
Developing Effective Alzheimer’s Disease Therapies: Clinical Experience and Future Directions
Abstract
Alzheimer’s disease (AD) clinical trials, focused on disease modifying drugs and conducted in patients with mild to moderate AD, as well as prodromal (early) AD, have failed to reach efficacy endpoints in improving cognitive function in most cases to date or have been terminated due to adverse events. Drugs that have reached clinical stage were reviewed using web resources (such as clinicaltrials.gov, alzforum.org, company press releases, and peer reviewed literature) to identify late stage (Phase II and Phase III) efficacy clinical trials and summarize reasons for their failure. For each drug, only the latest clinical trials and ongoing trials that aimed at improving cognitive function were included in the analysis. Here we highlight the potential reasons that have hindered clinical success, including clinical trial design and choice of outcome measures, heterogeneity of patient populations, difficulties in diagnosing and staging the disease, drug design, mechanism of action, and toxicity related to the long-term use. We review and suggest approaches for AD clinical trial design aimed at improving our ability to identify novel therapies for this devastating disease.
BACKGROUND
Treatments to prevent or slow down cognitive decline in Alzheimer’s disease (AD) remain an unmet therapeutic need. The drugs that have been approved to date for treatment of mild-to-moderate AD are acetylcholine esterase inhibitors, including tacrine (first approved treatment, withdrawn soon after because of reports of liver toxicity), donepezil (also approved for severe AD), rivastigmine (also approved for severe AD), and galantamine [1, 2]. These drugs increase levels of available acetylcholine, associated with memory and learning, during synaptic transmission and thus compensate for the diminished function of cholinergic neurons. Though the established mechanism is symptomatic, Dubois et al. recently reported a reasonably powered multicenter trial of donepezil versus placebo in mild cognitive impairment (MCI) that demonstrated significant effect of the drug in preserving hippocampal and whole brain volume at one year versus placebo [3]. In addition, memantine, an uncompetitive (open-channel) N-methyl-D-aspartate (NMDA) antagonist with neuroprotective properties [4], is approved for treatment of moderate to severe AD.
Numerous clinical trials have explored agents that could potentially not only interfere with the physiology of the disease but also provide short-term symptomatic improvement. However, to date none of the AD modifying treatments has reached regulatory approval.
While the mechanism of the onset of AD is still not fully understood, it is recognized that a strong genetic risk component is usually involved [5–11]. In addition to subjects’ genetic make up, multiple other lifestyle factors including physical and mental exercise, heart disease, diabetes, lower education, and mental diseases are thought to accelerate AD progression [12, 13].
Two primary neuropathologies in the brains of AD patients have been recognized and studied extensively over the last two decades, including amyloid plaques formation comprising aggregated amyloid-β (Aβ) and neurofibrillary tangles (NFTs) formed by aggregation of hyperphosphorylated tau protein. The aggregation and extracellular deposition of Aβ oligomers, drives neuronal death and AD pathogenesis [14, 15]. While pathologic concentrations of Aβ (nanomolar and micromolar) are considered neurotoxic, soluble Aβ protein present in low (picomolar) concentrations in normal brains, is thought to have multiple functions including modulating synaptic activity, memory formation, neuronal survival, antioxidant activity, effects on Ca transport, and maintenance of blood-brain barrier (BBB) integrity [16]. The sequential cleavage of native trans-membrane amyloid-β protein precursor (AβPP), thought to have a role in neurodevelopment, synaptogenesis, cell adhesion, and memory formation [17–20], is known to be the critical step in formation of Aβ peptides. It is believed that accumulation of Aβ oligomers hampers synaptic transmission and causes irreversible AD progression through an imbalance in production and clearance in neuronal synapses [21].
Several approaches to AD treatment have been tested in clinical trials, including agonists and antagonists of neurotransmitter receptors, β-secretase (β-site AβPP cleaving enzyme - BACE) or γ-secretase inhibitors, therapies targeting Aβ clearance, prevention of Aβ aggregation, modulation of phosphorylation and clearance of tau protein, as well as anti-inflammation compounds and multiple immunotherapy agents directed against Aβ and tau (Fig. 1). Among those, the BACE inhibitors, thought to improve cognitive and functional performance by suppressing Aβ production, are the widely chosen targets.
Fig.1
Schematic Representation of Therapeutic Strategies. 1) Enhancement of neurotransmission; 2) Reduction of Aβ production and aggregation; 3) Enhancement of Aβ clearance; 4) Prevention of tau aggregation; 5) Anti-inflammatory agents; 6) Enhancement of microglial phagocytosis.
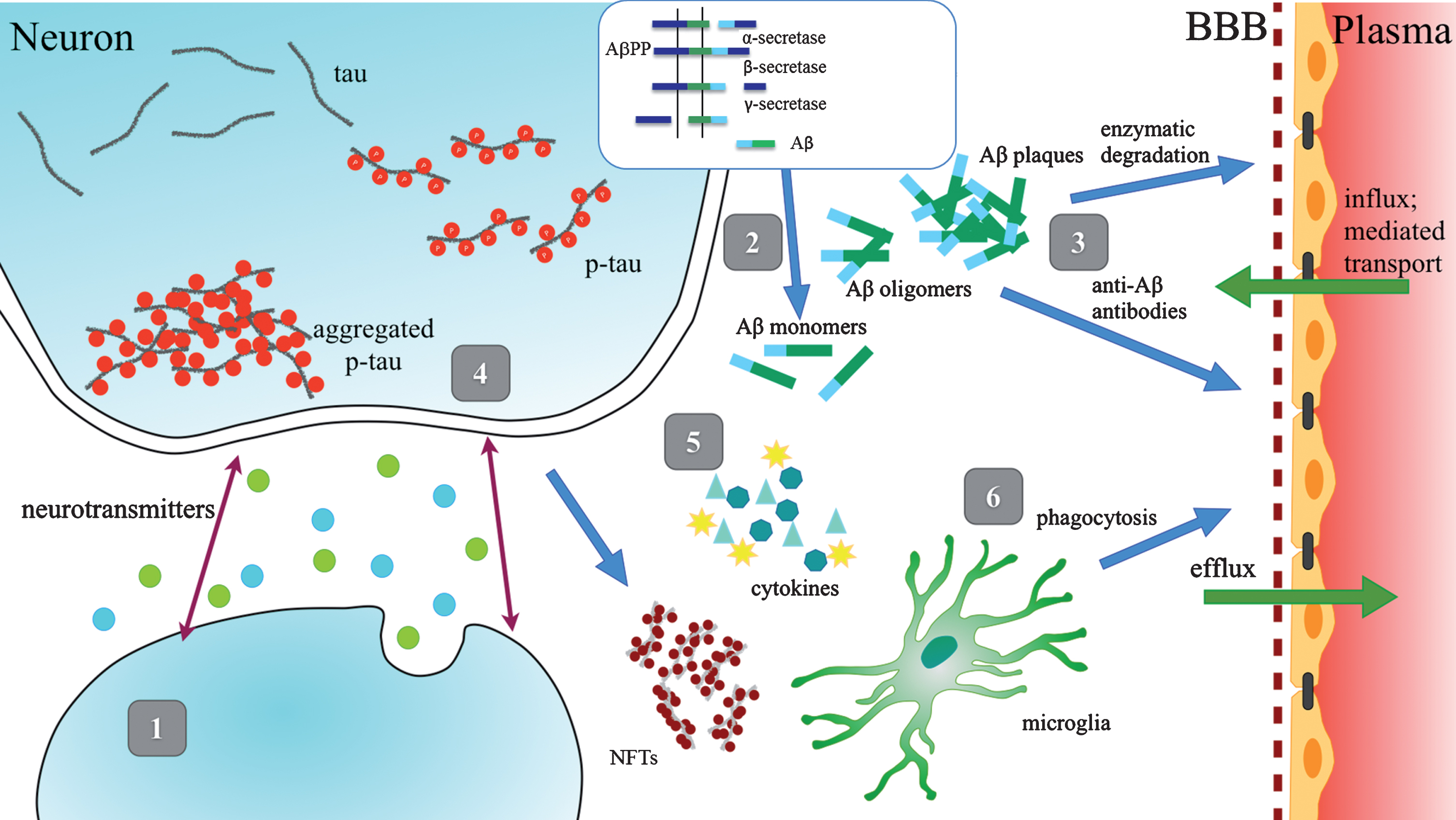
However, Aβ deposits in the brain start to accumulate years before cognitive symptoms appear [22]. While prevailing opinion supported by the stronger association of Aβ with genetic predisposition is that deposition of Aβ drives the pathology of the disease, NFTs are more closely associated with the cognitive decline [23, 24]. While transient and reversible hyperphosphorylation of tau has been reported to occur in normal brains during fetal development, anesthesia, and hypothermia, it is believed that in AD, the irreversible hyperphosphorylation of tau leads to neuronal loss and consequently to cognitive impairment [25].
Recent studies with a three-dimensional (3D) in vitro model of AD showed that APP and PSEN1 gene mutations induce extracellular deposition of Aβ, and plaque formation, as well as tau pathology [26]. Further, these studies suggest that phosphorylated tau (p-tau) accumulations are induced by Aβ accumulation. Other studies of Aβ and tau pathology in the course of AD in human samples showed that soluble Aβ oligomers were abundantly present in early stages of AD, while p-tau did not increase until late stages of the disease [27]. Tau imaging studies using 18F T807 PET in patients with MCI and AD dementia, reported high levels of tau in neocortex correlate with high Aβ burden [28]. It has been reported that NFTs may form independently of Aβ burden due to other neural death pathways [25, 29].
Therapeutic strategies postulate that preventing tau hyperphosphorylation and aggregation can decrease formation of NFTs. Research has identified several potential therapeutic approaches: modulation of tau phosphorylation, prevention of tau aggregation, and promotion of tau clearance by intracellular and extracellular proteolysis and phagocytosis, as well as anti-tau directed immunotherapies [30, 31]. Only a few drugs that target tau phosphorylation and aggregation have reached late stage clinical trials. In part, this may be due to differences in structure, conformation, and complexity of changes during AD of tau protein compared to Aβ (Table 1). While Aβ consists of 36–42 amino acids, the human central nervous system expresses six tau isoforms that comprise from 352 to 441 amino acids with four sequence repeats in normal as compared to three sequence repeats in AD [32, 33]. Further, changes in Aβ and tau during the progression of AD are very different. Extracellular Aβ modifications during AD progression involve slow polymerization into oligomers that further aggregate. Initial tau modifications in AD progression are intracellular. Therefore, targeting tau protein as the therapeutic approach poses more complex challenges than targeting Aβ.
Table 1
Comparison of properties of Tau and Aβ proteins
| Tau | Aβ | |
| Structure | ||
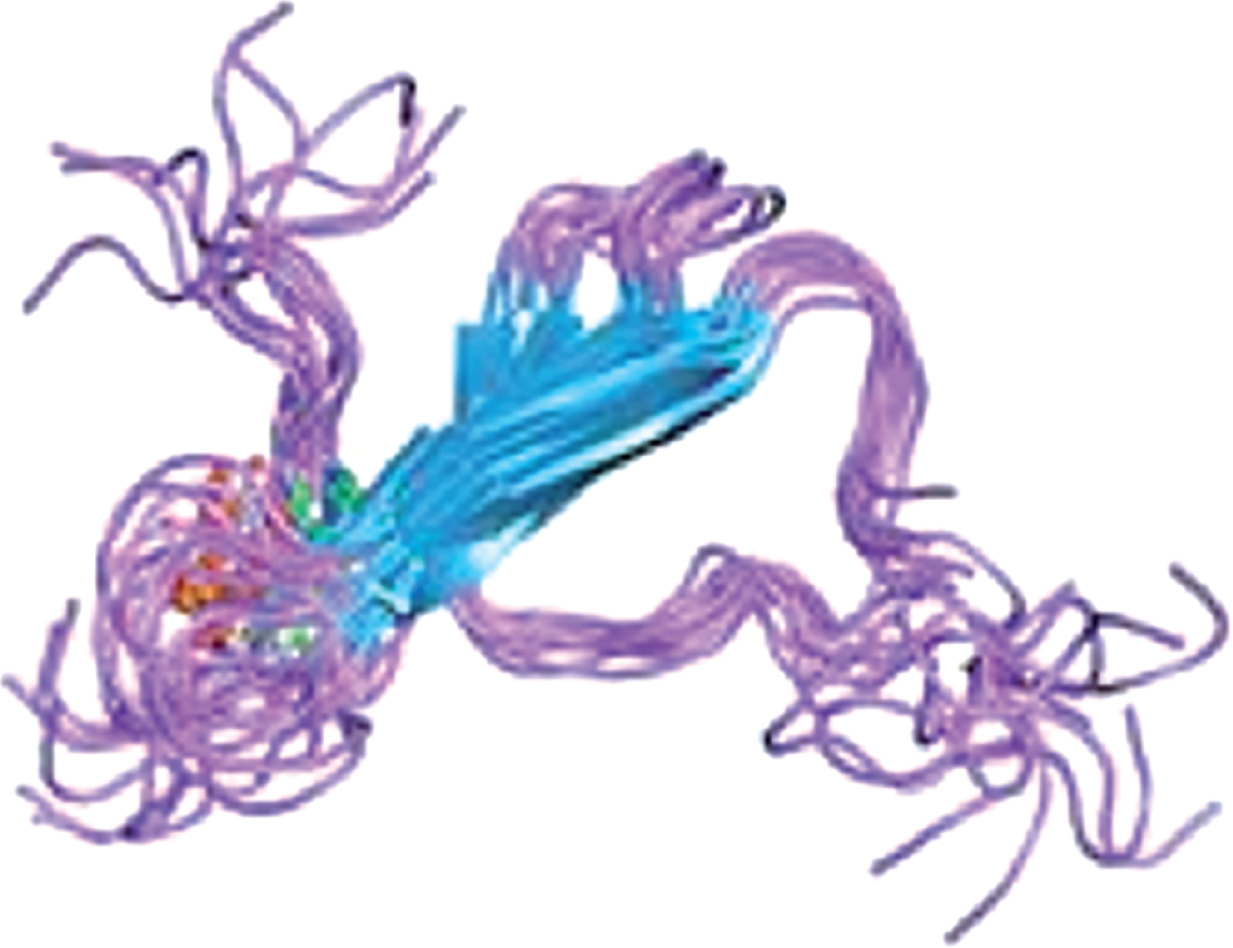 | 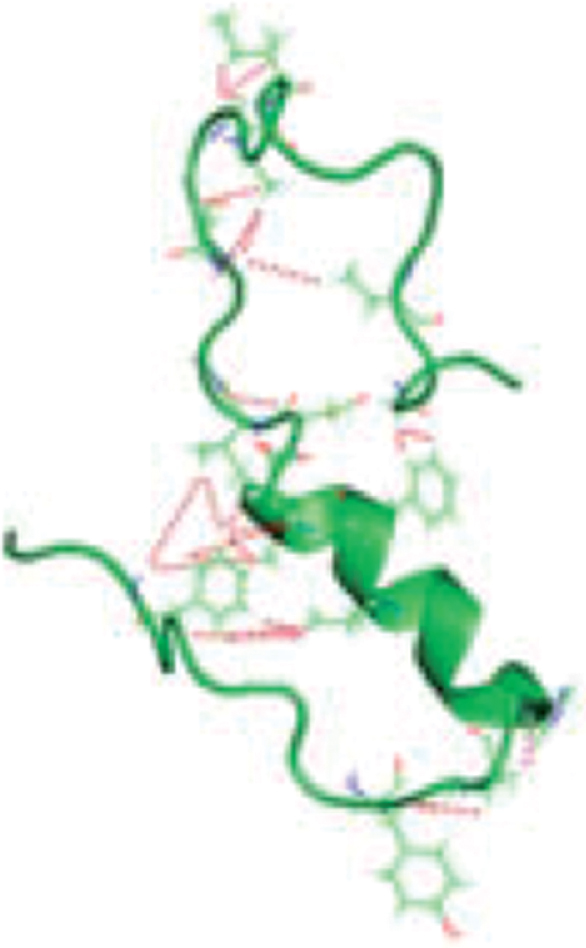 | |
| Role | Tabular stabilizer | Signal transduction /other |
| Size (MW) | 55,000–62,000 | Aβ42 = 45 |
| # amino acids | 352–441 | 36–43 |
| Normal structures | Six isoforms, 4 sequence repeats | Soluble/single |
| Slow polymerization | ||
| Normal washout | ||
| Structures in AD | 3 sequence repeats | Oligomers and tangles |
| Phosphorylation & tangles | ||
| AD tangles | Intra neuron | Synapse/interneuron |
Inflammation triggered by innate immunity has also been reported to play an important role during both the early and late stages of the disease and contribution of glia to AD pathology and Aβ metabolism has been widely studied [34–38]. Microglia are implicated in propagation of hyperphosphorylated tau between neurons via exosomes [39]. In addition to pro-inflammatory response, microglial activation to phagocytic states is believed to have neuroprotective properties [37, 38]. Katsel et al. showed using genetic and protein expression studies that development and progression of dementia depend on the age at onset and are different in demented younger and old aging populations; these features highlight the importance of the immune system in preventing cognitive decline [40].
Therefore, it is hypothesized that neuroinflammation plays a major role in AD progression and that activation and modulation of the innate immune system may lead to new approaches to treatment and prevention of cognitive decline in progression of AD.
While a number of anti-inflammatory drugs have been tested in therapeutic controlled clinical trials, none have been shown to slow the progression of cognitive symptoms in patients with mild to moderate AD [41–50]. For example, early epidemiological studies of NSAIDs, such as ibuprofen, reported lower rates of AD among individuals who had been taking these drugs for chronic treatment of inflammatory conditions [51, 52]. However, controlled clinical trials of ibuprofen at a dose of 400 mg/day showed no cognitive improvement and had known side effects [53].
Clinical trials that targeted Aβ plaque clearance in mild to moderate AD were potentially doomed to fail because at the onset of cognitive symptoms the brain has already been compromised with massive neuronal death. There is a general agreement that Aβ plaque removal cannot compensate for neural dysfunction and death. For mild to moderate AD, stabilizing AD progression by slowing down or inhibiting its pathology is the only viable treatment option. Novel approaches to treat AD at the prodromal stage, before significant neural damage has occurred, attempting to slow down and prevent disease progression are being explored.
Other pathogenic mechanisms have been reported to be associated with the progression of AD [54, 55], and these potential treatments have been studied in animal models and clinical trials. These approaches include antioxidants, drugs that target oxidative stress damage and mitochondrial dysfunction, iron deregulation, and abnormal cholesterol metabolism.
Given the complexity of AD progression and associated immune response, new approaches targeting multiple AD pathologies are being studied. Further, new understanding of the multiple roles of microglia and factors that affect their function in the progression of AD [10, 11, 35, 56, 57], has potential to open vastly new therapeutic options and targets.
SYMPTOMATIC AND DISEASE MODIFYING APPROACHES: CLINICAL EXPERIENCE
The drugs currently used to treat cognitive decline (i.e., cholinesterase inhibitors, NMDA receptor antagonist), and drugs that treat behavioral and psychological symptoms (such as antidepressants and antipsychotic drugs, despite the controversies surrounding their use) have limited therapeutic value. However, despite the large number of approaches tested in clinical trials, cholinesterase inhibitor and NMDA receptor antagonist remain the only approved treatments for AD. Several reviews already provide comprehensive summaries of ongoing clinical and pre-clinical efforts to treatment of AD [54, 55, 58, 59]. Here, we focus on reviewing late stage clinical trials that did not reach efficacy endpoints and ongoing clinical trials, highlighting possible reasons for failures based on the information that has emerged from the body of clinical research.
As we noted earlier all approaches to treat AD targeted one mechanism of action associated with disease pathology and, further, the majority of research of validation of new targets evolved around Aβ. Expanding treatment options to combination therapy addressing several mechanisms is an important direction with potential to impact treatment as has been shown to be successful for other complex diseases (e.g., HIV and cancer treatment). This multi-faceted approach is even more reasonable given the complex aging brain, where immune surveillance and increased vulnerability to inflammatory response can accelerate disease progression. Recently, Eli Lilly started a Phase II study of LY3202626, a small molecule BACE inhibitor, administered together with monoclonal antibody LY3002813 targeting Aβ(p3-42), a pyroglutamate form of Aβ localized to aggregated Aβ in amyloid plaques. However, the combination arm in this trial was halted due to concerns about impact of the BACE inhibition on general cognition.
Other largely unsuccessful attempts to improve cognitive decline by enhancing neurotransmission are shown in Table 2 [60–71]. Despite encouraging preclinical and Phase II clinical results, the number of agents thought to improve cognitive decline by enhancing cholinergic neurotransmission, using serotonin 6 (5-HT6) receptor antagonists, Intepirdine [60], Idalopirdine [61], and PF-05212377 [62], failed to reach efficacy endpoints. Similarly, previous attempts to enhance acetylcholine response using H3 receptor antagonists, ABT-288, GSK239512, and S 38093, did not show sufficient cognitive improvement [64–66]. Other clinical trials discontinued due to the lack of efficacy include Xaliproden (5HT1-A receptor antagonist) [63], Atomoxetine (norepinephrine uptake inhibitor approved for treatment of ADHD) [68], Dimebon (anti-histamine) [69], S47445 (agonist of AMPA receptors for glutamate) [70], and Sembragiline [71]. Encenicline (α7nAChR agonist) Phase III trial was discontinued after adverse events were noted [67].
Table 2
Late stage clinical trials: Neurotransmitters
| Drug | Treatment/Target | Clinical Trial Information | Results |
| Intepirdine | Antagonist of the serotonin 6 (5-HT6) receptor | Phase III (mild to moderate AD) | Lack of efficacy [60] |
| Idalopirdine | Antagonist of the serotonin 6 (5-HT6) receptor | Phase II (mild to moderate AD) | Lack of efficacy [61] |
| PF-05212377 | Antagonist of the serotonin 6 (5-HT6) receptor | Phase II (mild to moderate AD) | Lack of efficacy [62] |
| Xaliproden | Serotonin 1A (5HT1-A) receptor antagonist | Phase III (mild to moderate AD) | Lack of efficacy [63] |
| ABT-288 | Histamine H3 receptor antagonist | Phase II (mild to moderate AD) | Lack of efficacy [64] |
| GSK239512 | Histamine H3 receptor antagonist | Phase II (mild to moderate AD) | Lack of efficacy [65] |
| S 38093 | Histamine H3 receptor antagonist | Phase II (mild to moderate AD) | Lack of efficacy [66] |
| Encenicline | α7 nicotinic acetylcholine receptor (α7nAChR) agonist | Phase III (mild to moderate AD) | Gastrointestinal side effects [67] |
| Atomoxetine | Norepinephrine uptake inhibitor approved for Attention Deficit/Hyperactivity Disorder | Phase II/III (mild to moderate AD) | Lack of efficacy [68] |
| BI 425809 | Glycine transporter inhibitor; Modulate NMDA receptor function | Phase II (early AD) | Ongoing |
| Latrepirdine (Dimebon) | Anti-histamine | Phase III (mild to moderate AD) | Lack of efficacy [69] |
| S47445 | AMPARs positive allosteric modulator | Phase II (mild to moderate AD) | Lack of efficacy [70] |
| Sembragiline | Monoamine oxidase B (MAOB) inhibitor | Phase II (moderate to severe AD) | Lack of efficacy [71] |
The majority of drugs in development are focused on Aβ hypothesis, attempting to improve cognitive function through modulation of Aβ and tau levels. (Table 3) [72–87]. The γ-secretase inhibitors, i.e., Avagacestat and Semagacestat, as well as some BACE inhibitors (i.e., Atabecestat) induced serious side effects [73, 77, 79]. Development of other BACE inhibitors, such as Lanabecestat and Verubecestat, has been suspended due to lack of efficacy [74, 76].
Table 3
Late stage clinical trials: Disease modifying treatments targeting Aβ and tau proteins
| Drug | Treatment/Target | Clinical Trial Information | Results |
| Etazolate | α-secretase | Phase II (mild to moderate AD) | No significant differences between treatment groups during 3 months of treatment [72] |
| Atabecestat | BACE inhibitor | Phase II/III (asymptomatic people at risk of developing Alzheimer’s dementia) | Liver toxicity [73] |
| CNP520 | BACE inhibitor | Phase II/III (cognitively normal, homozygous ApoE4 carriers) | Ongoing |
| Elenbecestat | BACE inhibitor | Two Phase III trials (biomarker-confirmed MCI due to AD/prodromal AD) | Ongoing |
| Lanabecestat | BACE inhibitor | Phase III (early AD; mild dementia) | Lack of efficacy [74] |
| LY3202626 | BACE inhibitor | Phase II (mild AD) | Lack of efficacy [75] |
| Phase II (early AD) in combination with LY3002813 | Terminated (results not published) | ||
| Verubecestat | BACE inhibitor | Phase III (mild to moderate AD) | Lack of efficacy in both mild to moderate AD and in prodromal patients; Dermatological and behavioral side effects [76] |
| Phase III (prodromal AD) | |||
| Scyllo-inositol (ELND005) | Aβ aggregation | Phase II (mild to moderate AD) | Serious adverse events at higher doses [55] |
| Avagacestat | γ-secretase inhibitor | Phase II (mild to moderate AD) | Serious side effects [77] |
| r-flurbiprofen (Flurizan) | γ-secretase inhibitor | Phase III (mild to moderate AD) | Lack of efficacy; Poor bioavailability [78] |
| Semagacestat | γ-secretase inhibitor | Phase III (mild to moderate) | Increased risk of skin cancer and infections; Lack of efficacy [79] |
| Aducanumab | Aggregated Aβ | Two Phase III (MCI due to AD or mild AD ascertained by a positive amyloid PET scan) | Terminated due to lack of efficacy [80]. |
| BAN2401 | Aβ clearance | Phase III (MCI due to AD) | Did achieve primary endpoint at 18 months, but question regarding APOE genotype difference between placebo and drug groups [81] |
| Bapineuzumab | Aβ clearance | Phase III (mild to moderate AD) | Lack of efficacy; ARIA vasogenic edema in APOE4 carriers [82] |
| Crenezumab | Aggregated Aβ | Phase III (prodromal to mild AD) | Terminated due to lack of efficacy [83] |
| Phase II (prevention trial in presymptomatic PSEN1 E280A mutation carriers) | Ongoing | ||
| IVIg (Gammagard) | Aβ clearance | Phase III (mild to moderate AD) | Lack of efficacy [46] |
| Gantenerumab | Aβ clearance | Phase II/III (prodromal AD) | Lack of efficacy; Dose and APOE ɛ4 -dependent ARIA [84] |
| Phase II/III (asymptomatic and very mildly symptomatic carriers of PSEN1, and PSEN2) | Higher dose Phase III trial in mild AD and prevention trial are ongoing | ||
| Phase III (mild AD) | |||
| LY3002813 | Aggregated Aβ | Phase II (early AD; mild dementia) | Ongoing |
| Solanezumab | Soluble Aβ | Three Phase III (mild AD) | Lack of efficacy [85] |
| Phase III (prodromal AD) | Lack of efficacy [86] | ||
| Two Phase II/III prevention trials (asymptomatic or very mildly symptomatic people and asymptomatic and very mildly symptomatic carriers of PSEN1, and PSEN2) | A4 trial completed (results not published) | ||
| CAD106 | Aβ | Phase III (asymptomatic ApoE4 carriers) | Ongoing |
| ACI-24 | Aβ | Phase I/II | Ongoing |
| BIIB092 | Tau | Phase II (mild cognitive impairment due to AD or mild AD) | Ongoing |
| C2N 8E12 | Tau | Phase II (early AD) | Ongoing |
| AADvac-1 | Tau | Phase II (mild to moderate AD) | Ongoing |
| LY3303560 | Tau | Phase II (early AD) | Ongoing |
| Methylene Blue (LMTM) | Tau | Phase III (mild to moderate AD) | Lack of efficacy [87] |
| RO7105705 | Tau | Phase II (prodromal to mild AD) | Ongoing |
Several passive immunotherapy drugs are undergoing clinical testing with mixed results. Bapineuzumab development was terminated because of the lack of efficacy and risk of adverse effects including microhemorrhages in the brain [82]. Intravenous immunoglobulin (IVIg) showed significant reductions in plasma Aβ42 levels compared with placebo but no benefits for improving cognition in patients with mild-to-moderate AD dementia, according to the results of a Phase III trial [46].
Tau-targeting approaches to date have been sparse (Table 3). LMTM (Methylene Blue, Tau aggregation inhibitor) was tested in a Phase III study in patients with mild to moderate AD, and failed to show clinical improvement [87]. Similarly, late stage clinical trials, aiming to treat inflammation related to AD including a number of NSAIDs have also had poor efficacy results (Table 4).
Table 4
Late stage clinical trials: Disease modifying treatments targeting inflammation
| Drug | Description/Target | Clinical Trial Information | Results |
| Cromolyn sodium and ibuprofen (ALZT-OP1) | Aβ aggregation and inflammation | Phase III (early AD) | Ongoing |
| Azeliragon | RAGE inhibitor (Aβ and inflammation) | Phase III (mild to moderate AD) | Lack of efficacy; Tolerance issues at higher doses in a Phase II trial [41] |
| CHF 5074 | γ-secretase | Two Phase II studies (amnestic or non-amnestic MCI) | Discontinued |
| Celecoxib (Celebrex) | NSAID | Prevention trial (70 years and older with family history of dementia) | Risk of cardiovascular side effects; Lack of efficacy [42] |
| Etanercept (Enbrel) | TNF-α inhibitor | Phase II (mild to moderate) | Known side effects (gastroenteritis, respiratory and urinary-tract infections, pharyngitis, and cellulitis; Lack of efficacy [43] |
| r-flurbiprofen (Flurizan) | NSAID | Phase II/III (probable AD) | Lack of efficacy [44] |
| Sargramostim | Hematopoietic growth factor | Phase II (mild to moderate AD) | Lack of efficacy [45] |
| GRF6019 | Young adult plasma | Phase II (mild and moderate AD) | Ongoing |
| IVIg (Gammagard) | Human plasma antibodies | Phase III (mild to moderate AD) | Lack of efficacy [46] |
| Ibuprofen | NSAID | Phase II (mild to moderate AD) | Known side effects at high chronic dose. Lack of efficacy [53] |
| Lornoxicam | NSAID | Phase II (mild to moderate AD) | Terminated; data not available. |
| Minocycline | Inflammation and tau | Phase II (MCI and AD) | Data not available |
| Naproxen | NSAID | Prevention trial (people 70 with family history of dementia) | Risk of cardiovascular side effects; Lack of efficacy [48] |
| Phase III (mild to moderate AD) | |||
| Neflamapimod | Inflammation | Phase II (MCI and mild AD) | Ongoing |
| IVIg (Octagam®10%) | Inflammation | Phase II (amnestic MCI due to AD) | Lack of efficacy; Transient cognitive improvement reported [49] |
| Pioglitazone | Inflammation | Phase III (cognitively normal participants with genetic predisposition) | Ongoing |
| Prednisone | Corticosteroid | Phase II (patients with AD) | Lack of efficacy; Behavioral symptoms and an increase in blood glucose levels reported [47] |
| Rofecoxib | NSAID | Phase II (mild to moderate AD) | Lack of efficacy [48] |
| Withdrawn from market due to increased risk of heart attack and stroke associated with long-term, high-dosage use | |||
| Thalidomide (Thalomid) | Aβ and inflammation | Phase II/III (mild to moderate AD) | High therapeutic dose known to cause serious birth defects. Poor safety and tolerability in AD patients [50] |
The ongoing trials generally target earlier stages of the disease and use in part available biomarkers. BI 425809, a glycine transporter inhibitor thought to modulate NMDA receptor function, is currently in Phase II clinical trial in early AD patients. The next generation of small molecule BACE inhibitors (CNP520, Elenbecestat) are still in ongoing clinical trials in early AD patients and asymptomatic patients. Elenbecestat is being evaluated in a Phase III study in early AD patients with confirmed brain amyloid using positron emission tomography (PET) and/or cerebrospinal fluid (CSF) assessment. CNP520 is being tested in subjects who are otherwise healthy but at increased risk of developing AD based on their age, genotype, and amyloid levels.
Several passive immunotherapies advanced to Phase III and prevention trials after mixed Phase II efficacy results in mild to moderate AD. Crenezumab Phase III trials in patients with MCI or prodromal AD with PET and CSF evidence of Aβ pathology using higher dose, were terminated due to lack of efficacy [83]. Solanezumab missed on the primary endpoint in a Phase III trials in patients with mild AD and prodromal AD [85, 86]. It is also tested in the A4 prevention trial in asymptomatic or mildly symptomatic patients at risk of developing AD-related cognitive impairment, with amyloid plaque buildup as evidenced by florbetapir PET scan, with the goal of slowing down cognitive and memory decline and AD progression. The trial uses Preclinical Alzheimer Cognitive Composite (PACC) as a primary outcome measure as well as number of cognitive and functional scales, imaging and CSF biomarkers as secondary outcome measures. The results of the A4 prevention trial have not been reported to date. Gantenerumab is also being investigated in a Phase II/III trial aimed at preventing dementia in subjects with an inherited autosomal-dominant mutation in APP, PSEN1, or P SEN2. Aducanumab, a human IgG1 monoclonal antibody targeting aggregated Aβ, failed Phase III trials in patients with MCI and mild AD confirmed by a positive amyloid PET scan [80].
Recently, positive results of the Phase II testing of BAN2401 have been reported at 18 months despite the fact that the results of interim analysis at 12 months missed the primary endpoint [81]. The Phase II trial results reported statistically significant slowing of the course of AD symptoms, as measured by a combination of cognitive assessments and dementia ratings (ADCOMS). However, the changes in the Clinical Dementia Rating Sum of Boxes (CDR-SB) were not statistically significant. Results were further compounded by exclusion of APOE4 patients form the highest dose group party way through the trial. The concern of the differences between the treatment and control groups remains. In addition, while the safety profile was reported to be acceptable (incidence of amyloid-related edema lower than 10% at any dose, and under 15% in patients with APOE4 taking the highest dose), the concern of effects of chronic administration remain and the reported results warrant periodical repeated monitoring of patients making disease management even more complex.
Active immunotherapy trials include CAD106, vaccine that induces immunity to Aβ without eliciting an inflammatory response, currently in a Phase II/III prevention trial in homozygous APOE4 subjects who are cognitively normal. Tau targeting approaches through passive (BIIB092, C2N 8E12, LY3303560, RO7105705) and active (AADvac-1, ACI-24) immunotherapy are currently in Phase II clinical trials.
Few anti-inflammatory agents remain in clinical testing: Pioglitazone, an insulin sensitizer that is approved for treatment of Type 2 diabetes mellitus, GRF6019 (young adult plasma), and Neflamapimod, which is a small molecule believed to shift microglial pro-inflammatory state to a phagocytic state.
AZTherapies reported research results of beneficial effects of a combination treatment, currently in Phase III clinical testing, that studies modified cromolyn sodium, an asthma therapeutic agent, in combination with newly formulated low dose ibuprofen. It was shown that the combination treatment may simultaneously affect Aβ aggregation and inflammation associated with AD in animal models of the disease [88–90].
DISCUSSION
A large number of AD clinical trials have failed despite significant advances in scientific understanding of the disease. The fact that over 400 trials testing over 200 therapeutics have been performed to date with a 99.6% failure rate (i.e., a success rate of only 0.4%) clearly illustrates the importance of the issue [2, 91]. In Tables 2–4, we highlight the late stage clinical trials, some of which are still ongoing, using various disease modifying and symptomatic approaches. An overview of recent clinical trial reports indicates that potential flaws include overall clinical trial design with utilized statistical measures that are perhaps not aligned with the studies’ objectives, heterogeneity of patient populations due to deficient inclusion criteria, and difficulties in diagnosing early disease for both treatment and placebo groups. Other flaws include single target mechanism, drug bioavailability, genetic making, and toxicity from chronic administration, which alone or in combination may have contributed to recent failures. These potential shortcomings are explored in detail below.
Study design
Most studies to date were performed in mild and moderate AD patients, with neuronal damage already present to a considerable extent. At this stage, the disease progression path and rate are uncertain and show individual variability in part due to variability in dementia rating and patient populations [92, 93]. In mild and moderate AD patient populations, it becomes apparent that the use of amyloid and tau modulation agents to inhibit generation and/or increase clearance may not compensate for the neuronal damage that is already present, thus challenging the clinical trial’s odds of success. Some published results show no or very slow cognitive decline in their placebo group, strongly suggesting a failure in clinical trial design [2]. Other trials have suffered from lack of sensitivity of standard measures of cognitive performance (i.e., CDR-SB) at early and prodromal stages of the disease [93, 94].
In addition, the widely used CDR-SB scale, while clinically meaningful, was not designed to be used in traditional statistical analysis of efficacy of treatment. Because the evaluation is subjective, additional variability may be introduced in multicenter trials, further hampering success. When used as a measure of disease progression in the evaluation of the primary endpoints, unequal size of the change between stabilization and/or improvement in the treatment group (a small change of a few points in this population) as compared to disease progression in the placebo group (a larger change). Since the typical annual change in CDR-SB reported for early AD patients is between 1-2 points [95, 96], the changes are small and require large number of subjects to reach statistical significance, even in cases when the drug is clinically effective in stabilizing cognitive decline.
As mentioned previously, to overcome the issues with measuring cognitive change early in AD progression, Phase II a clinical trial of BAN2401 used the combination measures, ADCOMS, which at 18 months showed statistically significant changes compared to placebo. However, the changes in CDR-SB were not statistically significant, which could be, in part, attributed to the selected patient population and disease staging in the trial. In prevention trials, the time to diagnosis of dementia has been used as an endpoint and it is likely to show similar large variability. Further, multiple trials have attempted to conduct subgroup analysis after failures to report a clinical benefit. These attempts have been criticized to provide inaccurate information as subgroups are often not properly randomized and do not have significant sample size [91].
Patient population
The majority of clinical trials conducted to date included patients aged from 50 to 90 years old, leading to large variability in the cognitive impairment and disease progression. Thus, heterogeneous mild to moderate AD patient populations require a large number of subjects to accommodate for age, gender, disease stage, genetic predisposition, dementia type (such as Lewy body, senile and vascular, alcohol abuse, injury, or a result of other diseases) and inevitably lead to obscuring observed treatment effects and to failure to show clinical efficacy. The need to identify homogeneous patient populations at risk of developing AD brain pathology requires the use of imaging, CSF, and blood for disease staging, in addition to cognition and functional measures.
Bioavailability and toxicity from chronic administration
It has been noted previously that many drugs in development suffer from inadequate brain bioavailability and appropriate dose selection to show clinical benefit due to toxicity from chronic administration [91]. This is true for small molecules that may be transported from the central nervous system, even after crossing the BBB to target enzyme inhibition or a biochemical pathway, and it is particularly important for large molecules, such as monoclonal antibodies and other biologic drugs. Data on clinical trials that failed due to toxicity issues are shown in Table 4 (drugs targeting inflammation) and Table 5 (drugs targeting Aβ [73, 77, 79, 97–102]) and illustrate the problem in development of disease modifying treatments. While the clinical development of most of these drugs was halted before reaching Phase III, some drugs (such as Bapineuzumab, Azeliragon, Verubecestat) advanced into Phase III with doses that were not sufficient to show benefit in cognitive function. The Atabecestat Phase II/III trial was recently discontinued due to issues with liver toxicity. Similarly, a number of anti-inflammatory agents (Table 4) failed due to increased risk of adverse events with chronic administration. The need to administer the treatment frequently for several years, with high risk of systemic toxicity, requires that an adequate dose is selected early in the drug development phase [91].
Table 5
Discontinued clinical trails targeting Aβ: Adverse events and toxicity
| Drug | Description/Target | Results |
| AAB-003 | Aβ | ARIA and microhemorrhages [97] |
| AN-1792 | Aβ | Brain inflammation [98] |
| Atabecestat | BACE inhibitor | Elevated liver enzymes [73] |
| Avagacestat | γ-secretase inhibitor | Gastrointestinal and dermatological side effects; Nonmelanoma skin cancers reported; ARIA [77] |
| Azeliragon | RAGE inhibitor | High dose was associated with confusion, falls, and greater ADAS-cog decline; Low dose was not effective in subsequent trials [99] |
| LY2886721 | BACE inhibitor | Abnormal liver biochemistry values [100] |
| Semagacestat | γ-secretase inhibitor | Increased risk of skin cancer and infections [79] |
| Bexarotene | Retinoid (Aβ) | Increased blood lipid levels, risk of cardiovascular side effects [101] |
| Bapineuzumab | Aβ clearance | Reversible vasogenic edema APOE ɛ4 carriers [102] |
Intervention is too late for disease modifying agents
Consistent with the observation that Aβ deposits in the brain start to accumulate years before cognitive symptoms appear [22], the clinical results from trials in mild to moderate AD patients, which attempt to modulate Aβ production and clearance, indicate that removing plaques will not reverse the neuronal damage or stop the AD. This limits the treatment targeting Aβ production and clearance to be effective only at early stages of the disease. Limiting Aβ production and aggregation may have limited benefit in mild to moderate AD. Similarly, stage-dependent efficacy of anti-inflammatory agents, suggests that these treatments may be useful early in AD [90, 103]. It is hypothesized, that once Aβ-induced inflammation initiates tauopathy, neurodegeneration progresses and leads to cognitive decline.
Biomarkers
The need to focus interventions on a homogeneous patient population at an appropriate disease stage, as well as the need to diagnose the disease before cognitive symptoms occur, has led to a renewed focus on identifying and validating biomarkers, including structural MRI signs of hippocampal and global brain atrophy, CSF, and blood analysis biomarkers as well as the usage of PET imaging agents. The ability to diagnose the disease at earlier stages while preserving normal brain function would significantly contribute to the potential to develop disease-modifying therapies. So far, no biomarker proposed for early diagnosis and monitoring treatments has been validated.
Currently, the most widely used CSF biomarkers, obtained through lumbar puncture, include those targeting amyloid-β (Aβ42 or Aβ42/Aβ40), total tau protein (T-tau) and phosphorylated tau (P-tau181) [104]. It has been shown that CSF Aβ42 is an indicator of early stages of the disease, while CSF tau indicates the extent of cognitive decline at later stages [23, 24]. CSF and blood levels of neurofilament light chain protein have been shown to correlate well with neurodegeneration.
The use of CSF biomarkers in research and clinical trials, in addition to clinical criteria is detailed in the revised diagnostic criteria of AD [24, 105], suggesting the use of CSF biomarkers in differentiating between AD and other types of dementia. The utility of neuro-inflammatory biomarkers collected from blood, may have limited use when patients are suffering from additional systemic inflammation. Burchell and Panegyres reviewed several other biomarkers and assays including those involving BBB integrity, mitochondrial DNA, vascular endothelial growth factor, as well as immunological factors involved in AD pathogenesis [106]. However, these are still in early development and have not been validated.
With the renewed interest in targeting multiple disease hallmarks, inclusion of other exploratory biomarkers in clinical trials may be a critical component of advancing the understanding of the role of neuroinflammation in AD and developing additional tools for early diagnosis and follow up of disease progression. The correlation of multiple indicators with disease progression, that may help to design improved patient selection related to brain pathophysiology in the future.
Several imaging techniques, including structural magnetic resonance imaging (MRI) and 18F-fluorodeoxyglucose (FDG) are commonly used in clinical research to assist in early diagnosis. There has been significant development in the PET imaging tracers binding to Aβ with the approval of Amyvid (18F-florbetapir), Vizamyl (18F-flutametamol), and Neuraceq (18F-florbetaben).
Similarly, recent results suggest that buildup of tau pathology could better predict future cognitive impairment than Aβ, and subsequently driving research toward new tau imaging agents. For example, increased flortaucipir binding has been shown to correlate with increased cognitive impairment in patients with Aβ plaque [107]. Other tau imaging tracers, including 18F- PI-2620 are currently in clinical testing.
While early clinical trials rarely used imaging and CSF biomarkers, recent clinical work includes more frequent use of PET imaging and CSF biomarkers, either as a secondary outcome measure and/or as inclusion criteria. However, in most of these studies, biomarkers are used in sub-studies with a much smaller number of subjects (i.e., clinical trials for Verubecestat, Lanabecestat, Bapineuzumab, Gantenerumab, and Semagacestat).
Genetic testing is particularly useful in identifying individuals at risk of developing AD and has gained momentum with the number of prevention trials. Some of the genetic testing targets genes with rare variations that cause inherited AD or those that are associated with increased risk of developing AD, such as APOE ɛ4.
New biomarkers and imaging agents as well as potentially combinations of biomarkers, that target specific AD pathology in asymptomatic patients at risk of developing AD, could potentially assist in identifying homogeneous patient populations and provide additional tools for monitoring efficacy of clinical trials.
Combination treatments
Experience to date points to the fact that AD is multi-target disease, and that approaches using one drug, focused on one target may not be sufficient to achieve improvement of clinical symptoms [55].
Azeliragon, a small-molecule RAGE inhibitor, has been thought to provide a combined treatment by lowering Aβ plaque deposition and inducing anti-inflammatory response, but it failed to reach efficacy endpoints in Phase III [41]. Other treatments proposed to mediate amyloid beta clearance and provide anti-inflammatory properties either failed later stage testing due to poor efficacy (e.g., GM-CSF Leukine, Intravenous immunoglobulin) or are still in ongoing clinical trials (e.g., Pioglitazone, GC 021109, GRF6019).
Other approaches targeting both Aβ accumulation and inflammation associated with AD may be needed to slow disease progression before symptoms occur.
CONCLUSIONS AND RECOMMENDATIONS
The number of clinical trials focused on early, mild, and moderate AD that did not achieve primary endpoints and failed to achieve cognitive improvement keeps growing. We believe that the scientific rationale behind all potential AD therapies tested is valid and supported by in vitro and in vivo animal model data and that this is not the main reason for these clinical trial failures. Trial design and analytical outcome measures are becoming especially important when attempting to treat a chronically progressive disease as AD. Brain neurons and synapses expand the brain network in early years and with aging, network remodeling shifts toward deterioration. This life-long process is associated with genetic predisposition, education, lifestyle, and the environment. It is unequivocally important that new potential therapies address the unique irreversible nature of the brain aging process. Unlike treatments of other diseases such as cancer, where short term aggressive treatments are used in doses that induce systemic toxicity, destroy normal cells, and/or impact the immune system, treatment of AD requires approaches that preserve the balance of Aβ and tau proteins that is essential for neuronal function and further stabilize the brain’s innate immune system.
To address multiple parameters affecting complex AD clinical trial design, we highlight some recommendations and potential new approaches.
Experience to date shows that clinical trial design and outcome measures have to be adjusted to allow monitoring of disease progression in the pre-symptomatic stage of AD with sufficient minimum clinically important differences. To eliminate other factors that were associated with failure, clinical trials should include in the study design adequate outcome measures and endpoint selections (see Early Alzheimer’s Disease: Developing Drugs for Treatment-Guidance for Industry). The challenge remains to devise methods to reliably identify patients with preclinical AD and to ensure that the methods have predictive value. As the duration, complexity, and cost of clinical trials increase, the clinical research would benefit from introducing appropriate interim analyses and adaptive clinical trial designs.
The need to eliminate heterogeneity of patient population, and especially to identify homogeneous patient populations at risk of developing AD will require the use of imaging, CSF, and blood biomarkers for disease staging, in addition to cognitive and functional measures. It will be necessary to minimize variations due to age (i.e., limit the age spread of the study population to patients between 55 to 70, to avoid younger patients that are far from AD onset and older patients who show faster AD progression), define proper cutoffs for cognitive performance (done in most cases with varying rationale, mainly based on published data), and limit the use of concomitant AD medication that affects brain function (such as strong antipsychotic drugs) at entry. Limiting the use of approved medications (i.e., for a period of more than six months or a year) during the trial, unless the test drug is an adjuvant therapy, should also contribute to smaller variation in cognitive performance during the trial. In addition, it will be necessary to reflect underlying AD pathology when selecting patient population using validated CSF and/or blood biomarkers as well as proper imaging techniques with adequate ranges for the targeted disease stage.
Because the treatment is expected to last many years, systemic toxicity is an expected outcome with aggressive therapies at high doses. To address this concern, new preclinical methods need to be explored to adequately evaluate potential in vitro or in vivo toxicity, not only for small molecules but also for biologic treatments, where effect of chronic treatment are typically not easily estimated. In slow progressing AD, the risk of chronic toxicity when using high doses of drugs does not provide benefit rationale as in treatment of other diseases (i.e., cancer). The lowest effective dose needs to be determined and validated based on brain pathophysiology, to provide treatment at Aβ and tau levels typically present in the brains of patients with early AD. Prolonged brain titration with the lowest effective doses, sufficient to treat small daily Aβ and tau changes (reported at pico and nano molar levels) should provide measurable changes that can affect species production and clearance as well as treat the associated neuroinflammation. These changes are not easily predictable and are hard to achieve even with low doses of potentially effective but highly toxic drugs.
AD is a neurodegenerative aging-related disease with no known cure, and neuronal damage cannot be fully reversed even if the mechanisms underlying disease progression are targeted. As a strategy, the goal of AD treatment should be slowing down neural degeneration. Therefore, early treatment options, before the symptoms of AD occur, appear to be the most viable current approach. However, early diagnosis of AD, before cognitive symptoms occur, is still a challenge, because of the lack of appropriate biomarkers and diagnostic criteria for this pre-symptomatic stage of AD. As the focus of current research moves toward prevention trials, the aim of intervention becomes delaying the symptoms and slowing down progression of the disease, especially in subjects who are potentially vulnerable to early disease onset, due to genetic profile, environmental conditions associated with lifestyle, and other contributing diseases.
It is clear that new approaches to treatment of AD will require the identification and validation of new targets and will need to target multiple mechanisms of action that may slow disease progression, when used before clinical symptoms appear (Fig. 1). In addition, performance of drugs that showed limited efficacy and failed to achieve statistical significance when used as a single treatment, may be improved by exploring their combination with anti-inflammatory mechanisms of action. As an example, Solanezumab, an Aβ removal antibody with some degree of success in clinical studies, could be combined with low dose anti-inflammatory agent, a mast cell stabilizer as part of innate immune system, or with an inhibitor of amyloid peptide oligomerization and polymerization.
Identifying a suitable pool of asymptomatic patients and following the rate of progression of the disease is likely to require the use of multiple biomarkers in addition to effective cognition and function measures. Thus, the development and validation of new CSF and blood biomarkers, as well as highly specific PET agents, becomes critical for future clinical research. To ensure sufficient brain bioavailability, brain or CSF drug uptake should be evaluated in the CSF and blood by labeled molecules or their close analogs using biodistribution and pharmacokinetics studies in experimental animals or by brain imaging in humans. This approach should provide proof of bioavailability and has potential to increase success of clinical studies and to reduce the cost and waste of resources associated with unsuccessful clinical trials.
Proposed selection of homogeneous patient population, with narrow age range and with early stages of the disease, as well as the use of CSF and blood biomarkers in addition to stricter cognition and functional measures, would likely increase the number of screening failures resulting in higher recruitment cost and prolong duration of the trials. To address these issues, we recommend the use of adaptive and interim analyses, per FDA guidelines, and additional measures of clinical utility of the treatment. Cognitive and functional performance scales (such as CDR-SB), are fairly acceptable scales recommended by the FDA (see Early Alzheimer’s Disease: Developing Drugs for Treatment-Guidance for Industry). While useful in diagnosis and staging of the disease, they have limited value as a measure of disease progression. The CDR-SB scale (as well as other cognition and function tests) was not designed to be used in traditional statistical analysis of efficacy of treatment. The evaluation of the primary endpoints using CDR-SB mean change from baseline in early AD may potentially be challenged by the unequal size of the change between stabilization and improvement (a small change of a few points in this population) as compared to disease progression (a larger change). Due to these potential challenges, we believe that an exploratory responder analysis may alleviate the issues and provide additional information on the efficacy of clinical trials.
Adding CDR-SB analysis at the end of a study for determining response to drug treatment (stable or improvement; responder or non-responder) may be an additional useful tool as an efficacy measure. Adaptive and interim analyses, per FDA guidelines, as well as, exploring responder analysis in the interim and final analysis, should be considered. Additionally, validated biomarkers may provide more accurate correlation with the progression of the disease, especially when used in combination with cognitive and functional outcomes.
Advances in understanding the role of the brain’s innate immune system in the development of AD, and specifically its genetic regulation, have potential to open new therapeutic solutions. Experience to date points out that AD is a multi-target disease, and that approaches using one drug, focused on one target may not be sufficient to achieve improvement of clinical symptoms. Combination treatments with multiple targets may potentially lead to effective therapies.
ACKNOWLEDGMENTS
We acknowledge AZTherapies for their support.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/19-0507r2).
REFERENCES
[1] | Farlow M ((2002) ) A clinical overview of cholinesterase inhibitors in Alzheimer’s disease. Int Psychogeriatr 14: (Suppl 1), 93–126. |
[2] | Cummings JL , Morstorf T , Zhong K ((2014) ) Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimers Res Ther 6: , 37–43. |
[3] | Dubois B , Chupin M , Hampel H , Lista S , Cavedo E , Croisile B , Louis Tisserand G , Touchon J , Bonafe A , Ousset PJ , Ait Ameur A , Rouaud O , Ricolfi F , Vighetto A , Pasquier F , Delmaire C , Ceccaldi M , Girard N , Dufouil C , Lehericy S , Tonelli I , Duveau F , Colliot O , Garnero L , Sarazin M , Dormont D , Hippocampus Study Group ((2015) ) Donepezil decreases annual rate of hippocampal atrophy in suspected prodromal Alzheimer’s disease. Alzheimers Dement 11: , 1041–1049. |
[4] | Molinuevo JL , Lladó A , Rami L ((2005) ) Memantine: Targeting glutamate excitotoxicity in Alzheimer’s disease and other dementias. Am J Alzheimers Dis Other Demen 20: , 77–85. |
[5] | Tanzi RE , Bertram L ((2005) ) Twenty years of the Alzheimer’s disease amyloid hypothesis: A genetic perspective. Cell 120: , 545–555. |
[6] | Bertram L , Lill CM , Tanzi RE ((2010) ) The genetics of Alzheimer disease: Back to the future. Neuron 68: , 270–281. |
[7] | Tanzi RE ((2012) ) The genetics of Alzheimer disease. Cold Spring Harb Perspect Med 2: , a006296. |
[8] | Tanzi RE ((2013) ) A brief history of Alzheimer’s disease gene discovery. J Alzheimers Dis 33: (Suppl 1), S5–S13. |
[9] | Bertram L , Lange C , Mullin K , Parkinson M , Hsiao M , Hogan MF , Schjeide BM , Hooli B , Divito J , Ionita I , Jiang H , Laird N , Moscarillo T , Ohlsen KL , Elliott K , Wang X , Hu-Lince D , Ryder M , Murphy A , Wagner SL , Blacker D , Becker KD , Tanzi RE ((2008) ) Genome-wide association analysis reveals putative Alzheimer’s disease susceptibility loci in addition to APOE. Am J Hum Genet 83: , 623–632. |
[10] | Guerreiro R , Wojtas A , Bras J , Carrasquillo M , Rogaeva E , Majounie E , Cruchaga C , Sassi C , Kauwe JS , Younkin S , Hazrati L , Collinge J , Pocock J , Lashley T , Williams J , Lambert JC , Amouyel P , Goate A , Rademakers R , Morgan K , Powell J , St George-Hyslop P , Singleton A , Hardy J ; Alzheimer Genetic Analysis Group ((2013) ) TREM2 variants in Alzheimer’s disease. N Engl J Med 368: , 117–127. |
[11] | Jonsson T , Stefansson H , Steinberg S , Jonsdottir I , Jonsson PV , Snaedal J , Bjornsson S , Huttenlocher J , Levey AI , Lah JJ , Rujescu D , Hampel H , Giegling I , Andreassen OA , Engedal K , Ulstein I , Djurovic S , Ibrahim-Verbaas C , Hofman A , Ikram MA , van Duijn CM , Thorsteinsdottir U , Kong A , Stefansson K ((2013) ) Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med 368: , 107–116. |
[12] | Akter K , Lanza EA , Martin SA , Myronyuk N , Rua M , Raffa RB ((2011) ) Diabetes mellitus and Alzheimer’s disease: Shared pathology and treatment? Br J Clin Pharmacol 71: , 365–376. |
[13] | Sando SB , Melquist S , Cannon A , Hutton M , Sletvold O , Saltvedt I , White LR , Lydersen S , Aasly J ((2008) ) Risk-reducing effect of education in Alzheimer’s disease. Int J Geriatr Psychiatry 23: , 1156–1162. |
[14] | Selkoe DJ ((2001) ) Alzheimer’s disease results from the cerebral accumulation and cytotoxicity of amyloid β -protein. J Alzheimers Dis 3: , 75–80. |
[15] | Townsend M , Shankar GM , Mehta T , Walsh DM , Selkoe DJ ((2006) ) Effects of secreted oligomers of amyloid β -protein on hippocampal synaptic plasticity: A potent role for trimers. J Physio 572: , 477–492. |
[16] | Cardenas-Aguayo M del C , Silva-Lucero M del C , Cortes-Ortiz M , Jimenez-Ramos B , Gomez-Virgilio L , Ramirez-Rodriguez G , Vera-Arroyo E , Fiorentino-Perez R , García U , Luna-Munoz J , Meraz-Rios MA ((2014) ) Physiological role of amyloid beta in neural cells: The cellular trophic activity. In Neurochemistry, Heinbockel T, ed. IntechOpen, London. |
[17] | Luo Y , Sunderland T , Roth GS , Wolozin B ((1996) ) Physiological levels of beta-amyloid peptide promote PC12 cell proliferation. Neurosci Lett 217: , 125–128. |
[18] | Moya KL , Benowitz LI , Schneider GE , Allinquant B ((1994) ) The amyloid precursor protein is developmentally regulated and correlated with synaptogenesis. Dev Biol 1612: , 597–603. |
[19] | Mileusnic R , Lancashire CL , Johnston AN , Rose SP ((2000) ) APP is required during an early phase of memory formation. Eur J Neurosci 12: , 4487–4495. |
[20] | Selkoe DJ ((2001) ) Alzheimer’s disease: Genes, proteins, and therapy. Physiol Rev 81: , 741–766. |
[21] | Hardy J , Selkoe DJ ((2002) ) The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 297: , 353–356. |
[22] | Jansen WJ , Ossenkoppele R , Knol DL , Tijms BM , Scheltens P , Verhey FR , Visser PJ , Amyloid Biomarker Study Group ((2015) ) Prevalence of cerebral amyloid pathology in persons without dementia: A meta-analysis. JAMA 313: , 1924–1938. |
[23] | Jack CR Jr , Holtzman DM ((2013) ) Biomarker modeling of Alzheimer’s disease. Neuron 80: , 1347–1358. |
[24] | Albert MS , DeKosky ST , Dickson D , Dubois B , Feldman HH , Fox NC , Gamst A , Holtzman DM , Jagust WJ , Petersen RC , Snyder PJ , Carrillo MC , Thies B , Phelps CH ((2011) ) The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7: , 270–279. |
[25] | Iqbal K , Liu F , Gong CX , Alonso AC , Iqbal IG ((2009) ) Mechanisms of tau-induced neurodegeneration. Acta Neuropathol 118: , 53–69. |
[26] | Choi SH , Kim YH , Hebisch M , Sliwinski C , Lee S , D’Avanzo C , Chen H , Hooli B , Asselin C , Muffat J , Klee JB , Zhang C , Wainger BJ , Peitz M , Kovacs DM , Woolf CJ , Wagner SL , Tanzi RE , Kim DY ((2014) ) A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 515: , 274–278. |
[27] | Bilousova T , Miller CA , Poon WW , Vinters HV , Corrada M , Kawas C , Hayden EY , Teplow DB , Glabe C , Albay R 3rd , Cole GM , Teng E , Gylys KH ((2016) ) Synaptic amyloid-beta oligomers precede p-tau and differentiate high pathology control cases. Am J Pathol 186: , 185–198. |
[28] | Johnson KA , Schultz A , Betensky RA , Becker JA , Sepulcre J , Rentz D , Mormino E , Chhatwal J , Amariglio R , Papp K , Marshall G , Albers M , Mauro S , Pepin L , Alverio J , Judge K , Philiossaint M , Shoup T , Yokell D , Dickerson B , Gomez-Isla T , Hyman B , Vasdev N , Sperling R ((2016) ) Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann Neurol 79: , 110–119. |
[29] | Braak H , Braak E ((1997) ) Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging 18: , 351–357. |
[30] | Schneider A , Mandelkow E ((2008) ) Tau-based treatment strategies in neurodegenerative diseases. Neurotherapeutics 5: , 443–457. |
[31] | Šimić G , Babić Leko M , Wray S , Harrington C , Delalle I , Jovanov-Milošević N , Bažadona D , Buée L , de Silva R , Di Giovanni G , Wischik C , Hof PR ((2016) ) Tau protein hyperphosphorylation and aggregation in Alzheimer’s disease and other tauopathies, and possible neuroprotective strategies. Biomolecules 6: , 6–33. |
[32] | Ow SY , Dunstan DE ((2014) ) A brief overview of amyloids and Alzheimer’s disease. Protein Sci 23: , 1315–1331. |
[33] | Lee G , Leugers CJ ((2012) ) Tau and tauopathies. Prog Mol Biol Transl Sci 107: , 263–293. |
[34] | Hong S , Beja-Glasser VF , Nfonoyim BM , Frouin A , Li S , Ramakrishnan S , Merry KM , Shi Q , Rosenthal A , Barres BA , Lemere CA , Selkoe DJ , Stevens B ((2016) ) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352: , 712–716. |
[35] | Griciuc A , Serrano-Pozo A , Parrado AR , Lesinski AN , Asselin CN , Mullin K , Hooli B , Choi SH , Hyman BT , Tanzi RE ((2013) ) Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 78: , 631–643. |
[36] | Park J , Wetzel I , Marriott I , Dréau D , D’Avanzo C , Kim DY , Tanzi RE , Cho H ((2018) ) A 3D human triculture system modeling neurodegeneration and neuroinflammation in Alzheimer’s disease. Nat Neurosci 21: , 941–951. |
[37] | Wilcock DM ((2012) ) A changing perspective on the role of neuroinflammation in Alzheimer’s disease. Int J Alzheimers Dis 2012: , 495243. |
[38] | McGeer PL , McGeer EG ((2015) ) Targeting microglia for the treatment of Alzheimer’s disease. Expert Opin Ther Targets 19: , 497–506. |
[39] | Asai H , Ikezu S , Tsunoda S , Medalla M , Luebke J , Haydar T , Wolozin B , Butovsky O , Kügler S , Ikezu T ((2015) ) Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci 18: , 1584–1593. |
[40] | Katsel P , Tan W , Haroutunian V ((2009) ) Gain in brain immunity in the oldest-old differentiates cognitively normal from demented individuals. PLoS One 4: , e7642. |
[41] | Burstein AH , Sabbagh M , Andrews R , Valcarce C , Dunn I , Altstiel L ((2018) ) Development of azeliragon, an oral small molecule antagonist of the receptor for advanced glycation endproducts, for the potential slowing of loss of cognition in mild Alzheimer’s disease. J Prev Alzheimers Dis 5: , 149–154. |
[42] | Soininen H , West C , Robbins J , Niculescu L ((2007) ) Long-term efficacy and safety of celecoxib in Alzheimer’s disease. Dement Geriatr Cogn Disord 23: , 8–21. |
[43] | Butchart J , Brook L , Hopkins V , Teeling J , Püntener U , Culliford D , Sharples R , Sharif S , McFarlane B , Raybould R , Thomas R , Passmore P , Perry VH , Holmes C ((2015) ) Etanercept in Alzheimer disease: A randomized, placebo-controlled, double-blind, Phase 2 trial. Neurology 84: , 2161–2168. |
[44] | Wan HI , Jacobsen JS , Rutkowski JL , Feuerstein GZ ((2009) ) Translational medicine lessons from flurizan’s failure in Alzheimer’s disease (AD) trial: Implication for future drug discovery and development for AD. Clin Transl Sci 2: , 242–247. |
[45] | Potter H , Woodcock JH , Boyd T , Sillau SH , Bettcher BM , Daniels J , Heffernan KS , Gray H ((2017) ) Interim report of Phase 2 pilot safety and efficacy trial of GM-CSF/Leukine in mild-to-moderate Alzheimer’s disease. Alzheimers Dement 13: , P1572. |
[46] | Relkin NR , Thomas RG , Rissman RA , Brewer JB , Rafii MS , van Dyck CH , Jack CR , Sano M , Knopman DS , Raman R , Szabo P , Gelmont DM , Fritsch S , Aisen PS ; Alzheimer’s Disease Cooperative Study ((2017) ) A Phase 3 trial of IV immunoglobulin for Alzheimer disease. Neurology 88: , 1–8. |
[47] | Aisen PS , Davis KL , Berg JD , Schafer K , Campbell K , Thomas RG , Weiner MF , Farlow MR , Sano M , Grundman M , Thal LJ ((2000) ) A randomized controlled trial of prednisone in Alzheimer’s disease. Alzheimer’s Disease Cooperative Study. Neurology 54: , 588–593. |
[48] | Aisen PS , Schafer KA , Grundman M , Pfeiffer E , Sano M , Davis KL , Farlow MR , Jin S , Thomas RG , Thal LJ ; Alzheimer’s Disease Cooperative Study ((2003) ) Effects of rofecoxib or naproxen vs placebo on Alzheimer’s disease progression: A randomized controlled trial. JAMA 289: , 2819–2826. |
[49] | Kile S , Au W , Parise C , Rose K , Donnel T , Hankins A , Chan M , Ghassemi A ((2017) ) IVIG treatment of mild cognitive impairment due to Alzheimer’s disease: A randomised double-blinded exploratory study of the effect on brain atrophy, cognition and conversion to dementia. J Neurol Neurosurg Psychiatry 88: , 106–112. |
[50] | Decourt B , Drumm-Gurnee D , Wilson J , Jacobson S , Belden C , Sirrel S , Ahmadi M , Shill H , Powell J , Walker A , Gonzales A , Macias M , Sabbagh MN ((2017) ) Poor safety and tolerability hamper reaching a potentially therapeutic dose in the use of thalidomide for Alzheimer’s disease: Results from a double-blind, placebo-controlled trial. Curr Alzheimer Res 14: , 403–411. |
[51] | McGeer PL , Schulzer M , McGeer EG ((1996) ) Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer’s disease: A review of 17 epidemiologic studies. Neurology 47: , 425–432. |
[52] | in ‘t Veld BA , Launer LJ , Hoes AW , Ott A , Hofman A , Breteler MM , Stricker BH ((1998) ) NSAIDs and incident Alzheimer’s disease. The Rotterdam Study. Neurobiol Aging 19: , 607–611. |
[53] | Pasqualetti P , Bonomini C , Dal Forno G , Paulon L , Sinforiani E , Marra C , Zanetti O , Rossini PM ((2009) ) A randomized controlled study on effects of ibuprofen on cognitive progression of Alzheimer’s disease. Aging Clin Exp Res 21: , 102–110. |
[54] | Galimberti D , Scarpini E ((2011) ) Disease-modifying treatments for Alzheimer’s disease. Ther Adv Neurol Disord 4: , 203–216. |
[55] | Mangialasche F , Solomon A , Winblad B , Mecocci P , Kivipelto M ((2010) ) Alzheimer’s disease: Clinical trials and drug development. Lancet Neurol 9: , 702–716. |
[56] | Hansen DV , Hanson JE , Sheng M ((2018) ) Microglia in Alzheimer’s disease. J Cell Biol 217: , 459–472. |
[57] | Huang KL , Marcora E , Pimenova AA , Di Narzo AF , Kapoor M , Jin SC , Harari O , Bertelsen S , Fairfax BP , Czajkowski J , Chouraki V , Grenier-Boley B , Bellenguez C , Deming Y , McKenzie A , Raj T , Renton AE , Budde J , Smith A , Fitzpatrick A , Bis JC , DeStefano A , Adams HH , Ikram MA , van der Lee S , Del-Aguila JL , Fernandez MV , Ibañez L , International Genomics of Alzheimer’s Project, Alzheimer’s Disease Neuroimaging Initiative, Sims R , Escott-Price V , Mayeux R , Haines JL , Farrer LA , Pericak-Vance MA , Lambert JC , van Duijn C , Launer L , Seshadri S , Williams J , Amouyel P , Schellenberg GD , Zhang B , Borecki I , Kauwe JS , Cruchaga C , Hao K , Goate AM ((2017) ) A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer’s disease. Nat Neurosci 20: , 1052–1061. |
[58] | Hung SY and Fu WM ((2017) ) Drug candidates in clinical trials for Alzheimer’s disease. J Biomed Sci 24: , 47–58. |
[59] | Vassar R ((2014) ) BACE1 inhibitor drugs in clinical trials for Alzheimer’s disease. Alzheimers Res Ther 6: , 89–102. |
[60] | Axovant, Axovant Announces Negative Topline Results of Intepirdine Phase 3 MINDSET Trial in Alzheimer’s Disease, http://investors.axovant.com/node/7286/pdf, Posted September 26, 2017, Accessed February 13, 2019. |
[61] | Atri A , Frölich L , Ballard C , Tariot PN , Molinuevo JL , Boneva N , Windfeld K , Raket LL , Cummings JL ((2018) ) Effect of idalopirdine as adjunct to cholinesterase inhibitors on change in cognition in patients with Alzheimer disease: Three randomized clinical trials. JAMA 319: , 130–142. |
[62] | Fullerton T , Binneman B , David W , Delnomdedieu M , Kupiec J , Lockwood P , Mancuso J , Miceli J , Bell J ((2018) ) A Phase 2 clinical trial of PF-05212377 (SAM-760) in subjects with mild to moderate Alzheimer’s disease with existing neuropsychiatric symptoms on a stable daily dose of donepezil. Alzheimers Res Ther 10: , 38–47. |
[63] | Sanofi, Clinical Trial Report: SR57746 (xaliproden), https://www.sanofi.com/-/media/Project/One-Sanofi-Web/Websites/Global/Sanofi-COM/Home/common/docs/clinical-study-results/efc2946-summary.pdf, Posted April 4, 2016, Accessed February 13, 2019. |
[64] | Haig GM , Pritchett Y , Meier A , Othman AA , Hall C , Gault LM , Lenz RA ((2014) ) A randomized study of H3 antagonist ABT-288 in mild-to-moderate Alzheimer’s dementia. J Alzheimers Dis 42: , 959–971. |
[65] | Grove RA , Harrington CM , Mahler A , Beresford I , Maruff P , Lowy MT , Nicholls AP , Boardley RL , Berges AC , Nathan PJ , Horrigan JP ((2014) ) A randomized, double-blind, placebo-controlled, 16-week study of the H3 receptor antagonist, GSK239512 as a monotherapy in subjects with mild-to-moderate Alzheimer’s disease. Curr Alzheimer Res 11: , 47–58. |
[66] | Servier, Clinical Study Report: S 38093, http://clinicaltrials.servier.com/wp-content/uploads/CL2-38093-009_synopsis_report.pdf, Posted March 9, 2012, Accessed February 13, 2019. |
[67] | BusinessWire, FORUM Pharmaceuticals Inc. Updates Encenicline Phase 3 Clinical Trial Programs in Alzheimer’s Disease and Cognitive Impairment in Schizophrenia, https://www.businesswire.com/news/home/20150914005855/en/FORUM-Pharmaceuticals-Updates-Encenicline-Phase-3-Clinical, Posted September 14, 2015, Accessed February 13, 2019. |
[68] | Mohs RC , Shiovitz TM , Tariot PN , Porsteinsson AP , Baker KD , Feldman PD ((2009) ) Atomoxetine augmentation of cholinesterase inhibitor therapy in patients with Alzheimer disease: 6-month, randomized, double-blind, placebo-controlled, parallel-trial study. Am J Geriatr Psychiatry 17: , 752–759. |
[69] | Cano-Cuenca N , Solís-García Del Pozo JE , Jordán J ((2014) ) Evidence for the efficacy of latrepirdine (dimebon) treatment for improvement of cognitive function: A meta-analysis. J Alzheimers Dis 38: , 155–164. |
[70] | Servier, Clinical Study Report: S 47445, https://clinicaltrials.servier.com/wp-content/uploads/CL2-47445-011-synopsis-report.pdf, Posted June 8, 2018, Accessed February 13, 2019. |
[71] | Nave S , Doody RS , Boada M , Grimmer T , Savola JM , Delmar P , Pauly-Evers M , Nikolcheva T , Czech C , Borroni E , Ricci B , Dukart J , Mannino M , Carey T , Moran E , Gilaberte I , Muelhardt NM , Gerlach I , Santarelli L , Ostrowitzki S , Fontoura P ((2017) ) Sembragiline in moderate Alzheimer’s disease: Results of a randomized, double-blind, placebo-controlled phase II trial (MAyflOwer RoAD). J Alzheimers Dis 58: , 1217–1228. |
[72] | Vellas B , Sol O , Snyder PJ , Ousset PJ , Haddad R , Maurin M , Lemarié JC , Désiré L , Pando MP ; EHT0202/002 study group ((2011) ) EHT0202 in Alzheimer’s disease: A 3-month, randomized, placebo-controlled, double-blind study. Curr Alzheimer Res 8: , 203–212. |
[73] | Janssen, Update on Janssen’s BACE Inhibitor Program, https://www.janssen.com/update-janssens-bace-inhibitor-program, Posted May 17, 2018, Accessed February13, 2019. |
[74] | Lilly Press Release, Update on https://investor.lilly.com/node/39051/pdf 3 Clinical Trials of Lanabecestat for Alzheimer’s Disease. https://investor.lilly.com/node/39051/pdf, Posted June 12, 2018, Accessed February13, 2019. |
[75] | Evaluate, Eli Lilly backs away from Bace but not from novel Alzheimer’s targets, http://www.evaluate.com/vantage/articles/analysis/spotlight/eli-lilly-backs-away-bace-not-novel-alzheimers-targets, Posted November 14, 2018, Accessed March 29, 2019. |
[76] | Merck Press Release, Merck Announces EPOCH Study of Verubecestat for the Treatment of People with Mild to Moderate Alzheimer’s Disease to Stop for Lack of Efficacy, https://investors.merck.com/news/press-release-details/2017/Merck-Announces-EPOCH-Study-of-Verubecestat-for-the-Treatment-of-People-with-Mild-to-Moderate-Alzheimers-Disease-to-Stop-for-Lack-of-Efficacy/, Posted February 14, 2017, Accessed February 13, 2019. |
[77] | Coric V , van Dyck CH , Salloway S , Andreasen N , Brody M , Richter RW , Soininen H , Thein S , Shiovitz T , Pilcher G , Colby S , Rollin L , Dockens R , Pachai C , Portelius E , Andreasson U , Blennow K , Soares H , Albright C , Feldman HH , Berman RM ((2012) ) Safety and tolerability of the γ-secretase inhibitor avagacestat in a phase 2 study of mild to moderate Alzheimer disease. Arch Neurol 3: , 1–12. |
[78] | Green RC , Schneider LS , Amato DA , Beelen AP , Wilcock G , Swabb EA , Zavitz KH ((2009) ) Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: A randomized controlled trial. JAMA 302: , 2557–2564. |
[79] | Doody RS , Raman R , Farlow M , Iwatsubo T , Vellas B , Joffe S , Kieburtz K , He F , Sun X , Thomas RG , Aisen PS , Alzheimer’s Disease Cooperative Study Steering Committee, Siemers E , Sethuraman G , Mohs R , Semagacestat Study Group ((2013) ) A Phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N Engl J Med 369: , 341–350. |
[80] | Biogen Press Release, Biogen and Eisai to Discontinue Phase 3 ENGAGE and EMERGE Trials of Aducanumab in Alzheimer’s Disease, http://investors.biogen.com/news-releases/news-release-details/biogen-and-eisai-discontinue-phase-3-engage-and-emerge-trials, Posted March 21, 2019, Accessed March 29, 2019. |
[81] | Biogen Press Release, Eisai and Biogen Announce Positive Topline Results of the Final Analysis for BAN2401 at 18 Months, http://investors.biogen.com/news-releases/news-release-details/eisai-and-biogen-announce-positive-topline-results-final, Posted July 5, 2018, Accessed February 13, 2019. |
[82] | Vandenberghe R , Rinne JO , Boada M , Katayama S , Scheltens P , Vellas B , Tuchman M , Gass A , Fiebach JB , Hill D , Lobello K , Li D , McRae T , Lucas P , Evans I , Booth K , Luscan G , Wyman BT , Hua L , Yang L , Brashear HR , Black RS , Bapineuzumab 3000 and 3001 Clinical Study Investigators ((2016) ) Bapineuzumab for mild to moderate Alzheimer’s disease in two global, randomized, Phase 3 trials. Alzheimers Res Ther 8: , 18–30. |
[83] | Release, Roche to discontinue Phase III CREAD 1 and 2 clinical studies of crenezumab in early Alzheimer’s disease (AD) - other company programmes in AD continue, https://www.roche.com/media/releases/med-cor-2019-01-30.htm, Posted January 30, 2019, Accessed March 29, 2019. |
[84] | Ostrowitzki S , Lasser RA , Dorflinger E , Scheltens P , Barkhof F , Nikolcheva T , Ashford E , Retout S , Hofmann C , Delmar P , Klein G , Andjelkovic M , Dubois B , Boada M , Blennow K , Santarelli L , Fontoura P , SCarlet RoAD Investigators ((2017) ) A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimers Res Ther 9: , 95–109. |
[85] | Lilly Press Release, Lilly Announces Top-Line Results of Solanezumab Phase 3 Clinical Trial, https://investor.lilly.com/static-files/9117f89a-0f6a-4dd8-a73e-97c108cf613d, Posted November 23, 2016, Accessed February 13, 2019. |
[86] | BioSpace, Eli Lilly Washes Its Hands of Another Phase 3 Study After the Failure of Solanezumab, https://www.biospace.com/article/eli-lilly-washes-its-hands-of-another-phase-3-study-after-the-failure-of-solanezumab-/ Posted February 2, 2017, Accessed March 29, 2019. |
[87] | Gauthier S , Feldman HH , Schneider LS , Wilcock G , Frisoni GB , Hardlund J , Kook K , Wischik DJ , Schelter BO , Storey JM , Harrington CR , Wischik CM ((2016) ) Phase 3 trial of the tau aggregation inhibitor leuco-methylthionium-bis (hydropethanesulfonate) (LMTM) in mild to moderate Alzheimer’s disease. Alzheimers Dement 12: (7 Suppl), P351–P352. |
[88] | Zhang C , Griciuc A , Hudry E , Wan Y , Quinti1 L , Ward J , Forte AM , Shen X , Ran CZ , Elmaleh DR and Tanzi RE ((2018) ) Cromolyn reduces levels of the Alzheimer’s disease-associated amyloid β-protein by promoting microglial phagocytosis. Sci Rep 8: , 1–9. |
[89] | Hori Y , Takeda S , Cho H , Wegmann S , Shoup TM , Takahashi K , Irimia D , Elmaleh DR , Hyman BT , Hudry E ((2015) ) A Food and Drug Administration-approved asthma therapeutic agent impacts amyloid beta in the brain in a transgenic model of Alzheimer disease. J Biol Chem 290: , 1966–1978. |
[90] | Elmaleh DR. Combination Therapies for Treatment of Alzheimer’s Disease and Related Disorders. US 9,855,276 B2, (2018) . |
[91] | Cummings JL ((2018) ) Lessons learned from Alzheimer disease: Clinical trials with negative outcomes. Clin Transl Sci 11: , 147–152. |
[92] | Gamberger D , Lavrac N , Srivatsa S , Tanzi RE , Doraiswamy PM ((2017) ) Identification of clusters of rapid and slow decliners among subjects at risk for Alzheimer’s disease. Sci Rep 7: , 1–12. |
[93] | Becker RE , Greig NH , Giacobini E ((2008) ) Why do so many drugs for Alzheimer’s disease fail in development? Time for new methods and new practices? J Alzheimers Dis 15: , 303–325. |
[94] | Aisen PS ((2015) ) Cognitive/clinical endpoints for pre-dementia AD trials. J Prev Alzheimers Dis 2: , 82–84. |
[95] | Doody RS , Ferris SH , Salloway S , Sun Y , Goldman R , Watkins WE , Xu Y , Murthy AK ((2009) ) Donepezil treatment of patients with MCI: A 48-week randomized, placebo-controlled trial. Neurology 72: , 1555–1561. |
[96] | Williams MM , Storandt M , Roe CM , Morris JC ((2013) ) Progression of Alzheimer’s disease as measured by Clinical Dementia Rating Sum of Boxes scores. Alzheimers Dement 9: (1 Suppl), S39–S44. |
[97] | Delnomdedieu M , Duvvuri S , Li DJ , Atassi N , Lu M , Brashear HR , Liu E , Ness S , Kupiec JW ((2016) ) First-In-Human safety and long-term exposure data for AAB-003 (PF-05236812) and biomarkers after intravenous infusions of escalating doses in patients with mild to moderate Alzheimer’s disease. Alzheimers Res Ther 8: , 12–21. |
[98] | Nicoll JA , Wilkinson D , Holmes C , Steart P , Markham H , Weller RO ((2003) ) Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: A case report. Nat Med 9: , 448–452. |
[99] | Galasko D , Bell J , Mancuso JY , Kupiec JW , Sabbagh MN , van Dyck C , Thomas RG , Aisen PS , Alzheimer’s Disease Cooperative Study ((2014) ) A randomized clinical trial of an inhibitor of RAGE-Aβ interactions in patients with mild to moderate AD. Neurology 82: , 1536–1542. |
[100] | Lilly Press Release, Lilly Voluntarily Terminates Phase II Study for LY2886721, a Beta Secretase Inhibitor, Being Investigated as a Treatment for Alzheimer’s Disease, https://investor.lilly.com/news-releases/news-release-details/lilly-voluntarily-terminates-phase-ii-study-ly2886721-beta, Posted June 13, 2016, Accessed February 13, 2019. |
[101] | Cummings JL , Zhong K , Kinney JW , Heaney C , Moll-Tudla J , Joshi A , Pontecorvo M , Devous M , Tang A , Bena J ((2016) ) Double-blind, placebo-controlled, proof-of-concept trial of bexarotene X in moderate Alzheimer’s disease. Alzheimers Res Ther 8: , 4–12. |
[102] | Salloway S , Sperling R , Gilman S , Fox NC , Blennow K , Raskind M , Sabbagh M , Honig LS , Doody R , van Dyck CH , Mulnard R , Barakos J , Gregg KM , Liu E , Lieberburg I , Schenk D , Black R , Grundman M ((2009) ) A Phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology 73: , 2061–2070. |
[103] | Heneka MT , Carson MJ , E Khoury J , Landreth GE , Brosseron F , Feinstein DL , Jacobs AH , Wyss-Coray T , Vitorica J , Ransohoff RM , Herrup K , Frautschy SA , Finsen B , Brown GC , Verkhratsky A , Yamanaka K , Koistinaho J , Latz E , Halle A , Petzold GC , Town T , Morgan D , Shinohara ML , Perry VH , Holmes C , Bazan NG , Brooks DJ , Hunot S , Joseph B , Deigendesch N , Garaschuk O , Boddeke E , Dinarello CA , Breitner JC , Cole GM , Golenbock DT , Kummer MP ((2015) ) Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14: , 388–405. |
[104] | Niemantsverdriet E , Valckx S , Bjerke M , Engelborghs S ((2017) ) Alzheimer’s disease CSF biomarkers: Clinical indications and rational use. Acta Neurol Belg 117: , 591–602. |
[105] | McKhann GM , Knopman DS , Chertkow H , Hyman BT , Jack CR Jr , Kawas CH , Klunk WE , Koroshetz WJ , Manly JJ , Mayeux R , Mohs RC , Morris JC , Rossor MN , Scheltens P , Carrillo MC , Thies B , Weintraub S , Phelps CH ((2011) ) The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7: , 263–269. |
[106] | Burchell JT , Panegyres PK ((2017) ) New cerebrospinal fluid biomarkers in Alzheimer’s disease. Future Neurol 12: , 53–57. |
[107] | Pontecorvo MJ , Devous MD , Navitsky M , Lu M , Salloway S , Schaerf FW , Jennings D , Arora AK , McGeehan A , Lim NC , Xiong H , Joshi AD , Siderowf A , and Mintun MA for the 18F-AV-1451-A05 investigators ((2017) ) Relationships between flortaucipir PET tau binding and amyloid burden, clinical diagnosis, age and cognition. Brain 140: , 748–763. |


