
A Comparative Study of Pathological Outcomes in The University of Manchester Longitudinal Study of Cognition in Normal Healthy Old Age and Brains for Dementia Research Cohorts
Abstract
In the present study, we have characterized and compared individuals whose brains were donated as part of The University of Manchester Longitudinal Study of Cognition in Normal Healthy Old Age (UoM) with those donated through the Manchester arm of the UK Brains for Dementia Research (BDR) program. The aim of this study was to investigate how differences in study recruitment may affect final pathological composition of cohort studies. The UoM cohort was established as a longitudinal study of aging and cognition whereas the BDR program was established, prima facie, to collect brains from both demented and non-demented individuals for the purpose of building a tissue research resource. Consequently, the differences in recruitment patterns generated differences in demographic, clinical, and neuropathological characteristics. There was a higher proportion of recruits with dementia [mostly Alzheimer’s disease (AD)] within the BDR cohort than in the UoM cohort. In pathological terms, the BDR cohort was more ‘polarized’, being more composed of demented cases with high Braak pathology scores and non-demented cases with low Braak scores, and fewer non-AD pathology cases, than the UoM cohort. In both cohorts, cerebral amyloid angiopathy tended to be greater in demented than non-demented individuals. Such observations partly reflect the recruitment of demented and non-demented individuals into the BDR cohort, and also that insufficient study time may have elapsed for disease onset and development in non-demented individuals to take place. Conversely, in the UoM cohort, where there had been nearly 30 years of study time, a broader spread of AD-type pathological changes had ‘naturally’ evolved in the brains of both demented and non-demented participants.
INTRODUCTION
Longitudinal community or population-based studies with an end-point of brain donation offer a chance to examine correlations between pathology and cognitive function, and are therefore important for the field of dementia research. Nonetheless, truly unbiased community-based longitudinal studies are rare. It is common in epidemiological studies of dementia to use cohorts selected according to their cognitive status, age, gender, or ethnicity. Notwithstanding this, there have been several longitudinal studies of brain aging and dementia that have included brain autopsy end points such as The Medical Research Council Cognitive Function and Ageing Studies (CFAS) [1, 2], the Nun Study [3], the Religious Orders Study [4], and many others [5–10]. Table 1 outlines a selection of these studies and examines their limitations. Most of these studies commenced in the late 1980 s or early 1990 s and were based either on healthy volunteers of all ages from local communities, or selected cohorts based either on age specification or particular lifestyle which included both cognitively normal and cognitively impaired individuals. Cohort size has ranged from a few hundred to many thousand individuals with brain donations ranging from 180-500 (at time of last publication). Most are ongoing, though the Honolulu-Asia Aging Study (HAAS) [7] and Oxford Project to Investigate Memory and Ageing (OPTIMA) [8] studies have now closed.
Table 1
Overview and possible limitations of a selection of longitudinal studies with brain donation end-points
| Study | Commenced | Cohort size | Recruitment age | Cognitive status of participants | Limitations of study |
| MRC CFAS [1, 2] | 1989 | 18000 | 65+ | Normal/C.I. | Sampling strategy led to cohort of older and more cognitively impaired individuals when compared with the general population |
| Nun study [3] | 1986 | 678 | 75+ | Random | Selected cohort (female only) |
| Religious Orders Study [4] | 1994 | 1000+ | Aged | Normal | Selected cohort (male only) |
| BLSA [5] | 1958 | 1400 | 20 – 90 | Normal | Sampling strategy led to cohort of predominantly male, white well educated individuals |
| HAAS [7] | 1991-2012 | 3734 | 71 – 93 | Random | Selected cohort (Japanese-American males) |
| Vantaa 85 + [9] | 1991 | 601 | 85+ | Random | Selectively population-based (South Finland) |
| ACT [10] | 1994 | 2581 | 65+ | Random | Selected cohort (King’s County, WA) |
The present investigation compares neuropathological findings in brains donated as part of The University of Manchester Longitudinal Study of Cognition in Normal Healthy Old Age (UoM) with those donated through the Manchester arm of the UK Brains for Dementia Research (BDR) program. The UoM study [11] began in 1983 and recruited, via local advertisement, 6,542 healthy individuals aged between 42 and 92 years from the Manchester and Newcastle regions of UK. People with evidence of cognitive decline/dementia at the time of recruitment were not eligible for the study. The brain donation program started 20 years later with 312 individuals (less than 5% of the original group) giving consent to brain donation. To date 119 of these 312 individuals have died and donated their brains to the study. Thirty eight of the 119 donors had cognitive impairment/dementia at time of death whereas 81 donors remained cognitively intact. This study represents one of a number of long running studies in which cognitively healthy individuals at the outset have been followed up for periods of 30 years or more. On the other hand, the BDR program began in 2009 across 5 centers within UK, Bristol, London (King’s College), Manchester, Newcastle, and Oxford, and collectively, again through local advertising, national press, and media coverage, has recruited 3,257 individuals to date, 534 of these from the North of England, encompassing Greater Manchester and Merseyside regions, Cumbria, West Yorkshire, and parts of Derbyshire, Nottinghamshire, and Lincolnshire. People both with and without evidence of cognitive decline/dementia at the time of recruitment were eligible for the study. To date, 139 of these 534 individuals have died and donated their brains to the study. One hundred and two of the donors had cognitive impairment/dementia whereas 37 donors remained cognitively normal at time of death.
Using the BDR and UoM cohorts, the aim of this study was to investigate how differences in recruitment strategy might affect the distribution of pathological conditions in cohort studies where brain donation was the end point, and in doing so, determine their suitability for clinical and laboratory research. Although a proportion of the UoM data has already been published [12], it is important to note that in the present study the UoM data is used as a comparison to the BDR data.
MATERIAL AND METHODS
Cognitive assessments
Participants within the UoM study had demographic, education, lifestyle, and health information collected through study-specific self-report questionnaires. Information regarding educational level were standardized using the International Standard Classification of Education (ISCED) guidelines [13]. Over five waves between 2004 and 2017, surviving participants underwent assessment by the modified Telephone Instrument for Cognitive Status (TICSm) which contains 13 questions testing orientation, concentration, immediate and delayed memory, naming, calculation, comprehension, and reasoning. The TICSm test had a maximum score of 39 [14] and the cut-off point, which was used to define cognitive impairment in the present study, was a score of 21 [15]. Cognitive status at death was ascertained using a combination of last TICSm score, patient notes obtained via participants’ general practitioner, cause of death as recorded on the death certificate and information gained from the Brain Bank Coordinator (SCG). Using cognitive status at death and neuropathological findings, diagnostic accuracy was approximately 74% within the UoM cohort.
Participants within the BDR underwent assessments either via telephone interview (for those individuals without memory problems, participants without a significant hearing impairment, study partners for control participants, for follow up and retrospective interviews of control participants), or via a visit to the participant’s home (for initial control visit, people with existing diagnosis of dementia, and controls with a significant hearing problem). Standardized assessments included past medical history from Cambridge Mental Disorders of the Elderly Examination (CAMDEX), clinical dementia rating worksheet (study partner), clinical dementia rating worksheet (participant), Bristol activities of daily living scale (BADLS), lifestyle questionnaire, neuropsychiatric inventory (NPI), Cornell scale for depression in dementia, Geriatric Depression Scale (GDS-15 point), Mini-Mental State Examination (MMSE), TICSm (on healthy controls only), Montreal Cognitive Assessment (MOCA), MOCA-blind (telephone interviews), hearing and eyesight impairment, physical parameters (blood pressure, waist and hip measurement), Global Deterioration Scale, and Hachinski ischemic scoring system. Using cognitive status at death and neuropathological findings, diagnostic accuracy was approximately 71% within the BDR cohort.
Pathological methods
One fresh hemi-brain was fixed in 10% neutral buffered formalin for 3-4 weeks with the other hemi-brain frozen at –80°C. Standard blocks of frontal (mid frontal and superior frontal gyri), cingulate, temporal (including superior and middle temporal gyrus), inferior parietal and occipital cortex, entorhinal cortex, and hippocampus, amygdala, corpus striatum (caudate nucleus, putamen, and globus pallidus), thalamus, midbrain (to include substantia nigra), brainstem (to include locus coeruleus and dorsal vagus), and cerebellum with dentate nucleus were cut from the fixed tissue and processed into wax blocks. In the first instance, paraffin sections (6μm) from all blocks were stained with hematoxylin and eosin. Further sections from all blocks were then immunostained for amyloid-β (Aβ) (Cambridge Bioscience, monoclonal antibody 4G8, 1 : 3000) and tau proteins phosphorylated at Ser202 and Thr205 (P-tau) (Innogenetics, monoclonal antibody AT8, 1 : 750). Sections from hippocampus, midbrain, superior frontal gyrus, amygdala, pons, and medulla were immunostained for phosphorylated α-synuclein (rabbit polyclonal antibody #1175, 1 : 1000; kind gift of Dr. Masato Hasegawa at Tokyo Metropolitan Institute of Medical Science, Japan). Sections from hippocampus and amygdala were immunostained for n-terminal TDP-43 (polyclonal antibody, 10782-2-AP, Proteintech, Manchester, 1 : 1000) which recognizes the intact 45 kDa protein as well as post translationally modified and truncated forms. For antigen retrieval, sections were either immersed in 70% formic acid for 20 min (for Aβ only), or (for all other antibodies) by microwaving or pressure cooking (for 30 min, reaching 120°C and > 15 kPa pressure) in 0.1 M citrate buffer, pH 6.0, prior to incubation with primary antibody. Thal phase for amyloid deposition [16] was determined from the Aβ immunostained sections and Braak stage for tau deposition [17, 18] was assigned using the AT8 immunostained sections. The presence/absence of TDP-43 pathology was assessed in the frontal cortex, temporal cortex, fusiform gyrus, and dentate gyrus of the hippocampus. Phosphorylated α-synuclein pathology was assessed in the cingulate gyrus, amygdala, entorhinal cortex, substantia nigra, locus coeruleus, and dorsal motor nucleus of the vagus nerve and Braak Lewy body stage was determined [19].
All neuropathological diagnoses were assigned by experienced neuropathologists (DM, FR, DDP, and PP). Pathological diagnosis of Alzheimer’s disease (AD) was made according to established criteria [20] whereby a low, intermediate, or high probability of AD is ascribed according to relative density and distribution of amyloid plaques (Thal phase), neurofibrillary tangles (Braak tau stage), and Aβ neuritic plaques (Consortium to Establish a Registry for Alzheimer’s Disease – CERAD). Diagnosis of Dementia with Lewy bodies (DLB)/Parkinson’s Disease dementia (PDD) was made according to McKeith et al criteria [21–23]. Other pathological diagnoses were made in accordance with published criteria or descriptions for frontotemporal lobar degeneration (FTLD) [24, 25], corticobasal degeneration (CBD) [26], progressive supranuclear palsy (PSP) [27], multiple system atrophy (MSA) [28], argyrophilic grain disease (AGD) [29], primary age-related tauopathy (PART) [30], age related tau astrogliopathy (ARTAG) [31], and cerebral amyloid angiopathy (CAA) [32]. The likelihood that cerebrovascular disease had contributed to the development of dementia was assessed using the three key elements developed in the Vascular Cognitive Impairment Neuropathology Guidelines (VCING) [33]. These involve the presence of brain infarction within cortical or subcortical regions, the degree of small vessel disease (SVD) in basal ganglia and the extent of CAA in the occipital lobe. Examination of vascular pathology in these three regions proved to be clinical predictive for cognitive impairment.
Tissue infarction in basal ganglia was assessed as:
0 = no apparent tissue changes.
1 = single area of microinfarction or multiple areas of perivascular lacunar change.
2 = multiple areas of microinfarction or a single large (>10 mm) infarction.
3 = multiple large infarctions.
In accordance with Olichney et al. [32], CAA in occipital lobe was assessed as:
0 = No CAA in blood vessel walls in leptomeninges or brain parenchyma.
1 = Occasional blood vessels with CAA in leptomeninges and/or within brain parenchyma, usually not occupying the full thickness of the wall.
2 = A moderate number of blood vessels with CAA in leptomeninges or brain parenchyma in leptomeninges or within brain parenchyma, some occupying the full thickness of the wall.
3 = Many or all blood vessels with CAA in leptomeninges or brain parenchyma, most occupying the full thickness of the wall.
SVD in basal ganglia was assessed as:
0 = no changes in vessel wall.
1 = mild thickening of vessel wall.
2 = moderate thickening of vessel wall.
3 = severe thickening of vessel wall.
Scores from each of these assessments were summated to provide an overall rating for vascular pathology on a scale from 0 to 9.
Genetic analysis
DNA was extracted from frozen brain tissue using REDExtract-N-Amp™ Tissue PCR Kit (Sigma) or from previously obtained blood samples (3 cases). The APOE genotype was determined using routine polymerase chain reaction (PCR) methods [34]. APOE could not be determined for 2 UoM and 5 BDR participants because of lack of blood samples or frozen brain tissue.
Statistical analyses
T-test was used to compare age demographics (age at death, age at onset and duration of disease (where applicable), age when recruited, time spent on study from recruitment to death, years of illness before recruitment and years on study before onset of illness) between the two cohorts both as a whole, and stratified by cognitive status. Chi-squared test was used to analyze whether there were differences in gender ratio, frequency of presence of dementia among cases, pathological causes of dementia, and severity grades according to pathological status. Differences in frequency of APOE ɛ2 or ɛ4 alleles according dementia status was also analyzed by Chi-squared test. Where significant, logistic regression was used to investigate if pathological and genetic differences remained after adjustment for gender, age at death, and presence of APOE ɛ4 allele(s).
Pearson correlations were used to assess relationships between MMSE/TICSm scores, cognitive status and pathology.
A p value of < 0.05 was considered significant for all tests.
RESULTS
Demographics
Of the 139 participants in the BDR study, 74 were male (53%) and 65 (47%) female (gender ratio 1.1 : 1), whereas of the 119 participants in the UoM study 38 were males (32%) and 81 (68%) were females (gender ratio 0.45 : 1) (Table 2). Consequently, the male-to-female ratio was significantly higher in the BDR than the UoM cohort (χ2 = 11.8; p = 0.001). In the BDR study, 102 participants (73%) (54 males and 48 females) had cognitive impairment/dementia, whereas 37 (27%) (20 males and 17 females) remained cognitively normal. In the UoM study, 38 participants (32%) had cognitive impairment/dementia, whereas 81 (68%) remained cognitively normal at time of death. The ratio of cognitively impaired to non-impaired individuals was significantly higher in the BDR study than in the UoM study (χ2 = 44.4; p < 0.001).
Table 2
Demographics of participants in the Brains for Dementia Research (BDR) and University of Manchester Longitudinal Study of Cognition in Normal Healthy Old Age (UoM) cohorts stratified by gender and cognitive impairment. *** denotes proportion of patients with neurodegenerative disease significantly greater in BDR cohort compared to MH cohort, p < 0.001. +++ denotes proportion of females significantly greater than that of males, p < 0.001
| BDR | UoM | |||||
| All | Normal | Dementia | All | Normal | Dementia | |
| Gender | ||||||
| All | 139 | 37 (27%) | 102 (73%)*** | 119 | 81 (68%) | 38 (32%) |
| Males | 74 (53%) | 20 (27%) | 54 (73%)*** | 38 (32%) | 24 (63%) | 14 (37%) |
| Females | 65 (47%) | 17 (26%) | 48 (74%)*** | 81 (68%)+++ | 57 (70%) | 24 (30%) |
In the BDR study, MMSE scores significantly declined in those individuals who were assessed more than once (n = 35) during the course of the study (p < 0.001). There was a strong correlation between final MMSE score before death and cognitive status (n = 54; p < 0.001). Likewise, in the UoM study, there was a strong correlation between final TICSm score before death and cognitive status (n = 110; p < 0.001).
Age demographics
Demographic details concerning age at death, age at onset, and duration of disease (where applicable), age when recruited, time spent on study from recruitment to death, years of illness before recruitment, and years on study before onset of illness (where applicable) for both BDR and UoM cohorts are shown in Table 3. Overall, mean recruitment age in the UoM cohort (62.8±5.0 years) was significantly younger (p < 0.001) than that of the BDR cohort (79.8±10.4 years), and they had spent considerably longer (p < 0.001) on the study (26.1±4.0 years) than the BDR cohort (1.7±1.9 years). By contrast, mean age at death in the BDR cohort (81.5±10.5 years) was significantly earlier (p < 0.001) than that in the UoM cohort (88.5±6.0 years).
Table 3
Demographic details concerning age at death, age at onset and duration of disease (where applicable), age when recruited, time spent on study from recruitment to death, years of illness before recruitment, and years on study before onset of illness (where applicable) for both BDR and UoM cohorts
| BDR | UoM | p | |||
| Recruitment age (all) | All (n = 139) | 79.8±10.4 | All (n = 118) | 62.8±5.0 | 0.000 |
| Males (n = 74) | 77.4±8.9 | Males (n = 37) | 62.8±4.6 | 0.000 | |
| Females (n = 65) | 82.5±11.3 | Females (n = 81) | 62.8±5.2 | 0.000 | |
| Recruitment age (normal) | All (n = 37) | 79.8±12.5 | All (n = 81) | 62.8±5.2 | 0.000 |
| Males (n = 20) | 79.7±11.4 | Males (n = 24) | 63.2±4.4 | 0.000 | |
| Females (n = 17) | 80.7±14.0 | Females (n = 57) | 62.6±5.6 | 0.000 | |
| Recruitment age (demented) | All (n = 102) | 79.8±9.6 | All (n = 37) | 62.8±4.6 | 0.000 |
| Males (n = 54) | 76.5±7.7 | Males (n = 13) | 62.1±5.2 | 0.000 | |
| Females (n = 48) | 83.2±10.2 | Females (n = 24) | 63.2±4.2 | 0.000 | |
| Time on study (all) | All (n = 139) | 1.7±1.9 | All (n = 118) | 26.1±4.0 | 0.000 |
| Males (n = 74) | 1.8±2.0 | Males (n = 37) | 25.7±4.6 | 0.000 | |
| Females (n = 65) | 1.6±1.8 | Females (n = 81) | 26.2±3.6 | 0.000 | |
| Time on study (normal) | All (n = 37) | 2.8±2.3 | All (n = 81) | 25.9±4.3 | 0.000 |
| Males (n = 20) | 3.2±2.3 | Males (n = 24) | 25.1±4.9 | 0.000 | |
| Females (n = 17) | 2.5±2.2 | Females (n = 57) | 26.3±4.0 | 0.000 | |
| Time on study (demented) | All (n = 102) | 1.3±1.6 | All (n = 37) | 26.5±3.2 | 0.000 |
| Males (n = 54) | 1.4±1.7 | Males (n = 13) | 26.8±4.1 | 0.000 | |
| Females (n = 48) | 1.2±1.5 | Females (n = 24) | 26.3±2.7 | 0.000 | |
| Age at onset (demented) | All (n = 94) | 72.3±10.6 | All (n = 20) | 83.0±4.4 | 0.000 |
| Males (n = 49) | 69.0±8.8 | Males (n = 7) | 82.4±6.0 | 0.000 | |
| Females (n = 45) | 75.8±11.0 | Females (n = 13) | 83.3±3.5 | 0.000 | |
| Age at death (all) | All (n = 139) | 81.5±10.5 | All (n = 119) | 88.5±6.0 | 0.000 |
| Males (n = 74) | 79.2±9.3 | Males (n = 38) | 88.5±5.1 | 0.000 | |
| Females (n = 65) | 84.1±11.3 | Females (n = 81) | 88.6±6.4 | 0.000 | |
| Age at death (normal) | All (n = 37) | 82.5±13.0 | All (n = 81) | 88.3±6.5 | 0.013 |
| Males (n = 20) | 83.0±12.1 | Males (n = 24) | 88.3±5.0 | 0.078 | |
| Females (n = 17) | 82.6±14.4 | Females (n = 57) | 88.4±7.2 | 0.094 | |
| Age at death (demented) | All (n = 102) | 81.2±9.5 | All (n = 38) | 88.9±4.8 | 0.000 |
| Males (n = 54) | 77.9±7.8 | Males (n = 14) | 88.7±5.5 | 0.000 | |
| Females (n = 48) | 84.7±9.9 | Females (n = 24) | 89.0±4.4 | 0.017 | |
| Duration of illness (demented) | All (n = 94) | 8.6±3.6 | All (n = 20) | 5.6±2.6 | 0.000 |
| Males (n = 49) | 8.4±3.4 | Males (n = 7) | 4.9±3.4 | 0.014 | |
| Females (n = 45) | 8.6±3.6 | Females (n = 13) | 5.9±2.2 | 0.010 | |
| Years of illness before recruitment (demented) | All (n = 94) | 7.1±3.4 | All (n = 38) | 0.0±0.0 | na |
| Males (n = 49) | 7.1±3.2 | Males (n = 14) | 0.0±0.0 | na | |
| Females (n = 45) | 7.0±3.4 | Females (n = 24) | 0.0±0.0 | na | |
| Years of recruitment before onset of disease (demented) | All (n = 94) | 0.0±0.0 | All (n = 20) | 20.9±3.0 | na |
| Males (n = 49) | 0.0±0.0 | Males (n = 7) | 21.9±3.9 | na | |
| Females (n = 45) | 0.0±0.0 | Females (n = 13) | 20.4±2.6 | na | |
In those individuals with cognitive impairment/dementia, age at onset in BDR cohort (72.3±10.6 years) was also significantly earlier (p < 0.001) than that in the UoM cohort (83.0±4.4 years), and duration of disease in the BDR cohort (8.6±3.6 years) was significantly longer (p < 0.001) than that in the UoM cohort (5.6±2.6 years). On average, those individuals with cognitive impairment/dementia in the BDR cohort were 79.6±9.8 years old when recruited but had already suffered from their illness for 7.1±3.4 years before recruitment, and therefore only spent 1.3±1.6 years on the study before their death. Conversely, in the UoM cohort, no individuals had been suffering from cognitive impairment/dementia at the time of recruitment. Those who ultimately developed cognitive impairment/dementia at 83.0±4.4 years had spent 26.5±3.2 years on the study before their death at 88.9±4.8 years. Those individuals who remained free from cognitive impairment/dementia in the BDR cohort were 79.8±12.5 years when recruited, were 82.5±13.0 years at death, and therefore only spent 2.8±2.3 years on the study. On the other hand, those individuals in the UoM cohort who remained free from cognitive impairment/dementia were 62.8±5.2 years when recruited, 88.3±6.5 years at death and therefore spent 25.9±4.3 years on the study before death. When analyzed separately, there were no significant differences between males and females for either cohort in respect of any of these various demographic features.
Clinical characteristics
Of the 102 participants in the BDR cohort who were diagnosed clinically with dementia/cognitive impairment, 53 were clinically ascribed a diagnosis of AD, 11 had frontotemporal dementia (FTD) [7 with behavioral variant FTD (bvFTD), 1 with progressive non-fluent aphasia (PNFA), 2 with bvFTD/PNFA, and 1 with semantic dementia (SD)], 10 had vascular dementia (VaD), 10 had DLB/PDD, 16 had unspecified dementia, 1 had corticobasal syndrome, and 1 had MSA. Of the 38 participants in the UoM cohort who were clinically diagnosed with dementia, 7 were ascribed a diagnosis of AD, 1 had bvFTD, 5 had VaD, 2 had DLB/PDD, 22 had unspecified dementia, and 1 had MSA.
Pathological characteristics
The pathological characteristics of those individuals exhibiting cognitive impairment/dementia in both BDR and UoM cohorts can be found in Table 4. Although primary neuropathological diagnoses were used in analyses, comorbid pathologies were not uncommon; especially in those with a primary diagnosis of AD. In both cohorts, there were incidences of AD+DLB pathology (BDR 14 cases; UoM 5 cases), AD+vascular pathology (BDR 18 cases; UoM 11 cases), and AD+TDP43 pathology (BDR 11 cases; UoM 7 cases). There were no significant differences between the BDR and UoM cohorts with respect to the frequency of pathological AD (p = 0.459), DLB (p = 0.988), or the combined frequency of other disorders (p = 0.420).
Table 4
Pathological characteristics of those individuals exhibiting cognitive impairment/dementia in the BDR and UoM cohorts. AD, Alzheimer’s disease (*includes posterior cortical atrophy variant of AD: 3 in BDR and 1 in UoM); DLB, dementia with Lewy bodies; PD, Parkinson’s disease; FTLD, frontotemporal lobar degeneration; CBD, corticobasal degeneration; PSP, progressive supranuclear palsy; MSA, multiple system atrophy; AGD, argyrophilic grain disease; ARTAG, age related tau astrogliopathy
| Principle pathology | Cohort | Total | ||||
| BDR | UoM | |||||
| n | % | n | % | n | % | |
| AD | 68* | 67 | 24* | 63 | 92 | 66 |
| DLB | 16 | 16 | 6 | 16 | 22 | 16 |
| PD | 2 | 2 | 0 | 0 | 2 | 1 |
| FTLD | 3 | 3 | 0 | 0 | 3 | 2 |
| Vascular | 4 | 4 | 4 | 11 | 8 | 6 |
| CBD | 3 | 3 | 2 | 5 | 5 | 4 |
| PSP | 1 | 1 | 0 | 0 | 1 | 1 |
| MSA | 1 | 1 | 0 | 0 | 1 | 1 |
| AGD | 2 | 2 | 0 | 0 | 2 | 1 |
| ARTAG | 0 | 0 | 1 | 3 | 1 | 1 |
| No specific changes | 2 | 2 | 1 | 3 | 3 | 2 |
Braak tau staging
The proportion of individuals at each Braak tau stage, stratified by cognitive status and cohort, can be found in Fig. 1a. The proportion of individuals with neurodegenerative disease in the BDR cohort at Braak stages V or VI (57/92, 62%) was significantly higher (p < 0.001) than that in the UoM cohort (20%). In contrast, the proportion of individuals with neurodegenerative disease in the BDR cohort at Braak stages III-IV (24/91, 26%) was significantly lower (p < 0.001) than that in the UoM cohort (60%). There was no difference in the proportion of individuals at Braak stages 0-II (11/91, 12% in BDR, 7/35, 20.0% in UoM; p = 0.350). Differences in Braak tau staging between the cohorts remained significant when controlling for age, gender, and presence of APOE ɛ4 allele(s) (OR = 0.485, 95% CI: 0.324 – 0.725; p < 0.001).
Fig.1
Proportions of individuals exhibiting Alzheimer pathology, stratified by cognitive status and cohort. (Braak tau stage is shown in panel a; Thal phase is shown in panel b).
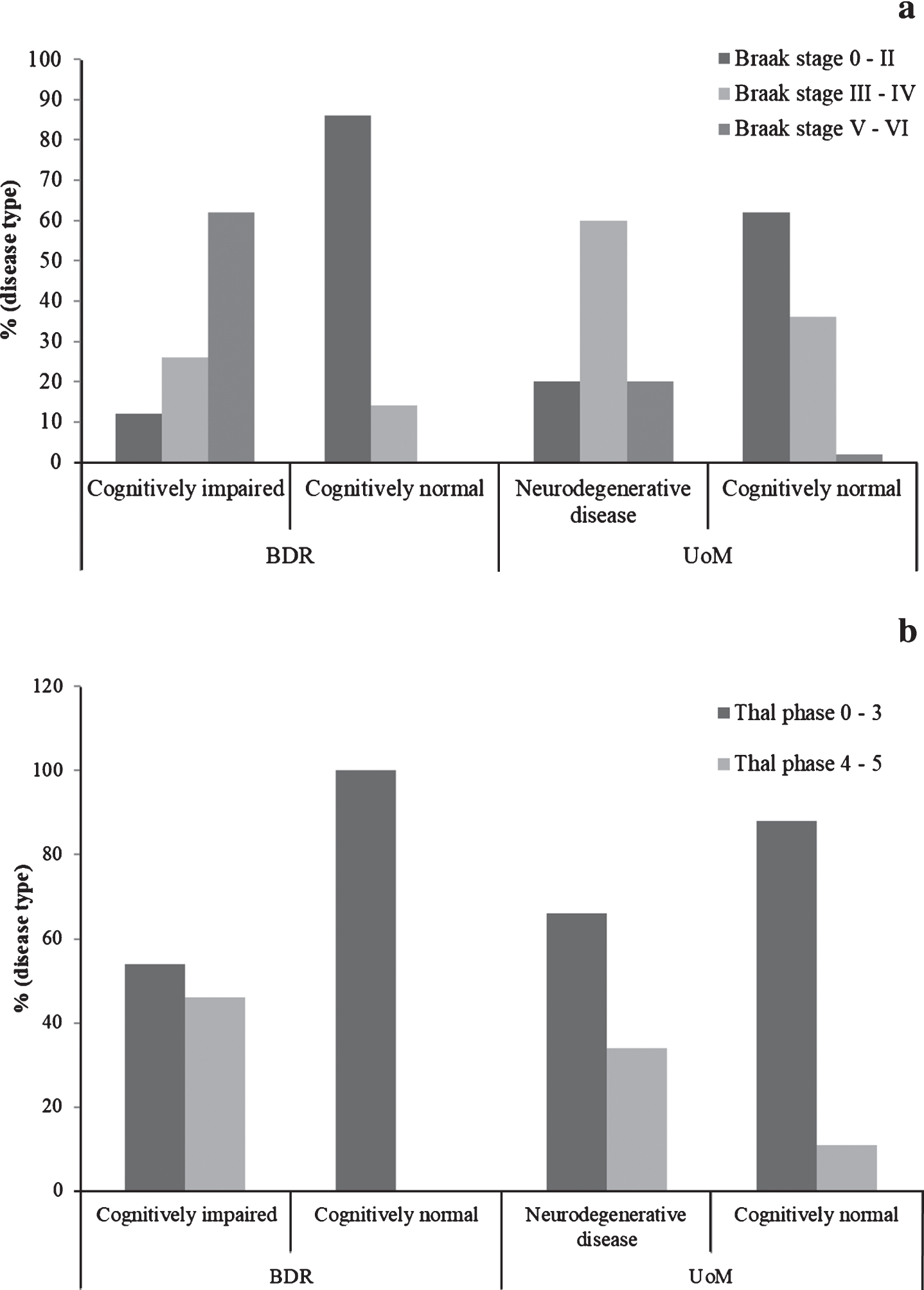
In the cognitively normal individuals, the proportion at Braak stages III-IV in the UoM cohort (36%) was significantly higher (p = 0.013) than that in the BDR cohort (14%), while the proportion of individuals at Braak stages 0-II in the UoM cohort (62%) was significantly lower (p = 0.007) than that in the BDR cohort (86%). Curiously, there were two cognitively normal individuals in the UoM cohort at Braak stages V-VI, but none such as these were present in the BDR cohort. Differences in Braak tau staging between cognitively normal individuals in both cohorts remained significant when controlling for age, gender, and presence of APOE ɛ4 allele(s) (OR = 4.551, 95% CI: 1.480 – 13.990; p = 0.008).
In the BDR study, there was a strong negative correlation between last MMSE scores and Braak tau stage (r=-0.781, n = 54, p < 0.001). Similarly, in the UoM study, there was a strong negative correlation between last TICSm score and Braak tau stage (r=-0.298, n = 107, p = 0.002).
Thal phase
The proportion of individuals at each Thal phase, stratified by cognitive status and cohort, can be found in Fig. 1b. There was no difference (p = 0.207) in the proportion of individuals with neurodegenerative disease in the BDR cohort at Thal stages 4 or 5 (47/102, 46%) and the UoM cohort (34%). Similarly, there was no difference (p = 0.207) in the proportion of individuals with neurodegenerative disease in the BDR cohort at Thal phases 3 or less (54%) and the UoM cohort (66%).
In the cognitively normal individuals, no individuals exceeded Thal phase 3 in the BDR cohort, but, conversely, 9 cognitively intact individuals in the UoM cohort were found to be either Thal stage 4 or 5.
In the BDR study, there was a strong negative correlation between last MMSE scores and Thal phase (r=-0.554, n = 54, p < 0.001). Similarly, in the UoM study, there was a negative correlation between last TICSm score and Thal phase (r=-0.243, n = 110, p = 0.011).
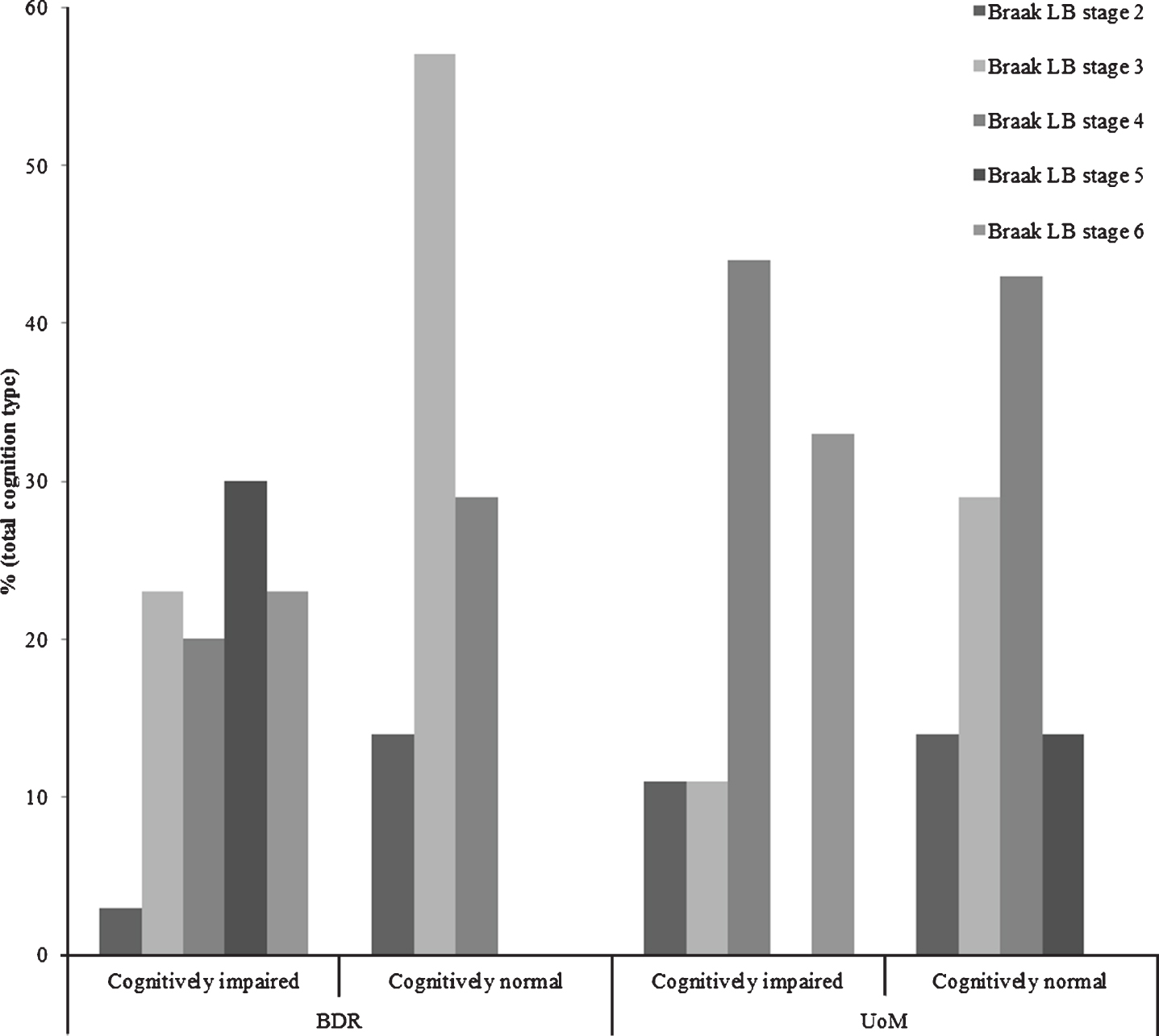
Lewy body pathology
The proportion of individuals exhibiting Lewy body pathology at each Braak Lewy body stage, stratified by cognitive status and cohort, can be found in Fig. 2. Thirty-seven of the 139 (27%) participants in the BDR cohort displayed some degree of Lewy body pathology, 30 with, and 7 without, cognitive impairment/dementia. In the UoM cohort, Lewy body pathology occurred in 16/119 (13%) participants; 9 with, and 7 without, cognitive impairment/dementia. The proportion of individuals with cognitive impairment/dementia in the BDR cohort with LBD at Braak Lewy body stages 5 or 6 (53%) was not significantly higher (p = 0.292) than that in the UoM cohort (33%).
Fig.2
Proportions of individuals exhibiting Lewy body pathology at each Braak Lewy body stage, stratified by cognitive status and cohort.
TDP-43 proteinopathy
Overall, TDP-43 proteinopathy was present in the BDR cohort in 21/139 (15%) individuals; all had neurodegenerative disease (2 with FTLD-TDP type A pathology,13 with AD, 4 with DLB, 1 with CBD, and 1 with PSP). In the UoM cohort, TDP-43 proteinopathy was present in 19/119 (16%) individuals. When restricted to individuals with cognitive impairment/dementia, TDP-43 proteinopathy was present in 21/102 (20%) individuals in the BDR cohort and in 11/38 (29%) individuals in the UoM cohort. In two of these individuals in the BDR cohort with FTD, this was severe and widespread throughout the cerebral cortex and conformed to FTLD-TDP type A pathology. Of the remaining 19 individuals, 13 had AD, 4 had DLB, 1 had CBD, and 1 had PSP. In the UoM cohort, 6 individuals had AD, 2 had DLB, 1 had ARTAG, and 2 had cerebrovascular disease. In these, TDP-43 pathology was restricted to cells of dentate gyrus, fusiform gyrus and, to variable but limited extents, inferior and middle temporal gyri, rarely occurring in frontal cortex. No individuals without neurodegenerative disease in the BDR cohort showed TDP-43 pathological changes in any brain region examined whereas 1 individual with pathology considered ‘normal for age’ showed TDP-43 inclusions in the UoM cohort. The overall proportion of individuals with cognitive impairment/dementia in the BDR cohort with TDP-43 proteinopathy did not differ from that in the UoM cohort (21/102, 21% versus 11/38, 29%, respectively; p = 0.295). Similarly, when only those individuals with AD were considered, there was a similar proportion of individuals with TDP-43 proteinopathy in the UoM cohort compared to the BDR cohort (6/23, 26% versus 13/68, 19%, respectively; p = 0.477).
Vascular pathology
There was no significant difference in the degree of vascular pathology when all participants in the BDR and UoM cohorts were compared for total scores (p = 0.612), degree of CAA (p = 0.314), SVD (p = 0.353), or degree of tissue infarction (p = 0.220). Similarly, there were no significant differences in the degree of vascular pathology (infarction, p = 0.647; degree of CAA, p = 0.984; SVD, p = 0.842; total scores, p = 0.567) when all demented participants in the BDR and UoM cohorts were compared, or when all non-demented participants in the BDR and UoM cohorts were compared (infarction, p = 0.435; degree of CAA, p = 0.760; SVD, p = 0.760; total scores, p = 0.612). When demented participants in the UoM cohort were compared with non-demented participants, the degree of CAA tended to be greater in the demented participants (p = 0.064) though there were no significant differences in the extent of infarction (p = 0.611), SVD (p = 0.462), or overall scores (p = 0.561). Similarly, when demented participants in the BDR cohort were compared with non-demented participants, the degree of CAA again tended to be greater in the demented participants (p = 0.068), and again there were no significant differences in the extent of infarction (p = 0.746), SVD (p = 0.413), or overall scores (p = 0.484). When analyzed separately, there were no significant differences in either cohort between males and females or demented and non-demented, in respect of any of these vascular pathology measures. Similarly, there were no differences with regards any of the vascular measures between the BDR or UoM cohorts with respect to males or females or demented or non-demented individuals.
APOE genotypes
Full details regarding APOE genotypes are presented in Table 5. Overall, APOE ɛ4 allele frequency was significantly higher (p = 0.003) in the BDR cohort than in the UoM cohort. Although the age and gender composition of the cohorts seemed to have a small effect on this outcome, the difference remained when controlling for the effects of age and gender (OR = 0.546, 95% CI: 0.327–1.015; p = 0.056). When participants were stratified into individuals with or without neurodegenerative disease, no such differences were seen either for normal individuals (p = 0.605) or those with neurodegenerative disease (p = 0.542). The higher frequency of APOE ɛ4 alleles in the BDR cohort was due to the higher proportion of individuals with neurodegenerative disease (mostly AD) for which possession of APOE ɛ4 allele is a known risk factor. Conversely, APOE ɛ2 allele frequency was significantly higher (p = 0.022) in the UoM cohort than in the BDR cohort, and this difference remained the case when controlling for the effects of age and gender (OR = 3.696, 95% CI: 1.334–10.244; p = 0.012). Similarly, when only normal individuals were considered, this was also the case (p = 0.050) due to the higher proportion of individuals without neurodegenerative disease in the UoM than the BDR cohorts, more of whom bore APOE ɛ2 allele. Again, gender and age at death had no influence on this outcome (OR = 5.094, 95% CI: 0.990–26.201; p = 0.051). No such difference between each cohort was seen for those individuals with neurodegenerative disease (p = 0.764).
Table 5
APOE allele and genotype numbers (percentage frequency in parentheses) in BDR and UoM cohorts, in total and when stratified into normal individuals and those with dementia. *, **, and *** denotes significantly different from respective BDR group, p < 0.05, <0.01, and < 0.001, respectively
| BDR | UoM | |||||
| All (n = 134) | Normal (n = 36) | Demented (n = 98) | All (n = 116) | Normal (n = 77) | Demented (n = 39) | |
| ɛ2/ɛ2 | 0 (0) | 0 (0) | 0 (0) | 2 (1.7) | 2 (2.6) | 0 (0) |
| ɛ2/ɛ3 | 6 (4.5) | 1 (2.8) | 5 (5.1) | 12 (10.3) | 12 (15.6) | 0 (0) |
| ɛ2/ɛ4 | 2 (1.5) | 1 (2.8) | 1 (1.0) | 2 (1.7) | 0 (0) | 2 (5.1) |
| ɛ3/ɛ3 | 62 (48.6) | 26 (72.2) | 36 (36.7) | 66 (56.9) | 45 (58.4) | 21 (53.8) |
| ɛ3/ɛ4 | 48 (36.1) | 7 (19.4) | 42 (42.9) | 32 (27.6) | 18 (23.4) | 14 (35.9) |
| ɛ4/ɛ4 | 15 (11.3) | 1 (2.8) | 14 (14.3) | 2 (1.7) | 0 (0) | 2 (5.1) |
| ɛ2 | 8 (3.0) | 2 (2.8) | 6 (3.1) | 18 (7.8)** | 16 (10.4)* | 2 (2.6) |
| ɛ3 | 179 (66.8) | 60 (83.3) | 119 (60.7) | 176 (75.8) | 120 (77.9) | 56 (71.8) |
| ɛ4 | 81 (30.2) | 10 (13.9) | 71 (36.2) | 38 (16.4)*** | 18 (11.7) | 20 (25.6)*** |
DISCUSSION
In the present study we have compared the demographic, clinical, and neuropathological characteristics of individuals whose brains were donated as part of The University of Manchester Longitudinal Study of Cognition in Normal Healthy Old Age (UoM) with donors recruited through the Manchester arm of the UK Brains for Dementia Research (BDR) program. In contrast to the recent study [35] looking at the BDR cohorts at two different centers (Cardiff and London), clinicopathological correlations were not the principal focus of this work. However, we too found similar distribution of pathological features in the UoM and BDR cohorts and an extent of disagreement between clinical and pathological diagnoses.
Community and population-based studies with brain donation end-points are becoming more common [1–9]. The UoM cohort was initially established as a longitudinal study of aging and cognition through voluntary recruitment of healthy persons and as such avoided many of the selection criteria inherent in other studies, such as cognitive status, age, gender, or ethnicity [3, 4]. It is typical and representative when compared with other community-based, population-based, or clinico-pathological cohorts [12]. Unlike the UoM, the BDR program was established, prima facie, to collect brains from both demented and non-demented individuals for the purpose of building a tissue research resource and, as such, does not mirror other population-based longitudinal cognitive studies with autopsy outcomes. Clinical and demographic details were incorporated into the study, though given that many of the demented individuals had suffered for many years before recruitment and were at the end-stages of their illness when accessioned, these largely provided only a ‘snapshot’ of their decline and could not be considered to truly map the course of their illness. Of the non-demented individuals recruited, all had died without developing overt neurodegenerative disease. Hence, the BDR cohort largely represents a cross-sectional rather than a longitudinal study. As a consequence, there was a much higher proportion of recruits with dementia (mostly with AD) within the BDR cohort than in the UoM cohort, and this elevated the overall APOE ɛ4 allele frequency in the BDR cohort compared to the UoM cohort. Nonetheless, APOE ɛ4 allele frequency did not differ between demented or non-demented participants of either cohort suggesting that although the method of recruitment may have differed between the two cohorts, the genetic characteristics of the participants in each may be similar.
There are some general limitations of this investigation. In the UoM study, brain donation was only introduced in 2004 and was not in the original scope of the study. Thus, a number of potential donors were lost due to withdrawal from the study or death before 2004. In the BDR study, only 13 donations were missed due to various circumstances. These included absence of staff during holiday periods, donor’s family deciding against brain donation after death and, most commonly, the Brain Bank not being informed of a donor’s death in a timely manner. This highlights the need for study coordinators to have good communication and relationships with donor family members as well as the donors themselves. Similar to all studies of this kind, sample size is a limitation imposed on both the BDR and UoM cohorts, and the fact that both cohorts were self-selected suggests that the study samples may not be representative of the general population. In addition, the geographical areas covered by the BDR (North of England) and UoM (Greater Manchester and Newcastle) may not reflect society as a whole. Although a diagnosis of dementia was not confirmed by a diagnostic clinical interview, consensus was reached by experts using a wide range of in-life measures. Time from last examination until death is also a limiting factor, especially for the UoM cohort. For example, the two cognitively normal individuals who exhibited Braak V-VI pathology were last examined many years before death when they were still cognitively intact (as ascertained by TICSm score). A lack of follow up closer to death means that cognitive decline may have been missed. It is of note that no dementing illness was listed as a cause of death for either individual meaning that either they were cognitively intact at death or that dementia was present but not recognized as a primary or secondary cause of death; a problem which persists and has been highlighted previously [36, 37].
Although there was a marginally higher frequency of pathologically confirmed AD cases among the BDR demented individuals compared to the UoM cohort (67% versus 61%), the converse was true for other disorders, particularly small vessel disease which was more common in the UoM cohort. As would be anticipated, given the higher proportion of cases with AD, there was a higher proportion of cases at Braak stages V-VI among the BDR dementia cases compared to the UoM cohort, but the converse was seen for cases at Braak stages III-IV which is higher in the UoM cases. Among the non-demented cases, there was higher proportion of cases at Braak stages 0-II in the BDR cohort compared to the UoM cohort, but converse was seen for Braak stages III-IV which was higher in the UoM cases. Such observations again reflect the polar recruitment of demented and non-demented individuals in the BDR cohort where insufficient study time may have elapsed for disease onset and development in the non-demented individuals to take place, whereas in the UoM cohort there had been nearly 30 years of study time for most individuals still enrolled at the point of brain donation during which there had been sufficient time for a much broader spread of AD-type pathological changes to have evolved in the brains of both demented and non-demented participants.
Although Lewy body pathology was found in both cohorts, the greater overall prevalence in the BDR cohort probably reflects the targeted recruitment of demented individuals in this cohort over the natural evolution of disease in initially healthy individuals recruited into the UoM cohort, though the proportions of people clinically affected by Lewy body disease within each cohort did not differ.
Similarly, TDP-43 pathology was found in both cohorts, mainly as a comorbid pathology. Excluding those 2 individuals in the BDR cohort with FTLD-TDP type A, TDP-43 pathology was restricted in all other 38 individuals, to cells of dentate gyrus, fusiform gyrus, and, to variable but limited extents, inferior and middle temporal gyri, rarely occurring in frontal cortex. This histological pattern of TDP-43 pathology has been widely reported previously in both late onset AD [38], DLB [39, 40], and PSP/CBD [41] and has been considered to be a ‘secondary’ pathology as opposed to the primary pathology associated with FTLD-TDP. Again, the higher overall prevalence of TDP-43 proteinopathy in the BDR cohort compared to the UoM cohort reflects the targeted recruitment of individuals into this cohort with those dementing disorders in which this kind of TDP-43 proteinopathy is commonly seen. Although the overall proportion of individuals with cognitive impairment/dementia in the BDR cohort with TDP-43 proteinopathy did not differ from that in the UoM cohort, it was seen that there was a higher proportion of individuals with TDP-43 proteinopathy in the UoM cohort compared to the BDR cohort when only those individuals with AD were considered. In AD, the prevalence of TDP-43 proteinopathy is greater in late onset AD compared to early onset AD [38] and its presence is considered to be an age-associated phenomenon. The higher proportion of individuals with AD showing TDP-43 proteinopathy in the UoM cohort compared to the BDR cohort may therefore reflect the later age at death of those individuals in the UoM cohort.
Vascular pathology, as assessed by VCING guidelines [33], was commonly seen in both cohorts. Demented participants were more likely to exhibit moderate to severe CAA when compared with their cognitively normal counterparts. This is unsurprising as CAA is a known contributor to cognitive decline and intracerebral hemorrhage stroke in the elderly [42] However, this was not the case when examining SVD or tissue infarction. It is possible that SVD in isolation or tissue infarction was not sufficient to produce the clinical manifestation of cognitive impairment.
In conclusion, this study highlights the importance of an appropriate recruitment strategy when conducting this type of research. Case-control studies are possibly more suited to a recruitment strategy similar to that employed by the BDR whereas community or population based clinico-pathological studies should perhaps opt for a recruitment strategy similar to the UoM and add longitudinal aspects to the study. In pathological terms, the BDR cohort seems to be more ‘polarized’, being more likely to be composed of demented cases with high Braak pathology scores and non-demented cases with low Braak scores, and fewer non-Alzheimer pathology cases than the UoM cohort. The latter seems to be more mixed in pathology with more demented cases showing combined Alzheimer/vascular/other pathologies, each of which in isolation is less likely to meet established pathological diagnostic criteria for AD and other disorders, but in combination with age-related reductions in ‘cerebral reserve’ will generate cognitive failure/dementia. Dementia in the oldest old seems to be a feature of combined pathologies whereas in less elderly persons a single overwhelming cause is usual.
Research Ethics Committee Approval
The study was approved by Manchester Brain Bank Management Committee (REC reference 09/H0906/52 + 5). Under conditions agreed with the Research Ethics Committee, The Manchester Brain Bank can supply tissue or data to researchers, without requirement for researchers to apply individually to the REC for approval.
ACKNOWLEDGMENTS
Longitudinal Cognitive studies were funded by Medical Research Council, Economic and Social Research Council, The Wellcome Trust (grant reference number 003889) and Unilever PLC. The work of Manchester Brain Bank is supported by Alzheimer’s Research UK and Alzheimer’s Society through the Brains for Dementia Research (BDR) Programme, which also kindly allowed AR to undertake a PhD studentship.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/19-0580r2).
REFERENCES
[1] | Matthews FE , Brayne C , Lowe J , McKeith I , Wharton SB , Ince P ((2009) ) Epidemiological pathology of dementia: Attributable-risks at death in the medical research council cognitive function and ageing study. PLoS Med. 6: , e1000180. |
[2] | Neuropathology Group of the Medical Research Council Cognitive Function and Aging Study ((2001) ) Pathological correlates of late-onset dementia in a multicentre, community-based population in England and Wales. Neuropathology Group of the Medical Research Council Cognitive Function and Ageing Study (MRC CFAS). Lancet 357: , 169–175. |
[3] | Snowden DA , Nun Study ((2003) ) Healthy aging and dementia: Findings from the nun study. Ann Intern Med 139: , 450–454. |
[4] | Bennett DA , Schneider JA , Arvanitakis Z , Wilson RS ((2012) ) Overview and findings from the religious orders study. Curr Alzheimer Res 9: , 628–645. |
[5] | National Institute on Ageing ((2013) ) Baltimore Longitudinal Study of Ageing. https://www.blsa.nih.gov. Accessed January 19, 2018. |
[6] | Cambridge City over 75 Cohort Study ((2005) ) Cambridge City over 75 Cohort Study, http://www.cc75c.group.cam.ac.uk. Accessed January 19, 2018. |
[7] | Gelber RP , Launer LJ , White LR ((2012) ) The Honolulu-Asia aging study: Epidemiologic and neuropathologic research on cognitive impairment. Curr Alzheimer Res 9: , 664–672. |
[8] | Oxford Project to Investigate Memory and Ageing ((2016) ) OPTIMA. http://www.ndcn.ox.ac.uk/research/centre-prevention-stroke-dementia/resources/optima-oxford-project-to-investigate-memory-and-ageing. Accessed January 19, 2018. |
[9] | University of Helsinki ((2006) ) The Vantaa 85+study. http://www.hi.helsinki.fi/english/research/groups/pathology/vantaa85study.html. Accessed January 19, 2018. |
[10] | Kukull WA , Higdon R , Bowen JD , McCormick WC , Teri L , Schellenberg GD , van Belle G , Jolley L , Larson EB ((2002) ) Dementia and Alzheimer disease incidence: A prospective cohort study. Arch Neurol 59: , 1737–1746. |
[11] | Rabbitt PMA , McInnes L , Diggle P , Holland F , Bent N , Abson V , Pendleton N , Horan M ((2004) ) The University of Manchester longitudinal study of cognition in normal healthy old age, 1983 through 2003. Aging Neuropsychol Cogn 11: , 245–279. |
[12] | Robinson AC , Davidson YS , Horan MA , Pendleton N , Mann DMA ((2018) ) Pathological correlates of cognitive impairment in the university of manchester longitudinal study of cognition in normal healthy old age. J Alzheimers Dis 64: , 483–496. |
[13] | UNESCO ((2006) ) International Standard Classification of Education guidelines. http://www.uis.unesco.org/Library/Documents/isced97-en.pdf. Accessed March 21, 2018. |
[14] | Plassman BL , Welsh KA , Helms M , Brandt J , Page WF , Breitner JC ((1995) ) Intelligence and education as predictors of cognitive state in late life: A 50 year follow-up. Neurology 45: , 1446–1450. |
[15] | Evans O , Singleton N , Meltzer D , Stewart R , Prince M ((2003) ) The mental health of older people: Report based on the analysis of the ONS survey of psychiatric morbidity among adults in Great Britain. Office of National Statistics (IBSN 0116216603). |
[16] | Thal DR , Rüb U , Orantes M , Braak H ((2002) ) Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 58: , 1791–1800. |
[17] | Braak H , Alafuzoff I , Arzberger T , Kretzschmar H , Del Tredici K ((2006) ) Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112: , 389–404. |
[18] | Braak H , Braak E ((1991) ) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82: , 239–259. |
[19] | Braak H , Tredici KD , Rüb U , de Vos RAI , Jansen Steur ENH , Braak E ((2003) ) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24: , 197–211. |
[20] | Montine TJ , Phelps CH , Beach TG , Bigio EH , Cairns NJ , Dickson DW , Duyckaerts C , Frosch MP , Masliah E , Mirra SS , Nelson PT , Schneider JA , Thal DR , Trojanowski JQ , Vinters HV , Hyman BT ; National Institute on Aging; Alzheimer’s Association ((2012) ) National Institute on Aging-Alzheimer’s association guidelines for the neuropathologic assessment of Alzheimer’s disease: A practical approach. Acta Neuropathol 123: , 1–11. |
[21] | McKeith IG , Galasko D , Kosaka K , Perry EK , Dickson DW , Hansen LA , Salmon DP , Lowe J , Mirra SS , Byrne EJ , Lennox G , Quinn NP , Edwardson JA , Ince PG , Bergeron C , Burns A , Miller BL , Lovestone S , Collerton D , Jansen EN , Ballard C , de Vos RA , Wilcock GK , Jellinger KA , Perry RH ((1996) ) Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): Report of the consortium on DLB international workshop. Neurology 47: , 1113–1124. |
[22] | McKeith IG , Dickson DW , Lowe J , Emre M , O’Brien JT , Feldman H , Cummings J , Duda JE , Lippa C , Perry EK , Aarsland D , Arai H , Ballard CG , Boeve B , Burn DJ , Costa D , Del Ser T , Dubois B , Galasko D , Gauthier S , Goetz CG , Gomez-Tortosa E , Halliday G , Hansen LA , Hardy J , Iwatsubo T , Kalaria RN , Kaufer D , Kenny RA , Korczyn A , Kosaka K , Lee VM , Lees A , Litvan I , Londos E , Lopez OL , Minoshima S , Mizuno Y , Molina JA , Mukaetova-Ladinska EB , Pasquier F , Perry RH , Schulz JB , Trojanowski JQ , Yamada M ; Consortium on DLB ((2005) ) Consortium on DLB. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 65: , 1863–1872. |
[23] | McKeith IG , Boeve BF , Dickson DW , Halliday G , Taylor JP , Weintraub D , Aarsland D , Galvin J , Attems J , Ballard CG , Bayston A , Beach TG , Blanc F , Bohnen N , Bonanni L , Bras J , Brundin P , Burn D , Chen-Plotkin A , Duda JE , El-Agnaf O , Feldman H , Ferman TJ , Ffytche D , Fujishiro H , Galasko D , Goldman JG , Gomperts SN , Graff-Radford NR , Honig LS , Iranzo A , Kantarci K , Kaufer D , Kukull W , Lee VMY , Leverenz JB , Lewis S , Lippa C , Lunde A , Masellis M , Masliah E , McLean P , Mollenhauer B , Montine TJ , Moreno E , Mori E , Murray M , O’Brien JT , Orimo S , Postuma RB , Ramaswamy S , Ross OA , Salmon DP , Singleton A , Taylor A , Thomas A , Tiraboschi P , Toledo JB , Trojanowski JQ , Tsuang D , Walker Z , Yamada M , Kosaka K ((2017) ) Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology 89: , 88–100. |
[24] | Mackenzie IR , Neumann M , Bigio EH , Cairns NJ , Alafuzoff I , Kril J , Kovacs GG , Ghetti B , Halliday G , Holm IE , Ince PG , Kamphorst W , Revesz T , Rozemuller AJ , Kumar-Singh S , Akiyama H , Baborie A , Spina S , Dickson DW , Trojanowski JQ , Mann DM ((2009) ) Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: Consensus recommendations. Acta Neuropathol 117: , 15–18. |
[25] | Mackenzie IR , Neumann M , Baborie A , Sampathu DM , du Plessis D , Jaros E , Perry RH , Trojanowski JQ , Mann DM , Lee VM ((2011) ) A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol 122: , 111–113. |
[26] | Dickson DW , Bergeron C , Chin SS , Duyckaerts C , Horoupian D , Ikeda K , Jellinger K , Lantos PL , Lippa CF , Mirra SS , Tabaton M , Vonsattel JP , Wakabayashi K , Litvan I ; Office of Rare Diseases of the National Institutes of Health ((2002) ) Office of rare diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol 61: , 935–946. |
[27] | Hauw JJ , Daniel SE , Dickson D , Horoupian DS , Jellinger K , Lantos PL , McKee A , Tabaton M , Litvan I ((1994) ) Preliminary NINDS neuropathologic criteria for Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy). Neurology 44: , 2015–2019. |
[28] | Trojanowski JQ , Revesz T , Neuropathology Working Group on MSA ((2007) ) Proposed neuropathological criteria for the post mortem diagnosis of multiple system atrophy. Neuropathol Appl Neurobiol 33: , 615–620. |
[29] | Braak H , Braak E ((1998) ) Argyrophilic grain disease: frequency of occurrence in different age categories and neuropathological diagnostic criteria. J Neural Transm 105: , 801–819. |
[30] | Crary JF , Trojanowski JQ , Schneider JA , Abisambra JF , Abner EL , Alafuzoff I , Arnold SE , Attems J , Beach TG , Bigio EH , Cairns NJ , Dickson DW , Gearing M , Grinberg LT , Hof PR , Hyman BT , Jellinger K , Jicha GA , Kovacs GG , Knopman DS , Kofler J , Kukull WA , Mackenzie IR , Masliah E , McKee A , Montine TJ , Murray ME , Neltner JH , Santa-Maria I , Seeley WW , Serrano-Pozo A , Shelanski ML , Stein T , Takao M , Thal DR , Toledo JB , Troncoso JC , Vonsattel JP , White CL 3rd , Wisniewski T , Woltjer RL , Yamada M , Nelson PT ((2014) ) Primary age-related tauopathy (PART): A common pathology associated with human aging. Acta Neuropathol 128: , 755–766. |
[31] | Kovacs GG , Ferrer I , Grinberg LT , Alafuzoff I , Attems J , Budka H , Cairns NJ , Crary JF , Duyckaerts C , Ghetti B , Halliday GM , Ironside JW , Love S , Mackenzie IR , Munoz DG , Murray ME , Nelson PT , Takahashi H , Trojanowski JQ , Ansorge O , Arzberger T , Baborie A , Beach TG , Bieniek KF , Bigio EH , Bodi I , Dugger BN , Feany M , Gelpi E , Gentleman SM , Giaccone G , Hatanpaa KJ , Heale R , Hof PR , Hofer M , Hortobágyi T , Jellinger K , Jicha GA , Ince P , Kofler J , Kövari E , Kril JJ , Mann DM , Matej R , McKee AC , McLean C , Milenkovic I , Montine TJ , Murayama S , Lee EB , Rahimi J , Rodriguez RD , Rozemüller A , Schneider JA , Schultz C , Seeley W , Seilhean D , Smith C , Tagliavini F , Takao M , Thal DR , Toledo JB , Tolnay M , Troncoso JC , Vinters HV , Weis S , Wharton SB , White CL 3rd , Wisniewski T , Woulfe JM , Yamada M , Dickson DW ((2016) ) Aging-related tau astrogliopathy (ARTAG): Harmonized evaluation strategy. Acta Neuropathol 131: , 87–102. |
[32] | Olichney JM , Hansen LA , Galasko D , Saitoh T , Hofstetter CR , Katzman R , Thal LJ ((1996) ) The apolipoprotein E epsilon 4 allele is associated with increased neuritic plaques and cerebral amyloid angiopathy in Alzheimer’s disease and Lewy body variant. Neurology 47: , 190–196. |
[33] | Skrobot OA , Attems J , Esiri M , Hortobágyi T , Ironside JW , Kalaria RN , King A , Lammie GA , Mann D , Neal J , Ben-Shlomo Y , Kehoe PG , Love S. ((2016) ) Vascular cognitive impairment neuropathology guidelines (VCING): The contribution of cerebrovascular pathology to cognitive impairment. Brain 139: , 2957–2969. |
[34] | Wenham PR , Price WH , Blandell G ((1991) ) Apolipoprotein E genotyping by one-stage PCR. Lancet 337: , 1158–1159. |
[35] | Selvackadunco S , Langford K , Shah Z , Hurley S , Bodi I , King A , Aarsland D , Troakes C , Al-Sarraj S ((2019) ) Comparison of clinical and neuropathological diagnoses of neurodegenerative diseases in two centres from the Brains for Dementia Research (BDR) cohort. J Neural Transm (Vienna) 126: , 327–337. |
[36] | Ganguli M , Rodríguez EG ((1999) ) Reporting of dementia on death certificates: A community study. J Am Geriatr Soc 47: , 842–849. |
[37] | Romero JP , Benito-León J , Mitchell AJ , Trincado R , Bermejo-Pareja F ((2014) ) Under reporting of dementia deaths on death certificates using data from a population-based study (NEDICES). J Alzheimers Dis 39: , 741–748. |
[38] | Davidson YS , Raby S , Foulds PG , Robinson A , Thompson JC , Sikkink S , Yusuf I , Amin H , du Plessis D , Troakes C , Al-Sarraj S , Sloan C , Esiri MM , Prasher VP , Allsop D , Neary D , Pickering-Brown SM , Snowden JS , Mann DM ((2011) ) TDP-43 pathological changes in early onset familial and sporadic Alzheimer’s disease, late onset Alzheimer’s disease and Down’s Syndrome: association with age, hippocampal sclerosis and clinical phenotype. Acta Neuropathol 122: , 703–713. |
[39] | Arai T , Mackenzie IR , Hasegawa M , Nonoka T , Niizato K , Tsuchiya K , Iritani S , Onaya M , Akiyama H ((2009) ) Phosphorylated TDP-43 in Alzheimer’s disease and dementia with Lewy bodies. Acta Neuropathol 117: , 125–136. |
[40] | Higashi S , Iseki E , Yamamoto R , Minegishi M , Hino H , Fujisawa K , Togo T , Katsuse O , Uchikado H , Furukawa Y , Kosaka K , Arai H ((2007) ) Concurrence of TDP-43, tau and alpha-synuclein pathology in brains of Alzheimer’s disease and dementia with Lewy bodies. Brain Res 1184: , 284–294. |
[41] | Yokota O , Davidson Y , Bigio EH , Ishizu H , Terada S , Arai T , Hasegawa M , Akiyama H , Sikkink S , Pickering-Brown S , Mann DM ((2010) ) Phosphorylated TDP-43 pathology and hippocampal sclerosis in progressive supranuclear palsy. Acta Neuropathol 120: , 55–66. |
[42] | Viswanathan A , Greenberg SM ((2011) ) Cerebral amyloid angiopathy in the elderly. Ann Neurol 70: , 871–880. |


