up to hexameric species of Aβ1-42 could be detected using SDS-PAGE with little propensity to develop into ThT positive fibrils upon prolonged incubation [394]. These Aβ1-40 and Aβ1-42 oligomers demonstrated prolonged disruption of phospholipid vesicles which is one of the proposed cytotoxic mechanisms of Aβ oligomers [394]. However, the precise aggregation number of such Aβ oligomers is difficult to pin down using most standard biophysical and biochemical techniques as a result of their heterogeneous, dynamic, and interconverting nature.
Interaction of Aβ with mitochondrial factors
Several mechanistically indirect pathways have been suggested by which means Aβ can influence mitochondrial function.
By interaction of Aβ with mitochondrial alcohol dehydrogenase
Yeast-two-hybrid based screening of the human brain and a HeLa cell model demonstrated that Aβ and mitochondrial alcohol dehydrogenase may interact [183]. This interaction was shown to be specific involving residues 12–24 of Aβ1-40 and disturbs the NAD-binding pocket and the catalytic triad of the enzyme leading to functional inhibition of nicotinamide dinucleotide binding required for the function of the enzyme [183, 197, 395, 396], resulting in oxidative stress and neurodegeneration [183]. Given the fact that Aβ is primarily an extracellularly generated peptide it is questionable whether Aβ and mitochondrial alcohol dehydrogenase may ever reside in close proximity of each other. However, by means of co-immunoprecipitation assays, immunogold electron microscopy and confocal microscopy it was demonstrated that Aβ can be detected in the mitochondria within the cerebral cortex of AD patients and mutant APP (mAPP) as well as mAPP/ABAD transgenic mice. Also, in these in vivo animal models Aβ was reported to co-localize with the N-terminus (residues 98–101 and 108–110) of alcohol dehydrogenase while mutations within this site abrogated Aβ binding to alcohol dehydrogenase [197]. Strengthening the suggestion that alcohol dehydrogenase may play a role in AD, degenerating neurons in the brains of AD patients were found to express up-regulated levels of alcohol dehydrogenase [197], particularly those in close proximity to Aβ deposits [183]. Although it cannot be ruled out that this observation represents an Aβ-unrelated compensatory mechanism to counteract the impaired energy homeostasis generally observed in AD neurons [313, 397]. The role of alcohol dehydrogenase in AD has been more extensively covered in a review [398].
By disruption of energy generation from mitochondria
The brain requires ATP and its intermediates for the formation of the neurotransmitter acetylcholine [399], and the critical membrane component cholesterol [374]. To accommodate these requirements it was recognized more than a century ago that the vascular system exerts some degree of plasticity to ensure sustained local activity of the neuronal network [400]. Further, astrocytes, expressing GLUT1 type glucose transporters [401], play a central role in neuronal energy supply [402] supporting the notion that the multicellular context of the brain is highly supportive of neuronal energy-consuming activities. Current PET and fMRI functional brain imaging techniques are based on the assumption that brain function is associated with brain energy consumption and from such techniques a wealth of information has been acquired over recent years on brain energy homeostasis in a variety of neurodegenerative disorders. The association between oxidative stress and energy production becomes apparent when considering that the regeneration of GSH from GSSG is an NADPH dependent process, where NADPH is mainly obtained through glucose metabolism. In various neurodegenerative disorders, including AD, a decrease in brain ATP generation is observed. Membrane fluidity changes observed upon iodoacetic acid-induced inhibition of ATP production could be rescued by treatment with anti-oxidants tirilazad and gossypol [403] suggesting that reduced ATP availability may result in oxidative stress and membrane damage. Upon aging, glucose metabolism derails progressively as a result of changes in brain insulin [404], and cortisol [405] levels. Also, activity of synaptic ATPases was significantly decreased in rat frontal cerebral cortex upon aging [406]. This condition seems to worsen in AD patients as it was shown in patients with incipient late-onset AD type dementia, that cerebral glucose utilization had reduced with 45% which progressed to 55% in advanced stages of the disease [407]. Oxidative stress, induced by NO was shown to transiently reduce ATP generation in rat astrocytes while increasing glycolysis rate in an F1F0-ATPase and adenosine nucleotide translocase dependent manner specifically in primary astrocytes to rescue this ATP-depleted phenotype [408]. At the same time, rat primary neurons exposed to NO were shown to progressively become ATP depleted which ultimately lead to cell death [408]. A number of metabolism-related enzymes have been identified to be affected in a progressive manner in AD, including pyruvate dehydrogenase, ATP-citrate lyase and acetoacetyl-CoA thiolase [409]. Activities of glycolysis and citric acid cycle related enzymes aldolase, triose phosphate isomerase, phosphoglycerate kinase, and phosphoglycerate mutase are affected in AD as a result of oxidation or nitration [410–414]. Inconsistent results have been published on phosphofructokinase activity in AD brains, with some researchers suggesting no significant reductions [415]. Subcortical regions of the brain further displayed increased activities of hexokinase, an enzyme involved in the initiating step of glycolysis, and lactate dehydrogenase in AD patient brains while activities of these enzymes in cortical regions were unaffected [416]. As the authors already suggest [416], increased activity of lactate dehydrogenase is suggestive of a metabolic need for anaerobic respiration to compensate for lost ability to generate ATP via aerobic metabolic routes. Taken together, oxidation and nitration processes of many of the enzymes involved in cellular metabolism may reduce activity of such enzymes sufficiently to explain the significantly reduced ATP generation observed in AD brains while the bioenergetic adaptation of the cell towards anaerobic routes for obtaining sufficient quantities of ATP to sustain high levels of ATP generation cannot be sufficiently compensated for. The question remains whether tau or Aβ only play an indirect role in activity reduction by inducing oxidation or nitration of these enzymes or whether a direct role, for example by activity-reducing interaction such as demonstrated for ABAD causes loss of metabolic rate. A study using co-immunoprecipitation assays combined with tandem mass tag multiplexed quantitative mass spectrometry identified glycolysis enriched proteins such as pyruvate kinase and aldolase as potential interactors with tau [417]. Using ELISA and gel filtration assays, it was reported that synthesized Aβ42 and Aβ1-28 can interact in vitro with a KD of 5 nM with rat phosphofructokinase, but not lactate dehydrogenase [418]. It is still debatable what the triad of factors oxidative stress, Aβ or tau and cellular metabolism exactly comprises in terms of molecular interactions and how they reciprocatively interact with each other. One attempt to address this question was published by Casley and colleagues who incubated isolated rat brain mitochondria either with Aβ25-35 or Aβ1-42 with or without NO to determine the relative impact of each of these factors on mitochondrial respiration using oxygen sensitive electrodes [419]. Both Aβ peptides significantly inhibited mitochondrial respiration by specifically affecting the activity of complex IV, while exposure of the mitochondria to NO substantially worsened respirational outcome.
Interaction with RAGE
RAGE is a transmembrane receptor widely expressed in all tissue types including the brain [420, 421]. One diverse group of ligands known to interact with this receptor are advanced glycation end products, AGEs. AGEs are the product of non-enzymatic aldose-mediated glycated or oxidized proteins [422], and accumulation of AGEs was shown to be aging-related [423, 424] and accelerates in conditions such as diabetes [425]. Potential clinical relevance of RAGE to AD was demonstrated using ELISA of AD brain homogenates showing a 2.5 fold increase in expression of RAGE compared to age-matched control subjects [176]. The roles of RAGE and AGEs in AD pathogenesis have been covered in a number of reviews [426, 427]. The offspring of a transgenic RAGE overexpressing mouse model crossed with Tg APP animals showed neuronal perturbation already at 3-4 months of age, and astrogliosis and reactive microglia at 14–18 months [428]. One of the general observations is that Aβ can interact with RAGE. For example, Yan and co-workers have shown that endothelial or PC12 RAGE provides a binding site for 125I-labeled synthetic Aβ on cellular surfaces. This interaction was found to result in cellular perturbation and dose-dependent generation of TBARS, NF-κB-mediated microglial activation, and cytotoxicity. TBAR generation could be blocked by pre-treatment of cultures with antioxidants probucol or N-acetylcysteine [176] demonstrating a direct or indirect association with oxidative stress. Indeed, interaction of Aβ with RAGE on the cell-surface exerted localized oxidant activity. The authors further observed that stimulation of RAGE by other ligands which do not themselves generate ROS induces intracellular generation of oxidants in target cells [176] suggesting that it is the interaction between RAGE and its ligand that induces the oxidation effect and not the ligand by itself. Not only Aβ as AD hallmark peptide appeared to show an association with RAGE-induced oxidative stress: using immunostaining it was shown that tau paired helical filaments colocalize with AGEs in AD temporal cortex tissue [116]. Also, tau was shown to be amenable to ribose-mediated glycation and exposure of SH-SY5Y cells to these glycated tau species resulted in oxidative stress without affecting cell viability [116]. It appears that AGE-RAGE interaction with AD hallmark partners results in a variety of cellular responses that can mediate oxidative stress.
Disruption of calcium homeostasis
ROS generation and Ca2+ signaling – a bidirectional paradigm
One important step in the process of synaptic transmission is the Ca2+-regulated fusion of neurotransmitter containing synaptic vesicles with the pre-synaptic membrane which results in the release of neurotransmitters in the synaptic cleft. Apart from neurotransmitter release a plethora of other cellular mechanisms are regulated by Ca2+-dependent signaling, for example regulation of membrane excitability, neuronal growth and differentiation as well as expression of a wide range of genes that are activity-induced and apoptotic processes. Extracellular and endoplasmic reticular Ca2+ levels largely exceed intracellular levels and this sustained Ca2+ homeostasis is an important factor required for cellular functioning. A number of reviews that extensively cover neuronal Ca2+ homeostasis mechanisms have been published [429–432]. To aid Ca2+ homeostasis, Ca2+ levels in neuronal cells are strictly regulated by controlled release from various intracellular and extracellular pools. Release of Ca2+ from the extracellular pools into the cell is mediated by factors such as voltage-gated Ca2+ channels, N-methylo-D-aspartate (NMDA) receptors and transient receptor potential channels while intracellular Ca2+, stored in the endoplasmic reticulum, is released via the inositol triphosphate receptor (Ins[1,4,5]P3 R) and ryanodine receptor (RyRs) [429]. These factors act in a concerted manner to regulate intracellular Ca2+ levels in a temporal and spatial manner. Reports show that dyshomeostasis of Ca2+ levels and oxidative stress amplify each other in a bidirectional manner. For example, ROS species superoxide anion, hydrogen peroxide and hydroxyl radicals are known to regulate signaling pathways involving Ca2 + . Mitochondrial Ca2+ mediates activity of a number of mitochondrial enzymes involved in the tricarboxylic acid cycle and oxidative phosphorylation and, as such, increased Ca2+ levels were reported to elevate cellular metabolic rate [433–435]. In turn, cellular metabolic rate was shown to be directly proportional to ROS generation in rat and porcine lung [436], and hepatomas [437]. Bidirectionality of the ROS-generation/Ca2+ signaling paradigm was observed by functional impairment of membrane-bound receptors and channels that regulate influx or efflux of Ca2+ mediated by oxidative stress-induced lipid peroxidation. Ca2+ dyshomeostasis was reported in neurodegenerative disorders in general, including AD. For example, whereas wild type presenilin was reported to form Ca2+ permeable channels allowing Ca2+ leakage from the endoplasmic reticulum [438, 439], some, but not all FAD related mutations in Psen1 and 2, show deficiencies in Ca2+ leak function in a mouse embryonic fibroblast model, primary fibroblasts obtained from patients and a planar lipid bilayer [439, 440]. Apart from neurons, astrocytes also play an ion regulating role supporting neuronal activity. Astrocytes act in a concerted effort to regulate Ca2+ levels via release of ATP which subsequently binds to purine receptors on the membrane of adjacent astrocytes [441]. ATP binding then results in release of intracellular Ca2+ via the phospholipase C beta/inositol triphosphate (IP3) pathway [442]. Using a microarray analysis, it was reported that the expression of 32 Ca2+ signaling pathway-related genes were altered in astrocytes at various stages of AD pathology [443]. Aβ was reported to perturb astrocytic Ca2+ regulation indirectly by inducing oxidative stress which impairs membrane Ca2+ pumps and enhances Ca2+ influx through voltage-dependent channels and ionotropic glutamate receptors [444–446].
By potentiating mitochondrial permeability transition pore formation
The mitochondrial permeability transition pore (mPTP) plays an important role in mitochondrial Ca2+ homeostasis [447]. Its molecular composition and mechanism of action have been under debate in the last years. Initially, the mPTP was thought to consist of the voltage-dependent anion channel (VDAC) in the outer membrane, the adenine nucleotide translocase (ANT) in the inner membrane and Cyclophilin D (CypD) in the matrix. In this way, when CypD translocates to the inner mitochondrial membrane to interact with ANT and VDAC [448], the mPTP opens allowing non-selective exchange of calcium [449–451]. More recent findings dismiss this initial idea and indicate that VDAC is not a key component of the mPTP [452, 453] and ANT has only a regulatory function rather than being a core unit of the mPTP [454]. Only CypD, a peptidyl-prolyl isomerase F located in the mitochondrial matrix, remains a critical molecule in the mPTP in both postulations. Consistent with this, studies in animal models have shown that mPTP formation can be efficiently blocked by the addition of a cyclophilin D inhibitor, cyclosporine A (CSA) or by depletion of CypD [448, 455, 456]. Short and transient mPTP opening has been shown to occur under physiological conditions, allowing rapid passage of protons, Ca2+ and other substances of a size up to 1.5 kDa [457, 458]. However, prolonged mPTP leaking may result in mitochondrial disruption. Apart from a high concentration of Ca2+ in the mitochondrial matrix [459], ROS can trigger mPTP opening. Moreover, cytosolic ROS was observed to induce signals leading to mPTP opening resulting in further ROS release and this phenomenon is referred to as “ROS-induced ROS release (RIRR)” [460]. Several lines of investigation have proposed that Aβ-induced ROS and increased Ca2+ levels could well link with mPTP formation. In this regard, increased CypD expression has been identified in neurons of the hippocampus and temporal lobe of AD patients [461]. CypD was shown to specifically bind to Aβ oligomers both in vitro and in vivo in a dose-dependent manner facilitating permeability transition and ROS formation causing mitochondrial dysfunction [461–463]. Removal of CypD improved cognitive and synaptic function in a mouse model for AD [462], protecting cells from Ca2+, oxidative stress or Aβ-induced cell death [448, 464, 465], and restored oligomeric Aβ42-mediated loss of axonal mitochondrial transport in neurons. The role of ROS was highlighted by demonstrating rescue of Aβ-induced loss of axonal mitochondrial movement upon administration of Probucol, an anti-oxidant agent [466]. As a result of these observations, various studies embarked on targeting CypD/mPTP inhibition as potential treatment for neurodegenerative diseases [467, 468].
CONCLUSIONS
Oxidative stress is an early clinical feature of AD, as well as other neurodegenerative disorders. AD-related oxidative stress arises as a result of increased generation of ROS, induced by mitochondrial failure, but also decreased levels of endogenous antioxidants are commonly observed in AD. Many of the biomacromolecules that are part of normal cellular physiology are susceptible to oxidative modification altering their function. Also, Aβ and tau, two AD hallmark proteins are subject to oxidation, while Aβ itself was reported to induce the formation of ROS. A number of genetic factors have been identified to play a role in AD-related failure to maintain physiological ROS levels within strict limits. For example, clusterin, apolipoprotein E, klotho, and enzymes involved in the AβPP processing machinery regulating Aβ generation are related, either directly or indirectly, to oxidative stress. Various molecular mechanisms explaining the association between oxidative stress and AD have been identified. Aβ and its potent interaction with Cu2+ was identified as key feature in AD-related oxidative stress. But also various other mitochondria-associated factors play a role, including mitochondrial permeability transition pore formation, regulation of cellular metabolic pathways and Ca2+ signaling. The association between oxidative stress and AD appears complex, bi-directional, and self-reinforcing.
ACKNOWLEDGMENTS
Work related to oxidative stress in the group of the corresponding author is financially supported by a ZonMw Memorabel grant number 733050304.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/19-0863r1).
REFERENCES
[1] | Scheltens P , Blennow K , Breteler MMB , de Strooper B , Frisoni GB , Salloway S , Van der Flier WM ((2016) ) Alzheimer’s disease. Lancet 388: , 505–517. |
[2] | Matsuoka Y , Picciano M , La Francois J , Duff K ((2001) ) Fibrillar β-amyloid evokes oxidative damage in a transgenic mouse model of Alzheimer’s disease. Neuroscience 104: , 609–613. |
[3] | Moreira PI , Carvalho C , Zhu X , Smith MA , Perry G ((2010) ) Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim Biophys Acta 1802: , 2–10. |
[4] | Butterfield DA , Poon HF , Clair DS , Keller JN , Pierce WM , Klein JB , Markesbery WR ((2006) ) Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: Insights into the development of Alzheimer’s disease. Neurobiol Dis 22: , 223–232. |
[5] | Mecocci P , Polidori MC ((2012) ) Antioxidant clinical trials in mild cognitive impairment and Alzheimer’s disease. Biochim Biophys Acta 1822: , 631–638. |
[6] | Mariani E , Polidori MC , Cherubini A , Mecocci P ((2005) ) Oxidative stress in brain aging, neurodegenerative and vascular diseases: An overview. J Chromatogr B 827: , 65–75. |
[7] | Mangialasche F , Polidori MC , Monastero R , Ercolani S , Camarda C , Cecchetti R , Mecocci P ((2009) ) Biomarkers of oxidative and nitrosative damage in Alzheimer’s disease and mild cognitive impairment. Ageing Res Rev 8: , 285–305. |
[8] | Yao J , Irwin RW , Zhao L , Nilsen J , Hamilton RT , Brinton RD ((2009) ) Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 106: , 14670–14675. |
[9] | Shah K , Kumar RG , Verma S , Dubey RS ((2001) ) Effect of cadmium on lipid peroxidation, superoxide anion generation and activities of antioxidant enzymes in growing rice seedlings. Plant Sci 161: , 1135–1144. |
[10] | Mattson MP ((2004) ) Pathways towards and away from Alzheimer’s disease. Nature 430: , 631–639. |
[11] | Akbar H , Duan X , Saleem S , Davis AK , Zheng Y ((2016) ) RhoA and Rac1 GTPases differentially regulate agonist-receptor mediated reactive oxygen species generation in platelets. PLoS One 11: , e0163227. |
[12] | Persson T , Popescu BO , Cedazo-Minguez A ((2014) ) Oxidative stress in Alzheimer’s disease: Why did antioxidant therapy fail? Oxid Med Cell Longev 2014: , 427318. |
[13] | Ramassamy C , Averill D , Beffert U , Bastianetto S , Theroux L , Lussier-Cacan S , Cohn JS , Christen Y , Davignon J , Quirion R , Poirier J ((1999) ) Oxidative damage and protection by antioxidants in the frontal cortex of Alzheimer’s disease is related to the apolipoprotein E genotype. Free Radic Biol Med 27: , 544–553. |
[14] | Selek S , Bulut M , Ocak AR , Kalenderoğlu A , Savaş HA ((2012) ) Evaluation of total oxidative status in adult attention deficit hyperactivity disorder and its diagnostic implications. J Psychiatr Res 46: , 451–455. |
[15] | Milonski J , Zielinska-Blizniewska H , Olszweski J , Majsterek I , Mrowicka M ((2015) ) DNA damage and oxidant-antioxidant status in blood of patients with head and neck cancer. DNA Cell Biol 34: , 213–219. |
[16] | Isobe C , Abe T , Terayama Y ((2010) ) Levels of reduced and oxidized coenzyme Q-10 and 8-hydroxy-2’-deoxyguanosine in the cerebrospinal fluid of patients with living Parkinson’s disease demonstrate that mitochondrial oxidative damage and/or oxidative DNA damage contributes to the neurodegenerative process. Neurosci Lett 469: , 159–163. |
[17] | Lapenna D , de Gioia S , Ciofani G , Mezzetti A , Ucchino S , Calafiore AM , Napolitano AM , Di Ilio C , Cuccurullo F ((1998) ) Glutathione-related antioxidant defenses in human atherosclerotic plaques. Circulation 97: , 1930–1934. |
[18] | Ide T , Tsutsui H , Hayashidani S , Kang D , Suematsu N , Nakamura K , Utsumi H , Hamasaki N , Takeshita A ((2001) ) Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circulation Res J Am Heart Assoc 88: , 529–535. |
[19] | Walter PB , Fung EB , Killilea DW , Jiang Q , Hudes M , Madden J , Porter J , Evans P , Vichinsky E , Harmatz P ((2006) ) Oxidative stress and inflammation in iron-overloaded patients with β-thalassaemia or sickle cell disease. Brit J Haemat 135: , 254–263. |
[20] | Jovanovic SV , Clements D , MacLeod K ((1998) ) Biomarkers of oxidative stress are significantly elevated in Down syndrome. Free Radic Biol Med 25: , 1044–1048. |
[21] | Chung CP , Schmidt D , Stein CM , Morrow JD , Salomon RM ((2013) ) Increased oxidative stress in patients with depression and its relationship to treatment. Psychiatry Res 206: , 213–216. |
[22] | Greenman IC , Gomez E , Moore CEJ , Herbert TP ((2007) ) Distinct glucose-dependent stress responses revealed by translational profiling in pancreatic β-cells. J Endocrinol 192: , 179–187. |
[23] | Rosa R De , Spinozzi F , Itri R ((2018) ) Hydroperoxide and carboxyl groups preferential location in oxidized biomembranes experimentally determined by small angle X-ray scattering: Implications in membrane structure. Biochim Biophys Acta 1860: , 2299–2307. |
[24] | Wong-Ekkabut J , Xu Z , Triampo W , Tang IM , Tieleman DP , Monticelli L ((2007) ) Effect of lipid peroxidation on the properties of lipid bilayers: A molecular dynamics study. Biophys J 93: , 4225–4236. |
[25] | Haluska CK , Baptista MS , Fernandes AU , Schroder AP , Marques CM , Itri R ((2012) ) Photo-activated phase separation in giant vesicles made from different lipid mixtures. Biochim Biophys Acta 1818: , 666–672. |
[26] | Cutler RG , Kelly J , Storie K , Pedersen WA , Tammara A , Hatanpaa K , Troncoso JC , Mattson MP ((2004) ) Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc Natl Acad Sci U S A 101: , 2070–2075. |
[27] | Sayre LM , Zelasko DA , Harris PLR , Perry G , Salomon RG , Smith MA ((1997) ) 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer’s disease. J Neurochem 68: , 2092–2097. |
[28] | Echtay KS , Esteves TC , Pakay JL , Jekabsons MB , Lambert AJ , Portero-Otín M , Pamplona R , Vidal-Puig AJ , Wang S , Roebuck SJ , Brand MD ((2003) ) A signalling role for 4-hydroxy-2-nonenal in regulation of mitochondrial uncoupling. EMBO J 22: , 4103–4110. |
[29] | Takeda A , Smith MA , Avilá J , Nunomura A , Siedlak SL , Zhu X , Perry G , Sayre LM ((2000) ) In Alzheimer’s disease, heme oxygenase is coincident with Alz50, an epitope of τ induced by 4-hydroxy-2-nonenal modification. J Neurochem 75: , 1234–1241. |
[30] | Zhu X , Rottkamp CA , Boux H , Takeda A , Perry G , Smith MA ((2000) ) Activation of p38 kinase links tau phosphorylation, oxidative stress, and cell cycle-related events in Alzheimer disease. J Neuropathol Exp Neurol 59: , 880–888. |
[31] | Giraldo E , Lloret A , Fuchsberger T , Viña J ((2014) ) Aβ and tau toxicities in Alzheimer’s are linked via oxidative stress-induced p38 activation: Protective role of vitamin E. Redox Biol 2: , 873–877. |
[32] | Martinet W , Knaapen MWM , De Meyer GRY , Herman AG , Kockx MM ((2001) ) Oxidative DNA damage and repair in experimental atherosclerosis are reversed by dietary lipid lowering. Circ Res 88: , 733–739. |
[33] | Misiaszek R , Crean C , Joffe A , Geacintov NE , Shafirovich V ((2004) ) Oxidative DNA damage associated with combination of guanine and superoxide radicals and repair mechanisms via radical trapping. J Biol Chem 279: , 32106–32115. |
[34] | Mecocci P , MacGarvey U , Kaufman AE , Koontz D , Shoffner JM , Wallace DC , Beal MF ((1993) ) Oxidative damage to mitochondrial DNA shows marked age-dependent increases in human brain. Ann Neurol 34: , 609–616. |
[35] | Martinet W , De Meyer GRY , Herman AG , Kockx MM ((2004) ) Reactive oxygen species induce RNA damage in human atherosclerosis. Eur J Clin Invest 34: , 323–327. |
[36] | Nunomura A , Perry G , Pappolla MA , Wade R , Hirai K , Chiba S , Smith MA ((1999) ) RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer’s disease. J Neurosci 19: , 1959–1964. |
[37] | Liu J , Head E , Gharib AM , Yuan W , Ingersoll RT , Hagen TM , Cotman CW , Ames BN ((2002) ) Memory loss in old rats is associated with brain mitochondrial decay and RNA/DNA oxidation: Partial reversal by feeding acetyl-L-carnitine and/or R-α-lipoic acid. Proc Natl Acad Sci U S A 99: , 2356–2361. |
[38] | Weingarten MD , Lockwood AH , Hwo SY , Kirschner MW ((1975) ) A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A 72: , 1858–1862. |
[39] | Heston LL , White J ((1978) ) Pedigrees of 30 families with Alzheimer disease: Associations with defective organization of microfilaments and microtubules. Behav Genet 8: , 315–331. |
[40] | Drum BML , Yuan C , Li L , Liu Q , Wordeman L , Santana LF ((2016) ) Oxidative stress decreases microtubule growth and stability in ventricular myocytes. J Mol Cell Cardiol 93: , 32–43. |
[41] | Lee CF , Liu CY , Hsieh RH , Wei YH ((2005) ) Oxidative stress-induced depolymerization of microtubules and alteration of mitochondrial mass in human cells. Ann NY Acad Sci 1042: , 246–254. |
[42] | Valen G , Sondén A , Vaage J , Malm E , Kjellström BT ((1999) ) Hydrogen peroxide induces endothelial cell atypia and cytoskeleton depolymerization. Free Radic Biol Med 26: , 1480–1488. |
[43] | Hinshaw DB , Miller MT , Omann GM , Beals TF , Hyslop PA ((1993) ) A cellular model of oxidant-mediated neuronal injury. Brain Res 615: , 13–26. |
[44] | Hu WG , Lu QP ((2014) ) Impact of oxidative stress on the cytoskeleton of pancreatic epithelial cells. Exp Ther Med 8: , 1438–1442. |
[45] | Neely MD , Sidell KR , Graham DG , Montine TJ ((1999) ) The lipid peroxidation product 4-hydroxynonenal inhibits neurite outgrowth, disrupts neuronal microtubules, and modifies cellular tubulin. J Neurochem 72: , 2323–2333. |
[46] | Giulivi C , Traaseth NJ , Davies KJA ((2003) ) Tyrosine oxidation products: Analysis and biological relevance. Amino Acids 25: , 227–232. |
[47] | Lovell MA , Robertson JD , Teesdale WJ , Campbell JL , Markesbery WR ((1998) ) Copper, iron and zinc in Alzheimer’s disease senile plaques. J Neurol Sci 158: , 47–52. |
[48] | Dong J , Atwood CS , Anderson VE , Siedlak SL , Smith MA , Perry G , Carey PR ((2003) ) Metal binding and oxidation of amyloid-β within isolated senile plaque cores: Raman microscopic evidence. Biochemistry 42: , 2768–2773. |
[49] | Al-Hilaly YK , Williams TL , Stewart-Parker M , Ford L , Skaria E , Cole M , Bucher WG , Morris KL , Sada AA , Thorpe JR , Serpell LC ((2013) ) A central role for dityrosine crosslinking of Amyloid-β in Alzheimer’s disease. Acta Neuropathol Commun 1: , 83. |
[50] | Aeschbach R , Amadò R , Neukom H ((1976) ) Formation of dityrosine cross-links in proteins by oxidation of tyrosine residues. Biochim Biophys Acta 439: , 292–301. |
[51] | Gu M , Bode DC , Viles JH ((2018) ) Copper redox cycling inhibits Aβ fibre formation and promotes fibre fragmentation, while generating a dityrosine Aβ dimer. Sci Rep 8: , 16190. |
[52] | Hureau C , Faller P ((2009) ) Aβ-mediated ROS production by Cu ions: Structural insights, mechanisms and relevance to Alzheimer’s disease. Biochimie 91: , 1212–1217. |
[53] | Sarell CJ , Syme CD , Rigby SEJ , Viles JH ((2009) ) Copper (II) binding to amyloid-β fibrils of Alzheimer’s disease reveals a picomolar affinity: Stoichiometry and coordination geometry are independent of Aβ oligomeric form. Biochemistry 48: , 4388–4402. |
[54] | Atwood CS , Moir RD , Huang X , Scarpa RC , Bacarra NME , Romano DM , Hartshorn MA , Tanzi RE , Bush AI ((1998) ) Dramatic aggregation of Alzheimer Aβ by Cu (II) is induced by conditions representing physiological acidosis. J Biol Chem 273: , 12817–12826. |
[55] | Atwood CS , Scarpa RC , Huang X , Moir RD , Jones WD , Fairlie DP , Tanzi RE , Bush AI ((2000) ) Characterization of dopper interactions with Alzheimer amyloid β peptides: Identification of an attomolar-affinity copper binding site on amyloid β1-42. J Neurochem 75: , 1219–1233. |
[56] | La Penna G , Hureau C , Andreussi O , Faller P ((2013) ) Identifying, by first-principles simulations, Cu [amyloid-β] species making fenton-type reactions in Alzheimer’s disease. J Phys Chem B 117: , 16455–16467. |
[57] | Barnham KJ , Haeffner F , Ciccotosto GD , Curtain CC , Tew D , Mavros C , Beyreuther K , Carrington D , Masters CL , Cherny RA , Cappai R , Bush AI ((2004) ) Tyrosine gated electron transfer is key to the toxic mechanism of Alzheimer’s disease β-amyloid. FASEB J 18: , 1427–1429. |
[58] | Kok WM , Cottam JM , Ciccotosto GD , Miles LA , Karas JA , Scanlon DB , Roberts BR , Parker MW , Cappai R , Barnham KJ , Hutton CA ((2013) ) Synthetic dityrosine-linked β-amyloid dimers form stable, soluble, neurotoxic oligomers. Chem Sci 4: , 4449–4454. |
[59] | O’Malley TT , Oktaviani NA , Zhang D , Lomakin A , O’Nuallain B , Linse S , Benedek GB , Rowan MJ , Mulder FAA , Walsh DM ((2014) ) Aβ dimers differ from monomers in structural propensity, aggregation paths and population of synaptotoxic assemblies. Biochem J 461: , 413–426. |
[60] | Mukherjee S , Kapp EA , Lothian A , Roberts AM , Vasil’ev YV , Boughton BA , Barnham KJ , Kok WM , Hutton CA , Masters CL , Bush AI , Beckman JS , Dey SG , Roberts BR ((2017) ) Characterization and identification of dityrosine cross-linked peptides using tandem mass spectrometry. Anal Chem 89: , 6136–6145. |
[61] | Sarell CJ , Wilkinson SR , Viles JH ((2010) ) Substoichiometric levels of Cu2+ ions accelerate the kinetics of fiber formation and promote cell toxicity of amyloid-β from Alzheimer disease. J Biol Chem 285: , 41533–41540. |
[62] | Matheou CJ , Younan ND , Viles JH ((2015) ) Cu2+ accentuates distinct misfolding of Aβ (1-40) and Aβ (1-42) peptides, and potentiates membrane disruption. Biochem J 466: , 233–242. |
[63] | Näslund J , Schierhorn A , Hellman U , Lannfelt L , Roses AD , Tjernberg LO , Silberring J , Gandy SE , Winblad B , Greengard P ((1994) ) Relative abundance of Alzheimer A beta amyloid peptide variants in Alzheimer disease and normal aging. Proc Natl Acad Sci U S A 91: , 8378–8382. |
[64] | Kuo YM , Kokjohn TA , Beach TG , Sue LI , Brune D , Lopez JC , Kalback WM , Abramowski D , Sturchler-Pierrat C , Staufenbiel M , Roher AE ((2001) ) Comparative analysis of amyloid-β chemical structure and amyloid plaque morphology of transgenic mouse and Alzheimer’s disease brains. J Biol Chem 276: , 12991–12998. |
[65] | Sharov VS , Schöneich C ((2000) ) Diastereoselective protein methionine oxidation by reactive oxygen species and diastereoselective repair by methionine sulfoxide reductase. Free Radic Biol Med 29: , 986–994. |
[66] | Gabbita SP , Aksenov MY , Lovell MA , Markesbery WR ((1999) ) Decrease in peptide methionine sulfoxide reductase in Alzheimer’s disease brain. J Neurochem 73: , 1660–1666. |
[67] | Moskovitz J , Du F , Bowman CF , Yan SS ((2016) ) Methionine sulfoxide reductase A affects β-amyloid solubility and mitochondrial function in a mouse model of Alzheimer’s disease. Am J Physiol Metab 310: , E388–E393. |
[68] | Misiti F , Clementi ME , Giardina B ((2010) ) Oxidation of methionine 35 reduces toxicity of the amyloid beta-peptide (1-42) in neuroblastoma cells (IMR-32) via enzyme methionine sulfoxide reductase A expression and function. Neurochem Int 56: , 597–602. |
[69] | Friedemann M , Helk E , Tiiman A , Zovo K , Palumaa P , Tõugu V ((2015) ) Effect of methionine-35 oxidation on the aggregation of amyloid-β peptide. Biochem Biophys Rep 3: , 94–99. |
[70] | Gu M , Viles JH ((2016) ) Methionine oxidation reduces lag-times for amyloid-β (1-40) fiber formation but generates highly fragmented fibers. Biochim Biophys Acta 1864: , 1260–1269. |
[71] | Hou L , Kang I , Marchant RE , Zagorski MG ((2002) ) Methionine 35 oxidation reduces fibril assembly of the amyloid Aβ-(1–42) peptide of Alzheimer’s disease. J Biol Chem 277: , 40173–40176. |
[72] | Palmblad M , Westlind-Danielsson A , Bergquist J ((2002) ) Oxidation of methionine 35 attenuates formation of amyloid β-peptide 1-40 oligomers. J Biol Chem 277: , 19506–19510. |
[73] | Anantharamaiah GM , Hughes TA , Iqbal M , Gawish A , Neame PJ , Medley MF , Segrest JP ((1988) ) Effect of oxidation on the properties of apolipoproteins AI and A-II. J Lipid Res 29: , 309–318. |
[74] | Hardas SS , Sultana R , Clark AM , Beckett TL , Szweda LI , Murphy MP , Butterfield DA ((2013) ) Oxidative modification of lipoic acid by HNE in Alzheimer disease brain. Redox Biol 1: , 80–85. |
[75] | Zhu X , Castellani RJ , Moreira PI , Aliev G , Shenk JC , Siedlak SL , Harris PLR , Fujioka H , Sayre LM , Szweda PA , Szweda LI , Smith MA , Perry G ((2012) ) Hydroxynonenal-generated crosslinking fluorophore accumulation in Alzheimer disease reveals a dichotomy of protein turnover. Free Radic Biol Med 52: , 699–704. |
[76] | Williams TI , Lynn BC , Markesbery WR , Lovell MA ((2006) ) Increased levels of 4-hydroxynonenal and acrolein, neurotoxic markers of lipid peroxidation, in the brain in mild cognitive impairment and early Alzheimer’s disease. Neurobiol Aging 27: , 1094–1099. |
[77] | Esterbauer H , Benedetti A , Lang J , Fulceri R , Fauler G , Comporti M ((1986) ) Studies on the mechanism of formation of 4-hydroxynonenal during microsomal lipid peroxidation. Biochim Biophys Acta 876: , 154–166. |
[78] | Pryor WA , Porter NA ((1990) ) Suggested mechanisms for the production of 4-hydroxy-2-nonenal from the autoxidation of polyunsaturated fatty acids. Free Radic Biol Med 8: , 541–543. |
[79] | Murray IVJ , Liu L , Komatsu H , Uryu K , Xiao G , Lawson JA , Axelsen PH ((2007) ) Membrane-mediated amyloidogenesis and the promotion of oxidative lipid damage by amyloid β proteins. J Biol Chem 282: , 9335–9345. |
[80] | Tamagno E , Guglielmotto M , Aragno M , Borghi R , Autelli R , Giliberto L , Muraca G , Danni O , Zhu X , Smith MA , Perry G , Jo DG , Mattson MP , Tabaton M ((2008) ) Oxidative stress activates a positive feedback between the γ-and β-secretase cleavages of the β-amyloid precursor protein. J Neurochem 104: , 683–695. |
[81] | Tamagno E , Bardini P , Obbili A , Vitali A , Borghi R , Zaccheo D , Pronzato MA , Danni O , Smith MA , Perry G , Jo DG , Mattson MP , Tabaton M ((2002) ) Oxidative stress increases expression and activity of BACE in NT2 neurons. Neurobiol Dis 10: , 279–288. |
[82] | Ellis G , Fang E , Maheshwari M , Roltsch E , Holcomb L , Zimmer D , Martinez D , Murray IVJ ((2010) ) Lipid oxidation and modification of amyloid-β (Aβ) in vitro and in vivo. J Alzheimers Dis 22: , 593–607. |
[83] | Maheshwari M , Roberts JK , DeSutter B , Duong KT , Tingling J , Fawver JN , Schall HE , Kahle M , Murray IVJ ((2010) ) Hydralazine modifies Aβ fibril formation and prevents modification by lipids in vitro. Biochemistry 49: , 10371–10380. |
[84] | Montine KS , Olson SJ , Amarnath V , Whetsell WO Jr , Graham DG , Montine TJ ((1997) ) Immunohistochemical detection of 4-hydroxy-2-nonenal adducts in Alzheimer’s disease is associated with inheritance of APOE4. Am J Pathol 150: , 437. |
[85] | Ando Y , Brännström T , Uchida K , Nyhlin N , Näsman B , Suhr O , Yamashita T , Olsson T , Salhy MEL , Uchino M , Ando M ((1998) ) Histochemical detection of 4-hydroxynonenal protein in Alzheimer amyloid. J Neurol Sci 156: , 172–176. |
[86] | Castro JP , Jung T , Grune T , Siems W ((2017) ) 4-Hydroxynonenal (HNE) modified proteins in metabolic diseases. Free Radic Biol Med 111: , 309–315. |
[87] | Atamna H , Frey II WH ((2007) ) Mechanisms of mitochondrial dysfunction and energy deficiency in Alzheimer’s disease. Mitochondrion 7: , 297–310. |
[88] | Brown KR , Allan BM , Do P , Hegg EL ((2002) ) Identification of novel hemes generated by heme A synthase: Evidence for two successive monooxygenase reactions. Biochemistry 41: , 10906–10913. |
[89] | Wielburski A , Nelson BD ((1983) ) Evidence for the sequential assembly of cytochrome oxidase subunits in rat liver mitochondria. Biochem J 212: , 829–834. |
[90] | Atamna H , Frey WH ((2004) ) A role for heme in Alzheimer’s disease: Heme binds amyloid β and has altered metabolism. Proc Natl Acad Sci U S A 101: , 11153–11158. |
[91] | Howlett D , Cutler P , Heales S , Camilleri P ((1997) ) Hemin and related porphyrins inhibit β-amyloid aggregation. FEBS Lett 417: , 249–251. |
[92] | Sankar SB , Donegan RK , Shah KJ , Reddi AR , Wood LB ((2018) ) Heme and hemoglobin suppress amyloid β–mediated inflammatory activation of mouse astrocytes. J Biol Chem 293: , 11358–11373. |
[93] | Atwood CS , Perry G , Zeng H , Kato Y , Jones WD , Ling KQ , Huang X , Moir RD , Wang D , Sayre LM , Smith MA , Chen SG , Bush AI ((2004) ) Copper mediates dityrosine cross-linking of Alzheimer’s amyloid-β. Biochemistry 43: , 560–568. |
[94] | Lu N , Li J , Tian R , Peng YY ((2014) ) Key roles of Arg5, Tyr10 and His residues in Aβ-heme peroxidase: Relevance to Alzheimer’s disease. Biochem Biophys Res Commun 452: , 676–681. |
[95] | Thiabaud G , Pizzocaro S , Garcia-Serres R , Latour JM , Monzani E , Casella L ((2013) ) Heme binding induces dimerization and nitration of truncated β-amyloid peptide Aβ16 under oxidative stress. Angew Chemie Int Ed 52: , 8041–8044. |
[96] | Zhou Y , Wang J , Liu L , Wang R , Lai X , Xu M ((2013) ) Interaction between amyloid-β peptide and heme probed by electrochemistry and atomic force microscopy. ACS Chem Neurosci 4: , 535–539. |
[97] | Atamna H , Boyle K ((2006) ) Amyloid-β peptide binds with heme to form a peroxidase: Relationship to the cytopathologies of Alzheimer’s disease. Proc Natl Acad Sci U S A 103: , 3381–3386. |
[98] | Pramanik D , Ghosh C , Dey SG ((2011) ) Heme-Cu bound Aβ peptides: Spectroscopic characterization, reactivity, and relevance to Alzheimer’s disease. J Am Chem Soc 133: , 15545–15552. |
[99] | Chiziane E , Telemann H , Krueger M , Adler J , Arnhold J , Alia A , Flemmig J ((2018) ) Free heme and amyloid-β: A fatal liaison in Alzheimer’s disease. J Alzheimers Dis 61: , 963–984. |
[100] | Smith MA , Hirai K , Hsiao K , Pappolla MA , Harris PL , Siedlak SL , Tabaton M , Perry G ((1998) ) Amyloid-beta deposition in Alzheimer transgenic mice is associated with oxidative stress. J Neurochem 70: , 2212–2215. |
[101] | MacAllister RJ , Parry H , Kimoto M , Ogawa T , Russell RJ , Hodson H , Whitley GSJ , Vallance P ((1996) ) Regulation of nitric oxide synthesis by dimethylarginine dimethylaminohydrolase. Br J Pharmacol 119: , 1533–1540. |
[102] | Calingasan NY , Uchida K , Gibson GE ((1999) ) Protein-bound acrolein: A novel marker of oxidative stress in Alzheimer’s disease. J Neurochem 72: , 751–756. |
[103] | Jicha GA , Bowser R , Kazam IG , Davies P ((1997) ) Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res 48: , 128–132. |
[104] | Carmel G , Mager EM , Binder LI , Kuret J ((1996) ) The structural basis of monoclonal antibody Alz50’s selectivity for Alzheimer’s disease pathology. J Biol Chem 271: , 32789–32795. |
[105] | Barone E , Di Domenico F , Sultana R , Coccia R , Mancuso C , Perluigi M , Butterfield DA ((2012) ) Heme oxygenase-1 posttranslational modifications in the brain of subjects with Alzheimer disease and mild cognitive impairment. Free Radic Biol Med 52: , 2292–2301. |
[106] | Troncoso JC , Costello A , Watson AL Jr , Johnson GVW ((1993) ) In vitro polymerization of oxidized tau into filaments. Brain Res 613: , 313–316. |
[107] | Schweers O , Mandelkow EM , Biernat J , Mandelkow E ((1995) ) Oxidation of cysteine-322 in the repeat domain of microtubule-associated protein tau controls the in vitro assembly of paired helical filaments. Proc Natl Acad Sci U S A 92: , 8463–8467. |
[108] | Li X , Du X , Ni J ((2019) ) Zn2+ aggravates tau aggregation and neurotoxicity. Int J Mol Sci 20: , E487. |
[109] | Du X , West MB , Cheng W , Ewert DL , Li W , Saunders D , Towner RA , Floyd RA , Kopke RD ((2016) ) Ameliorative effects of antioxidants on the hippocampal accumulation of pathologic tau in a rat model of blast-induced traumatic brain injury. Oxid Med Cell Longev 2016: , 4159357. |
[110] | Vana L , Kanaan NM , Hakala K , Weintraub ST , Binder LI ((2011) ) Peroxynitrite-induced nitrative and oxidative modifications alter tau filament formation. Biochemistry 50: , 1203–1212. |
[111] | Santa-Maria I , Hernández F , Martín CP , Avila J , Moreno FJ ((2004) ) Quinones facilitate the self-assembly of the phosphorylated tubulin binding region of tau into fibrillar polymers. Biochemistry 43: , 2888–2897. |
[112] | Mattson MP , Fu W , Waeg G , Uchida K ((1997) ) 4-Hydroxynonenal, a product of lipid peroxidation, inhibits dephosphorylation of the microtubule-associated protein tau. Neuroreport 8: , 2275–2281. |
[113] | Vitek MP , Bhattacharya K , Glendening JM , Stopa E , Vlassara H , Bucala R , Manogue K , Cerami A , Selkoe DJ , Guénette S ((1994) ) Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc Natl Acad Sci U S A 91: , 4766–4770. |
[114] | Ko L , Ko EC , Nacharaju P , Liu WK , Chang E , Kenessey A , Yen SHC ((1999) ) An immunochemical study on tau glycation in paired helical filaments. Brain Res 830: , 301–313. |
[115] | Yan S Du , Yan SF , Chen X , Fu J , Chen M , Kuppusamy P , Smith MA , Perry G , Godman GC , Nawroth P , Zweier JL , Stern D ((1995) ) Non-enzymatically glycated tau in Alzheimer’s disease induces neuronal oxidant stress resulting in cytokine gene expression and release of amyloid β-peptide. Nat Med 1: , 693–699. |
[116] | Yan SD , Chen X , Schmidt AM , Brett J , Godman G , Zou YS , Scott CW , Caputo C , Frappier T , Smith MA ((1994) ) Glycated tau protein in Alzheimer disease: A mechanism for induction of oxidant stress. Proc Natl Acad Sci U S A 91: , 7787–7791. |
[117] | Lüth HJ , Ogunlade V , Kuhla B , Kientsch-Engel R , Stahl P , Webster J , Arendt T , Münch G ((2004) ) Age-and stage-dependent accumulation of advanced glycation end products in intracellular deposits in normal and Alzheimer’s disease brains. Cereb Cortex 15: , 211–220. |
[118] | Wong A , Lüth HJ , Deuther-Conrad W , Dukic-Stefanovic S , Gasic-Milenkovic J , Arendt T , Münch G ((2001) ) Advanced glycation endproducts co-localize with inducible nitric oxide synthase in Alzheimer’s disease. Brain Res 920: , 32–40. |
[119] | Baloyannis SJ ((2009) ) Dendritic pathology in Alzheimer’s disease. J Neurol Sci 283: , 153–157. |
[120] | Manczak M , Calkins MJ , Reddy PH ((2011) ) Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum Mol Genet 20: , 2495–2509. |
[121] | Gomes LC , Di Benedetto G , Scorrano L ((2011) ) During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 13: , 589–598. |
[122] | Twig G , Graf SA , Wikstrom JD , Mohamed H , Haigh SE , Elorza A , Deutsch M , Zurgil N , Reynolds N , Shirihai OS ((2006) ) Tagging and tracking individual networks within a complex mitochondrial web with photoactivatable GFP. Am J Physiol Physiol 291: , C176–C184. |
[123] | Brand CS , Tan VP , Brown JH , Miyamoto S ((2018) ) RhoA regulates Drp1 mediated mitochondrial fission through ROCK to protect cardiomyocytes. Cell Signal 50: , 48–57. |
[124] | Lee JY , Kapur M , Li M , Choi MC , Choi S , Kim HJ , Kim I , Lee E , Taylor JP , Yao TP ((2014) ) MFN1 deacetylation activates adaptive mitochondrial fusion and protects metabolically challenged mitochondria. J Cell Sci 127: , 4954–4963. |
[125] | Garcia I , Innis-Whitehouse W , Lopez A , Keniry M , Gilkerson R ((2018) ) Oxidative insults disrupt OPA1-mediated mitochondrial dynamics in cultured mammalian cells. Redox Rep 23: , 160–167. |
[126] | MacVicar TDB , Lane JD ((2014) ) Impaired OMA1-dependent cleavage of OPA1 and reduced DRP1 fission activity combine to prevent mitophagy in cells that are dependent on oxidative phosphorylation. J Cell Sci 127: , 2313–2325. |
[127] | Meister A , Tate SS ((1976) ) Glutathione and related γ-glutamyl compounds: Biosynthesis and utilization. Annu Rev Biochem 45: , 559–604. |
[128] | Tong J , Fitzmaurice PS , Moszczynska A , Mattina K , Ang LC , Boileau I , Furukawa Y , Sailasuta N , Kish SJ ((2016) ) Do glutathione levels decline in aging human brain? Free Radic Biol Med 93: , 110–117. |
[129] | Chai YC , Ashraf SS , Rokutan K , Johnston RB , Thomas JA ((1994) ) S-thiolation of individual human neutrophil proteins including actin by stimulation of the respiratory burst: Evidence against a role for glutathione disulfide. Arch Biochem Biophys 310: , 273–281. |
[130] | Bermejo P , Martín-Aragón S , Benedí J , Susín C , Felici E , Gil P , Manuel Ribera J , Villar ÁM ((2008) ) Peripheral levels of glutathione and protein oxidation as markers in the development of Alzheimer’s disease from mild cognitive impairment. Free Radic Res 42: , 162–170. |
[131] | Perry TL , Yong VW , Bergeron C , Hansen S , Jones K ((1987) ) Amino acids, glutathione, and glutathione transferase activity in the brains of patients with Alzheimer’s disease. Ann Neurol 21: , 331–336. |
[132] | Adams JD Jr , Klaidman LK , Odunze IN , Shen HC , Miller CA ((1991) ) Alzheimer’s and Parkinson’s disease. Brain levels of glutathione, glutathione disulfide, and vitamin E. Mol Chem Neuropathol 14: , 213–226. |
[133] | Calabrese V , Sultana R , Scapagnini G , Guagliano E , Sapienza M , Bella R , Kanski J , Pennisi G , Mancuso C , Stella AMG , Butterfield DA ((2006) ) Nitrosative stress, cellular stress response, and thiol homeostasis in patients with Alzheimer’s disease. Antioxid Redox Signal 8: , 1975–1986. |
[134] | Lloret A , Badia MC , Mora NJ , Pallardó FV , Alonso MD , Vina J ((2009) ) Vitamin E paradox in Alzheimer’s disease: It does not prevent loss of cognition and may even be detrimental. J Alzheimers Dis 17: , 143–149. |
[135] | Liu RM , Choi J ((2000) ) Age-associated decline in γ-glutamylcysteine synthetase gene expression in rats. Free Radic Biol Med 28: , 566–574. |
[136] | Kang Y , Qiao X , Jurma OP , Knusel B , Andersen JK ((1997) ) Cloning of cDNA encoding the mouse glutamyl cysteine synthetase heavy subunit and localization of expression in the brain. Neuroreport 8: , 2053–2060. |
[137] | Kang Y , Viswanath V , Jha N , Qiao X , Mo JQ , Andersen JK ((1999) ) Brain γ-glutamyl cysteine synthetase (GCS) mRNA expression patterns correlate with regional-specific enzyme activities and glutathione levels. J Neurosci Res 58: , 436–441. |
[138] | Hardeland R ((2005) ) Antioxidative protection by melatonin. Endocrine 27: , 119–130. |
[139] | Bucana CD , Nadakavukaren MJ , Frehn JL ((1974) ) Novel features of hamster pinealocyte ultrastructure. Tissue Cell 6: , 85–93. |
[140] | Calvo J , Boya J ((1984) ) Ultrastructure of the pineal gland in the adult rat. J Anat 138: , 405–409. |
[141] | Manchester LC , Coto-Montes A , Boga JA , Andersen LPH , Zhou Z , Galano A , Vriend J , Tan DX , Reiter RJ ((2015) ) Melatonin: An ancient molecule that makes oxygen metabolically tolerable. J Pineal Res 59: , 403–419. |
[142] | Reiter RJ , Mayo JC , Tan DX , Sainz RM , Alatorre-Jimenez M , Qin L ((2016) ) Melatonin as an antioxidant: Under promises but over delivers. J Pineal Res 61: , 253–278. |
[143] | Rodriguez C , Mayo JC , Sainz RM , Antolín I , Herrera F , Martín V , Reiter RJ ((2004) ) Regulation of antioxidant enzymes: A significant role for melatonin. J Pineal Res 36: , 1–9. |
[144] | Acuna Castroviejo D , Lopez LC , Escames G , López A , Garcia JA , Reiter RJ ((2011) ) Melatonin-mitochondria interplay in health and disease. Curr Top Med Chem 11: , 221–240. |
[145] | Teixeira A , Morfim MP , de Cordova CAS , Charão CCT , de Lima VR , Creczynski-Pasa TB ((2003) ) Melatonin protects against pro-oxidant enzymes and reduces lipid peroxidation in distinct membranes induced by the hydroxyl and ascorbyl radicals and by peroxynitrite. J Pineal Res 35: , 262–268. |
[146] | Liu RY , Zhou JN , van Heerikhuize J , Hofman MA , Swaab DF ((1999) ) Decreased melatonin levels in postmortem cerebrospinal fluid in relation to aging, Alzheimer’s disease, and apolipoprotein E-epsilon4/4 genotype. J Clin Endocrinol Metab 84: , 323–327. |
[147] | Kraft AD , Johnson DA , Johnson JA ((2004) ) Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. J Neurosci 24: , 1101–1112. |
[148] | Shih AY , Johnson DA , Wong G , Kraft AD , Jiang L , Erb H , Johnson JA , Murphy TH ((2003) ) Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J Neurosci 23: , 3394–3406. |
[149] | Ahlgren-Beckendorf JA , Reising AM , Schander MA , Herdler JW , Johnson JA ((1999) ) Coordinate regulation of NAD (P) H: Quinone oxidoreductase and glutathione-S-transferases in primary cultures of rat neurons and glia: Role of the antioxidant/electrophile responsive element. Glia 25: , 131–142. |
[150] | Rushmore TH , Morton MR , Pickett CB ((1991) ) The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J Biol Chem 266: , 11632–11639. |
[151] | Hayes JD , McMahon M , Chowdhry S , Dinkova-Kostova AT ((2010) ) Cancer chemoprevention mechanisms mediated through the Keap1-Nrf2 pathway. Antioxid Redox Signal 13: , 1713–1748. |
[152] | Suzuki T , Motohashi H , Yamamoto M ((2013) ) Toward clinical application of the Keap1-Nrf2 pathway. Trends Pharmacol Sci 34: , 340–346. |
[153] | Itoh K , Chiba T , Takahashi S , Ishii T , Igarashi K , Katoh Y , Oyake T , Hayashi N , Satoh K , Hatayama I , Yamamoto M , Nabeshima Y ((1997) ) An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun 236: , 313–322. |
[154] | Itoh K , Wakabayashi N , Katoh Y , Ishii T , Igarashi K , Engel JD , Yamamoto M ((1999) ) Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev 13: , 76–86. |
[155] | Rahman MM , Sykiotis GP , Nishimura M , Bodmer R , Bohmann D ((2013) ) Declining signal dependence of N rf2-M af S-regulated gene expression correlates with aging phenotypes. Aging Cell 12: , 554–562. |
[156] | Suh JH , Shenvi SV , Dixon BM , Liu H , Jaiswal AK , Liu RM , Hagen TM ((2004) ) Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc Natl Acad Sci U S A 101: , 3381–3386. |
[157] | von Otter M , Landgren S , Nilsson S , Zetterberg M , Celojevic D , Bergström P , Minthon L , Bogdanovic N , Andreasen N , Gustafson DR , Skoog I , Wallin A , Tasa G , Blennow K , Nilsson M , Hammarsten O , Zetterberg H ((2010) ) Nrf2-encoding NFE2L2 haplotypes influence disease progression but not risk in Alzheimer’s disease and age-related cataract. Mech Ageing Dev 131: , 105–110. |
[158] | Kanninen K , Heikkinen R , Malm T , Rolova T , Kuhmonen S , Leinonen H , Ylä-Herttuala S , Tanila H , Levonen AL , Koistinaho M , Koistanaho J ((2009) ) Intrahippocampal injection of a lentiviral vector expressing Nrf2 improves spatial learning in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 106: , 16505–16510. |
[159] | Joshi G , Gan KA , Johnson DA , Johnson JA ((2015) ) Increased Alzheimer’s disease-like pathology in the APP/PS1ΔE9 mouse model lacking Nrf2 through modulation of autophagy. Neurobiol Aging 36: , 664–679. |
[160] | Rojo AI , Pajares M , Rada P , Nuñez A , Nevado-Holgado AJ , Killik R , Van Leuven F , Ribe E , Lovestone S , Yamamoto M , Cuadrado A ((2017) ) NRF2 deficiency replicates transcriptomic changes in Alzheimer’s patients and worsens APP and TAU pathology. Redox Biol 13: , 444–451. |
[161] | Fragoulis A , Siegl S , Fendt M , Jansen S , Soppa U , Brandenburg LO , Pufe T , Weis J , Wruck CJ ((2017) ) Oral administration of methysticin improves cognitive deficits in a mouse model of Alzheimer’s disease. Redox Biol 12: , 843–853. |
[162] | Jimenez-Blasco D , Santofimia-Castaño P , Gonzalez A , Almeida A , Bolaños JP ((2015) ) Astrocyte NMDA receptors’ activity sustains neuronal survival through a Cdk5-Nrf2 pathway. Cell Death Differ 22: , 1877–1889. |
[163] | Bell KFS , Al-Mubarak B , Martel MA , McKay S , Wheelan N , Hasel P , Márkus NM , Baxter P , Deighton RF , Serio A , Bilican B , Chowdhry S , Meakin PJ , Ashford ML , Wyllie DJ , Scannevin RH , Chandran S , Hayes JD , Hardingham GE ((2015) ) Neuronal development is promoted by weakened intrinsic antioxidant defences due to epigenetic repression of Nrf2. Nat Commun 6: , 7066. |
[164] | Trombley PQ , Shepherd GM ((1996) ) Differential modulation by zinc and copper of amino acid receptors from rat olfactory bulb neurons. J Neurophysiol 76: , 2536–2346. |
[165] | Kardos J , Kovács I , Hajós F , Kálmán M , Simonyi M ((1989) ) Nerve endings from rat brain tissue release copper upon depolarization. A possible role in regulating neuronal excitability. Neurosci Lett 103: , 139–144. |
[166] | Muñoz P , Humeres A ((2012) ) Iron deficiency on neuronal function. Biometals 25: , 825–835. |
[167] | Gunshin H , Mackenzie B , Berger UV , Gunshin Y , Romero MF , Boron WF , Nussberger S , Gollan JL , Hediger MA ((1997) ) Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 388: , 482–488. |
[168] | Garrick MD , Singleton ST , Vargas F , Kuo HC , Zhao L , Knöpfel M , Davidson T , Costa M , Paradkar P , Roth JA , Garrick LM ((2006) ) DMT1: Which metals does it transport? Biol Res 39: , 79–85. |
[169] | Haeger P , Alvarez A , Leal N , Adasme T , Núnez MT , Hidalgo C ((2010) ) Increased hippocampal expression of the divalent metal transporter 1 (DMT1) mRNA variants 1B and+ IRE and DMT1 protein after NMDA-receptor stimulation or spatial memory training. Neurotox Res 17: , 238–247. |
[170] | Loeffler DA , Connor JR , Juneau PL , Snyder BS , Kanaley L , DeMaggio AJ , Nguyen H , Brickman CM , LeWitt PA ((1995) ) Transferrin and iron in normal, Alzheimer’s disease, and Parkinson’s disease brain regions. J Neurochem 65: , 710–716. |
[171] | Deibel MA , Ehmann WD , Markesbery WR ((1996) ) Copper, iron, and zinc imbalances in severely degenerated brain regions in Alzheimer’s disease: Possible relation to oxidative stress. J Neurol Sci 143: , 137–142. |
[172] | Squitti R , Lupoi D , Pasqualetti P , Dal Forno G , Vernieri F , Chiovenda P , Rossi L , Cortesi M , Cassetta E , Rossini PM ((2002) ) Elevation of serum copper levels in Alzheimer’s disease. Neurology 59: , 1153–1161. |
[173] | Smith MA , Zhu X , Tabaton M , Liu G , McKeel DW Jr , Cohen ML , Wang X , Siedlak SL , Dwyer BE , Hayashi T , Nakamura M , Nunomura A , Perry G ((2010) ) Increased iron and free radical generation in preclinical Alzheimer disease and mild cognitive impairment. J Alzheimers Dis 19: , 363–372. |
[174] | Bush AI , Pettingell WH , Multhaup G , d Paradis M , Vonsattel JP , Gusella JF , Beyreuther K , Masters CL , Tanzi RE ((1994) ) Rapid induction of Alzheimer A beta amyloid formation by zinc. Science 265: , 1464–1467. |
[175] | Kuchibhotla KV , Goldman ST , Lattarulo CR , Wu HY , Hyman BT , Bacskai BJ ((2008) ) Aβ plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron 59: , 214–225. |
[176] | Yan SD , Chen X , Fu J , Chen M , Zhu H , Roher A , Slattery T , Zhao L , Nagashima M , Morser J , Migheli A , Nawroth P , Stern D , Schmidt AM ((1996) ) RAGE and amyloid-β peptide neurotoxicity in Alzheimer’s disease. Nature 382: , 685–691. |
[177] | Kawakami K , Scheidereit C , Roeder RG ((1988) ) Identification and purification of a human immunoglobulin-enhancer-binding protein (NF-kappa B) that activates transcription from a human immunodeficiency virus type 1 promoter in vitro. Proc Natl Acad Sci U S A 85: , 4700–4704. |
[178] | Lenardo MJ , Kuang A , Gifford A , Baltimore D ((1988) ) NF-kappa B protein purification from bovine spleen: Nucleotide stimulation and binding site specificity. Proc Natl Acad Sci U S A 85: , 8825–8829. |
[179] | Baeuerle PA , Baltimore D ((1988) ) Activation of DNA-binding activity in an apparently cytoplasmic precursor of the NF-κB transcription factor. Cell 53: , 211–217. |
[180] | Toledano MB , Leonard WJ ((1991) ) Modulation of transcription factor NF-kappa B binding activity by oxidation-reduction in vitro. Proc Natl Acad Sci U S A 88: , 4328–4332. |
[181] | Schreck R , Rieber P , Baeuerle PA ((1991) ) Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J 10: , 2247–2258. |
[182] | Zhong H , SuYang H , Erdjument-Bromage H , Tempst P , Ghosh S ((1997) ) The transcriptional activity of NF-κB is regulated by the IκB-associated PKAc subunit through a cyclic AMP–independent mechanism. Cell 89: , 413–424. |
[183] | Du Yan S , Fu J , Soto C , Chen X , Zhu H , Al-Mohanna F , Collison K , Zhu A , Stern E , Saido T , Tohyama M , Ogawa S , Roher A , Stern D ((1997) ) An intracellular protein that binds amyloid-β peptide and mediates neurotoxicity in Alzheimer’s disease. Nature 389: , 689–695. |
[184] | Di Domenico F , Sultana R , Tiu GF , Scheff NN , Perluigi M , Cini C , Butterfield DA ((2010) ) Protein levels of heat shock proteins 27, 32, 60, 70, 90 and thioredoxin-1 in amnestic mild cognitive impairment: An investigation on the role of cellular stress response in the progression of Alzheimer disease. Brain Res 1333: , 72–81. |
[185] | Mota SI , Costa RO , Ferreira IL , Santana I , Caldeira GL , Padovano C , Fonseca AC , Baldeiras I , Cunha C , Letra L , Oliveira CR , Pereira CM , Rego AC ((2015) ) Oxidative stress involving changes in Nrf2 and ER stress in early stages of Alzheimer’s disease. Biochim Biophys Acta 1852: , 1428–1441. |
[186] | Mecocci P , Polidori MC , Ingegni T , Cherubini A , Chionne F , Cecchetti R , Senin U ((1998) ) Oxidative damage to DNA in lymphocytes from AD patients. Neurology 51: , 1014–1017. |
[187] | Te Koppele JM , Lucassen PJ , Sakkee AN , Van Asten JG , Ravid R , Swaab DF , Van Bezooijen CFA ((1996) ) 8OHdG levels in brain do not indicate oxidative DNA damage in Alzheimer’s disease. Neurobiol Aging 17: , 819–826. |
[188] | Ansari MA , Scheff SW ((2010) ) Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J Neuropathol Exp Neurol 69: , 155–167. |
[189] | Markesbery WR , Kryscio RJ , Lovell MA , Morrow JD ((2005) ) Lipid peroxidation is an early event in the brain in amnestic mild cognitive impairment. Ann Neurol 58: , 730–735. |
[190] | Boström K , Hassler O ((1966) ) A pilot microradiological investigation on the occurrence of calcifications in various arteries of the human body. J Gerontol 21: , 97–102. |
[191] | Bell MA , Ball MJ ((1981) ) Morphometric comparison of hippocampal microvasculature in ageing and demented people: Diameters and densities. Acta Neuropathol 53: , 299–318. |
[192] | Mancardi GL , Perdelli F , Rivano C , Leonardi A , Bugiani O ((1980) ) Thickening of the basement membrane of cortical capillaries in Alzheimer’s disease. Acta Neuropathol 49: , 79–83. |
[193] | Perry G , Smith MA , McCann CE , Siedlak SL , Jones PK , Friedland RP ((1998) ) Cerebrovascular muscle atrophy is a feature of Alzheimer’s disease. Brain Res 791: , 63–66. |
[194] | Li Y , Choi WJ , Wei W , Song S , Zhang Q , Liu J , Wang RK ((2018) ) Aging-associated changes in cerebral vasculature and blood flow as determined by quantitative optical coherence tomography angiography. Neurobiol Aging 70: , 148–159. |
[195] | Gutierrez-Jimenez E , Angleys H , Rasmussen PM , West MJ , Catalini L , Iversen NK , Jensen MS , Frische S , Østergaard L ((2018) ) Disturbances in the control of capillary flow in an aged APPswe/PS1ΔE9 model of Alzheimer’s disease. Neurobiol Aging 62: , 82–94. |
[196] | Nagata K , Kondoh Y , Atchison R , Sato M , Satoh Y , Watahiki Y , Hirata Y , Yokoyama E ((2000) ) Vascular and metabolic reserve in Alzheimer’s disease. Neurobiol Aging 21: , 301–307. |
[197] | Lustbader JW , Cirilli M , Lin C , Xu HW , Takuma K , Wang N , Caspersen C , Chen X , Pollak S , Chaney M , Trinchese F , Liu S , Gunn-Moore F , Lue LF , Walker DG , Kuppusamy P , Zewier ZL , Arancio O , Stern D , Yan SS , Wu H ((2004) ) ABAD directly links Aβ to mitochondrial toxicity in Alzheimer’s disease. Science 304: , 448–452. |
[198] | Snyder B , Shell B , Cunningham JT , Cunningham RL ((2017) ) Chronic intermittent hypoxia induces oxidative stress and inflammation in brain regions associated with early-stage neurodegeneration. Physiol Rep 5: , e13258. |
[199] | Zhang J , Piantadosi CA ((1992) ) Mitochondrial oxidative stress after carbon monoxide hypoxia in the rat brain. J Clin Invest 90: , 1193–1199. |
[200] | Katz LM , Young AS , Frank JE , Wang Y , Park K ((2004) ) Regulated hypothermia reduces brain oxidative stress after hypoxic-ischemia. Brain Res 1017: , 85–91. |
[201] | Troussière AC , Charley CM , Salleron J , Richard F , Delbeuck X , Derambure P , Pasquier F , Bombois S ((2014) ) Treatment of sleep apnoea syndrome decreases cognitive decline in patients with Alzheimer’s disease. J Neurol Neurosurg Psychiatry 85: , 1405–1408. |
[202] | Young T , Palta M , Dempsey J , Skatrud J , Weber S , Badr S ((1993) ) The occurrence of sleep-disordered breathing among middle-aged adults. N Engl J Med 328: , 1230–1235. |
[203] | Durán J , Esnaola S , Rubio R , Iztueta Á ((2001) ) Obstructive sleep apnea-hypopnea and related clinical features in a population-based sample of subjects aged 30 to 70 yr. Am J Respir Crit Care Med 163: , 685–689. |
[204] | Row BW , Liu R , Xu W , Kheirandish L , Gozal D ((2003) ) Intermittent hypoxia is associated with oxidative stress and spatial learning deficits in the rat. Am J Respir Crit Care Med 167: , 1548–1553. |
[205] | Wang GL , Jiang BH , Rue EA , Semenza GL ((1995) ) Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A 92: , 5510–5514. |
[206] | Maxwell PH , Wiesener MS , Chang GW , Clifford SC , Vaux EC , Cockman ME , Wykoff CC , Pugh CW , Maher ER , Ratcliffe PJ ((1999) ) The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399: , 271–275. |
[207] | Bruick RK , McKnight SL ((2001) ) A conserved family of prolyl-4-hydroxylases that modify HIF. Science 294: , 1337–1340. |
[208] | Epstein ACR , Gleadle JM , McNeill LA , Hewitson KS , O’Rourke J , Mole DR , Mukherji M , Metzen E , Wilson MI , Dhanda A , Tian YM , Masson N , Hamilton DL , Jaakkola P , Barstead R , Hodgkin J , Maxwell PH , Pugh CW , Schofield CJ , Ratcliffe PJ ((2001) ) C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107: , 43–54. |
[209] | Ivan M , Kondo K , Yang H , Kim W , Valiando J , Ohh M , Salic A , Asara JM , Lane WS , Kaelin WG Jr ((2001) ) HIFα targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 292: , 464–468. |
[210] | Jaakkola P , Mole DR , Tian YM , Wilson MI , Gielbert J , Gaskell SJ , von Kriegsheim A , Hebestreit HF , Mukherji M , Schofield CJ , Maxwell PH , Pugh CW , Ratcliffe PJ ((2001) ) Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292: , 468–472. |
[211] | Bell EL , Emerling BM , Chandel NS ((2005) ) Mitochondrial regulation of oxygen sensing. Mitochondrion 5: , 322–332. |
[212] | Bell EL , Klimova TA , Eisenbart J , Moraes CT , Murphy MP , Budinger GRS , Chandel NS ((2007) ) The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J Cell Biol 177: , 1029–1036. |
[213] | Bergeron M , Yu AY , Solway KE , Semenza GL , Sharp FR ((1999) ) Induction of hypoxia-inducible factor-1 (HIF-1) and its target genes following focal ischaemia in rat brain. Eur J Neurosci 11: , 4159–4170. |
[214] | West MD , Pereira-Smith OM , Smith JR ((1989) ) Replicative senescence of human skin fibroblasts correlates with a loss of regulation and overexpression of collagenase activity. Exp Cell Res 184: , 138–147. |
[215] | Ogawa S , Ishibashi Y , Sakamoto Y , Kitamura K , Kubo M , Sakai T , Inoue K ((1997) ) The glycoproteins that occur in the colloids of senescent porcine pituitary glands are clusterin and glycosylated albumin fragments. Biochem Biophys Res Commun 234: , 712–718. |
[216] | Weinstein G , Beiser AS , Preis SR , Courchesne P , Chouraki V , Levy D , Seshadri S ((2016) ) Plasma clusterin levels and risk of dementia, Alzheimer’s disease, and stroke. Alzheimers Dement (Amst) 3: , 103–109. |
[217] | Lambert JC , Heath S , Even G , Campion D , Sleegers K , Hiltunen M , Combarros O , Zelenika D , Bullido MJ , Tavernier B , Letenneur L , Bettens K , Berr C , Pasquier F , Fiévet N , Barberger-Gateau P , Engelborghs S , De Deyn P , Mateo I , Franck A , Helisalmi S , Porcellini E , Hanon O , de Pancorbo MM , Lendon C , Dufouil C , Jaillard C , Leveillard T , Alvarez V , Bosco P , Mancuso M , Panza F , Nacmias B , Bossú P , Piccardi P , Annoni G , Seripa D , Galimberti D , Hannequin D , Licastro F , Soininen H , Ritchie K , Blanché H , Dartigues JF , Tzourio C , Gut I , Van Broeckhoven C , Alpérovitch A , Lathrop M , Amouyel P , Amouyel P ((2009) ) Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet 41: , 1094–1099. |
[218] | Thambisetty M , Simmons A , Velayudhan L , Hye A , Campbell J , Zhang Y , Wahlund LO , Westman E , Kinsey A , Güntert A , Priotsi P , Powell J , Causevic M , Killick R , Lunnon K , Lynham S , Broadstock M , Choudhry F , Howlett DR , Williams RJ , Sharp SI , Mitchelmore C , Tunnard C , Leung R , Foy C , O’Brien D , Breen G , Furney SJ , Ward M , Kloszeska I , Mecocci P , Soininen H , Tsolaki M , Vellas B , Hodges A , Murphy DG , Parkins S , Richardson JC , Resnick SM , Ferrucci L , Wong DF , Zhou Y , Muehlboeck S , Evans A , Francis PT , Spenger C , Lovestone S ((2010) ) Association of plasma clusterin concentration with severity, pathology, and progression in Alzheimer disease. Arch Gen Psychiatry 67: , 739–748. |
[219] | Thambisetty M , An Y , Kinsey A , Koka D , Saleem M , Güntert A , Kraut M , Ferrucci L , Davatzikos C , Lovestone S , Resnick SM ((2012) ) Plasma clusterin concentration is associated with longitudinal brain atrophy in mild cognitive impairment. Neuroimage 59: , 212–217. |
[220] | Michel D , Chatelain G , Herault Y , Brun G ((1995) ) The expression of the avian clusterin gene can be driven by two alternative promoters with distinct regulatory elements. Eur J Biochem 229: , 215–223. |
[221] | Jun HO , Kim D , Lee SW , Lee HS , Seo JH , Kim JH , Kim JH , Yu YS , Min BH , Kim KW ((2011) ) Clusterin protects H9c2 cardiomyocytes from oxidative stress-induced apoptosis via Akt/GSK-3β signaling pathway. Exp Mol Med 43: , 53–61. |
[222] | Zlokovic BV , Martel CL , Matsubara E , McComb JG , Zheng G , McCluskey RT , Frangione B , Ghiso J ((1996) ) Glycoprotein 330/megalin: Probable role in receptor-mediated transport of apolipoprotein J alone and in a complex with Alzheimer disease amyloid beta at the blood-brain and blood-cerebrospinal fluid barriers. Proc Natl Acad Sci U S A 93: , 4229–4234. |
[223] | Eferl R , Wagner EF ((2003) ) AP-1: A double-edged sword in tumorigenesis. Nat Rev Cancer 3: , 859–868. |
[224] | Kwon HS , Kim TB , Lee YS , Jeong SH , Bae YJ , Moon KA , Bang BR , Moon HB , Cho YS ((2014) ) Clusterin expression level correlates with increased oxidative stress in asthmatics. Ann Allergy Asthma Immunol 112: , 217–221. |
[225] | Sol IS , Kim YH , Lee KE , Hong JY , Kim MN , Kim YS , Oh MS , Kim MJ , Yoon SH , Park YA , Kim KW , Sohn MH , Kim KE ((2016) ) Serum clusterin level in children with atopic dermatitis. Allergy Asthma Proc 37: , 335–339. |
[226] | Trougakos IP , Poulakou M , Stathatos M , Chalikia A , Melidonis A , Gonos ES ((2002) ) Serum levels of the senescence biomarker clusterin/apolipoprotein J increase significantly in diabetes type II and during development of coronary heart disease or at myocardial infarction. Exp Gerontol 37: , 1175–1187. |
[227] | Trougakos IP , So A , Jansen B , Gleave ME , Gonos ES ((2004) ) Silencing expression of the clusterin/apolipoprotein j gene in human cancer cells using small interfering RNA induces spontaneous apoptosis, reduced growth ability, and cell sensitization to genotoxic and oxidative stress. Cancer Res 64: , 1834–1842. |
[228] | Chen QM , Prowse KR , Tu VC , Purdom S , Linskens MHK ((2001) ) Uncoupling the senescent phenotype from telomere shortening in hydrogen peroxide-treated fibroblasts. Exp Cell Res 265: , 294–303. |
[229] | Strocchi P , Smith MA , Perry G , Tamagno E , Danni O , Pession A , Gaiba A , Dozza B ((2006) ) Clusterin up-regulation following sub-lethal oxidative stress and lipid peroxidation in human neuroblastoma cells. Neurobiol Aging 27: , 1588–1594. |
[230] | Rohne P , Prochnow H , Wolf S , Renner B , Koch-Brandt C ((2014) ) The chaperone activity of clusterin is dependent on glycosylation and redox environment. Cell Physiol Biochem 34: , 1626–1639. |
[231] | Shin YJ , Kim JH , Seo JM , Lee SM , Hyon JY , Yu YS , Wee WR ((2009) ) Protective effect of clusterin on oxidative stress-induced cell death of human corneal endothelial cells. Mol Vis 15: , 2789–2795. |
[232] | Lee YN , Shim YJ , Kang BH , Park JJ , Min BH ((2012) ) Over-expression of human clusterin increases stress resistance and extends lifespan in Drosophila melanogaster. Biochem Biophys Res Commun 420: , 851–856. |
[233] | Wang X , Wang W , Li L , Perry G , Lee H , Zhu X ((2014) ) Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim Biophys Acta 1842: , 1240–1247. |
[234] | Trougakos IP ((2013) ) The molecular chaperone apolipoprotein J/clusterin as a sensor of oxidative stress: Implications in therapeutic approaches-a mini-review. Gerontology 59: , 514–523. |
[235] | Oda T , Wals P , Osterburg HH , Johnson SA , Pasinetti GM , Morgan TE , Rozovsky I , Stine WB , Snyder SW , Holzman TF , Krafft GA , Finch CE ((1995) ) Clusterin (apoJ) alters the aggregation of amyloid β-peptide (Aβ1-42) and forms slowly sedimenting Aβ complexes that cause oxidative stress. Exp Neurol 136: , 22–31. |
[236] | Farrer LA , Cupples LA , Haines JL , Hyman B , Kukull WA , Mayeux R , Myers RH , Pericak-Vance MA , Risch N , Van Duijn CM ((1997) ) Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease: A meta-analysis. JAMA 278: , 1349–1356. |
[237] | Corder EH , Saunders AM , Strittmatter WJ , Schmechel DE , Gaskell PC , Small Gw , Roses AD , Haines JL , Pericak-Vance MA ((1993) ) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261: , 921–923. |
[238] | Poirier J , Bertrand P , Kogan S , Gauthier S , Davignon J , Bouthillier D ((1993) ) Apolipoprotein E polymorphism and Alzheimer’s disease. Lancet 342: , 697–699. |
[239] | Strittmatter WJ , Saunders AM , Schmechel D , Pericak-Vance M , Enghild J , Salvesen GS , Roses AD ((1993) ) Apolipoprotein E: High-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A 90: , 1977–1981. |
[240] | Ma J , Yee A , Brewer HB Jr , Das S , Potter H ((1994) ) Amyloid-associated proteins α1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer β-protein into filaments. Nature 372: , 92–94. |
[241] | Deane R , Sagare A , Hamm K , Parisi M , Lane S , Finn MB , Holtzman DM , Zlokovic BV ((2008) ) apoE isoform-specific disruption of amyloid β peptide clearance from mouse brain. J Clin Invest 118: , 4002–4013. |
[242] | Chang S , ran Ma T , Miranda RD , Balestra ME , Mahley RW , Huang Y ((2005) ) Lipid-and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc Natl Acad Sci U S A 102: , 18694–18699. |
[243] | Veinbergs I , Everson A , Sagara Y , Masliah E ((2002) ) Neurotoxic effects of apolipoprotein E4 are mediated via dysregulation of calcium homeostasis. J Neurosci Res 67: , 379–387. |
[244] | Ramassamy C , Averill D , Beffert U , Theroux L , Lussier-Cacan S , Cohn JS , Christen Y , Schoofs A , Davignon J , Poirier J ((2000) ) Oxidative insults are associated with apolipoprotein E genotype in Alzheimer’s disease brain. Neurobiol Dis 7: , 23–37. |
[245] | Hamanaka H , Katoh-Fukui Y , Suzuki K , Kobayashi M , Suzuki R , Motegi Y , Nakahara Y , Takeshita A , Kawai M , Ishiguro K , Yokoyama M , Fujita SC ((2000) ) Altered cholesterol metabolism in human apolipoprotein E4 knock-in mice. Hum Mol Genet 9: , 353–361. |
[246] | Chen Y , Durakoglugil MS , Xian X , Herz J ((2010) ) ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proc Natl Acad Sci U S A 107: , 12011–12016. |
[247] | Ramassamy C , Krzywkowski P , Averill D , Lussier-Cacan S , Theroux L , Christen Y , Davignon J , Poirier J ((2001) ) Impact of apoE deficiency on oxidative insults and antioxidant levels in the brain. Mol Brain Res 86: , 76–83. |
[248] | Tamaoka A , Miyatake F , Matsuno S , Ishii K , Nagase S , Sahara N , Ono S , Mori H , Wakabayashi K , Tsuji S , Takahashi H , Shoji S ((2000) ) Apolipoprotein E allele–dependent antioxidant activity in brains with Alzheimer’s disease. Neurology 54: , 2319–2321. |
[249] | Kharrazi H , Vaisi-Raygani A , Rahimi Z , Tavilani H , Aminian M , Pourmotabbed T ((2008) ) Association between enzymatic and non-enzymatic antioxidant defense mechanism with apolipoprotein E genotypes in Alzheimer disease. Clin Biochem 41: , 932–936. |
[250] | Miyata M , Smith JD ((1996) ) Apolipoprotein E allele-specific antioxidant activity and effects on cytotoxicity by oxidative insults and β-amyloid peptides. Nat Genet 14: , 55–61. |
[251] | Hayek T , Oiknine J , Brook JG , Aviram M ((1994) ) Increased plasma and lipoprotein lipid peroxidation in apo E-deficient mice. Biochem Biophys Res Commun 201: , 1567–1574. |
[252] | Lauderback CM , Kanski J , Hackett JM , Maeda N , Kindy MS , Butterfield DA ((2002) ) Apolipoprotein E modulates Alzheimer’s Aβ (1-42)-induced oxidative damage to synaptosomes in an allele-specific manner. Brain Res 924: , 90–97. |
[253] | Saitoh M , Nishitoh H , Fujii M , Takeda K , Tobiume K , Sawada Y , Kawabata M , Miyazono K , Ichijo H ((1998) ) Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J 17: , 2596–2606. |
[254] | Machado AK , Morgan BA , Merrill GF ((1997) ) Thioredoxin reductase-dependent inhibition of MCB cell cycle box activity in Saccharomyces cerevisiae. J Biol Chem 272: , 17045–17054. |
[255] | Akterin S , Cowburn RF , Miranda-Vizuete A , Jiménez A , Bogdanovic N , Winblad B , Cedazo-Minguez A ((2006) ) Involvement of glutaredoxin-1 and thioredoxin-1 in β-amyloid toxicity and Alzheimer’s disease. Cell Death Differ 13: , 1454–1465. |
[256] | Lovell MA , Xie C , Gabbita SP , Markesbery WR ((2000) ) Decreased thioredoxin and increased thioredoxin reductase levels in Alzheimer’s disease brain. Free Radic Biol Med 28: , 418–427. |
[257] | Persson T , Lattanzio F , Calvo-Garrido J , Rimondini R , Rubio-Rodrigo M , Sundström E , Maioli S , Sandebring-Matton A , Cedazo-Mínguez Á ((2017) ) Apolipoprotein E4 elicits lysosomal cathepsin D release, decreased thioredoxin-1 levels, and apoptosis. J Alzheimers Dis 56: , 601–617. |
[258] | TaniguchiY, Taniguchi-UedaY, MoriK, YodoiJ ((1996) ) A novel promoter sequence is involved in the oxidative stress-induced expression of the adult T-cell leukemia-derived factor (ADF)/human thioredoxin (Trx) gene. Nucleic Acids Res 24: , 2746–2752. |
[259] | Mateos L , Persson T , Kathozi S , Gil-Bea FJ , Cedazo-Minguez A ((2012) ) Estrogen protects against amyloid-β toxicity by estrogen receptor α-mediated inhibition of Daxx translocation. Neurosci Lett 506: , 245–250. |
[260] | Bimonte-Nelson HA , Hunter CL , Nelson ME , Granholm ACE ((2003) ) Frontal cortex BDNF levels correlate with working memory in an animal model of Down syndrome. Behav Brain Res 139: , 47–57. |
[261] | Granholm ACE , Sanders LA , Crnic LS ((2000) ) Loss of cholinergic in basal forebrain coincides with cognitive decline in a mouse model of Down’s syndrome. Exp Neurol 161: , 647–663. |
[262] | Leong MPY , Bala U , Lim CL , Rosli R , Cheah PS , Ling KH ((2018) ) Transcriptomic profiling of skeletal muscle from the Ts1Cje mouse model of Down syndrome suggests dysregulation of trisomic genes associated with neuromuscular junction signaling, oxidative stress and chronic inflammation. Neurosci Res Notes 1: , 21–41. |
[263] | Busciglio J , Yankner BA ((1995) ) Apoptosis and increased generation of reactive oxygen species in Down’s syndrome neurons in vitro. Nature 378: , 776–779. |
[264] | Peled-Kamar M , Lotem J , Wirguin I , Weiner L , Hermalin A , Groner Y ((1997) ) Oxidative stress mediates impairment of muscle function in transgenic mice with elevated level of wild-type Cu/Zn superoxide dismutase. Proc Natl Acad Sci U S A 94: , 3883–3887. |
[265] | Roizen NJ , Patterson D ((2003) ) Down’s syndrome. Lancet 361: , 1281–1289. |
[266] | Di Domenico F , Coccia R , Cocciolo A , Murphy MP , Cenini G , Head E , Butterfield DA , Giorgi A , Schinina ME , Mancuso C , Cini C , Perluigi M ((2013) ) Impairment of proteostasis network in Down syndrome prior to the development of Alzheimer’s disease neuropathology: Redox proteomics analysis of human brain. Biochim Biophys Acta 1832: , 1249–1259. |
[267] | Campos C , Guzmán R , López-Fernández E , Casado Á ((2011) ) Evaluation of urinary biomarkers of oxidative/nitrosative stress in adolescents and adults with Down syndrome. Biochim Biophys Acta 1812: , 760–768. |
[268] | Strydom A , Dickinson MJ , Shende S , Pratico D , Walker Z ((2009) ) Oxidative stress and cognitive ability in adults with Down syndrome. Prog Neuropsychopharmacol Biol Psychiatry 33: , 76–80. |
[269] | Corrales A , Parisotto EB , Vidal V , García-Cerro S , Lantigua S , Diego M , Wilhem Filho D , Sanchez-Barceló EJ , Martínez-Cué C , Rueda N ((2017) ) Pre-and post-natal melatonin administration partially regulates brain oxidative stress but does not improve cognitive or histological alterations in the Ts65Dn mouse model of Down syndrome. Behav Brain Res 334: , 142–154. |
[270] | Odetti P , Angelini G , Dapino D , Zaccheo D , Garibaldi S , Dagna-Bricarelli F , Piombo G , Perry G , Smith M , Traverso N , Tabaton M ((1998) ) Early glycoxidation damage in brains from Down’s syndrome. Biochem Biophys Res Commun 243: , 849–851. |
[271] | Perrone S , Longini M , Bellieni CV , Centini G , Kenanidis A , De Marco L , Petraglia F , Buonocore G ((2007) ) Early oxidative stress in amniotic fluid of pregnancies with Down syndrome. Clin Biochem 40: , 177–180. |
[272] | Paola D , Domenicotti C , Nitti M , Vitali A , Borghi R , Cottalasso D , Zaccheo D , Odetti P , Strocchi P , Marinari UM , Tabaton M , Pronzato MA ((2000) ) Oxidative stress induces increase in intracellular amyloid β-protein production and selective activation of βI and βII PKCs in NT2 cells. Biochem Biophys Res Commun 268: , 642–646. |
[273] | Misonou H , Morishima-Kawashima M , Ihara Y ((2000) ) Oxidative stress induces intracellular accumulation of amyloid β-protein (Aβ) in human neuroblastoma cells. Biochemistry 39: , 6951–6959. |
[274] | Tamagno E , Parola M , Bardini P , Piccini A , Borghi R , Guglielmotto M , Santoro G , Davit A , Danni O , Smith MA , Perry G , Tabaton M ((2005) ) β-Site APP cleaving enzyme up-regulation induced by 4-hydroxynonenal is mediated by stress-activated protein kinases pathways. J Neurochem 92: , 628–636. |
[275] | Frederikse PH , Garland D , Zigler JJS , Piatigorsky J ((1996) ) Oxidative stress increases production of beta-amyloid precursor protein and beta-amyloid (Abeta) in mammalian lenses, and Abeta has toxic effects on lens epithelial cells. J Biol Chem 271: , 10169–10174. |
[276] | Li F , Calingasan NY , Yu F , Mauck WM , Toidze M , Almeida CG , Takahashi RH , Carlson GA , Flint Beal M , Lin MT , Gouras GK ((2004) ) Increased plaque burden in brains of APP mutant MnSOD heterozygous knockout mice. J Neurochem 89: , 1308–1312. |
[277] | Sturchler-Pierrat C , Abramowski D , Duke M , Wiederhold KH , Mistl C , Rothacher S , Ledermann B , Burki K , Frey P , Paganetti PA , Waridel C , Calhoun ME , Jucker M , Probst A , Staufenbiel M , Sommer B ((1997) ) Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci U S A 94: , 13287–13292. |
[278] | Li L , Zhang X , Yang D , Luo G , Chen S , Le W ((2009) ) Hypoxia increases Aβ generation by altering β-and γ-cleavage of APP. Neurobiol Aging 30: , 1091–1098. |
[279] | Roher AE , Lowenson JD , Clarke S , Woods AS , Cotter RJ , Gowing E , Ball MJ ((1993) ) Beta-amyloid-(1-42) is a major component of cerebrovascular amyloid deposits: Implications for the pathology of Alzheimer disease. Proc Natl Acad Sci U S A 90: , 10836–10840. |
[280] | Muche A , Arendt T , Schliebs R ((2017) ) Oxidative stress affects processing of amyloid precursor protein in vascular endothelial cells. PLoS One 12: , e0178127. |
[281] | Zetterberg H , Mörtberg E , Song L , Chang L , Provuncher GK , Patel PP , Ferrell E , Fournier DR , Kan CW , Campbell TG , Meyer R , Rivnak AJ , Pink BA , Minnehan KA , Piech T , Rissin DM , Duffy DC , Rubertsson S , Wilson DH , Blennow K ((2011) ) Hypoxia due to cardiac arrest induces a time-dependent increase in serum amyloid β levels in humans. PLoS One 6: , e28263. |
[282] | Chafekar SM , Baas F , Scheper W ((2008) ) Oligomer-specific Aβ toxicity in cell models is mediated by selective uptake. Biochim Biophys Acta 1782: , 523–531. |
[283] | Pauwels K , Williams TL , Morris KL , Jonckheere W , Vandersteen A , Kelly G , Schymkowitz J , Rousseau F , Pastore A , Serpell LC , Broersen K ((2012) ) Structural basis for increased toxicity of pathological aβ42: Aβ40 ratios in Alzheimer disease. J Biol Chem 287: , 5650–5660. |
[284] | Kuperstein I , Broersen K , Benilova I , Rozenski J , Jonckheere W , Debulpaep M , Vandersteen A , Segers-Nolten I , Van Der Werf K , Subramaniam V , Braeken D , Callewaert G , Bartic C , D’Hooge R , Martins IC , Rousseau F , Schymkowitz J , De Strooper B ((2010) ) Neurotoxicity of Alzheimer’s disease Aβ peptides is induced by small changes in the Aβ42 to Aβ40 ratio. EMBO J 29: , 3408–3420. |
[285] | Gabuzda D , Busciglio J , Chen LB , Matsudaira P , Yankner BA ((1994) ) Inhibition of energy metabolism alters the processing of amyloid precursor protein and induces a potentially amyloidogenic derivative. J Biol Chem 269: , 13623–13628. |
[286] | Annaert W , De Strooper B ((2002) ) A cell biological perspective on Alzheimer’s disease. Annu Rev Cell Dev Biol 18: , 25–51. |
[287] | Ertekin-Taner N ((2007) ) Genetics of Alzheimer’s disease: A centennial review. Neurol Clin 25: , 611–667. |
[288] | Bentahir M , Nyabi O , Verhamme J , Tolia A , Horré K , Wiltfang J , Esselmann H , De Strooper B ((2006) ) Presenilin clinical mutations can affect γ-secretase activity by different mechanisms. J Neurochem 96: , 732–742. |
[289] | Suzuki N , Cheung TT , Cai XD , Odaka A , Otvos L , Eckman C , Golde TE , Younkin SG ((1994) ) An increased percentage of long amyloid beta protein secreted by familial amyloid beta protein precursor (beta APP717) mutants. Science 264: , 1336–1340. |
[290] | Duff K , Eckman C , Zehr C , Yu X , Prada CM , Perez-Tur J , Hutton M , Buee L , Harigaya Y , Yager D , Morgan D , Gordon MN , Holcomb L , Refolo L , Zenk B , Hardy J , Younkin S ((1996) ) Increased amyloid-β42 (43) in brains of mice expressing mutant presenilin 1. Nature 383: , 710–713. |
[291] | Scheuner D , Eckman C , Jensen M , Song X , Citron M , Suzuki N , Bird TD , Hardy J , Hutton M , Kukull W , Larson E , Levy-Lahad E , Viitanen M , Peskind E , Poorkaj P , Schellenberg G , Tanzi R , Wasco W , Lannfelt L , Selkoe D , Younkin S ((1996) ) Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med 2: , 864–870. |
[292] | Nik M , Hani S , Wilson L , Newman M , Croft K , Mori TA , Musgrave I , Lardelli M ((2012) ) The BACE1-PSEN-AβPP regulatory axis has an ancient role in response to low oxygen/oxidative stress. J Alzheimers Dis 28: , 515–530. |
[293] | De Gasperi R , Sosa MAG , Dracheva S , Elder GA ((2010) ) Presenilin-1 regulates induction of hypoxia inducible factor-1α: Altered activation by a mutation associated with familial Alzheimer’s disease. Mol Neurodegener 5: , 38. |
[294] | Wang R , Zhang YW , Zhang X , Liu R , Zhang X , Hong S , Xia K , Xia J , Zhang Z , Xu H ((2006) ) Transcriptional regulation of APH-1A and increased γ-secretase cleavage of APP and Notch by HIF-1 and hypoxia. FASEB J 20: , 1275–1277. |
[295] | Van Uden P , Kenneth NS , Rocha S ((2008) ) Regulation of hypoxia-inducible factor-1α by NF-κB. Biochem J 412: , 477–484. |
[296] | Picone P , Nuzzo D , Caruana L , Messina E , Barera A , Vasto S , Di Carlo M ((2015) ) Metformin increases APP expression and processing via oxidative stress, mitochondrial dysfunction and NF-κB activation: Use of insulin to attenuate metformin’s effect. Biochim Biophys Acta 1853: , 1046–1059. |
[297] | Savage MJ , Lin YG , Ciallella JR , Flood DG , Scott RW ((2002) ) Activation of c-Jun N-terminal kinase and p38 in an Alzheimer’s disease model is associated with amyloid deposition. J Neurosci 22: , 3376–3385. |
[298] | Tamagno E , Guglielmotto M , Bardini P , Santoro G , Davit A , Di Simone D , Danni O , Tabaton M ((2003) ) Dehydroepiandrosterone reduces expression and activity of BACE in NT2 neurons exposed to oxidative stress. Neurobiol Dis 14: , 291–301. |
[299] | Yang H , Xie Z , Wei L , Ding M , Wang P , Bi J ((2018) ) Glutathione-mimetic D609 alleviates memory deficits and reduces amyloid-β deposition in an AβPP/PS1 transgenic mouse model. Neuroreport 29: , 833–838. |
[300] | Tamagno E , Guglielmotto M , Giliberto L , Vitali A , Borghi R , Autelli R , Danni O , Tabaton M ((2009) ) JNK and ERK1/2 pathways have a dual opposite effect on the expression of BACE1. Neurobiol Aging 30: , 1563–1573. |
[301] | Guglielmotto M , Monteleone D , Giliberto L , Fornaro M , Borghi R , Tamagno E , Tabaton M ((2011) ) Amyloid-β 42 activates the expression of BACE1 through the JNK pathway. J Alzheimers Dis 27: , 871–883. |
[302] | Tong Y , Zhou W , Fung V , Christensen MA , Qing H , Sun X , Song W ((2005) ) Oxidative stress potentiates BACE1 gene expression and Aβ generation. J Neural Transm 112: , 455–469. |
[303] | Mouton-Liger F , Paquet C , Dumurgier J , Bouras C , Pradier L , Gray F , Hugon J ((2012) ) Oxidative stress increases BACE1 protein levels through activation of the PKR-eIF2α pathway. Biochim Biophys Acta 1822: , 885–896. |
[304] | Quiroz-Baez R , Rojas E , Arias C ((2009) ) Oxidative stress promotes JNK-dependent amyloidogenic processing of normally expressed human APP by differential modification of α-, β-and γ-secretase expression. Neurochem Int 55: , 662–670. |
[305] | Kao SC , Krichevsky AM , Kosik KS , Tsai LH ((2004) ) BACE1 suppression by RNA interference in primary cortical neurons. J Biol Chem 279: , 1942–1949. |
[306] | Kwak YD , Wang R , Li JJ , Zhang YW , Xu H , Liao FF ((2011) ) Differential regulation of BACE1 expression by oxidative and nitrosative signals. Mol Neurodegener 6: , 17. |
[307] | Tan JL , Li QX , Ciccotosto GD , Crouch PJ , Culvenor JG , White AR , Evin G ((2013) ) Mild oxidative stress induces redistribution of BACE1 in non-apoptotic conditions and promotes the amyloidogenic processing of Alzheimer’s disease amyloid precursor protein. PLoS One 8: , e61246. |
[308] | Fukumoto H , Cheung BS , Hyman BT , Irizarry MC ((2002) ) β-Secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch Neurol 59: , 1381–1389. |
[309] | Holsinger RMD , McLean CA , Beyreuther K , Masters CL , Evin G ((2002) ) Increased expression of the amyloid precursor β-secretase in Alzheimer’s disease. Ann Neurol 51: , 783–786. |
[310] | Yang LB , Lindholm K , Yan R , Citron M , Xia W , Yang XL , Beach T , Sue L , Wong P , Price D , Li R , Shen Y ((2003) ) Elevated β-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat Med 9: , 3–4. |
[311] | Christensen MA , Zhou W , Qing H , Lehman A , Philipsen S , Song W ((2004) ) Transcriptional regulation of BACE1, the β-amyloid precursor protein β-secretase, by Sp1. Mol Cell Biol 24: , 865–874. |
[312] | Huang P , Sun J , Wang F , Luo X , Zhu H , Gu Q , Sun X , Liu T , Sun X ((2018) ) DNMT1 and Sp1 competitively regulate the expression of BACE1 in A2E-mediated photo-oxidative damage in RPE cells. Neurochem Int 121: , 59–68. |
[313] | Zhu X , Rottkamp CA , Hartzler A , Sun Z , Takeda A , Boux H , Shimohama S , Perry G , Smith MA ((2001) ) Activation of MKK6, an upstream activator of p38, in Alzheimer’s disease. J Neurochem 79: , 311–318. |
[314] | Xiantao W , Martindale JL , Yusen LIU , Holbrook NJ ((1998) ) The cellular response to oxidative stress: Influences of mitogen-activated protein kinase signalling pathways on cell survival. Biochem J 333: , 291–300. |
[315] | Le-Niculescu H , Bonfoco E , Kasuya Y , Claret FX , Green DR , Karin M ((1999) ) Withdrawal of survival factors results in activation of the JNK pathway in neuronal cells leading to Fas ligand induction and cell death. Mol Cell Biol 19: , 751–763. |
[316] | Webster NJ , Green KN , Peers C , Vaughan PFT ((2002) ) Altered processing of amyloid precursor protein in the human neuroblastoma SH-SY5Y by chronic hypoxia. J Neurochem 83: , 1262–1271. |
[317] | Kimberly WT , Zheng JB , Town T , Flavell RA , Selkoe DJ ((2005) ) Physiological regulation of the β-amyloid precursor protein signaling domain by c-Jun N-terminal kinase JNK3 during neuronal differentiation. J Neurosci 25: , 5533–5543. |
[318] | Hatfield CF , Herbert J , Van Someren EJW , Hodges JR , Hastings MH ((2004) ) Disrupted daily activity/rest cycles in relation to daily cortisol rhythms of home-dwelling patients with early Alzheimer’s dementia. Brain 127: , 1061–1074. |
[319] | Prinz PN , Peskind ER , Vitaliano PP , Raskind MA , Eisdorfer C , Zemcuznikov HN , Gerber CJ ((1982) ) Changes in the sleep and waking EEGs of nondemented and demented elderly subjects. J Am Geriatr Soc 30: , 86–92. |
[320] | Ju YES , McLeland JS , Toedebusch CD , Xiong C , Fagan AM , Duntley SP , Morris JC , Holtzman DM ((2013) ) Sleep quality and preclinical Alzheimer disease. JAMA Neurol 70: , 587–593. |
[321] | Witting W , Kwa IH , Eikelenboom P , Mirmiran M , Swaab DF ((1990) ) Alterations in the circadian rest-activity rhythm in aging and Alzheimer’s disease. Biol Psychiatry 27: , 563–572. |
[322] | Ramanathan L , Gulyani S , Nienhuis R , Siegel JM ((2002) ) Sleep deprivation decreases superoxide dismutase activity in rat hippocampus and brainstem. Neuroreport 13: , 1387–1390. |
[323] | Li T , Cao R , Xia R , Xia Z ((2017) ) Effects of 72 hours sleep deprivation on liver circadian clock gene expression and oxidative stress in rats. Yangtze Med 1: , 194–201. |
[324] | Edgar RS , Green EW , Zhao Y , van Ooijen G , Olmedo M , Qin X , Xu Y , Pan M , Valekunja UK , Feeney KA , Maywood ES , Hastings MH , Baliga NS , Merrow M , Millar AJ , Johnson CH , Kyriacou CP , O’Neill JS , Reddy AB ((2012) ) Peroxiredoxins are conserved markers of circadian rhythms. Nature 485: , 459–464. |
[325] | Lopez-Molina L , Conquet F , Dubois-Dauphin M , Schibler U ((1997) ) The DBP gene is expressed according to a circadian rhythm in the suprachiasmatic nucleus and influences circadian behavior. EMBO J 16: , 6762–6771. |
[326] | Murai Y , Ishibashi H , Koyama S , Akaike N ((1997) ) Ca2+-activated}+ currents in rat locus coeruleus neurons induced by experimental ischemia, anoxia, and hypoglycemia. J Neurophysiol 78: , 2674–2681. |
[327] | Zhu Y , Fenik P , Zhan G , Mazza E , Kelz M , Aston-Jones G , Veasey SC ((2007) ) Selective loss of catecholaminergic wake-active neurons in a murine sleep apnea model. J Neurosci 27: , 10060–10071. |
[328] | Panossian L , Fenik P , Zhu Y , Zhan G , McBurney MW , Veasey S ((2011) ) SIRT1 regulation of wakefulness and senescence-like phenotype in wake neurons. J Neurosci 31: , 4025–4036. |
[329] | Aston-Jones G , Bloom FE ((1981) ) Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep-waking cycle. J Neurosci 1: , 876–886. |
[330] | Zhang J , Zhu Y , Zhan G , Fenik P , Panossian L , Wang MM , Reid S , Lai D , Davis JG , Baur JA , Veasey S ((2014) ) Extended wakefulness: Compromised metabolics in and degeneration of locus ceruleus neurons. J Neurosci 34: , 4418–4431. |
[331] | Someya S , Yu W , Hallows WC , Xu J , Vann JM , Leeuwenburgh C , Tanokura M , Denu JM , Prolla TA ((2010) ) Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 143: , 802–812. |
[332] | Yu W , Dittenhafer-Reed KE , Denu JM ((2012) ) SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J Biol Chem 287: , 14078–14086. |
[333] | Jacobs KM , Pennington JD , Bisht KS , Aykin-Burns N , Kim HS , Mishra M , Sun L , Nguyen P , Ahn BH , Leclerc J , Deng CX , Spitz DR , Gius D ((2008) ) SIRT3 interacts with the daf-16 homolog FOXO3a in the mitochondria, as well as increases FOXO3a dependent gene expression. Int J Biol Sci 4: , 291–299. |
[334] | Qiu X , Brown K , Hirschey MD , Verdin E , Chen D ((2010) ) Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab 12: , 662–667. |
[335] | Rutter J , Reick M , Wu LC , McKnight SL ((2001) ) Regulation of clock and NPAS2 DNA binding by the redox state of NAD cofactors. Science 293: , 510–514. |
[336] | Rudic RD , McNamara P , Curtis AM , Boston RC , Panda S , Hogenesch JB , FitzGerald GA ((2004) ) BMAL1 and CLOCK, two essential components of the circadian clock, are involved in glucose homeostasis. PLoS Biol 2: , e377. |
[337] | Kondratov RV , Kondratova AA , Gorbacheva VY , Vykhovanets OV , Antoch MP ((2006) ) Early aging and age-related pathologies in mice deficient in BMAL1, the core componentof the circadian clock. Genes Dev 20: , 1868–1873. |
[338] | Wyse CA , Coogan AN ((2010) ) Impact of aging on diurnal expression patterns of CLOCK and BMAL1 in the mouse brain. Brain Res 1337: , 21–31. |
[339] | Bunger MK , Wilsbacher LD , Moran SM , Clendenin C , Radcliffe LA , Hogenesch JB , Simon MC , Takahashi JS , Bradfield CA ((2000) ) Mop3 is an essential component of the master circadian pacemaker in mammals. Cell 103: , 1009–1017. |
[340] | Kondratova AA , Dubrovsky YV , Antoch MP , Kondratov RV ((2010) ) Circadian clock proteins control adaptation to novel environment and memory formation. Aging (Albany NY) 2: , 285–297. |
[341] | Musiek ES , Lim MM , Yang G , Bauer AQ , Qi L , Lee Y , Roh JH , Ortiz-Gonzalez X , Dearborn JT , Culver JP , Herzog ED , Hogenesch JB , Wozniak DF , Dikranian K , Giasson BI , Weaver DR , Holtzman DM , Fitzgerald GA ((2013) ) Circadian clock proteins regulate neuronal redox homeostasis and neurodegeneration. J Clin Invest 123: , 5389–5400. |
[342] | Peek CB , Affinati AH , Ramsey KM , Kuo HY , Yu W , Sena LA , Ilkayeva O , Marcheva B , Kobayashi Y , Omura C , Levine DC , Bacsik DJ , Gius D , Newgard CB , Goetzman E , Chandel NS , Denu JM , Mrksich M , Bass J ((2013) ) Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science 342: , 1243417. |
[343] | Desvergne A , Ugarte N , Petropoulos I , Friguet B ((2014) ) Circadian modulation of proteasome activities and removal of carbonylated proteins. Free Radic Biol Med 75: , S18. |
[344] | Matsumura Y , Aizawa H , Shiraki-Iida T , Nagai R , Kuro-o M , Nabeshima Y ((1998) ) Identification of the human klotho gene and its two transcripts encoding membrane and secreted klotho protein. Biochem Biophys Res Commun 242: , 626–630. |
[345] | Chen CD , Podvin S , Gillespie E , Leeman SE , Abraham CR ((2007) ) Insulin stimulates the cleavage and release of the extracellular domain of Klotho by ADAM10 and ADAM17. Proc Natl Acad Sci U S A 104: , 19796–19801. |
[346] | Kuro-o M , Matsumura Y , Aizawa H , Kawaguchi H , Suga T , Utsugi T , Ohyama Y , Kurabayashi M , Kaname T , Kume E , Iwasaki H , Iida A , Shiraki-Iida T , Nishikawa S , Nagai R , Nabeshima YI ((1997) ) Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 390: , 45–51. |
[347] | Kurosu H , Yamamoto M , Clark JD , Pastor JV , Nandi A , Gurnani P , McGuinness OP , Chikuda H , Yamaguchi M , Kawaguchi H , Shimomura I , Takayama Y , Herz J , Kahn CR , Rosenblatt KP , Kuro-o M ((2005) ) Suppression of aging in mice by the hormone Klotho. Science 309: , 1829–1833. |
[348] | Arking DE , Krebsova A , Macek M , Arking A , Mian IS , Fried L , Hamosh A , Dey S , McIntosh I , Dietz HC ((2002) ) Association of human aging with a functional variant of klotho. Proc Natl Acad Sci U S A 99: , 856–861. |
[349] | Duce JA , Podvin S , Hollander W , Kipling D , Rosene DL , Abraham CR ((2008) ) Gene profile analysis implicates Klotho as an important contributor to aging changes in brain white matter of the rhesus monkey. Glia 56: , 106–117. |
[350] | Yamamoto M , Clark JD , Pastor JV , Gurnani P , Nandi A , Kurosu H , Miyoshi M , Ogawa Y , Castrillon DH , Rosenblatt KP , Kuro-o M ((2005) ) Regulation of oxidative stress by the anti-aging hormone klotho. J Biol Chem 280: , 38029–38034. |
[351] | Han DH , Piao SG , Song JH , Ghee JY , Hwang HS , Choi BS , Kim J , Yang CW ((2010) ) Effect of sirolimus on calcineurin inhibitor-induced nephrotoxicity using renal expression of KLOTHO, an antiaging gene. Transplantation 90: , 135–141. |
[352] | Ikushima M , Rakugi H , Ishikawa K , Maekawa Y , Yamamoto K , Ohta J , Chihara Y , Kida I , Ogihara T ((2006) ) Anti-apoptotic and anti-senescence effects of Klotho on vascular endothelial cells. Biochem Biophys Res Commun 339: , 827–832. |
[353] | Zeldich E , Chen CD , Colvin TA , Bove-Fenderson EA , Liang J , Zhou TBT , Harris DA , Abraham CR ((2014) ) The neuroprotective effect of Klotho is mediated via regulation of members of the redox system. J Biol Chem 289: , 24700–24715. |
[354] | German DC , Khobahy I , Pastor J , Kuro-o M , Liu X ((2012) ) Nuclear localization of Klotho in brain: An anti-agingrotein. Neurobiol Aging 33: , 1483-e25–e30. |
[355] | Salech F , Varela-Nallar L , Arredondo SB , Bustamante DB , Andaur GA , Cisneros R , Ponce DP , Ayala P , Inestrosa NC , Valdés JL , I Behrens M , Couve A ((2019) ) Local Klotho enhances neuronal progenitor proliferation in the adult hippocampus. J Gerontol A Biol Sci Med Sci 74: , 1043–1051. |
[356] | Nagai T , Yamada K , Kim HC , Kim YS , Noda Y , Imura A , Nabeshima Y , Nabeshima T ((2003) ) Cognition impairment in the genetic model of aging klotho gene mutant mice: A role of oxidative stress. FASEB J 17: , 50–52. |
[357] | Brunet A , Bonni A , Zigmond MJ , Lin MZ , Juo P , Hu LS , Anderson MJ , Arden KC , Blenis J , Greenberg ME ((1999) ) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96: , 857–868. |
[358] | Colton CA , Snell J , Chernyshev O , Gilbert DL ((1994) ) Induction of superoxide anion and nitric oxide production in cultured microglia. Ann NY Acad Sci 738: , 54–63. |
[359] | Opazo C , Huang X , Cherny RA , Moir RD , Roher AE , White AR , Cappai R , Masters CL , Tanzi RE , Inestrosa NC , Bush AI ((2002) ) Metalloenzyme-like activity of Alzheimer’s disease β-amyloid Cu-dependent catalytic conversion of dopamine, cholesterol, and biological reducing agents to neurotoxic H2O2. J Biol Chem 277: , 40302–40308. |
[360] | Smith DP , Ciccotosto GD , Tew DJ , Fodero-Tavoletti MT , Johanssen T , Masters CL , Barnham KJ , Cappai R ((2007) ) Concentration dependent Cu 2+ induced aggregation and dityrosine formation of the Alzheimer’s disease amyloid-β peptide. Biochemistry 46: , 2881–2891. |
[361] | Atwood CS , Huang X , Khatri A , Scarpa RC , Kim YS , Moir RD , Tanzi RE , Roher AE , Bush AI ((2000) ) Copper catalyzed oxidation of Alzheimer Abeta. Cell Mol Biol (Noisy-le-grand) 46: , 777–783. |
[362] | Atamna H , Frey II WH , Ko N ((2009) ) Human and rodent amyloid-β peptides differentially bind heme: Relevance to the human susceptibility to Alzheimer’s disease. Arch Biochem Biophys 487: , 59–65. |
[363] | Kontush A , Berndt C , Weber W , Akopyan V , Arlt S , Schippling S , Beisiegel U ((2001) ) Amyloid-β is an antioxidant for lipoproteins in cerebrospinal fluid and plasma. Free Radic Biol Med 30: , 119–128. |
[364] | Sayre LM , Perry G , Harris PLR , Liu Y , Schubert KA , Smith MA ((2000) ) In situ oxidative catalysis by neurofibrillary tangles and senile plaques in Alzheimer’s disease: A central role for bound transition metals. J Neurochem 74: , 270–279. |
[365] | Smith MA , Harris PLR , Sayre LM , Perry G ((1997) ) Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc Natl Acad Sci U S A 94: , 9866–9868. |
[366] | Mukherjee S , Ghosh C , Seal M , Dey SG ((2017) ) Copper induced spin state change of heme-Aβ associated with Alzheimer’s disease. Dalt Trans 46: , 13171–13175. |
[367] | Grasso G , Komatsu H , Axelsen PH ((2017) ) Covalent modifications of the amyloid beta peptide by hydroxynonenal: Effects on metal ion binding by monomers and insights into the fibril topology. J Inorg Biochem 174: , 130–136. |
[368] | Cheignon C , Faller P , Testemale D , Hureau C , Collin F ((2016) ) Metal-catalyzed oxidation of Aβ and the resulting reorganization of Cu binding sites promote ROS production. Metallomics 8: , 1081–1089. |
[369] | Boyd-Kimball D , Sultana R , Mohmmad-Abdul H , Butterfield DA ((2004) ) Rodent Aβ (1-42) exhibits oxidative stress properties similar to those of human Aβ (1-42): Implications for proposed mechanisms of toxicity. J Alzheimers Dis 6: , 515–525. |
[370] | Parker WD , Parks J , Filley CM , Kleinschmidt-DeMasters BK ((1994) ) Electron transport chain defects in Alzheimer’s disease brain. Neurology 44: , 1090–1096. |
[371] | Mutisya EM , Bowling AC , Beal MF ((1994) ) Cortical cytochrome oxidase activity is reduced in Alzheimer’s disease. J Neurochem 63: , 2179–2184. |
[372] | Girvan P , Teng X , Brooks NJ , Baldwin GS , Ying L ((2018) ) Redox kinetics of the amyloid-β-Cu complex and its biological implications. Biochemistry 57: , 6228–6233. |
[373] | Whitson JS , Selkoe DJ , Cotman CW ((1989) ) Amyloid beta protein enhances the survival of hippocampal neurons in vitro. Science 243: , 1488–1490. |
[374] | Zou K , Gong JS , Yanagisawa K , Michikawa M ((2002) ) A novel function of monomeric amyloid β-protein serving as an antioxidant molecule against metal-induced oxidative damage. J Neurosci 22: , 4833–4841. |
[375] | Stamer K , Vogel R , Thies E , Mandelkow E , Mandelkow EM ((2002) ) Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol 156: , 1051–1063. |
[376] | Montine TJ , Amarnath V , Martin ME , Strittmatter WJ , Graham DG ((1996) ) E-4-hydroxy-2-nonenal is cytotoxic and cross-links cytoskeletal proteins in P19 neuroglial cultures. Am J Pathol 148: , 89–93. |
[377] | Greenberg SG , Davies P , Schein JD , Binder LI ((1992) ) Hydrofluoric acid-treated tau PHF proteins display the same biochemical properties as normal tau. J Biol Chem 267: , 564–569. |
[378] | Hyman BT , Elvhage TE , Reiter J ((1994) ) Extracellular signal regulated kinases. Localization of protein and mRNA in the human hippocampal formation in Alzheimer’s disease. Am J Pathol 144: , 565–572. |
[379] | Guyton KZ , Liu Y , Gorospe M , Xu Q , Holbrook NJ ((1996) ) Activation of mitogen-activated protein kinase by ho role in cell survival following oxidant injury. J Biol Chem 271: , 4138–4142. |
[380] | Gómez-Ramos A , Díaz-Nido J , Smith MA , Perry G , Avila J ((2003) ) Effect of the lipid peroxidation product acrolein on tau phosphorylation in neural cells. J Neurosci Res 71: , 863–870. |
[381] | Kuhla B , Haase C , Flach K , Lüth HJ , Arendt T , Münch G ((2007) ) Effect of pseudophosphorylation and cross-linking by lipid peroxidation and advanced glycation end product precursors on tau aggregation and filament formation. J Biol Chem 282: , 6984–6991. |
[382] | Hensley K , Carney JM , Mattson MP , Aksenova M , Harris M , Wu JF , Floyd RA , Butterfield DA ((1994) ) A model for beta-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: Relevance to Alzheimer disease. Proc Natl Acad Sci U S A 91: , 3270–3274. |
[383] | Liang JJ , Gu CL , Kacher ML , Foote CS ((1983) ) Chemistry of singlet oxygen. 45. Mechanism of the photooxidation of sulfides. J Am Chem Soc 105: , 4717–4721. |
[384] | Smith LM , Strittmatter SM ((2017) ) Binding sites for amyloid-β oligomers and synaptic toxicity.a. Cold Spring Harb Perspect Med 7: , 024075. |
[385] | Sengupta U , Nilson AN , Kayed R ((2016) ) The role of amyloid-β oligomers in toxicity, propagation, and immunotherapy. EBioMedicine 6: , 42–49. |
[386] | Khodarahmi R , Ashrafi-Kooshk MR ((2017) ) Is there correlation between Aβ-heme peroxidase activity and the peptide aggregation state? A literature review combined with hypothesis. Int J Biol Macromol 100: , 18–36. |
[387] | Broersen K , Rousseau F , Schymkowitz J ((2010) ) The culprit behind amyloid beta peptide related neurotoxicity in Alzheimer’s disease: Oligomer size or conformation? Alzheimers Res Ther 2: , 12. |
[388] | Gong Y , Chang L , Viola KL , Lacor PN , Lambert MP , Finch CE , Krafft GA , Klein WL ((2003) ) Alzheimer’s disease-affected brain: Presence of oligomeric Aβ ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci U S A 100: , 10417–10422 . |
[389] | Kayed R , Head E , Thompson JL , McIntire TM , Milton SC , Cotman CW , Glabe CG ((2003) ) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300: , 486–489. |
[390] | Mc Donald JM , Savva GM , Brayne C , Welzel AT , Forster G , Shankar GM , Selkoe DJ , Ince PG , Walsh DM ((2010) ) The presence of sodium dodecyl sulphate-stable Aβ dimers is strongly associated with Alzheimer-type dementia. Brain 133: , 1328–1341. |
[391] | Lesné S , Koh MT , Kotilinek L , Kayed R , Glabe CG , Yang A , Gallagher M , Ashe KH ((2006) ) A specific amyloid-β protein assembly in the brain impairs memory. Nature 440: , 352–357. |
[392] | Tamagno E , Bardini P , Guglielmotto M , Danni O , Tabaton M ((2006) ) The various aggregation states of β-amyloid 1-42 mediate different effects on oxidative stress, neurodegeneration, and BACE-1 expression. Free Radic Biol Med 41: , 202–212. |
[393] | Tabner BJ , El-Agnaf OMA , Turnbull S , German MJ , Paleologou KE , Hayashi Y , Cooper LJ , Fullwood NJ , Allsop D ((2005) ) Hydrogen peroxide is generated during the very early stages of aggregation of the amyloid peptides implicated in Alzheimer disease and familial British dementia. J Biol Chem 280: , 35789–35792. |
[394] | Williams TL , Serpell LC , Urbanc B ((2016) ) Stabilization of native amyloid β-protein oligomers by copper and hydrogen peroxide induced cross-linking of unmodified proteins (CHICUP). Biochim Biophys Acta 1864: , 249–259. |
[395] | Oppermann UCT , Salim S , Tjernberg LO , Terenius L , Jörnvall H ((1999) ) Binding of amyloid β-peptide to mitochondrial hydroxyacyl-CoA dehydrogenase (ERAB): Regulation of an SDR enzyme activity with implications for apoptosis in Alzheimer’s disease. FEBS Lett 451: , 238–242. |
[396] | Du Yan S , Shi Y , Zhu A , Fu J , Zhu H , Zhu Y , Gibson L , Stern E , Collison K , Al-Mohanna F , Ogawa S , Roher A , Clarke SG , Stern DM ((1999) ) Role of ERAB/L-3-hydroxyacyl-coenzyme A dehydrogenase type II activity in Aβ-induced cytotoxicity. J Biol Chem 274: , 2145–2156. |
[397] | Hoyer S , Oesterreich K , Wagner O ((1988) ) Glucose metabolism as the site of the primary abnormality in early-onset dementia of Alzheimer type? J Neurol 235: , 143–148. |
[398] | Yan S Du , Stern DM ((2005) ) Mitochondrial dysfunction and Alzheimer’s disease: Role of amyloid-β peptide alcohol dehydrogenase (ABAD). Int J Exp Pathol 86: , 161–171. |
[399] | Gibson GE , Jope R , Blass JP ((1975) ) Decreased synthesis of acetylcholine accompanying impaired oxidation of pyruvic acid in rat brain minces. Biochem J 148: , 17–23. |
[400] | Roy CS , Sherrington CS ((1890) ) On the regulation of the blood-supply of the brain. J Physiol 11: , 85–158. |
[401] | Morgello S , Uson RR , Schwartz EJ , Haber RS ((1995) ) The human blood-brain barrier glucose transporter (GLUT1) is a glucose transporter of gray matter astrocytes. Glia 14: , 43–54. |
[402] | Andriezen WL ((1893) ) On a system of fibre-cells surrounding the blood-vessels of the brain of man and mammals and its physiological significance. Int Monschr Anat Physiol 10: , 532–540. |
[403] | Wu Y , Sun FF , Tong DM , Taylor BM ((1996) ) Changes in membrane properties during energy depletion-induced cell injury studied with fluorescence microscopy. Biophys J 71: , 91–100. |
[404] | Frölich L , Blum-Degen D , Bernstein HG , Engelsberger S , Humrich J , Laufer S , Muschner D , Thalheimer A , Türk A , Hoyer S , Zöchling R , Boissl KW , Jellinger K , Riederer P ((1998) ) Brain insulin and insulin receptors in aging and sporadic Alzheimer’s disease. J Neural Transm 105: , 423–438. |
[405] | Swaab DF , Raadsheer FC , Endert E , Hofman MA , Kamphorst W , Ravid R ((1994) ) Increased cortisol levels in aging and Alzheimer’s disease in postmortem cerebrospinal fluid. J Neuroendocrinol 6: , 681–687. |
[406] | Ferrari F , Viscardi P , Gorini A , Villa RF ((2019) ) Synaptic ATPases system of rat frontal cerebral cortex during aging. Neurosci Lett 694: , 74–79. |
[407] | Hoyer S , Nitsch R , Oesterreich K ((1991) ) Predominant abnormality in cerebral glucose utilization in late-onset dementia of the Alzheimer type: A cross-sectional comparison against advanced late-onset and incipient early-onset cases. J Neural Transm Dis Dement Sect 3: , 1–14. |
[408] | Almeida A , Almeida J , Bolaños JP , Moncada S ((2001) ) Different responses of astrocytes and neurons to nitric oxide: The role of glycolytically generated ATP in astrocyte protection. Proc Natl Acad Sci U S A 98: , 15294–15299. |
[409] | Perry EK , Perry RH , Tomlinson BE , Blessed G , Gibson PH ((1980) ) Coenzyme A-acetylating enzymes in Alzheimer’s disease: Possible cholinergic ‘compartment’ of pyruvate dehydrogenase. Neurosci Lett 18: , 105–110. |
[410] | Castegna A , Thongboonkerd V , Klein JB , Lynn B , Markesbery WR , Butterfield DA ((2003) ) Proteomic identification of nitrated proteins in Alzheimer’s disease brain. J Neurochem 85: , 1394–1401. |
[411] | Korolainen MA , Auriola S , Nyman TA , Alafuzoff I , Pirttilä T ((2005) ) Proteomic analysis of glial fibrillary acidic protein in Alzheimer’s disease and aging brain. Neurobiol Dis 20: , 858–870. |
[412] | Korolainen MA , Goldsteins G , Nyman TA , Alafuzoff I , Koistinaho J , Pirttilä T ((2006) ) Oxidative modification of proteins in the frontal cortex of Alzheimer’s disease brain. Neurobiol Aging 27: , 42–53. |
[413] | Reed T , Perluigi M , Sultana R , Pierce WM , Klein JB , Turner DM , Coccia R , Markesbery WR , Butterfield DA ((2008) ) Redox proteomic identification of 4-hydroxy-2-nonenal-modified brain proteins in amnestic mild cognitive impairment: Insight into the role of lipid peroxidation in the progression and pathogenesis of Alzheimer’s disease. Neurobiol Dis 30: , 107–120. |
[414] | Sultana R , Boyd-Kimball D , Poon HF , Cai J , Pierce WM , Klein JB , Merchant M , Markesbery WR , Butterfield DA ((2006) ) Redox proteomics identification of oxidized proteins in Alzheimer’s disease hippocampus and cerebellum: An approach to understand pathological and biochemical alterations in AD. Neurobiol Aging 27: , 1564–1576. |
[415] | Sims NR , Blass JP , Murphy C , Bowen DM , Neary D ((1987) ) Phosphofructokinase activity in the brain in Alzheimer’s disease. Ann Neurol 21: , 509–510. |
[416] | Liguri G , Taddei N , Nassi P , Latorraca S , Nediani C , Sorbi S ((1990) ) Changes in Na+,}+-ATPase, Ca2+-ATPase and some soluble enzymes related to energy metabolism in brains of patients with Alzheimer’s disease. Neurosci Lett 112: , 338–342. |
[417] | Liu C , Song X , Nisbet R , Götz J ((2016) ) Co-immunoprecipitation with tau isoform-specific antibodies reveals distinct protein interactions and highlights a putative role for 2N tau in disease. J Biol Chem 291: , 8173–8188. |
[418] | Bigl M , Eschrich K ((1995) ) Interaction of rat brain phosphofructokinase with Alzheimer’s βA4-amyloid. Neurochem Int 26: , 69–75. |
[419] | Casley CS , Canevari L , Land JM , Clark JB , Sharpe MA ((2002) ) β-Amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J Neurochem 80: , 91–100. |
[420] | Brett J , Schmidt AM , Yan SD , Zou YS , Weidman E , Pinsky D , Nowygrod R , Neeper M , Przysiecki C , Shaw A , Migheli A , Stern D ((1993) ) Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am J Pathol 143: , 1699–1712. |
[421] | Neeper M , Schmidt AM , Brett J , Yan SD , Wang F , Pan YC , Elliston K , Stern D , Shaw A ((1992) ) Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J Biol Chem 267: , 14998–15004. |
[422] | Sell DR , Monnier VM ((1989) ) Structure elucidation of a senescence cross-link from human extracellular matrix. Implication of pentoses in the aging process. J Biol Chem 264: , 21597–215602. |
[423] | Makita Z , Vlassara H , Cerami A , Bucala R ((1992) ) Immunochemical detection of advanced glycosylation end products in vivo. J Biol Chem 267: , 5133–5138. |
[424] | Dunn JA , McCance DR , Thorpe SR , Lyons TJ , Baynes JW ((1991) ) Age-dependent accumulation of N. epsilon.-(carboxymethyl) lysine and N. epsilon.-(carboxymethyl) hydroxylysine in human skin collagen. Biochemistry 30: , 1205–1210. |
[425] | Makita Z , Radoff S , Rayfield EJ , Yang Z , Skolnik E , Delaney V , Friedman EA , Cerami A , Vlassara H ((1991) ) Advanced glycosylation end products in patients with diabetic nephropathy. N Engl J Med 325: , 836–842. |
[426] | Srikanth V , Maczurek A , Phan T , Steele M , Westcott B , Juskiw D , Münch G ((2011) ) Advanced glycation endproducts and their receptor RAGE in Alzheimer’s disease. Neurobiol Aging 32: , 763–777. |
[427] | Cai Z , Liu N , Wang C , Qin B , Zhou Y , Xiao M , Chang L , Yan LJ , Zhao B ((2016) ) Role of RAGE in Alzheimer’s disease. Cell Mol Neurobiol 36: , 483–495. |
[428] | Arancio O , Zhang HP , Chen X , Lin C , Trinchese F , Puzzo D , Liu S , Hegde A , Yan SF , Stern A , Luddy JS , Lue LF , Walker DG , Roher A , Buttini M , Mucke L , Li W , Schmidt AM , Kindy M , Hyslop PA , Stern DM , Du Yan SS ((2004) ) RAGE potentiates Aβ-induced perturbation of neuronal function in transgenic mice. EMBO J 23: , 4096–4105. |
[429] | Bezprozvanny I , Mattson MP ((2008) ) Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci 31: , 454–463. |
[430] | Berridge MJ , Bootman MD , Lipp P ((1998) ) Calcium-a life and death signal. Nature 395: , 645–648. |
[431] | Mattson MP , Chan SL ((2003) ) Neuronal and glial calcium signaling in Alzheimer’s disease. Cell Calcium 34: , 385–397. |
[432] | Pchitskaya E , Popugaeva E , Bezprozvanny I ((2018) ) Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 70: , 87–94. |
[433] | Gellerich FN , Gizatullina Z , Trumbekaite S , Korzeniewski B , Gaynutdinov T , Seppet E , Vielhaber S , Heinze HJ , Striggow F ((2012) ) Cytosolic Ca2+ regulates the energization of isolated brain mitochondria by formation of pyruvate through the malate-aspartate shuttle. Biochem J 443: , 747–755. |
[434] | Denton RM , Randle PJ , Martin BR ((1972) ) Stimulation by calcium ions of pyruvate dehydrogenase phosphate phosphatase. Biochem J 128: , 161–163. |
[435] | Siess EA , Wieland OH ((1972) ) Purification and characterization of pyruvate-dehydrogenase phosphatase from pig-heart muscle. Eur J Biochem 26: , 96–105. |
[436] | Turrens JF , Freeman BA , Levitt JG , Crapo JD ((1982) ) The effect of hyperoxia on superoxide production by lung submitochondrial particles. Arch Biochem Biophys 217: , 401–410. |
[437] | Konstantinov AA , Peskin AV , Popova EY , Khomutov GB , Ruuge EK ((1987) ) Superoxide generation by the respiratory chain of tumor mitochondria. Biochim Biophys Acta 894: , 1–10. |
[438] | Nelson O , Supnet C , Tolia A , Horré K , De Strooper B , Bezprozvanny I ((2011) ) Mutagenesis mapping of the presenilin 1 calcium leak conductance pore. J Biol Chem 286: , 22339–22347. |
[439] | Tu H , Nelson O , Bezprozvanny A , Wang Z , Lee SF , Hao YH , Serneels L , De Strooper B , Yu G , Bezprozvanny I ((2006) ) Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell 126: , 981–993. |
[440] | Nelson O , Tu H , Lei T , Bentahir M , De Strooper B , Bezprozvanny I ((2007) ) Familial Alzheimer disease-linked mutations specifically disrupt Ca 2+ leak function of presenilin 1. J Clin Invest 117: , 1230–1239. |
[441] | Simard M , Nedergaard M ((2004) ) The neurobiology of glia in the context of water and ion homeostasis. Neuroscience 129: , 877–896. |
[442] | Salter MW , Hicks JL ((1995) ) ATP causes release of intracellular Ca2+ via the phospholipase C beta/IP3 pathway in astrocytes from the dorsal spinal cord. J Neurosci 15: , 2961–2971. |
[443] | Simpson JE , Ince PG , Shaw PJ , Heath PR , Raman R , Garwood CJ , Gelsthorpe C , Baxter L , Forster G , Matthews FE , Brayne C , Wharton SB , MRC Cognitive Function and Ageing Neuropathology Study Group ((2011) ) Microarray analysis of the astrocyte transcriptome in the aging brain: Relationshito Alzheimer’s pathology and APOE genotype. Neurobiol Aging 32: , 1795–1807. |
[444] | Keller JN , Pang Z , Geddes JW , Begley JG , Germeyer A , Waeg G , Mattson MP ((1997) ) Impairment of glucose and glutamate transport and induction of mitochondrial oxidative stress and dysfunction in synaptosomes by amyloid beta-peptide: Role of the lipid peroxidation product 4-hydroxynonenal. J Neurochem 69: , 273–284. |
[445] | Blanc EM , Keller JN , Fernandez S , Mattson MP ((1998) ) 4-Hydroxynonenal, a lipid peroxidation product, impairs glutamate transport in cortical astrocytes. Glia 22: , 149–160. |
[446] | Mark RJ , Pang Z , Geddes JW , Uchida K , Mattson MP ((1997) ) Amyloid beta-peptide impairs glucose transport in hippocampal and cortical neurons: Involvement of membrane lipid peroxidation. J Neurosci 17: , 1046–1054. |
[447] | Briston T , Roberts M , Lewis S , Powney B , M Staddon J , Szabadkai G , Duchen MR ((2017) ) Mitochondrial permeability transition pore: Sensitivity to opening and mechanistic dependence on substrate availability. Sci Rep 7: , 10492. |
[448] | Baines CP , Kaiser RA , Purcell NH , Blair NS , Osinska H , Hambleton MA , Brunskill EW , Sayen MR , Gottlieb RA , Dorn GW , Robbins J , Molkentin JD ((2005) ) Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434: , 658–662. |
[449] | Crompton M , Virji S , Ward JM ((1998) ) Cyclophilin-D binds strongly to complexes of the voltage-dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. Eur J Biochem 258: , 729–735. |
[450] | Szabó I , Zoratti M ((1993) ) The mitochondrial permeability transition pore may comprise VDAC molecules. I. Binary structure and voltage dependence of the pore. FEBS Lett 330: , 201–205. |
[451] | Vyssokikh MY , Katz A , Rueck A , Wuensch C , Dörner A , Zorov DB , Brdiczka D ((2001) ) Adenine nucleotide translocator isoforms 1 and 2 are differently distributed in the mitochondrial inner membrane and have distinct affinities to cyclophilin D. Biochem J 358: , 349–358. |
[452] | Baines CP ((2007) ) The mitochondrial permeability transition pore as a target of cardioprotective signaling. Am J Physiol Circ Physiol 293: , H903–H904. |
[453] | Krauskopf A , Eriksson O , Craigen WJ , Forte MA , Bernardi P ((2006) ) Properties of the permeability transition in VDAC1-/- mitochondria. Biochim Biophys Acta 1757: , 590–595. |
[454] | Kokoszka JE , Waymire KG , Levy SE , Sligh JE , Cai J , Jones DP , MacGregor GR , Wallace DC ((2004) ) The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 427: , 461–465. |
[455] | Halestrap AP , Woodfield KY , Connern CP ((1997) ) Oxidative stress, thiol reagents, and membrane potential modulate the mitochondrial permeability transition by affecting nucleotide binding to the adenine nucleotide translocase. J Biol Chem 272: , 3346–3354. |
[456] | Khaspekov L , Friberg H , Halestrap A , Viktorov I , Wieloch T ((1999) ) Cyclosporin A and its nonimmunosuppressive analogue N-Me-Val-4-cyclosporin A mitigate glucose/oxygen deprivation-induced damage to rat cultured hippocampal neurons. Eur J Neurosci 11: , 3194–3198. |
[457] | Ichas F , Jouaville LS , Mazat JP ((1997) ) Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell 89: , 1145–1153. |
[458] | Petronilli V , Miotto G , Canton M , Brini M , Colonna R , Bernardi P , Di Lisa F ((1999) ) Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophys J 76: , 725–734. |
[459] | Duchen MR ((2000) ) Mitochondria and Ca2+ in cell physiology and pathophysiology. Cell Calcium 28: , 339–348. |
[460] | Zorov DB , Filburn CR , Klotz LO , Zweier JL , Sollott SJ ((2000) ) Reactive oxygen species (Ros-Induced) Ros release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med 192: , 1001–1014. |
[461] | Du H , Guo L , Fang F , Chen D , Sosunov AA , McKhann GM , Yan Y , Wang C , Zhang H , Molkentin JD , Gunn-Moore FJ , Vonsattel JP , Arancio O , Chen JX , Yan SD ((2008) ) Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer s disease. Nat Med 14: , 1097–1105. |
[462] | Du H , Guo L , Zhang W , Rydzewska M , Yan S ((2011) ) Cyclophilin D deficiency improves mitochondrial function and learning/memory in aging Alzheimer disease mouse model. Neurobiol Aging 32: , 398–406. |
[463] | Du H , Yan SS ((2010) ) Mitochondrial permeability transition pore in Alzheimer’s disease: Cyclophilin D and amyloid beta. Biochim Biophys Acta 1802: , 198–204. |
[464] | Basso E , Fante L , Fowlkes J , Petronilli V , Forte MA , Bernardi P ((2005) ) Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J Biol Chem 280: , 18558–18561. |
[465] | Schinzel AC , Takeuchi O , Huang Z , Fisher JK , Zhou Z , Rubens J , Hetz C , Danial NN , Moskowitz MA , Korsmeyer SJ ((2005) ) Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci U S A 102: , 12005–12010. |
[466] | Guo L , Du H , Yan S , Wu X , McKhann GM , Chen JX , Yan SS ((2013) ) Cyclophilin D deficiency rescues axonal mitochondrial transport in Alzheimer’s neurons. PLoS One 8: , e54914. |
[467] | Elkamhawy A , Park J , Hassan AHE , Ra H , Pae AN , Lee J , Park BG , Moon B , Park HM , Roh EJ ((2017) ) Discovery of 1-(3-(benzyloxy) pyridin-2-yl)-3-(2-(piperazin-1-yl) ethyl) urea: A new modulator for amyloid beta-induced mitochondrial dysfunction. Eur J Med Chem 128: , 56–69. |
[468] | Rao VK , Carlson EA , Yan SS ((2014) ) Mitochondrial permeability transition pore is a potential drug target for neurodegeneration. Biochim Biophys Acta 1842: , 1267–1272. |


