
Long-Term Trazodone Use and Cognition: A Potential Therapeutic Role for Slow-Wave Sleep Enhancers
Abstract
Background:
Recent studies reveal an association between slow-wave sleep (SWS), amyloid-β aggregation, and cognition.
Objective:
This retrospective study examines whether long-term use of trazodone, an SWS enhancer, is associated with delayed cognitive decline.
Methods:
We identified 25 regular trazodone users (mean age 75.4±7.5; 9 women, 16 men) who carried a diagnosis of Alzheimer’s dementia, mild cognitive impairment, or normal cognition, and 25 propensity-matched trazodone non-users (mean age 74.5±8.0; 13 women, 12 men), accounting for age, sex, education, type of sleep deficit (hypersomnia, insomnia, parasomnia), diagnosis, and baseline Mini-Mental State Examination (MMSE). Longitudinal group differences in cognitive testing were evaluated through repeated measures tests over an average inter-evaluation interval of four years.
Results:
Trazodone non-users had 2.6-fold faster decline MMSE (primary outcome) compared to trazodone users, 0.27 (95% confidence interval [CI]: 0.07–0.48) versus 0.70 (95% CI: 0.50–0.90) points per year (p = 0.023). The observed effects were especially associated with subjective improvement of sleep complaints in post-hoc analyses (p = 0.0006). Secondary outcomes of other cognitive and functional scores had variable worsening in non-users and varied in significance when accounting for co-administered medications and multiple comparisons. Trazodone effects on MMSE remained significant within participants with AD-predicted pathology, with 2.4-fold faster decline in non-users (p = 0.038).
Conclusions:
These results suggest an association between trazodone use and delayed cognitive decline, adding support for a potentially attractive and cost-effective intervention in dementia. Whether the observed relationship of trazodone to cognitive function is causal or an indirect marker of other effects, such as treated sleep disruption, and if such effects are mediated through SWS enhancement requires confirmation through prospective studies.
INTRODUCTION
Sleep disorders increase in frequency with aging, and their manifestation, even in cognitively non-impaired individuals, can be a harbinger of subsequent cognitive decline [1]. Sleep changes with aging include decreased total sleep time and efficiency, increased waking after sleep onset, and decreased slow wave sleep (SWS) [2]. SWS in humans, as part of non-rapid eye movement (NREM) sleep, mediates episodic memory consolidation [3, 4]. Recent studies in transgenic mouse models of Alzheimer’s disease (AD) reveal that SWS restriction accelerates activity-dependent amyloid-β aggregation, whereas SWS enhancement delays aggregation [5]. Furthermore, patients with AD have less SWS, and cognitively non-impaired adults with decreased SWS exhibit increased amyloid-β burden and worse sleep-mediated episodic memory consolidation [6, 7]. Several double-blind randomized placebo-controlled trials for sleep consolidation in AD have tested the effectiveness of commonly used hypnotic agents: melatonin, ramelteon, mirtazapine, and trazodone [8, 9]. While melatonin, ramelteon, or mirtazapine use did not produce significant improvement on sleep measures, trazodone, previously shown to enhance SWS by 50–56% on polysomnography in younger and older adults [10, 11], increased total sleep time by 42.5 minutes on actigraphy in patients with AD [12, 13]. Furthermore, there were no cognitive side effects or daytime somnolence after a 2-week intervention period, thus diminishing concerns for possible cholinergic, α-adrenergic, or histaminergic antagonistic effects in sleep-promoting doses [12, 13]. Although these studies did not reveal a change in cognitive performance, improvement or worsening, they were too short in duration to observe possible SWS-mediated effects on course of neurodegeneration, and did not specifically test overnight sleep-mediated memory consolidation that may have been a more sensitive test for sleep-mediated synaptic potentiation [14]. To date, no studies have assessed long-term effects of trazodone on cognitive performance.
We thus hypothesized that trazodone, a SWS enhancer, may correlate with delayed cognitive decline over prolonged use [15, 16]. To test whether such an association exists, we retrospectively analyzed longitudinal cognitive decline trajectories dependent on prolonged use of trazodone in volunteers from our research cohort.
METHODS
Participant selection
We identified participants from the UCSF Memory and Aging Center cohort who satisfied the following criteria: 1) presence of sleep disturbances (insomnia, hypersomnia, or parasomnia) as documented on the National Alzheimer’s Coordinating Center assessment form, 2) longitudinal clinical, medication, and neuropsychological data, 3) and diagnostically grouped as AD, mild cognitive impairment (MCI), or cognitively non-impaired (CN) on their first visit (Fig. 1) [17]. All participants provided informed consent according to the Declaration of Helsinki. Presence of insomnia, hypersomnia, or parasomnia was documented by a physician in an ordinal manner as absent, mild (i.e., present and not interfering significantly with daily function), or severe (i.e., interfering significantly with daily function) (Table 1). Since only two participants had severe sleep symptoms, sleep variables were binarized as present or absent at each visit for further analysis. The diagnoses of AD, amnestic MCI, and non-amnestic MCI were made using NINCDS-ADRDA criteria for probable AD, and the MCI criteria developed by Petersen et al. [18, 19]. Molecular AD biomarkers were available for 14 participants (amyloid PET performed in eleven and CSF amyloid-β/tau levels obtained in five; two participants underwent both tests). From the eligible pool of volunteers, we identified 25 participants who reported regular trazodone use for at least two consecutive annual visits. Detailed longitudinal dosage and indication for trazodone use was available for 16 out of 25 trazodone users, all of which listed insomnia as the reason for trazodone use. The median prescribed dosage was 50 mg per day before bedtime (25th percentile 50 mg; 75th percentile 100-125 mg), consistent with insomnia, rather than antidepressant, dosing. Trazodone users were propensity matched to 25 participants who did not use trazodone, according to age, sex, education, diagnostic group, type, and severity of sleep deficit, and baseline MMSE (Fig. 1). These measures were similar between trazodone users and non-users, indicating successful propensity matching (Table 1). Furthermore, the two groups did not differ on longitudinal subjective change in sleep quality (improvement: stability: worsening [trazodone users –10:9:6 versus non-users –11:4:10]; χ2 = 2.97, p = 0.23), even though this factor was not included for propensity matching the two groups. We purposely did not include a third comparison group of another hypnotic to evaluate whether observed effects are specific to trazodone or are class dependent, since available studies have shown that agents commonly recommended as hypnotics are not effective in prolonging sleep duration in AD [8, 9], or have significant carry-over cognitive side effects (e.g., zolpidem) [20, 21]. Hypnotics that have no or minimal cognitive side effects the following day and prolong SWS, such as sodium oxybate or orexin receptor antagonists, have not been tested in AD yet and are not used by participants in our cohort. This is a limitation in our study that will be addressed as our cohort includes participants on hypnotics without known cognitive side effects the following day.
Fig.1
Participant selection. Flow chart on participant selection from the UCSF Memory and Aging Center research volunteer cohort based on reported sleep disturbances (insomnia, hypersomnia, or parasomnia), diagnostic group, available medication data, and reported trazodone use. Propensity matching was based on age, sex, education, type of sleep disturbance, diagnostic group, and baseline MMSE.
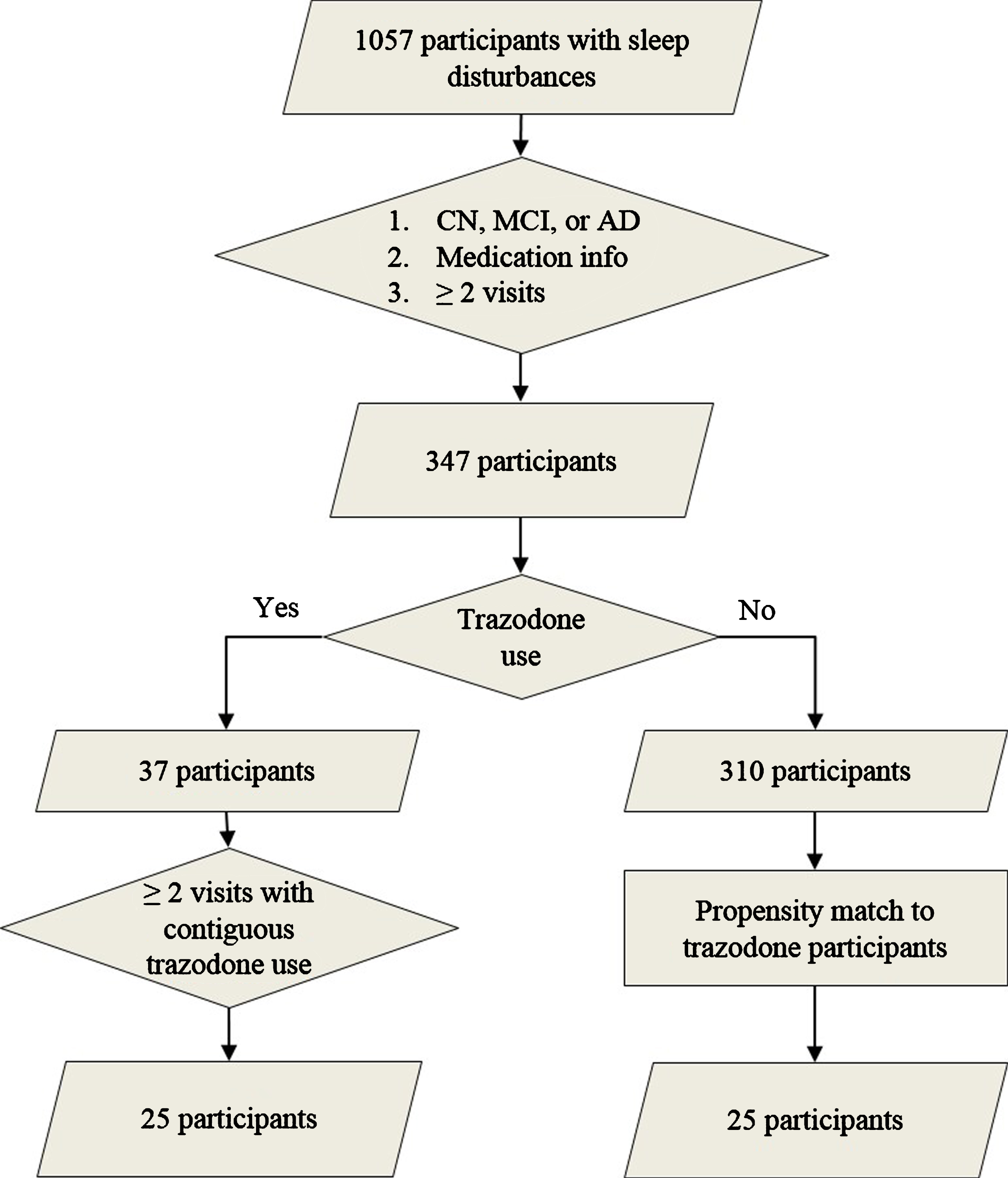
Table 1
Demographics at baseline visit
| Trazodone users (N = 25) | Trazodone non-users (N = 25) | pa | |
| Age | 75.4 | 74.5 | 0.69 |
| mean (SD) | (7.5) | (8.0) | |
| Women: Men | 9: 16 | 13: 12 | 0.25 |
| Education in years | 17.3 | 17.8 | 0.85 |
| mean (SD) | (4.0) | (2.7) | |
| Insomnia | Absent 13 | Absent 17 | 0.28 |
| Mild 10 | Mild 8 | ||
| Severe 2 | Severe 0 | ||
| Hypersomnia | Absent 20 | Absent 18 | 0.46 |
| Mild 5 | Mild 7 | ||
| Parasomnia | Absent 22 | Absent 22 | 0.91 |
| Mild 3 | Mild 3 | ||
| Diagnosis | CN 7 | CN 7 | 0.73 |
| MCI 13 | MCI 15 | ||
| AD 5 | AD 3 | ||
| Baseline MMSE | 29 | 30 | 0.64 |
| median (range) | (20–30) | (23–30) |
CN, cognitively non-impaired; MCI, mild cognitive impairment; AD, Alzheimer’s dementia; MMSE, Mini-Mental State Examination. aPearson χ2 and Mann-Whitney tests for non-parametric measures, t-test for parametric measures.
We further wanted to account for moderation effects on outcome measures of inter-evaluation interval secondary to natural disease progression. For trazodone users, baseline and final evaluations reflected the first and last visits with reported trazodone use. For trazodone non-users, these time points were the first and last visits with available medication data. We anticipated that follow-up intervals would not be identical for paired participants in the two groups to allow for propensity matching of inter-evaluation intervals, and thus planned our analysis a-priori with inter-evaluation intervals as a covariate. The interval between baseline and final visits was indeed different between the two groups (mean±SD; trazodone users: 3.1±1.9 years, non-users: 5.1±2.8 years; p = 0.013) and interval effects were accounted for in all analyses. Nonetheless, to address possible non-linear effects of inter-evaluation interval differences between groups, we selected shorter follow-up periods for the trazodone non-users post-hoc, making intervals comparable between groups, and repeated the analysis.
Data selection on primary and secondary outcomes
Our a priori defined primary outcome was the change in MMSE between baseline and final visits. Given increasing evidence on the association between SWS enhancement and improved sleep-mediated and awake episodic memory consolidation [22], we further pursued secondary outcomes of longitudinal changes in cognitive testing of visual and verbal episodic memory through 10-minute delayed recognition of the Benson Complex Figure and the California Verbal Learning Test (CVLT) and the CVLT Second Edition (CVLT-II) [4, 23].24-26 Furthermore, considering that improved sleep also allows for improved executive function and working memory further mediated through prefrontal cortex engagement, we also tested longitudinal performance on Modified Trail-Making B, Design Fluency, Calculations, Digit-Span Forward and Backward, phonemic and semantic Verbal Fluency, and Stroop Color-Naming and Interference [24]. We finally wanted to evaluate whether such effects translated to better disability scores through the Clinical Dementia Rating Scale Sum of Boxes (CDR-SB) [25]. Values for each of the variables were included as long as medication data were also available during the respective research visits. We did not impute data for our analyses.
Statistical analyses
Comparisons on primary and secondary outcomes between the two groups followed repeated-measures analysis of variance while accounting for inter-evaluation intervals, i.e., the length of time between baseline and final visits. Cognitive and functional assessment scores were treated as dependent variables, and trazodone use as a fixed factor. Significance level was set at 0.05, and one-tailed significance testing was performed given the a priori hypothesis that trazodone is associated with delayed cognitive decline. Significance testing on secondary outcomes and post-hoc analyses accounted for multiple comparisons by applying Bonferroni correction.
Table 2
Trazodone effects on primary outcome (Mini-Mental State Examination)
| Participants included (users: non-users) | Inter-evaluation interval (avg. years)a | Mean baseline and final scores in users versus non-users | Average decline in users versus non-users (95% CI) in points/year | Percent (%) of relative decline in users compared to non-users (95% CI) | Fdf, p | |
| MMSE across | 25: 25 | 4.12 | 28.6 → 27.5 | 0.27 versus 0.70 | 39.0 | F1,47 = 14.23, |
| participants | versus | (0.07–0.48 versus | (14.0–52.8) | p = 0.023 | ||
| (primary outcome) | 28.1 → 25.2 | 0.50–0.90) | ||||
| MMSE accounting for | 25: 25 | 4.12 | 28.2 → 26.9 | 0.33 versus 0.64 | 51.5 | F1,46 = 3.28, |
| sedative and stimulant | versus | (0.17–0.49 versus | (35.7–61.1) | p = 0.038 | ||
| use | 28.5 → 25.8 | 0.48–0.80) | ||||
| MMSE accounting for | 25: 25 | 4.12 | 28.3 → 26.8 | 0.37 versus 0.60 | 61.9 | F1,46 = 1.32, |
| ChEi use | versus | (0.20–0.55 versus | (46.3–70.5) | p = 0.128 | ||
| 28.4 → 25.9 | 0.43–0.77) | |||||
| MMSE in | 13: 16 | 3.75 | 28.1 → 26.5 | 0.42 versus 1.02 | 41.6 | F1,25 = 3.41, |
| AD-predicted | versus | (0.15–0.69 versus | (20.0–54.7) | p = 0.038 | ||
| pathologyb | 27.9 → 24.1 | 0.77–1.27) |
CI, confidence interval; MMSE, Mini-Mental State Examination; ChEi, cholinesterase inhibitor. aInter-evaluation intervals differ according to subsets of participants included. bIn addition to CN participants, MCI participants without AD predicted primary pathology were excluded.
Additional analyses tested trazodone effects on MMSE only in participants who had AD-predicted pathology based on clinical judgment, and while accounting for concomitant sedative and stimulant medication effects. A sedative medication binary variable represented use of the following: benzodiazepines, non-benzodiazepine hypnotics, narcotics, atypical antipsychotics, antihistamines, or anticholinergic medications. A stimulant medication binary variable represented use of the following: cholinesterase inhibitors (ChEi), dopaminergic, noradrenergic, or serotoninergic antidepressant medications. A final group comparison of trazodone effects on MMSE accounted specifically for the concomitant use of ChEi, because they represent the main medication class with an established cognitive benefit in AD. Six participants in each group used ChEi. Furthermore, to test whether trazodone use was correlated with ChEi use, a possible confounder for observed trazodone effects, we calculated the mean square contingency coefficient (φ). Finally, to assess whether the observed trazodone effects were mediated through improvement of sleep, we performed post-hoc exploratory repeated measures analysis of variance while accounting, first, for presence or absence of sleep problems (insomnia or hypersomnia) at the baseline visit and, second, for longitudinal changes in sleep complaints between baseline and follow-up evaluations accounting for multiple comparisons. Analyses were performed using the Statistical Package for the Social Sciences.
RESULTS
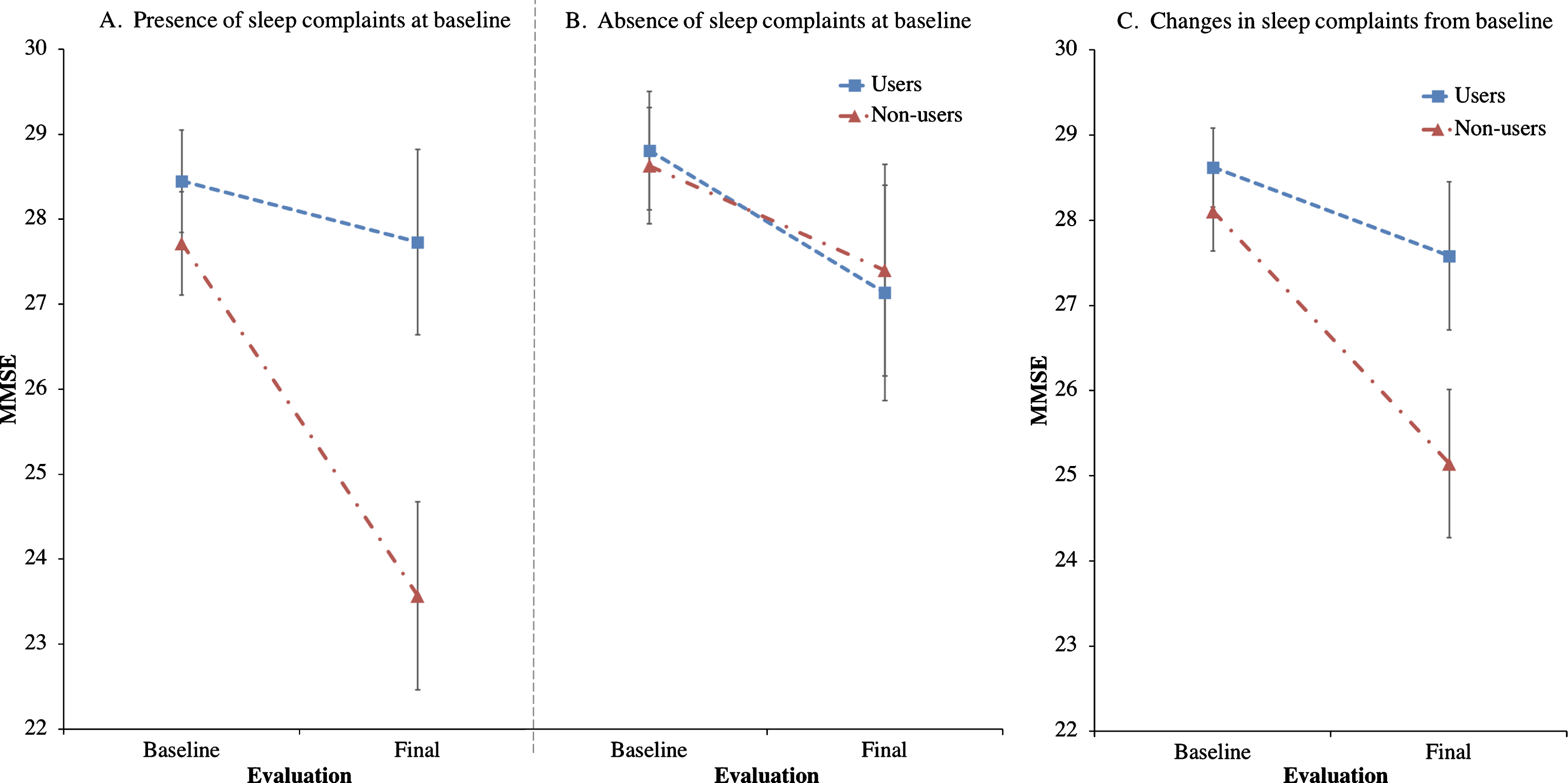
Trazodone longitudinal effects on primary and secondary outcomes are listed in Tables 2 and 3. Trazodone non-users declined 2.6-fold faster on the MMSE than trazodone users, at an estimated inter-evaluation interval for both groups averaging 4.12 years (Fig. 2). Trazodone effects on MMSE remained significant even when only participants with AD-predicted pathology were included, with non-users declining 2.4-fold faster than trazodone users across an average of 3.75 years. These effects varied in significance when accounting for co-administered medications, retaining significance when accounting for overall concomitant sedative and stimulant use, with non-users declining 1.94-fold faster than trazodone users. Trazodone effects were not significant when accounting only for ChEi use. This latter finding did not reflect confounding effects from correlated administration of trazodone and ChEi (φ= –0.131; p = 0.355). We further performed additional post-hoc analysis to address a possible non-linear decline in MMSE associated with the longer inter-evaluation interval available for trazodone non-users. Still, trazodone non-users declined 2.0-fold faster on the MMSE than trazodone users, at an estimated inter-evaluation interval averaging 3.59 years, with each group retaining the same annual rate of decline (0.3 versus 0.7 points per year on MMSE for trazodone users versus non-users). These effects of trazodone on MMSE were marginally non-significant (p = 0.054, compare to Table 1). When evaluating, however, for sleep mediating effects of trazodone to longitudinal MMSE performance, significant effects were observed for both presence of sleep problems at the baseline visit (p = 0.006) and changes in sleep from baseline (p = 0.006) (Fig. 3), the latter being driven by improvement in sleep on the follow up visit (p = 0.0006) but not for stable (p = 0.326) or worsening of sleep (p = 0.176) in the follow up visit.
Fig.2
Effects of trazodone use on primary outcome (MMSE). Effects of trazodone on MMSE performance between 25 trazodone users and 25 trazodone non-users over an inter-evaluation interval of 4.12 years. Error bars indicate standard error of the mean.
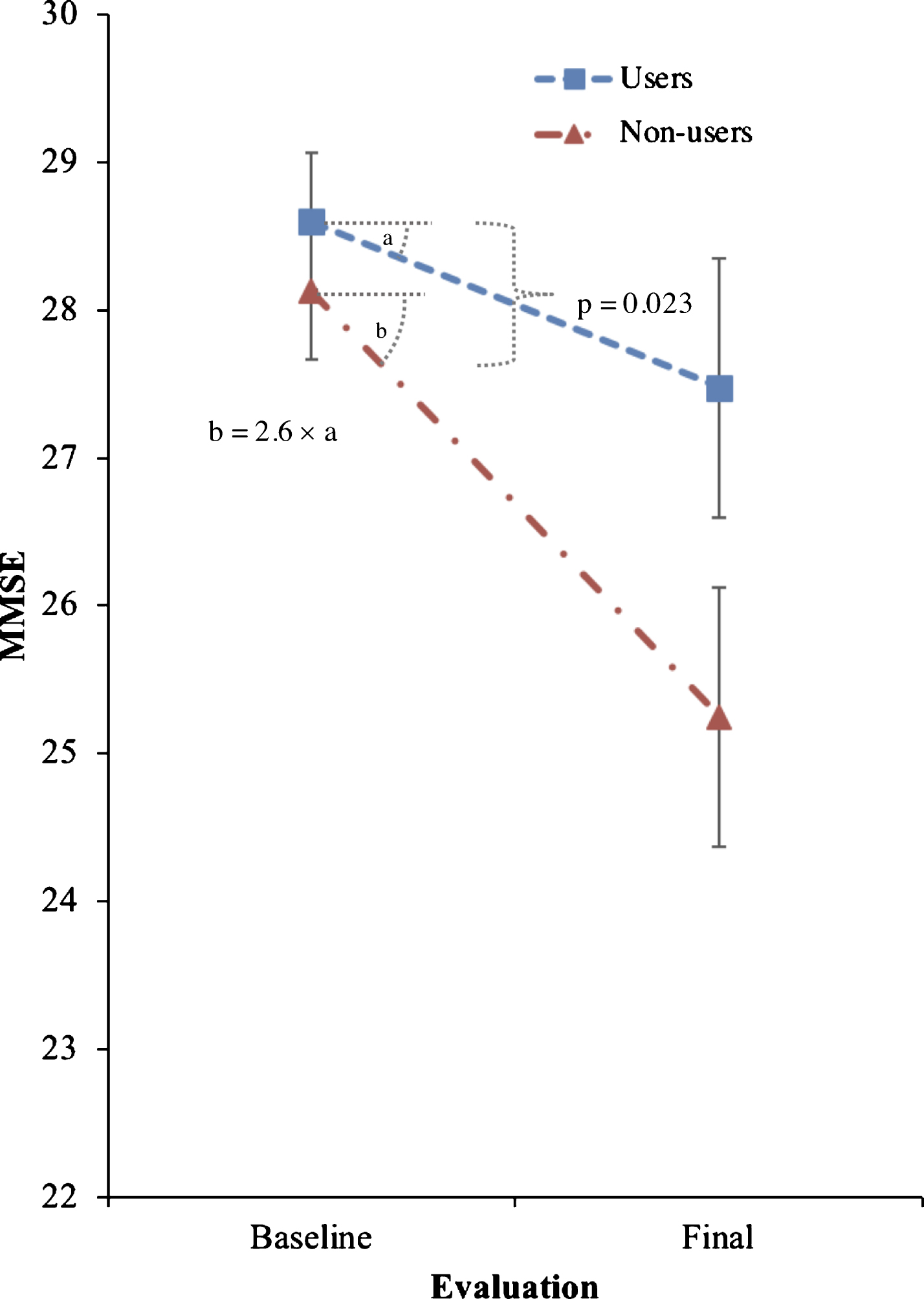
Table 3
Trazodone effects on secondary outcomes
| Cognitive Domain | Task | Participants included interval (users: non-users) | Inter-evaluation (avg. years) | Baseline → final task scores in users versus non-users [metric] | Percent (%) improvement (↑) or decline (↓) compared to baseline in users versus non-users (±SEM) | Fdf, pa |
| Visual Episodic Memory | Benson Complex (10-min delay recognition) | 24: 22 | 4.10 | 10.2 → 11.1 versus 8.7 → 6.8 [correct points] | ↑ 9.2 (±9.9) versus ↓ 21.8 (±11.6) | F1,43 = 7.37, p = 0.005 |
| Verbal Episodic Memoryb | CVLTII (recognition) | 7: 6 | 4.23 | 15.1 → 15.6 versus 15.6 → 15.3 [# correct words] | ↑ 3.6 (±2.6) versus ↓ 1.9 (±2.7) | F1,10 = 3.07, p = 0.055 |
| CVLTII (trial 5 learning) | 7: 6 | 4.23 | 14.6 → 14.0 versus 13.3 → 13.5 [# correct words] | ↓ 3.8 (±7.6) versus ↑ 1.0 (±9.1) | F1,10 = 0.48, p = 0.252 | |
| CVLT (trial 4 learning) | 10: 6 | 2.43 | 6.5 → 6.0 versus 6.1 → 5.3 [# correct words] | ↓ 7.5 (±9.1) versus ↓ 14.0 (±12.5) | F1,13 = 0.22, p = 0.324 | |
| CVLT (10-min delay) | 10: 6 | 2.43 | 5.8 → 3.5 versus 2.4 → 0.8 [# correct words] | ↓ 39.2 (±16.0) versus ↓ 66.0 (±51.1) | F1,13 = 0.36, p = 0.278 | |
| CVLT (recognition) | 10: 6 | 2.43 | 8.1 → 7.8 versus 7.7 → 7.5 [# correct words] | ↓ 3.7 (±6.5) versus ↓ 2.2 (±8.9) | F1,13 = 0.03, p = 0.435 | |
| Executive Function | Stroop Color-Naming | 23: 23 | 4.09 | 81.2 → 80.2 versus 84.5 → 72.1 [# correct color identification] | ↓ 1.2 (±5.7) versus ↓ 14.7 (±5.4) | F1,42 = 5.00, p = 0.015 |
| Stroop Interference | 23: 23 | 4.13 | 45.4 → 44.6 versus 46.6 → 39.6 [# correct color identification] | ↓ 1.7 (±8.6) versus ↓ 15.1 (±8.3) | F1,43 = 2.28, p = 0.069 | |
| Calculations | 24: 22 | 3.83 | 4.6 → 4.5 versus 4.6 → 4.1 [# correct calculations] | ↓ 1.2 (±5.0) versus ↓ 11.5 (±5.2) | F1,43 = 3.04, p = 0.044 | |
| Phonemic Verbal Fluency | 23: 24 | 4.15 | 14.0 → 14.5 versus 16.1 → 14.1 [# unique correct words] | ↑ 3.3 (±10.5) versus ↓ 12.3 (±9.0) | F1,44 = 3.05, p = 0.044 | |
| Semantic Verbal Fluency | 24: 23 | 4.09 | 20.6 → 20.0 versus 19.3 → 15.3 [# unique correct words] | ↓ 3.0 (±7.2) versus ↓ 20.6 (±7.9) | F1,44 = 2.81, p = 0.051 | |
| Design Fluency | 24: 24 | 3.99 | 9.3 → 8.7 versus 9.7 → 10.1 [# unique shapes] | ↓ 6.6 (±10.9) versus ↑ 3.8 (± 10.4) | F1,45 = 0.73, p = 0.198 | |
| Modified Trail-Making B | 23: 23 | 4.00 | 43.6 → 35.0 versus 44.8 → 47.2 [seconds] | ↑ 19.8 (±15.9) versus ↓ 5.4 (±15.9) | F1,44 = 2.25, p = 0.070 | |
| Working Memory | Digit-Span Forward | 21: 12 | 3.22 | 6.8 → 6.8 versus 6.9 → 6.5 [sequence length correctly repeated] | ↑ 0.5 (±4.6) versus ↓ 6.9 (±6.0) | F1,30 = 1.95, p = 0.086 |
| Digit-Span Backward | 24: 22 | 3.98 | 5.0 → 4.4 versus 5.2 → 4.9 [sequence length correctly repeated] | ↓ 13.1 (±6.7) versus ↓ 6.2 (±6.8) | F1,43 = 0.49, p = 0.244 | |
| Disability Score | CDR-SB | 25: 23 | 4.28 | 2.0 → 2.2 versus 2.0 → 3.6 [units] | ↓ 9.9 (±43.1) versus ↓ 78.4 (±43.8) | F1,45 = 2.62, p = 0.056 |
Abbreviations: CVLTII, California Verbal Learning Test – Second Edition; CVLT, California Verbal Learning Test; CDR-SB, Clinical Dementia Rating Scale Sum of Boxes; SEM, Standard Error of the Mean. aBetween-group statistical significance before Bonferroni correction for multiple comparisons (all non-significant after correction). bCVLT administered to participants with MCI or AD diagnoses, and CVLTII to CN participants.
Fig.3
Effects of trazodone use on MMSE are dependent on sleep symptom severity at baseline and on their longitudinal improvement. Post-hoc analyses of trazodone effects on longitudinal MMSE performance in 25 trazodone users and 25 trazodone non-users when accounting for (A) presence or (B) absence of sleep complaints at baseline evaluation, and (C) changes (improvement, worsening or stability) in sleep complaints between baseline and final evaluations. MMSE inter-evaluation interval was 4.12 years. Error bars indicate standard error of the mean. See text for details.
Secondary outcomes on processing speed, disability scores, and visual recall also worsened faster in trazodone non-users, though none of the results were significant after correcting for multiple comparisons. All but three secondary outcomes revealed a trend that trazodone was beneficial in delaying cognitive decline. Prior to correcting for multiple comparisons, most notable were the apparent beneficial effects of trazodone in short-term visual memory, processing speed, calculations, and phonemic fluency (Table 3).
DISCUSSION
This study was conducted on the premise that SWS enhancement delays amyloid-β aggregation in AD models and improves sleep-mediated episodic memory consolidation through synaptic potentiation, both of which may delay long-term cognitive decline. Indeed, our results, by suggesting an association between longitudinal trazodone use and delayed cognitive decline, are supportive of this hypothesis, since the rate of decline in trazodone users was less than half (39.01%) of that of non-users on global measures of cognition over an estimated four-year period. Notably, it was participants treated with trazodone with concomitant baseline sleep complaints, especially those who reported improvement in sleep quality over time, who had delayed cognitive decline compared to patients with sleep disruption that were not on trazodone. After post-hoc analyses focusing on shorter inter-evaluation intervals, rates of decline remained relatively unchanged for each group, verifying a quasi-linear effect of inter-evaluation intervals to our primary outcome. However, effects were marginally non-significant, likely indicating a slightly underpowered study in revealing possible trazodone benefits for shorter follow-up periods. Overall, the above findings support for the first time the consideration of trazodone for clinical use, not only toward improving quality of life in older adults by increasing sleep duration [10], but also as an intervention against cognitive decline. The findings are also in keeping with the therapeutic concept of consolidating brain rhythms of sleep and wakefulness toward delaying neurodegeneration and optimizing synaptic plasticity [16]. On this basis, sleep consolidation, as defined by integrity of sleep continuity and architecture, can be directly achieved through medications that consolidate sleep rhythms [26], as well as indirectly by consolidating rhythms of wakefulness that in turn allow deeper rhythms in subsequent sleep [27, 28]. Through this paradigm, SWS enhancers are only part of a gestalt approach toward optimizing sleep-wake rhythm separation, in which light therapy and daytime activities may also be useful [29].
Our findings partially contrast with previous results on AD, in which short-term trazodone use was not associated with cognitive benefits despite increased sleep duration [13]. The most likely explanation of such a discrepancy is the difference in duration of trazodone use and follow-up between those studies and ours, i.e., two weeks versus four years. This discrepancy also negates the argument that the cognitive benefits observed in our study were the result of a single or a few nights of better sleep allowing people to be more vigilant the following day, and instead reflect a longitudinal effect that is associated with chronic trazodone use. Further support for this argument are our results indicating delayed decline across cognitive domains, beyond executive and vigilance tasks that are typically improved after a good night’s sleep [30]. One explanation on why trazodone cognitive benefits present longitudinally, and not after only few weeks of use, is that it may have protective effects on pathology progression. Specifically, it is expected that significant amyloid-β aggregation or degeneration-dependent effects will be reflected through progression of cognitive decline over longer intervals than a couple of weeks. Similarly, it is likely that a longer inter-evaluation interval is required to observe cognitive benefits mediated by synaptic plasticity during sleep. To date, direct evaluation of overnight sleep-mediated memory consolidation through SWS enhancers has not been performed in AD.
Based on previous studies that indicate a relationship between decreased SWS, cognitive deficits, and amyloid-β pathology as revealed through molecular PET even in cognitively non-impaired individuals, we included a CN group in addition to the two patient groups [6]. Longitudinal trazodone use was associated with delayed cognitive decline across diagnostic groups in our study, supporting its possible utility as a treatment from cognitively non-impaired to mildly-impaired patients. None of our patients had moderate or severe dementia to allow for inferences of trazodone use on more advanced disease.
Trazodone group comparative delay in cognitive decline persisted when accounting for concomitant use of medications that modulate brain rhythms, such as stimulants and sedatives, which may signify trazodone’s contribution to sleep-wake and ultradian rhythm consolidation toward delayed cognitive decline [16]. However, when specifically accounting for ChEi use, trazodone users did not significantly benefit in delayed cognitive decline compared to non-users. This particular finding does not fully detract from our hypothesis that trazodone use may delay cognitive decline, considering that there may be 1) decreased statistical power when accounting for ChEi, 2) common mechanistic effects on sleep-wake rhythm consolidation between trazodone and ChEi, or 3) ceiling effects on cognitive outcomes for ChEi users. Specifically, ChEi users benefit from improved attention and alertness through enhancement of cholinergic transmission and may thus perform at ceiling throughout the follow-up period [31]. Indeed, the observed rate of decline in MMSE for all non-users when accounting for ChEi use was slower than anticipated, at 0.60 points per year (Table 2), compared to the one-point decline per year observed in longitudinal trials on ChEi use in mild AD [32, 33]. Furthermore, for 25–40% of patients with mild to moderate AD who suffer from excessive daytime somnolence, daytime administration of ChEi can help consolidate sleep-wake rhythms by directly promoting daytime wakefulness [34, 35]. In turn, direct wakefulness consolidation indirectly enhances subsequent SWS, and may thus facilitate the hypothesized mechanism for trazodone’s benefits [27–29, 36]. Even if ChEi have shared effects with trazodone toward delaying cognitive decline, trazodone has certain additional useful clinical qualities. In addition to directly improving sleep consolidation, the current results indicate a potential cognitive benefit for patients with MCI that is not verified in ChEi trials, where benefits are more definitively shown for mild to moderate AD. Whether trazodone’s beneficial effects are indeed mediated through neuroprotection, however, remains to be clarified. Additionally, trazodone does not have the prevalent side effects of ChEi, such as insomnia, diarrhea, or bradycardia, making it even more favorable for patients who also do not tolerate ChEi.
Effects of trazodone in all secondary outcomes were non-significant after correction for multiple comparisons but reveal a trend of delayed cognitive decline for almost all measures. We specifically tested trazodone effects on short-term memory and executive tests, considering known involvement of hippocampal and prefrontal areas in sleep-mediated effects via SWS enhancement on these cognitive domains [3, 37]. Even though the results were not significant, the observed trend is promising in pursuing tailored studies that are powered to identify a potential beneficial effect in the specific cognitive domains. It is worth keeping in mind that cognitive benefits via SWS enhancement may vary depending on a patient’s specific syndrome (e.g., late age-of-onset AD, early age-of-onset AD, logopenic variant primary progressive aphasia, posterior cortical atrophy). Additionally, they may be dependent on a person’s age, with younger patients potentially benefitting most in executive function versus older ones in short-term memory. In our cohort of predominantly older amnestic patients, a trend was observed in both executive and short-term memory tasks.
There are also inherent limitations to our study, primarily due to its retrospective nature. Ideally, we would prospectively standardize medication dosage and timing, after randomized allocation between groups, and monitor medication compliance across a predefined time period. For example, benefits observed in the trazodone group may reflect better overall medical management of participants by their physicians and delayed cognitive decline due to unaccounted factors. Possible adverse effects of trazodone were not explicitly reported during the assessment of participants’ medication regimen. It is thus possible there is a biased selection against people who tried but did not tolerate trazodone for inclusion in the trazodone group, but who could have been included in the trazodone non-user group. Such participants may be more resistant in achieving better sleep consolidation through SWS enhancers. Alternatively, there may be different metabolism of trazodone in these participants, given that prior work indicates its active metabolite may have different effects on alertness compared to trazodone itself [38], a possibility that personalized medicine may be able to address. Eventually, optimal accounting for confounders necessitates prospective double-blind randomized trials to confirm that differential rates of cognitive decline are directly caused by trazodone. Such prospective studies, ideally incorporating interval cognitive evaluations, could also answer whether potential long-term trazodone benefits are due to continuous modulation of brain networks, or whether they primarily reflect early treatment period effects. Even though there are no prospective long-term studies of trazodone for insomnia in elders, long-term persistent benefits have been observed in depression [39].
Another limitation pertains to whether the observed trazodone effects are primarily mediated through SWS enhancement. In this study, quantified data of SWS were not available for correlating to rate of cognitive decline. Additionally, it is possible that trazodone effects could be mediated through its antidepressant benefits. We do not believe the latter is the case, since trazodone is rarely, if ever, given as an antidepressant in our cohort. Moreover, antidepressant doses of trazodone are usually much higher than doses for insomnia that were prescribed for our participants, and often relate to carry-over cognitive deficits the following day, albeit less severe than those observed with certain other antidepressants [40–43]. Nonetheless, a prospective study would be the optimal approach in controlling potential antidepressant-mediated cognitive benefits of trazodone. An alternative possibility of the observed trazodone benefits could be explained by its possible neuroprotective effects via inhibition of the phosphorylated eukaryotic initiation factor 2α signaling pathway of the unfolded protein response (eIF2α-P/UPR) [44]. Specifically, in the MAPT P301L mouse model of tauopathy, trazodone use was associated with less memory deficits, preserved hippocampal neuronal counts, delayed gross brain atrophy, and prolonged survival [44]. Interestingly, the eIF2α-P/UPR pathway is also induced during sleep deprivation of wild type mice, providing an additional mechanistic explanation through which improving disturbances in sleep may be neuroprotective [45]. This latter inference, that potentially beneficial trazodone effects on cognition may be mediated by reducing sleep disturbance, is supported by the results from our post-hoc exploratory analyses indicating delayed cognitive decline in trazodone users with baseline sleep problems and, even more, in trazodone users who improved their sleep on follow-up evaluation (Fig. 3). A fine distinction should be highlighted on the above observation in that, although trazodone effects on cognition were mediated by improvement in sleep, which in turn raises the possibility that cognitive benefits may relate to improved sleep alone irrespective to trazodone use, the post-hoc analysis also indicates that participants who longitudinally improved their sleep while on trazodone had proportionally less cognitive decline compared to participants who improved their sleep but were not on trazodone (Fig. 3c). Given that the current retrospective study and group matching were not structured to answer mechanisms of trazodone benefits, nor answer whether longitudinally improving sleep in general delays cognitive decline, the above post-hoc observations should be interpreted with caution until prospective intervention studies are completed. Nonetheless, speculating on the current findings, it is possible that the improvement in sleep symptoms could serve as a proxy for trazodone effects in general. One such effect to consider is the well-established SWS enhancement achieved through trazodone [12, 15], which in turn shows promise towards neuroprotection [16]. If this finding replicates in future prospective studies, then the entire class of medications decreasing the degree of sleep disturbance, and especially those promoting SWS, can be considered in the prevention of cognitive decline. Such beneficial effects may be achieved mechanistically via modulation of both amyloid-β and tau pathologic burden, as well as optimization of sleep-mediated synaptic potentiation.
In summary, the pervasiveness and consistency of the observed trazodone effects, especially after considering disease severity and concomitant medication use, encourages further investigation into its effectiveness on preserving cognition via sleep-wake rhythm consolidation. Implications on clinical practice involve improving quality of life for patients who experience sleep disturbances that, if untreated, may promote future amyloid-β deposition. Furthermore, potential cognitive benefits of trazodone may be applicable to both cognitively intact older adults and mildly impaired patients with dementia. The effectiveness of trazodone, possibly achieved by decreasing amyloid-β aggregation and pathologic tau burden, as well as by improving sleep-mediated memory consolidation, can be further explored through controlled clinical trials.
ACKNOWLEDGMENTS
This study was supported by the American Brain Foundation, the Alzheimer’s Association, the J.D. French Foundation, the Tau Consortium, and the National Institute on Aging grants P01 AG1972403 and P50 AG023501.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/18-1145).
REFERENCES
[1] | Yaffe K , Falvey CM , Hoang T ((2014) ) Connections between sleep and cognition in older adults. Lancet Neurol 13: , 1017–1028. |
[2] | Ohayon MM , Carskadon MA , Guilleminault C , Vitiello MV ((2004) ) Meta-analysis of quantitative sleep parameters from childhood to old age in healthy individuals: Developing normative sleep values across the human lifespan. Sleep 27: , 1255–1273. |
[3] | Fogel SM , Smith CT , Beninger RJ ((2009) ) Evidence for 2-stage models of sleep and memory: Learning-dependent changes in spindles and theta in rats. Brain Res Bull 79: , 445–451. |
[4] | Ribeiro S , Gervasoni D , Soares ES , Zhou Y , Lin SC , Pantoja J , Lavine M , Nicolelis MA ((2004) ) Long-lasting novelty-induced neuronal reverberation during slow-wave sleep in multiple forebrain areas. PLoS Biol 2: , E24. |
[5] | Roh JH , Jiang H , Finn MB , Stewart FR , Mahan TE , Cirrito JR , Heda A , Snider BJ , Li M , Yanagisawa M , de Lecea L , Holtzman DM ((2014) ) Potential role of orexin and sleep modulation in the pathogenesis of Alzheimer’s disease. J Exp Med 211: , 2487–2496. |
[6] | Mander BA , Marks SM , Vogel JW , Rao V , Lu B , Saletin JM , Ancoli-Israel S , Jagust WJ , Walker MP ((2015) ) beta-amyloid disrupts human NREM slow waves and related hippocampus-dependent memory consolidation. Nat Neurosci 18: , 1051–1057. |
[7] | Guarnieri B , Adorni F , Musicco M , Appollonio I , Bonanni E , Caffarra P , Caltagirone C , Cerroni G , Concari L , Cosentino FI , Ferrara S , Fermi S , Ferri R , Gelosa G , Lombardi G , Mazzei D , Mearelli S , Morrone E , Murri L , Nobili FM , Passero S , Perri R , Rocchi R , Sucapane P , Tognoni G , Zabberoni S , Sorbi S ((2012) ) Prevalence of sleep disturbances in mild cognitive impairment and dementing disorders: A multicenter Italian clinical cross-sectional study on 431 patients. Dement Geriatr Cogn Disord 33: , 50–58. |
[8] | McCleery J , Cohen DA , Sharpley AL ((2014) ) Pharmacotherapies for sleep disturbances in Alzheimer’s disease. Cochrane Database Syst Rev 3: , CD009178. |
[9] | Scoralick FM , Louzada LL , Quintas JL , Naves JO , Camargos EF , Nobrega OT ((2017) ) Mirtazapine does not improve sleep disorders in Alzheimer’s disease: Results from a double-blind, placebo-controlled pilot study. Psychogeriatrics 17: , 89–96. |
[10] | Suzuki H , Yamadera H , Nakamura S , Endo S ((2002) ) Effects of trazodone and imipramine on the biological rhythm: An analysis of sleep EEG and body core temperature. J Nippon Med Sch 69: , 333–341. |
[11] | Montgomery I , Oswald I , Morgan K , Adam K ((1983) ) Trazodone enhances sleep in subjective quality but not in objective duration. Br J Clin Pharmacol 16: , 139–144. |
[12] | Camargos EF , Louzada LL , Quintas JL , Naves JO , Louzada FM , Nobrega OT ((2014) ) Trazodone improves sleep parameters in Alzheimer disease patients: A randomized, double-blind, and placebo-controlled study. Am J Geriatr Psychiatry 22: , 1565–1574. |
[13] | Camargos EF , Quintas JL , Louzada LL , Naves JO , Furioso AC , Nobrega OT ((2015) ) Trazodone and cognitive performance in Alzheimer disease. J Clin Psychopharmacol 35: , 88–89. |
[14] | Watson BO , Buzsáki G ((2015) ) Sleep, memory & brain rhythms. Daedalus 144: , 67–82. |
[15] | Walsh JK ((2009) ) Enhancement of slow wave sleep: Implications for insomnia. J Clin Sleep Med 5: , S27–S32. |
[16] | Karageorgiou E , Vossel KA ((2017) ) Brain rhythm attractor breakdown in Alzheimer’s disease: Functional and pathologic implications. Alzheimers Dement 13: , 1054–1067. |
[17] | Beekly DL , Ramos EM , Lee WW , Deitrich WD , Jacka ME , Wu J , Hubbard JL , Koepsell TD , Morris JC , Kukull WA , NIA Alzheimer’s Disease Centers ((2007) ) The National Alzheimer’s Coordinating Center (NACC) database: The Uniform Data Set. Alzheimer Dis Assoc Disord 21: , 249–258. |
[18] | McKhann GM , Knopman DS , Chertkow H , Hyman BT , Jack CR , Kawas CH , Klunk WE , Koroshetz WJ , Manly JJ , Mayeux R , Mohs RC , Morris JC , Rossor MN , Scheltens P , Carrillo MC , Thies B , Weintraub S , Phelps CH ((2011) ) The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7: , 263–269. |
[19] | Petersen RC ((2004) ) Mild cognitive impairment as a diagnostic entity. J Intern Med 256: , 183–194. |
[20] | Glass J , Lanctot KL , Herrmann N , Sproule BA , Busto UE ((2005) ) Sedative hypnotics in older people with insomnia: Meta-analysis of risks and benefits. BMJ 331: , 1169. |
[21] | Clay E , Falissard B , Moore N , Toumi M ((2013) ) Contribution of prolonged-release melatonin and anti-benzodiazepine campaigns to the reduction of benzodiazepine and Z-drugs consumption in nine European countries. Eur J Clin Pharmacol 69: , 1–10. |
[22] | Yeh AY , Pressler SJ , Giordani BJ , Pozehl BJ , Berger AM ((2018) ) Integrative review of the relationship between sleep disturbances and episodic memory in older adults. Biol Res Nurs 20: , 440–451. |
[23] | Woods SP , Delis DC , Scott JC , Kramer JH , Holdnack JA ((2006) ) The California Verbal Learning Test–second edition: Test-retest reliability, practice effects, and reliable change indices for the standard and alternate forms. Arch Clin Neuropsychol 21: , 413–420. |
[24] | Kramer JH , Jurik J , Sha SJ , Rankin KP , Rosen HJ , Johnson JK , Miller BL ((2003) ) Distinctive neuropsychological patterns in frontotemporal dementia, semantic dementia, and Alzheimer disease. Cogn Behav Neurol 16: , 211–218. |
[25] | Berg L ((1988) ) Clinical Dementia Rating (CDR). Psychopharmacol Bull 24: , 637–639. |
[26] | Walsh CM , Blackwell T , Tranah GJ , Stone KL , Ancoli-Israel S , Redline S , Paudel M , Kramer JH , Yaffe K ((2014) ) Weaker circadian activity rhythms are associated with poorer executive function in older women. Sleep 37: , 2009–2016. |
[27] | Huber R , Ghilardi MF , Massimini M , Tononi G ((2004) ) Local sleep and learning. Nature 430: , 78–81. |
[28] | Frase L , Piosczyk H , Zittel S , Jahn F , Selhausen P , Krone L , Feige B , Mainberger F , Maier JG , Kuhn M , Kloppel S , Normann C , Sterr A , Spiegelhalder K , Riemann D , Nitsche MA , Nissen C ((2016) ) Modulation of total sleep time by transcranial direct current stimulation (tDCS). Neuropsychopharmacology 41: , 2577–2586. |
[29] | Hanford N , Figueiro M ((2013) ) Light therapy and Alzheimer’s disease and related dementia: Past, present, and future. J Alzheimers Dis 33: , 913–922. |
[30] | Alhola P , Polo-Kantola P ((2007) ) Sleep deprivation: Impact on cognitive performance. Neuropsychiatr Dis Treat 3: , 553–567. |
[31] | Birks J ((2006) ) Cholinesterase inhibitors for Alzheimer’s disease. Cochrane Database Syst Rev 1: , CD005593. |
[32] | Wattmo C , Minthon L , Wallin AK ((2016) ) Mild versus moderate stages of Alzheimer’s disease: Three-year outcomes in a routine clinical setting of cholinesterase inhibitor therapy. Alzheimers Res Ther 8: , 7–016. |
[33] | Droogsma E , van Asselt D , Diekhuis M , Veeger N , van der Hooft C , De Deyn PP ((2015) ) Initial cognitive response to cholinesterase inhibitors and subsequent long-term course in patients with mild Alzheimer’s disease. Int Psychogeriatr 27: , 1323–1333. |
[34] | Song HR , Woo YS , Wang HR , Jun TY , Bahk WM ((2013) ) Effect of the timing of acetylcholinesterase inhibitor ingestion on sleep. Int Clin Psychopharmacol 28: , 346–348. |
[35] | Moran M , Lynch CA , Walsh C , Coen R , Coakley D , Lawlor BA ((2005) ) Sleep disturbance in mild to moderate Alzheimer’s disease. Sleep Med 6: , 347–352. |
[36] | Van Dort CJ , Baghdoyan HA , Lydic R ((2009) ) Adenosine A(1) and A(2A) receptors in mouse prefrontal cortex modulate acetylcholine release and behavioral arousal. J Neurosci 29: , 871–881. |
[37] | Nofzinger EA , Buysse DJ , Miewald JM , Meltzer CC , Price JC , Sembrat RC , Ombao H , Reynolds CF , Monk TH , Hall M , Kupfer DJ , Moore RY ((2002) ) Human regional cerebral glucose metabolism during non-rapid eye movement sleep in relation to waking. Brain 125: , 1105–1115. |
[38] | Lawlor BA , Radcliffe J , Martinez R , Sunderland T , Murphy DL ((1997) ) Plasma levels of m-chlorophenylpiperazine following single oral dose administration of m-chlorophenylpiperazine and trazodone in human volunteers. Biol Psychiatry 41: , 756–757. |
[39] | Fabre LF , Feighner JP ((1983) ) Long-term therapy for depression with trazodone. J Clin Psychiatry 44: , 17–21. |
[40] | Burns M , Moskowitz H , Jaffe J ((1986) ) A comparison of the effects of trazodone and amitriptyline on skills performance by geriatric subjects. J Clin Psychiatry 47: , 252–254. |
[41] | Moskowitz H , Burns MM ((1986) ) Cognitive performance in geriatric subjects after acute treatment with antidepressants. Neuropsychobiology 15: (Suppl 1), 38–43. |
[42] | Rakel RE ((1987) ) The greater safety of trazodone over tricyclic antidepressant agents: 5-year experience in the United States. Psychopathology 20: (Suppl 1), 57–63. |
[43] | Gerner RH ((1987) ) Geriatric depression and treatment with trazodone. Psychopathology 20: (Suppl 1), 82–91. |
[44] | Halliday M , Radford H , Zents KAM , Molloy C , Moreno JA , Verity NC , Smith E , Ortori CA , Barrett DA , Bushell M , Mallucci GR ((2017) ) Repurposed drugs targeting eIF2alpha-P-mediated translational repression prevent neurodegeneration in mice. Brain 140: , 1768–1783. |
[45] | Naidoo N , Giang W , Galante RJ , Pack AI ((2005) ) Sleep deprivation induces the unfolded protein response in mouse cerebral cortex. J Neurochem 92: , 1150–1157. |


