
Cofilin, a Master Node Regulating Cytoskeletal Pathogenesis in Alzheimer’s Disease
Abstract
The defining pathological hallmarks of Alzheimer’s disease (AD) are proteinopathies marked by the amyloid-β (Aβ) peptide and hyperphosphorylated tau. In addition, Hirano bodies and cofilin-actin rods are extensively found in AD brains, both of which are associated with the actin cytoskeleton. The actin-binding protein cofilin known for its actin filament severing, depolymerizing, nucleating, and bundling activities has emerged as a significant player in AD pathogenesis. In this review, we discuss the regulation of cofilin by multiple signaling events impinging on LIM kinase-1 (LIMK1) and/or Slingshot homolog-1 (SSH1) downstream of Aβ. Such pathophysiological signaling pathways impact actin dynamics to regulate synaptic integrity, mitochondrial translocation of cofilin to promote neurotoxicity, and formation of cofilin-actin pathology. Other intracellular signaling proteins, such as β-arrestin, RanBP9, Chronophin, PLD1, and 14-3-3 also impinge on the regulation of cofilin downstream of Aβ. Finally, we discuss the role of activated cofilin as a bridge between actin and microtubule dynamics by displacing tau from microtubules, thereby destabilizing tau-induced microtubule assembly, missorting tau, and promoting tauopathy.
INTRODUCTION
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder and the most common form of dementia, accounting for ∼65% of all dementia cases [1, 2]. Two pathological hallmarks define AD brains, namely the accumulation of senile plaques composed of the amyloid-β (Aβ) peptide and neurofibrillary tangles/neuropil threads composed of hyperphosphorylated tau. Aβ is a peptide derived from the amyloid-β protein precursor (AβPP) via two sequential proteolytic cleavages by BACE1 (β-secretase) and the presenilin complex (γ-secretase) [3]. Neurofibrillary tangles and neuropil threads are intracellular inclusions principally composed of the microtubule-associated protein tau in hyperphosphorylated form [4], which are ultrastructurally seen as paired helical filament [5]. In addition to Aβ and tau, multiple other proteinopathies are found in brains of AD and related dementias, including as α-synuclein-containing Lewy bodies [6] and TDP-43 inclusions [7], as well as Hirano bodies [8, 9] and cofilin-actin rods [10], the latter two being associated with the actin cytoskeleton.
Aβ42 AND TAU IN AD PATHOGENESIS
Evidence of an early causal role of Aβ (i.e., Aβ cascade hypothesis) [11] is supported by multiple lines of genetic evidence, such as mutations in APP and PSEN1 or PSEN2 that co-segregate with early-onset familial AD. All APP FAD mutations identified thus far are concentrated near the β-secretase or γ-secretase cleavage sites in AβPP, which lead to increases in total Aβ or the more pathogenic Aβ42 peptide [12]. PSEN1 or PSEN2 mutations also increase the ratio of Aβ42 to shorter Aβ peptides [13]. Aβ42, while generated at less than 15% of Aβ40 from wild type AβPP in experimental cell models, aggregates faster and seeds the aggregation of Aβ40 [14]. Transgenic mice engineered to produce only Aβ40 or Aβ42 cleaved from the familial British and Danish Dementia-related BRI protein demonstrates that Aβ40 alone cannot form aggregates into plaques even by 18 months of age, while a lesser concentration of Aβ42 induces robust plaque formation even at 12 months of age. Furthermore, the BRI-Aβ42 mice crossed with APP Tg2576 mice bearing the “Swedish” mutation exponentially exacerbates parenchymal amyloid burden [15].
Aβ exists as soluble monomers, dimers, trimers, and higher order oligomers prior to assembly into protofibrils and insoluble amyloid fibrils [14]. Indeed, the early folding properties of Aβ42 and Aβ40 differ in that Aβ42 populates a more stable structured oligomeric state than Aβ40 [16]. Soluble Aβ oligomers can induce synaptic dysfunction at picomolar concentrations, and SDS-stable dimers and trimers impair long term potentiation (LTP) in rats in vivo at subnanomolar concentrations [17]. Furthermore, soluble SDS-resistant Aβ dimers derived from AD brains promote hyperphosphorylation of tau and neuritic degeneration in primary hippocampal neurons at picomolar concentrations [18]. A mutation in APP identified in a Japanese family with dementia [deletion of residue 22 glutamic acid of the Aβ peptide (E22Δ)] produces an Aβ peptide that is more resistant to degradation, unable to form fibrils, but is far more prone to self-association as Aβ oligomers [19]. Expression of this APP mutation in transgenic mice leads to learning and memory deficits associated with impaired LTP, enhanced neuroinflammation, and tau hyperphosphorylation in the absence of thioflavin S-positive amyloid plaques. However, intracellular E22Δ Aβ oligomers accumulate in an age-dependent fashion, indicating that Aβ oligomers are sufficient and fibrillar amyloid deposition is not necessary for Aβ-induced neurotoxicity and memory deficits [20].
Numerous studies have shown that Aβ promotes the hyperphosphorylation of tau in vitro and in vivo [21, 22]. Aβ also enhances tauopathy in transgenic mice engineered to express a frontotemporal dementia (FTDP-17) tau mutation [23]. Depletion of Aβ by injection of an antibody directed against Aβ reduces not only Aβ pathology but also tau pathology in the APP/tau/presenilin-1 mutant (3xTg) mice [24]. However, despite the tauopathy promoting effects of Aβ, the toxic effects of Aβ require the presence of tau. For example, neurite retraction and progressive neuronal atrophy are seen when neurons are treated with Aβ but not in neurons derived from tau knockout mice [18, 25]. Learning and memory impairment as well as high sensitivity to excitotoxin treatment are present in mutant APP transgenic mice but not in the same transgenic mice on a tau-knockout background, even though the level of Aβ deposition is unaffected by tau [26]. While Aβ induces impairments in LTP and axonal transport of mitochondria, such impairments are lost in tau knockout neurons [27], indicating that tau is required for multiple facets of Aβ-induced neurotoxicity.
One of the earliest changes in tau observed in AD is the mislocalization of tau from somatoaxonal to somatodendritic compartments [28, 29]. Hyperphosphorylation of tau is linked to reduced affinity for microtubules and mislocalization to dendritic spines, where tau promotes the removal of surface AMPA and NMDA receptors critical for excitatory synaptic transmission [30]. When expressed in neurons, tau lacking multiple phosphorylation sites fails to mislocalize tau to dendritic spines and affect excitatory synaptic transmission [30]. Notably, like soluble Aβ oligomers, soluble hyperphosphorylated tau rather than insoluble tau aggregates may be the toxic species, since turning off FTD mutant tau expression in an inducible transgenic model does not remove insoluble PHF-1 positive tangle-like structures over several months but improves learning and memory [31].
These results all indicate that Aβ-induced toxic signals are transmitted via tau. However, what might be a mechanism of transmitting Aβ-induced neurotoxic signals from the cell surface? Aβ oligomers rapidly promote the disassembly of microtubules but only in cells expressing tau [32]. Moreover, the actin-binding protein cofilin is also required for Aβ oligomer-induced neurotoxicity [33]. Multiple neuronal surface receptors for Aβ oligomers have been identified, including PrPc, NMDARs, mGluR5, IR/IGFR, LilrB2, α7nAchR, p75NTR, LRP1, integrins, and others [34–37]. These receptors appear to function discretely as well as in coordination with each other to transmit Aβ oligomer signals via multiple signaling cascades. In this review, we will highlight some downstream signaling events, focusing on the actin and microtubule cytoskeletal network.
ACTIN AND MICROTUBULE NETWORKS IN NEURONS
The highly polarized nature of neurons dictates their reliance on the cytoskeletal networks to define their morphology and the distribution of various organelles and proteins to specific polarized regions. These cytoskeletal elements are composed of the actin (microfilament), neurofilament (intermediate filaments), and microtubule networks distributed in overlapping and discrete patterns throughout neurons. While the actin and microtubule networks have been well studied, the neurofilament network is the least well understood. Nonetheless, these intermediate neurofilaments are known to contribute to neuronal morphology and vesicular mobility [38].
Actin filaments (F-actin) are concentrated in leading edge of non-neuronal cells [39]. In a similar manner, F-actin is enriched in growth cones of immature neurons and in dendritic spines and presynaptic zones of mature neurons [40, 41]. In dendritic spines, F-actin crosslinks surface receptors as well as integral postsynaptic proteins such as drebrin and PSD95 [7]. In presynaptic zones, F-actin acts as a scaffold to mediate vesicular trafficking and regulates neurotransmitter release [42]. Actin dynamics (polymerization, depolymerization, severing, bundling, etc.) continually regulate synaptic remodeling at both sides of the synapse and hence mediate changes in synaptic plasticity [43]. Actin dynamics are regulated by several different pathways including the Rho family of small GTPase (RhoA, Rac1, cdc42), and various actin-binding proteins (ADF/cofilin, drebrin, profilin, Arp2/3, etc). Of note, Rho family GTPases generally promote actin polymerization at leading edge of cells by cycling between the inactive GDP-bound to the active GTP-bound state, thereby regulating cell migration and growth cone motility [44, 45]. On the other hand, ADF/cofilin enhances actin dynamics through F-actin severing [46].
The neuronal organization and distribution of microtubules are distinct from the actin network [47]. Microtubules are polar structures polymerized from α- and β-tubulin heterodimers that assemble into hollow tubules via the binding and hydrolysis of GTP [48]. The fast-growing end is termed the plus end, whereas the slow-growing end is termed the minus end. In axons, the plus end is directed away from the cell body toward the presynaptic terminal, whereas dendrites possess mixed populations of plus and minus ends pointing away from the cell body [49]. Unlike the actin network, microtubules are not enriched at synaptic sites but can transiently assemble inside dendritic spines in an activity-dependent manner [47, 50, 51]. The microtubule-associated proteins (MAPs), which include MAP1A, MAP1B, MAPT2, MAP4, and tau, regulate the assembly, stability, and disassembly of the microtubule network [48, 52]. Microtubule dynamics play important roles in neurite extension, arborization, and dendritic spine morphogenesis [47, 51]. In addition, microtubules in neurons function as ‘tracks’ for transport of proteins and organelles along axons and dendrites. These transport activities are mediated by motor proteins such as kinesin and dynein [54]. This function is particularly important in long axons that require the long-distance anterograde transport of organelles such as mitochondria and synaptic vesicles as well as retrograde transport of dysfunctional mitochondria and misfolded proteins for autophagic clearance [55]. As extensively illustrated in tauopathies, disruption of axonal transport is thought to play a key role in synaptic dysfunction in these neurodegenerative diseases [56, 57].
Aβ, TAU, AND THE MICROTUBULE NETWORK
Tauopathy is induced by the detachment of tau from microtubules, associated with tau hyperphosphorylation [53]. While Aβ is not required to induce tauopathy, as evidenced by multiple tauopathies lacking Aβ pathology (FTLD-tau, PSP, CBD, etc.) [58], studies with experimental transgenic models clearly demonstrate that Aβ drives tauopathy [23, 24]. As outlined above, Aβ oligomers promote the hyperphosphorylation of tau, leading to microtubule disassembly. Multiple kinases mediating this neurotoxic action of Aβ have been studied, including GSK-3β, MAPK, PKA, CamKII, Cdk-5, JNK, Fyn, AMPK, and MARK [53, 59–61]. Of at least 85 phosphorylation sites on tau [62], Ser262 phosphorylation has been particularly associated with reduced affinity of tau for microtubules [63] and tau stability in microtubule unbound form [64]. MARK, AMPK, PKA, and CaMKII have been shown to phosphorylate tau on Ser262 [53]. In addition, phosphorylation of tau on Ser214 and Thr231 are also associated with detachment of tau from microtubules [53]. These tau phosphorylation sites are situated within the proline-rich domain or the adjacent microtubule binding repeat 1. Notably, the phosphorylation of Thr231, which undergoes trans-to-cis isomerization, is significantly increased in mild cognitive impairment [53, 65]. These and other phosphorylation sites play key roles in tauopathy. However, it is important to note that hyperphosphorylation of tau in AD brains is far more extensive than that induced by Aβ in neurons. It is still unclear whether the bulk of tau hyperphosphorylation seen in AD occurs before or after its detachment from microtubules.
COFILIN AND THE ACTIN CYTOSKELETON
The assembly and disassembly of G-actin to F-actin is a process critical to many cellular processes, including cell motility, migration, dendritic spine morphogenesis, as well as provision of physical force for membrane bending needed for endocytosis and exocytosis [66–69]. F-actin is composed of two stranded helical polymers derived from the assembly of G-actin in a head to tail configuration, giving rise to its polarity (barbed and pointed ends). Actin assembly is initiated by the rate-limiting nucleation step in which actin forms dimers and trimers. The actin oligomer serves as a seed for elongation of filaments by the incorporation of ATP-actin monomers, after which ATP is hydrolyzed to ADP with subsequent release of inorganic phosphate (Pi). ADF/Cofilin, a family of actin-binding protein, is one of the key regulators of actin dynamics via its F-Actin severing, depolymerizing, nucleating, and bundling activities [70]. Cofilin is inactivated by phosphorylation on Ser3 by LIM kinase1 (LIMK1) [71], whereas its dephosphorylation by Slingshot Homolog-1 (SSH1) activates cofilin [72]. In addition to phosphorylation, cofilin activity is also regulated by its interaction with the membrane polyphosphoinositide PI(4,5)P2, which inhibits cofilin binding to F-actin [73, 74]. Activated cofilin differentially modulates actin dynamics depending on the ratio of cofilin to actin. At regions of high cofilin/actin ratios, cofilin can bind and stabilize F-actin in a twisted form, thereby promoting the nucleation of actin rather than severing [75]. However, at regions of low cofilin/actin ratios, cofilin does not bind ADP-actin fast enough to saturate F-actin but induces persistent severing to create new barbed and pointed ends [75], which may enhance filament depolymerization from pointed ends (–) and/or filament growth from barbed ends (+) [76]. These processes mediated by activated cofilin greatly contribute to dendritic spine remodeling in neurons [77]. Cofilin activity is facilitated by other actin-binding proteins, such as coronin 1A and Aip1, which can enhance cofilin recruitment to F-actin, increase cofilin severing activity, or accelerate monomer dissociation from F-actin [78–81].
Aβ IN COFILIN DEREGULATION
Table 1 summarizes findings from various model systems and human brains regarding cofilin deregulation with direct relevance for AD pathogenesis (Table 1). Figure 1 also illustrates a schematic model incorporating mechanistic findings from a survey of the literature. These points are specifically discussed in the sections below.
Table 1
Cofilin deregulation and associated pathogenesis
| Model | Treatment | Active cofilin | Overall findings | References |
| AD and aging brains | N/A | N/A | Cofilin in Hirano bodies increase with age and AD | [8, 9] |
| AD brains, rat neurons | ATP depletion or oxidative stress | Increased | Increased cofilin-actin rods | [82–85] |
| Primary neurons | Natural Aβ oligomers, inflammatory cytokines | Increased | Increased cofilin-actin rods via Nox & PrPc | [86, 115] |
| Primary neurons | Natural Aβ oligomers | Increased | Cofilin-actin rods increased w/ SSH1 & decreased w/ LIMK1 | [87] |
| AD patients, Tg2576 mice, mouse neurons | Aβ1-42 oligomers | Increased | Reduced PAK & Drebrin in AD and Tg2676 mice; increased pPAK surrounding Aβ deposits | [100] |
| 3xTg mice | N/A | Increased | Reduced PAK; dominant-negative PAK results in memory deficits in 3xTg mice | [101] |
| Primary neurons | Aβ1-40 & Aβ1-25 fibrils | Decreased | Increased LIMK1 activation in dystrophic neurites | [102] |
| Rat brain | Aβ1-40 fibrils | Increased | Reduced PSD95 & GluR1 and decreased silent synapses | [103] |
| AD brains, APP/PS1 x RanBP9 + /–mice | Aβ1-42 oligomers | Increased | APP/PS1 mice with increased cofilin activation; RanBP9 promotes SSH1 stability; RanBP9 reduction mitigates cofilin-actin pathology synaptic deficits in APP/PS1 mice | [82, 96, 107] |
| APP/PS1 x RanBP9 Tg | N/A | Increased | RanBP9 Tg promotes cofilin activation in synaptosomes | [108] |
| APP/PS1 x cofilin+/–mice, primary neurons | Aβ1-42 oligomers | Increased | Integrin conformers found to mediates Aβ oligomer-induced cofilin activation & translocation to mitochondria; cofilin+/–mitigates synaptic plasticity deficits in APP/PS1 mice | [33] |
| AD brains, LilrB2 ko mice and primary neurons | Aβ1-42 oligomers | Increased | LilrB2, an Aβ oligomer receptor that mediates cofilin activation and synaptic plasticity deficits | [110] |
| Primary neurons | Natural Aβ oligomers | Increased | Aβ-induced loss of dendritic spines mediated by calcineurin & cofilin; calcineurin activates SSH1 | [111, 112] |
| AD brains, APP/PS1 mice, primary neurons | Aβ1-42 oligomers | Decreased | Increased cofilin phosphorylation in PSD fraction of AD and APP/PS1 mouse brains | [116] |
| Cholinergic neurons | Aβ1-42 oligomers | Decreased | Increased cofilin phosphorylation and actin stabilization selectively in cholinergic neurons via p75 | [117] |
| APP/PS1 mice | N/A | Biphasic | Cofilin activation increased at 4 months and decreased at 10 months of APP/PS1 mice | [118] |
| Cell line | PAR-2 | Increased | Cofilin activation requires β-arrestin-mediated scaffolding of CIN & cofilin | [121, 122] |
| Brain slices & neurons | Aβ1-42 oligomers | Increased | β-arrestin2 translocates activated cofilin to spines; β-arrestin2-/- neurons are resistant to Aβ oligomer-induced spine loss | [77] |
| Primary neurons | ATP depletion | Increased | Activation of CIN by ATP depletion promotes cofilin-actin rod formation | [125] |
| APP/PS1 x cofilin+/–mice | N/A | Increased | Cofilin displaces tau from microtubules by increasing cofilin-microtubule complex in APP/PS1 mice; Cofilin+/–rescues imbalance of complexes | [132] |
| TauP301 S x cofilin+/–mice | N/A | N/A | Cofilin+/–rescues tauopathy in TauP301 S mice; Activated cofilin (S3A) selectively promotes tauopathy & microtubule instability | [132] |
| Primary neurons | Aβ1-42 oligomers | Increased | Aβ oligomers promote cofilin activation and F-actin dynamics at the axon initial segment (AIS), thereby mediating tau missorting to somatodendritic compartments | [133] |
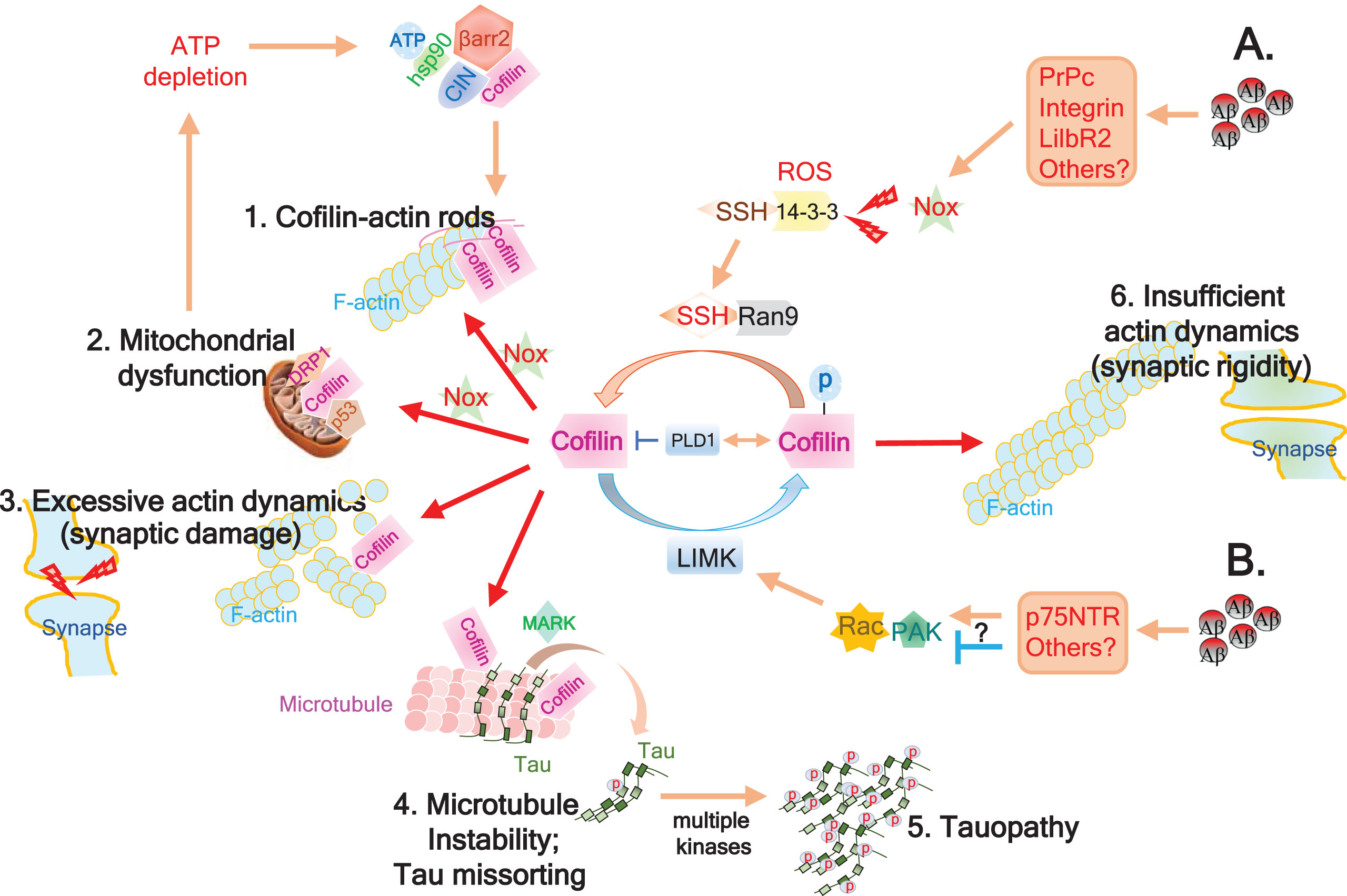
Fig.1
Schematic model of Aβ-induced cofilin deregulation in AD. A) Accumulation of Aβ oligomers impinge on various surface receptors (PrPc, Integrins, LilbR2, etc.), impacting several signaling cascades including Nox activation. Nox-mediated generation of reactive oxygen species (ROS) promotes 14-3-3 oxidation, thereby releasing and activating SSH1. Scaffolding protein RanBP9 (Ran9) stabilizes and promotes SSH1-mediated cofilin dephosphorylation and activation. Excessive cofilin activation can then result in several downstream consequences: 1. Together with increased ADP-actin, activated cofilin, & ROS, intermolecular disulfide bridging of cofilin induces cofilin-actin rod formation, potentially impeding axonal transport and depleting cofilin; 2. Intramolecular disulfide bridging of oxidized and activated cofilin loses affinity for actin and translocates to mitochondria to promotes mitochondrial dysfunction together with p53 and Drp1. This causes ATP depletion, which releases chronophin (CIN) from hsp90 inhibitory control and further promotes CIN-mediated cofilin activation, which is enhanced by β-arrestin-mediated scaffolding of CIN and cofilin; 3. Non-oxidized activated cofilin severs F-actin, increasing actin dynamics and potentially damaging synapses by loss of drebrin and PSD95; and 4. Activated cofilin displaces tau from microtubules with the aid of MARK-mediated tau phosphorylation on Ser262, which when coupled to dissolution of the tau diffusion barrier by F-actin severing at the AIS, induces missorting of tau to somatodendritic compartments. Displacement of tau from microtubules also destabilizes microtubules; 5. Hyperphosphorylation of microtubule-displaced tau by multiple kinases promotes tauopathy. B) In some cases, Aβ species impinge on p75NTR or other surface receptors, either activating or inhibiting Rac-Pak signaling, thereby enhancing or limiting LIMK-mediated inactivation / phosphorylation of cofilin, respectively. PLD1 plays a role in the negative regulation of cofilin by either inhibiting activated cofilin or increasing inactive/phosphorylated cofilin. Inhibition of LIMK feeds into the SSH1-mediated cofilin activation pathway (1–5), whereas excessive activation of LIMK together with PLD1 may override the SSH1 pathway and promote deregulated cofilin inactivation, which can result in insufficient actin dynamics and synaptic rigidity (6).
Cofilin-actin pathology
Actin-based pathologies including Hirano bodies [8, 9] and cofilin-actin rods/aggregates are significantly increased in AD and animal models of AD [82–85]. Specifically, a study by Rahman and colleagues reported a 4-fold increase in cofilin rods/aggregates in AD versus age-matched controls, which correlates with the extent of tauopathy [85]. Previous studies have also shown that bioactive Aβ dimers/trimers at subnanomolar concentrations promote cofilin-actin rod formation in a subset of neurons associated with activation of cofilin and NADPH oxidase (NOX) [33, 70, 82, 86]. While it is not clear whether cofilin-actin pathology plays an essential role in AD pathogenesis, it is certainly a pathology saliently present in AD brains and clearly serves as evidence for the deregulation of cofilin and actin in AD. The formation of cofilin-actin pathology (rods and aggregates) requires several key events. First, cofilin must be in the activated form (dephosphorylated), as SSH1 or CIN-mediated cofilin dephosphorylation and LIMK1-mediated cofilin phosphorylation promotes and inhibits cofilin-actin rod generation, respectively [83, 87]. Notably, phospholipase-mediated hydrolysis of phosphatidylinositol 4,5-biphosphate (PI(4,5)P2), which locally releases active cofilin from the membrane, also dynamically contributes to cofilin activation [88, 89]. Second, activated cofilin levels must rise to saturate local regions of F-actin in the presence abnormally high levels of ADP-actin, which preferentially binds to cofilin [75, 83, 90]. Third, cofilin must undergo intermolecular disulfide linkages via oxidation of several key cysteine residues [91]. These cofilin-actin inclusions are generally reversible and can form under conditions of heat shock, osmotic stress, ATP depletion, excitotoxicity, ischemia, and oxidative stress, potentially interfering with axonal/neuritic transport and depleting activated cofilin needed for actin remodeling [70]. For example, ischemia-induced cofilin-actin pathology interferes with mitochondrial transport and leads to synaptic failure, which could be partially rescued by LIMK1 or its upstream activator Rho [92].
Mitochondrial translocation of cofilin
Activated cofilin also plays an important role in mitochondrial dysfunction via direct translocation to mitochondria. Upon oxidative stress, cofilin becomes oxidized on several cysteine residues, thereby promoting intramolecular disulfide bridging of cofilin [93]. This causes cofilin to lose affinity for actin and translocate to mitochondria, where it induces swelling, drop in mitochondrial membrane potential, and cytochrome c release by promoting the opening of the permeability transition pore [93–95] Interestingly, this occurs independently of Bax. In addition to cofilin oxidation, dephosphorylation (or activation) of cofilin is required for its translocation to the mitochondria and oxidant-induced apoptosis. When oxidation of cofilin is prevented by cysteine mutagenesis, oxidant-induced apoptosis is also inhibited. Furthermore, knockdown of endogenous cofilin by siRNA also inhibits both oxidant and staurosporine-induced apoptosis, indicating that cofilin is critical for mitochondria-mediated apoptosis [93–95].
Woo and colleagues first demonstrated that Aβ1-42 oligomers promote the translocation of cofilin to mitochondria, which induces a drop in mitochondrial membrane potential, increase in mitochondrial superoxide, and cell death. These events are largely abolished by siRNA-mediated knockdown of cofilin [96] or SSH1 [33]. Liu and colleagues showed that activated cofilin forms a complex with the tumor suppressor protein p53, which promotes the translocation of p53 to both mitochondria and nucleus [97]. Cofilin interaction with the mitochondrial fission protein Drp1 has also been reported to regulate both mitochondrial morphology and apoptosis [98, 99]. In brains of AD patients, the level of mitochondrial cofilin is strongly increased compared to healthy age-matched controls [33], indicating a role for cofilin in mitochondrial dysfunction in AD.
LIMK1 and SSH1 pathways
Aβ has been shown to play important roles in cofilin deregulation and synaptic dysfunction via both LIMK1 and SSH1 pathways. LIMK1-mediated phosphorylation of cofilin generally involves upstream Rac-PAK signaling, which leads to cofilin inactivation. PAK1 and PAK3 levels and activity are depleted in AD brains [100], which leads to the activation of cofilin and loss of drebrin, a postsynaptic actin-stabilizing protein. Similar loss of drebrin is found in APP/PS1, Tg2576, and 3xTg transgenic models of AD [33, 100, 101]. Zhao and colleagues found that Aβ1-42 oligomers can directly induce PAK signaling deficits in primary neurons [100]. Despite a general loss of PAK levels and signaling in AD brains, pPAK is intensely increased surrounding Aβ deposits together with cofilin pathology [100]. Arsenault et al. confirmed the loss of PAK in AD brains and 3xTg mice and showed that expression of a dominant-negative form of PAK results in memory deficits in 3xTg mice [101]. In contrast, Heredia and colleagues showed that Aβ1-40 and Aβ25-35 fibrils induce the activation of LIMK and resultant cofilin inactivation (phosphorylation), associated with dystrophic neurites in primary neurons [102]. This finding may help to explain the intense pPAK staining surrounding the presumably fibrillar Aβ deposits in AD brains [100], although another recent study found that injection of Aβ1-40 fibrils into rat brains results in increased activation of cofilin rather than inactivation [103]. Indeed, Ariadna and colleague showed the activation LIMK1 by Aβ1-42 fibrils, but this was paradoxically associated with increased cofilin activation, suggesting that Aβ1-42 fibrils may act via bifurcating mechanisms of Rac-LIMK1 activation and perhaps also activation of the SSH1 pathway, the latter perhaps via Rac-mediated activation of NOX and oxidation of 14-3-3 releasing SSH1 [104, 105].
RanBP9 is a scaffolding protein known to promote Aβ production [106] and is highly elevated in brains of AD patients [107] and APP transgenic mice [82, 96]. RanBP9 was shown to promote cofilin activation via enhancing SSH1 in primary neurons and in brain [82]. RanBP9 transgenic mice also contain increased activated cofilin in synaptosomes [108], where cofilin-mediated synaptic remodeling is well known [109]. Conversely, genetic reduction of RanBP9 mitigates both amyloid and cofilin-actin pathology in APP/PS1 transgenic mice [82], indicating a role for the RanBP9-SSH1 pathway in cofilin-actin pathology. Moreover, Kim and colleagues observed increased levels of cofilin activation in AD brains with no apparent changes in phospho-LIMK1 [110]. In the same study, the Aβ oligomer receptor LilrB2 was shown to mediate the activation of cofilin by Aβ1-42 oligomers [110], while Woo and colleagues found that β1-integrin conformers mediate Aβ1-42 oligomer-induced cofilin activation via the activation of SSH1 [33]. The neurotoxic effects of Aβ1-42 oligomers could be abolished by knockdown of SSH1, which also mitigates mitochondrial translocation of activated cofilin [33]. In the same study, genetic reduction of cofilin was shown to mitigate the loss of drebrin and other F-actin-associated synaptic proteins as well as deficits in synaptic plasticity (i.e., LTP) and memory in APP/PS1 transgenic mice [33]. The role of SSH1 in Aβ-induced cofilin activation is supported by a previous study showing that Aβ-induced dendritic spine loss is mediated by calcineurin and cofilin [111]. This is likely via calcineurin-mediated dephosphorylation of SSH1, which activates cofilin [112], by allowing SSH1 to escape inhibition by 14-3-3 proteins [105, 113]. The role of reactive oxygen species (ROS) in both SSH1 and cofilin activation is highlighted by observations that oxidation of 14-4-3 releases both SSH1 [105] and cofilin [114] from 14-3-3-mediated inhibitory control. Indeed, NOX-mediated ROS production is required for Aβ-induced cofilin activation [33] and cofilin-actin rod formation [86, 115]. Furthermore, direct oxidation of cofilin is also required for both mitochondrial translocation of cofilin [93] and cofilin-actin rod assembly [91].
Despite the strong evidence for a role of cofilin activation and oxidation in AD pathogenesis, other studies have shown that cofilin inactivation may also play a role in AD pathogenesis. In the postsynaptic density (PSD) fraction of AD and APP/PS1 mouse brains, phospho-cofilin is increased, and short duration (30 min) Aβ1-42 oligomer treatment promotes cofilin phosphorylation together with F-actin stabilization in dendritic spines [116], which decreases synaptic plasticity. Another study showed that Aβ oligomers increase cofilin phosphorylation and actin polymerization selectively in basal forebrain cholinergic neurons but not in non-cholinergic neurons via a p75-dependent mechanism [117]. In APP/PS1 transgenic mouse brains, phospho-cofilin is reduced at 4 months of age (early pathology) and then increased at 10 months of age (mid-late pathology) [118], indicating a biphasic regulation of cofilin during Aβ pathogenic progression. Hence, different Aβ species and conformations appear to act on cofilin in differing ways, depending on the locality, age, and neuronal type. This may not be entirely surprising, given the differences in the localization, affinity, and downstream signaling cascades coupled to perhaps a dozen known Aβ receptors expressed in different neuronal populations [34–37]. In cultured hippocampal primary neurons, bioactive Aβ dimers/trimers induce cofilin-actin rod formation (which requires cofilin activation) in neurites of only ∼20–30% of neurons, while organotypic hippocampal slices treated with Aβ dimers/trimers preferentially increase cofilin-actin rods in the dentate gyrus and the mossy fiber track but not in CA regions [87]. Hence, the binding of Aβ species to different neuronal Aβ receptors may promote cofilin activation, while other receptors might respond in the opposite direction depending on Aβ concentration, type, and locality. Despite the changes in phospho-cofilin seen by Aβ and in AD models, it is important to emphasize that the phosphorylation state of cofilin is not the sole determinant of cofilin activation status, as the local hydrolysis of PI(4,5)P2 releases active cofilin from the membrane [88, 89]. This pool of cofilin is far more difficult to measure and has largely been ignored in prior studies. Nevertheless, the observation that genetic reduction of cofilin rescues neurotoxicity [96] as well as synaptic plasticity and memory deficits in APP/PS1 transgenic mice [33] strongly supports the notion that cofilin (whether via activation, inactivation, or other mechanisms) mediates neurotoxic signaling induced by Aβ.
β-ARRESTIN, CHRONOPHIN, AND PLD1
While β-arrestins are traditionally known for their roles in G-protein coupled receptor (GPCR) fine-tuning and desensitization, it is now widely accepted that β-arrestins act as multifunctional adaptor proteins that regulate multiple signaling pathways [119, 120]. For example, Protease-activated receptor-2 (PAR-2)-mediated activation of cofilin requires β-arrestin to scaffold together cofilin and chronophin (CIN), a cofilin activating phosphatase [121, 122]. This action appears to be localized to the leading edge of cells [121]. Intriguingly, both β-arrestin1 and β-arrestin2 are significantly elevated in brains of AD patients, both of which promote γ-secretase activity to enhance Aβ production in brain [123, 124]. β-arrestin2 also plays an important role in dendritic spines and synapses via translocating activated cofilin to dendritic spines [77]. Hence, β-arrestin2-deficient neurons are resistant to Aβ-induced dendritic spine loss [77]. The CIN/Hsp90 complex functions as an ATP sensor, and upon ATP depletion, CIN dissociates from Hsp90 [125]. This leads to CIN activation and dephosphorylation of cofilin, thereby promoting cofilin-actin rod formation under conditions of ATP depletion [125].
Phospholipase D1 (PLD1), an enzyme of the phospholipase superfamily, catalyzes the hydrolysis of phosphatidylcholine (PC) into phosphatidic acid (PA) and choline in response to various stimuli [126]. PI(4,5)P2, which can be produced by PA-mediated activation of phosphatidylinositol-4-phosphate 5-kinase (PIP5K), functions to both activate PLD1 and inactivate cofilin [127], while phospho-cofilin activates PLD1 upon extracellular stimulation by cabachol [128]. Conversely, PLD1 reciprocally promotes cofilin phosphorylation / inactivation and inhibits cofilin-mediated mitochondrial toxicity [97]. Hence, PLD1 and cofilin reciprocally regulate each other, adding another layer of cofilin regulation. Interestingly, a previous study reported that PLD1 antagonizes Aβ production by regulating the γ-secretase complex [129], raising the specter that cofilin activation status may also impact Aβ production.
COFILIN IN TAUOPATHY
Early studies examining rod-like cofilin aggregates in primary neurons demonstrated 12E8 antibody immunoreactivity in a subset of cofilin-positive rods resembling neuropil threads. The 12E8 antibody recognizes the pSer262/pSer356 of tau and other phosphorylated MAPs, suggesting that cofilin might coprecipitate together with tau and/or other MAPs in cofilin-actin rods [130]. FTDP-17 mutant tau overexpression in transgenic mice and Drosophila has been shown to promote F-actin bundling. These transgenic animal brains contain hyperphosphorylated tau in F-actin-containing rod-like structures, although the presence of tau in cofilin-containing rods was not examined [131]. In AD brains, however, phospho-tau containing neuropil threads and cofilin-actin aggregates do not appear to colocalize, despite the abundance of both structures in the same brains [85], raising a quandary as to the relationship between cofilin and tau in AD.
Both tau and cofilin are required for Aβ-induced mitochondrial and synaptic dysfunction in primary neurons, and either cofilin or tau reduction also rescues defects in synaptic plasticity and memory in APP transgenic mice [18, 25–27, 33]. So how might cofilin and tau signaling downstream of Aβ be reconciled? Woo and colleagues recently showed that tau-microtubule complexes are decreased, and cofilin-microtubule complexes are increased in APP/PS1 transgenic mice, both of which are prevented by genetic reduction of cofilin. Experiments in vitro, cells, and in vivo suggest a role for cofilin in displacing tau from tubulin/microtubules, which inhibits tau-induced microtubule assembly and axonal transport [132]. Remarkably, genetic reduction of cofilin strongly mitigates tauopathy (tau hyperphosphorylation & insolubility) and synaptic plasticity deficits in Tau-P301 S (PS19) mice, in which the ‘activated’ but not ‘inactive’ form of cofilin was found to mediate tauopathy, microtubule instability, and synaptic deregulation in the Tau-P301 S model [132]. Hence, these findings show that activated cofilin can displace tau from microtubules, resulting not only in inhibition of tau-mediated microtubule dynamics but also tau hyperphosphorylation and tauopathy. Intriguingly, Zempel and colleagues showed that the axonal initial segment (AIS) of neurons contains a tau diffusion barrier composed of actin filaments that normally keeps tau in axons by preventing its retrograde traffic back to the soma [133]. Aβ oligomer-induced activation of cofilin at the AIS was found to dissolve the tau diffusion barrier, which allows missorting of tau to somatodendritic compartments [133]. Hence, it is plausible that Aβ-induced activation of cofilin, resulting in simultaneous displacement of tau from microtubules and severing of actin filaments (i.e., removing tau diffusion barrier), may underlie the mislocalization of tau from the somato-axonal to somatodendritic compartments, an early feature of tauopathy [28, 29]. It also raises the intriguing possibility that the local actions (i.e., dendritic spines, presynaptic boutons, cell body, and/or AIS) of Aβ oligomers or other neurotoxic insults produce defined and diverse consequences that could mark different components of Aβ-cofilin-tau-mediated signaling in AD pathogenesis.
CONCLUDING REMARKS
The findings generated from multiple studies implicate the critical role of cofilin in AD pathogenesis. These may come in the form of cofilin-actin pathology, deregulated F-actin dynamics, mitochondrial translocation of cofilin to coordinate cell death pathways, and displacement of tau from microtubules, the latter which couples actin dynamics with tau-regulated microtubule dynamics. The regulation of cofilin by multiple pathways interconnect different facets of AD pathogenesis, including Aβ, tau, and the cytoskeleton, suggesting that the machinery of cofilin activation and inactivation cycle represents a key node regulating cytoskeletal pathogenesis in AD. Hence, modulating cofilin activity could be a therapeutic strategy to slow multiple AD pathologies and mitigate synaptic dysfunction. This may be achieved by regulating upstream effectors, such as LIMK1, SSH1, chronophin, and/or β-arrestin as an alternative to directly targeting Aβ or tau.
ACKNOWLEDGMENTS
This work was supported in part by grants from the VA (1 I01 BX002478-01A1, D.E.K.), NIH (R01 AG053060-01A1, D.E.K.) and Florida department of Health (8AZ29, D.E.K.).
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/19-0585r1).
REFERENCES
[1] | Small GW , Rabins PV , Barry PP , Buckholtz NS , DeKosky ST , Ferris SH , Finkel SI , Gwyther LP , Khachaturian ZS , Lebowitz BD , McRae TD , Morris JC , Oakley F , Schneider LS , Streim JE , Sunderland T , Teri LA , Tune LE ((1997) ) Diagnosis and treatment of Alzheimer disease and related disorders. Consensus statement of the American Association for Geriatric Psychiatry, the Alzheimer’s Association, and the American Geriatrics Society. JAMA 278: , 1363–1371. |
[2] | Kemle K , Ackermann RJ ((2018) ) Issues in geriatric care: Alzheimer disease. FP Essent 468: , 26–34. |
[3] | Holtzman DM , Mandelkow E , Selkoe DJ ((2012) ) Alzheimer disease in 2020. Cold Spring Harb Perspect Med 2: , a011585. |
[4] | Goedert M ((1993) ) Tau protein and the neurofibrillary pathology of Alzheimer’s disease. Trends Neurosci 16: , 460–465. |
[5] | Crowther RA , Wischik CM ((1985) ) Image reconstruction of the Alzheimer paired helical filament. EMBO J 4: , 3661–3665. |
[6] | Kosaka K , Yoshimura M , Ikeda K , Budka H ((1984) ) Diffuse type of Lewy body disease: Progressive dementia with abundant cortical Lewy bodies and senile changes of varying degree–a new disease? Clin Neuropathol 3: , 185–192. |
[7] | Josephs KA , Whitwell JL , Tosakulwong N , Weigand SD , Murray ME , Liesinger AM , Petrucelli L , Senjem ML , Ivnik RJ , Parisi JE , Petersen RC , Dickson DW ((2015) ) TAR DNA-binding protein 43 and pathological subtype of Alzheimer’s disease impact clinical features. Ann Neurol 78: , 697–709. |
[8] | Galloway PG , Perry G , Gambetti P ((1987) ) Hirano body filaments contain actin and actin-associated proteins. J Neuropathol Exp Neurol 46: , 185–199. |
[9] | Maciver SK , Harrington CR ((1995) ) Two actin binding proteins, actin depolymerizing factor and cofilin, are associated with Hirano bodies. Neuroreport 6: , 1985–1988. |
[10] | Bamburg JR , Bloom GS ((2009) ) Cytoskeletal pathologies of Alzheimer disease. Cell Motil Cytoskeleton 66: , 635–649. |
[11] | Hardy J , Allsop D ((1991) ) Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci 12: , 383–388. |
[12] | Goate AM ((1998) ) Monogenetic determinants of Alzheimer’s disease: APP mutations. Cell Mol Life Sci 54: , 897–901. |
[13] | De Strooper B , Iwatsubo T , Wolfe MS ((2012) ) Presenilins and gamma-secretase: Structure, function, and role in Alzheimer disease. Cold Spring Harb Perspect Med 2: , a006304. |
[14] | Glabe CG ((2008) ) Structural classification of toxic amyloid oligomers. J Biol Chem 283: , 29639–29643. |
[15] | McGowan E , Pickford F , Kim J , Onstead L , Eriksen J , Yu C , Skipper L , Murphy MP , Beard J , Das P , Jansen K , DeLucia M , Lin WL , Dolios G , Wang R , Eckman CB , Dickson DW , Hutton M , Hardy J , Golde T ((2005) ) Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron 47: , 191–199. |
[16] | Chen YR , Glabe CG ((2006) ) Distinct early folding and aggregation properties of Alzheimer amyloid-beta peptides Abeta40 and Abeta42: Stable trimer or tetramer formation by Abeta42. J Biol Chem 281: , 24414–24422. |
[17] | Walsh DM , Klyubin I , Fadeeva JV , Cullen WK , Anwyl R , Wolfe MS , Rowan MJ , Selkoe DJ ((2002) ) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416: , 535–539. |
[18] | Jin M , Shepardson N , Yang T , Chen G , Walsh D , Selkoe DJ ((2011) ) Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci U S A 108: , 5819–5824. |
[19] | Tomiyama T , Nagata T , Shimada H , Teraoka R , Fukushima A , Kanemitsu H , Takuma H , Kuwano R , Imagawa M , Ataka S , Wada Y , Yoshioka E , Nishizaki T , Watanabe Y , Mori H ((2008) ) A new amyloid beta variant favoring oligomerization in Alzheimer’s-type dementia. Ann Neurol 63: , 377–387. |
[20] | Nishitsuji K , Tomiyama T , Ishibashi K , Ito K , Teraoka R , Lambert MP , Klein WL , Mori H ((2009) ) The E693Delta mutation in amyloid precursor protein increases intracellular accumulation of amyloid beta oligomers and causes endoplasmic reticulum stress-induced apoptosis in cultured cells. Am J Pathol 174: , 957–969. |
[21] | Avila J , Lucas JJ , Perez M , Hernandez F ((2004) ) Role of tau protein in both physiological and pathological conditions. Physiol Rev 84: , 361–384. |
[22] | Gendron TF , Petrucelli L ((2009) ) The role of tau in neurodegeneration. Mol Neurodegener 4: , 13. |
[23] | Lewis J , McGowan E , Rockwood J , Melrose H , Nacharaju P , Van Slegtenhorst M , Gwinn-Hardy K , Paul Murphy M , Baker M , Yu X , Duff K , Hardy J , Corral A , Lin WL , Yen SH , Dickson DW , Davies P , Hutton M ((2000) ) Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet 25: , 402–405. |
[24] | Oddo S , Billings L , Kesslak JP , Cribbs DH , LaFerla FM ((2004) ) Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron 43: , 321–332. |
[25] | Rapoport M , Dawson HN , Binder LI , Vitek MP , Ferreira A ((2002) ) Tau is essential to beta -amyloid-induced neurotoxicity. Proc Natl Acad Sci U S A 99: , 6364–6369. |
[26] | Roberson ED , Scearce-Levie K , Palop JJ , Yan F , Cheng IH , Wu T , Gerstein H , Yu GQ , Mucke L ((2007) ) Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 316: , 750–754. |
[27] | Shipton OA , Leitz JR , Dworzak J , Acton CE , Tunbridge EM , Denk F , Dawson HN , Vitek MP , Wade-Martins R , Paulsen O , Vargas-Caballero M ((2011) ) Tau protein is required for amyloid beta-induced impairment of hippocampal long-term potentiation. J Neurosci 31: , 1688–1692. |
[28] | Ballatore C , Lee VM , Trojanowski JQ ((2007) ) Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci 8: , 663–672. |
[29] | Biernat J , Mandelkow EM ((1999) ) The development of cell processes induced by tau protein requires phosphorylation of serine 262 and 356 in the repeat domain and is inhibited by phosphorylation in the proline-rich domains. Mol Biol Cell 10: , 727–740. |
[30] | Hoover BR , Reed MN , Su J , Penrod RD , Kotilinek LA , Grant MK , Pitstick R , Carlson GA , Lanier LM , Yuan LL , Ashe KH , Liao D ((2010) ) Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68: , 1067–1081. |
[31] | Santacruz K , Lewis J , Spires T , Paulson J , Kotilinek L , Ingelsson M , Guimaraes A , DeTure M , Ramsden M , McGowan E , Forster C , Yue M , Orne J , Janus C , Mariash A , Kuskowski M , Hyman B , Hutton M , Ashe KH ((2005) ) Tau suppression in a neurodegenerative mouse model improves memory function. Science 309: , 476–481. |
[32] | King ME , Kan HM , Baas PW , Erisir A , Glabe CG , Bloom GS ((2006) ) Tau-dependent microtubule disassembly initiated by prefibrillar beta-amyloid. J Cell Biol 175: , 541–546. |
[33] | Woo JA , Zhao X , Khan H , Penn C , Wang X , Joly-Amado A , Weeber E , Morgan D , Kang DE ((2015) ) Slingshot-Cofilin activation mediates mitochondrial and synaptic dysfunction via Abeta ligation to beta1-integrin conformers. Cell Death Differ 22: , 1069–1070. |
[34] | Rowan MJ , Klyubin I , Wang Q , Hu NW , Anwyl R ((2007) ) Synaptic memory mechanisms: Alzheimer’s disease amyloid beta-peptide-induced dysfunction. Biochem Soc Trans 35: , 1219–1223. |
[35] | Smith LM , Strittmatter SM ((2017) ) Binding sites for amyloid-beta oligomers and synaptic toxicity. Cold Spring Harb Perspect Med 7: , a024075. |
[36] | Patel AN , Jhamandas JH ((2012) ) Neuronal receptors as targets for the action of amyloid-beta protein (Abeta) in the brain. Expert Rev Mol Med 14: , e2. |
[37] | Kang DE , Roh SE , Woo JA , Liu T , Bu JH , Jung AR , Lim Y ((2011) ) The interface between cytoskeletal aberrations and mitochondrial dysfunction in Alzheimer’s disease and related disorders. Exp Neurobiol 20: , 67–80. |
[38] | Yuan A , Rao MV , Veeranna , Nixon RA ((2017) ) Neurofilaments and neurofilament proteins in health and disease. Cold Spring Harb Perspect Biol 9: , a018309. |
[39] | Ridley AJ ((2011) ) Life at the leading edge. Cell 145: , 1012–1022. |
[40] | Omotade OF , Pollitt SL , Zheng JQ ((2017) ) Actin-based growth cone motility and guidance. Mol Cell Neurosci 84: , 4–10. |
[41] | Urbanska M , Swiech L , Jaworski J ((2012) ) Developmental plasticity of the dendritic compartment: Focus on the cytoskeleton. Adv Exp Med Biol 970: , 265–284. |
[42] | Doussau F , Augustine GJ ((2000) ) The actin cytoskeleton and neurotransmitter release: An overview. Biochimie 82: , 353–363. |
[43] | Fu AK , Ip NY ((2017) ) Regulation of postsynaptic signaling in structural synaptic plasticity. Curr Opin Neurobiol 45: , 148–155. |
[44] | Schelski M , Bradke F ((2017) ) Neuronal polarization: From spatiotemporal signaling to cytoskeletal dynamics. Mol Cell Neurosci 84: , 11–28. |
[45] | Lawson CD , Ridley AJ ((2018) ) Rho GTPase signaling complexes in cell migration and invasion. J Cell Biol 217: , 447–457. |
[46] | Bamburg JR , Wiggan OP ((2002) ) ADF/cofilin and actin dynamics in disease. Trends Cell Biol 12: , 598–605. |
[47] | Coles CH , Bradke F ((2015) ) Coordinating neuronal actin-microtubule dynamics. Curr Biol 25: , R677–691. |
[48] | Goodson HV , Jonasson EM ((2018) ) Microtubules and microtubule-associated proteins. Cold Spring Harb Perspect Biol 10: , a022608. |
[49] | Kelliher MT , Saunders HA , Wildonger J ((2019) ) Microtubule control of functional architecture in neurons. Curr Opin Neurobiol 57: , 39–45. |
[50] | Gu J , Firestein BL , Zheng JQ ((2008) ) Microtubules in dendritic spine development. J Neurosci 28: , 12120–12124. |
[51] | Miller KE , Suter DM ((2018) ) An integrated cytoskeletal model of neurite outgrowth. Front Cell Neurosci 12: , 447. |
[52] | Brouhard GJ , Rice LM ((2018) ) Microtubule dynamics: An interplay of biochemistry and mechanics. Nat Rev Mol Cell Biol 19: , 451–463. |
[53] | Wang Y , Mandelkow E ((2016) ) Tau in physiology and pathology. Nat Rev Neurosci 17: , 5–21. |
[54] | Lu W , Gelfand VI ((2017) ) Moonlighting motors: Kinesin, dynein, and cell polarity. Trends Cell Biol 27: , 505–514. |
[55] | Tammineni P , Cai Q ((2017) ) Defective retrograde transport impairs autophagic clearance in Alzheimer disease neurons. Autophagy 13: , 982–984. |
[56] | Kanaan NM , Pigino GF , Brady ST , Lazarov O , Binder LI , Morfini GA ((2013) ) Axonal degeneration in Alzheimer’s disease: When signaling abnormalities meet the axonal transport system. Exp Neurol 246: , 44–53. |
[57] | De Vos KJ , Grierson AJ , Ackerley S , Miller CC ((2008) ) Role of axonal transport in neurodegenerative diseases. Annu Rev Neurosci 31: , 151–173. |
[58] | Saha P , Sen N ((2019) ) Tauopathy: A common mechanism for neurodegeneration and brain aging. Mech Ageing Dev 178: , 72–79. |
[59] | Hasegawa M , Morishima-Kawashima M , Takio K , Suzuki M , Titani K , Ihara Y ((1992) ) Protein sequence and mass spectrometric analyses of tau in the Alzheimer’s disease brain. J Biol Chem 267: , 17047–17054. |
[60] | Morishima-Kawashima M , Hasegawa M , Takio K , Suzuki M , Yoshida H , Titani K , Ihara Y ((1995) ) Proline-directed and non-proline-directed phosphorylation of PHF-tau. J Biol Chem 270: , 823–829. |
[61] | Henriques AG , Oliveira JM , Carvalho LP , da Cruz ESOAB ((2015) ) Abeta influences cytoskeletal signaling cascades with consequences to Alzheimer’s disease. Mol Neurobiol 52: , 1391–1407. |
[62] | Wang JZ , Xia YY , Grundke-Iqbal I , Iqbal K ((2013) ) Abnormal hyperphosphorylation of tau: Sites, regulation, and molecular mechanism of neurofibrillary degeneration. J Alzheimers Dis 33 Suppl 1: , S123–139. |
[63] | Thies E , Mandelkow EM ((2007) ) Missorting of tau in neurons causes degeneration of synapses that can be rescued by the kinase MARK2/Par-1. J Neurosci 27: , 2896–2907. |
[64] | Ando K , Maruko-Otake A , Ohtake Y , Hayashishita M , Sekiya M , Iijima KM ((2016) ) Stabilization of microtubule-unbound tau via tau phosphorylation at Ser262/356 by Par-1/MARK contributes to augmentation of AD-related phosphorylation and Abeta42-induced tau toxicity. PLoS Genet 12: , e1005917. |
[65] | Wang JZ , Zhang Y ((2015) ) Configuration-specific immunotherapy targeting cis pThr231-Pro232 tau for Alzheimer disease. J Neurol Sci 348: , 253–255. |
[66] | Senju Y , Lappalainen P ((2019) ) Regulation of actin dynamics by PI(4,5)P2 in cell migration and endocytosis. Curr Opin Cell Biol 56: , 7–13. |
[67] | Porat-Shliom N , Milberg O , Masedunskas A , Weigert R ((2013) ) Multiple roles for the actin cytoskeleton during regulated exocytosis. Cell Mol Life Sci 70: , 2099–2121. |
[68] | Inagaki N , Katsuno H ((2017) ) Actin waves: Origin of cell polarization and migration? Trends Cell Biol 27: , 515–526. |
[69] | Konietzny A , Bar J , Mikhaylova M ((2017) ) Dendritic actin cytoskeleton: Structure, functions, and regulations. Front Cell Neurosci 11: , 147. |
[70] | Bamburg JR , Bernstein BW ((2016) ) Actin dynamics and cofilin-actin rods in Alzheimer disease. Cytoskeleton (Hoboken) 73: , 477–497. |
[71] | Arber S , Barbayannis FA , Hanser H , Schneider C , Stanyon CA , Bernard O , Caroni P ((1998) ) Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature 393: , 805–809. |
[72] | Niwa R , Nagata-Ohashi K , Takeichi M , Mizuno K , Uemura T ((2002) ) Control of actin reorganization by Slingshot, a family of phosphatases that dephosphorylate ADF/cofilin. Cell 108: , 233–246. |
[73] | Yonezawa N , Nishida E , Iida K , Yahara I , Sakai H ((1990) ) Inhibition of the interactions of cofilin, destrin, and deoxyribonuclease I with actin by phosphoinositides. J Biol Chem 265: , 8382–8386. |
[74] | Gorbatyuk VY , Nosworthy NJ , Robson SA , Bains NP , Maciejewski MW , Dos Remedios CG , King GF ((2006) ) Mapping the phosphoinositide-binding site on chick cofilin explains how PIP2 regulates the cofilin-actin interaction. Mol Cell 24: , 511–522. |
[75] | Andrianantoandro E , Pollard TD ((2006) ) Mechanism of actin filament turnover by severing and nucleation at different concentrations of ADF/cofilin. Mol Cell 24: , 13–23. |
[76] | Bernstein BW , Bamburg JR ((2010) ) ADF/cofilin: A functional node in cell biology. Trends Cell Biol 20: , 187–195. |
[77] | Pontrello CG , Sun MY , Lin A , Fiacco TA , DeFea KA , Ethell IM ((2012) ) Cofilin under control of beta-arrestin-2 in NMDA-dependent dendritic spine plasticity, long-term depression (LTD), and learning. Proc Natl Acad Sci U S A 109: , E442–451. |
[78] | Lin MC , Galletta BJ , Sept D , Cooper JA ((2010) ) Overlapping and distinct functions for cofilin, coronin and Aip1 in actin dynamics in vivo. J Cell Sci 123: , 1329–1342. |
[79] | Kueh HY , Charras GT , Mitchison TJ , Brieher WM ((2008) ) Actin disassembly by cofilin, coronin, and Aip1 occurs in bursts and is inhibited by barbed-end cappers. J Cell Biol 182: , 341–353. |
[80] | Mikati MA , Breitsprecher D , Jansen S , Reisler E , Goode BL ((2015) ) Coronin enhances actin filament severing by recruiting cofilin to filament sides and altering F-Actin conformation. J Mol Biol 427: , 3137–3147. |
[81] | Nadkarni AV , Brieher WM ((2014) ) Aip1 destabilizes cofilin-saturated actin filaments by severing and accelerating monomer dissociation from ends. Curr Biol 24: , 2749–2757. |
[82] | Woo JA , Boggess T , Uhlar C , Wang X , Khan H , Cappos G , Joly-Amado A , De Narvaez E , Majid S , Minamide LS , Bamburg JR , Morgan D , Weeber E , Kang DE ((2015) ) RanBP9 at the intersection between cofilin and Abeta pathologies: Rescue of neurodegenerative changes by RanBP9 reduction. Cell Death Dis 6: , 1676. |
[83] | Minamide LS , Striegl AM , Boyle JA , Meberg PJ , Bamburg JR ((2000) ) Neurodegenerative stimuli induce persistent ADF/cofilin-actin rods that disrupt distal neurite function. Nat Cell Biol 2: , 628–636. |
[84] | Bamburg JR , Bernstein BW , Davis RC , Flynn KC , Goldsbury C , Jensen JR , Maloney MT , Marsden IT , Minamide LS , Pak CW , Shaw AE , Whiteman I , Wiggan O ((2010) ) ADF/Cofilin-actin rods in neurodegenerative diseases. Curr Alzheimer Res 7: , 241–250. |
[85] | Rahman T , Davies DS , Tannenberg RK , Fok S , Shepherd C , Dodd PR , Cullen KM , Goldsbury C ((2014) ) Cofilin rods and aggregates concur with tau pathology and the development of Alzheimer’s disease. J Alzheimers Dis 42: , 1443–1460. |
[86] | Walsh KP , Kuhn TB , Bamburg JR ((2014) ) Cellular prion protein: A co-recetor mediating neuronal cofilin-actin rod formation induced by beta-amyloid and proinflammatory cytokines. Prion 8: , 375–380. |
[87] | Davis RC , Marsden IT , Maloney MT , Minamide LS , Podlisny M , Selkoe DJ , Bamburg JR ((2011) ) Amyloid beta dimers/trimers potently induce cofilin-actin rods that are inhibited by maintaining cofilin-phosphorylation. Mol Neurodegener 6: , 10. |
[88] | van Rheenen J , Song X , van Roosmalen W , Cammer M , Chen X , Desmarais V , Yip SC , Backer JM , Eddy RJ , Condeelis JS ((2007) ) EGF-induced PIP2 hydrolysis releases and activates cofilin locally in carcinoma cells. J Cell Biol 179: , 1247–1259. |
[89] | Leyman S , Sidani M , Ritsma L , Waterschoot D , Eddy R , Dewitte D , Debeir O , Decaestecker C , Vandekerckhove J , van Rheenen J , Ampe C , Condeelis J , Van Troys M ((2009) ) Unbalancing the phosphatidylinositol-4,5-bisphosphate-cofilin interaction impairs cell steering. Mol Biol Cell 20: , 4509–4523. |
[90] | Chen H , Bernstein BW , Sneider JM , Boyle JA , Minamide LS , Bamburg JR ((2004) ) In vitro activity differences between proteins of the ADF/cofilin family define two distinct subgroups. Biochemistry 43: , 7127–7142. |
[91] | Bernstein BW , Shaw AE , Minamide LS , Pak CW , Bamburg JR ((2012) ) Incorporation of cofilin into rods depends on disulfide intermolecular bonds: Implications for actin regulation and neurodegenerative disease. J Neurosci 32: , 6670–6681. |
[92] | Shu L , Chen B , Chen B , Xu H , Wang G , Huang Y , Zhao Y , Gong H , Jiang M , Chen L , Liu X , Wang Y ((2018) ) Brain ischemic insult induces cofilin rod formation leading to synaptic dysfunction in neurons. J Cereb Blood Flow Metab, doi: 10.1177/0271678X18785567 |
[93] | Klamt F , Zdanov S , Levine RL , Pariser A , Zhang Y , Zhang B , Yu LR , Veenstra TD , Shacter E ((2009) ) Oxidant-induced apoptosis is mediated by oxidation of the actin-regulatory protein cofilin. Nat Cell Biol 11: , 1241–1246. |
[94] | Wang C , Zhou GL , Vedantam S , Li P , Field J ((2008) ) Mitochondrial shuttling of CAP1 promotes actin- and cofilin-dependent apoptosis. J Cell Sci 121: , 2913–2920. |
[95] | Chua BT , Volbracht C , Tan KO , Li R , Yu VC , Li P ((2003) ) Mitochondrial translocation of cofilin is an early step in apoptosis induction. Nat Cell Biol 5: , 1083–1089. |
[96] | Woo JA , Jung AR , Lakshmana MK , Bedrossian A , Lim Y , Bu JH , Park SA , Koo EH , Mook-Jung I , Kang DE ((2012) ) Pivotal role of the RanBP9-cofilin pathway in Abeta-induced apoptosis and neurodegeneration. Cell Death Differ 19: , 1413–1423. |
[97] | Liu T , Wang F , LePochat P , Woo JA , Bukhari MZ , Hong KW , Trotter C , Kang DE ((2017) ) Cofilin-mediated neuronal apoptosis via p53 translocation and PLD1 regulation. Sci Rep 7: , 11532. |
[98] | Li G , Zhou J , Budhraja A , Hu X , Chen Y , Cheng Q , Liu L , Zhou T , Li P , Liu E , Gao N ((2015) ) Mitochondrial translocation and interaction of cofilin and Drp1 are required for erucin-induced mitochondrial fission and apoptosis. Oncotarget 6: , 1834–1849. |
[99] | Rehklau K , Hoffmann L , Gurniak CB , Ott M , Witke W , Scorrano L , Culmsee C , Rust MB ((2017) ) Cofilin1-dependent actin dynamics control DRP1-mediated mitochondrial fission. Cell Death Dis 8: , e3063. |
[100] | Zhao L , Ma QL , Calon F , Harris-White ME , Yang F , Lim GP , Morihara T , Ubeda OJ , Ambegaokar S , Hansen JE , Weisbart RH , Teter B , Frautschy SA , Cole GM ((2006) ) Role of p21-activated kinase pathway defects in the cognitive deficits of Alzheimer disease. Nat Neurosci 9: , 234–242. |
[101] | Arsenault D , Dal-Pan A , Tremblay C , Bennett DA , Guitton MJ , De Koninck Y , Tonegawa S , Calon F ((2013) ) PAK inactivation impairs social recognition in 3xTg-AD Mice without increasing brain deposition of tau and Abeta. J Neurosci 33: , 10729–10740. |
[102] | Heredia L , Helguera P , de Olmos S , Kedikian G , Sola Vigo F , LaFerla F , Staufenbiel M , de Olmos J , Busciglio J , Caceres A , Lorenzo A ((2006) ) Phosphorylation of actin-depolymerizing factor/cofilin by LIM-kinase mediates amyloid beta-induced degeneration: A potential mechanism of neuronal dystrophy in Alzheimer’s disease. J Neurosci 26: , 6533–6542. |
[103] | Bie B , Wu J , Foss JF , Naguib M ((2018) ) Amyloid fibrils induce dysfunction of hippocampal glutamatergic silent synapses. Hippocampus 28: , 549–556. |
[104] | Mendoza-Naranjo A , Contreras-Vallejos E , Henriquez DR , Otth C , Bamburg JR , Maccioni RB , Gonzalez-Billault C ((2012) ) Fibrillar amyloid-beta1-42 modifies actin organization affecting the cofilin phosphorylation state: A role for Rac1/cdc42 effector proteins and the slingshot phosphatase. J Alzheimers Dis 29: , 63–77. |
[105] | Kim JS , Huang TY , Bokoch GM ((2009) ) Reactive oxygen species regulate a slingshot-cofilin activation pathway. Mol Biol Cell 20: , 2650–2660. |
[106] | Lakshmana MK , Yoon IS , Chen E , Bianchi E , Koo EH , Kang DE ((2009) ) Novel role of RanBP9 in BACE1 processing of amyloid precursor protein and amyloid beta peptide generation. J Biol Chem 284: , 11863–11872. |
[107] | Lakshmana MK , Chung JY , Wickramarachchi S , Tak E , Bianchi E , Koo EH , Kang DE ((2010) ) A fragment of the scaffolding protein RanBP9 is increased in Alzheimer’s disease brains and strongly potentiates amyloid-beta peptide generation. FASEB J 24: , 119–127. |
[108] | Wang H , Lewsadder M , Dorn E , Xu S , Lakshmana MK ((2014) ) RanBP9 overexpression reduces dendritic arbor and spine density. Neuroscience 265: , 253–262. |
[109] | Rust MB ((2015) ) ADF/cofilin: A crucial regulator of synase physiology and behavior. Cell Mol Life Sci 72: , 3521–3529. |
[110] | Kim T , Vidal GS , Djurisic M , William CM , Birnbaum ME , Garcia KC , Hyman BT , Shatz CJ ((2013) ) Human LilrB2 is a beta-amyloid receptor and its murine homolog PirB regulates synaptic plasticity in an Alzheimer’s model. Science 341: , 1399–1404. |
[111] | Shankar GM , Bloodgood BL , Townsend M , Walsh DM , Selkoe DJ , Sabatini BL ((2007) ) Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci 27: , 2866–2875. |
[112] | Wang Y , Shibasaki F , Mizuno K ((2005) ) Calcium signal-induced cofilin dephosphorylation is mediated by Slingshot via calcineurin. J Biol Chem 280: , 12683–12689. |
[113] | Eiseler T , Doppler H , Yan IK , Kitatani K , Mizuno K , Storz P ((2009) ) Protein kinase D1 regulates cofilin-mediated F-actin reorganization and cell motility through slingshot. Nat Cell Biol 11: , 545–556. |
[114] | Gohla A , Bokoch GM ((2002) ) 14-3-3 regulates actin dynamics by stabilizing phosphorylated cofilin. Curr Biol 12: , 1704–1710. |
[115] | Walsh KP , Minamide LS , Kane SJ , Shaw AE , Brown DR , Pulford B , Zabel MD , Lambeth JD , Kuhn TB , Bamburg JR ((2014) ) Amyloid-beta and proinflammatory cytokines utilize a prion protein-dependent pathway to activate NADPH oxidase and induce cofilin-actin rods in hippocampal neurons. PLoS One 9: , e95995. |
[116] | Rush T , Martinez-Hernandez J , Dollmeyer M , Frandemiche ML , Borel E , Boisseau S , Jacquier-Sarlin M , Buisson A ((2018) ) Synaptotoxicity in Alzheimer’s disease involved a dysregulation of actin cytoskeleton dynamics through cofilin 1 phosphorylation. J Neurosci 38: , 10349–10361. |
[117] | Gu Z , Cheng J , Zhong P , Qin L , Liu W , Yan Z ((2014) ) Abeta selectively impairs mGluR7 modulation of NMDA signaling in basal forebrain cholinergic neurons: Implication in Alzheimer’s disease. J Neurosci 34: , 13614–13628. |
[118] | Barone E , Mosser S , Fraering PC ((2014) ) Inactivation of brain Cofilin-1 by age, Alzheimer’s disease and gamma-secretase. Biochim Biophys Acta 1842: , 2500–2509. |
[119] | Lefkowitz RJ , Rajagopal K , Whalen EJ ((2006) ) New roles for beta-arrestins in cell signaling: Not just for seven-transmembrane receptors. Mol Cell 24: , 643–652. |
[120] | Lefkowitz RJ ((2013) ) Arrestins come of age: A personal historical perspective. Prog Mol Biol Transl Sci 118: , 3–18. |
[121] | Zoudilova M , Kumar P , Ge L , Wang P , Bokoch GM , DeFea KA ((2007) ) Beta-arrestin-dependent regulation of the cofilin pathway downstream of protease-activated receptor-2. J Biol Chem 282: , 20634–20646. |
[122] | Zoudilova M , Min J , Richards HL , Carter D , Huang T , DeFea KA ((2010) ) beta-Arrestins scaffold cofilin with chronophin to direct localized actin filament severing and membrane protrusions downstream of protease-activated receptor-2. J Biol Chem 285: , 14318–14329. |
[123] | Thathiah A , Horre K , Snellinx A , Vandewyer E , Huang Y , Ciesielska M , De Kloe G , Munck S , De Strooper B ((2013) ) beta-arrestin 2 regulates Abeta generation and gamma-secretase activity in Alzheimer’s disease. Nat Med 19: , 43–49. |
[124] | Liu X , Zhao X , Zeng X , Bossers K , Swaab DF , Zhao J , Pei G ((2013) ) beta-arrestin1 regulates gamma-secretase complex assembly and modulates amyloid-beta pathology. Cell Res 23: , 351–365. |
[125] | Huang TY , Minamide LS , Bamburg JR , Bokoch GM ((2008) ) Chronophin mediates an ATP-sensing mechanism for cofilin dephosphorylation and neuronal cofilin-actin rod formation. Dev Cell 15: , 691–703. |
[126] | Exton JH ((2002) ) Phospholipase D-structure, regulation and function. Rev Physiol Biochem Pharmacol 144: , 1–94. |
[127] | Kusano K , Abe H , Obinata T ((1999) ) Detection of a sequence involved in actin-binding and phosphoinositide-binding in the N-terminal side of cofilin. Mol Cell Biochem 190: , 133–141. |
[128] | Han L , Stope MB , de Jesus ML , Oude Weernink PA , Urban M , Wieland T , Rosskopf D , Mizuno K , Jakobs KH , Schmidt M ((2007) ) Direct stimulation of receptor-controlled phospholipase D1 by phospho-cofilin. EMBO J 26: , 4189–4202. |
[129] | Cai D , Netzer WJ , Zhong M , Lin Y , Du G , Frohman M , Foster DA , Sisodia SS , Xu H , Gorelick FS , Greengard P ((2006) ) Presenilin-1 uses phospholipase D1 as a negative regulator of beta-amyloid formation. Proc Natl Acad Sci U S A 103: , 1941–1946. |
[130] | Whiteman IT , Gervasio OL , Cullen KM , Guillemin GJ , Jeong EV , Witting PK , Antao ST , Minamide LS , Bamburg JR , Goldsbury C ((2009) ) Activated actin-depolymerizing factor/cofilin sequesters phosphorylated microtubule-associated protein during the assembly of alzheimer-like neuritic cytoskeletal striations. J Neurosci 29: , 12994–13005. |
[131] | Fulga TA , Elson-Schwab I , Khurana V , Steinhilb ML , Spires TL , Hyman BT , Feany MB ((2007) ) Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat Cell Biol 9: , 139–148. |
[132] | Woo JA , Liu T , Fang CC , Cazzaro S , Kee T , LePochat P , Yrigoin K , Penn C , Zhao X , Wang X , Liggett SB , Kang DE ((2019) ) Activated cofilin exacerbates tau pathology by impairing tau-mediated microtubule dynamics. Commun Biol 2: , 112. |
[133] | Zempel H , Dennissen FJA , Kumar Y , Luedtke J , Biernat J , Mandelkow EM , Mandelkow E ((2017) ) Axodendritic sorting and pathological missorting of Tau are isoform-specific and determined by axon initial segment architecture. J Biol Chem 292: , 12192–12207. |


