
Alzheimer’s Amyloidopathy: An Alternative Aspect
Abstract
The ‘amyloid hypothesis’ dominates Alzheimer’s disease (AD) research but has failed to deliver effective therapies. Amyloid precursor protein (APP) and presenilin-1 (PSEN1) genetic mutations are undoubtedly pathogenic, albeit by unclear mechanisms. Conversely, high dose B-vitamins convincingly slow brain atrophy in a pre-stage state of sporadic AD. Here we suggest a link between sporadic and genetic AD: 1) Increased serum homocysteine, a marker of B-vitamin deficiencies, is a significant risk factor for sporadic AD. It also correlates with elevated levels of antichymotrypsin, a serine protease inhibitor. 2) Family members with codon 717 APP mutations and dementia have low serum vitamin B12 values. Overexpression of the APP domain coding for a Kunitz type serine protease inhibitor might explain this. 3) PSEN1 mutations disrupt lysosomal function due to reduced proteolytic activity. They also trap cobalamin (B12) within lysosomes, leading to intracellular deficiency of the vitamin. In summary, APP and PSEN1 mutations both confer a risk for reduced protease activity and B12 bio-availability. Comparably, sporadic AD features a constellation of increased protease inhibition and B-vitamin deficiencies, the central part of which is believed to be B12. These concordant observations in three disparate AD etiologies suggest a common neuropathogenic pathway. This hypothesis is evaluable in laboratory and clinical trials.
INTRODUCTION
In 1991, amyloid precursor protein (APP) gene mutations were found in families with early-onset Alzheimer’s disease (AD). Three different mutations at codon 717 implied pathogenicity; amyloid became a prime focus of AD research. Amyloid-β protein precursor (AβPP) is a transmembrane protein cleaved by two proteases (β- and γ-secretase) into amyloid-β (Aβ). This is regarded as a central pathogenic event in AD [1]. According to the ‘amyloid hypothesis’ Aβ is neurotoxic, its deposition leading to amyloid plaques, neuronal death, and ultimately dementia. This idea was strengthened by finding γ-secretase (presenilin) mutations in other families with early-onset AD, and from the utility of cerebrospinal fluid Aβ levels as a marker of AD diagnosis. In a whirl of optimism, the hypothesis prompted development of anti-amyloid drugs to treat all patients with AD, regardless of age or the presence of such mutations.
Sadly, more than 25 years later, this hypothesis has not delivered effective therapies. Perhaps its focus was misguided [2, 3]? Aβ itself is barely toxic [4]; even its capacity to induce oxidative stress remains controversial [5]. Importantly, AβPP has trophic functions and is involved in neuronal survival, neurite outgrowth, and neurorepair in the brain [6]. We suggest that Aβ deposition does not drive the pathogenic process but is a consequence of the disease state. APP and presenilin mutations are undoubtedly pathogenic but the resultant increased Aβ deposition is likely a relatively innocent bystander—the mutations leading to AD by other mechanisms.
VITAMIN B12 AND FAMILIAL AD
A serendipitous finding was made in one of the families with an APP gene mutation; it was associated with lower serum vitamin B12 values [7]. We suggested this might result from systemic protease inhibition [8]. An insert in the APP sequence has striking homology to the Kunitz family of serine protease inhibitors. These are expressed in most tissues and inhibit the activity of proteases such as trypsin and chymotrypsin. If overexpressed, they could interfere with protein-bound transfer of vitamin B12 posing a risk for its deficiency.
Presenilin-1 (PSEN1) is required for lysosomal proteolysis, and lysosomal function is disrupted by AD-related PSEN1 mutations [9]. Lysosomes are a key component for endocytosis of protein-bound molecules; in defective proteolysis proteins accumulate detrimentally in lysosomes. This was recently explored by an Australian group [10]. Firstly, in vitro neuroblastoma cells were transfected with the ‘Swedish’ APP mutation and then treated with a protease inhibitor. Secondly, wild type mice were compared with transgenic (APPxPSEN1) mice. Radio-labelled cobalamin (B12) was used to study lysosomal function. Both experiments showed that cobalamin became trapped within lysosomes, i.e., intracellular cobalamin availability is reduced. This is analogous to the cobalamin F group of hereditary B12 deficiency (see Fig. 1). In infancy this leads to failure to thrive and mild to severe developmental delay, although there is a good response to B12 supplementation if started at an early stage [11].
Fig.1
Simplified scheme of entry and utilization of vitamin B12 by mammalian cells. TC is transcobalamin degraded in the lysosome; Cbl-F is the site causing the Cobalamin F group of hereditary B12 deficiency; (Co1 +) indicates valency of the cobalt atom in the cobalamin/B12 molecule; X is substrate transmethylated by a methyltransferase.
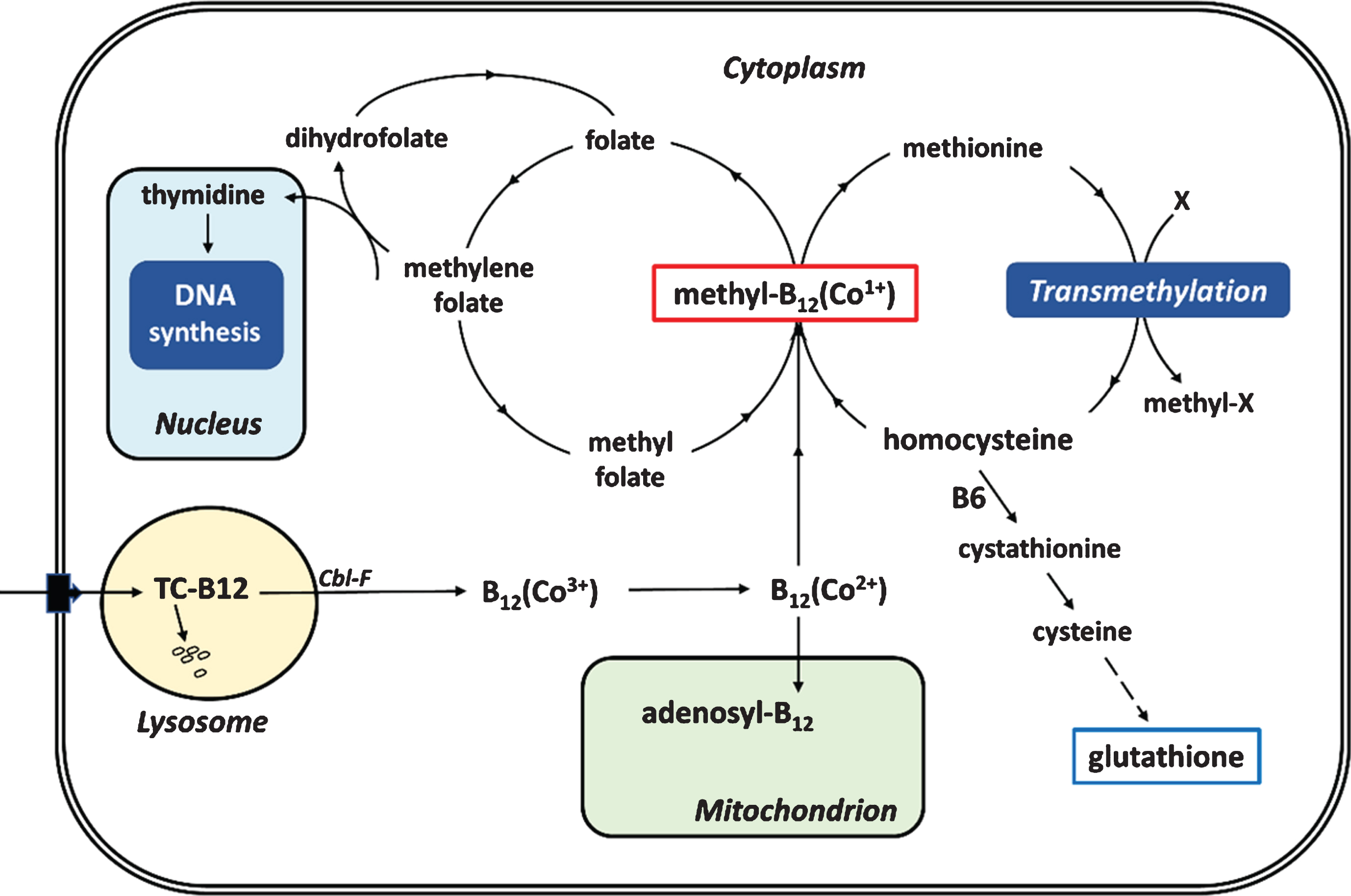
In summary, APP and PSEN1 mutations confer a risk for B12 deficiency, presumably due to reduced proteolytic activity.
Of course, the vast majority of AD patients have neither APP nor PSEN1 mutations; the disease occurs sporadically, with a relatively late age of onset. Elevated antichymotrypsin and homocysteine blood levels are consistently reported in sporadic AD. In a meta-analysis of 13,000 AD patients and a similar number of healthy controls, antichymotrypsin and homocysteine were significantly elevated [12]. However, these have never been considered to be directly related.
HOMOCYSTEINE AND SPORADIC AD
Reports of low vitamin B12 and folate blood levels in AD prompted investigation of the utility of serum homocysteine as a marker of such deficiencies in dementia. Several long-term prospective studies in healthy populations yielded startling results: elevated homocysteine predicts dementia up to several decades before its onset [13–15]. ‘Treating’ AD with B vitamins became an interesting option, but a demanding challenge. Established AD is too advanced to achieve evident effects, and it is difficult to evaluate how such treatment modifies brain function in a clinical trial. Erroneous methodology can give false negative outcomes [16, 17].
The Oxford-based OPTIMA group formulated a careful design to overcome such pitfalls. They focused on patients with mild cognitive impairment (MCI), regarded as a ‘pre-stage’ state with a highly increased risk for developing AD. They also used magnetic resonance imaging as a highly sensitive and accurate tool to detect ‘brain atrophy rate changes’ as a primary outcome measure.
In 2010, OPTIMA reported on their 24-month randomized double-blind controlled clinical trial in which MCI patients (n = 168; age > 70 years) were given either placebo or vitamins B12, B6 and folic acid—all necessary for optimal homocysteine metabolism [18]. As part of normal aging, our brains lose volume of around 0.5% annually [19]. In the OPTIMA cohort, brain volume of the placebo group diminished by 1.08% per year, compared with 0.76% for the treatment group (p < 0.001); i.e., brain volume reduction was 30% slower in treated patients. In other words, MCI patients treated with B vitamins significantly slowed their rate of brain volume loss. Global cognition, episodic and semantic memory also improved in treated patients with supra-median homocysteine levels [20]. Directed acyclic graph analysis showed that vitamin B12 was the main driver in the protective effect of B vitamins in slowing both brain atrophy and cognitive decline in this study [21].
We believe this trial is ‘epoch-making’ in AD prevention strategies. Effective homocysteine metabolism is crucial for all cells (Fig. 1). B vitamin adequacy supports a chain of vital reactions: methyl-folate is de-methylated to support DNA synthesis (its methyl group is transferred to B12); methyl-B12 re-methylates homocysteine to methionine which, in its activated form S-adenosyl-methionine, is the trans-methylating agent for hundreds of different methyl acceptors. Moreover, vitamin B6 enables a proportion of homocysteine to be metabolized to cysteine, a precursor of the key cellular antioxidant glutathione.
If any of these reactions are compromised, cells undergo cytostatic involution: in the brain, an increasing number of neurons collapse [22]. Dementia is the ultimate consequence. Essentially, this is why insufficiency of these particular B-vitamins is more closely related to AD brain cell death than is amyloid deposition.
A recent meta-analysis included almost 22,000 cognitively healthy elderly individuals treated with B vitamins, including B12, for up to 7 years. It was concluded that although B vitamins did not improve global cognitive function, they did improve specific subdomains (information processing, sensorimotor speed and short term memory) when administered for over 3 years. However, no clear conclusion can yet be drawn as to whether B vitamins can ‘treat’ AD if administered sufficiently early [16].
Other important questions remain. How is increased homocysteine, and thus the need for B-vitamins, explicable in relation to sporadic AD? And how does protease inhibition exactly link familial and sporadic AD? Perhaps oxidative stress or elevated serine protease inhibition, such as by antichymotrypsin, or both, are key pieces of the missing picture in sporadic AD?
ANTICHYMOTRYPSIN AND SPORADIC AD
Antichymotrypsin is an acute-phase protein induced during inflammation. Of hepatic origin, it inhibits serine proteases and protects tissues from excess proteolytic damage. High levels occur in a substantial proportion of patients with sporadic AD, albeit with unexplained consequences. The largest study (n = 359) found that antichymotrypsin increases in parallel with disease progression [23].
Antichymotrypsin is expressed in astrocytes, and found in amyloid plaques, tightly bound to Aβ [24]. Interestingly, a secreted form of Kunitz type serine protease inhibitor, nexin-II, is a potent antichymotrypsin showing identity to AβPP [25]. The exact origin of antichymotrypsin in plaques has, to our knowledge, not been determined [26].
Elevated plasma antichymotrypsin in sporadic AD might reflect its reactivity towards chronic low-grade inflammation. Increased homocysteine levels, in turn, may either arise from an independent oxidative stress reaction caused by such inflammation, or from antichymotrypsin disrupting cleavage of bonds between B12 and its several carrier proteins. Other than as a transient phenomenon following injection, B12 is not found in the ‘free’ state. Like all co-enzymes, it is attached to protein in its natural form. Released from food by saliva, B12 and other corrinoids can be considered as ‘rough diamonds’ to be carefully sorted, selected and finally delivered as ‘crown jewels’ for co-enzyme functions in their target cells (see Fig. 1). This complex process includes at least 14 proteins, functioning as carriers, receptors or enzymes, for which separate genes or complementation groups have been identified in patients with inherited B12 disorders [27, 28].
Importantly, this intricate B12 trafficking is a multistep process in terms of repeated protein binding and proteolysis (Table 1). For each repetition, B12 is preferentially selected and attached to yet another binder protein and then carried to a location where the binder is degraded and B12 is released. For a minireview, see Gherasim et al. [29].
Table 1
Binding proteins and their degradation as a multistep process necessary for delivery of vitamin B12 to its ultimate target cells
| Compartments | Processing events |
| In the mouth and stomach | B12 is released from food and bound by Haptocorrin, a salivary glycoprotein with high affinity and broad specificity |
| The upper intestinal tract of duodenum and jejunum | Pancreatic proteases degrade Haptocorrin and release B12, which is subsequently bound to Intrinsic factor, a second glycoprotein with high specificity |
| In the enterocytes of ileum intestine mucosa | Intrinsic factor is degraded in the lysosome, and B12 is released into the blood stream |
| In the blood stream | B12 is associated with two separate carriers in the blood: Haptocorrin for passive storage in the circulating blood, and Transcobalamin as an active carrier that binds B12 avidly and mediates its transport into target cells |
| In the target cell | Transcobalamin is degraded in the lysosome, and B12 is released to be further processed intracellularly (see Fig. 1) |
Our hypothesis is based on the concept that impaired proteolysis in any of the compartments in Table 1 predisposes to the development of B12 deficiency. Some proof already exists. For example, trafficking events in the upper gastrointestinal tract are defective in pancreatic insufficiency, since three pancreatic proteases (trypsin, chymotrypsin, and elastase) are required to partially degrade haptocorrin and release B12 [30, 31].
Elevated antichymotrypsin is therefore a candidate for causing B12 deficiency due to insufficient proteolysis and aberrant protein binding at the blood and/or the target cell/lysosomal level, and its potential to interfere with carriage and delivery of B12 into the brain requires further investigation.
CONCLUSION
AβPP is important in maintaining the aging brain. Genetic mutations in APP and PSEN1 lead to pre-senile AD. However, solely focusing on ’anti-amyloid therapy’ might be counter-productive in discovering a panacea for dementia.
Conversely B12 deficiency, as part of a broader scheme of deficiencies relating to homocysteine and folate metabolism, is a feature of both genetic and sporadic AD; it perhaps reflects reduced proteolytic activity.
Our hypothesis makes testable predictions and suggestions for future research. Additional confirmatory studies of the effects of APP and PSEN1 mutations on B12 intracellular processing would be helpful. It would also be valuable to assess the vitamin B12 and homocysteine status of a large cohort of patients with familial AD, and to conduct clinical trials of B vitamins on such patients.
It is worth noting additional ‘downstream’ effects attributable to this hypothesis and the resulting elevated homocysteine levels. There is clinical and experimental evidence linking hyperhomocysteinemia with tau hyperphosphorylation [32–34]. Tau hyperphosphorylation and its subsequent aggregation results in the other key pathological feature of AD, neurofibrillary tangles. Elevated homocysteine might also contribute to protein aggregation and subsequent neurodegeneration. This can occur by a mechanism involving protein N-homocysteinylation of lysine residues by homocysteine thiolactone [35–37] or, more speculatively, by undergoing self-fibril formation to induce seeding of other protein aggregates, including Aβ polypeptide itself [38].
Regardless of such possible underlying pathogenic mechanisms, it is reassuring to know that high dose B-vitamin supplements convincingly reduce brain atrophy and cognitive decline in a pre-stage state of sporadic AD. This is an excellent starting-point for finding the best preventive means of this devastating disorder.
DISCLOSURE STATEMENT
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/18-1007r1).
REFERENCES
[1] | Hardy J , Allsop D ((1991) ) Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci 12: , 383–388. |
[2] | Regland B , Gottfries CG ((1992) ) The role of amyloid beta-protein in Alzheimer’s disease. Lancet 340: , 467–469. |
[3] | Lee HG , Castellani RJ , Zhu X , Perry G , Smith MA ((2005) ) Amyloid-beta in Alzheimer’s disease: The horse or the cart? Pathogenic or protective? Int J Exp Pathol 86: , 133–138. |
[4] | Benilova I , Karran E , De Strooper B ((2012) ) The toxic Aβ oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat Neurosci 15: , 349–357. |
[5] | Rottkamp CA , Raina AK , Zhu X , Gaier E , Bush AI , Atwood CS , Chevion M , Perry G , Smith MA ((2001) ) Redox-active iron mediates amyloid-beta toxicity. Free Radic Biol Med 30: , 447–450. |
[6] | Dawkins E , Small DH ((2014) ) Insights into the physiological function of the β-amyloid precursor protein: Beyond Alzheimer’s disease. J Neurochem 129: , 756–769. |
[7] | McCaddon A , Kelly CL ((1994) ) Familial Alzheimer’s disease and vitamin B12 deficiency. Age Ageing 23: , 334–337. |
[8] | McCaddon A , Regland B , Fear CF ((1995) ) Trypsin inhibition: A potential cause of cobalamin deficiency common to the pathogenesis of Alzheimer-type dementia and AIDS dementia complex? Med Hypotheses 45: , 200–204. |
[9] | Lee JH , Yu WH , Kumar A , Lee S , Mohan PS , Peterhoff CM , Wolfe DM , Martinez-Vicente M , Massey AC , Sovak G , Uchiyama Y , Westaway D , Cuervo AM , Nixon RA ((2010) ) Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 141: , 1146–1158. |
[10] | Zhao H , Li H , Ruberu K , Garner B ((2015) ) Impaired lysosomal cobalamin transport in Alzheimer’s disease. J Alzheimers Dis 43: , 1017–1030. |
[11] | Froese DS , Gravel RA ((2010) ) Genetic disorders of vitamin B12 metabolism: Eight complementation groups – eight genes. Expert Rev Mol Med 12: , e37. |
[12] | Lai KSP , Liu CS , Rau A , Lanctôt KL , Köhler CA , Pakosh M , Carvalho AF , Herrmann N ((2017) ) Peripheral inflammatory markers in Alzheimer’s disease: A systematic review and meta-analysis of 175 studies. J Neurol Neurosurg Psychiatry 88: , 876–882. |
[13] | McCaddon A , Hudson P , Davies G , Hughes A , Williams JH , Wilkinson C ((2001) ) Homocysteine and cognitive decline in healthy elderly. Dement Geriatr Cogn Disord 12: , 309–313. |
[14] | Seshadri S , Beiser A , Selhub J , Jacques PF , Rosenberg IH , D’Agostino RB , Wilson PW , Wolf PA ((2002) ) Plasma homocysteine as a risk factor for dementia and Alzheimer’s disease. N Engl J Med 346: , 476–483. |
[15] | Zylberstein DE , Lissner L , Björkelund C , Mehlig K , Thelle DS , Gustafson D , Ostling S , Waern M , Guo X , Skoog I ((2011) ) Midlife homocysteine and late-life dementia in women. A prospective population study. Neurobiol Aging 32: , 380–386. |
[16] | McCaddon A , Miller JW ((2015) ) Assessing the association between homocysteine and cognition: Reflections on Bradford Hill, meta-analyses, and causality. Nutr Rev 73: , 723–735. |
[17] | Smith AD , Refsum H , Bottiglieri T , Fenech M , Hooshmand B , McCaddon A , Miller JW , Rosenberg IH , Obeid R ((2018) ) Homocysteine and dementia: An international consensus statement. J Alzheimers Dis 62: , 561–570. |
[18] | Smith AD , Smith SM , de Jager CA , Whitbread P , Johnston C , Agacinski G , Oulhaj A , Bradley KM , Jacoby R , Refsum H ((2010) ) Homocysteine-lowering by B vitamins slows the rate of accelerated brain atrophy in mild cognitive impairment: A randomized controlled trial. PLoS One 5: , e12244. |
[19] | Jack CR Jr , Shiung MM , Gunter JL , O’Brien PC , Weigand SD , Knopman DS , Boeve BF , Ivnik RJ , Smith GE , Cha RH , Tangalos EG , Petersen RC ((2004) ) Comparison of different MRI brain atrophy rate measures with clinical disease progression in AD. Neurology 62: , 591–600. |
[20] | de Jager CA , Oulhaj A , Jacoby R , Refsum H , Smith AD ((2012) ) Cognitive and clinical outcomes of homocysteine-lowering B-vitamin treatment in mild cognitive impairment: A randomized controlled trial. Int J Geriatr Psychiatry 27: , 592–600. |
[21] | Douaud G , Refsum H , de Jager CA , Jacoby R , Nichols TE , Smith SM , Smith AD ((2013) ) Preventing Alzheimer’s disease-related gray matter atrophy by B-vitamin treatment. Proc Natl Acad Sci U S A 110: , 9523–9528. |
[22] | Smith AD , Refsum H ((2016) ) Homocysteine, B vitamins, and cognitive impairment. Annu Rev Nutr 36: , 211–239. |
[23] | DeKosky ST , Ikonomovic MD , Wang X , Farlow M , Wisniewski S , Lopez OL , Becker JT , Saxton J , Klunk WE , Sweet R , Kaufer DI , Kamboh MI ((2003) ) Plasma and cerebrospinal fluid alpha1-antichymotrypsin levels in Alzheimer’s disease: Correlation with cognitive impairment. Ann Neurol 53: , 81–90. |
[24] | Pasternack JM , Abraham CR , Van Dyke BJ , Potter H , Younkin SG ((1989) ) Astrocytes in Alzheimer’s disease gray matter express alpha 1-antichymotrypsin mRNA. Am J Pathol 135: , 827–834. |
[25] | Van Nostrand WE , Wagner SL , Suzuki M , Choi BH , Farrow JS , Geddes JW , Cotman CW , Cunningham DD ((1989) ) Protease nexin-II, a potent antichymotrypsin, shows identity to amyloid beta-protein precursor. Nature 341: , 546–549. |
[26] | Gopalan S , Kasza A , Xu W , Kiss DL , Wilczynska KM , Rydel RE , Kordula T ((2005) ) Astrocyte- and hepatocyte-specific expression of genes from the distal serpin subcluster at 14q32.1 associates with tissue-specific chromatin structures. J Neurochem 94: , 763–773. |
[27] | Banerjee R , Gherasim C , Padovani D ((2009) ) The tinker, tailor, soldier in intracellular B12 trafficking. Curr Opin Chem Biol 13: , 484–491. |
[28] | Hannibal L , Jacobsen DW ((2017) ) Intracellular processing and utilization of cobalamins. In Vitamin B12: Advances and Insights, Obeid R, ed. CRC Press/Taylor and Francis Group, pp. 46–93. |
[29] | Gherasim C , Lofgren M , Banerjee R ((2013) ) Navigating the B(12) road: Assimilation, delivery, and disorders of cobalamin. J Biol Chem 288: , 13186–13193. |
[30] | Allen RH , Seetharam B , Podell E , Alpers DH ((1978) ) Effect of proteolytic enzymes on the binding of cobalamin to R protein and intrinsic factor. In vitro evidence that a failure to partially degrade R protein is responsible for cobalamin malabsorption in pancreatic insufficiency. J Clin Invest 61: , 47–54. |
[31] | Marcoullis G , Parmentier Y , Nicolas JP , Jimenez M , Gerard P ((1980) ) Cobalamin malabsorption due to nondegradation of R proteins in the human intestine. Inhibited cobalamin absorption in exocrine pancreatic dysfunction. J Clin Invest 66: , 430–440. |
[32] | Obeid R , Schlundt J , Umanskaya N , Herrmann W , Herrmann M ((2011) ) Folate is related to phosphorylated neurofilament-H and P-tau (Ser396) in rat brain. J Neurochem 117: , 1047–1054. |
[33] | Wei W , Liu YH , Zhang CE , Wang Q , Wei Z , Mousseau DD , Wang JZ , Tian Q , Liu GP ((2011) ) Folate/vitamin-B12 prevents chronic hyperhomocysteinemia-induced tau hyperphosphorylation and memory deficits in aged rats. J Alzheimers Dis 27: , 639–650. |
[34] | Hooshmand B , Polvikoski T , Kivipelto M , Tanskanen M , Myllykangas L , Erkinjuntti T , Mäkelä M , Oinas M , Paetau A , Scheltens P , van Straaten EC , Sulkava R , Solomon A ((2013) ) Plasma homocysteine, Alzheimer and cerebrovascular pathology: A population-based autopsy study. Brain 136: , 2707–2716. |
[35] | Jakubowski H ((1999) ) Protein homocysteinylation: Possible mechanism underlying pathological consequences of elevated homocysteine levels. FASEB J 13: , 2277–2283. |
[36] | Stroylova YY , Chobert JM , Muronetz VI , Jakubowski H , Haertlé T ((2012) ) N-homocysteinylation of ovine prion protein induces amyloid-like transformation. Arch Biochem Biophys 526: , 29–37. |
[37] | Akchiche N , Bossenmeyer-Pourié C , Kerek R , Martin N , Pourié G , Koziel V , Helle D , Alberto JM , Ortiou S , Camadro JM , Léger T , Guéant JL , Daval JL ((2012) ) Homocysteinylation of neuronal proteins contributes to folate deficiency-associated alterations of differentiation, vesicular transport, and plasticity in hippocampal neuronal cells. FASEB J 26: , 3980–3992. |
[38] | Sade D , Shaham-Niv S , Arnon ZA , Tavassoly O , Gazit E ((2018) ) Seeding of proteins into amyloid structures by metabolite assemblies may clarify certain unexplained epidemiological associations. Open Biol 8: , 170229. |


