
A Clinical Trial of Transcranial Electromagnetic Treatment in Alzheimer’s Disease: Cognitive Enhancement and Associated Changes in Cerebrospinal Fluid, Blood, and Brain Imaging
Abstract
Background:
Small aggregates (oligomers) of the toxic proteins amyloid-β (Aβ) and phospho-tau (p-tau) are essential contributors to Alzheimer’s disease (AD). In mouse models for AD or human AD brain extracts, Transcranial Electromagnetic Treatment (TEMT) disaggregates both Aβ and p-tau oligomers, and induces brain mitochondrial enhancement. These apparent “disease-modifying” actions of TEMT both prevent and reverse memory impairment in AD transgenic mice.
Objective:
To evaluate the safety and initial clinical efficacy of TEMT against AD, a comprehensive open-label clinical trial was performed.
Methods:
Eight mild/moderate AD patients were treated with TEMT in-home by their caregivers for 2 months utilizing a unique head device. TEMT was given for two 1-hour periods each day, with subjects primarily evaluated at baseline, end-of-treatment, and 2 weeks following treatment completion.
Results:
No deleterious behavioral effects, discomfort, or physiologic changes resulted from 2 months of TEMT, as well as no evidence of tumor or microhemorrhage induction. TEMT induced clinically important and statistically significant improvements in ADAS-cog, as well as in the Rey AVLT. TEMT also produced increases in cerebrospinal fluid (CSF) levels of soluble Aβ1-40 and Aβ1-42, cognition-related changes in CSF oligomeric Aβ, a decreased CSF p-tau/Aβ1-42 ratio, and reduced levels of oligomeric Aβ in plasma. Pre- versus post-treatment FDG-PET brain scans revealed stable cerebral glucose utilization, with several subjects exhibiting enhanced glucose utilization. Evaluation of diffusion tensor imaging (fractional anisotropy) scans in individual subjects provided support for TEMT-induced increases in functional connectivity within the cognitively-important cingulate cortex/cingulum.
Conclusion:
TEMT administration to AD subjects appears to be safe, while providing cognitive enhancement, changes to CSF/blood AD markers, and evidence of stable/enhanced brain connectivity.
INTRODUCTION
For almost two decades, researchers in both academia and the pharmaceutical industry have been searching for a “disease-modifying” drug that could arrest or reverse the severe memory impairment of AD. This failure so far may not be due to starting drug therapy too late in the disease process (as many believe), but rather because: 1) current drugs being clinically tested (especially proteins) have difficulty in crossing the blood-brain barrier and entering the brain’s neurons [1], and 2) the majority of AD drugs in clinical trials target either the large aggregates of amyloid-β (Aβ) protein outside of neurons (neuritic plaques) or intraneuronal neurofibrillary tangles (NFTs) made up of large insoluble phospho-tau (p-tau) protein deposits [2, 3]. However, both monomeric Aβ and p-tau are produced inside neurons, where they aggregate into soluble toxic “oligomers”—oligomers that most recent studies suggest are the primary culprits in AD (not the insoluble Aβ plaques or NFTs) [4–10]. These “intraneuronal” Aβ and tau oligomers initiate and promote AD development by causing neuronal dysfunction and death through: 1) mitochondrial dysfunction/ATP suppression [11–13], 2) microtubule destruction [14–16], 3) dendritic spine regression [8, 17], and 4) synaptic dysfunction [7, 18, 19]. Thus, any effective AD therapeutics or combination of therapeutics directed at Aβ or tau will probably need to penetrate the brain’s neurons and be capable of disaggregating both Aβ and tau oligomers inside neurons.
In view of the inability of drugs to slow or reverse the cognitive impairment of AD thus far, investigating non-pharmaceutic interventions against AD as a possible alternative is now clearly warranted. Neuromodulatory approaches have consequently emerged and are currently being clinically tested in AD subjects. These approaches include transcranial magnetic stimulation (tMS), transcranial direct current stimulation, and deep brain stimulation. All three approaches provide a generalized stimulatory/inhibitory effect on neuronal activity. The most recent and largest clinical studies involving long-term tMS (Phase III clinical trial) or deep brain stimulation (Phase II clinical trial) in AD subjects have reported minimal or no cognitive benefits [20–22]. As the newest neuromodulatory approach against AD, Transcranial Electromagnetic Treatment (TEMT) is very different from tMS because TEMT involves perpendicular magnetic and electric waves emanating away from an antenna/emitter source (rather than tMS’s magnetic waves radiating from and returning to a conductor). For our studies, these “electromagnetic waves” are actually within the radiofrequency range (around 1 GHz), which can easily penetrate the human cranium and underlying brain areas.
In a number of pre-clinical studies involving AD transgenic mice [11, 23–27], we have administered TEMT daily (1 h in early morning and 1 h in late afternoon) for up to 8 months, at a frequency of 918 MHz, power (SAR) level of approximately 1 W/kg, and pulse repetition rate of 217 Hz. Utilizing these parameters and protocol, we have demonstrated the ability of TEMT to prevent/reverse both oligomeric and insoluble Aβ aggregation—both inside and outside neurons [11, 23]. These TEMT-induced reductions in brain Aβ aggregation are accompanied by brain mitochondrial enhancement [11] and prevention or reversal of cognitive impairment in AD transgenic mice at multiple ages [23, 24, 27]. Our most recent studies involving brain homogenates from human AD subjects provide evidence that TEMT not only disaggregates Aβ oligomers, but also disaggregates tau and α-synuclein oligomers [Cao et al., unpublished data]. Oligomeric tau, although downstream from Aβ in AD pathogenesis, may be the main propagator of AD [9, 28], and increasing evidence suggests α-synuclein aggregation plays a role in AD as well [29, 30]. The general disaggregating ability of TEMT against toxic oligomers may be critical because any effective AD therapeutics will probably need to be capable of disaggregating both Aβ and tau oligomers within neurons; targeting only Aβ oligomers/aggregation may not be enough, especially after the disease has become established [9]. Thus, TEMT would appear to provide a cocktail of disease-modifying mechanisms by directly targeting brain aggregation of three toxic proteins, as well as the mitochondrial dysfunction of AD [11, 23, 24, 27, Cao et al., unpublished data].
Any “disease-modifying” therapeutic against AD must also be safe to administer over a long period of time. It is therefore important to note that we found no deleterious behavioral effects of daily TEMT administration for as long as 8 months in AD mice, as well as no changes in brain markers of oxidative damage or antioxidant enzymes/compounds [23, 26]. Moreover, measurement of brain temperature at intervals during long-term TEMT revealed no significant increases in brain temperature during “ON” periods [24]; thus, cognitive benefits of TEMT occur without brain heating (e.g., through ‘non-thermal” mechanisms). This is important because essentially all reported deleterious effects of electromagnetic treatment on biologic functions/processes in animals or cell cultures involved induction of tissue hyperthermia [31–33] and/or involved only acute treatment. Indeed, many human studies involving short- or long-term exposure to electromagnetic fields similar to those in our pre-clinical studies (and in this open-label clinical study) have found no deleterious effects on general health, cognitive function, or a variety of physiologic measures [26]. Just as importantly, numerous recent large epidemiologic studies involving hundreds of thousands of individuals have shown that electromagnetic field (EMF) exposure (at frequencies and power levels similar to those in this study) does not increase the risk of any type of cancer [see 25,26 for discussion and references].
In view of our extensive pre-clinical platform and the aforementioned wide spectrum of human safety studies, clinical trials of TEMT technology in AD were clearly warranted. Therefore, we designed and built a first-of-its-kind head device (the MemorEMTM) for administration of TEMT to human subjects in their homes and by their caregivers. The present study reports on safety and efficacy endpoints in an open-label clinical trial (ClinicalTrials.gov Protocol NCT0295830) utilizing MemorEMTM head devices to provide daily TEMT to AD subjects over a 2-month period, as well as evaluation at two weeks following completion of treatment. The same TEMT parameters (frequency, power level) and protocol (twice daily 1-h treatments) were utilized in this clinical trial as used in our pre-clinical studies that demonstrated cognitive protection and reversal of impairment in AD mice. For efficacy endpoints, a battery of cognitive tasks (including the Alzheimer’s Disease Assessment Scale–Cognitive Subscale, ADAS-cog) was administered, blood/cerebrospinal fluid (CSF) was analyzed for AD markers (e.g., soluble Aβ, oligomeric Aβ, tau), and both FDG-PET and functional MRI (diffusion tensor imaging, DTI) brain imaging were performed. Given the relatively small number of subjects in this study (all of whom received TEMT), the significant treatment-induced effects being reported should be interpreted proportionally until results from a larger pivotal trial are available.
METHODS
Subjects
A total of eight (8) subjects with mild-moderate AD were enrolled in this clinical trial over a rolling enrollment period of late 2017 through mid-2018 at the University of South Florida Health/Byrd Alzheimer’s Institute (Tampa, FL), with all subjects completing the 2-month treatment study by the end of 2018 at the Byrd Institute. Subjects had to be diagnosed with mild or moderate AD, according to the National Institute of Neurological and Communicative Disorders and Stroke-Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) criteria. They had to be at least 63 years of age and exhibit a Mini-Mental State Examination (MMSE) score of 16–26 at screening. All subjects had a minimum of an 8th grade education and a head circumference between 53–60 cm (to minimize variability in head RF antenna/emitter locations). If they were being medicated with a cholinesterase inhibitor and/or memantine, subjects needed to be on such medication for at least 3 months prior to screening, on a stable dose for at least 60 days prior to screening, and maintained on that dose throughout the period of this study.
Table 1 shows demographics and characteristics for each of the eight subjects participating in this study. In addition to their AD diagnosis from cognitive assessment at screening/baseline, the diagnosis of AD was further established by three indices included in this table. First, anatomic MRI analysis at screening indicated the presence of frontal/parietal lobe atrophy, hippocampal/temporal lobe atrophy, and/or global cortical atrophy. Second, quantitative data from baseline FDG-PET scans were evaluated for glucose hypometabolism. Although there is no established cut-off that would definitely differentiate AD subjects from normal controls, an “AD signature meta-ROI” has been developed that evaluates the averaged cerebral metabolic rate for glucose (CMRgl) in three brain areas (angular gyrus, posterior cingulate, and inferior temporal cortex) [34]. The mean AD signature meta-ROI for young adults (aged 30–49) has been reported to be 1.87 and for aged control (>70 years) to be 1.56 [34]. An abnormal AD ratio has been defined as ≤1.32, which corresponds to 90% sensitivity for AD dementia [35, 36]. Averaging “baseline” quantitative CMRgl measures from the same three brain regions from each of the eight subjects of this study provided a similar AD signature meta-ROI index of brain hypometabolism for each subject. The results of this analysis (Table 1) indicate a mean of 1.20 for all subjects collectively, with only one subject having a ratio (1.40) a little above the abnormal AD ratio of 1.32 (Table 1). Third, because of its use in identifying and confirming clinically-diagnosed AD, the CSF Aβ1-42/total tau (t-tau) ratio at baseline was calculated for each subject and entered into Table 1. The mean ratio of 0.90 is close to the mean ratio of 1.30 reported for diagnosed AD subjects by Niemantsverdriet et al. [37], and far from the mean ratio of 3.96 for aged controls in that same study. Further supporting the diagnosis of AD for this study’s subjects, another study determined a cut-off of 1.14 in the CSF Aβ1-42/t-tau ratio between mild cognitive impairment and AD subjects, with AD subjects typically being below this value [38]. Therefore, the cognitive assessment, anatomic MRI analysis, hypometabolism analysis, and CSF Aβ1-42/t-tau ratios from the subjects of this study are all consistent with the diagnosis of AD. It should be mentioned that this study involved indices of AD diagnosis (i.e., PET AD signature analysis of energy utilization, anatomic MRI analysis of brain atrophy, spinal taps for CSF marker analysis) that are often not performed together to firmly establish AD diagnosis.
Table 1
Subject Demographics/Characteristics
| Subject | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | Mean |
| Age | 63 | 66 | 82 | 65 | 74 | 63 | 77 | 76 | 70.8 |
| Gender (M,F) | F | M | M | F | F | F | F | F | − |
| APOE genotype | 3/3 | 3/3 | 2/3 | 2/3 | 3/3 | 3/3 | 2/3 | 3/3 | − |
| ADAS-cog score | 62.0 | 30.3 | 44.0 | 37.3 | 38.7 | 30.7 | 24.0 | 26.7 | 36.7 |
| MMSE score | 18 | 21 | 20 | 19 | 16 | 17 | 26 | 19 | 19.5 |
| GDS Rating | 4 | 3 | 4 | 4 | 3 | 5 | 4 | 4 | 3.9 |
| Education (yrs) | 14 | 15 | 12 | 14 | 16 | 13 | 19 | 16 | 14.9 |
| Anat. MRI Analysis | a | b | b,c | b | a,b | c | b,c | b,c | − |
| PET AD Sign. ROI | 1.32 | 1.10 | 1.06 | 1.40 | 1.25 | 1.32 | 1.10 | 1.06 | 1.20 |
| Aβ1-42/t-tau ratio | 0.30 | 0.93 | 0.98 | − | 0.99 | 0.61 | 1.48 | 1.02 | 0.90 |
APOE, apolipoprotein; ADAS-Cog, Alzheimer’s Disease Assessment Scale–Cognitive Subscale; MMSE, Mini-Mental State Examination; GDS, Global Deterioration Scale; a, frontal/parietal lobe atrophy; b, hippocampal/temporal lobe atrophy; c, global cortical atrophy.
Exclusion criteria for this study included having more than 5 microhemorrhages evident in anatomic MRI at screening, a score of 4 or higher on the Hachinski Test, and a score of 2 or less on the Global Deterioration Scale (the latter as rated by the caregiver). Additional exclusion criteria included participation in a pharmaceutic clinical trial within 6 months of study enrollment and a history of immunotherapy research participation, uncontrolled major depression, a history of any clinically-defined neurological/psychiatric disorder (other than AD), including but not limited to: stroke, brain lesions, cerebrovascular condition, other neurodegenerative disease, significant head trauma, multiple sclerosis, or personal history of previous neurosurgery or brain radiation. All patients agreed to be in this study and gave written informed consent. For each subject, a caregiver (spouse, family member, etc.) needed to be identified who agreed to be responsible for the participation of the patient in the study (e.g., keeping a diary of health measures they collect on the patient at home, logging the patient’s condition daily, and assuming responsibility for administering daily in-home treatment). Caregivers were required to have non-impaired mental abilities and normal motor skills, as determined by the clinical investigators at screening.
Investigational device
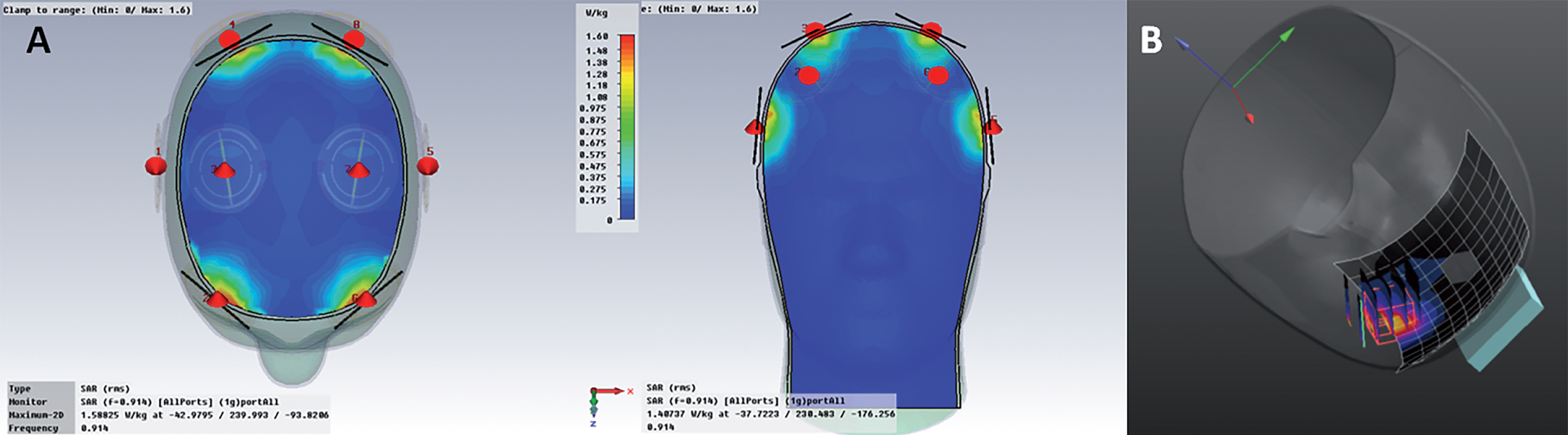
The MemorEMTM head device is self-contained and has been designed for in-home daily treatment, allowing for complete mobility and comfort in performing daily activities during treatment (Fig. 1A). The device has a custom-engineered circuit board and a rechargeable battery inside the box housing, as well as a control panel on the outside of the housing for treatment control. This control panel/battery box is worn on the upper arm and wired via a cable to eight specialized antennas/emitters embedded between a double-layered head cap worn by the subject (Fig. 1B). The device is designed to allow no more than two 1-h treatments within a given 24-h period and requires at least a 7-h interval between those two daily treatments. When running a treatment, the device transmits electromagnetic waves in a pulsed fashion and sequentially through the 8 emitters at 915 MHz carrier frequency every 4.6 ms (e.g., a pulse repetition rate to each antenna of 217 Hz). Power levels (specific absorption rate, SAR) for each emitter were set at an average of 1.6 W/kg. At this frequency and power level, FDTD human head computer simulations (IEEE Model 1528 phantom) show that the eight emitters collectively provide both global and penetrating TEMT to the human forebrain, including the cerebral cortex and underlying structures (Fig. 2A). A very similar calculated SAR distribution is arrived at from actual electric field measurements taken under individual emitters and within brain gel “in situ” (inside a human head phantom), utilizing a robotic probe system and grid measurement pattern (Fig. 2B). The MemorEMTM head device and this clinical trial protocol were both approved as “non-significant risk” by the Western Institutional Review Board.
Fig.1
A) A MemorEMTM head device being worn by a subject. B) Position of the eight electromagnetic emitters embedded between the device’s two-layered head cap. Emitters collectively provide global forebrain TEMT via rapid sequential emitter activation.
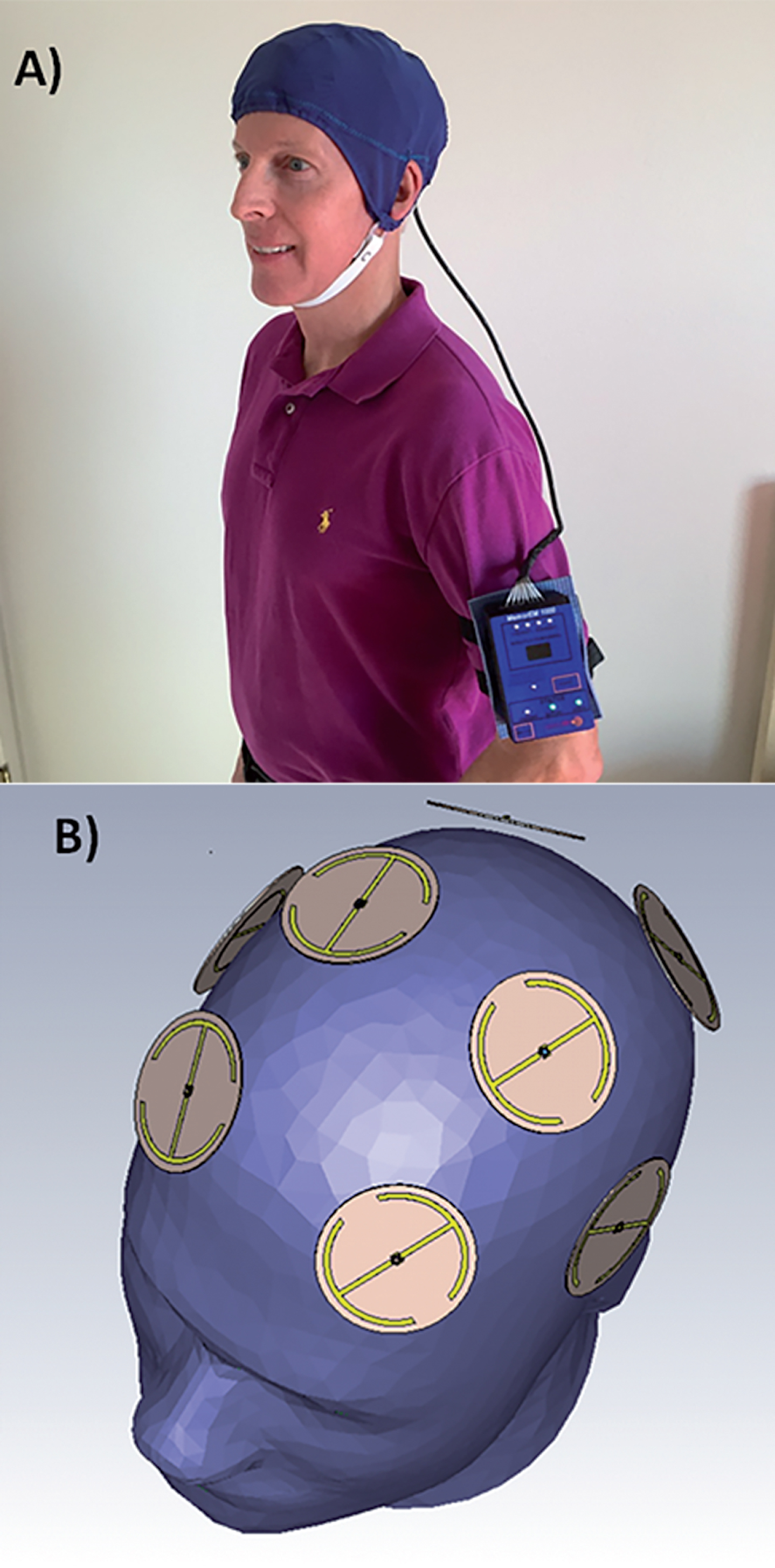
Fig.2
A) FDTD simulations showing electric field distribution underneath all 8 emitters. Note there is no overlap in brain specific absorption rate (SAR) distribution between any two emitters, even when they are all on at once. B) Real “in situ” brain SAR distribution under an emitter, as calculated from volumetric grid electric field measurements.
General protocol
This clinical study was an open-label within-patient (single arm) single center study that was intended to evaluate the safety and efficacy of 2 months of daily TEMT administration in patients with mild-to-moderate AD. All screening events occurred within two weeks of treatment initiation, and all baseline measures were attained within one week of treatment initiation. Following the baseline clinical visits, succeeding visits occurred on the first day of treatment (D1), D2, D7, D14, D30, D60, and at two weeks following completion of treatment on D60. The protocol for this study is detailed at ClinicalTrials.gov (NCT02958930).
Screening
Potential subjects were given both a physical exam and a neurologic exam at the Byrd Institute, as well as a MMSE, the Hachinski Test, and Global Deterioration Scale assessment. A 3-Tesla “anatomic” MRI brain scan (specifically susceptibility-weighted imaging or SWI and Axial Flair sequences) was taken at University Diagnostic Institute, an MRI facility on the USF campus; this was done for determining any pre-existing brain microhemorrhages (and to be the baseline for any treatment effects on MRI-related endpoints). This SWI/Axial Flair scan also served to rule out other neurological conditions that are exclusionary to the trial, such as previous cerebrovascular disease/infarction, tumor, demyelinating diseases, etc. All of the above screening events necessitated several visits to the nearby Byrd Institute or facilities.
Baseline
Baseline consisted of three to four separate days of evaluation/testing, all within one week of treatment Day 1 and all scheduled during the morning hours. One day involved an FDG-PET scan being done at the Byrd Institute, while another day involved functional MRI scanning (DTI) for functional connectivity done at the University Diagnostic Institute. A third day entailed an office visit at the Byrd Institute during which a comprehensive battery of cognitive tasks was administered to establish baseline cognitive performance. These tasks included the principal measure of efficacy, the ADAS-Cog13 (maximum poor score of 85 points), and secondary measures including: Rey Auditory Verbal Learning Test (Rey AVLT), Alzheimer’s Disease Cooperative Study - Activities of Daily Living, Trails A & B, Digit span, and clock draw tasks. In addition, baseline Adverse Event Assessment and a baseline suicide ideation score (via the Columbia Suicide Severity Rating Scale) were obtained. On one of the above three days, or an additional day, a 20 ml blood sample and a 15 ml CSF sample (via spinal tap) was taken for later AD marker analyses and APOE genotyping.
Two-month treatment period
Subjects were given their first “morning” TEMT treatment in the clinic (Day 1), during which time their caregivers were instructed on the proper procedure for administering TEMT to the subject at home. A complete, yet easy-to-understand “Instructions for Use” manual was provided to each caregiver, who was also instructed on when and how to take blood pressure measurement with a supplied fully-automated BP device, as well as when and how to take body temperature with a supplied thermometer. Caregivers were also given a “Patient Daily Diary” for them to enter each day’s blood pressure and temperature reading, check off daily activities (e.g., eating/drinking) as normal or different, and comment on any different behaviors or undesirable side effects that occurred to the patient during or after that day’s treatments. Upon completion of the initial “morning” treatment at the Byrd Alzheimer’s Institute, a blood sample was drawn for later analysis. The second “late afternoon” TEMT administration was administered at the patient’s home by the caregiver. On Day 2, another clinical visit occurred following morning in-home TEMT administration. This D2 visit was to confirm the caregivers were using proper TEMT procedures and to collect a blood sample. For this clinical visit, and all succeeding visits during the trial, caregivers/patients arrived within 1 h of the end of that morning’s in-home treatment.
Throughout the following 2-month period, subjects were given twice-daily TEMT treatment for 1 h each (early morning and late afternoon), as administered and supervised by the caregiver. Subjects returned to the clinic on Days 7, 14, 30, and 60 into TEMT treatment (within 1 h of morning treatment ending) for adverse event assessment and 20 ml blood withdrawal. Cognitive assessment in the battery of cognitive tasks was done during clinical visits on 14, 30, and 60 days into treatment, as well as 2 weeks post-treatment. Day 60 consisted of three to four separate days of clinical evaluation/testing with a window of ± 5 days in order to collect all end-of-treatment (Day 60) data, which included not only cognitive testing/blood withdrawal, but also a spinal tap, FDG-PET scan, and a functional MRI scan (DTI). In addition, Day 60 Adverse Event Assessment and a suicide ideation score (via the Columbia Suicide Severity Rating Scale) were obtained to compare to baseline values.
Post-treatment period
After completion of the 2-month treatment period, a clinical visit occurred at 2 weeks post-treatment at which time the full cognitive battery was administered and a blood sample taken. On one day during this 2-week post-treatment period, a follow-up MRI “anatomic” susceptibility-weighted imaging scan (SWI and Axial Flair sequences) was done at the University Diagnostic Institute to determine any induction of brain microhemorrhages or any other brain structural abnormalities by the treatment.
Patient monitoring and safety
The primary safety measure was an Adverse Event Assessment performed during every clinical visit at the Byrd Institute beginning with baseline. Secondary safety measures also collected at the same clinical visit time points were vitals (e.g., blood pressure, temperature). The secondary safety measure of suicide tendencies was assessed at baseline, halfway through the 2-month treatment period, and at the completion of treatment. During the entire 2-month TEMT administration period, subjects were monitored daily by their caregivers for any undesirable side-effects of treatment, including any different behaviors during/after treatment.
Blood and CSF processing/analysis
All 20 ml blood samples (collected at baseline and treatment Days 1, 2, 7, 14, 30, 60, and post-treatment Day 14) were divided into two 10 ml BD k2-EDTA tubes and centrifuged at 300×g for 10 min. The plasma (upper layer) for each tube was transferred into a new 15 ml tube, then centrifuged again at 2000 g for 10 min. One ml volumes of the top plasma layer were aliquoted into 1.5 ml ml tubes and stored at –80°C for future analysis. Also, 0.2 ml of the first EDTA blood sample was used for DNA preparation for APOE genotyping/DNA analysis. The two 15-ml samples of CSF collected at baseline and on Day 60 were each aliquoted into 1.5 ml tubes, then frozen and stored at –80°C until analysis of the same AD markers as indicated for plasma. At the end of the study, plasma/CSF samples were thawed completely on ice, then mixed well on vortex and centrifuged at 2,000 g for 10 min to precipitate any debris for determination of the following AD biomarkers in duplicate: soluble/monomeric Aβ1-40 and Aβ1-42, oligomeric Aβ, total tau (t-tau), and p-tau.
Human total tau determination
Instructions were followed according to those provided for the Thermo Fisher Human Tau (total) kit (Cat: KHB0041). Standard, Streptavidin-HRP, and wash buffer solutions were prepared according to the menu. For each well, 100 μl of standard and plasma or CSF sample (undiluted) were added, incubated overnight at 4°C with shaking, then washed 4 times with wash buffer. Detection antibody (100 μl/well) was then added, followed by incubation for 1 h at room temperature. Plates were washed 4 times with wash buffer, then 100 μl of diluted streptavidin-PE was added to each well, followed by incubation for 1 h at room temperature with shaking. Next, plates were washed 4 times, followed by addition of 100 μl of stabilized chromogen to each well. The reaction was allowed to occur for 10 min, then 100 μl of stop solution was added to each well, followed by plate reading on the BioTek Synergy H4 reader.
Human phospho-tau (p-tau) determination
Instructions were followed according to those provided for the Thermo Fisher Human p-tau (pT231) phosphoELISA kit (Cat: KHB8051). Standard, anti-rabbit IgG HRP, and wash buffer solutions were prepared according to the menu. For each well, 100 μl of standard and plasma or CSF sample (undiluted) was added, incubated overnight at 4°C with shaking, then washed 4 times with wash buffer. Detection antibody (100 μl /well) was then added, followed by incubation for 1 h at room temperature. Plates were washed 4 times, then 100 μl of diluted anti-rabbit IgG HRP was added to each well, followed by incubation for 1 h at room temperature with shaking. Next, plates were washed 4 times, followed by addition of 100 μl of stabilized chromogen to each well to allow reaction to occur for 10 min. Then 100 μl of stop solution was added to each well, followed by plate reading on the BioTek Synergy H4 reader.
Human Aβ1-40, Aβ1-42, and oligomeric Aβ determination (A8)
Antibodies (goat anti- human Aβ1-42, and Goat anti-human Aβ31 - 40 specific antibody were purchased from Biomer Tech. Inc., CA) and A8 antibody was prepared by our collaborator (Zhang HYBRIDOMA Volume 28, Number 5, 2009 DOI: 10.1089 = hyb.2009.0015). Instructions were followed according to those provided Standard, detection antibody, anti-rabbit IgG HRP, and wash buffer were all prepared according to the menu. For each well, 50 μl standard and plasma or CSF (1:100 diluted for Aβ determinations) sample were added to appropriate wells, then 50 μl of detection antibody for Aβ1-40, Aβ1-42, or A8 (Aβ oligomers) was added to each well. Incubation occurred overnight at 4°C with shaking. After 4 washes, 100 μl of diluted anti-rabbit IgG HRP was added to each well, followed by incubation for 1 h at room temperature with shaking. This was followed by 4 washes with wash buffer, then addition of 100 μl of stabilized chromogen to each well to allow reaction for 10 min. Stop solution (100 μl /well) was then added and plates read on the BioTeck Synergy H4 reader.
FDG-PET analysis
For FDG-PET analysis, “anatomic” MRI scans (SWI and Axial Flair sequences) from pre-treatment (Baseline) and post-treatment (Day 60) were selected. Pre-treatment PET images were first co-registered to MRI, followed by post-treatment PET images being co-registered to pre-treatment PET images. The MRI images were then spatially normalized to standard space. This normalization transformation was then combined with transformation co-registration of pre-treatment PET images to MRI, which was then applied to the pre-treatment PET images to bring them into standard space. Normalization transformation from the earlier spatial normalization of MRI to standard space was then combined with transformations from pre- and post-treatment PET images that had been co-registered to MRI and pre-treatment PET images, respectively. Finally, atlas volumes of interest (VOIs) were applied and quantitative data extracted. In the above PET analysis, Invicro’s standard PNEURO pipeline was used, which performs PET to MRI affine registration, spatially normalizes MRI to standard template (MNI) space, and applies the normalization transformation to the matched PET scan. In this PNEURO pipeline, a secondary PET data extraction was performed in template space utilizing the ADNI meta ROI; these were not intersected with the g/w matter maps. For each brain analyzed, quantitative data was obtained from 109 brain areas, which included a total of 40 sub-areas from all four lobes of both the left and right cortices (20 areas on each side). The pons/vermis was used as reference for normalizing each brain area’s value.
Functional MRI imaging
DTI is a variant of fMRI that measures the diffusion of water molecules in brain tissue and is particularly sensitive to changes in white matter integrity [39, 40]. Fractional anisotropy (FA) is a widely used DTI measure in AD clinical research wherein higher values are associated with greater axonal integrity and lower values indicate a loss of neuronal connectivity/axonal damage. DTI/FA analysis was carried out using fsl DTI tools (https://fsl.fmib.ox.ac.uk/fsl/fslwiki/FDT). Briefly, data was motion corrected and eddy-current de-warped. A DTI model was fit to the data and FA maps created. Non-linear registration was carried out to align an FA white matter skeleton to atlas for each individual FA map. Subtraction of baseline from post-treatment FA maps were computed in a voxel-by-voxel manner and thresholded to a minimum change of 0.05 (a.u.) for each individual. The skeletonized FA was used for analysis instead of the voxel-based FA maps. Region of Interest (ROI) analysis was performed in 12 predetermined structures (identified from atlases provided within the fsl package). The structures included are listed in Table 3. Baseline versus post-treatment FA values were compared pair-wise using a non-parametric robust permutation test as implemented in fsl (https://fsl.fmib.ox.ac.uk/fsl/fslwiki/Randomise). A multiple-comparison corrected “p” value of 0.05 was used to assess significant differences.
Statistical analysis
The study’s primary time points were Baseline (BL), Day 60 (D60; end of treatment), and Day 14 Post-treatment (14D Post) for analysis of differences in cognitive performance, blood/CSF levels of Aβ and tau isoforms, cerebral metabolic rate for glucose (via 2DG-PET), and fMRI (DTI) imaging. Baseline values were statistically compared to D60 and/or 14D Post values to determine effects immediately following the 2-month treatment period and any residual effects two weeks thereafter, respectively. Given the limited number of subjects in this study (n = 8), it is appropriate to report clinical results in terms of Effect Size (ES) [41], which measures the “magnitude of the difference between groups” or the minimal difference that is clinically important/meaningful. For determination of ES, the following established scale was utilized for signifying a “clinically important effect” based on Cohen’s “d” [42]: Moderate effect (>0.5), Large effect (>0.8), Very Large effect (>1.2), Huge effect (>2.0). In addition to calculation of ES, paired t-tests were utilized to assess group differences between BL and either D60 or D14 Post that are “statistically significant” at p < 0.05 or lower (despite the low sample size, which predisposes the results to bias towards Type II Error). Wherever a significant ES was present, the associated p-value was also reported. Though rare and usually involving plasma or CSF samples, data from a subject on a single measure was sometimes omitted due to undetectable baseline readings, inconsistent duplicate values, or as a clear outlier (Grubb’s single outlier test).
RESULTS
TEMT administration does not induce behavioral/physiologic side effects or brain abnormalities in AD subjects
Over the 2-month TEMT period, subjects received two 1-h daily treatments (a total of approximately 120 in-home treatments as administered by their caregiver). Through daily diary records kept by the caregiver, no subject exhibited any recurrent changes in eating/drinking, daily movement activities, or anxiety level/mood. As well, subjects did not complain of headaches, brain sensations, or any other side effects of TEMT during or following treatment. Caregiver recordings of blood pressure and temperature before, during, and 30-min following every treatment also did not show consistent changes in these physiologic parameters associated with TEMT administration. Adverse Event Assessment performed during every clinical visit indicated no adverse events of treatment and clinical assessment of suicide tendencies during/following the 2-month treatment period revealed no suicide tendencies from treatment. Supportive of their diagnosis of AD, pre-treatment “anatomic” MRI scans (SWI/Flair sequences) from all eight subjects indicated that they had significant hippocampal/temporal lobe atrophy, frontal/parietal lobe atrophy, and/or global (diffuse) cerebral cortical atrophy. In post-treatment anatomic MRI scans taken between 2½–3 months thereafter, no visible induction of tumors or brain microhemorrhages by the 2 months of daily TEMT administration was evident.
The aforementioned safety endpoints collectively indicate that global brain TEMT administration (at currently used parameters) should be a safe therapeutic for AD subjects when given daily and long-term.
TEMT enhances cognitive performance of AD subjects
For reference in this and ensuing section, “clinically important” effects are indicated by ES using the following scale established by Cohen (Cohen’s d) [42]: Moderate effect (>0.5), Large effect (>0.8), Very Large effect (>1.2), Huge effect (>2.0). “p” values are also indicated wherever an ES is significant.
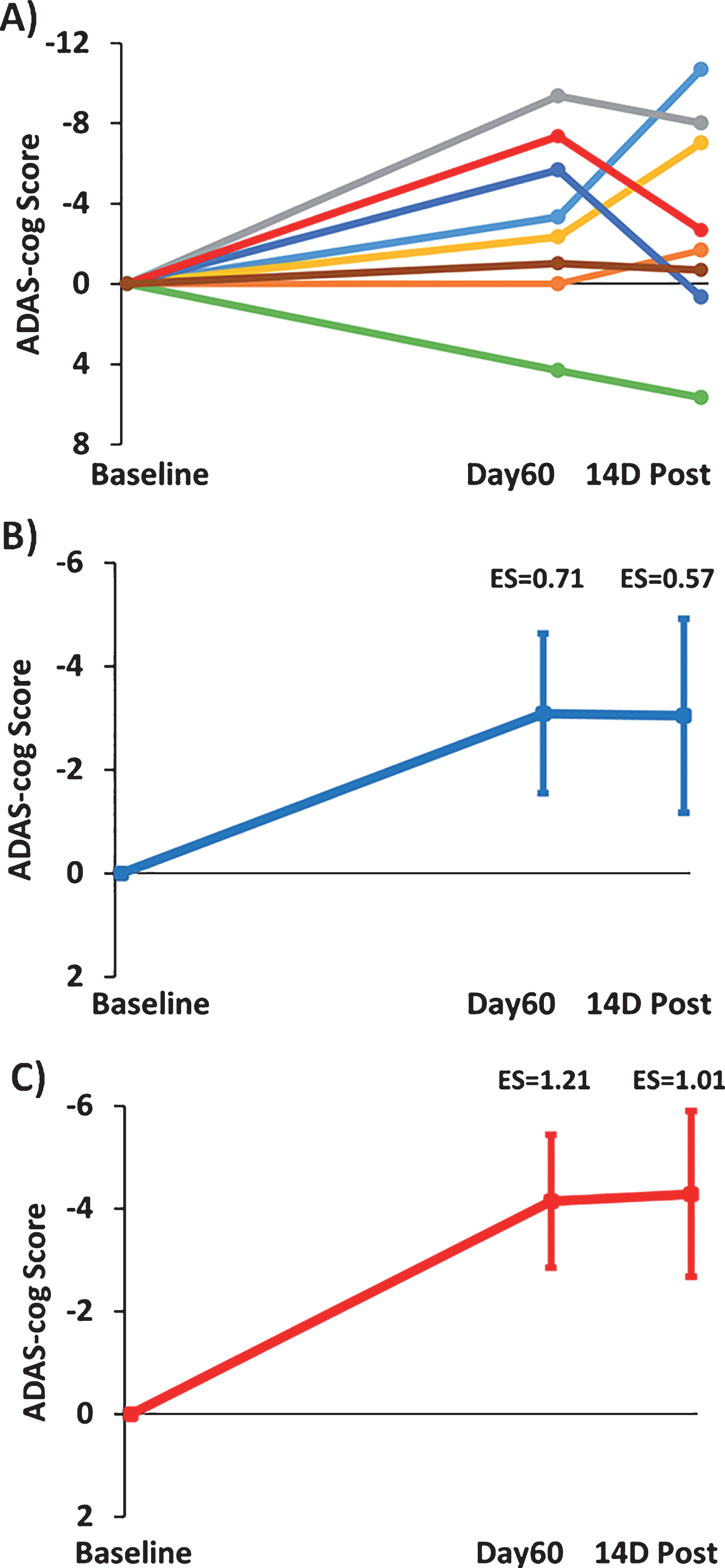
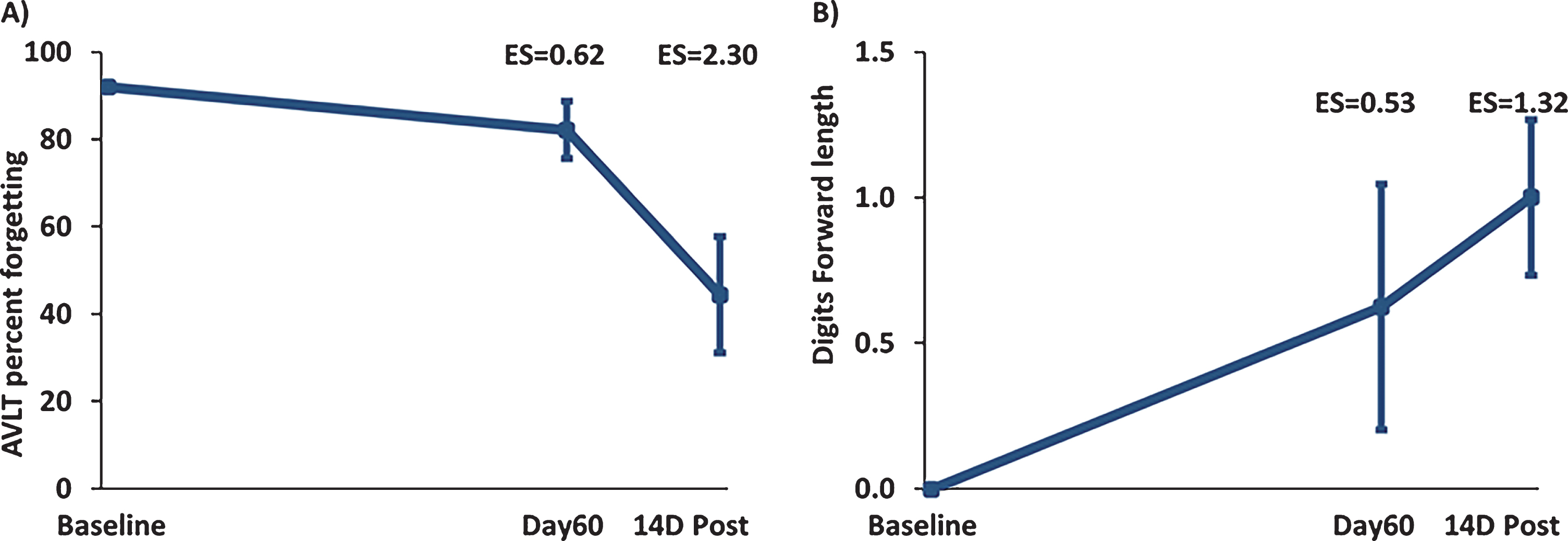
Figure 3A shows the change in individual subject ADAS-cog overall scores after 2-months of TEMT (Day60) and 14 days following completion of TEMT (14D Post) compared to Baseline performance. Seven of the 8 subjects improved their performance, with a single subject displaying the typical decrease in ADAS-cog performance reported in numerous clinical trials for untreated (control) AD subjects over a similar time period [43]. The combined ADAS-cog performance of all 8 subjects (Fig. 3B) revealed a clinically important improvement following 2 months of TEMT (ES = 0.71; p = 0.09) that was generally maintained at 14 days following completion of TEMT (ES = 0.57; p = 0.15). Figure 3C shows that removal of the one non-responder resulted in a greater clinically important improvement in ADAS-cog for both Day60 (ES = 1.21; p < 0.02) and 14D Post testing (ES = 1.01; p < 0.05), with average reductions of 4.1 and 4.3 points in ADAS-cog score, respectively. Thus, removal of this one non-responder in a relatively small group of subjects revealed a considerably stronger ES.
Fig.3
Effects of TEMT administration on change in ADAS-cog overall scores after 2 months of treatment (Day60) and 14 days following completion of treatment (14D Post) compared to Baseline performance. A) Change in individual ADAS-cog scores for all eight subjects. B) Improvement in combined ADAS-cog scores for all eight subjects on both Day60 and 14D Post. C) Greater improvement in combined ADAS-cog scores with omission of the one non-responder (green line subject in “A”). For (B) and (C), means±SEMs are presented. Negative scores indicate improved performance.
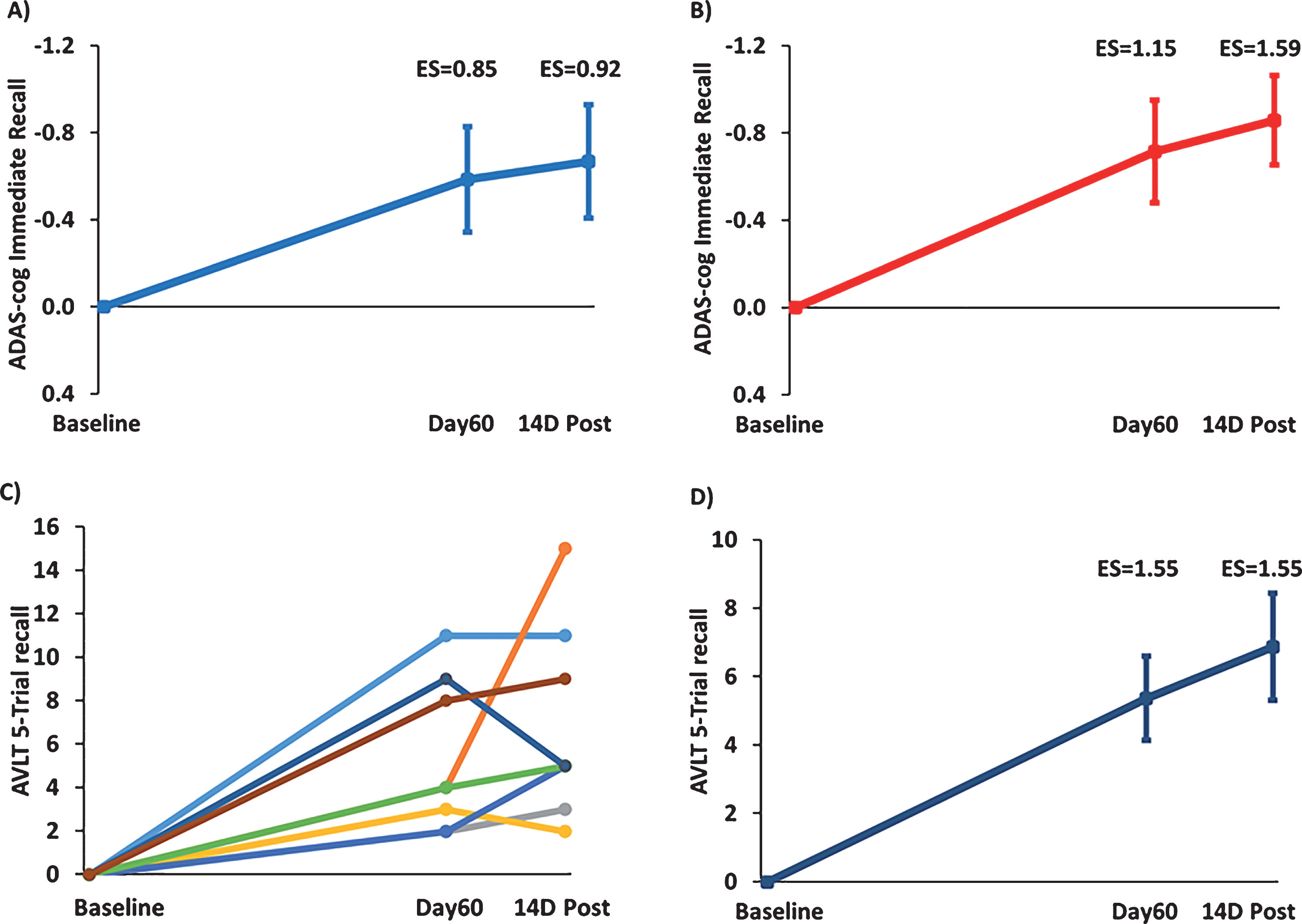
“Immediate recall” is an important component of both the ADAS-cog and the Rey AVLT. For the immediate recall measure of ADAS-cog and with all 8 subjects included (Fig. 4A), TEMT provided a clinically important increase in the number of words recalled at both Day60 (ES = 0.85; p < 0.05) and 14D Post (ES = 0.92; p < 0.05) compared to Baseline. With the one non-responder in overall ADAS-cog performance omitted (Fig. 4B), the clinically important increase in ADAS-cog immediate recall was further enhanced at both Day60 (ES = 1.15; p < 0.025) and 14D Post (ES = 1.59; p < 0.01). For the “5-trial recall” measure of Rey AVLT, the individual scores of all 8 subjects are shown in Fig. 4C and their combined performance depicted in Fig. 4D. TEMT induced a substantial clinically important increase in word recall at both Day60 (ES = 1.55; p < 0.005) and 14D Post (ES = 1.55; p < 0.005) compared to BL.
Fig.4
TEMT administration effects on the “Immediate Recall” component of ADAS-cog (A,B) and on the “5-Trial Recall” measure of the Rey AVLT (C,D). Increase in number of words recalled at Day60 (end of treatment) and 14D Post versus Baseline for (A) all eight subjects or (B) with omission of the one non-responder whose overall ADAS-cog performance is indicated by the green line in Fig. 3A. C) Improvement in individual scores of all eight subjects in 5-Trial Recall of the Rey AVLT immediately following 2 months of TEMT (Day60) and 14 days thereafter (14D Post) compared to Baseline. D) Combined performance of all subjects in (C) for Rey AVLT 5-Trail Recall showing substantial enhancement of word recall following 2 months of TEMT administration. For (A), (B), and (D), means±SEMs are presented.
Although word recall in the “Proactive Interference” component of the Rey AVLT did not show any effect of TEMT administration (Table 2), the ensuing “Percent Forgetting” component of Rey AVLT showed clinically important effects of treatment to reduce forgetting at both Day60 (ES = 0.53; p = 0.179) and 14D Post (ES = 1.27; p < 0.01) versus BL (Fig. 5A). In Digit Forward/Backward testing, Digit Forward Length was increased by TEMT administration at both Day60 (ES = 0.53; p = 1.79) and 14D Post (ES = 1.32; p < 01) versus BL (Fig. 5B). Compared to BL performance, the remaining cognitive measures showed stable performance following 2 months of TEMT administration on both Day60 and 14D Post (Table 2).
Table 2
Effects of TEMT on other cognitive measures evaluated after 2 months of treatment (D60) and 14 days following completion of treatment (14D Post) compared to baseline performance
| Task | Baseline | Day60 | 14D Post |
| Rey AVLT Proactive Interference (Number of words recalled) | 2.4±0.3 | 2.6±0.3 (n.s.) | 2.4±0.5 (n.s.) |
| MMSE Score | 19.5±1.1 | 19.1±1.3 (n.s.) | Not Administered |
| Trails A Time (s) | 59±9 | 56±10 (n.s.) | 71±14 (n.s.) |
| Trails B Time (s) | 220±30 | 192±48 (n.s.) | 198±35 (n.s.) |
| Activities of Daily Living Score | 62.2±2.4 | 61.8±2.0 (n.s.) | 58.9±2.3 (n.s.) |
| Digit Backward Length | 4.0±0.6 | 4.1±0.6 (n.s.) | 4.1±0.4 (n.s.) |
| Clock Drawing (# Correct out of 5) | 2.8±0.6 | 3.1±0.4 (n.s.) | 2.8±0.5 (n.s.) |
| Clock Copy (# Correct out of 5) | 3.9±0.5 | 4.1±0.3 (n.s.) | 3.9±0.3 (n.s.) |
All values are mean±SEM; n.s., not significantly different from baseline by both Effect Size (>0.5 threshold) and “p” value (p < 0.05) analyses. Rey AVLT, Rey Auditory Verbal Learning Test; MMSE, Mini-Mental State Examination.
Fig.5
A) “Percent Forgetting” in the Rey AVLT for all eight subjects combined. TEMT resulted in a reduction in forgetting at both Day60 and 14D Post versus Baseline (improved memory is indicated by a decrease in percentage). B) Increased Digit Forward Length for all eight subjects combined at Day60 (end of treatment) and 14D Post compared to Baseline. Daily TEMT administration for 2 months resulted in an increase in digits remembered at both Day60 and 14 days thereafter. Means±SEMs are presented.
TEMT induces changes in Aβ and tau in both CSF and blood of AD subjects
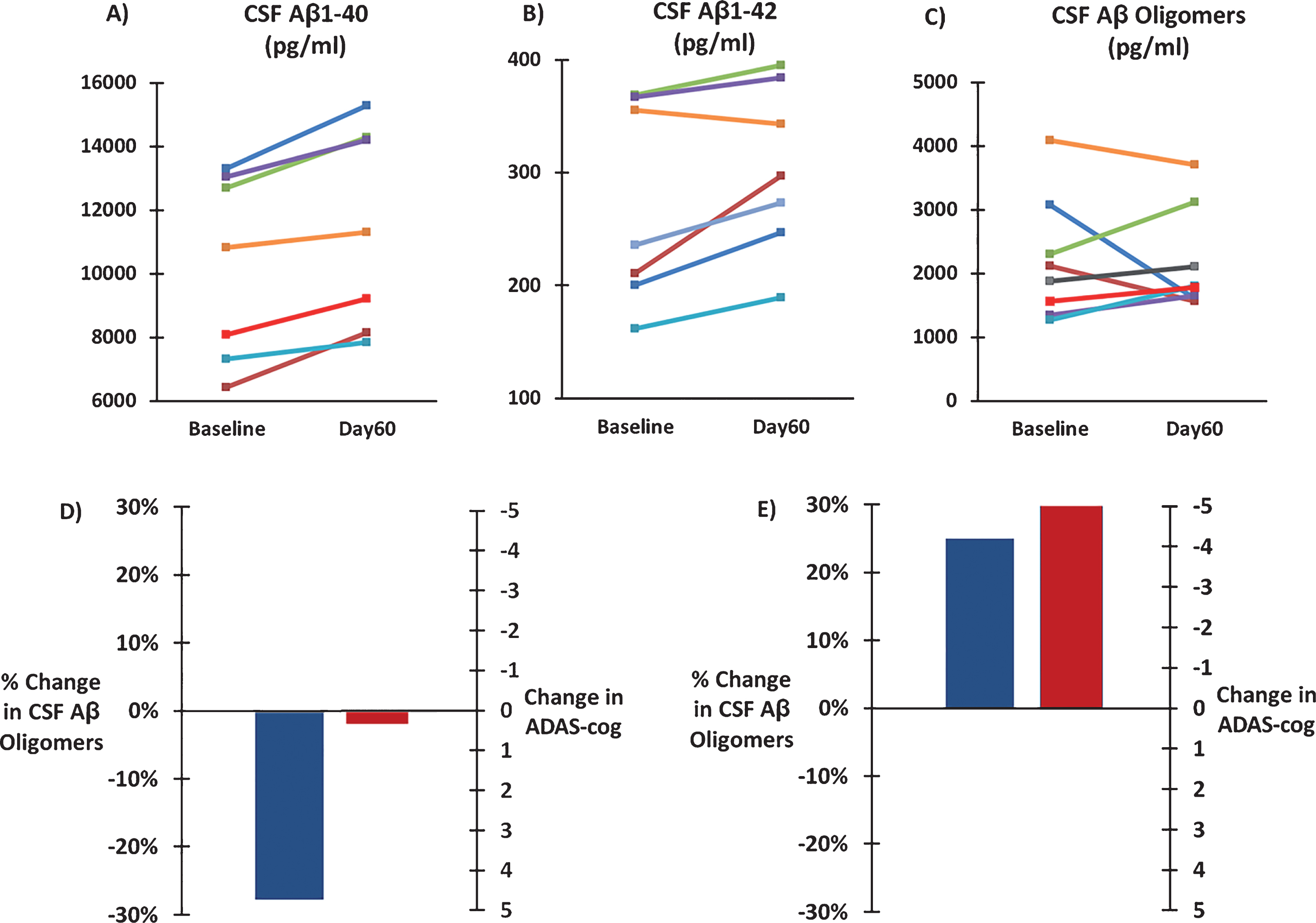
CSF levels of soluble Aβ1-40, soluble Aβ1-42, and oligomeric Aβ were analyzed from spinal tap samples taken at Baseline and Day60 (end of 2-month treatment period). Two months of daily TEMT administration induced a 12% clinically important and significant increase in CSF soluble Aβ1-40 levels compared to Baseline (10,234±1,103 versus 11,458±1,184 pg/ml; ES = 2.10; p < 0.001). Similarly, a 12% clinically important increase in CSF soluble Aβ1-42 levels was induced by TEMT administration compared to BL (272±34 versus 304±28 pg/ml; ES = 1.06; p < 0.05). For soluble Aβ1-40 and Aβ1-42, BL and Day60 values for individual subjects are shown in Fig. 6A and B. By contrast, CSF levels of Aβ oligomers were not significantly changed by 2 months of TEMT administration (2163±283 pg/ml) compared to BL (2203±339 pg/ml). BL and Day60 values of CSF oligomeric Aβ levels for individual subjects are shown in Fig. 6C. When subjects were assigned to one of two groups based on whether 2 months of TEMT resulted in a decrease (n = 3) or increase (n = 5) in CSF oligomeric Aβ, decreased oligomeric Aβ (averaging ↓28%) was associated with stable ADAS-cog performance between Day60 and BL (Fig. 6D). By contrast, a treatment-induced increase in CSF oligomeric Aβ (averaging ↑25%) was associated with over a 5-point improvement in ADAS-cog for Day60 versus BL (Fig. 6E).
Fig.6
Effects of TEMT administration on CSF levels of soluble Aβ1-40 (A), soluble Aβ1-42 (B), and oligomeric Aβ (C) following 2 months of TEMT (Day60) compared to levels at Baseline for all individual subjects. D) For subjects that showed a decrease in CSF Aβ oligomers following 2 months of TEMT (n = 3), combined performance on the ADAS-cog was stable on Day60 compared to Baseline. For subjects that showed an increase in CSF Aβ oligomer levels after 2 months of TEMT (n = 5), a sizable 5+ point improvement in their combined ADAS-cog score was present on Day60 versus Baseline.
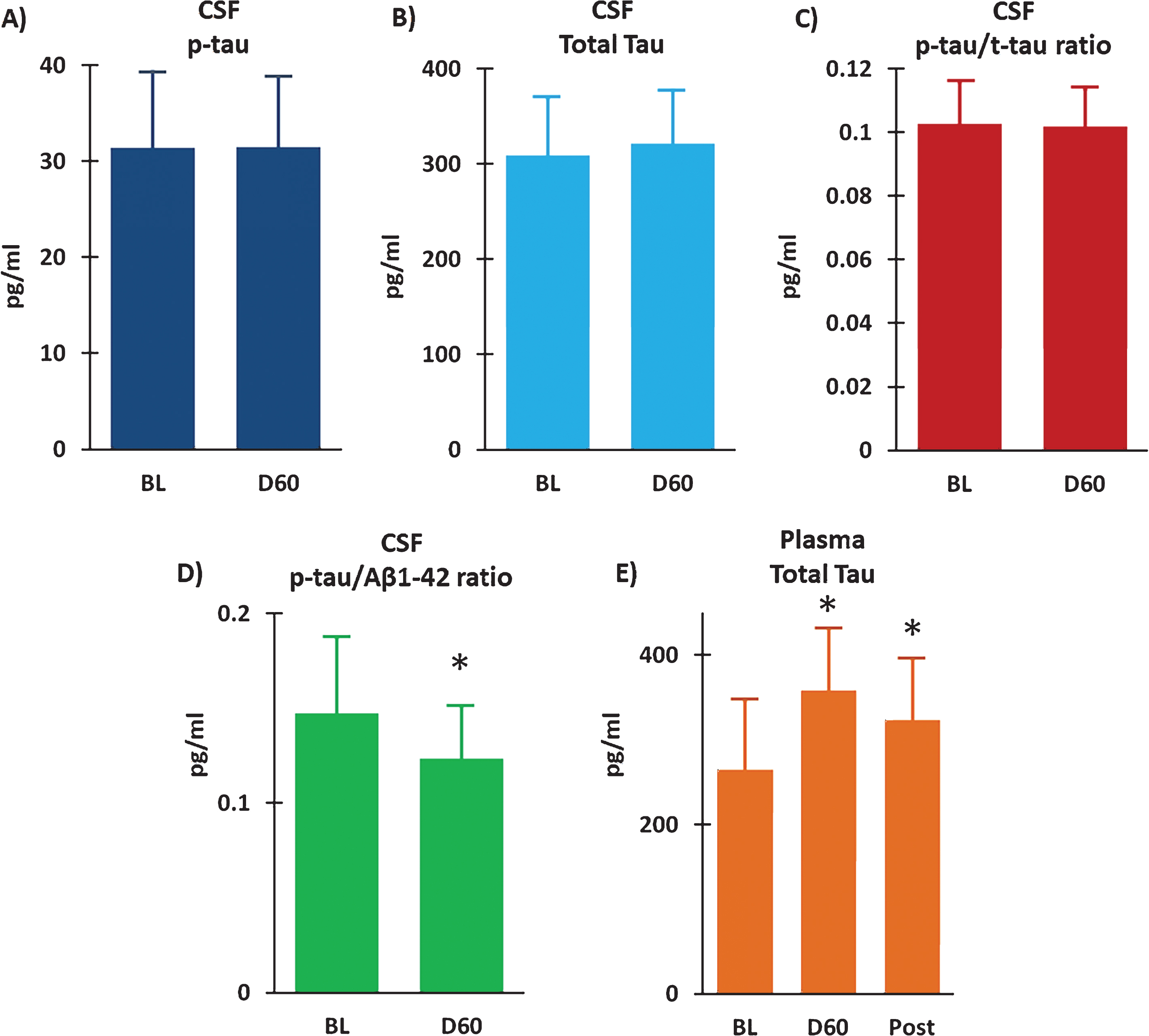
Measurement of t-tau and p-tau in CSF at BL and following 2 months of TEMT indicated no effect of treatment on either measure (Fig. 7A, B). As well, there was no change in the p-tau to t-tau ratio in CSF following 2 months of daily TEMT administration (Fig. 7C). However, there was a modest, but clinically important, reduction in both the CSF p-tau to Aβ1-42 ratio (ES = 0.61; p = 0.161) (Fig. 7D) and the CSF t-tau to Aβ1-42 ratio (ES = 0.52; p = 0.202) following TEMT. Similarly, measurement of t-tau levels in plasma revealed modest, but clinically important, increases in t-tau at both Day60 (ES = 0.56; p = 0.161) and 14D Post (ES = 0.51) (Fig. 7E).
Fig.7
Following 2 months of daily TEMT (D60), CSF levels of p-tau (A) and total tau (B) were unchanged compared to their Baseline (BL) levels. As well, there was no change in the p-tau to t-tau ratio in CSF as a result of TEMT administration (C). However, the p-tau to Aβ1-42 ratio was reduced (D) and total tau in plasma was increased (E) following 2 months of daily TEMT. Means±SEMs are presented. Post = 14Days Post-treatment. *ES significant at > 0.5 level versus Baseline.
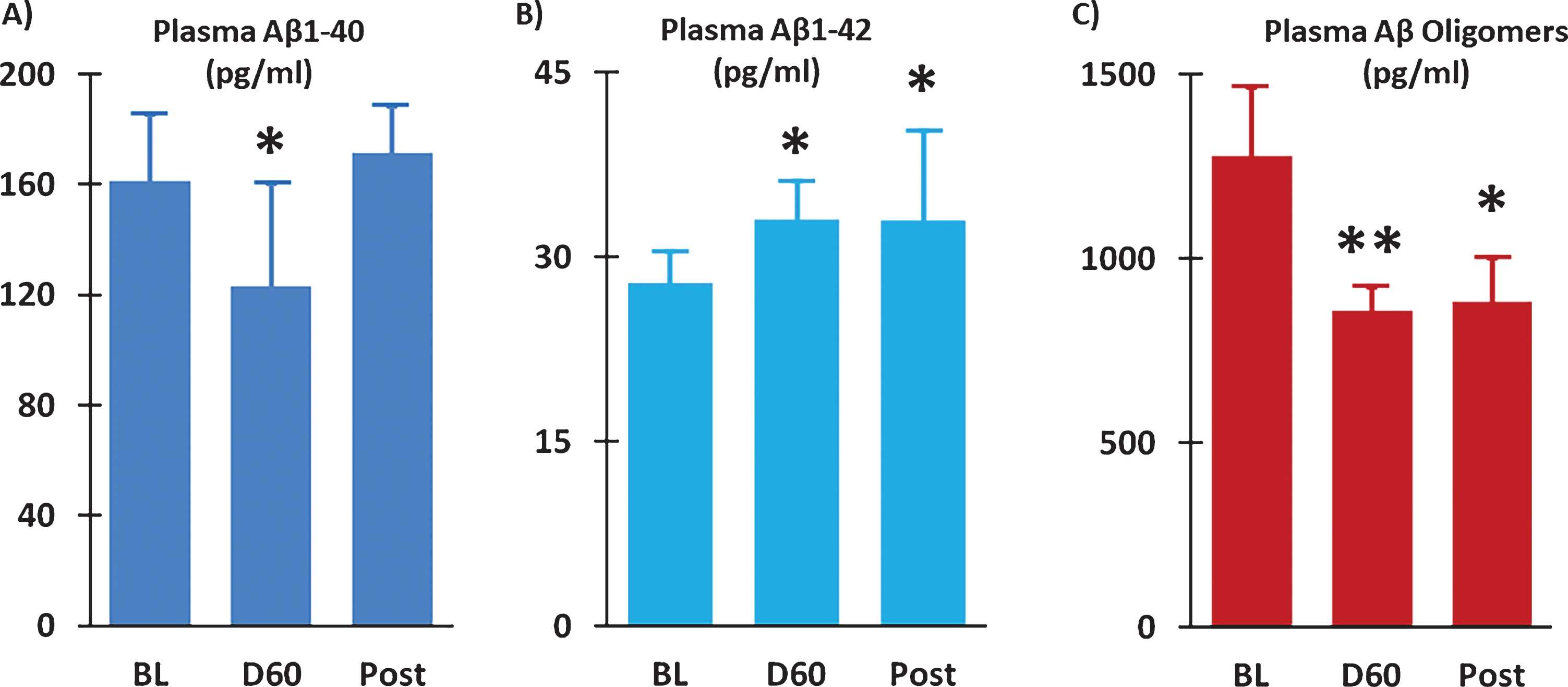
Two months of TEMT induced a 24% reduction in plasma soluble Aβ1-40 versus BL (ES = 0.54; p = 0.241) (Fig. 8A), with the percent reduction in individual subjects positively correlated with their improvement in ADAS-cog performance (r = 0.72; p < 0.05). Modest increases in plasma Aβ1-42 were present at both Day 60 (ES = 0.60; p = 0.166) and 14D Post (ES = 0.60; p = 0.205) compared to BL (Fig. 8B). A prominent effect of TEMT on plasma Aβ oligomers was evident, wherein a large clinically important 33% reduction was observed after 2 months of TEMT administration (ES = 1.16; p < 0.05) and maintained 14 days after treatment termination (ES = 0.73; p = 0.177).
Fig.8
Plasma Aβ1-40 levels were reduced (A) and plasma Aβ1-42 levels were increased (B) following 2 months of TEMT administration (D60) compared to Baseline (BL). Levels of Aβ oligomers in plasma were substantially reduced after TEMT (D60), as well as 14 days thereafter (Post). Means±SEMs are presented. *ES significant at > 0.5 level versus BL; **ES significant at > 0.8 level versus BL.
Separate from the above TEMT-induced effects within CSF and plasma were the presence of large concentration differences between CSF versus plasma for the various Aβ isoforms—this, irrespective of whether evaluated at BL or Day60. For soluble Aβ1-40, approximately 50-fold higher levels were present in CSF versus plasma. Similarly, a 6-fold higher level of soluble Aβ1-42 was evident in CSF versus plasma. By contrast, oligomeric Aβ levels were twice as high in plasma compared to CSF. For p-tau, levels were always higher in CSF because p-tau levels were essentially zero in plasma. Subjects exhibited no clear concentration difference for t-tau, with half of them having higher levels in CSF and the other half having higher levels in plasma.
AD subjects given 2 months of TEMT have stable cerebral metabolic rates for glucose
Progressive decreases in glucose utilization, often referred to as Cerebral Metabolic Rate for Glucose (CMRgl), are consistently seen in AD subjects in longitudinal scans separated by 12 months [44, 45]. However, we are not aware of any published FDG-PET study involving AD subjects with only a 2-month interval between scans (as in the present study), much less a change in CMRgl within that interval. With stability in CMRgl thus assumed to be the case in AD subjects over any given 2-month period, we anticipated a priori that only a decrease or increase in CMRgl (not simply stabilization) observed over the 2-month TEMT period would indicate a treatment effect. As such, FDG-PET scans were quantitatively evaluated in 109 forebrain areas for BL versus Day 60 (end of treatment) differences in CMRgl for each subject.
Analysis of BL versus Day 60 (end of treatment) CMRgl involving all 109 brain areas collectively and for all subjects combined indicated essentially no treatment-induced change in CMRgl (average percent change of –0.74±0.95% for Day 60 versus BL). Similarly, there was no treatment-induced change in CMRgl when only the 40 bilateral cortical regions (frontal, parietal, temporal, and occipital) were included (average percent change of –1.16±1.19% for Day 60 versus BL). Both of these percent changes are well below the 6% threshold set for a significant change in CMRgl at p < 0.05. For 7 of the 8 subjects, post-treatment FDG-PET brain scans revealed stable or enhanced (>6%) glucose utilization in at least 102 of the 109 brain regions.
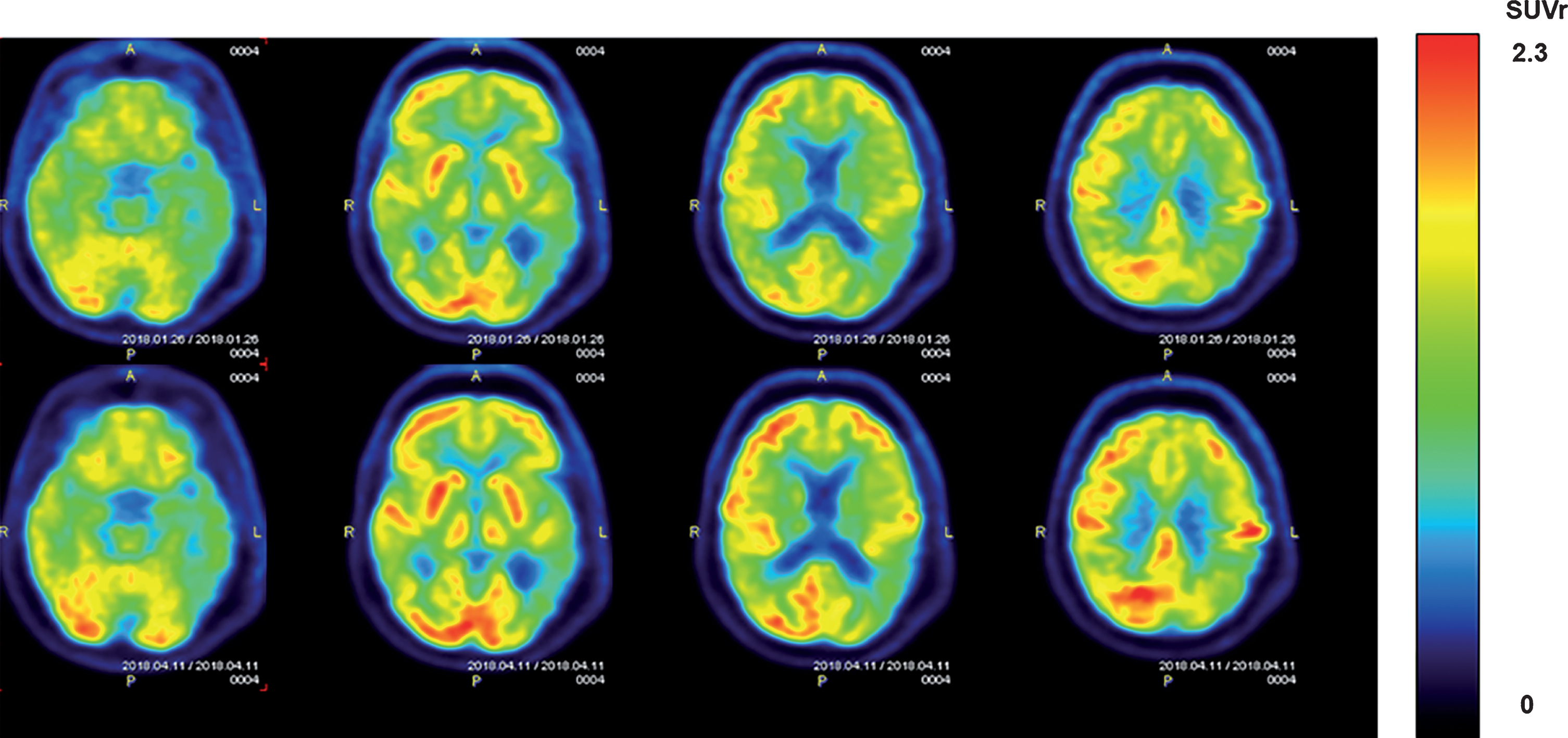
Visual comparison of pre- versus post-treatment FDG-PET horizontal sections for individual subjects revealed that most subjects exhibited no clear pre- versus post-treatment visual differences in CMRgl. This is consistent with the aforementioned quantification of forebrain/cortical areas and underscores stabilization of CMRgl over the 2-month TEMT period for most subjects in this trial. However, several subjects did exhibit a visual enhancement in CMRgl throughout the forebrain following 2 months of TEMT. Horizontal sections from one such subject are shown in Fig. 9. This subject also exhibited a “quantitative” increase of ≥6% in CMRgl within 45 of the 109 brain areas evaluated.
Fig.9
FDG-PET scans from an AD subject showing cerebral metabolic rate for glucose (CMGgl) in four horizontal levels. The upper scans were taken at baseline, with corresponding lower scans taken at the same level following 2 months of daily TEMT. Note higher FDG-PET intensity after TEMT throughout the forebrain (especially in the left hemisphere), as evidenced by more prevalent red/orange areas.
Although pre- versus post-treatment quantitative assessment of CMGgl did not indicate any treatment effects when CMGgl values from all subjects were combined, a significant correlation was present for individual subjects between their percent change in CMGgl for the 40 cortical regions collectively and performance in Rey AVLT 5-Trial Recall at 14 Days following completion of treatment (r = 0.802; p < 0.01); more positive percent changes in CMGgl following treatment were correlated with greater cognitive performance.
Areas of increased neuronal connectivity (DTI/FA) are present in the cingulate/cingulum following 2 months of TEMT in individual AD subjects
Tract-based Spatial Statistics (TBSS) provided a skeleton containing all major tracts common to all eight subjects (Fig. 10). Track-based statistical analysis for group differences between BL and Day60 (end of treatment) FA values indicated FA stability during the treatment period. Only a small group of voxels in the posterior cingulate/cingulate and corpus callosum exhibited significant FA enhancement or reduction (p < 0.05-corrected) collectively for the entire group of eight subjects (Fig. 10). This FA stability was also evident in sagittal, frontal, and horizontal planes of group results for voxel-by-voxel differences between BL and Day60; not a single voxel for all eight subjects collectively showed a significant enhancement or reduction in FA at p < 0.05 (data not shown). Further supportive of this overall stability in FA across all eight subjects was an ROI-based comparison of BL versus Day60 mean FA values in 12 a priori-selected brain ROIs (Table 3). Only one of the 12 brain ROIs showed a significant change in FA intensity for all subjects collectively at the end of the 2-month TEMT administration period.
Fig.10
Co-registration of DTI-derived fractional anisotropy (FA) images via tract-based spatial statistics (TBSS) resulted in a skeleton containing all major FA tracts common to all eight subjects, as shown in mid-sagittal, coronal, and horizontal sections. FA values were then compared in a voxel-by-voxel analysis for group differences between Baseline and Day60 (end of treatment). FA stability was evident during the treatment period, with only a small group of voxels in the posterior cingulate/cingulate and corpus callosum (arrows) exhibiting significant FA enhancement (red voxels) or FA reduction (blue voxels) for all eight subjects collectively (p < 0.05).
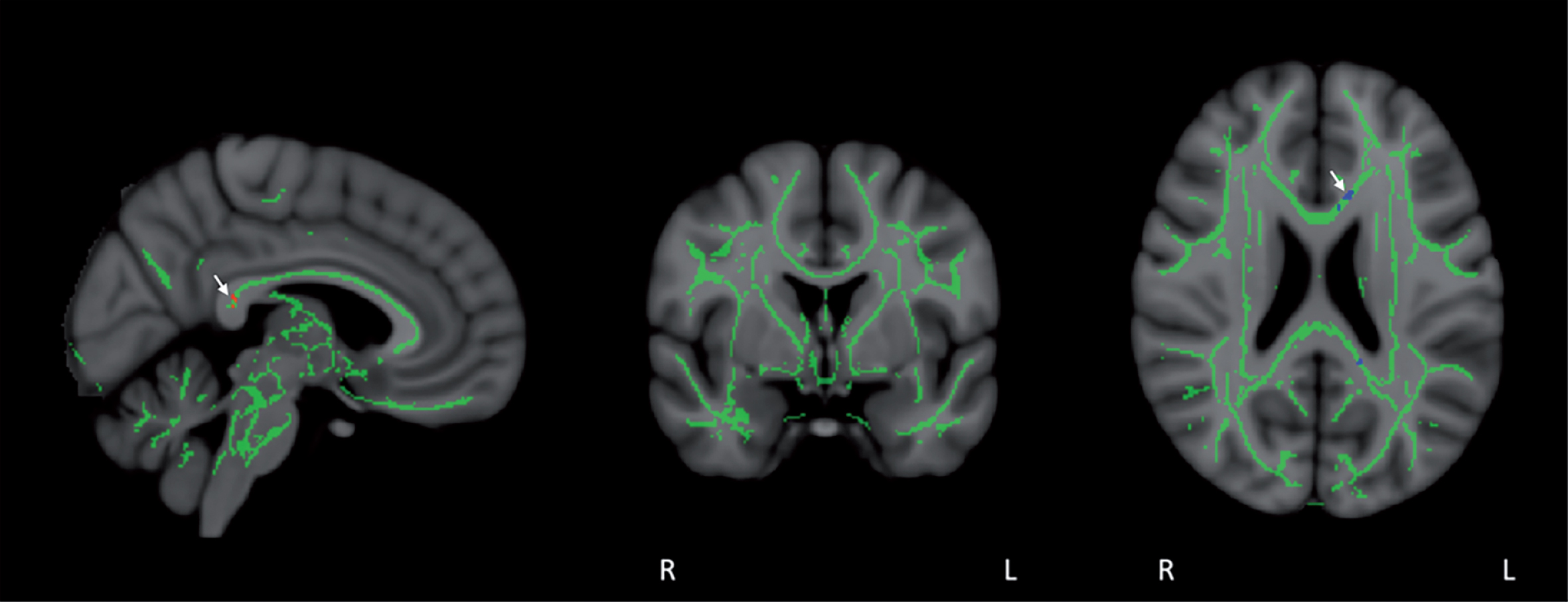
Table 3
Comparison of pre-treatment versus post-treatment mean fractional anisotropy (FA) fMRI in 12 a priori-selected brain regions of interest (ROI)
| Brain ROI | Pre-Treatment | Post-Treatment | “p” Value |
| Right Anterior Cingulate/Cingulum | 0.391±0.012 | 0.397±0.013 | 0.358 (n.s.) |
| Left Anterior Cingulate/Cingulum | 0.399±0.011 | 0.402±0.009 | 0.531 (n.s.) |
| Right Posterior Cingulate/Cingulum | 0.410±0. | 0.416±0.007 | 0.414 (n.s.) |
| Left Posterior Cingulate/Cingulum | 0.438±0.014 | 0.439±0.015 | 0.849 (n.s.) |
| Right Cingulum/Hippocampus | 0.319±0.007 | 0.321±0.007 | 0.650 (n.s.) |
| Left Cingulum/Hippocampus | 0.295±0.024 | 0.298±0.023 | 0.528 (n.s.) |
| Corpus Callosum | 0.551±0.015 | 0.546±0.016 | 0.389 (n.s.) |
| Fornix | 0.188±0.013 | 0.187±0.013 | 0.826 (n.s.) |
| Right Superior Long. Fasciculus | 0.442±0.006 | 0.439±0.006 | 0.239 (n.s.) |
| Left Superior Long. Fasciculus | 0.433±0.007 | 0.427±0.008 | 0.034 |
| Right Inferior Long. Fasciculus | 0.470±0.007 | 0.465±0.009 | 0.268 (n.s.) |
| Left Inferior Long. Fasciculus | 0.429±0.010 | 0.426±0.011 | 0.445 (n.s.) |
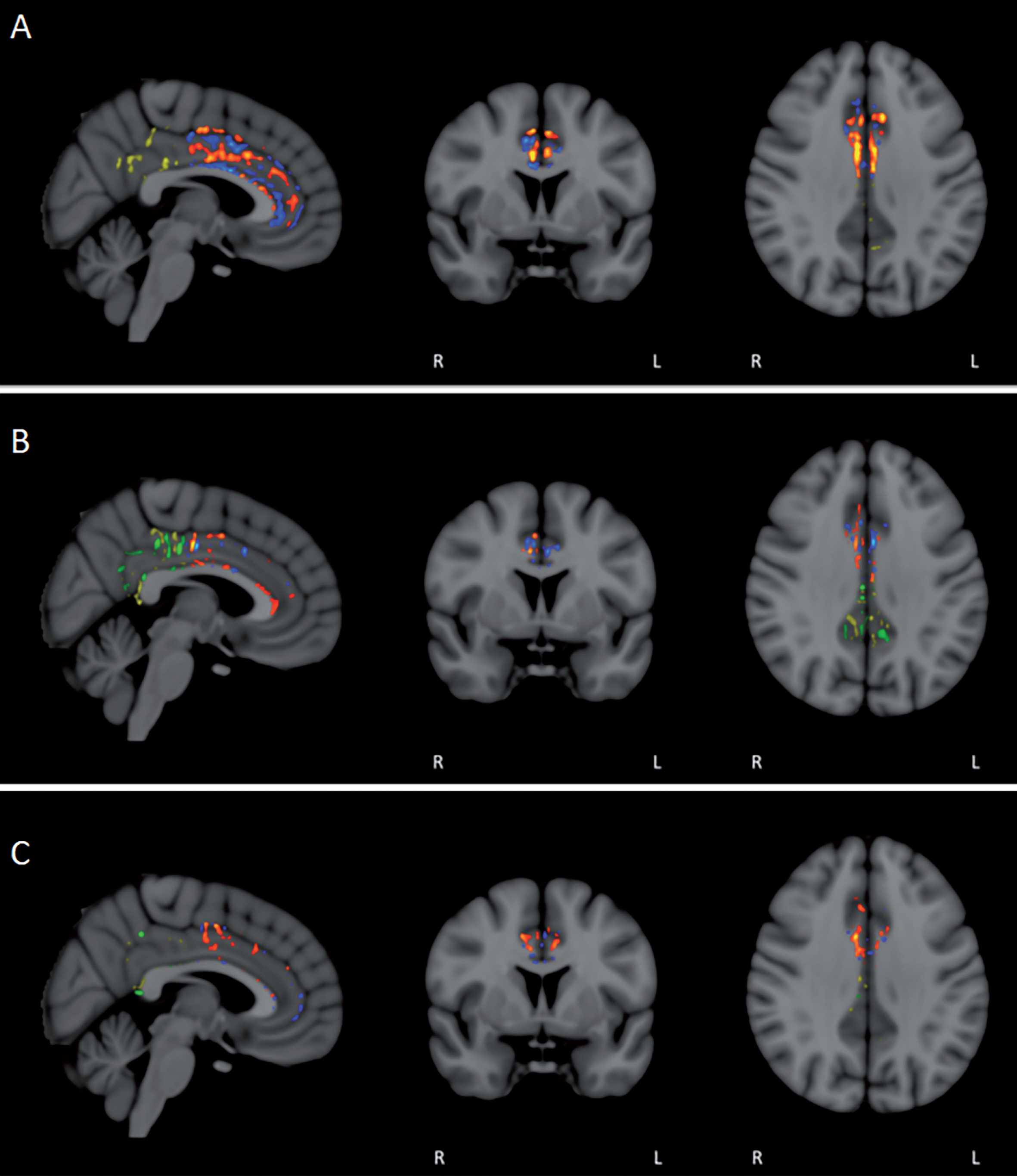
In contrast to the above FA analysis involving combined pre- versus post-treatment FA comparisons from all eight subjects, clear differences in FA were revealed when subjects were evaluated individually. Because of its important role for integrating cognitive processes and involvement in AD [46], the cingulate cortex/cingulum was pre-selected for doing this voxel-by-voxel comparison of baseline versus Day60 FA for each of the eight subjects. As anticipated from numerous studies reporting progressive reductions in FA throughout the brains of AD subjects even across only 3 months [47], areas of decreased FA (pre- versus post-treatment) were seen in both anterior and posterior cingulate cortex/cingulum (Fig. 11). However, all subjects also had unexpected regions of “enhanced” FA within the cingulate cortex/cingulum at the end of TEMT administration (Fig. 11). Most subjects exhibited prominent areas of enhanced FA in both the anterior and posterior cingulate cortex/cingulum (Fig. 11A, B), while a few subjects showed more pronounced FA enhancement in either the anterior (Fig. 11C) or posterior region.
Fig.11
FA/DTI analysis in individual subjects for pre- versus post-treatment differences in the anterior and posterior cingulate cortex/cingulum. Voxel-by-voxel analysis for significant differences (p < 0.05) revealed not only the expected areas of decreased FA in individual subjects (blue/green in anterior/posterior), but also prominent area of enhanced FA (red/yellow in anterior/posterior). For most subjects, enhanced FA was observed in both the anterior and posterior cingulate cortex/cingulum (A and B), although several subjects exhibited more pronounced FA enhancement in either the anterior (C) or posterior regions.
DISCUSSION
In view of the inability thus far of drugs to stabilize or reverse the memory impairment of AD, investigating non-pharmacologic interventions against the disease are now clearly warranted. Using a novel neuromodulatory approach against AD, Transcranial Electromagnetic Treatment (TEMT), we have consistently shown in our pre-clinical studies the ability of TEMT to prevent and reverse cognitive impairment in AD transgenic mice [23, 24, 27]. Based on our extensive pre-clinical data, the primary mechanisms for these cognitive benefits appear to be disaggregation of toxic Aβ and tau oligomers, along with associated mitochondrial enhancement [9, 23, Cao et al., unpublished data]. Utilizing a first-of-its-kind head device (the MemorEMTM), the present clinical study provides evidence for both the safety and efficacy of daily “in-home” TEMT administration to mild/moderate AD subjects by their caregivers. Results indicate no deleterious side effects during the 2-month treatment period and reversal of cognitive impairment in key tasks (e.g., ADAS-cog, Rey AVLT). In addition, changes in CSF levels of soluble/oligomeric Aβ following 2 months of TEMT are consistent with a TEMT-induced disaggregation of Aβ in the brain and brain imaging results suggest that TEMT may have the capacity to enhance brain/neuronal functionality. Although these results need to be confirmed and extended in a controlled ‘pivotal” trial, they suggest that TEMT may be a safe, non-invasive therapeutic to stabilize or reverse the memory impairment of AD.
Safety analysis
This is the first study to administer TEMT to the entire human brain over an extended period of time. As such, a primary goal of this open-label clinical trial was to determine the safety of daily, long-term, and global TEMT—specifically in AD subjects. No recurrent adverse/side effects were reported over the 2-month treatment period involving a total of 120 1-h treatments administered twice daily. Daily diaries kept by the subjects’ caregivers indicated no consistent changes in blood pressure or body temperature associated with TEMT, as well as no recurrent changes in daily activities or anxiety level/mood. Moreover, no adverse effects were ascertained at every clinical visit and no visible induction of tumors/microhemorrhages was evident when comparing pre- versus post-treatment anatomic MRI scans. Therefore, TEMT appears to be a safe therapeutic intervention for chronic treatment of AD, at least for the parameters and treatment regime utilized in this study. This is consistent with a large body of epidemiologic literature indicating that humans exposed to similar EMF frequencies/power levels via mobile phones exhibit no deleterious behavioral/physiologic effects and no induction of brain cancer, even after many years of EMF exposure ([26] for review).
Cognitive analysis
A battery of established AD cognitive tasks was utilized to evaluate the effects of 2-month daily TEMT administration on cognitive function in the eight AD subjects of this study at both completion of treatment and two weeks thereafter. Compared to baseline, averaged performance in the ADAS-cog was improved by over 4 points following 2 months of TEMT for 7 of the 8 subjects. Since a typical decline in ADAS-cog expected for AD subjects is around 4 points over a 12- to 15-month period [48], 2 months of TEMT appears to have reversed cognitive decline (as measured by the ADAS-cog) of responding AD subjects as a group, perhaps back to the cognitive level subjects had 12 to 15 months earlier. Cognitive improvement after treatment was particularly apparent for the “immediate recall” component of ADAS-cog. Similarly, performance in the analogous “5-trial recall” measure of the Rey AVLT was greatly improved following TEMT administration. Results from both of these measures indicate the ability of TEMT to reverse short-term/working memory impairment.
Of the two other components comprising the Rey AVLT, “Percent Forgetting” (or Retroactive Interference) was improved following TEMT, but not Proactive Interference. These improvements in Rey AVLT 5-trial recall and Retroactive Interference (but not Proactive Interference) were predicted from our prior AD transgenic mouse studies, wherein the same results were seen in the same three measures of an analogous cognitive interference task utilizing essentially the same TEMT parameters and testing regime [23]. A third task wherein improvement was seen following TEMT was in repeating digits. MMSE scores remained stable during/following treatment, which is not surprising in view of this task being used primarily as an AD diagnostic screen and not for assessment of AD progression. By contrast, since the ADAS-cog and Rey AVLT are benchmark tasks that are often used as indices of AD progression and/or therapeutic efficacy, the TEMT-induced improvements (not just stabilization) shown by AD subjects in these tasks are particularly noteworthy. For example, there is a strong relationship between impairment in both Rey AVLT 5-trial Recall and Percent Forgetting with extent of brain atrophy in AD [49]. Moreover, impairment in both of these Rey AVLT measures (which was reversed by TEMT) has been shown to be a reliable index for AD diagnosis [50–52].
It is important to indicate that cognitive testing was not done immediately after morning treatments at home, but rather starting around 2 hours thereafter and in the clinic. This extended beneficial period for hours following TEMT is consistent with our multiple studies in AD transgenic mice (utilizing essentially the same TEMT treatment regime) in which we demonstrated cognitive protection and impairment reversal with testing done 2–8 hours after morning treatment [23, 24, 27]. Underscoring an even more protracted cognitive benefit of TEMT in the present study, cognitive performance of AD subjects at 14 days following completion of TEMT often resulted in a similarly enhanced level of performance as immediately following TEMT completion. Thus, a general maintenance of cognitive improvement for weeks, even without continued daily treatment, may be resulting from the TEMT regime presently utilized.
The cognitive enhancements seen with TEMT administration are not likely due to repeated testing for a number of reasons. First, weekly ADAS-cog testing of control AD subjects for up to 5 weeks in each of 17 AD treatment studies usually showed a progressive decline or stabilization in ADAS-cog performance over those weeks (i.e., no improvement) [42]. Secondly, performance in Rey AVLT gets worse with progression of AD [49]. The Rey AVLT task, which targets episodic memory—an area particularly vulnerable to AD-related deficits—is likely to be especially resistant to practice effects, yet showed clear treatment benefits for the AD subjects of this study Third, repeated testing effects should have manifest themselves at the first repeat [53–55], but that was not the case for ADAS-cog repeated testing in this study. And fourthly, the tasks most likely to show a repeated testing-induced improvement (clock draw, Trails A and B) did not do so. Instead, stability in cognitive performance (versus baseline) was observed in those tasks, and in all other cognitive measures that did not exhibit cognitive improvement. Thus, either stability or cognitive improvement was observed for all cognitive tasks administered in this study. It should be noted that cognitive testing involving the Uniform Data Set (UDS), which was incorporated into all Alzheimer’s Disease Research Centers in 2005, showed that while both cognitively normal and mild cognitive impairment subjects exhibited significant practice effects on repeated cognitive testing, no significant practice effects were present for AD subjects [56].
Parenthetically, none of the eight subjects in the present study wanted to return their head device to the USF Health/Byrd Alzheimer’s Institute following completion of treatment because they felt TEMT was providing benefit to them. Based on the encouraging cognitive benefits observed in this clinical trial and the subjects’ collective enthusiasm to continue treatment, an extension study (ClinicalTrials.gov Protocol NCT03927040) is now in progress providing these same AD subjects with TEMT over an extended number of months. It is noteworthy that two academic groups in Italy have published uncontrolled clinical studies reporting that electromagnetic treatment quite different from the present study, nonetheless provided significant cognitive improvement to AD subjects in multiple standard tests [57, 58]. From a similar (pre-clinical) perspective, the cognitive benefits of TEMT that we initially reported in AD transgenic mice have since been confirmed by three international laboratories utilizing similar or different EMF parameters [59–61].
CSF/blood analysis
In CSF, Aβ1–42, t-tau, and p-tau have shown to be consistently altered in AD versus controls, while Aβ1-40 levels appear much less affected if at all by AD. In this regard, the current diagnostic criteria for AD includes a CSF profile of low Aβ1-42 in combination with high t-tau and/or high p-tau levels compared to controls, a pattern commonly referred to as the “AD signature” [62–65]. CSF levels of Aβ1-42 appear to be the earliest CSF marker to reflect AD-related pathology taking place in the brain [66], while t-tau has been suggested to be the best single biomarker for AD diagnosis based on meta-analysis studies [65]. Indeed, recent studies have concluded that CSF biomarkers can identify early AD with the same high accuracy as amyloid PET scans [67]. As another potential AD biomarker, high CSF levels of oligomeric Aβ have also been associated with AD [68–70], although some studies report significant overlap with controls [71]. These higher levels of CSF oligomeric Aβ in AD, in concert with the lower CSF levels of soluble (monomeric) Aβ1-42, have been suggested to be due to increased oligomerization of monomeric Aβ1-42 in AD [72].
In the present study, 2 months of TEMT resulted in increased CSF levels of soluble (almost entirely monomeric) Aβ1-40 and Aβ1-42, while CSF levels of oligomeric Aβ were unchanged overall compared to baseline. However, those subjects with increased oligomeric Aβ in CSF following TEMT administration had an average 5+ point improvement in their ADAS-cog scores. This TEMT-induced overall increase in soluble (monomeric) Aβ and selective increase in oligomeric Aβ in CSF (for the best cognitive responders) is consistent with a TEMT-induced dissociation of both oligomeric (soluble) and insoluble Aβ aggregates in the brain. Such dissociation would include not only dissociation of oligomeric Aβ inside neurons, but also dissociation of insoluble Aβ deposits in neuritic plaques into soluble oligomers/monomers with the resultant increase in brain Aβ monomers/oligomers making its way to the CSF. Supportive of this premise, we have shown that long-term daily TEMT administration to AD transgenic mice results in increased soluble (monomeric) Aβ in their brains [11, 23].
CSF levels of t-tau, p-tau, and the p-tau/t-tau ratio were not affected by 2 months of TEMT administration in the present study. This could indicate a greater stability of soluble p-tau aggregates in the brain (compared to Aβ aggregates), resulting in limited disaggregation by TEMT in the present study, and thus minimal/no change in the flux of soluble brain tau into the CSF. Along this line, our work involving TEMT administration to human AD brain homogenates suggests that higher EMF power may be necessary to impact p-tau aggregates [Cao et al., unpublished data]. Nonetheless, modest reductions in p-tau/ Aβ1-42 and t-tau/Aβ1-42 ratios occurred in the CSF following 2 months of TEMT. If confirmed in future studies, these reductions could be important in view of studies showing that both ratios are increased in early AD [67], that a higher CSF tau/Aβ1-42 ratio in aged controls results in increased risk of later AD [73], and that such ratios may be more effective than standalone markers in predicting brain Aβ deposition detected by PET imaging in AD subjects [67, 74]. Thus, the presently-reported reduction in CSF tau/Aβ1-42 ratios following TEMT administration may signify, to some extent, an effect on the AD pathologic process.
Concentrations of tau, p-tau, Aβ1-42, and Aβ1-40 in plasma are 10- and 100-fold lower than in CSF [64]. Subjects in the present study exhibited these large concentration differences between CSF and plasma for p-tau, Aβ1-42, and Aβ1-40, but not for t-tau. Such concentration differences have called into question the accuracy of conventional ELISAs that are routinely used, in part due to the 1000x greater concentration of proteins in plasma versus CSF that can interfere with ELISA-based assays and cause decreased assay sensitivity [64, 75, 76]. Not surprisingly then, plasma Aβ and tau levels have been inconsistent for demonstrating AD-related changes versus controls [77–80]. In the present study, plasma Aβ1-40 levels were modestly reduced following 2 months of TEMT, with the percent reduction in individual subjects correlated with their improvement in ADAS-cog. This suggests that plasma Aβ1-40 may provide at least a general index of cognitive benefit provided by TEMT. Importantly, plasma Aβ oligomer levels were reduced by 33% after 2 months of TEMT, which is noteworthy in view of multiple studies that have reported increased plasma levels of Aβ oligomers in AD subjects [81–83]. Exactly how TEMT provided this lowering effect on plasma Aβ oligomers is unknown, but could be related to the 2-fold higher level of Aβ oligomers in plasma versus CSF in the same AD subjects of this study.
Comparison of plasma t-tau levels in AD versus controls have been contradictory, with studies showing elevated levels in AD [64, 84], no difference [85], or reduced levels in AD [86]. Unlike CSF tau’s high diagnostic accuracy for AD [87], plasma tau alone would appear to be an inadequate AD biomarker. Nonetheless, the present study found that 2 months of TEMT resulted in a modest increase in plasma t-tau levels. Since soluble t-tau is almost totally monomeric tau, this increase may reflect a treatment-induced increase in monomeric t-tau within plasma.
It should be noted that CSF levels of p-tau for AD subjects in the present study were determined with the p-Tau231 detection kit. This kit reports considerably lower p-tau levels in AD subjects compared to the p-Tau181 detection kit. In that context, CSF p-tau levels for AD subjects in the present study (around 32 pg/ml at baseline) are very consistent with levels in AD subjects reported in other studies utilizing p-Tau231 detection [88] and well above the CSF p-tau levels for aged controls using p-Tau231 detection [89]. Similarly, the baseline average t-tau level in CSF for the present study’s AD subjects was 310±60 pg/ml. This is very consistent with CSF t-tau levels for AD subjects reported in other studies wherein the same t-tau ELISA kit was utilized [65, 90] and well above the 100 pg/ml or less seen for aged controls in these same studies. Thus, CSF levels of both p-tau and t-tau for the subjects of this study were consistent with the diagnosis of AD. Because various detection kits are utilized for both p-tau and t-tau, it is important to recognize that there are no hard cut offs or thresholds for p-tau or t-tau levels in CSF that identify AD subjects.
To summarize the blood/CSF results, 2 months of TEMT to AD subjects appears to have affected multiple AD markers in CSF and plasma. Although the meaning of these effects is presently open to interpretation, impacting such AD markers by TEMT suggests an effect on the AD pathologic process and reduces any chance of placebo effects in this open-label trial.
FDG-PET (glucose metabolism) analysis
This is the first study evaluating the effects of TEMT to the entire human forebrain on glucose (energy) metabolism therein. Brain glucose (energy) metabolism, or CMRgl, decreases years before the onset of AD cognitive impairment [91, 92] and continues to decline after AD diagnosis, as indicated by longitudinal FDG-PET scanning over intervals of 12 months or longer [44, 45]. However, we are aware of no studies that evaluated FDG-PET in established AD subjects over the relatively short 2-month interval of this study. Nonetheless, three prior studies suggested that we might observe enhanced CMRgl with TEMT administration to the AD subjects of the present study: 1) Daily electromagnetic field treatment to AD transgenic mice for 8 months increased brain FDG-PET signaling [61], 2) A single 50-min mobile phone exposure in normal humans (at EMF parameters similar to those in the present study) increased FDG-PET signaling in brain regions closest to the mobile phone’s ear location [93], and 3) Enhanced mitochondrial function was seen in cerebral cortex and hippocampus in aged AD Tg mice given 1 month of daily TEMT at the same treatment parameters as in the present study [11].
In the current study, however, stabilization (not enhancement) of CMRgl was observed in most subjects—not only for 109 forebrain areas evaluated collectively, but also for the 40 cortical areas evaluated collectively. Nonetheless, several AD subjects did have “visually” increased CMRgl throughout the forebrain following 2 months of TEMT, with one of these subjects also showing quantitative CMRgl increases (≥6%) in almost half of the 109 forebrain areas evaluated. The stabilization of CMGgl observed in most AD subjects could indicate either: 1) stabilization that would have been present in any event over the 2-month period, or 2) a treatment-induced stabilization that may have continued with a more protracted treatment period. In either case, the strong correlation between cortical CMRgl and Rey AVLT performance at 14 Days post-treatment is supportive that greater positive effects of TEMT on brain glucose utilization (CMGgl) were associated with better cognitive performance weeks thereafter.
DTI/FA analysis
In functional MRI, Fractional Anisotropy (FA) is a widely used measure of Diffusion Tensor Imaging (DTI) that is sensitive to changes in white matter integrity and neuronal connectivity [94]. Multiple studies have shown that brain FA consistently decreases as AD progresses [46, 95, 96], indicating a progressive decline in neuronal integrity/connectivity. Along this line, longitudinal studies have shown a progressive decline in brain FA with DTI scans taken every 12 months for 3 years [96] and FA decreases within posterior cingulum and corpus callosum have also been reported over a 12-month interval [85]. Even over a relatively short 3-month interval between DTI scans, decreases in FA within cingulate cortex/cingulum, fornix, and splenium have been reported for AD versus control subjects [47].
In the present study, DTI/FA was analyzed for TEMT effects in Tract-based Spatial Statistics (TBSS), wherein a skeleton was created that contains all major tracts common to all eight subjects. A voxel-by-voxel analysis within these major tracts indicated stabilization (no change) in FA for all eight subjects collectively across the 2-month treatment period. Similarly for all subjects combined, there was stabilization of FA across the treatment period for 12 pre-selected brain areas that were quantitatively analyzed. These results suggest either a TEMT-induced stabilization of FA or too short an interval (2 months) between DTI/FA scans to see spontaneous or treatment-induced changes in FA for all subjects collectively. However, when focusing on the cingulate cortex/cingulum and comparing each individual’s pre- versus post-treatment scans in a voxel-by-voxel comparison, regional enhancements in FA were seen in all subjects. Such FA enhancements following 2 months of TEMT, which have not been reported to occur spontaneously during AD progression, were observed in both the anterior and posterior cingulate cortex/cingulum. This is noteworthy because: 1) both the anterior and posterior cingulate cortex/cingulum are important for memory integration, working memory, and/or executive function [46], and 2) DTI/FA imaging of the cingulate cortex/cingulum shows progressive decreases in AD over periods as short as 3 months [47]. FA enhancements seen in individual subjects following TEMT suggest greater neuronal integrity/connectivity induced by treatment within discrete and subject-specific areas of this critical brain region for cognitive integration. These visually-evident enhancements in FA within sub-regions of cingulate/cingulum were apparently masked by evaluating the entire anterior or posterior cingulate/cingulum for all subjects collectively.
Mechanisms of TEMT action
The primary mechanism of TEMT action for providing cognitive benefits to AD subjects appears to be disaggregation of toxic “intraneuronal” Aβ and tau oligomers, which most recent studies show to initiate and promote AD development, not the insoluble Aβ in neuritic plaques or insoluble p-tau in NFTs) [4–10]. Our and others’ pre-clinical studies have repeatedly demonstrated the ability of TEMT to prevent Aβ aggregation and to disaggregate existing soluble/insoluble Aβ deposits in the brains of AD transgenic mice [11, 23, 24, 60] using essentially the same TEMT treatment parameters/regime as in the current clinical study. In our most recent studies using human AD brain homogenates, we have evidence that TEMT not only directly disaggregates oligomeric Aβ, but also can directly disaggregate both oligomeric tau and oligomeric α-synuclein [Cao et al., unpublished data]. Mounting evidence indicates that such disaggregation occurs through a destabilization of H-bonds between oligomer monomers through dipole-dipole inter-actions, vibration, and/or resonance phenomena [31–33, 97]. In this regard, electromagnetic/radiofrequency waves in the range used in our studies have been shown to cause reduced dipole-dipole interactions (dielectric loss), which leads to a decrease in inter-molecular H-bonding [98]. Indeed, the toxic protein β-sheet aggregates of Aβ, tau, and α-synuclein have a common backbone polarization that is stabilized via “two-electron” interactions of H-bond [99], a backbone that appears to be selectively disrupted by radiofrequency waves.
In addition to toxic protein disaggregation, we have also shown in AD transgenic mice that TEMT induces mitochondrial enhancement (e.g., increased ATP levels, membrane potential, and respiratory rates) through not only removal of toxic intra-mitochondrial Aβ, but also directly through Complex IV activation [11]. Moreover, we have shown that TEMT induces increases in neuronal activity within the entorhinal cortex of AD transgenic mice [27]. Unlike pharmacologic interventions against AD, TEMT can penetrate the blood-brain barrier and enter the brain’s neurons to provide toxic protein disaggregation, mitochondrial enhancement, and increased neuronal activity. Indeed, the ability of TEMT to directly disaggregate both Aβ and tau protein oligomers, and to also enhance mitochondrial function, provides a combination (cocktail) of AD therapeutic actions that has thus far not been possible for drug-based therapeutics. As was the case for AIDS, a multi-targeted therapeutic approach (such as that provided by TEMT alone or in combination with other therapies) will likely provide the best chance to stabilize or reverse the cognitive impairment of AD.
Study limitations
The main limitation of the present study, as with most open-label studies, is that all subjects received treatment (single-arm) without inclusion of an untreated/placebo group. However, the improvements in multiple cognitive measures observed with TEMT would have been highly unlikely to occur spontaneously in AD subjects, even with repeated testing (as discussed above). Moreover, it is difficult to explain away the CSF/plasma changes and DTI/FA localized enhancements, which are consistent with TEMT’s primary mechanisms of action to disaggregate toxic brain oligomers and enhance mitochondrial function.
A second limitation is the relatively small number of AD subjects (eight) in this study. In this regard, a small number of subjects (less than 10) has been typical of initial studies involving neuromodulatory approaches against AD such as tMS [100] and deep brain stimulation [101]. It is important to underscore that the diversity of safety, cognitive, CSF/blood AD marker, and brain imaging endpoints evaluated in the present study has not yet occurred for other neuromodulatory approaches being evaluated against AD. This study’s comprehensive design and the presence of both cognitive and non-cognitive benefits/effects strengthen the premise that TEMT had a real and meaningful impact in the study’s AD subjects.
A third limitation of this study is the relatively short (2-month) treatment period. Although 120 treatments were given over that period, the actual treatment period may have been too short for sizable decreases in glucose utilization (CGMgl) or DTI/FA to occur. As such, the stability in these brain imaging endpoints may be due to a short pre- versus post period rather than to TEMT-induced stabilization. Parenthetically, we were unable to find any longitudinal FDG-PET or DTI study involving AD subjects that had a 2-month interval between brain scans.
A final limitation of this study is that the degree of TEMT penetration into the brain is probably not as great as needed to more profoundly affect the forebrain and the end-points evaluated (particularly FDG-PET and DTI). Although the current MemorEMTM head device’s power levels does provide sub-cortical EMF penetration, a more powerful head device would likely provide more robust benefits on cognition, blood/CSF markers, and functional brain imaging in AD subjects.
Conclusion
In summary, TEMT administration to AD subjects appears to be safe, while providing cognitive enhancement, changes to CSF/blood markers, and evidence of stable/enhanced brain functionality. Although these promising results need to be replicated in controlled clinical trials, they suggest that TEMT may provide a vertical leap to an entirely new therapeutic intervention against AD, an intervention that is “disease-modifying”, non-invasive without apparent side effects, and is easily administered in-home.
CONFLICT OF INTEREST
The University of South Florida has a financial interest in NeuroEM Therapeutics, a company that provided all of the financial support for this clinical trial. The interest has been reviewed and managed by the University in accordance with its Institutional Conflict of Interest policy.
ACKNOWLEDGMENTS
NeuroEM Therapeutics, Inc. gratefully acknowledges the financial support of the Glass Charitable Foundation, Angel Investor Forum, and NINDS for this study and studies related to its scientific foundation. As well, we thank the clinical staff at the USF Health/Byrd Alzheimer’s Institute for their expert behavioral evaluation of this study’s subjects. We are also most appreciative of Dr. Richard Caselli, Dr. Huntington Potter, Dr. Ed Goodwin, and Eric Knight for their helpful review of this manuscript prior to submission.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/19-0367r1).
REFERENCES
[1] | Pardridge W ((2009) ) Alzheimer’s disease drug development and the problem of the blood-brain barrier. Alzheimers Dement 5: , 427–432. |
[2] | Gerson J , Kayed R ((2013) ) Formation and propagation of tau oligomeric seeds. Front Neurol 4: , 93. |
[3] | Cummings J , Lee G , Ritter A , Zhong K ((2018) ) Alzheimer’s disease drug development pipeline: 2018. Alzheimers Dement (N Y) 4: , 195–214. |
[4] | Pedersen J , Heegaard N ((2013) ) Analysis of protein aggregation in neurodegenerative disease. Anal Chem 85: , 4215–4227. |
[5] | Hayden E , Teplow D ((2013) ) Amyloid-β proteins and Alzheimer’s disease. Alzheimers Res Ther 5: , 60–71. |
[6] | Goure W , Krafft G , Jerecic J , Hefti F ((2014) ) Targeting the proper amyloid-beta neuronal toxins: A path forward for Alzheimer’s disease immunotherapeutics. Alzheimer Res Ther 6: , 42–56. |
[7] | Viola K , Klein W ((2015) ) Amyloid β oligomers in Alzheimer’s disease pathogenesis, treatment, and diagnosis. Acta Neuropathol 129: , 183–206. |
[8] | Guerrero-Muñoz M , Gerson J , Castillo-Carranza D ((2015) ) Tau oligomers: The toxic player at synapses in Alzheimer’s disease. Front Cell Neurosci 9: , 464. |
[9] | Fa M , Puzzo D , Piacentini R , Staniszewski A , Zhang H , Baltrons M , Li Puma D , Chatterjee I , Li J , Saeed F , Berman H , Ripoli C , Gulisano W , Gonzalez J , Tian H , Costa JA , Lopez P , Davidowitz E , Yu WH , Haroutunian V , Brown LM , Palmeri A , Sigurdsson EM , Duff KE , Teich AF , Honig LS , Sierks M , Moe JG , D’Adamio L , Grassi C , Kanaan NM , Fraser PE , Arancio O ((2016) ) Extracellular tau oligomers produce an immediate impairment of LTP and memory. Sci Rep 6: , 19393. |
[10] | Cline E , Bicca M , Viola K , Kirssten L ((2018) ) The amyloid-β oligomer hypothesis: Beginning of the third decade. J Alzheimers Dis 64: , S567–S610. |
[11] | Dragicevic N , Bradshaw P , Mamcartz M , Lin X , Wang L , Cao C , Arendash G ((2011) ) Long-term electromagnetic field treatment enhances brain mitochondrial function of both Alzheimer’s transgenic mice and normal mice: A mechanism for electromagnetic field-induced cognitive benefit? Neuroscience 185: , 135–149. |
[12] | Zussy C , Brureau A , Keller E , Marchal S , Blayo C , Delair B , Ixart G , Maurice T , Givalois L ((2013) ) Alzheimer’s Disease related markers, cellular toxicity, and behavioral deficits induced six weeks after oligomeric amyloid-beta peptide injection in rats. PloS One 8: , e53117. |
[13] | Park J , Choi H , Min J , Kim B , Lee S , Yun J , Choi M , Change K , Lee D ((2015) ) Loss of mitofusin 2 links beta-amyloid-mediated mitochondrial fragmentation and Cdk5-induced oxidative stress in neuron cells. J Neurochem 132: , 687–702. |
[14] | Zempel H , Thies E , Mankelkow E , Mandelkow E-M ((2010) ) Abeta oligomers cause localized calcium elevation, missorting of endogenous tau into dendrites, tau phosphorylation, and destruction of microtubules and spines. J Neurosci 30: , 11938–11950. |
[15] | Jin M , Shepardson N , Yang T , Chen G , Walsh D , Selkoe D ((2011) ) Soluble amyloid-beta protein dimers isolated from Alzheimer cortex directly induce tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci U S A 108: , 5819–5824. |
[16] | Zempel H , Mandelkow E ((2012) ) Linking amyloid-beta and tau: Amyloid-beta induced synaptic dysfunction via local wreckage of the neuronal cytoskeleton. Neurodegener Dis 10: , 64–72. |
[17] | Umeda T , Ramser E , Yamashita M , Nakajima K , Mori H , Silverman M , Tomiyama T ((2015) ) Intracellular amyloid-beta oligomers impair organelle transport and induce dendritic spine loss in primary neurons. Acta Neuropathol Commun 3: , 51–65. |
[18] | Takahashi R , Almeida G , Kearney F , Yu F , Lin M , Milner T , Gouras G ((2004) ) Oligomerization of Alzheimer’s beta-amyloid within processes and synapses of cultured neurons and brain. J Neurosci 24: , 3592–3599. |
[19] | Lasagna-Reeves C , Castillo-Carranza D , Kayed R ((2011) ) Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol Neurodegener 6: , 39–52. |
[20] | Tousi B , Pascual-Leone A , Sadowsky C , Sabbagh M , Agronin M , Alva G , Armon C , Bernick C , Keegan A , Karantzoulis S ((2017) ) Effects of a combined transcranial magnetic stimulation (TMS) and cognitive training in Alzheimer patients: Safety results of Medical Device Pivotal Multi-Center Study B. Neurodegener Dis 17: (Suppl 1), 526. |
[21] | Lozano A , Fosdick L , Chakravarty C , Leoutsako J-M , Munro C , Oh E , Drake K , Lyman C , Rosenberg P , Anderson W , Tang-Wai DF , Pendergrass JC , Salloway S , Asaad WF , Ponce FA , Burke A , Sabbagh M , Wolk DA , Baltuch G , Okun MS , Foote KD , McAndrews MP , Giacobbe P , Targum SD , Lyketsos CG , Smith GS ((2016) ) A Phase II study of fornix deep brain stimulation in mild Alzheimer’s disease. J Alzheimers Dis 54: , 777–787. |
[22] | Leoutsaka J , Yan H , Anderson W , Asaad WF , Baltuch G , Burke A ((2018) ) Deep brain stimulation targeting the fornix for mild Alzheimer dementia (the ADvance trial): A two year follow-up including results of delayed activation. J Alzheimers Dis 64: , 597–606. |
[23] | Arendash G , Sanchez-Ramos J , Mori T , Mamcarz M , Lin X , Runfeldt M , Wang L , Zhang G , Sava V , Tan J , Cao C ((2010) ) Electromagnetic field treatment protects against and reverses cognitive impairment in Alzheimer’s transgenic mice. J Alzheimers Dis 19: , 191–210. |
[24] | Arendash G , Mori T , Dorsey M , Gonzalez R , Tajiri N , Borlongan C ((2012) ) Electromagnetic treatment to old Alzheimer’s mice reverses β-amyloid deposition, modifies cerebral blood flow, and provides selected cognitive benefit. PLoS One 7: , e35751. |
[25] | Arendash G ((2012) ) Transcranial electromagnetic treatment against Alzheimer’s disease: Why it has the potential to trump Alzheimer’s disease drug development. J Alzheimers Dis 32: , 243–266. |
[26] | Arendash G ((2016) ) Review of the evidence that transcranial electromagnetic treatment will be a safe and effective therapeutic against Alzheimer’s disease. J Alzheimers Dis 53: , 753–771. |
[27] | Mori T , Arendash G ((2011) ) Electromagnetic field treatment enhances neuronal activity: Linkage to cognitive benefit and therapeutic implications for Alzheimer’s disease. J Alzheimers Dis Parkinsonism 1: , 2. |
[28] | Nisbet R , Polanco J-C , Ittner L , Gotz J ((2015) ) Tau aggregation and its interplay with amyloid-β. Acta Neuropathol 129: , 207–220. |
[29] | Larson M , Sherman M, Greimel S , Kuskowski M , Schneider J , Bennett D , Lesné S ((2012) ) Alpha-synuclein is a novel modulator of Alzheimer’s disease pathophysiology. J Neurosci 32: , 10253–10266. |
[30] | Robert H , Brown D ((2015) ) Seeking a mechanism for the toxicity of oligomeric alpha-synuclein. Biomolecules 5: , 282–305. |
[31] | English N , Solomentsev G , O’Brien P ((2009) ) Non-equilibrium molecular dynamics study of electric and low-frequency microwave fields on hen egg white lysozyme. J Chem Phys 131: , 035106. |
[32] | Gerner C , Haudek V , Schandl U, Bayer B , Gundacker N , Hutter H , Mosgoeller W ((2010) ) Increased protein synthesis by cells exposed to a 1800MHz RF mobile phone electromagnetic field. Int Arch Occup Environm Health 83: , 691–702. |
[33] | Todorova N , Bentveizen A , English N , Yarovsky I ((2016) ) Electromagnetic-field effects on structure and dynamics of amyloidogenic peptides. J Chem Physics 144: , 085101. |
[34] | Knopman D , Jack C , Wiste H , Lundt E , Weigand S , Vemuri P , Low V , Kantarci K , Gunter JL , Senjem ML , Mielke MM , Roberts RO , Boeve BF , Petersen RC ((2014) ) 18F-fluorodeoxyglucose positron emission tomography, aging, and apolipoprotein E genotype in cognitively normal persons. Neurobiol Aging 35: , 2096–2106. |
[35] | Jack C , Knopman D , Weigand S , Wiste H , Vemuri P , Lowe V , Kantarci K , Gunter J , Senjem M , Ivnik R , Roberts R , Rocca W , Boeve B , Petersen R ((2012) ) An operational approach to National Institute on Aging-Alzheimer’s Association criteria for preclinical Alzheimer disease. Ann Neurol 71: , 765–775. |
[36] | Jack C , Wiste H , Weigand S , Rocca W , Knopman D , Mielke M , Lowe V , Senjem M , Gunter J , Preboske G , Pankratz V , Vemuri P , Petersen R ((2014) ) Age-specific population frequencies of cerebral beta-amyloidosis and neurodegeneration among people with normal cognitive function aged 50-89 years: A cross-section study. Lancet Neurol 13: , 997–1005. |
[37] | Niemantsverdriet E , Ottoy J , Somers C , De Roeck E , Struyfs H , Soetewey F , Verhaeghe J , Van den Bossche T , Mossevelde S , Goeman J , De Deyn P , Marien P , Verijpt J , Sleegers K , Van Broeckhoven C , Wyffels L , Albert A , Cyssens S , Stoobants S , Staelens S , Bjerke M , Engelborghs S ((2017) ) The cerebrospinal fluid Aβ1-42/Aβ1-40 ratio improves concordance with amyloid-PET for diagnosing Alzheimer’s disease in a clinical setting. J Alzheimers Dis 60: , 561–576. |
[38] | Leuzy A , Carter S , Chiotis K , Aimkvist O , Wall A , Nordberg A ((2015) ) Concordance and diagnostic accuracy of [11C]PIB PET and cerebrospinal fluid biomarkers in a sample of patients with mild cognitive impairment and Alzheimer’s disease. J Alzheimers Dis 45: , 1077–1088. |
[39] | Xie S , Xiao J , Gong G , Zang Y , Wang Y , Wu H , Jiang X ((2006) ) Voxel-based detection of white matter abnormalities in mild Alzheimer disease. Neurology 66: , 1845–1849. |
[40] | Canu E , McLaren D , Fitzgerald M , Bendlin B , Zoccatelli G , Alessandrini F , Pizzini F , Ricciardi G , Beltramello A , Johnson S , Frisoni G ((2010) ) Microstructural diffusion changes are independent of macrostructural volume loss in moderate to severe Alzheimer’s disease. J Alzheimers Dis 19: , 963–976. |
[41] | Farivar S , Liu H , Hays R ((2004) ) Half standard deviastion estimate of the minimally important difference in HRQOL scores? Expert Res Pharmacoecon Outcomes Res 4: , 515–523. |
[42] | Sawilowsky S ((2009) ) New effect size rules of thumb. J Mod Appl Stat Methods 8: , 467–474. |
[43] | Rockwood K ((2004) ) Size of the treatment effect on cognition of cholinesterase inhibition in Alzheimer’s disease. J Neurol Neurosurg Psychiatry 75: , 677–685. |
[44] | Chen K , Langbaum J , Fleisher A , Ayutyanont N , Reschke C , Lee W , Liu X , Bandy D , Alexander GE , Thompson PM , Foster NL , Harvey DJ , de Leon MJ , Koeppe RA , Jagust WJ , Weiner MW , Reiman EM ((2010) ) Twelve-month metabolic declines in probable Alzheimer’s disease and amnestic mild cognitive impairment assessed using an empirically pre-defined statistical region-of-interest: Findings from the Alzheimer’s Disease Neuroimaging Initiative. Neuroimage 51: , 654–664. |
[45] | Landau S , Harvey D , Madison C , Koeppe R , Reiman E , Foster N , Weiner M , Jagusta W ((2011) ) Associations between cognitive, functional, and FDG-PET measures of decline in AD and MCI. Neurobiol Aging 32: , 1207–1218. |
[46] | Bubb E , Metzler-Baddeley C , Aggleton J ((2018) ) The cingulum bundle: Anatomy, function, and dysfunction. Neurosci Biobehav Rev 92: , 104–127. |
[47] | Nowrangi M , Lyketsos C , Leoutsakos J-M , Oishi K , Albert M , Mori S , Mielke M ((2013) ) Longitudinal, region-specific course of diffusion tensor imaging measures in mild cognitive impairment and Alzheimer’s disease. Alzheimers Dement 9: , 519–528. |
[48] | Podhorna J , Krahnke T , Shear M , Harrison J , Alzheimer’s Disease Neuroimaging Initiative ((2016) ) Alzheimer’s Disease assessment scale – Cognitive subscale variants in mild cognitive impairment and mild Alzheimer’s disease; change over time and the effect of enrichment strategies. Alzheimers Res Ther 8: , 8. |
[49] | Moradi E , Hallikainen I , Hanninen T , Tohka J ((2017) ) Rey’s Auditory Verbal Learning Test scores can be predicted from whole brain MRI in Alzheimer’s disease. Neuroimage Clin 13: , 415–427. |
[50] | Tierney M , Szalai J , Snow W , Fisher R , Nores A , Nadon G , Dunn E , George-Hyslop P ((1996) ) Prediction of probable Alzheimer’s disease in memory-impaired patients: A prospective longitudinal study. Neurology 46: , 661–665. |
[51] | Estévez-González A , Kulisevsky J , Boltes A , Otermín P , García-Sánchez C ((2003) ) Rey verbal learning test is a useful tool for differential diagnosis in the preclinical phase of Alzheimer’s disease: Comparison with mild cognitive impairment and normal aging. Int J Geriatr Psychiatry 18: , 1021–1028. |
[52] | Schoenberg M , Dawson K , Duff K , Patton D , Scott J , Adams R ((2006) ) Test performance and classification statistics for the Rey auditory verbal learning test in selected clinical samples. Arch Clin Neuropsychol 21: , 693–703. |
[53] | Vivot A , Power M , Glymour M , Mayeda E , Benitez A , Spiro A , Manly J , Proust-Lima C , Dufouil C , Gross A ((2016) ) Jump, hop, or skip: Modeling practice effects in studies of determinants of cognitive change in older adults. Am J Epidemiol 183: , 302–314. |
[54] | Benedict R , Zgalijardic D ((1998) ) Practice effects during repeated administrations of memory tests with and without alternate forms. J Clin Exp Neuropsychol 20: , 339–352. |
[55] | Jacobs D , Ard M , Salmon D , Galasko D , Bondi M , Edland S ((2017) ) Potential implications of practice effects in Alzheimer’s disease prevention trials. Alzheimers Dement (NY) 3: , 531–535. |
[56] | Mathews M , Abner E , Kryscio R , Jicha G , Cooper G , Smith C , Caban-Holt A , Schmitt F ((2014) ) Diagnostic accuracy and practice effects in the National Alzheimer’s Coordinating Center Uniform Data Set neuropsychological battery. Alzheimers Dement 10: , 675–683. |
[57] | Mannu P , Rinaldi S , Fontani V , Castagna A ((2011) ) Radio electric asymmetric brain stimulation in the treatment of behavioral and psychiatric symptoms in Alzheimer disease. Clin Interv Aging 6: , 207–211. |
[58] | Guerriero F , Botarelli E , Mele G , Polo L , Zoncu D , Renati P , Garlata C , Rollone M , Ricevuti G , Maurizi N , Francis M , Rondanelli M , Perna S , Guido D , Mannu P ((2015) ) An innovative intervention for the treatment of cognitive impairment – Emisymmetric bilateral stimulation improves cognitive functions in Alzheimer’s disease and mild cognitive impairment: An open-label study. Neuropsychiatr Dis Treat 11: , 2391–2404. |
[59] | Banaceur S , Banasr S , Sakly M , Abdelmelek H ((2013) ) Whole body exposure to 2.4GHz WIFI signals: Effects on cognitive impairment in adult triple transgenic mouse models of Alzheimer’s disease (3xTg-AD). Behav Brain Res 240: , 197–201. |
[60] | Jeong Y , Kang G , Kwon J , Choi H , Pack J , Kim N , Lee Y , Le H ((2015) ) 1950MHz electromagnetic fields ameliorate A-beta pathology in Alzheimer’s disease mice. Curr Alzheimer Res 12: , 481–492. |
[61] | Son Y , Kim J , Jeong Y , Jeong Y , Kwon J , Choi H-D ((2018) ) Long-term RF exposure on behavior and cerebral glucose metabolism in 5xFAD mice. Neurosci Lett 666: , 64–69. |
[62] | Blennow K , Dubois B , Fagan A , Lewczuk P , de Leon M , Hampel H ((2015) ) Clinical utility of cerebrospinal fluid biomarkers in the diagnosis of early Alzheimer’s disease. Alzheimers Dement 11: , 58–69. |
[63] | Lue L-F , Guerra A , Walker D ((2017) ) Amyloid beta and tau as Alzheimer’s disease blood biomarkers: Promise from new technologies. Neurol Ther 6: (Suppl 1), S25–S36. |
[64] | Lue L-F , Sabbagh M , Chiu M-J , Jing N , Snyder N , Schmitz C , Guerra A , Belden ((2017) ) Plasma levels of Aβ42 and tau identified probable Alzheimer’s dementia: Findings in two cohorts. Front Aging Neurosci 9: , 226. |
[65] | Han P , Serrano G , Beach T , Caselli R , Yin J , Zhuang N ((2017) ) A Quantitative analysis of brain soluble tau and the tau secretion factor. J Neuropathol Exp Neurol 76: , 44–51. |
[66] | Byerke M , Engelborghs ((2018) ) Cerebrospinal fluid biomarkers for early and differential Alzheimer’s disease diagnosis. J Alzheimers Dis 62: , 1199–1209. |
[67] | Palmgvist S , Zetterberg H, Mattsson N , Johansson P , Minthon L , Blennow K , Olsson M , Hansson O , Swedish BioFINDER Study Group ((2015) ) Detailed comparison of amyloid PET and CSF biomarkers for identifying early Alzheimer disease. Neurology 85: , 1240–1249. |
[68] | Fukumoto H , Tokuda T , Kasai T , Ishigami N , Hidaka H , Kondo M ((2010) ) High-molecular-weight β-amyloid oligomers are elevated in cerebrospinal fluid of Alzheimer patients. FASEB J 24: , 2716–2726. |
[69] | Gao C , Yam A , Wang X , Magdangal E , Salisbury C , Peretz D , Zuckermann RN , Connolly MD , Hansson O , Minthon L , Zetterberg H , Blennow K , Fedynyshyn JP , Allauzen S ((2010) ) Aβ40 oligomers identified as a potential biomarker for the diagnosis of Alzheimer’s disease. PLoS One 5: , e15725. |
[70] | Funke S ((2011) ) Detection of soluble amyloid-β oligomers and insoluble high-molecular-weight particles in CSF: Development of methods with potential for diagnosis and therapy monitoring of Alzheimer’s Disease. Int J Alzheimers Dis 2011: , 151645. |
[71] | Holtta M , Hansson O , Andreasson U , Hertze J , Minthon L , Naogga K ((2013) ) Evaluating amyloid-β oligomers in cerebrospinal fluid as a biomarker for Alzheimer’s disease. PLoS One 8: , e66381. |
[72] | Englund H , Gunnarsson M , Brundin R , Hedlund M , Kilander L , Lannfelt L , Pettersson FE ((2009) ) Oligomerization partially explains the lowering of Aβ42 in Alzheimer’s disease cerebrospinal fluid. Neurodegener Dis 6: , 139–147. |
[73] | Li G , Sokal I , Quinn J , Leverenz J , Brodey M , Schellenberg G , Kaye J , Raskind M ((2007) ) CSF tau/Abeta42 ratio for increased risk of mild cognitive impairment: A follow-up study. Neurology 69: , 631–639. |
[74] | Racine A , Koscik R , Nicholas C , Clark L , Okonkwo O , Oh J , Hillmer A , Murali D , Barnhart T , Betthauser T , Gallagher C , Rowley H , Dowling N , Asthana S , Bendlin B , Blennow K , Zetterberg H , Carlsson C , Christian B , Johnson S ((2016) ) Cerebrospinal fluid ratios with Abeta42 predict preclinical brain beta-amyloid accumulation. Alzheimers Dement (Amst) 2: , 27–38. |
[75] | Rissin D , Kan C , Song L , Rivnak A , Fishburn M , Shao Q , Piech T , Ferrell EP , Meyer RE , Campbell TG , Fournier DR , Duffy DC ((2013) ) Multiplexed single molecule immunoassays. Lab Chip 13: , 2902–2911. |
[76] | Blennow K , Zetterberg H ((2015) ) Understanding biomarkers of neurodegeneration: Ultrasensitive detection techniques pave the way for mechanistic understanding. Nat Med 21: , 217–219. |
[77] | Hampel H , Shen Y , Walsh D , Aisen P , Shaw L , Zetterberg H , Trojanowski JQ , Blennow K ((2010) ) Biological markers of amyloid beta-related mechanisms in Alzheimer’s disease. Exp Neurol 223: , 334–346. |
[78] | Chiu M , Yang S , Horng H , Yang C , Chen T , Chieh J , Chen HH , Chen TC , Ho CS , Chang SF , Liu HC , Hong CY , Yang HC ((2013) ) Combined plasma biomarkers for diagnosing mild cognition impairment and Alzheimer’s disease. ACS Chem Neurosci 4: , 1530–1536. |
[79] | Zetterberg H , Wilson D , Andreasson U , Minthon L , Blennow K , Randall J , Hansson O ((2013) ) Plasma tau levels in Alzheimer’s disease. Alzheimers Res Ther 5: , 9. |
[80] | Toledo J , Shaw L , Trojanowski J ((2013) ) Plasma amyloid beta measurements - a desired but elusive Alzheimer’s disease biomarker. Alzheimers Res Ther 5: , 8. |
[81] | Xia W , Yang T , Shankar G , Smith I , Shen Y , Walsh D , Selkoe D ((2009) ) A specific enzyme-linked immunosorbent assay for measuring β-amyloid protein oligomers in human plasma and brain tissue of patients with Alzheimer disease. Arch Neurol 66: , 190–199. |
[82] | Zhou L , Chan K , Chu L , Kwan J , Song Y , Chen L , Ho P , Cheng O , Ho J , Lam K ((2012) ) Plasma amyloid-β oligomers level is a biomarker for Alzheimer’s disease diagnosis. Biochem Biophys Res Commun 423: , 697–702. |
[83] | Wang M , Yi S , Han J-Y , Park S , Jang JJ-W , Chun I , Kim S ((2017) ) Oligomeric forms of amyloid-β protein in plasma as a potential blood-based biomarker for Alzheimer’s disease. Alzheimers Res Ther 9: , 98–107. |
[84] | Chiu M , Chen Y , Chen T , Yang SY , Yang FP , Tseng TW , Chieh JJ , Chen JC , Tzen KY , Hua MS , Horng HE ((2014) ) Plasma tau as a window to the brain-negative associations with brain volume and memory function in mild cognitive impairment and early Alzheimer’s disease. Hum Brain Mapp 35: , 3132–3142. |
[85] | Wang T , Xiao S , Liu Y , Lin Z , Su N , Li X , Li G , Zhang M , Fang Y ((2014) ) The efficacy of plasma biomarkers in early diagnosis of Alzheimer’s disease. Int J Geriatr Psychiatry 29: , 713–719. |
[86] | Sparks D , Kryscio R , Sabbagh M , Ziolkowski C , Lin Y , Sparks LM , Liebsack C , Johnson-Traver S ((2012) ) Tau is reduced in AD plasma and validation of employed ELISA methods. Am J Neurodegener Dis 1: , 99–106. |
[87] | Bloudek L , Spackman D , Blankenburg M , Sullivan S ((2011) ) Review and meta-analysis of biomarkers and diagnostic imaging in Alzheimer’s disease. J Alzheimers Dis 26: , 627–645. |
[88] | Spiegel J , Pirraglia E , Osorio R , Glodzik L , Li Y , Tsui W , Saint Lousi L , Randall C , Butler T , Xu J , Zinkowski R , Zetterberg H , Fortea J , Fossati S , Wisniewski T , Davies P , Blennow K , de Leon M ((2016) ) Greater specificity for cerebrospinal fluid P-tau231 over P-tau181 in the differentiation of healthy controls from Alzheimer’s disease. J Alzheimers Dis 49: , 93–100. |
[89] | Glodzik L , de Santi S , Tsui W , Mosconi L , Zinkowski R , Pirraglia E , Wang K , Li Y , Rich K , Zetterberg H , Blennow K , Mehta P , de Leon M ((2011) ) Phosphorylated tau 231, memory decline and medial temporal atrophy in normal elders. Neurobiol Aging 32: , 2131–2141. |
[90] | Takeda S , Commins C , DeVos S , Nobuhara C , Wegmann S , Roe A , Costantino I , Fan Z , Nichollis S , Sherman A , Trisini A , Scherzer C , Carlson G , Pitstick R , Peskind E , Raskind M , Li G , Montine T , Frosch M , Hyman B ((2016) ) Seed-competent HMW tau species accumulates in the cerebrospinal fluid of Alzheimer’s disease mouse model and human patients. Ann Neurol 80: , 355–367. |
[91] | Reiman E , Caselli R , Yun L , Chen K , Bandy D , Minoshima S , Thibodeau S , Osborne D ((1996) ) Preclinical evidence of Alzheimer’s disease in persons homozygous for the epsilon 4 allele for apolipoprotein. N Engl J Med 334: , 752–758. |
[92] | Reiman E , Chen K , Alexander G , Caselli R , Bandy D , Osborne D , Saunders A , Hardy J. ((2004) ) Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc Natl Acad Sci U S A 101: , 284–289. |
[93] | Volkow N , Tomasi D , Wang G , Vaska P , Fowler J , Telang F , Alexoff D , Logan J , Wong C ((2011) ) Effects of cell phone radiofrequency signal exposure on brain glucose metabolism. JAMA 305: , 808–813. |
[94] | Nir T , Jahanshada N , Villaon-Reina J , Toga A , Jack C , Weiner M ((2013) ) Effectiveness of regional DTI measures in distinguishing Alzheimer’s disease, MCI, and normal aging. Neuroimage Clin 3: , 180–195. |
[95] | Acosta-Cabronero J , Nestor P ((2014) ) Diffusion tensor imaging in Alzheimer’s disease: Insights into the limbic-diencephalic network and methodological considerations. Front Aging Neurosci 6: , 266. |
[96] | Chitra R , Bairavi K , Vinisha V , Kavitha A ((2017) ) Analysis of structural connectivity on progression of Alzheimer’s disease using diffusion tensor imaging. 2017 Fourth International Conference on Signal Processing, Communication and Networking (ICSCN). |
[97] | Blank M , Goodman R ((2004) ) Initial interactions in electromagnetic field-induced biosynthesis. J Cell Physiol 199: , 359–363. |
[98] | Segwa R , Kaur K ((1999) ) Microwave absorption in oligomers of ethylene glycol. Indian J Biochem Biophys 36: , 325–329. |
[99] | Ciepak A ((2017) ) Protein folding, misfolding, and aggregation: The importance of two-electron stabilizing interactions. PLoS One 12: , e0180905. |
[100] | Bentwich J , Dobronevsky E , Aichenbaum S , Shorer R , Peretz R , Khaigrekht M , Marton RG , Rabey JM ((2011) ) Beneficial effect of repetitive transcranial magnetic stimulation combined with cognitive training for the treatment of Alzheimer’s disease: A proof of concept study. J Neural Transm 118: , 463–471. |
[101] | Laxton A , Tang-Wai D , McAndrews M , Zumsteg D , Wennberg R , Keren R , Wherrett J , Naglie G , Hamani C , Smith G , Lozano A ((2010) ) A phase I trial of deep brain stimulation of memory circuits in Alzheimer’s disease. Ann Neurol 68: , 521–534. |


