
Calcilytic NPS 2143 Reduces Amyloid Secretion and Increases sAβPPα Release from PSEN1 Mutant iPSC-Derived Neurons
Abstract
Despite numerous efforts and studies over the last three decades, Alzheimer’s disease (AD) remains a disorder not fully understood and incurable so far. Development of induced pluripotent stem cell (iPSC) technology to obtain terminally differentiated neurons from adult somatic cells revolutionized the study of AD, providing a powerful tool for modelling the disease and for screening candidate drugs. Indeed, iPSC reprogramming allowed generation of neurons from both sporadic and familial AD patients with the promise to recapitulate the early pathological mechanisms in vitro and to identify novel targets. Interestingly, NPS 2143, a negative allosteric modulator of the calcium sensing receptor, has been indicated as a possible therapeutic for AD. In the present study, we assessed the potential of our iPSC-based familial AD cellular model as a platform for drug testing. We found that iPSC-derived neurons respond to treatment with γ-secretase inhibitor, modifying the physiological amyloid-β protein precursor (AβPP) processing and amyloid-β (Aβ) secretion. Moreover, we demonstrated the expression of calcium sensing receptor (CaSR) protein in human neurons derived from healthy and familial AD subjects. Finally, we showed that calcilytic NPS 2143 induced a changing of Aβ and sAβPPα secreted into conditioned media and modulation of CaSR and PSEN1 expression at the plasma membrane of AD neurons. Overall, our findings suggest that NPS 2143 affects important AD processes in a relevant in vitro system of familial AD.
INTRODUCTION
Alzheimer’s disease (AD) represents the most common cause of dementia with 50 million patients currently around the world. The lack of a cure and the failure of clinical trials over the years clearly demonstrate that the pathomechanism of AD is not yet completely understood. Moreover, evidence strongly indicates that AD is a multifactorial disease, with several aberrant processes contributing to it [1]. Amyloid accumulation and tau hyperphosphorylation are responsible for extracellular amyloid plaques and intracellular neurofibrillary tangles, respectively, both of which constitute the biochemical signatures of the disease [2–4]. Furthermore, oxidative stress, mitochondrial dysfunction, synaptic failure, and neuroinflammation widely characterize AD [5–8]. In addition to the multiplicity of pathological mechanisms concurring in AD, a main difficulty in studying the disease is represented by the limited access to human brain and the analyses of postmortem cortex samples which give information on the very late stage of the disease, making the understanding of the early pathogenic processes more challenging. Another problematic aspect is the limited relevance of in vitro and in vivo systems used to model AD. Indeed, use of transgenic models overexpressing the clinical mutations in the amyloid-β protein precursor (AβPP) or in the γ-secretase subunits PSEN1 and PSEN2, responsible for the familial form of AD (fAD), often lead to an overestimation of the efficacy of candidate drugs. This was the case for a group of nonsteroidal anti-inflammatory drugs, including flurbiprofen and indomethacin, which act as γ-secretase modulators. Such compounds were highly efficacious in lowering the production of Aβ42 in transgenic immortalized cell lines [9], but failed to exert any positive effect in human-derived neurons [10] and in clinical trials [11]. Moreover, transgenic animal models were ineffective to fully reproduce the pathological aspects of the human disease [12]. In this context, induced pluripotent stem cell (iPSC) technology provided a novel tool to overcome the limitations of transgenic systems and to alternatively approach the study of AD. In the last years, several groups, including ours, generated human iPSC-derived neurons from individuals with familial and sporadic AD. Compared to healthy iPSC neurons, neuronal lines generated from AD subjects presented overall augmented amyloid levels and increased Aβ42/Aβ40 ratios [13–16], which strongly resembles what happens in the brain of the patients. Interestingly, a study conducted on iPSC-three-dimensional (3D) neurons derived from AD subjects revealed a dysfunction of a number of proteins involved in axon growth, mitochondrial function, and oxidative stress, which were comparable to the alterations found in postmortem AD brain tissues [17]. In addition, a recent report established that the Aβ38/Aβ40 ratio measured in cerebrospinal fluid (CSF) from a patient with the AβPP V717I mutation was similar to the ratio observed in the cell culture medium of iPSC-neurons derived from the same subject, whereas the Aβ42/Aβ40 ratio of the patient’s CSF was lower than the ratio in the media from iPSC-neurons, which is consistent with deposition of amyloidogenic Aβ42 into Aβ plaques [18]. Such remarkable results provide the first correlation between CSF profiles from patients and their own Aβ secretome in the differentiated neuronal cultures, thus substantiating patient-derived cells as a reliable system to model the pathomechanisms of the disease [19, 20]. In addition to amyloid defects, tau hyperphosphorylation and increased GSK3β activity were also recapitulated in neurons differentiated from familial and sporadic AD patients [16, 21–23]. Therefore, iPSC-based systems promise to provide more suitable platforms for validation of candidate drugs and for identification of novel therapeutic targets.
In this regard, reports indicated the calcium sensing receptor (CaSR) as a potential key player in AD [24–26]. CaSR is a G-protein coupled receptor (GPCR) mainly expressed in the parathyroid glands where it exerts its primary role in sensing extracellular Ca2+ levels and in regulating parathyroid hormone secretion accordingly [27]. The receptor’s activity can be stimulated or inhibited by calcimimetics and calcilytics, which are positive and negative allosteric modulators, respectively [28]. In addition to Ca2+ ions, the receptor responds to several other agonists and plays different functions, other than maintaining Ca2+ homeostasis, in a tissue-dependent manner [29]. In the nervous system, CaSR is involved in physiological processes such as neurodevelopment [30] and synaptic transmission [31]. Moreover, a role for the receptor in mediating neuronal death upon hypoxia or ischemia [32] and in regulating amyloid production and secretion [33, 34] has been described. Relevant to AD, recent studies showed the capacity of synthetic amyloid-β (Aβ) to bind and activate the CaSR of non-tumorigenic cortical adult human astrocytes (NAHAs) [34, 35]. In particular, NAHAs increased the secreted amount of endogenous amyloid and hyperphosphorylated tau when exposed to synthetic Aβ. Noteworthy, co-treatment with calcilytic NPS 2143 fully inhibited such effects on amyloid and tau [34, 36], while it increased the release of soluble sAβPPα [37]. Based on these results, the authors hypothesized that Aβ/CaSR-activated signaling promoted de novo production and release of amyloid together with over-release of tau, thus feeding a vicious cycle. Accordingly, negative modulation of the receptor with calcilytic efficiently counteracted the Aβ/CaSR-mediated noxious effects by favoring the non-amyloidogenic pathway of AβPP and blocking the vicious cycle [37]. Consequently, inhibition of CaSR was proposed as a relevant approach for AD and the calcilytic NPS 2143 as potential therapeutic.
Considering this promising evidence, we set up a study aimed to assess the effect of NPS 2143 in iPSC-neurons differentiated from a patient with familial AD carrying a genetic mutation in PSEN1. Indeed, we previously characterized iPSC-neurons derived from healthy individuals and from patients with sporadic and familial AD [16]. We found that AD neurons secreted higher levels of amyloid compared to healthy cultures, whereas a significantly higher Aβ42/Aβ40 ratio with respect to control cells was detected only in fAD neurons [16].
To validate our in vitro system as a platform for the screening of potential anti-AD compounds, in the present study we first characterized the modulation of AβPP processing and amyloid secretion in control and fAD neurons, upon exposure to the potent γ-secretase inhibitor, DAPT. Next, we assessed CaSR expression in healthy and fAD differentiated neuronal cells as no evidence exists in this regard. Finally, we evaluated the impact of calcilytic NPS 2143 on amyloid secretion and on location at the plasma membrane of CaSR and proteins relevant for AβPP processing in human iPSC-derived neurons.
MATERIALS AND METHODS
The chemicals used were purchased from Sigma-Aldrich (St. Louis, MO, USA), and the cell culture reagents and culture plates were purchased from Thermo Fisher Scientific (Waltham, MA, USA), unless specified otherwise.
Neuronal differentiation
iPSCs were previously reprogrammed from a fAD patient with a genetic mutation in PSEN1 gene which leads to an amino-acid change (p.Val89Leu), as published in detail earlier [38]. Afterwards, neuronal progenitor cells (NPCs) were obtained and neuronal differentiation was characterized and published recently [16]. In the present study, we used two control NPCs (here named Ctrl-1 and Ctrl-2), derived from two healthy individuals, and the PSEN1 mutant patient iPSC-derived NPCs (named fAD-1), that we characterized in our previous study [16]. After thawing, control and fAD NPCs were plated on the poly-L-ornithine/laminin (POL/L)-coated dishes, and maintained in neural maintenance medium (NMM) (1:1 vol/vol mixture of DMEM/F12 and neurobasal medium, 1×N-2 supplement, 1×B-27 supplement, 1×NEAA, 2 mML-glutamine, 50 U/ml penicillin/streptomycin) supplemented with 10 ng/ml EGF and 10 ng/ml bFGF. To differentiate neuronal progenitors into neurons, NPCs were cultured for six weeks in NMM supplemented with 0.2 mM ascorbic acid and 25 μM β-mercaptoethanol, with medium changed every 3-4 days. Cells were plated on coverslips at a seeding density of 40.000 cells/cm2 for immunocytochemistry (ICC) and calcium imaging and on dishes at density of 100.000 cells/cm2 for ELISA, western blot (WB) and biotinylation experiments. To confirm differentiation into cells of neuronal lineage, neuronal expression markers and functional properties were assessed by ICC and calcium imaging respectively.
Calcium imaging
iPSC-derived neuronal cultures, differentiated for 4 weeks, were incubated in 1 μM fura-2-AM (30 min, 37°C). Coverslips were then placed in a perfusion chamber mounted upon an Olympus IX71 inverted microscope equipped with a Cairn monochromator-based epifluorescence system (Cairn Instruments, Faversham, UK). Solutions and agonists were locally applied to the neurons using a rapid solution changer (RSC160, Intracel RSC160, Intracel, Royston, UK) as follows: 60 mM KCl solution to determine expression of functional voltage-gated Ca2+ channels; 100 μM GABA in physiological extracellular solution (ECS), or in reduced Cl- solution (isosmotic replacement of NaCl with Na-isethionate in extracellular solution, resulting in 7.5 mM Cl- concentration) to test whether GABA induced an excitatory or inhibitory response, representative of immature or mature neuron respectively; 100 μM AMPA and 100 μM Kainic acid to assess expression of ionotropic glutamate receptors. Fura-2 was alternately excited with light of 340 and 380 nm and images were captured at 510 nm using an Orca CCD camera (Hamamatsu Photonics, Welwyn Garden City, UK). Following background subtraction of the emission intensities evoked by each excitation wavelength, emission ratios (340/380) were calculated offline.
Immunocytochemistry
iPSC-derived NPCs and 6-week-long differentiated neuronal cultures were fixed in 4% paraformaldehyde (PFA) (15 min, room temperature, RT), washed twice with phosphate buffered saline (PBS) and permeabilized with 0.2% Triton X-100 in PBS (15 min). Then, cells were blocked with 3% bovine serum albumin (BSA) in the presence of 0.2% Triton X-100 in PBS (1 h, RT). The respective primary antibodies were applied overnight at 4°C (Table 1). To detect the signal, cells were incubated (1 h, RT) with the appropriate secondary antibodies: Alexa Fluor 488 donkey anti-rabbit IgG (H + L) or Alexa Fluor 594 donkey anti-mouse IgG (H + L). Cells were analyzed under fluorescent microscope equipped with 3D imaging module (Axio Imager system with ApoTome; Carl Zeiss MicroImaging GmbH) controlled by AxioVision 4.8.1 software (Carl Zeiss). Alternatively, images were acquired with a Zeiss LSM 880 confocal laser scanning microscope with the ZEN Imaging Software (Carl Zeiss AG, Germany).
Table 1
Primary antibodies
| Application | Antibody | Dilution | Company |
| Immunocytochemistry | Mouse anti-NESTIN | 1:1000 | Merck Millipore |
| Rabbit anti-PAX6 | 1:500 | Covance | |
| Mouse anti-MAP2 | 1:1000 | Merck Millipore | |
| Rabbit anti-TAU | 1:1000 | Dako | |
| Rabbit anti-MAP2 | 1:1000 | Abcam | |
| Mouse anti-CaSR | 1:100 | Thermo Fisher Scientific | |
| Western Blotting | Rabbit anti-APP-CTF | 1:1000 | Thermo Fisher Scientific |
| Mouse anti-CaSR | 1:500 | Thermo Fisher Scientific | |
| Rabbit anti-PSEN1 | 1:1000 | Cell Signaling | |
| Mouse anti-APP | 1:1000 | Biolegend | |
| Goat anti-Integrin α7 | 1:1000 | Santa Cruz Biotechnology | |
| Rabbit anti-GAPDH | 1:10000 | Sigma- Aldrich | |
| Rabbit anti-ADAM10 | 1:1000 | Cell Signaling | |
| Rabbit anti-BACE | 1:1000 | Cell Signaling |
Immunoblotting
iPSC-derived neuronal cultures differentiated for 6 weeks were lysed with RIPA Lysis and Extraction Buffer supplemented with Halt™ Protease and Phosphatase Inhibitor Cocktail and Pierce™ Universal Nuclease for Cell Lysis. Lysed samples were sonicated and centrifugated at 13,000 rpm to collect the supernatants. Total protein concentration was determined using a Pierce BCA Protein Assay Kit. Cell lysates (5–15 μg) were separated on 12% or 7.5% precast gels and transferred to Immun-Blot® PVDF Membrane (Bio-Rad). The membranes were blocked by Tris-buffered saline with Tween 20, TBST (20 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.1% Tween-20) containing 5% BSA or non-fat milk, and then incubated with the respective primary antibody solution overnight at 4°C (Table 1). After washing with TBST, the membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (1 h, RT). Secondary antibodies were goat anti-mouse, goat anti-rabbit, or rabbit anti-goat, according to the host of the used primary antibodies. Signals were detected with Super Signal™ West Dura Extended Duration Substrate by KODAK Gel Logic 1500 Imaging System and Kodak MI SE imaging software. Densitometry measurement of protein bands intensity was carried out using Image Studio™ Lite software (LI-COR). To validate the presence of CaSR in human derived neurons, human hippocampal brain sample (provided by the Human Brain Tissue Bank, HBTB, Semmelweis University, Budapest, Hungary) of a control individual (without neurological disorders) and SH-SY5Y cells transiently transfected with the human HA-tagged-CaSR (hCaSR) DNA cloned into pcDNA5 FRT plasmid (provided by Prof. Hans Bräuner-Osborne, University of Copenhagen, Denmark) were lysed and processed for immunoblotting similarly to iPSC-derived neuronal cultures. Moreover, human kidney tissue lysate (Abcam) was included in western blot analyses as a further positive control of receptor’s expression.
Treatment with γ-secretase inhibitor DAPT and calcilytic NPS 2143
The iPSC-derived neuronal cultures differentiated for 6 weeks were treated with γ-secretase inhibitor DAPT (N-[N-(3,5-difluorophenacetyl)-Lalanyl]- S-phenylglycine t-butyl ester;) or with calcilytic NPS 2143 hydrochloride (2-chloro-6-[(2 R)-3-1,1-dimethyl-2-(2-naphtyl)ethylamino-2-hydroxypropoxy]-benzonitrile hydrochloride; Tocris Bioscience). Both DAPT and NPS 2143 were dissolved in DMSO and next diluted in NMM medium at a final concentration of 1 μM. In the experiments with the γ-secretase inhibitor, cells were treated with DAPT or vehicle for 48 h, followed by media collection and harvesting for protein. For the experiments with NPS 2143, cells were added with fresh media containing NPS 2143 or vehicle. After 24 h, the conditioned media were temporarily collected in sterile tubes and neuronal cultures were exposed for 30 min to either NPS 2143 or vehicle dissolved in fresh medium. Then, the previously cell-conditioned media, supplemented with a second pulse of calcilytic or vehicle, were added again to the plates. At the end of treatments (48 h), collection of conditioned media and cell lysates or biotinylation and isolation of cell surface proteins were performed.
ELISA of Aβ40 and Aβ42 secreted in conditioned media
After 48 h treatment with DAPT or NPS 2143 the conditioned media were collected. Extracellular Aβ40 and Aβ42 levels were measured using Human β-Amyloid (1–40) ELISA Kit and Human β-Amyloid (1–42) ELISA kit (FUJIFILM Wako Pure Chemical Corporation of Japan), according to the manufacturer’s instructions. The signal was detected with Varioskan Flash Multimode Reader (Thermo Fisher Scientific). The secreted Aβ levels were normalized to the total protein content of each cell lysate.
Biotinylation and isolation of plasmalemmal proteins
Following 48 h treatment with calcilytic as detailed above, samples were biotinylated and cell surface proteins were isolated using the Pierce TM Cell Surface Protein Isolation Kit. Briefly, after collection of cell culture media, cells were washed twice with ice-cold PBS and incubated with Sulfo-NHS-SS-Biotin in ice-cold PBS (30 min, 4°C) with gentle agitation. Biotinylation reaction was stopped by adding Quenching Solution. Cells were harvested by gentle scraping and pelleted by centrifugation (500×g, 5 min, 4°C). After washing with TBS, cell pellets were lysed using the provided Lysis Buffer (Pierce) containing a protease inhibitor cocktail (30 min) on ice with intermittent vortexing. Lysates were centrifuged (10,000×g, 2 min, 4°C) and supernatants were incubated for 1 h at RT to allow the biotinylated proteins to bind to the NeutrAvidin Gel. The unbound proteins, representing the intracellular fractions named “flow-throughs” (FT), were collected by centrifugation of the columns (at 1,000×g for 2 min). Finally, the biotinylated surface proteins were incubated with SDS-PAGE Sample Buffer (1 h, RT) and were collected by column centrifugation (1,000×g, 2 min) as “Eluate” fractions (E). FT and E samples were loaded on precast gels for WB analyses.
Statistical analysis
Data were analyzed using GraphPad Prism 5 software. Analysis of data was presented in the form of mean±S.E.M. (standard error of the mean). Statistical significance was tested by either Student’s t-test (two-tailed) or one-way ANOVA with Tukey’s post-test (p < 0.05).
RESULTS
Characterization of iPSC-derived neurons
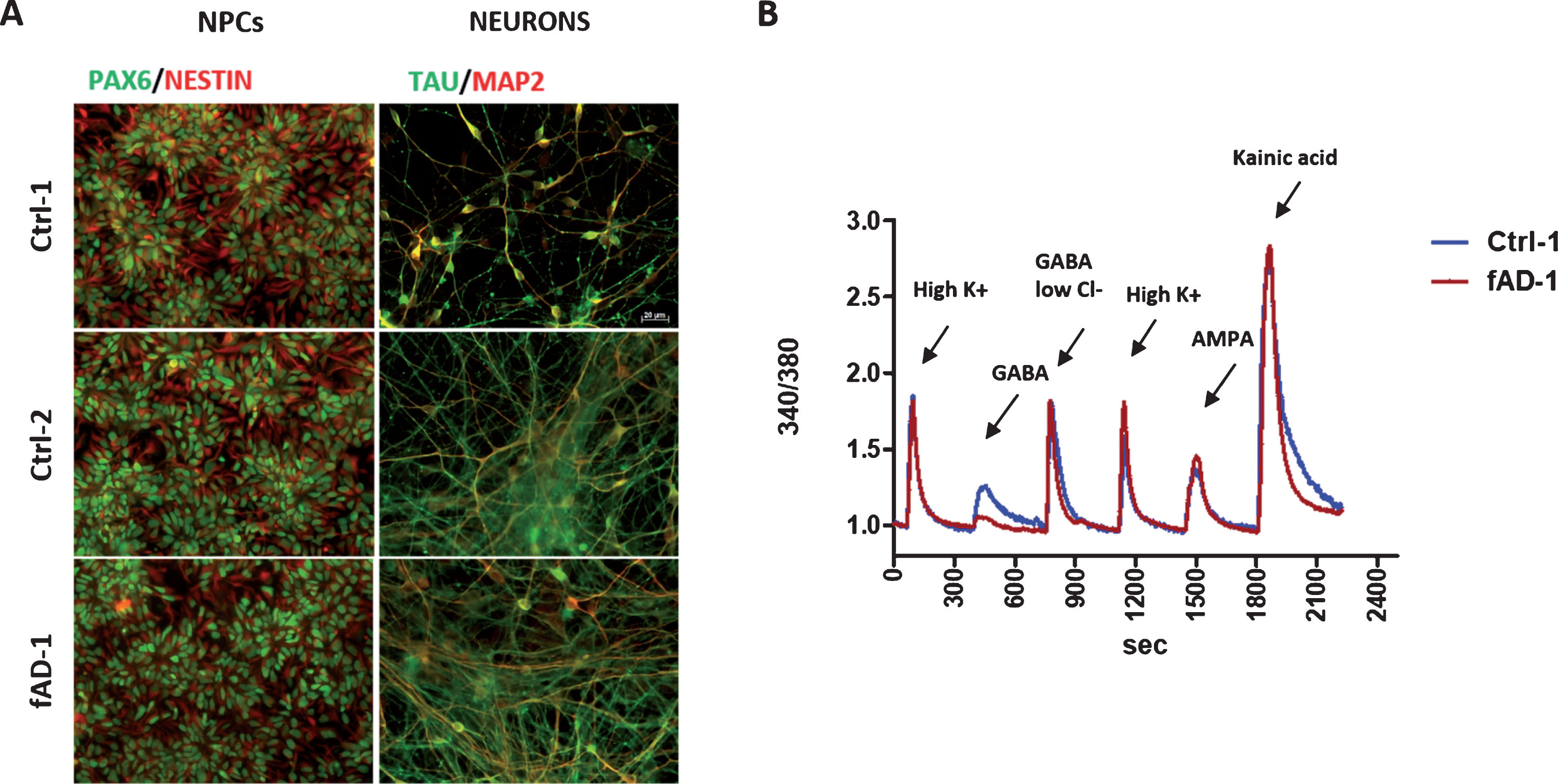
Human iPSC lines, reprogrammed from a fAD patient and from non-AD control individuals, were previously characterized by our group and successfully converted into neural progenitor cells [16, 38]. Here, we let control and fAD NPCs to terminally differentiate toward neuronal cells for 6 weeks. By immunostainings, we confirmed the differentiation of NPCs, stained with the specific markers PAX6 and Nestin, into neurons expressing MAP2 and tau (Fig. 1A). Moreover, Ca2+ imaging analyses conducted on 4-week differentiated control and fAD cultures revealed that cells positively respond to stimulation with high KCl, GABA, AMPA, and kainic acid, demonstrating that neurons express functional voltage gated ion channels, GABA receptors, and ionotropic glutamate receptors, already at this stage of differentiation (representative traces from control and fAD cells are shown in Fig. 1B). In accordance with our previous findings [16], no evident differences between neuronal cultures, differentiated from control and fAD iPSC were observed, which prompted us to use these cell lines for further experiments.
Fig.1
Neuronal differentiation of Ctrl and fAD iPSC-derived neural progenitors, NPCs. A) Representative immunocytochemical analyses showing the differentiation of NPCs, expressing the specific markers PAX6 in nuclei (green) and NESTIN cytoplasmic intermediate filaments (red), in 6-weeks old neurons, stained with MAP2 (red) and TAU (green); for color images, see the online version. Scale bar 20 μM. B) Calcium intracellular influx recorded from fura-2 loaded 4-weeks old neurons (Ctrl-1 and fAD-1 cell lines) in response to 60 mM KCl, 100 μM GABA in physiological extracellular chloride (GABA), 100 μM GABA in 7.5 mM chloride (GABA in low Cl-), 100 μM AMPA and 100 μM Kainic acid, demonstrating functional expression of voltage gated ion channels, GABA receptors and ionotropic glutamate receptors.
Modulation of AβPP processing and amyloid secretion by γ-secretase inhibition in iPSC-derived neuronal cells
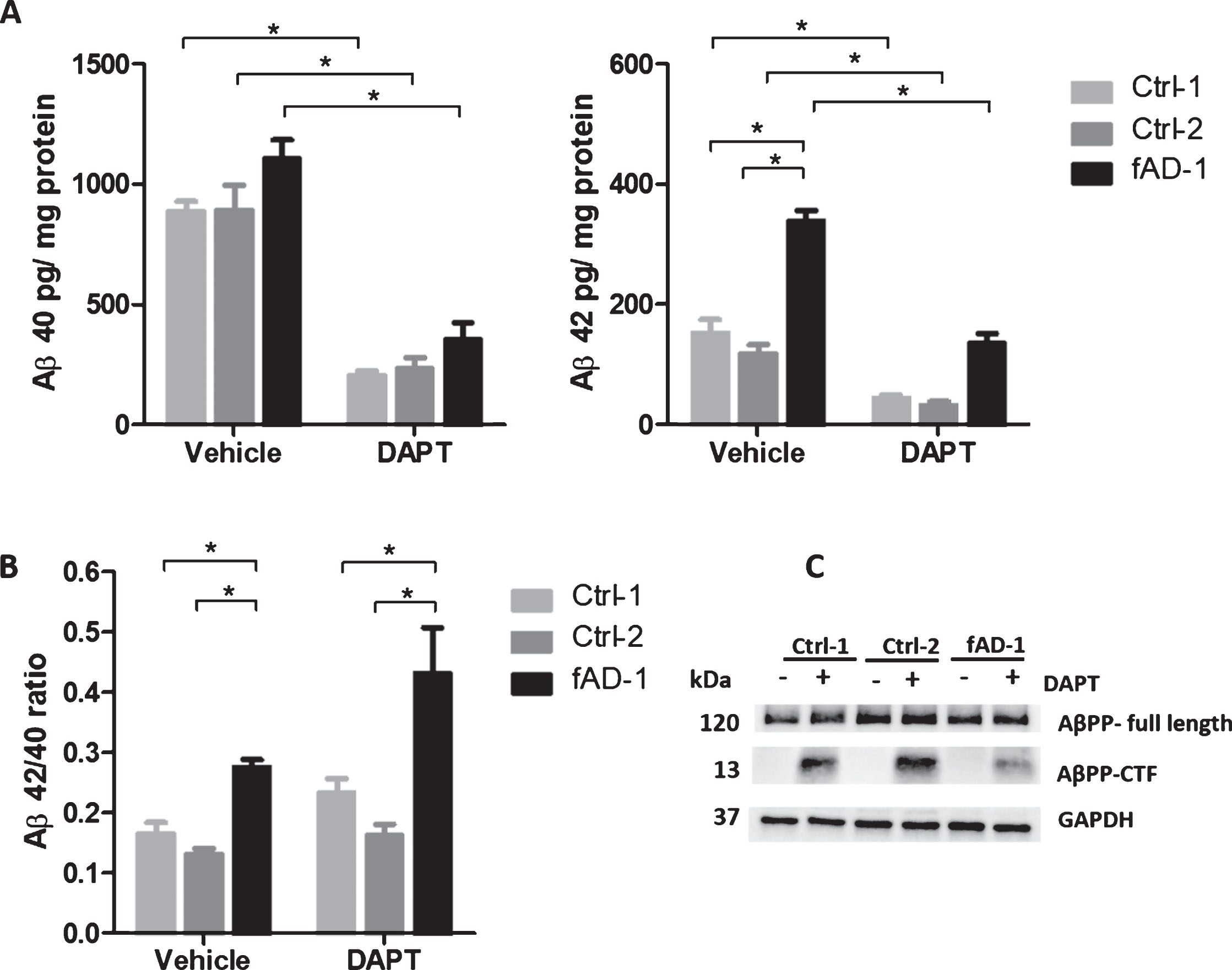
In order to use patient-derived neurons as a cellular system suitable for modeling AD-related physiological changes, we first assessed the effects of the potent γ-secretase inhibitor DAPT on AβPP physiological processing and Aβ secretion. In line with our previous report in which multiple control, sporadic, and familial AD cell lines were analyzed [16], ELISA of conditioned media confirmed that PSEN1 mutant neurons presented a higher Aβ42/Aβ40 ratio compared to control cells in this study (Fig. 2B, Vehicle condition). Data from each Aβ species showed that this ratio change was primarily due to an approximately 2-fold increase in the production of Aβ42 in fAD neurons compared to control cell lines (Fig. 2A, Vehicle condition). Importantly, 48 h treatment with DAPT (1 μM) drastically reduced the secretion of Aβ40 and Aβ42 from both control and fAD neurons (Fig. 2A, DAPT condition). Nevertheless, the ratio between Aβ42 and Aβ40 in fAD cells treated with DAPT remained higher than the ratio observed in control cells (Fig. 2B, DAPT condition). Consistent with inhibition of γ-secretase activity, WB analyses of lysates from healthy and PSEN1 mutant neurons demonstrated that DAPT treatment led to a strong accumulation of AβPP-C terminal fragment (AβPP-CTF), which constitutes the substrate of γ-secretase [39] (Fig. 2C). These observations demonstrated that AβPP and amyloid processing of control and patient-derived neurons efficiently respond to pharmacological modulation with γ-secretase inhibitor, thus allowing the use of this cellular model to explore the potential of calcilytic in these processes.
Fig.2
Pharmacological modulation of amyloid secretion and AβPP processing by DAPT treatment. A) ELISA analyses of Aβ40 and Aβ42 endogenously secreted into conditioned media from controls (Ctrl-1 and Ctrl-2) and fAD cell lines treated with vehicle (0.1% DMSO) or with 1 μM DAPT for 48 h. Data normalized to total mg of protein. B) Ratio between Aβ42 and Aβ40 secreted from controls (Ctrl-1 and Ctrl-2) and fAD cell lines±treatment with 1 μM DAPT for 48 h. C) Representative western blot analysis of total lysates showing the accumulation of AβPP-CTF upon treatment with 1 μM DAPT for 48 h; In A and B, two-tailed t-tests were performed when comparing different conditions (vehicle and DAPT) within the same cell line, while one-way ANOVA performed with Tukey’s multiple comparisons test was applied to find differences between the cell lines. *p < 0.05; Error bars = SEM; ELISA and WB measurements were performed at least as biological triplicates.
CaSR expression in human iPSC-derived neurons
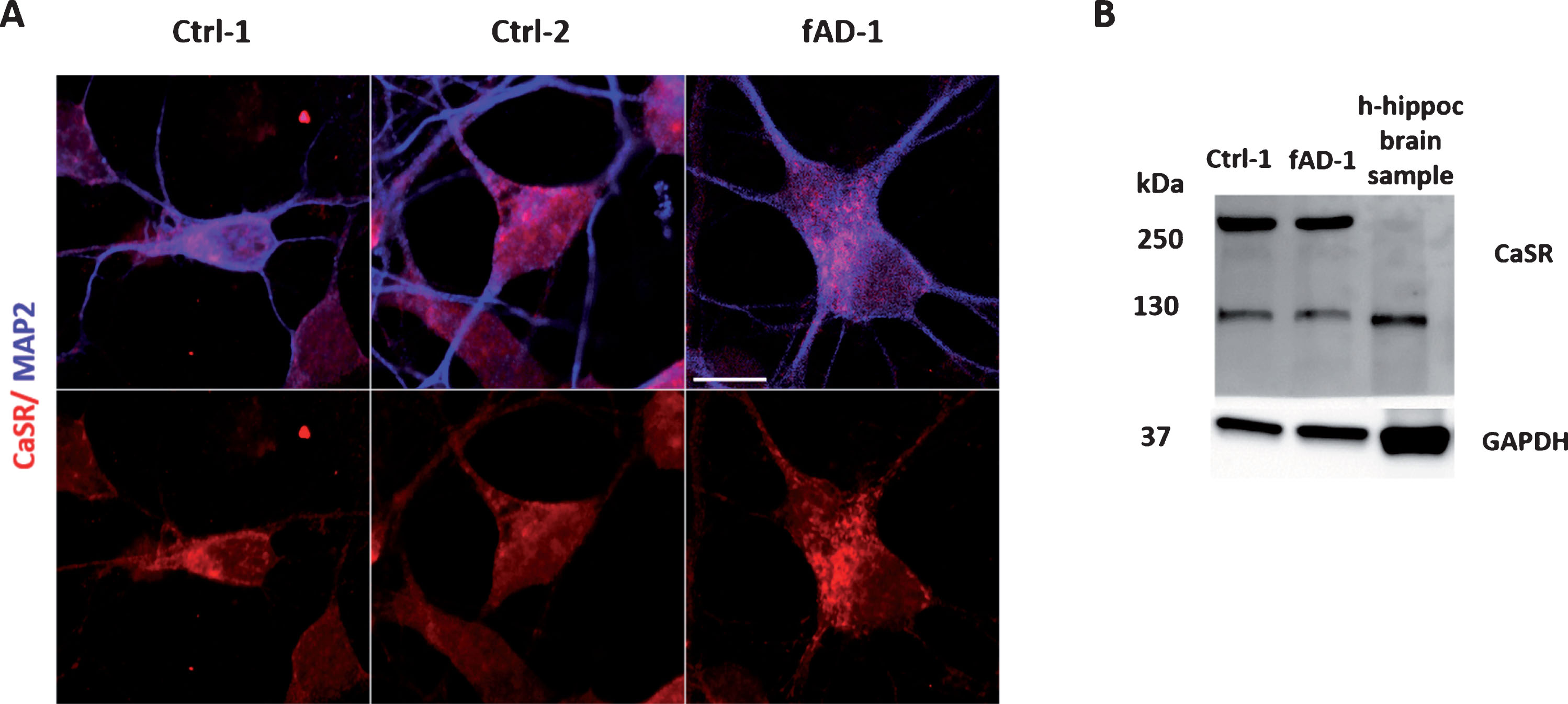
Although there is evidence showing that CaSR is expressed in several regions of human brain [40] and in human astrocytes [41, 42], no data is available in human iPSC-derived neuronal cultures. By ICC, we showed that CaSR is expressed in 6-week-old MAP2 positive neurons differentiated from control and fAD human NPCs (Fig. 3A). Furthermore, WB analyses confirmed that control and fAD cells present CaSR-specific proteins, representing both the monomeric form at ∼120–130 kDa and the dimeric form at ∼250–260 kDa (Fig. 3B). No evident differences were observed between healthy and PSEN1 mutant cells regarding the expression of the receptor. A lower level of receptor protein expression and only the monomeric form of CaSR was detected in control human hippocampal brain postmortem sample (Fig. 3B). Moreover, as positive controls, human neuroblastoma SH-SY5Y, transiently transfected with the HA-tagged human-CaSR, and human kidney lysate showed similar pattern of CaSR expression (Supplementary Figure 1). In conclusion, we detected the expression of CaSR on both control and fAD iPSC-derived human neurons for the first time.
Fig.3
CaSR expression in differentiated neuronal cultures. A) Immunocytochemical analysis showing the CaSR expression (in red) in neurons, co-stained with MAP2 (in blue); for color images see the online version. B) Representative western blot of control and fAD lysates demonstrating the receptor positive bands at ∼130 kDa and ∼260 kDa which should represent the monomeric and dimeric forms of CaSR. Human control adult hippocampal brain lysate has been loaded as positive control of CaSR expression in human brain.
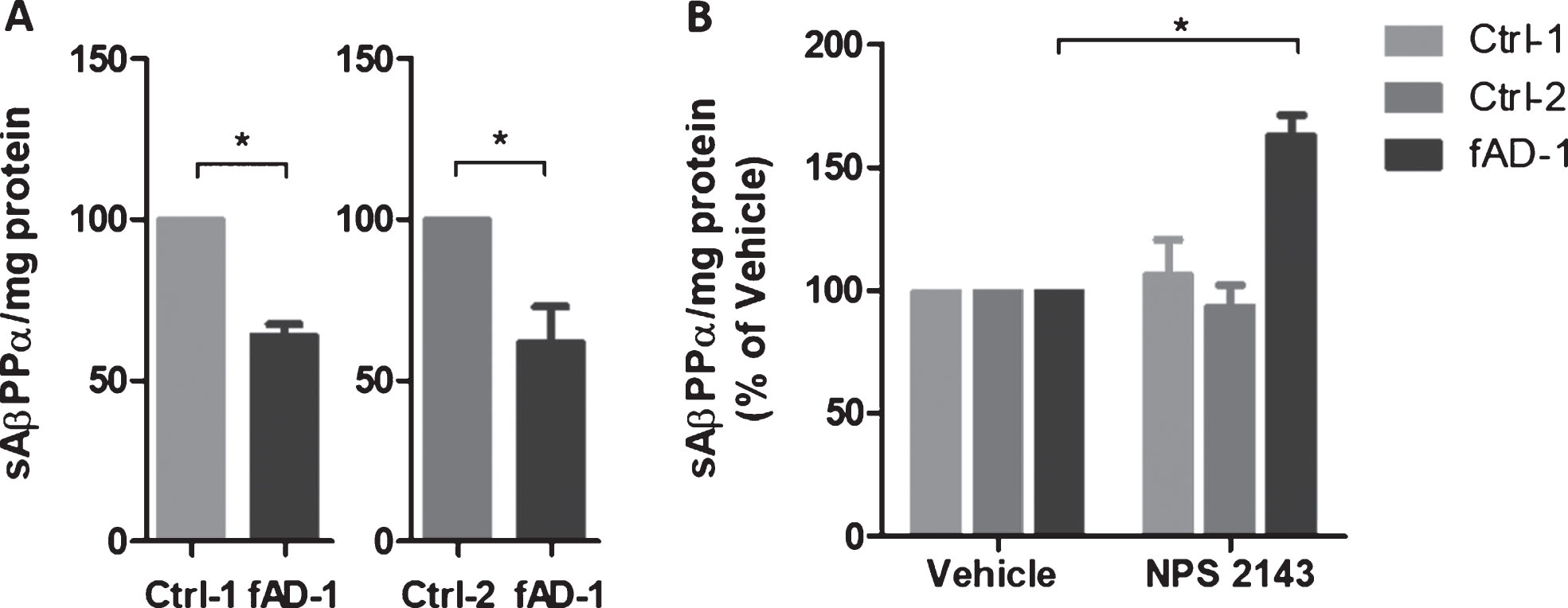
Reduced amyloid secretion and increased sAβPPα release in the media of fAD neural cells treated with calcilytic NPS 2143
Research reported that calcilytic promoted the sAβPPα release while it inhibited Aβ42 accumulation and secretion in human astrocytes treated with exogenous Aβ [34, 37]. To evaluate the effect of calcilytic in iPSC-derived neurons, we treated 6-week-old control and fAD cells with NPS 2143 for 48 h. ELISA analyses of conditioned media revealed that treatment with calcilytic had no significant effect on Aβ secretion in control cell lines (Fig. 4A). On the contrary, NPS 2143 reduced the levels of Aβ40 and Aβ42 in the conditioned media of fAD cells about 25% compared to the vehicle-treated cells (Fig. 4A). Moreover, as calcilytic caused a similar reduction of both amyloid species in fAD neurons, the resulting ratio between the Aβ42 and Aβ40 in PSEN1 mutant cells treated with NPS 2143 was not changed compared to the treatment with vehicle, remaining significantly higher than the ratio showed by the control cell lines (Fig. 4B). However, the levels of AβPP full-length and AβPP-CTF were not modified by calcilytic, which ruled out the possibility that NPS 2143 could act in a γ-secretase inhibitor fashion (Supplementary Figure 2). In addition, we assessed the sAβPPα release extracellularly. Interestingly, WB analyses of conditioned media demonstrated that fAD neurons secreted significantly lower amount of sAβPPα compared to the control cell lines (Fig. 5A). Interestingly, calcilytic strongly increased the release of sAβPPα from fAD cells, whereas no evident effect was observed in control cells (Fig. 5B). All together such results suggest that NPS 2143 favored the AβPP non-amyloidogenic α-pathway, while it decreased the amyloidogenic β-processing only in cells with PSEN1 mutation.
Fig.4
Modulation of amyloid secretion by NPS 2143 treatment. A) ELISA analyses of Aβ40 and Aβ42 endogenously secreted into conditioned media from controls (Ctrl-1 and Ctrl-2) and fAD cell lines treated with vehicle (0.1% DMSO) or with 1 μM NPS 2143 for 48 h. Vehicle treated samples were considered as 100%, while NPS 2143 treated samples were presented in the percentage of the vehicle. Two-tailed t-tests were performed. *p < 0.05; Error bars = SEM. B) Ratio between Aβ42 and Aβ40 endogenously secreted from controls and fAD cell lines upon the treatment with 1 μM NPS 2143 for 48 h. One-way ANOVA performed with Tukey’s multiple comparisons test was applied to find differences between the cell lines. *p < 0.05; Error bars = SEM.
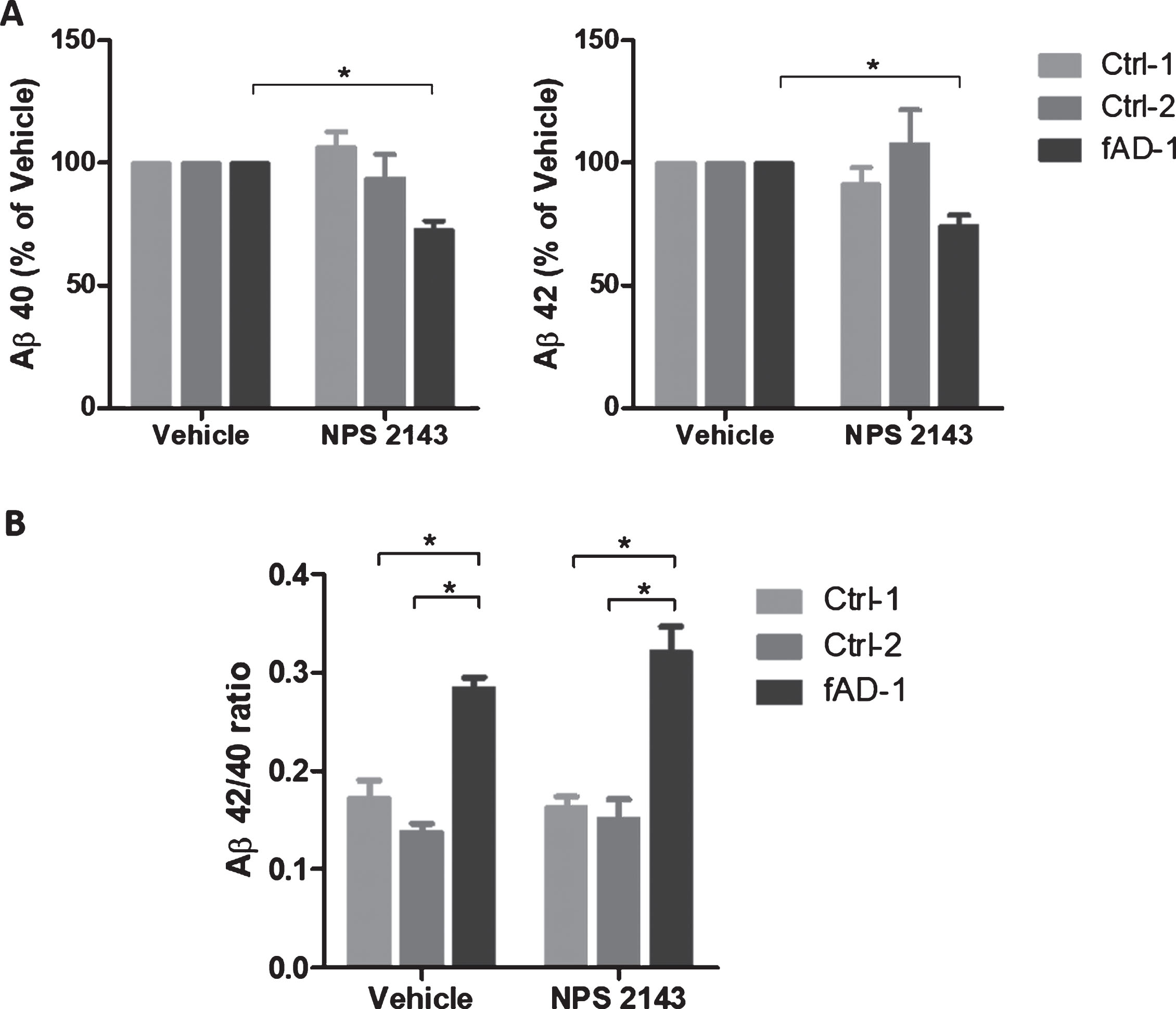
Fig.5
Modulation of sAβPPα secretion by NPS 2143 treatment. A) sAβPPα secreted by control (Ctrl-1 and Ctrl-2) and fAD neuronal cultures into conditioned medium detected by immunoblot with 6E10 antibody. Densitometric values were normalized to total mg of protein. B) sAβPPα secreted by control (Ctrl-1 and Ctrl-2) and fAD neuronal cultures treated with vehicle (0.1% DMSO) or with 1 μM NPS 2143 for 48 h. Vehicle treated samples were considered as 100%, while NPS 2143 treated samples were presented in the percentage of the vehicle. Two-tailed t-tests were performed. *p < 0.05; Error bars = SEM. Measurements were performed at least as biological triplicates.
Modulation of CaSR and PSEN1 expression at the plasma membrane of fAD neurons treated with calcilytic NPS 2143
To explore whether the effects of calcilytic on AβPP processing observed in PSEN1 mutant cells were due to changes in AβPP—or secretase—expression at plasma membrane, 6-week-old neuronal cultures were treated with NPS 2143 and studied for cell surface proteins localization measured by biotinylation assay. Integrin α7 was used as a positive control for the plasma membrane expression. WB analyses of FT and E samples confirmed that Integrin α7 was exclusively observed in the eluted fractions (Fig. 6A, lanes 2, 4, 6, 8). Based on our results, AβPP and CaSR were expressed at both intracellular and plasma membrane level (Fig. 6A). Moreover, we noted that in control and fAD cells, AβPP expression at the cell surface was not significantly changed following the treatment with calcilytic (Fig. 6A, lane 4 versus lane 2; lane 8 versus lane 6, and Fig. 6B). Interestingly, fAD cells treated with NPS 2143 presented significantly reduced levels of CaSR in the E fraction compared to vehicle treatment (Fig. 6A, lane 8 versus lane 6, and Fig. 6C). In contrast, cell surface expression of CaSR was not affected by calcilytic in the control line (Fig. 6A, lane 4 versus lane 2, and Fig. 6C). In addition, we observed that PSEN1 was mainly expressed intracellularly but it also presented discrete levels at the plasma membrane (Fig. 6A). Similar to what we observed for CaSR, cell surface expression of PSEN1 in fAD cultures incubated with calcilytic was decreased compared to treatment with vehicle (Fig. 6A, lane 8 versus lane 6, and Fig 6D). However, NPS 2143 had no evident effect on PSEN1 plasma membrane presence in control cells (Fig. 6A, lane 4 versus lane 2, and Fig. 6D). Despite the reduction of CaSR and PSEN1 at the cell surface of fAD cells treated with calcilytic, we did not detect evident changes in the levels of these proteins in the corresponding FT fractions (Supplementary Figure 3). Finally, levels of α-secretase ADAM10 and β-secretase BACE1 were assessed. The active form of ADAM10 (∼70 kDa) was mainly present in the E fractions in control cells (Supplementary Figure 4, lanes 1–4), while its expression in the FT and E fractions was similar in fAD neurons (Supplementary Figure 4, lanes 5–8). Instead BACE1 was expressed almost exclusively in the FT fractions of healthy and PSEN1 mutant cells (Supplementary Figure 4). However, cellular localization of ADAM10 and BACE1 was not affected by calcilytic treatment in both cell lines (Supplementary Figure 4).
Fig.6
Modulation of AβPP, CaSR, and PSEN1 at the plasma membrane by NPS 2143 treatment. A) Representative western blot bands representing AβPP, CaSR, and PSEN1 present in the intracellular (flow-through fractions, FT) and in the plasma membrane (Eluate, E) fractions of Ctrl-1 and fAD-1 biotinylated samples treated with vehicle (0.1% DMSO) or with 1 μM NPS 2143 for 48 h. The Integrin α7 is a plasma membrane marker and it has been used as a control of the efficiency of biotinylation; After the E fractions bands of (B) AβPP, (C) CaSR, and (D) PSEN1 were normalized to Integrin α7 (only present in the E fractions), the vehicle treated samples were considered as 100%, while NPS 2143 treated samples were presented in the percentage of the vehicle. Two-tailed t-tests were performed. *p < 0.05; Error bars = SEM. Western blot measurements were performed as biological triplicates.
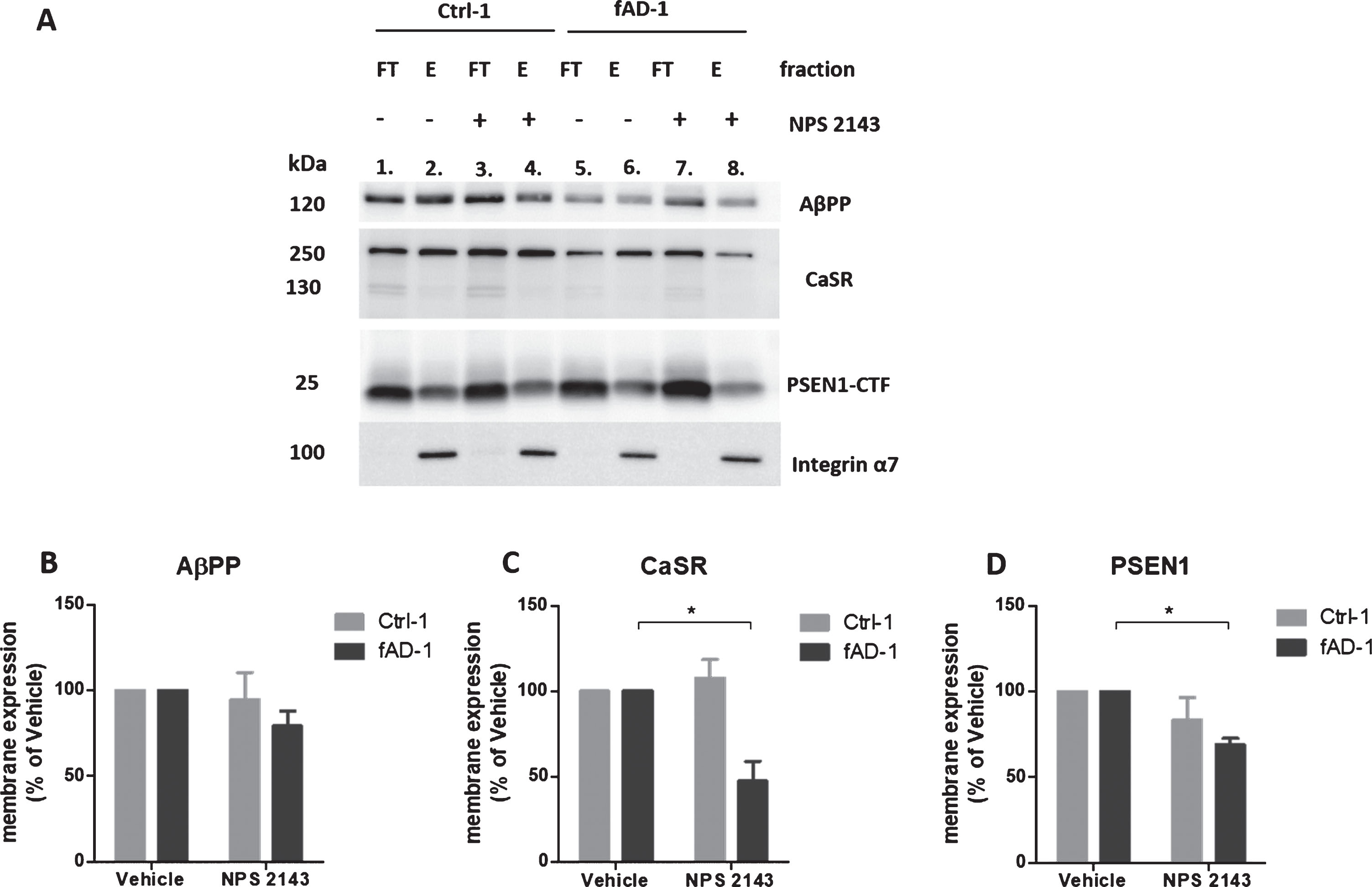
Based on the results of biotinylation studies, we can conclude that NPS 2143 modulated the presence of CaSR and PSEN1 at the plasma membrane of fAD neuronal cultures only.
DISCUSSION
In this study, we provided evidence for the relevance of iPSC-derived neurons as a suitable in vitro system for studying the cellular mechanisms of AD. Immunocytochemistry and calcium imaging analyses demonstrated expression of neuronal markers together with functional ion channels and neurotransmitter receptors in healthy and fAD iPSC-derived neurons. In agreement with our previous work [16], here we confirmed that iPSC-neurons derived from an early-onset fAD patient presented higher secretion of Aβ42 and higher Aβ42/Aβ40 ratio compared to neurons differentiated from two healthy individuals. Such findings are characteristic of AD phenotype and are in line with results from other groups, which also reported increased amyloid levels and amyloid ratios in iPSC-neurons from patients with mutations in PSEN1 and AβPP genes [13, 14, 22]. Human iPSC-neuronal cultures could represent a more relevant system for modeling complex diseases compared to the widely used cellular models, and it might be a promising tool for developing drug discovery platforms which could improve the translation of preclinical studies to patients. Remarkably, the iPSC technology enables the generation of specialized human cells which retain the genetic background of the individual derived from, thus patient-specific. Such unique characteristic allows to test potential personalized therapeutics while the cell lines derived from healthy individuals provide human-based, tissue-specific cell cultures which may possess more closely physiology to humans than transgenic systems derived cell cultures. Notably, we demonstrated that iPSC neurons from healthy and fAD donors drastically reduced endogenous amyloid secretion in response to treatment with the γ-secretase inhibitor DAPT, an effect also observed in other studies [14, 43]. On the other hand, DAPT induced an intracellular accumulation of AβPP-CTF, as expected [44]. Despite great potential, evidence showed that γ-secretase inhibitors tremendously failed as AD therapeutics so far. This poor outcome is due to several reasons [45]. A most problematic aspect is that γ-secretase is responsible for cleaving several substrates other than AβPP, such as Notch, AβPP-like proteins, and N- and E-cadherins [46], therefore its inhibition impacts other important signaling, producing serious side effects in vivo. In addition, accumulation of AβPP-CTF, consequent to γ-secretase blockage, represents another issue as these AβPP intermediates also become toxic when accumulated [48]. Although usage of γ-secretase inhibitors as AD therapeutics is almost out of further consideration nowadays, these compounds are still largely used as useful research tools for biological investigation. In our case, results mediated by DAPT confirmed that iPSC-neurons have a functional γ-secretase activity and a physiological AβPP processing, which allowed us to test the impact of calcilytic NPS 2143 in these cells.
In order to assess the effects of calcilytic in AD neurons, we first evaluated the presence of CaSR in human neuronal cultures. Interestingly, we demonstrated protein expression of CaSR in human iPSC-derived neurons for the first time. We observed that the receptor is expressed in both control and familial neurons at 6 weeks of differentiation. Although a recent study, which analyzed the receptor expression in transgenic AD mice, reported a higher CaSR expression in the hippocampus of AD animals compared to that of control rodents [49], we did not observe evident differences in receptor protein levels between control and AD neurons at the analyzed time of differentiation. Moreover, CaSR was present in both monomeric and dimeric forms in the iPSC-derived neuronal cultures, with predominance of dimers over monomers, while in the human brain hippocampal sample, only the monomeric form was observed. However, the presence of dimers found in the human derived neurons was also substantiated by comparison with HA-tag-hCaSR-transfected SH-SY5Y cells and human kidney tissue lysate. Considering the lack of studies about CaSR expression in iPSC-neurons to which to refer to, we can only speculate that the difference in CaSR dimeric species observed between derived neuronal cultures and hippocampal tissue could be attributable to the much higher complexity of the brain tissue in terms of kind of cells, which in turn might express different levels and forms of CaSR. Further studies are needed to draw a conclusion.
Importantly, we next reported the beneficial effects of calcilytic in counteracting the pathomechanism of AD. We found that calcilytic reduced levels of Aβ40 and Aβ42 secreted from PSEN1 mutant neurons, while amyloid levels were not affected in the control cell lines. Such NPS 2143-mediated results reported only in fAD neurons would indicate that calcilytic can modulate Aβ levels only in the presence of an AD phenotype. In our fAD model, the increased Aβ42 secretion compared to the healthy counterpart could explain the selective effect of NPS 2143 obtained in these cells. Indeed, according to the hypothesized role of Aβ as an activator of the CaSR [35] and due to the higher presence of Aβ42 in the medium of fAD cells, the receptor of AD cultures might be more activated compared to the receptor of healthy cells, thus justifying the evident effect of NPS 2143 observed only in fAD neurons. In agreement, when NAHAs were treated with NPS 2143 alone, it did not modify the basal levels of the intracellular and the secreted amyloid Aβ42 [34]. Only when cells were exposed to synthetic Aβ, which caused a de novo production and release of amyloid, thus mimicking AD condition, addition of calcilytic was beneficial in counteracting the Aβ-mediated noxious effects [34]. In the indicated study, cells were treated with 20 μM synthetic fAβ25 - 35, an Aβ42 proxy, to induce an AD phenotype. Indeed, following this treatment, NAHAs displayed an augmented accumulation and secretion of Aβ42 while in parallel, cells presented higher intracellular amounts of Aβ40 and unchanged extracellular Aβ40 levels [34]. The authors found that co-treatment of fAβ25 - 35 with NPS 2143 fully abolished the Aβ42 intracellular accumulation and release. Instead, co-treatment of fAβ25 - 35 and calcilytic only partially decreased the Aβ-induced intracellular increase of Aβ40, while it promoted its secretion at the same time [34], which suggested that calcilytic differently modulates the secretion of Aβ40 and Aβ42 from fAβ25 - 35 exposed astrocytes. In contrast with this report, we found that NPS 2143 induced a similar reduction of both Aβ40 and Aβ42 species in the medium of our fAD cellular system. Considering that NPS 2143 modified the endogenously secreted amyloid levels of iPSC-derived neurons without any exogenous amyloid treatment, the effects exerted by the calcilytic might be physiologically relevant.
In addition, as a new result, we found that PSEN1 mutant patient iPSC-derived neurons endogenously displayed lower levels of sAβPPα in the conditioned media compared to control cells. This observation represents a remarkable feature of AD phenotype which is recapitulated in our in vitro system. Soluble AβPPα is obtained from the α-secretase-mediated cleavage of AβPP, which initiates the non-amyloidogenic pathway of AβPP [50]. Interestingly, evidence reported the loss of sAβPPα during AD [51] and its beneficial role as neurotrophic factor [52], thus modulation of α-cleavage might be an alternative therapeutic approach [53]. Similar to what was observed for amyloid modulation, NPS 2143 was effective in modifying sAβPPα secretion only in fAD cells. Indeed, treatment with calcilytic significantly increased the release of soluble AβPPα from PSEN1 mutant cells, while it did not change the sAβPPα levels secreted from healthy neurons. Overall the amyloid reduction accompanied with the sAβPPα increase observed in PSEN1 mutant neurons treated with NPS 2143 would suggest a role of calcilytic in decreasing the β-amyloidogenic processing of AβPP and promoting the α-non-amyloidogenic pathway, substantiating the hypothesis corroborated by Chiarini and colleagues [37]. Indeed, in addition to the previously mentioned effect of calcilytic in modulating amyloid accumulation and secretion in cortical adult human astrocytes, NAHAs [34], a role for NPS 2143 has been recently reported in modifying the sAβPPα of NAHAs [37]. According to this study, exposure to 20 μM fAβ25 - 35 induced a reduced secretion while a concomitant intracellular accumulation of endogenous sAβPPα in human adult astrocytes. Interestingly, adding NPS 2143 to fAβ25 - 35 restored the physiological secretion of sAβPPα from the cells whereas it almost completely abolished its intracellular accumulation. The overall effects of NPS 2143 in reducing Aβ42 levels, and partially Aβ40, and increasing the sAβPPα shedding led authors to hypothesize that calcilytic might favor the non-amyloidogenic AβPP processing versus the amyloidogenic cleavage [37]. Chiarini and coworkers further demonstrated that this “switch” from amyloidogenic to non-amyloidogenic AβPP processing exerted by NPS 2143 was due to the capacity of calcilytic to drive the translocation of AβPP and ADAM10 from intracellular compartment to the plasma membrane in human adult astrocyte cultures. Indeed, the authors found that NPS 2143 increased the expression of AβPP and ADAM10 at cell surface of fAβ25 - 35 treated astrocytes [37]. In contrast with this evidence, NPS 2143 had no effect in modifying the cellular location of AβPP and ADAM10 in iPSC-derived neurons. However, we demonstrated that calcilytic significantly reduced the expression of CaSR and PSEN1 at the cell surface of fAD neurons, an effect which was not observed in healthy cells. Moreover, the intracellular levels of these proteins were not significantly modified. The calcilytic-mediated reduction of CaSR expression has been reported also by other groups. Huang and Breitweiser reported that allosteric modulators can regulate turnover of the CaSR, modifying the receptor expression levels [54]. Indeed, the authors found that in HEK293 cells transfected with a wild-type FLAG-CaSR, incubation with NPS 2143 for 12 h led to a reduction of CaSR protein expression compared to DMSO-treated cells. Such a decrease was partially rescued when the proteasomal inhibitor MG132 was added to NPS 2143, suggesting that calcilytic destabilizes CaSR and increases endoplasmic reticulum-associated degradation. Further, treatment with calcilytic modified CaSR function, by reducing the CaSR expression at cell surface and the receptor-mediated ERK1/2 phosphorylation [54]. Moreover, Armato and colleagues observed a transient decrease of the total CaSR protein at 24 h in astrocytes treated with NPS 2143 alone, which was rescued at 48 and 72 h. Instead, the receptor levels fell rapidly and were kept steadily reduced between 24 and 72 h in cells co-treated with NPS 2143 and fAβ25 - 35 [34]. Such effect of NPS 2143 on CaSR expression of human astrocytes, which became strongly evident when calcilytic was added to exogenous Aβ, is in line with the reduction of CaSR observed at the cell surface of fAD-neurons, and it might be connected to the anti-amyloidogenic capacity mediated by NPS 2143 only in the presence of the AD phenotype.
Moreover, the reduction of CaSR at the plasma membrane associated with the decrease of PSEN1 at the cell surface is intriguing and might indicate the probability of a physical interaction between the two proteins. Interestingly, several lines of evidence demonstrated the capability of some GPCRs to physically associate with γ-secretase, modifying its expression and activity. For instance, co-immunoprecipitation assays revealed a physical association of β2-adrenergic receptor (β2AR) with PSEN1 in transfected HEK293 cells [55]. This study reported that agonist of β2AR induced co-internalization of the receptor and PSEN1, which enhanced γ-secretase activity and Aβ production. Conversely, antagonizing of β2AR inhibited Aβ production and reduced cerebral plaques in a transgenic AD mouse model [55]. Similarly, coimmunoprecipitation assays demonstrated the interaction of the δ-opioid receptor (DOR) with PSEN1 and BACE1 which formed a secretases/receptor complex [55, 56]. Activation of DOR mediated the co-endocytic sorting of the GPCR/secretase complex together with an increased activity of secretases and Aβ production. Interestingly, knockdown or antagonization of DOR reduced secretase activities and ameliorated Aβ pathology and Aβ-dependent behavioral deficits, without affecting the processing of Notch, N-cadherin, or APLP, in AD model mice [56]. Although an interaction-based mechanism between CaSR and PSEN1 has not been demonstrated yet, it would represent a possibility to explain the calcilytic-mediated concomitant reduction of these proteins at the cell surface in fAD neurons. Based on our study and on evidence from the literature, we can only speculate that the beneficial effect of calcilytic in ameliorating the AD phenotype of PSEN1 mutant cells is correlated to the CaSR putative heterocomplex with secretases and/or to its internalization and potential degradation mediated by NPS 2143. However, further studies are needed to investigate such complex processes.
Altogether, here we provide evidence that calcilytic positively counteracts amyloid secretion and sAβPPα loss in a relevant model of fAD. It would be interesting to explore whether NPS 2143 produces similar effects in iPSC-neurons derived from individuals with sporadic AD. Indeed, these patients represent the majority of AD cases, totaling about 95% of all patients, whereas only up to 5% are familial cases. Therefore, testing the calcilytic in sporadic iPSC-neurons would be highly relevant and, in the case of a positive outcome, would represent an attractive therapeutic strategy against AD.
ACKNOWLEDGMENTS
We are grateful to Tamas Bellak (BioTalentum Ltd.) for his help with confocal imaging and to Annamária Téglási (BioTalentum Ltd.) for her help in NPC generation and culture. Moreover, we thank Prof. Hans Bräuner-Osborne and Iris Mos for providing the HA tagged CaSR (hCaSR) DNA. We are thankful for the Hungarian Brain Tissue Bank (HBTB) of Semmelweis University (Budapest, Hungary) for providing the human brain sample for this research.
This project has received funding from the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No 675228 (CaSR Biomedicine).
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/19-0602r2).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JAD-190602.
REFERENCES
[1] | Gong CX , Liu F , Iqbal K ((2018) ) Multifactorial hypothesis and multi-targets for Alzheimer’s disease. J Alzheimers Dis 64: , S107–S117. |
[2] | Polanco JC , Li C , Bodea LG , Martinez-Marmol R , Meunier FA , Götz J ((2017) ) Amyloid-β and tau complexity—towards improved biomarkers and targeted therapies. Nat Rev Neurol 14: , 22–39. |
[3] | Hardy J , Selkoe DJ ((2002) ) The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 297: , 353–356. |
[4] | Chong FP , Ng KY , Koh RY , Chye SM ((2018) ) Tau proteins and tauopathies in Alzheimer’s disease. Cell Mol Neurobiol 38: , 965–980. |
[5] | Guo L , Tian J , Du H ((2017) ) Mitochondrial dysfunction and synaptic transmission failure in Alzheimer’s disease. J Alzheimers Dis 57: , 1071–1086. |
[6] | Butterfield DA , Halliwell B ((2019) ) Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat Rev Neurosci 20: , 148–160. |
[7] | Chen Y , Fu AKY , Ip NY ((2019) ) Synaptic dysfunction in Alzheimer’s disease: Mechanisms and therapeutic strategies. Pharmacol Ther 195: , 186–198. |
[8] | Heppner FL , Ransohoff RM , Becher B ((2015) ) Immune attack: The role of inflammation in Alzheimer disease. Nat Rev Neurosci 16: , 358–372. |
[9] | Weggen S , Eriksen JL , Das P , Sagi SA , Wang R , Pietrzik CU , Findlay KA , Smith TE , Murphy MP , Bulter T , Kang DE , Marquez-Sterling N , Golde TE , Koo EH ((2001) ) A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature 414: , 212–216. |
[10] | Mertens J , Stüber K , Wunderlich P , Ladewig J , Kesavan JC , Vandenberghe R , Vandenbulcke M , van Damme P , Walter J , Brüstle O , Koch P ((2013) ) APP processing in human pluripotent stem cell-derived neurons is resistant to NSAID-based γ-secretase modulation. Stem Cell Reports 1: , 491–498. |
[11] | de Jong D , Jansen R , Hoefnagels W , Jellesma-Eggenkamp M , Verbeek M , Borm G , Kremer B ((2008) ) No effect of one-year treatment with Indomethacin on Alzheimer’s disease progression: A randomized controlled trial. PLoS One 3: , e1475. |
[12] | Mullane K , Williams M ((2019) ) Preclinical models of Alzheimer’s disease: Relevance and translational validity. Curr Protoc Pharmacol 84: , e57. |
[13] | Yagi T , Ito D , Okada Y , Akamatsu W , Nihei Y , Yoshizaki T , Yamanaka S , Okano H , Suzuki N ((2011) ) Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum Mol Genet 20: , 4530–4539. |
[14] | Muratore CR , Rice HC , Srikanth P , Callahan DG , Shin T , Benjamin LNP , Walsh DM , Selkoe DJ , Young-Pearse TL ((2014) ) The familial Alzheimer’s disease APPV717I mutation alters APP processing and tau expression in iPSC-derived neurons. Hum Mol Genet 23: , 3523–3536. |
[15] | Mahairaki V , Ryu J , Peters A , Chang Q , Li T , Park TS , Burridge PW , Talbot CC , Asnaghi L , Martin LJ , Zambidis ET , Koliatsos VE ((2014) ) Induced pluripotent stem cells from familial Alzheimer’s disease patients differentiate into mature neurons with amyloidogenic properties. Stem Cells Dev 23: , 2996–3010. |
[16] | Ochalek A , Mihalik B , Avci HX , Chandrasekaran A , Téglási A , Bock I , Giudice ML , Táncos Z , Molnár K , László L , Nielsen JE , Holst B , Freude K , Hyttel P , Kobolák J , Dinnyés A ((2017) ) Neurons derived from sporadic Alzheimer’s disease iPSCs reveal elevated TAU hyperphosphorylation, increased amyloid levels, and GSK3B activation. Alzheimers Res Ther 9: , 90. |
[17] | Chen M , Lee H-K , Moo L , Hanlon E , Stein T , Xia W ((2018) ) Common proteomic profiles of induced pluripotent stem cell-derived three-dimensional neurons and brain tissue from Alzheimer patients. J Proteomics 182: , 21–33. |
[18] | Arber C , Toombs J , Lovejoy C , Ryan NS , Paterson RW , Willumsen N , Gkanatsiou E , Portelius E , Blennow K , Heslegrave A , Schott JM , Hardy J , Lashley T , Fox NC , Zetterberg H , Wray S ((2019) ) Familial Alzheimer’s disease patient-derived neurons reveal distinct mutation-specific effects on amyloid beta. Mol Psychiatry, doi: 10.1038/s41380-019-0410-8 |
[19] | Liu Q , Waltz S , Woodruff G , Ouyang J , Israel MA , Herrera C , Sarsoza F , Tanzi RE , Koo EH , Ringman JM , Goldstein LSB , Wagner SL , Yuan SH ((2014) ) Effect of potent γ-secretase modulator in human neurons derived from multiple presenilin 1-induced pluripotent stem cell mutant carriers. JAMA Neurol 71: , 1481–1489. |
[20] | Kondo T , Imamura K , Funayama M , Tsukita K , Miyake M , Ohta A , Woltjen K , Nakagawa M , Asada T , Arai T , Kawakatsu S , Izumi Y , Kaji R , Iwata N , Inoue H ((2017) ) iPSC-based compound screening and in vitro trials identify a synergistic anti-amyloid β combination for Alzheimer’s disease. Cell Rep 21: , 2304–2312. |
[21] | Israel MA , Yuan SH , Bardy C , Reyna SM , Mu Y , Herrera C , Hefferan MP , Van Gorp S , Nazor KL , Boscolo FS , Carson CT , Laurent LC , Marsala M , Gage FH , Remes AM , Koo EH , Goldstein LSB ((2012) ) Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 482: , 216–220. |
[22] | Choi SH , Kim YH , Hebisch M , Sliwinski C , Lee S , D’Avanzo C , Chen H , Hooli B , Asselin C , Muffat J , Klee JB , Zhang C , Wainger BJ , Peitz M , Kovacs DM , Woolf CJ , Wagner SL , Tanzi RE , Kim DY ((2014) ) A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 515: , 274–278. |
[23] | Chang K-H , Lee-Chen G-J , Huang C-C , Lin J-L , Chen Y-J , Wei P-C , Lo Y-S , Yao C-F , Kuo M-W , Chen C-M ((2018) ) Modeling Alzheimer’s disease by induced pluripotent stem cells carrying APP D678H mutation. Mol Neurobiol 56: , 3972–3983. |
[24] | Chiarini A , Armato U , Liu D , Dal Prà I ((2016) ) Calcium-sensing receptors of human neural cells play crucial roles in Alzheimer’s disease. Front Physiol 7: , 134. |
[25] | Chiarini A , Armato U , Whitfield JF , Pra ID ((2018) ) Targeting human astrocytes’ calcium-sensing receptors for treatment of Alzheimer’s disease. Curr Pharm Des 23: , 4990–5000. |
[26] | Giudice M Lo , Mihalik B , Dinnyés A , Kobolák J ((2019) ) The nervous system relevance of the calcium sensing receptor in health and disease. Molecules 24: , 2546. |
[27] | Conigrave AD ((2016) ) The calcium-sensing receptor and the parathyroid: Past, present, future. Front Physiol 7: , 563. |
[28] | Nemeth EF ((2013) ) Allosteric modulators of the extracellular calcium receptor. Drug Discov Today Technol 10: , e277–e284. |
[29] | Hofer AM , Brown EM ((2003) ) Extracellular calcium sensing and signalling. Nat Rev Mol Cell Biol 4: , 530–538. |
[30] | Liu X-L , Lu Y-S , Gao J-Y , Marshall C , Xiao M , Miao D-S , Karaplis A , Goltzman D , Ding J ((2013) ) Calcium sensing receptor absence delays postnatal brain development via direct and indirect mechanisms. Mol Neurobiol 48: , 590–600. |
[31] | Phillips CG , Harnett MT , Chen W , Smith SM ((2008) ) Calcium-sensing receptor activation depresses synaptic transmission. J Neurosci 28: , 12062–12070. |
[32] | Kim JY , Ho H , Kim N , Liu J , Tu C-L , Yenari MA , Chang W ((2014) ) Calcium-sensing receptor (CaSR) as a novel target for ischemic neuroprotection. Ann Clin Transl Neurol 1: , 851–866. |
[33] | Bai S , Mao M , Tian L , Yu Y , Zeng J , Ouyang K , Yu L , Li L , Wang D , Deng X , Wei C , Luo Y ((2015) ) Calcium sensing receptor mediated the excessive generation of β-amyloid peptide induced by hypoxia in vivo and in vitro. Biochem Biophys Res Commun 459: , 568–573. |
[34] | Armato U , Chiarini A , Chakravarthy B , Chioffi F , Pacchiana R , Colarusso E , Whitfield JF , Dal Prà I ((2013) ) Calcium-sensing receptor antagonist (calcilytic) NPS 2143 specifically blocks the increased secretion of endogenous Aβ42 prompted by exogenous fibrillary or soluble Aβ25-35 in human cortical astrocytes and neurons—Therapeutic relevance to Alzheimer’s disease. Biochim Biophys Acta 1832: , 1634–1652. |
[35] | Dal Prà I , Chiarini A , Pacchiana R , Gardenal E , Chakravarthy B , Whitfield JF , Armato U ((2014) ) Calcium-sensing receptors of human astrocyte-neuron teams: Amyloid-β-driven mediators and therapeutic targets of Alzheimer’s disease. Curr Neuropharmacol 12: , 353–364. |
[36] | Chiarini A , Armato U , Gardenal E , Gui L , Dal Prà I ((2017) ) Amyloid β-exposed human astrocytes overproduce phospho-tau and overrelease it within exosomes, effects suppressed by calcilytic NPS 2143—further implications for Alzheimer’s therapy. Front Neurosci 11: , 217. |
[37] | Chiarini A , Armato U , Liu D , Dal Prà I ((2017) ) Calcium-sensing receptor antagonist NPS 2143 restores amyloid precursor protein physiological non-amyloidogenic processing in Aβ-exposed adult human astrocytes. Sci Rep 7: , 1277. |
[38] | Nemes C , Varga E , Táncos Z , Bock I , Francz B , Kobolák J , Dinnyés A ((2016) ) Establishment of PSEN1 mutant induced pluripotent stem cell (iPSC) line from an Alzheimer’s disease (AD) female patient. Stem Cell Res 17: , 69–71. |
[39] | Morohashi Y , Kan T , Tominari Y , Fuwa H , Okamura Y , Watanabe N , Sato C , Natsugari H , Fukuyama T , Iwatsubo T , Tomita T ((2006) ) C-terminal fragment of presenilin is the molecular target of a dipeptidic gamma-secretase-specific inhibitor DAPT (N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester). J Biol Chem 281: , 14670–14676. |
[40] | Kapoor A , Satishchandra P , Ratnapriya R , Reddy R , Kadandale J , Shankar SK , Anand A ((2008) ) An idiopathic epilepsy syndrome linked to 3q13.3-q21 and missense mutations in the extracellular calcium sensing receptor gene. Ann Neurol 64: , 158–167. |
[41] | Chattopadhyay N , Evliyaoglu C , Heese O , Carroll R , Sanders J , Black P , Brown EM ((2000) ) Regulation of secretion of PTHrP by Ca 2+ -sensing receptor in human astrocytes, astrocytomas, and meningiomas. Am J Physiol Physiol 279: , C691–C699. |
[42] | Dal Pra I , Chiarini A , Nemeth EF , Armato U , Whitfield JF ((2005) ) Roles of Ca2+ and the Ca2+-sensing receptor (CASR) in the expression of inducible NOS (nitric oxide synthase)-2 and its BH4 (tetrahydrobiopterin)-dependent activation in cytokine-stimulated adult human astrocytes. J Cell Biochem 96: , 428–438. |
[43] | Shi Y , Kirwan P , Smith J , MacLean G , Orkin SH , Livesey FJ ((2012) ) A human stem cell model of early Alzheimer’s disease pathology in Down syndrome. Sci Transl Med 4: , 124ra29. |
[44] | Koch P , Tamboli IY , Mertens J , Wunderlich P , Ladewig J , Stüber K , Esselmann H , Wiltfang J , Brüstle O , Walter J ((2012) ) Presenilin-1 L166P mutant human pluripotent stem cell-derived neurons exhibit partial loss of γ-secretase activity in endogenous amyloid-β generation. Am J Pathol 180: , 2404–2416. |
[45] | Wolfe MS ((2014) ) Unlocking truths of γ-secretase in Alzheimer’s disease: What is the translational potential? Future Neurol 9: , 419–429. |
[46] | Haapasalo A , Kovacs DM ((2011) ) The many substrates of presenilin/γ-secretase. J Alzheimers Dis 25: , 3–28. |
[47] | Searfoss GH , Jordan WH , Calligaro DO , Galbreath EJ , Schirtzinger LM , Berridge BR , Gao H , Higgins MA , May PC , Ryan TP ((2003) ) Adipsin, a biomarker of gastrointestinal toxicity mediated by a functional γ-secretase inhibitor. J Biol Chem 278: , 46107–46116. |
[48] | Mitani Y , Yarimizu J , Saita K , Uchino H , Akashiba H , Shitaka Y , Ni K , Matsuoka N ((2012) ) Differential effects between secretase inhibitors and modulators on cognitive function in amyloid precursor protein-transgenic and nontransgenic mice. J Neurosci 32: , 2037–2050. |
[49] | Gardenal E , Chiarini A , Armato U , Dal Prà I , Verkhratsky A , Rodríguez JJ ((2017) ) Increased calcium-sensing receptor immunoreactivity in the hippocampus of a triple transgenic mouse model of Alzheimer’s disease. Front Neurosci 11: , 81. |
[50] | Lammich S , Kojro E , Postina R , Gilbert S , Pfeiffer R , Jasionowski M , Haass C , Fahrenholz F ((1999) ) Constitutive and regulated alpha-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci U S A 96: , 3922–3927. |
[51] | Colciaghi F , Borroni B , Pastorino L , Marcello E , Zimmermann M , Cattabeni F , Padovani A , Di Luca M ((2002) ) [alpha]-secretase ADAM10 as well as [alpha]APPs is reduced in platelets and CSF of Alzheimer disease patients. Mol Med 8: , 67–74. |
[52] | Habib A , Sawmiller D , Tan J ((2017) ) Restoring soluble amyloid precursor protein α functions as a potential treatment for Alzheimer’s disease. J Neurosci Res 95: , 973–991. |
[53] | Postina R , Schroeder A , Dewachter I , Bohl J , Schmitt U , Kojro E , Prinzen C , Endres K , Hiemke C , Blessing M , Flamez P , Dequenne A , Godaux E , van Leuven F , Fahrenholz F ((2004) ) A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model. J Clin Invest 113: , 1456–1464. |
[54] | Huang Y , Breitwieser GE ((2007) ) Rescue of calcium-sensing receptor mutants by allosteric modulators reveals a conformational checkpoint in receptor biogenesis. J Biol Chem 282: , 9517–9525. |
[55] | Ni Y , Zhao X , Bao G , Zou L , Teng L , Wang Z , Song M , Xiong J , Bai Y , Pei G ((2006) ) Activation of β2-adrenergic receptor stimulates γ-secretase activity and accelerates amyloid plaque formation. Nat Med 12: , 1390–1396. |
[56] | Teng L , Zhao J , Wang F , Ma L , Pei G ((2010) ) A GPCR/secretase complex regulates β- and γ-secretase specificity for Aβ production and contributes to AD pathogenesis. Cell Res 20: , 138–153. |


