
Phospho-Tau Changes in the Human CA1 During Alzheimer’s Disease Progression
Abstract
Despite extensive studies regarding tau phosphorylation progression in both human Alzheimer’s disease cases and animal models, the molecular and structural changes responsible for neurofibrillary tangle development are still not well understood. Here, by using the antibodies AT100 (recognizes tau protein phosphorylated at Thr212 and Ser214 in the proline-rich region) and pS396 (recognizes tau protein phosphorylated at serine residue 396 in the C-terminal region), we examined phospho-tau immunostaining in neurons from the hippocampal CA1 region of 21 human cases with tau pathology ranging from Braak stage I to VI. Our results indicate that the AT100/pS396 ratio decreases in CA1 in accordance with the severity of the disease, along with its colocalization. We therefore propose the AT100/pS396 ratio as a new tool to analyze the tau pathology progression. Our findings also suggest a conformational modification in tau protein that may cause the disappearance of the AT100 epitope in the late stages of tau pathology, which may play a role in the toxic tangle aggregation. Thus, this study provides new insights underlying the stages for the formation of neurofibrillary tangles in Alzheimer’s disease.
INTRODUCTION
Neurofibrillary tangles (NFTs), together with amyloid-β (Aβ) plaques, are the major neuropathological hallmarks of Alzheimer’s disease (AD). Symptoms in early stages of the disease include loss of context, disorientation, and deficits in memory functions [1]. It has also been proposed that the early loss of episodic memory is closely associated with the progressive degeneration of the medial temporal lobe structures [2]. Damage to the hippocampus causes profound impairments in long-term episodic memory [3–5], suggesting a central role of the hippocampus in learning and memory. In particular, the CA1 field has been proposed as a relevant region for memory consolidation, since it represents a gateway of information between the hippocampus and cortico/subcortical areas [6, 7]. In support of this, in AD cases, the CA1 usually presents a clear atrophy when compared to the non-demented brains, as well as a reduction in the number of neurons and synapses, which correlates with cognitive decline [8–12]. Recently, it has been proposed that features of the CA1 are key indicators of memory dysfunction in amnestic mild cognitive impairment [13]. In this regard, it has been suggested that NFTs are most closely correlated with cognitive decline, promoting neuronal morphology impairment [14].
The main components of NFTs are the paired helical filaments (PHFs), composed of aggregates of abnormally hyperphosphorylated tau protein. In AD, anomalous tau phosphorylation first appears in the transentorhinal and entorhinal cortices; it then spreads to the hippocampus and, eventually, extends to the neocortex [15]. The monomeric form of tau protein promotes assembly and stabilization of microtubules [16], playing a crucial role in the cytoskeleton maintenance. However, once tau is hyperphosphorylated, it decreases microtubule binding affinity, inhibiting the assembly of microtubules and changing the interaction with plasma membranes. Some of the consequences of these modifications are the impairment of axonal transport, as well as differential dendritic and post-synaptic tau distribution, among others [17]. Recently, new harmful roles have been associated with pathological tau, such as targeting microglia to promote neuron phagocytosis [18] or the impairment of nucleocytoplasmic transport, which would affect the integrity and functioning of the nucleus of the neurons [19].
Tau protein contains over 80 potential tau phosphorylation sites that may alter the protein structure and promote conformational changes, which in turn may allow its aggregation [20, 21]. The microtubule binding domain (a repeat region at the C-terminus) is critical for PHF assembly while, in the proline-rich region, the phosphorylation of T212/S214 residues decreases the interaction with microtubules [22] and prevents PHF formation [23]. These two phosphorylated amino acids form the epitope of the antibody AT100 [24].
It has been proposed that the sequential phosphorylation of tau protein is linked to NFT development and the progression of AD [25]. Phosphorylation at Ser202/Thr205 (epitope recognized by the antibody AT8) represents an early degenerative change of the cytoskeleton [26, 27], which plays a central role in the successive tau phosphorylation [28] and has been associated with early stages of the AD [29, 30]. AT8 is used to classify the neurofibrillary degeneration into stages known as the Braak stages [31]. Conversely, phosphorylation at site Ser396 is commonly related to late stages, as it is mostly found in NFTs [29]. However, another study reported that tau phosphorylation at S396/S404 is one of the earliest events in AD [32], along with the truncation of the C-terminal region [33, 34]. In addition, Luna-Muñoz et al. have characterized the sequential appearance of specific tau phospho-dependent epitopes, revealing that AT100 appears after AT8 [34]. In general, although these studies suggest that toxic tau aggregates are formed by a cooperative and sequential tau hyperphosphorylation [35], further research is needed to understand which events are crucial to promote neurodegeneration.
In the present study, we have used the antibodies AT100 and pS396 which are directed against different phospho-tau sites to examine the phospho-tau staining in neurons from the human hippocampal CA1 region. For this purpose, we have studied human brain samples from individuals with tau pathology with a neurofibrillary tangle Braak stage ranging from I to VI. Our findings revealed that the AT100/pS396 ratio decreased in CA1 and that this decrease is in accordance with the severity of the disease. Furthermore, the lower AT100/pS396 ratio was in parallel with a decrease in the colocalization of immunostaining using AT100 and pS396 antibodies.
MATERIALS AND METHODS
Human brain tissue samples were obtained from two different sources: Banc de Teixits Neurologics from Hospital Universitari Clinic de Barcelona (Dr. I. Ferrer, Servicio de Anatomía Patológica, IBIDELL-Hospital Universitario de Bellvitge, Barcelona, Spain) and the Banco de Tejidos Fundación CIEN (Dr. A. R
Table 1
Pathological and technical information of the 21 cases from which the brain samples were obtained. We have used an internal code to ensure the confidentiality of each sample. LBD, Lewy body disease; HS, hippocampal sclerosis; Syn, synuclein; –, not available
| Cases | Age (y) | Gender | Postmortem delay (h:min) | Braak stage | CERAD stage | Evidence of cognitive impairment or dementia | Additional Neurological diagnosis |
| IF9 | 72 | Male | 3 : 30 | I | None | NO | Multi-infarct encephalopathy; Argyrophilic grain disease stage II |
| VK2 | 87 | Male | 4 | II | A | YES | Small Vessel Disease. Signs of ischemia-acute global anoxia, with infarction in the left hippocampus |
| IF8 | 91 | Male | 2 : 30 | III | A | NO | Argyrophilic grain disease stage III, neuronal ballooning |
| VK21 | 78 | Female | 3 : 30 | III | None | YES | Vascular alterations |
| BCN11 | 94 | Female | 5 : 45 | III | None | YES | Associated Parkinsonism |
| BCN15 | 64 | Female | 3 | III | None | YES | LBD; Small Vessel Disease |
| AB5 | 59 | Male | 4 | III | None | NO | – |
| AB6 | 92 | Female | 4 | III | A | NO | Aging-related tau astrogliopathy |
| BCN4 | 87 | Female | 5 : 30 | IV | B | YES | TDP43; HS |
| BCN2 | 82 | Female | 2 | V | C | YES | αSyn (amygdala); HS |
| BCN6 | 76 | Female | 5 | V | C | YES | TDP43 (limbic); HS |
| BCN10 | 80 | Female | 5 | V | C | YES | Meningioma |
| VK15 | 86 | Male | 2 : 30 | V | C | YES | Vascular alterations; HS |
| VK27 | 95 | Female | 4 | V | B | YES | LBD (amygdala) |
| VK28 | 86 | Female | 5 : 30 | V | C | YES | LBD (limbic) |
| TDP43 (hippocampus) | |||||||
| BCN1 | 90 | Male | 4 : 30 | VI | C | YES | Microinfarctions |
| BCN7 | 89 | Female | 4 : 15 | VI | C | YES | TDP43 (amygdala) |
| BCN9 | 84 | Female | 5 | VI | C | YES | TDP43 (limbic); αSyn (orbitofrontal); HS |
| BCN12 | 74 | Female | 3 : 30 | VI | C | YES | Small Vessel Disease |
| BCN13 | 83 | Male | 2 : 30 | VI | C | YES | Vascular encephalopathy; LBD (amygdala) |
| VK16 | 88 | Female | 2 | VI | C | YES | Vascular alterations; LBD; HS |
Tissue blocks were fixed in cold 4% paraformaldehyde (Sigma-Aldrich, St. Louis, MO, USA) in 0.1 M sodium phosphate buffer (PB; Panreac, 131965, Spain) pH 7.4 for 24-48 h. After fixation, the tissue was washed in PB, cryoprotected (30% sucrose solution for at least 24 h) and frozen at –20°C for longer preservation. Serial 50 μm thick coronal sections from the hippocampus were obtained using a microtome (MICROM, HM450; freeze unit, KS34, Thermo Fischer Scientific, Waltham, MA, USA).
Selected sections were first rinsed in 0.1 M PB, pretreated in 2% H2O2 for 30 min to remove endogenous peroxidase activity, and then incubated for 1 h at room temperature in a solution of 3% normal horse serum (Vector Laboratories Inc., Burlingame, CA) and 0.25% Triton-X (Merck, Darmstadt, Germany). Subsequently, sections were incubated for 48 h at 4°C in the same solution with mouse IgG1-anti-phospho-tau, clone AT8 (1 : 2000, MN1020, Thermo Scientific), mouse anti-human Aβ antibody (1 : 50, clone 6F/3D; Dako, Glostrup, Denmark) or rabbit anti phospho (409/410) TDP43 (1 : 500; 22309-1-AP; Proteintech, Chicago, IL, USA). Sections were then processed with a secondary biotinylated horse anti-mouse IgG antibody (1 : 200, Vector Laboratories), and then incubated for 1 h in an avidin-biotin peroxidase complex (Vectastain ABC Elite PK6100, Vector) and, finally, with the chromogen 3,3′-diaminobenzidine tetrahydrochloride (DAB; Sigma-Aldrich). Thereafter, sections were dehydrated, cleared with xylene and cover-slipped.
To study tau phosphorylation colocalization, double immunostainings were performed. Sections were first rinsed in 0.1 M PB, pretreated in 2% H2O2 for 30 min, and then incubated for 1 h at room temperature in a solution of 0.25% Triton-X (Merck) with 3% normal goat serum (Vector Laboratories Inc.). The sections were then incubated for 48 h at 4°C with the primary antibodies: mouse anti tau (phospho Thr212/Ser214; AT100) (1 : 500, MN1060, Thermo Scientific) and rabbit anti-Tau (PHF-Tau clone PHF1 pS396) (1 : 2000, 44752 G, Invitrogen Corp., Carlsbad, CA, USA). Afterwards, sections were rinsed in 0.1 M PB and incubated for 2 h at room temperature in different solutions containing a goat anti-rabbit antibody coupled to Alexa Fluor 594 and a goat anti-mouse coupled to Alexa Fluor 488 (1 : 1000; Molecular Probes, Eugene, OR, USA) along with DRAQ5 (1 : 5000; ab108410 Abcam, Cambridge, UK), a far-red fluorescent DNA dye used to label the cell nucleus [36]. The sections were finally washed and mounted with ProLong Gold Antifade Reagent (Invitrogen Corp.).
As fixation could affect the biochemical properties of proteins, such that the antigen of interest is masked, we performed antigen retrieval before immunostaining to confirm that the epitopes were still recognizable by our primary antibodies. For that purpose, several sections were incubated in a 10 mM sodium citrate buffer (pH 8.5) for 30 min at 80°C before incubation in 2% H2O2.
Image acquisition and analysis
Immunofluorescence sections were examined with a Zeiss LSM 710 confocal laser scanning system (Carl Zeiss Microscopy GmbH, Jena, Germany). They were recorded at 0.35 μm (63x objective) or 0.5 μm intervals (40x objective) through separate channels, and ZEN 2012 software (Zeiss) was then used to construct composite images from each optical series by combining the images recorded through the different channels (image resolution 1024×1024 pixels; pixel size 188 nm). Z depth in every confocal stack was 40 μm. We scanned at least 6 image stacks from the whole CA1 region from each case (at least one section for each tissue sample). We manually quantified the number of the colocalizing neurons (somas) by analyzing each confocal plane.
Measurements of Aβ plaques in CA1 area (mm2) were performed using Neurolucida software (MicroBrightField, Williston, USA). We have used one coronal section from each case. Normality and homoscedasticity of mean values obtained were analyzed with the Shapiro–Wilk and Levene tests, respectively. As normality and homoscedasticity criteria were satisfied, we used a paired t test to compare between the AT100/pS396 ratios and colocalization percentage mean values obtained in low (I−III) and high (V−VI) Braak Stage (GraphPad Prism version 5.00 for Windows, GraphPad Software, La Jolla, CA, USA, http://www.graphpad.com). The results are presented as mean±standard deviation of the mean (SD).
RESULTS
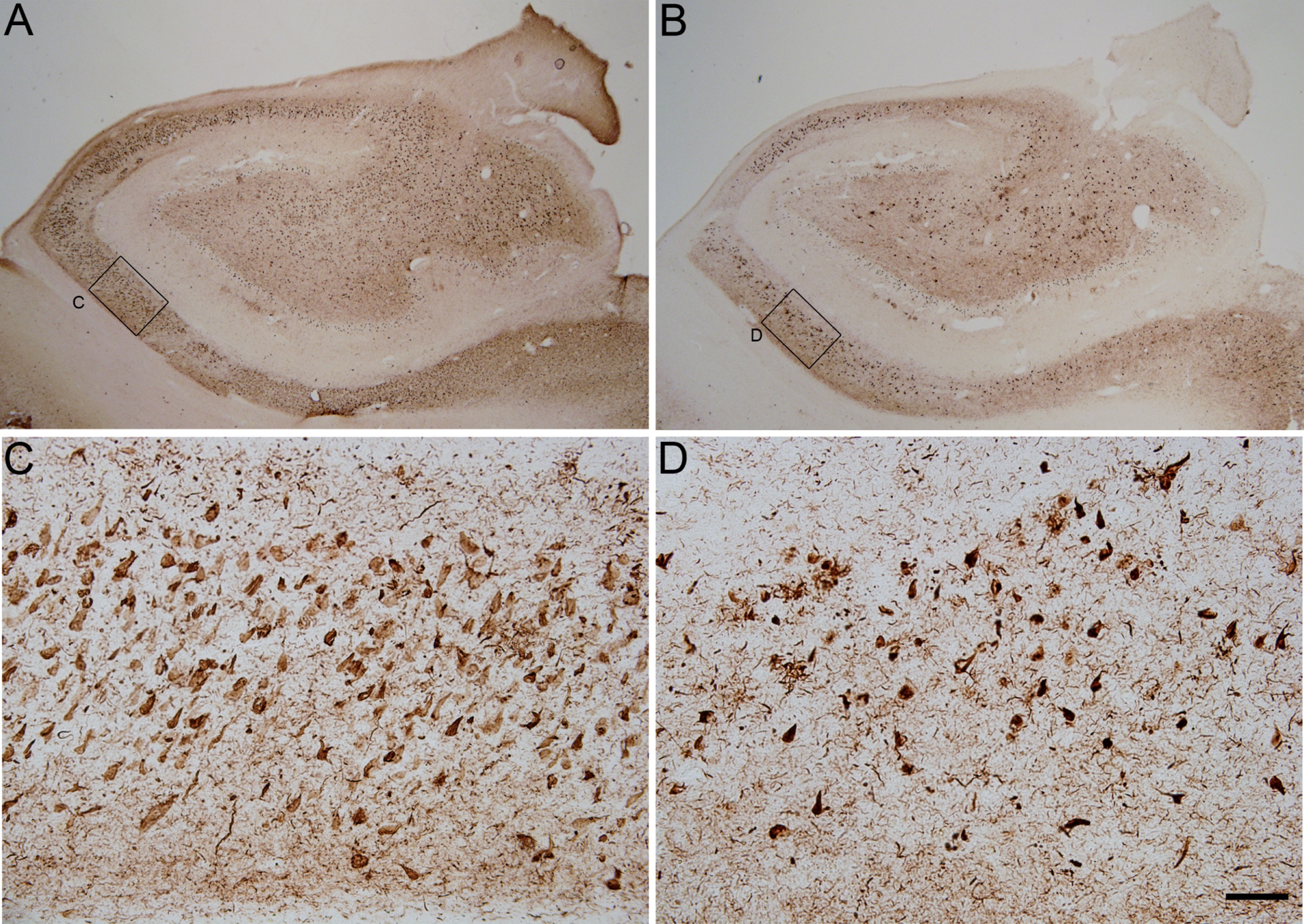
We have performed double immunofluorescences using the antibodies AT100 and pS396 to study the differential staining of the two markers depending on the severity of the disease associated with the neurofibrillary pathology. Our analyses were carried out using brain coronal sections containing the anterior portion of the CA1 region of the hippocampus from 21 autopsies of cases presenting different degrees of tau pathology, most of them diagnosed with AD (see Table 1). In order to classify the neurofibrillary pathology from the samples used in this study, we have considered the criteria provided by the donor Hospital or Foundation (see the Materials and Methods section), which is based on the profiles described by Braak et. al. (1994) using AT8 labeling (Braak staging). Figure 1 shows the differential pattern of tau phosphorylation (AT100 and pS396) from a Braak stage IV case.
Fig.1
Photomicrographs showing the pattern of hyperphosphorylated tau immunostaining using pS396 (A and C) and AT100 (B and D) antibodies in two adjacent coronal sections from the hippocampus of an AD case (BCN4). Small squared zones in A and B are shown at higher magnification in C–D (CA1 field of the hippocampus). Scale bar shown in D indicates 1824 μm in A–B and 114 μm in C–D.
Characterization of AT100 and pS396 staining and cell viability in CA1 neurons
The pattern of staining of the phospho-tau neurons was studied in detail. Figure 2A shows an example of a neuron from a Braak stage I case stained both with AT100 and pS396 that shows a granular (non-fibrillar) staining. This kind of staining may indicate an intermediate form of tau fibrils and has been described as an early event in the development of tau pathology [37]. Moreover, we found that in the cells showing a granular staining, AT100 labeling was also always present, either colocalizing with pS396 (Fig. 2A; empty yellow arrow) or alone (Fig. 2E; empty purple arrow). However, no granular AT100 negative but pS396 positive cells were found, supporting the notion that AT100 epitope appears prior to Serine 396 phosphorylation. In addition to the granular pattern of staining, we found cells in an intermediate stage between granular and NFT (Fig. 2C; empty yellow arrow). These cells were predominantly stained with AT100 and the majority of them showed isolated pS396 positive fibers or small aggregates in the process of forming (pre-tangles). Finally, it is worth noting that we found all possible phospho-tau combinations in the NFTs analyzed: AT100 positive (Fig. 2D; purple arrows), pS396 positive (Fig. 2C, E; white arrows), and AT100/pS396 positive (Fig. 2B, C; yellow arrows).
Fig.2
Photomicrographs belonging to cases with Braak stage I (A), III (B), and VI (C, D, and E) illustrating the different patterns of AT100 (green), pS396 (red), and DRAQ5 (grey) labeling found. Empty arrows indicate neurons with a granular staining—either AT100 positive (empty purple arrows) or both AT100 and pS396 positive (empty yellow arrows). Filled arrows indicate NFTs: AT100 positive (purple arrows), pS396 positive (white arrows), or AT100 and pS396 positive (yellow arrows). Higher magnification of panels in Supplementary Figs. 1–5. All scale bars: 40 μm.
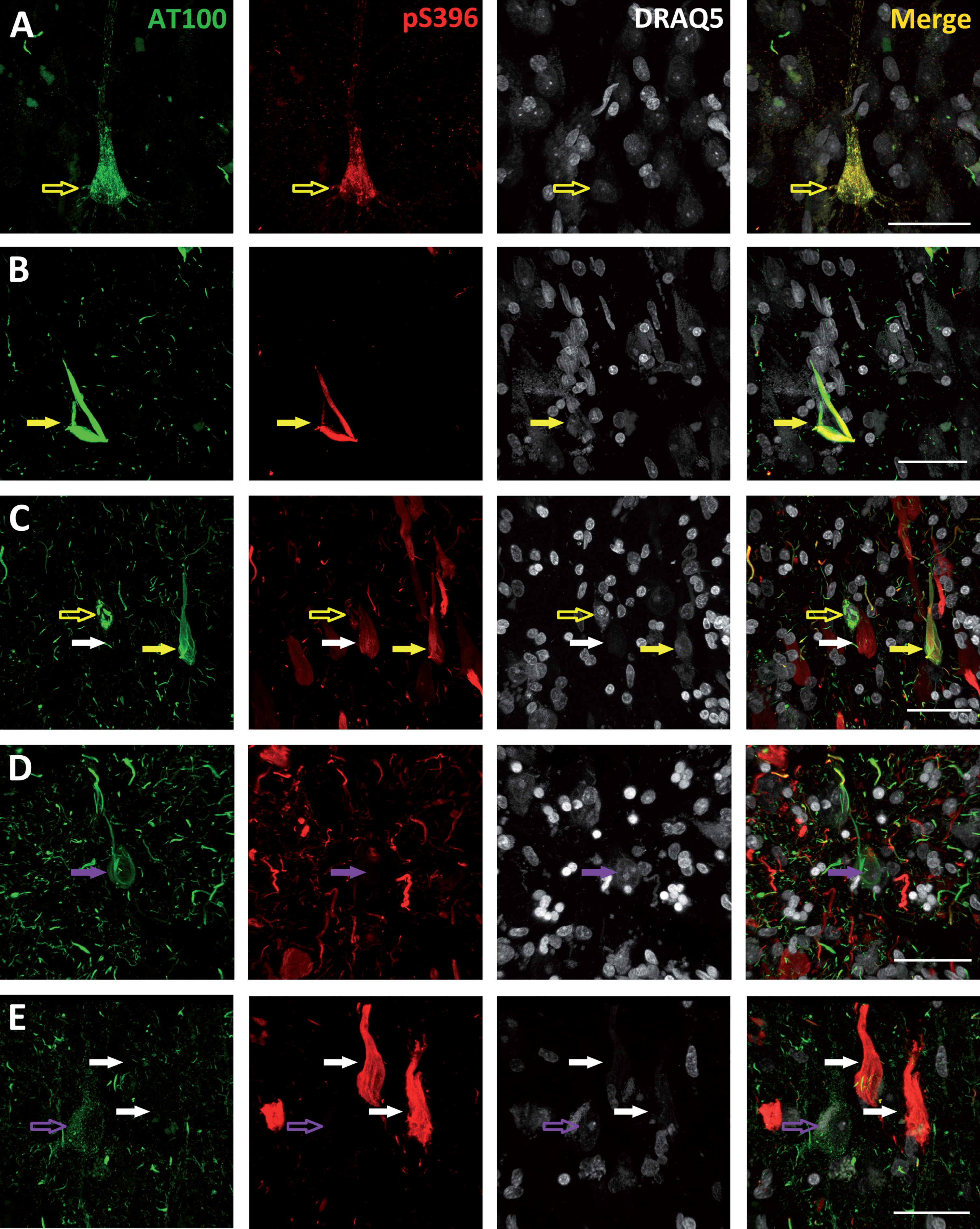
Based on the nuclear DRAQ5 staining, we have also analyzed the nucleus integrity of the phospho-tau labeled neurons. When a neuron dies, the intraneuronal aggregates (iNFTs) of hyperphosphorylated and misfolded tau become extracellular tangles (eNFTs, known as “ghost” tangles) [25]. Based on this, we found viable nuclei in all cells stained with a granular AT100 or AT100/pS396 (Fig. 2A; empty yellow arrows and Fig. 1E; empty purple arrows). Conversely, ghost tangles are only found in the pS396 positive NFTs (Fig. 2C, E; white arrows). Nonetheless, when analyzing cells showing either an AT100 NFT or an AT100/pS396 NFT, we did not find any alteration of their nuclei (Fig. 2B, C; yellow arrows). However, the nucleus was completely missing if pS396 immunoreactivity is predominant in NFTs labeled both with AT100 and pS396. Thus, the presence or absence of the nucleus appears to be dependent on the pS396 and AT100 immunoreactive elements, with AT100 epitope appearing to protect cell integrity while pS396 seems to promote cell death.
AT100/pS396 ratio and Braak stage
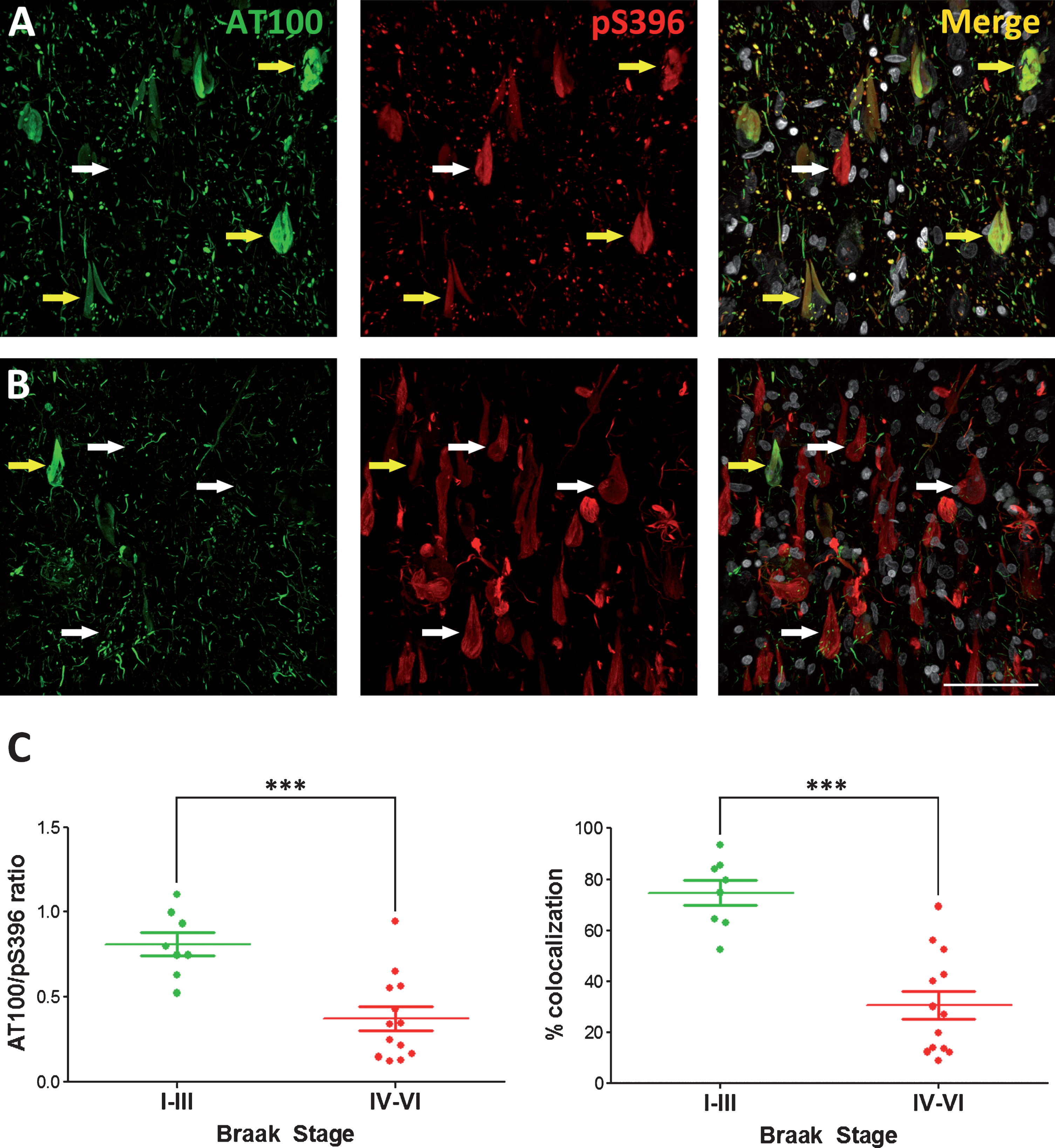
After characterizing the AT100 and pS396 labeling, we studied the ratios and colocalization values between these two markers, comparing early Braak stages (I−III) and late Braak stages (IV−VI). Our results indicate that the progression of tau pathology inversely correlates with the AT100/pS396 ratio (see Table 2). Cases with lower Braak stages display a higher ratio than cases with higher Braak stages. Figures 3A and 3B shows representative examples of the AT100 and pS396 immunostaining. Figure 3C shows that the differences found are statistically significant. In most of the analyzed cells, an intense labeling of pS396 was normally present in the NFT, which indicates a late state in the progression of PHF assembly. This reverse correlation was also observed when the colocalization percentage versus the Braak staging is compared (Fig. 3C). In the IV−VI Braak cases, the colocalization percentage was 30,8%, whereas in the I−III Braak cases, the colocalization was 74.8%. As for the AT100/pS936 ratio, the differences observed were statistically significant.
Table 2
Table showing the number of AT100 and pS396 ir-cells in each case, along as the volume analyzed, the ratio AT100/pS396 and the percentage of colocalization. In addition, the table shows the number of plaques per mm2 and if TDP43 cytoplasm inclusions were present. –, not available
| Cases | Braak | CERAD | Number of AT100 ir-cells | Number of pS396 ir-cells | Total volume analyzed (mm3) | Ratio AT100/pS396 | Percentage of colocalization | Number of Aβ plaques/mm2 | TDP43 cytoplasm inclusions |
| IF9 | I | None | 3 | 4 | 0.054 | 0.75 | 75 | 0 | NO |
| VK2 | II | A | 29 | 31 | 0.136 | 0.93 | 93.5 | – | NO |
| IF8 | III | A | 12 | 16 | 0.067 | 0.75 | 64.7 | 0 | NO |
| VK21 | III | A | 36 | 57 | 0.119 | 0.63 | 63.1 | 1.01 | NO |
| BCN11 | III | None | 20 | 25 | 0.102 | 0.80 | 80 | 0 | – |
| BCN15 | III | None | 31 | 28 | 0.153 | 1.10 | 84.3 | 0 | NO |
| AB5 | III | None | 13 | 13 | 0.067 | 1 | 85.7 | 0 | NO |
| AB6 | III | A | 10 | 19 | 0.071 | 0.53 | 52.6 | 2.03 | NO |
| BCN4 | IV | B | 81 | 231 | 0.119 | 0.35 | 27.3 | 4.61 | NO |
| BCN2 | V | C | 43 | 76 | 0.054 | 0.56 | 52.5 | 8.59 | – |
| BCN6 | V | C | 27 | 124 | 0.119 | 0.22 | 19.8 | 5.84 | YES |
| BCN10 | V | C | 33 | 96 | 0.102 | 0.34 | 30.3 | 23.11 | YES |
| VK15 | V | C | 38 | 40 | 0.102 | 0.95 | 69.5 | – | – |
| VK27 | V | B | 30 | 116 | 0.119 | 0.26 | 14 | 22.96 | NO |
| VK28 | V | C | 67 | 156 | 0.119 | 0.43 | 42.9 | 56.47 | YES |
| BCN1 | VI | C | 41 | 74 | 0.102 | 0.55 | 40.2 | 19.53 | YES |
| BCN7 | VI | C | 27 | 140 | 0.102 | 0.17 | 12.3 | 6.18 | – |
| BCN9 | VI | C | 4 | 32 | 0.102 | 0.12 | 12.5 | 9.35 | – |
| BCN12 | VI | C | 22 | 169 | 0.119 | 0.13 | 9.1 | 40.37 | YES |
| BCN13 | VI | C | 65 | 99 | 0.085 | 0.65 | 56.2 | 7.27 | YES |
| VK16 | VI | C | 37 | 246 | 0.102 | 0.15 | 13.6 | 10.49 | YES |
Fig.3
Representative photomicrographs showing the pattern of both AT100 and pS396 immunostaining in cases with low (I−III) (A) or high (V−VI) (B) Braak Stages. White arrows mark examples of neurons presenting no colocalization, whereas yellow arrows mark double-labeled neurons. (C) Histograms show the AT100/pS396 labeling ratio (left) or the colocalization percentage (right) in all the cases examined in this study grouped by low or high Braak Stage. A paired t test found significant differences in the comparisons between low and high Braak stages for both AT100/S396 ratio and colocalization (mean ± SD). ***p < 0.0005. Scale bar: 60 μm.
Furthermore, since TDP-43 accumulation is associated with cognitive impairment and dementia, we have determined the relationship between TDP43 and the progression of AD (see Supplementary Fig. 6 and Table 2). Our results are in accordance with previous reports which indicate that, in advanced AD cases, TDP43 positive cytoplasmic inclusions are found in around 75% of the cases [38]. Finally, we have measured the number of Aβ plaques in CA1 region for each case which, as expected, correlates with the severity of the disease (see Supplementary Fig. 6 and Table 2).
DISCUSSION
The main findings in the present study are: 1) the AT100/pS396 ratio decreases in line with the increase in the tau pathology of the samples analyzed and 2) the majority of the neurons in the CA1 area of human samples with a Braak stage of IV−VI do not show colocalization between the AT100 and pS396 tau antibodies (73%). We also demonstrated that, in the late Braak Stages, pS396 phosphorylation is predominant in most of the hippocampal neurons analyzed. Similar results have been reported when analyzing AT8 and pS396 tau immunostaining [39]. Therefore, the use of phospho-tau antibodies in immunohistochemistry may show an underrepresented population of positive phospho-tau cells, especially in advanced AD cases. Regarding the cases that were considered control but that after histopathological analysis of the brain samples showed NFTs with or without Aβ plaques in the hippocampal formation, it has been reported that AD affects about 80% of individuals over 65 years, but dementia only occurs in a small percentage of individuals at this age (the prevalence of dementia in AD increases to 25% in 80-year-old individuals). Consequently, it is possible that cases IF8, AB5 and AB6 represent a pre-dementia stage of AD (prodromal AD) [40].
The profiles of several phosphorylation sites have been recently analyzed by western blot to characterize the temporal and spatial phosphorylation patterns in AD tau pathology [41]. However, western blot studies with AD samples provide an average signal of mixed iNFTs and eNFTs, making interpretation of the results difficult. Therefore, we have used an immunocytochemical approach to study the possible tau pathology progression in single neurons.
In a native unfolded state, tau protein adopts a “paperclip” conformation [42]. Pseudo-phosphorylation experiments, targeting epitopes that are recognized by PHF-1 antibody (S396/S404), showed that the paperclip conformation is opened up [43]. The NFTs evolve in a sequential manner depending on the tau conformation adopted. Garcia-Sierra et al. demonstrated that Alz50 conformation, which has the N- and C-terminus intact, precedes the tau 66 conformation, characterized by truncations in the N- and C-terminus [44]. Thus, the order in which tau protein is phosphorylated may affect the tridimensional structure of tau. If this is the case, our results strongly suggest that the transition from T212/S214 to S396 phosphorylation promotes tau protein misfolding—a change that could be responsible for hiding the recognition sites for AT100 antibody. Further studies are needed to determine if this event is capable of triggering a conformational change that promotes transition to a toxic NFT. However, there are other possibilities that could explain the results obtained here: 1) serine at position 396 is mostly phosphorylated by GSK3 (via insulin receptor signaling), whereas AT100 recognition sites are phosphorylated by other kinases such as Dyrk1, PKA, JNK, or PKB/AKT (NMDA and calcium signaling) [21, 45–47]. Therefore, we could hypothesize that a specific neuron-dependent activity of one or more phosphatase/kinase is affecting the tau phosphorylation pattern in the progression of the disease. 2) Another possibility is that, after generation of truncated tau subproducts [44], the C-terminal aggregated region is not accessible to proteases but the proteolytic cleavage is feasible in the proline-rich region.
The analysis of the abnormal posttranslational modifications of tau may be dependent on the Aβ aggregates or other pathological hallmarks such as TDP43 cytoplasmic inclusions. Here, we have shown that the number of Aβ plaques or the TDP43 cytoplasm accumulation correlate, as expected, with the severity of the disease. Further studies are needed to study their possible role in the activation of signaling pathways involved in tau phosphorylation. Another issue to take into account is the influence of the postmortem delay on tau and other proteins’ structure/degradation [48, 49]. The postmortem delay in human brain samples usually ranges from around 2 hours to several days—a fact that should be taken into consideration when establishing relationships regarding tau phosphorylation and its impact on the development of AD. Our results come from the analysis of samples with very low postmortem intervals, minimizing the possible effects of tau degradation (see Table 1) thereby avoiding biased antibody recognition results.
Although PHF assembly is phospho-tau dependent, this tau modification does not necessarily have to be harmful. For example, it has been described that tau phosphorylation occurs naturally in hibernating mammals and is a non-pathological, reversible event [50]. The sequence recognized by AT100 and pS396 antibodies is conserved in human and in Syrian hamster—a small mammal with the capacity to hibernate under adverse conditions [51], which suggests that these phosphorylations could also be reversed in AD cases. In others studies, it has been described that phosphorylation of tau on threonine 205 may have a protective role by preventing Aβ-induced excitotoxicity [52] and that serine 305 phosphorylation inhibits tau aggregation [53]. In addition, in vitro experiments revealed that phosphorylation of threonine 212 may inhibit the toxic aggregation of tau [23]. Finally, it has been shown that tau phosphorylation at sites 202/205 alone is not sufficient to promote dendritic spine degeneration in human pyramidal neurons from AD cases [54].
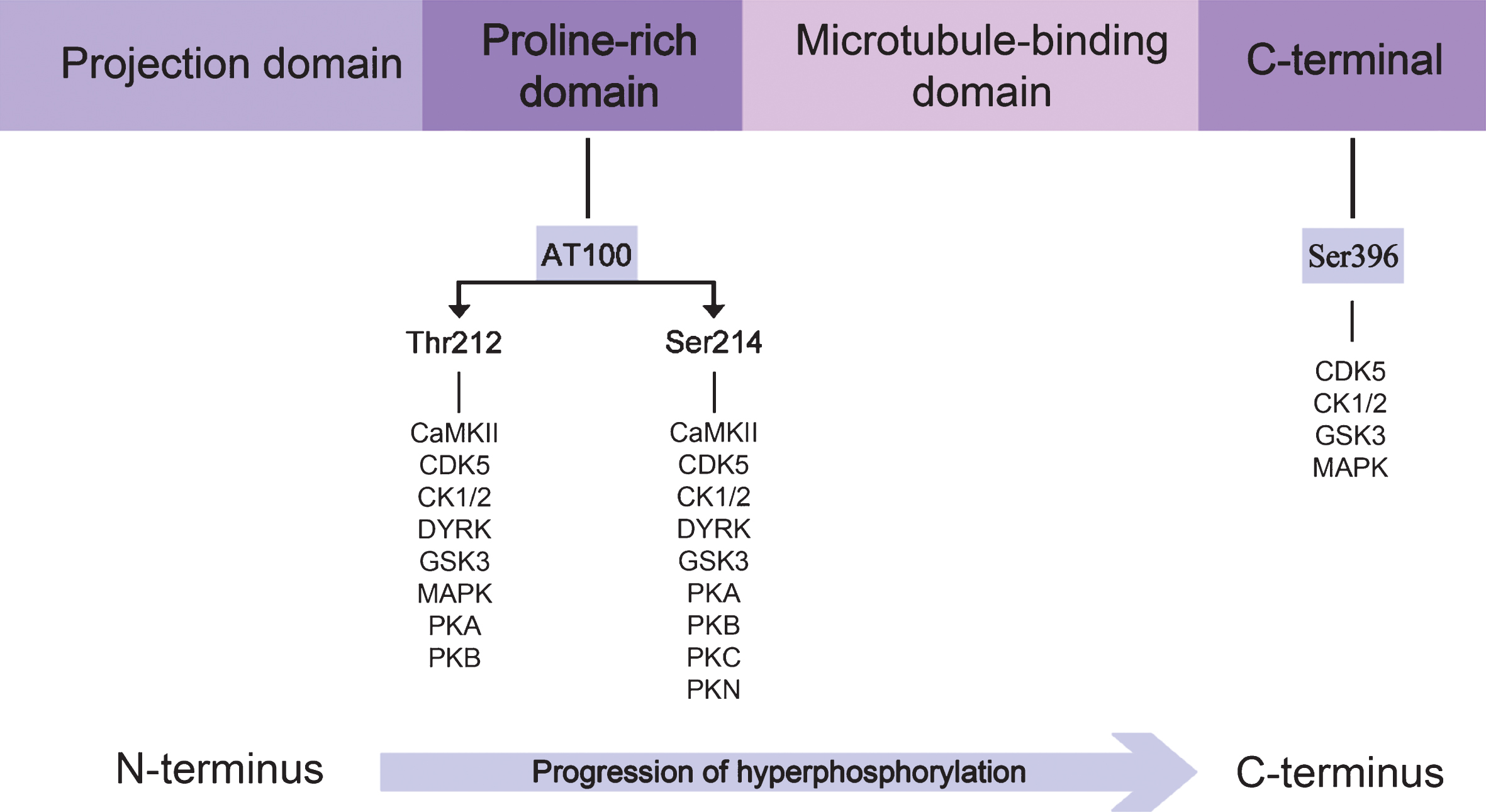
The development of neuronal cytopathology in AD reveals an interesting relationship: tau phosphorylation seems to sequentially advance from the N-terminal to the C-terminal domain, as illustrated in Fig. 4. We also highlight that tau phosphorylation, in the N-terminal or proline-rich regions, inhibits PHF formation and prevails during the early stages, whereas phosphorylation in the C-terminal is associated with PHF assembly and is the dominating feature during the late stages of the disease.
Fig.4
Direction of the progression of tau protein hyperphosphorylation. The proline-rich domain contains the epitope which is recognized by the AT100 antibody, whereas pS396 antibody recognizes the C-terminal region. Specific kinases phosphorylating each residue are illustrated. A blue arrow highlights the sequential tau hyperphosphorylation from the N-terminal to the C-terminal.
Conclusions
Our work represents a step forward in the understanding of NFT development and the progression of AD. We have characterized, using double immunofluorescence techniques, the staining of a proline rich region directed phospho-tau antibody (epitope AT100) and a C-terminal directed phospho-tau antibody (pS396). Our findings revealed that the AT100/pS396 ratio and the colocalization between these two phospho-tau markers decreases along with the severity of the disease in CA1, a hippocampal region linked to memory consolidation. This result may indicate that a conformational change in the tau protein hides the recognition site for the AT100 epitope during the late stages of the disease. As tau phosphorylation in the N-terminal or proline-rich regions inhibits PHF formation, this possible structural change could favor the aggregation of toxic tau and cell death.
In view of our results, the analysis of the CA1 region using double immunostaining with AT100 and anti-pS396 antibodies could provide more clues to correlate the tau phosphorylation pattern with the progression of AD.
AVAILABILITY OF DATA AND MATERIALS
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
ACKNOWLEDGMENTS
We would like to thank Carmen Álvarez, Miriam Marín, and Lorena Valdés for their helpful technical assistance. We are grateful to Alberto R
This work was supported by grants from the following entities: the Spanish Ministerio de Ciencia, Innovación y Universidades (Grants SAF 2015-66603-P and Cajal Blue Brain Project, Spanish partner of the Blue Brain Project initiative from EPFL, Switzerland to JDF and Grant BFU 2016-77885-P to JA and FH), Comunidad de Madrid (S2017/BMD-3700) to JA and FH, Centro de Investigación Biom
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/18-1263r1).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JAD-181263.
REFERENCES
[1] | Dickerson BC , Eichenbaum H ((2010) ) The episodic memory system: neurocircuitry and disorders. Neuropsychopharmacology 35: , 86–104. |
[2] | Serrano-Pozo A , Frosch MP , Masliah E , Hyman BT ((2011) ) Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med 1: , a006189. |
[3] | Bird CM , Burgess N ((2008) ) The hippocampus and memory: insights from spatial processing. Nat Rev Neurosci 9: , 182–194. |
[4] | Scoville WB , Milner B ((1957) ) Loss of recent memory after bilateral hippocampal lesions. J Neurol Neurosurg Psychiatry 20: , 11–21. |
[5] | Zola-Morgan S , Squire LR , Amaral DG ((1986) ) Human amnesia and the medial temporal region: enduring memory impairment following a bilateral lesion limited to field CA1 of the hippocampus. J Neurosci 6: , 2950–2967. |
[6] | Basu J , Siegelbaum SA ((2015) ) The corticohippocampal circuit, synaptic plasticity, and memory. Cold Spring Harb Perspect Biol 7: , a021733. |
[7] | van Strien NM , Cappaert NL , Witter MP ((2009) ) The anatomy of memory: an interactive overview of the parahippocampal-hippocampal network. Nat Rev Neurosci 10: , 272–282. |
[8] | West MJ , Kawas CH , Stewart WF , Rudow GL , Troncoso JC ((2004) ) Hippocampal neurons in pre-clinical Alzheimer’s disease. Neurobiol Aging 25: , 1205–1212. |
[9] | Andrade-Moraes CH , Oliveira-Pinto AV , Castro-Fonseca E , da Silva CG , Guimaraes DM , Szczupak D , Parente-Bruno DR , Carvalho LR , Polichiso L , Gomes BV , Oliveira LM , Rodriguez RD , Leite RE , Ferretti-Rebustini RE , Jacob-Filho W , Pasqualucci CA , Grinberg LT , Lent R ((2013) ) Cell number changes in Alzheimer’s disease relate to dementia, not to plaques and tangles. Brain 136: , 3738–3752. |
[10] | Scheff SW , Price DA , Schmitt FA , Mufson EJ ((2006) ) Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging 27: , 1372–1384. |
[11] | Hyman BT , Van Hoesen GW , Damasio AR ((1990) ) Memory-related neural systems in Alzheimer’s disease: an anatomic study. Neurology 40: , 1721–1730. |
[12] | Terry RD , Masliah E , Salmon DP , Butters N , DeTeresa R , Hill R , Hansen LA , Katzman R ((1991) ) Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30: , 572–580. |
[13] | Li H , Jia X , Qi Z , Fan X , Ma T , Pang R , Ni H , Li CR , Lu J , Li K ((2018) ) Disrupted functional connectivity of cornu ammonis subregions in amnestic mild cognitive impairment: a longitudinal resting-state fMRI study. Front Hum Neurosci 12: , 413. |
[14] | Nelson PT , Alafuzoff I , Bigio EH , Bouras C , Braak H , Cairns NJ , Castellani RJ , Crain BJ , Davies P , Del Tredici K , Duyckaerts C , Frosch MP , Haroutunian V , Hof PR , Hulette CM , Hyman BT , Iwatsubo T , Jellinger KA , Jicha GA , Kovari E , Kukull WA , Leverenz JB , Love S , Mackenzie IR , Mann DM , Masliah E , McKee AC , Montine TJ , Morris JC , Schneider JA , Sonnen JA , Thal DR , Trojanowski JQ , Troncoso JC , Wisniewski T , Woltjer RL , Beach TG ((2012) ) Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol 71: , 362–381. |
[15] | Braak H , Braak E ((1991) ) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82: , 239–259. |
[16] | Weingarten MD , Lockwood AH , Hwo SY , Kirschner MW ((1975) ) A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A 72: , 1858–1862. |
[17] | Ittner A , Ittner LM ((2018) ) Dendritic tau in Alzheimer’s disease. Neuron 99: , 13–27. |
[18] | Brelstaff J , Tolkovsky AM , Ghetti B , Goedert M , Spillantini MG ((2018) ) Living neurons with tau filaments aberrantly expose phosphatidylserine and are phagocytosed by microglia. Cell Rep 24: , 1939–1948 e1934. |
[19] | Eftekharzadeh B , Daigle JG , Kapinos LE , Coyne A , Schiantarelli J , Carlomagno Y , Cook C , Miller SJ , Dujardin S , Amaral AS , Grima JC , Bennett RE , Tepper K , DeTure M , Vanderburgh CR , Corjuc BT , DeVos SL , Gonzalez JA , Chew J , Vidensky S , Gage FH , Mertens J , Troncoso J , Mandelkow E , Salvatella X , Lim RYH , Petrucelli L , Wegmann S , Rothstein JD , Hyman BT ((2018) ) Tau protein disrupts nucleocytoplasmic transport in Alzheimer’s disease. Neuron 99: , 925–940 e927. |
[20] | Iqbal K , Liu F , Gong CX ((2016) ) Tau and neurodegenerative disease: the story so far. Nat Rev Neurol 12: , 15–27. |
[21] | Hanger DP , Anderton BH , Noble W ((2009) ) Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol Med 15: , 112–119. |
[22] | Illenberger S , Zheng-Fischhofer Q , Preuss U , Stamer K , Baumann K , Trinczek B , Biernat J , Godemann R , Mandelkow EM , Mandelkow E ((1998) ) The endogenous and cell cycle-dependent phosphorylation of tau protein in living cells: implications for Alzheimer’s disease. Mol Biol Cell 9: , 1495–1512. |
[23] | Schneider A , Biernat J , von Bergen M , Mandelkow E , Mandelkow EM ((1999) ) Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry 38: , 3549–3558. |
[24] | Zheng-Fischhofer Q , Biernat J , Mandelkow EM , Illenberger S , Godemann R , Mandelkow E ((1998) ) Sequential phosphorylation of Tau by glycogen synthase kinase-3beta and protein kinase A at Thr212 and Ser214 generates the Alzheimer-specific epitope of antibody AT100 and requires a paired-helical-filament-like conformation. Eur J Biochem 252: , 542–552. |
[25] | Augustinack JC , Schneider A , Mandelkow EM , Hyman BT ((2002) ) Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol 103: , 26–35. |
[26] | Goedert M , Jakes R , Vanmechelen E ((1995) ) Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neurosci Lett 189: , 167–169. |
[27] | Braak E , Braak H , Mandelkow EM ((1994) ) A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol 87: , 554–567. |
[28] | Bertrand J , Plouffe V , Senechal P , Leclerc N ((2010) ) The pattern of human tau phosphorylation is the result of priming and feedback events in primary hippocampal neurons. Neuroscience 168: , 323–334. |
[29] | Kimura T , Ono T , Takamatsu J , Yamamoto H , Ikegami K , Kondo A , Hasegawa M , Ihara Y , Miyamoto E , Miyakawa T ((1996) ) Sequential changes of tau-site-specific phosphorylation during development of paired helical filaments. Dementia 7: , 177–181. |
[30] | Su JH , Cummings BJ , Cotman CW ((1994) ) Early phosphorylation of tau in Alzheimer’s disease occurs at Ser-202 and is preferentially located within neurites. Neuroreport 5: , 2358–2362. |
[31] | Braak H , Alafuzoff I , Arzberger T , Kretzschmar H , Del Tredici K ((2006) ) Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112: , 389–404. |
[32] | Mondragon-Rodriguez S , Perry G , Luna-Munoz J , Acevedo-Aquino MC , Williams S ((2014) ) Phosphorylation of tau protein at sites Ser(396-404) is one of the earliest events in Alzheimer’s disease and Down syndrome. Neuropathol Appl Neurobiol 40: , 121–135. |
[33] | Fasulo L , Ugolini G , Visintin M , Bradbury A , Brancolini C , Verzillo V , Novak M , Cattaneo A ((2000) ) The neuronal microtubule-associated protein tau is a substrate for caspase-3 and an effector of apoptosis. J Neurochem 75: , 624–633. |
[34] | Luna-Munoz J , Chavez-Macias L , Garcia-Sierra F , Mena R ((2007) ) Earliest stages of tau conformational changes are related to the appearance of a sequence of specific phospho-dependent tau epitopes in Alzheimer’s disease. J Alzheimers Dis 12: , 365–375. |
[35] | Steinhilb ML , Dias-Santagata D , Fulga TA , Felch DL , Feany MB ((2007) ) Tau phosphorylation sites work in concert to promote neurotoxicity. Mol Biol Cell 18: , 5060–5068. |
[36] | Smith PJ , Wiltshire M , Davies S , Patterson LH , Hoy T ((1999) ) A novel cell permeant and far red-fluorescing DNA probe, DRAQ5, for blood cell discrimination by flow cytometry. J Immunol Methods 229: , 131–139. |
[37] | Maeda S , Sahara N , Saito Y , Murayama M , Yoshiike Y , Kim H , Miyasaka T , Murayama S , Ikai A , Takashima A ((2007) ) Granular tau oligomers as intermediates of tau filaments. Biochemistry 46: , 3856–3861. |
[38] | Chang XL , Tan MS , Tan L , Yu JT ((2016) ) The role of TDP-43 in Alzheimer’s disease. Mol Neurobiol 53: , 3349–3359. |
[39] | Furcila D , DeFelipe J , Alonso-Nanclares L ((2018) ) A study of amyloid-beta and phosphotau in plaques and neurons in the hippocampus of Alzheimer’s disease patients. J Alzheimers Dis 64: , 417–435. |
[40] | Ferrer I ((2012) ) Defining Alzheimer as a common age-related neurodegenerative process not inevitably leading to dementia. Prog Neurobiol 97: , 38–51. |
[41] | Neddens J , Temmel M , Flunkert S , Kerschbaumer B , Hoeller C , Loeffler T , Niederkofler V , Daum G , Attems J , Hutter-Paier B ((2018) ) Phosphorylation of different tau sites during progression of Alzheimer’s disease. Acta Neuropathol Commun 6: , 52. |
[42] | Jeganathan S , von Bergen M , Brutlach H , Steinhoff HJ , Mandelkow E ((2006) ) Global hairpin folding of tau in solution. Biochemistry 45: , 2283–2293. |
[43] | Jeganathan S , Hascher A , Chinnathambi S , Biernat J , Mandelkow EM , Mandelkow E ((2008) ) Proline-directed pseudo-phosphorylation at AT8 and PHF1 epitopes induces a compaction of the paperclip folding of Tau and generates a pathological (MC-1) conformation. J Biol Chem 283: , 32066–32076. |
[44] | Garcia-Sierra F , Ghoshal N , Quinn B , Berry RW , Binder LI ((2003) ) Conformational changes and truncation of tau protein during tangle evolution in Alzheimer’s disease. J Alzheimers Dis 5: , 65–77. |
[45] | Yoshida H , Goedert M ((2006) ) Sequential phosphorylation of tau protein by cAMP-dependent protein kinase and SAPK4/p38delta or JNK2 in the presence of heparin generates the AT100 epitope. J Neurochem 99: , 154–164. |
[46] | Ksiezak-Reding H , Pyo HK , Feinstein B , Pasinetti GM ((2003) ) Akt/PKB kinase phosphorylates separately Thr212 and Ser214 of tau protein. Biochim Biophys Acta 1639: , 159–168. |
[47] | Woods YL , Cohen P , Becker W , Jakes R , Goedert M , Wang X , Proud CG ((2001) ) The kinase DYRK phosphorylates protein-synthesis initiation factor eIF2Bepsilon at Ser539 and the microtubule-associated protein tau at Thr212: potential role for DYRK as a glycogen synthase kinase 3-priming kinase. Biochem J 355: , 609–615. |
[48] | Gartner U , Janke C , Holzer M , Vanmechelen E , Arendt T ((1998) ) Postmortem changes in the phosphorylation state of tau-protein in the rat brain. Neurobiol Aging 19: , 535–543. |
[49] | Gonzalez-Riano C , Tapia-Gonzalez S , Garcia A , Munoz A , DeFelipe J , Barbas C ((2017) ) Metabolomics and neuroanatomical evaluation of post-mortem changes in the hippocampus. Brain Struct Funct 222: , 2831–2853. |
[50] | Arendt T , Stieler J , Strijkstra AM , Hut RA , Rudiger J , Van der Zee EA , Harkany T , Holzer M , Hartig W ((2003) ) Reversible paired helical filament-like phosphorylation of tau is an adaptive process associated with neuronal plasticity in hibernating animals. J Neurosci 23: , 6972–6981. |
[51] | Leon-Espinosa G , Garcia E , Garcia-Escudero V , Hernandez F , DeFelipe J , Avila J ((2013) ) Changes in tau phosphorylation in hibernating rodents. J Neurosci Res 91: , 954–962. |
[52] | Ittner A , Chua SW , Bertz J , Volkerling A , van der Hoven J , Gladbach A , Przybyla M , Bi M , van Hummel A , Stevens CH , Ippati S , Suh LS , Macmillan A , Sutherland G , Kril JJ , Silva AP , Mackay JP , Poljak A , Delerue F , Ke YD , Ittner LM ((2016) ) Site-specific phosphorylation of tau inhibits amyloid-beta toxicity in Alzheimer’s mice. Science 354: , 904–908. |
[53] | Strang KH , Sorrentino ZA , Riffe CJ , Gorion KM , Vijayaraghavan N , Golde TE , Giasson BI ((2018) ) Phosphorylation of serine 305 in tau inhibits aggregation. Neurosci Lett 692: , 187–192. |


