
Altered Neural Networks in the Papez Circuit: Implications for Cognitive Dysfunction after Cerebral Ischemia
Abstract
Cerebral ischemia remains a leading cause of mortality worldwide. Although the incidence of death has decreased over the years, surviving patients may suffer from long-term cognitive impairments and have an increased risk for dementia. Unfortunately, research aimed toward developing therapies that can improve cognitive outcomes following cerebral ischemia has proved difficult given the fact that little is known about the underlying processes involved. Nevertheless, mechanisms that disrupt neural network activity may provide valuable insight, since disturbances in both local and global networks in the brain have been associated with deficits in cognition. In this review, we suggest that abnormal neural dynamics within different brain networks may arise from disruptions in synaptic plasticity processes and circuitry after ischemia. This discussion primarily concerns disruptions in local network activity within the hippocampus and other extra-hippocampal components of the Papez circuit, given their role in memory processing. However, impaired synaptic plasticity processes and disruptions in structural and functional connections within the Papez circuit have important implications for alterations within the global network, as well. Although much work is required to establish this relationship, evidence thus far suggests there is a link. If pursued further, findings may lead toward a better understanding of how deficits in cognition arise, not only in cerebral ischemia, but in other neurological diseases as well.
INTRODUCTION
Cerebral ischemia (CI), in the form of ischemic stroke or cardiac arrest, is a leading cause of death and major public health concern worldwide. Among survivors, CI may induce a vast array of physiological consequences that hinder important sensory, motor, and cognitive functions. On the molecular level, the reduction/loss of blood flow during an ischemic event depletes the supply of substrates, namely oxygen and glucose, necessary for energy production in the brain. A bioenergetic failure ensues following energy deprivation, which initiates a cascade of pathological molecular events. This results in neuronal cell damage and death due to several mechanisms (i.e., excitotoxicity, oxidative stress, free radical production, and inflammation).
CI may be categorized as focal if a brain artery is occluded, as occurs during ischemic stroke, or global, which is typically induced by cardiopulmonary or cardiac arrest (CA). Reduced energy levels during CI results in selective neuronal necrosis within the ischemic core in stroke or in the hippocampus, cortex, cerebellum, and striatum in CA [1–4]. Days following reperfusion, delayed neuronal death has been shown to occur in the ischemic penumbra after stroke [5] and in selective vulnerable cell populations after CA. Specifically, neurons located in the Cornu Ammonis 1 (CA1) subfield of the hippocampus and hilar region of the dentate gyrus are highly susceptible to ischemic injury in models of CA and transient forebrain ischemia [6, 7]. The hippocampus is a central part of the limbic system and plays a key role in memory processing. It is also an integral component of the Papez circuit— a closed neural circuit that is thought to be important for the transfer of information.
An ongoing field of research has been dedicated to understanding how cognitive deficits in patients with CI arise. The adverse effects on overall quality of life have prompted researchers to investigate treatments/therapies that can reduce cognitive burden. However, it is difficult to develop treatments when the cause of cognitive impairment after CI has not been clear. In order to fully understand the underlying mechanisms responsible for dysfunctional cognitive processes after CI, attention must be paid to impairments that occur at the network level. Defects in both synaptic plasticity processes and local circuits within the brain may alter neural network activity, which is essential for proper communication between distinct groups of neurons and the integration of information. CI has been shown to negatively affect synaptic transmission and circuitry; thus, research focused on providing a direct link between plasticity, circuitry, and network activity after CI is warranted.
To this end, we hope to establish the importance of synaptic plasticity and circuitry in relation to network activity within the brain. In this review, we will give special attention to structures within the Papez circuit, which have been attributed to cognitive deficits in other neurological disorders, such as Alzheimer’s disease (AD) [8, 9]. First, we will provide clinical evidence supporting the development of cognitive impairments in patients after CI, and highlight similarities in pathology with AD. Thereafter, we will outline dysfunctional plasticity processes that take place following CI, primarily in the hippocampus; highlight alterations observed in the Papez circuit after CI and their effect on network communication; and suggest novel approaches that may target plasticity processes and circuitry in order to restore network activity in CI patients. By fully understanding the nature of how deficits in cognition arise, not only in CI, but in other neurodegenerative diseases as well, more effective treatments targeting important aspects of neural circuitry and plasticity may be developed.
CLINICAL EVIDENCE OF COGNITIVE IMPAIRMENTS AFTER CEREBRAL ISCHEMIA
Cognitive deficits in patients after ischemic stroke
Cognitive deficits are common among patients after stroke attack. In a study that recruited patients with different subtypes of strokes, cognitive function as indicated by the Mini-Mental State Examination (MMSE) was significantly impaired 6 months following the initial insult [10]. The overall incidence of post-stroke dementia at 6 months is 12.2% [10]. Among different subtypes of strokes, 87% are ischemic and the incidence of dementia in these patients is a bit lower. At one year after ischemic stroke, the incidence of dementia is around 7% [11] to 8% [12].
Clinical studies found an increased risk of dementia after a stroke event. MMSE performance in post-stroke patients was significantly worse than that of the non-stroke control group [10]. After the first year of ischemic stroke, the risk for dementia is about twice the annual rate of the general population [11]. When compared to healthy controls, the risk increased to 3.83 times [12]. The long term risk of dementia in ischemic stroke survivors is also increased. In “first-ever” ischemic stroke patients, the 30-year risk of dementia is 1.72 times that of the population; however, the risk is even greater for hemorrhagic stroke survivors [13].
Among stroke survivors, the cumulative prevalence of a recurrent stroke at 3 years after the first event was estimated to be 26.4% [14]. Recurrent strokes increase the incidence of post-stroke dementia dramatically, with an estimated prevalence of 41.3% in patients with recurrent stroke [15]. Post-stroke dementia, in turn, is strongly related to an elevated risk for future stroke attack [16, 17]. Furthermore, post-stroke dementia is significantly associated with an increased mortality rate in ischemic stroke survivors [18, 19], which suggests the influence of cognitive impairments in ischemic stroke outcomes.
Cognitive deficits in patients after CA
CA is also associated with cognitive deficits, with longer episodes inducing a higher risk of impairments in short-term memory and recall [20, 21]. CA is characterized by a sudden stop of heart beating that results in the complete cessation of blood flow and subsequent interruption of blood supply to the brain. The reduced cerebral blood flow may contribute to future dementia in surviving patients as the incidence of cognitive impairment is significantly higher after sudden CA [21]. An increased long-term risk for memory impairment is also found in CA patients. Both in-hospital and outside hospital CA patients scored lower on the Rivermead Behavioral Memory Test (RBMT), an assessment of episodic memory, compared to the myocardial infarct control group eight months after the event [22]. Additionally, moderate or severe memory impairments were found in 26% and 38% of in-hospital and outside hospital CA patients, respectively [22].
Surprisingly, memory deficits persist 3 years following the event, as indicated by significantly decreased scores on cognitive assessments [23]. Specifically, CA patients had reduced scores (16.1) compared to age- and sex-matched controls (22.1) on the RBMT [23]. These patients also scored lower (5.8) compared to controls (10.8) on the Doors and People test (DPT)— an examination of recall and recognition memory [23]. Another study found that about 29% of CA survivors exhibited memory impairments four years after CA as indicated by low scores in both the Paired Associates Learning and Delayed Matching to Sample tests [24]. Interestingly, these patients were initially discharged from the hospital with good neurological scores as measured by the cerebral performance categories (CPC) test, which suggests it is not a robust tool for detecting subtle changes in cognitive function [24].
It should be noted that there exists heterogeneity among studies in measured cognitive outcomes and frequency of cognitive impairments following CA [25]. These discrepancies are attributed to differences in the number of patients studied, trial design, and tests used to assess cognitive impairments. Nonetheless, spatial memory [26], verbal memory [26], as well as recall [23, 27] and recognition [23] memory have been reported to be significantly affected.
Pathological changes after ischemic stroke and CA share similarities with AD-like pathology
Stroke has been shown to significantly increase the risk for AD. A meta-analysis estimated a 1.59 times increased risk for incident AD in patients after stroke [28]. In patients with ischemic stroke, the first year risk of AD is 1.5 times of the general population [11]. Abnormal phosphorylated tau and amyloid-β (Aβ) deposition are hallmarks of AD. Clinical evidence suggests AD-like pathology in the pathological changes of post ischemic stroke. Tau protein is found at high levels in both cerebrospinal fluid (CSF) and blood samples in patients with acute ischemic stroke [29]. The presence and level of tau protein is negatively associated with brain atrophy [29] and long-term outcome of ischemic stroke patients [29, 30]. In a three-year longitudinal study, cerebral Aβ deposition was found to be significantly associated with post ischemic stroke cognitive impairments [31]. In this study, ischemic stroke patients with or without brain Aβ deposition were assessed with neuropsychological tests annually up to 3 years after the event. Patients with brain Aβ deposition had a more severe and rapid decline in cognitive performance in global cognition and memory function than those patients without Aβ deposition [31].
Studies also suggest pathological overlap between CA and AD. Increased CSF tau and serum tau were observed in CA patients [32, 33]. At 2 weeks after the event, CSF tau levels in CA patients increased to 7 times of the control groups. Similar to the findings in ischemic stroke patients, CSF tau and serum tau levels are negatively associated with the outcome of CA survivors [32–34]. In addition to the pathological changes in tau, increased Aβ was also found in serum and brain tissue from CA patients. Postmortem brain tissue of CA patients revealed overexpression of Aβ [35]. An average 7–fold increase of serum Aβ was observed at about 10 hours after the event. The high levels of serum Aβ was related to poor outcome assessed at 6 months after CA [36].
It is clear that there is some overlap between CI and AD in terms of pathology. It seems likely that similar pathological mechanisms resulting in neuronal loss, Aβ accumulation, and tau hyperphosphorylation occur in both disease states; however, there has yet to be sufficient experimental evidence confirming this idea. More importantly, dysregulation of these disease-related proteins have also been attributed to impaired plasticity processes and synaptic dysfunction [37]. Thus, similar disruptions in network activity may also occur, which may account for deficits in cognition seen in both patients. Although we will solely focus on CI in this review, similar events leading up to altered network communication may also take place in AD and other neurodegenerative diseases.
SYNAPTIC PLASTICITY UNDERLIES COGNITIVE ABILITY AND FUNCTION
It has long been questioned what processes underlie the development of cognitive deficits in patients with neurological disorders. Although the degeneration of neurons presents itself as the main culprit, studies have revealed that synaptic dysfunction precedes neuronal loss in a number of neurodegenerative diseases and is a strong pathological correlate of cognitive decline [38–43]. As initially proposed by Ramón y Cajal, structural changes that strengthen existing connections between neurons may be the critical mechanism for learning and memory formation [44, 45]. This idea was later supported by Donald Hebb, who proposed that when two neurons fire simultaneously, the synaptic connection between them becomes strengthened [46, 47]. He theorized that in a strongly interconnected group of neurons, which he called neural “ensembles”, the activation of only a few members of the assembly is sufficient to activate the entire unit, either simultaneously or gradually by exhibiting well-timed activity patterns [48]. The nature of these ensembles is still not well understood and is not within the scope of this review; however, the idea that neural ensembles encode associative memory in the cortex is an important concept in terms of how other brain regions play a role. To elaborate, information is thought to circulate within certain brain regions in the form of short-term memory before being transferred for long term storage in the cortex. Hippocampal networks have been implicated in this temporary memory storage [49–51]. This explains why damage to different components of the Papez circuit (i.e., mammillary bodies, anterior thalamic nuclei, and cingulate gyrus), which will be discussed in detail later, can lead to anterograde amnesia in which patients lack the capability to form new episodic memories [51]. Thus, the process of modifying synapses by adjusting their strength or number, collectively referred to as synaptic plasticity, is an important mechanism involved in information encoding and storage in different structures such as the hippocampus.
It is now widely accepted that modulation of synaptic connectivity that increases synaptic efficacy within neural circuits is implicated in the production of complex behaviors and storing memories [52]. Thus, any form of synaptic dysfunction that impairs neuronal transmission and plasticity processes may be a common pathogenic mechanism among several neurological disorders. For example, studies have shown that different forms of CI can negatively affect synaptic transmission and structure [7, 53, 54] potentially inducing changes in neural circuity that can affect memory formation. It is important to understand the specific plasticity mechanisms that become disrupted during diseased states in order to understand how cognitive deficits arise. In this section, we will review the different forms of synaptic plasticity that exist in mammals and provide evidence supporting impairments in these processes during or after CI.
Long-term potentiation (LTP)
One of the most established models of activity-dependent synaptic plasticity is LTP. LTP can induce long-term changes in synaptic transmission and is considered to be a potential cellular substrate underlying certain forms of learning and memory, as implicated by studies showing that LTP can be induced in a subset of synapses during periods of learning [55–57]. While LTP exists in several brain regions, such as the cortex and striatum, it is most prominently studied in the hippocampus. In particular, synaptic connections between CA3 Schaffer collaterals and CA1 pyramidal cells, as illustrated in Fig. 1A, have been most widely investigated.
Notably, hippocampal LTP requires N-methyl-d-aspartate receptor (NMDAR) activation and follows Hebbian’s postulate of synaptic plasticity; thus, both postsynaptic and presynaptic elements are involved. LTP may be induced by giving one or more high frequency stimulus trains (tetanic stimulation) or short repetitive bursts of stimulation within the theta frequency range (theta burst stimulation or TBS). The latter is more physiological as it falls within normal activity patterns of the brain. LTP is reflected by changes in the amplitude or slope of excitatory postsynaptic potentials (EPSPs) or field EPSPs (fEPSPs). LTP is said to occur if there is a sustained increase in the amplitude or slope of the EPSP, referred to as potentiation, which can last up to hours, even days. These changes indicate increased efficacy of synaptic transmission.
Fig.1
Changes at excitatory synapses after ischemia. A) Simplified illustration of rodent hippocampal slice demonstrating the major excitatory pathways within the hippocampal formation. Perforant path fibers innervate dendrites of granule cells in the dentate gyrus, which project to CA3 neurons via mossy fibers. CA1 neurons receive input from CA3 neurons through Schaffer collaterals. Typical hippocampal field recordings involve electrical stimulation of Schaffer collaterals, which generate EPSPs that can be recorded from CA1 dendrites. B) Outline of long term synaptic plasticity changes after ischemia. Top: Induction of LTD prior to an ischemic insult may act as a neuroprotective intervention against CI as studies have shown decreased neuronal cell death following LTD preconditioning. Middle: Ischemia induces a pathological form of plasticity (iLTP) that prevents physiological LTP (pLTP). Bottom: Low frequency stimulation (LFS) can depotentiate iLTP and allow neurons to express pLTP following HFS or TBS.
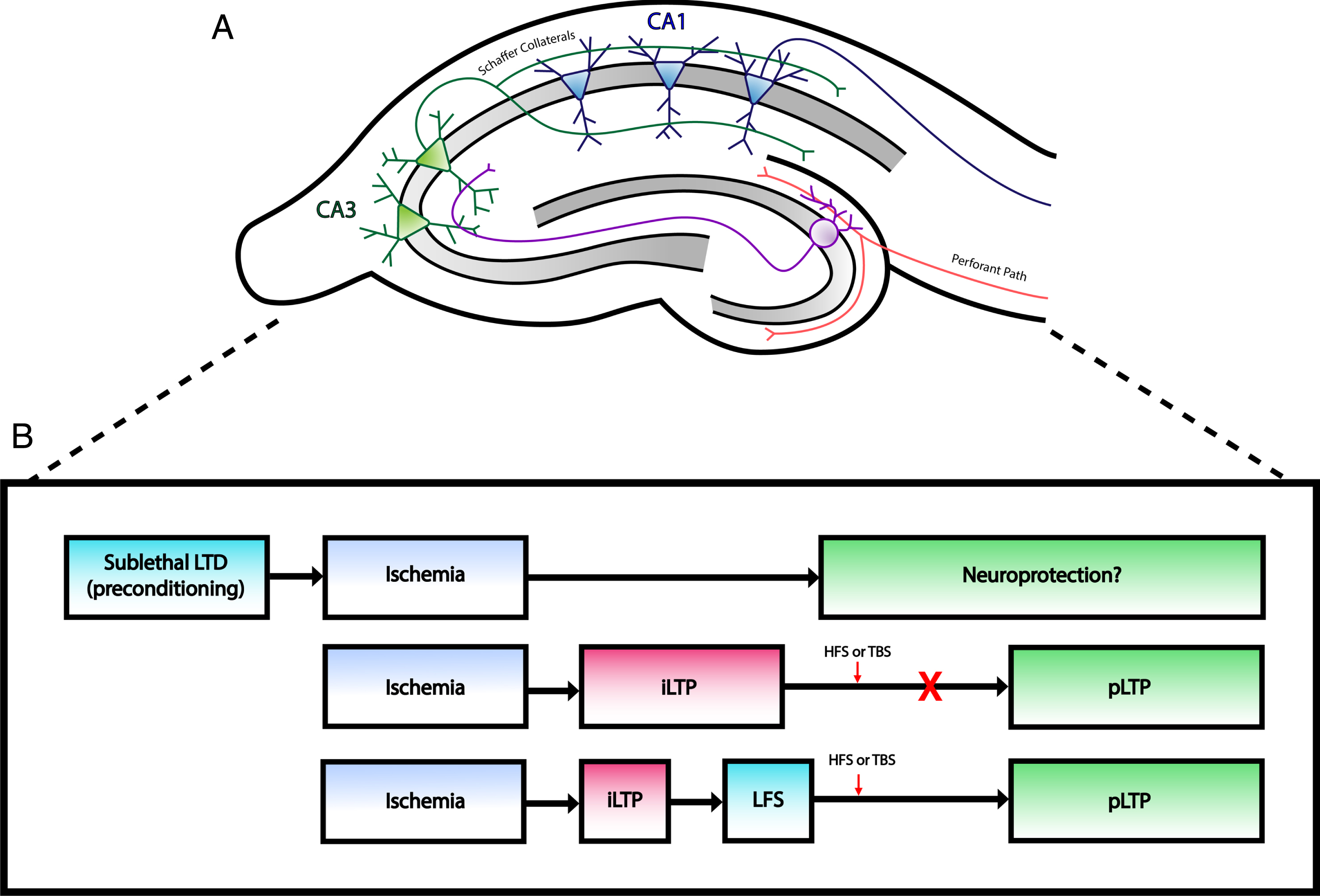
In the context of ischemia, LTP impairments have been observed in different models of CI [58–61]. For example, rats that underwent transient middle cerebral artery occlusion (tMCAO) exhibited impairments in hippocampal LTP and increased GABAergic neurotransmission 30 days after stroke [58]. This was correlated with deficits in spatial learning and memory [58]. Interestingly, molecular processes that become active during excitotoxicity are similar to those that induce physiological NMDAR-dependent LTP [62, 63]. For example, increased levels of extracellular glutamate lead to a rise in intracellular calcium due to prolonged activation of α-amino–3–hydroxy–5–methyl–4–isoxazolepropionic acid (AMPA) and NMDA receptors. Increased calcium influx can stimulate calcium/calmodulin-dependent protein kinase (CAMKII) signaling, which results in AMPAR phosphorylation and increased expression of surface AMPAR at the synapse. Thus, biochemical pathways that are activated during ischemia may be similar to those required for the induction of activity-dependent synaptic plasticity. As it turns out, studies have shed light on a novel form of LTP that may be induced by pathological stimuli. This form of plasticity was first characterized by Crepel et al. who demonstrated that anoxia combined with aglycemia could generate NMDAR-dependent LTP as evidenced by an upregulation of postsynaptic NMDAR-mediated currents [64, 65]. Within the field of ischemia, this form of synaptic plasticity is referred to as post-ischemic long-term potentiation (iLTP). iLTP has been observed within the ischemic penumbra and is due to the excessive release of glutamate during an ischemic event [66].
This pathological form of plasticity may have implications for altered protein expression in neurons due to the modulatory effects on nuclear transcription factors, which may have a large impact on neuronal function and circuitry [66]. More importantly, iLTP has been found to alter physiological LTP in numerous studies. In acute hippocampal slices, prior induction of synaptic transmission potentiation by anoxia significantly attenuated subsequent induction of tetanic LTP [67]. This finding is consistent with more recent studies showing delayed inhibition of LTP induction in models of ischemic stroke [68–70]. Using an endothelin–1–induced mini stroke model, Wang et al. investigated whether iLTP, measured 3 hours after onset of ischemia in rats, altered LTP induction in CA1 neurons of the hippocampus [69]. They found that LTP was completely abolished 24 hours following focal ischemia, suggesting that iLTP may increase the induction threshold of subsequent synaptic plasticity [69]. Local field potentials— a measure of brain activity reflecting the transmission of information through neural networks— were also suppressed 12 and 24 hours after ischemia [69, 71]. Similarly, iLTP occluded physiological LTP in a murine model of cardiac arrest and cardiopulmonary resuscitation (CA/CPR) [72]. The study also demonstrated that iLTP was reversible. Physiological LTP could be induced by depotentiating iLTP using low frequency stimulation prior to TBS, which suggests that synaptic function may be improved using therapies that target LTP deficits [72].
Overall, these findings suggest that ischemia may cause impairments in LTP induction via iLTP, which has detrimental consequences for the function of individual neurons within the affected region and entire neuronal networks involved in associative learning. Conversely, a beneficial role for iLTP has also been proposed. iLTP may influence post-lesional reorganization of surviving neural networks after ischemia by inducing new connections between neurons that did not previously interact [66]. This is supported by the fact that brief episodes of anoxia-hypoglycemia have been shown to modify the structure of synapses, thereby promoting structural organization of hippocampal synaptic networks [73]. This compensatory mechanism has important implications for the early stages of ischemia where neuronal network activity is perturbed. However, maintaining circuit excitability under pathological conditions can also be harmful. Existing plasticity may increase the susceptibility of neurons to exhibit hyperexcitability or seizure like activity if surviving lesioned networks undergo pathological rewiring [74, 75]. At this point in time, it is not clear whether iLTP exerts protective or detrimental effects. Given the fact that iLTP is dependent on NMDAR activation, it is important for future studies to investigate the potential role of iLTP in later stages of the disease to elucidate whether sustained iLTP contributes to excitotoxicity and delayed neuronal death.
Long-term depression (LTD)
LTD is another form of Hebbian long lasting plasticity and shares similar characteristics with LTP, such as the activation of NMDARs. Notably, LTD induction is determined by the magnitude of calcium influx, whereby a moderate increase results in LTD and a large increase in LTP. Unlike LTP, activity-dependent LTD arises from the activation of calcium dependent phosphatases that remove phosphate groups from target proteins, such as AMPARs, and potentially other molecules that also become phosphorylated during the late phase of LTP [76]. As such, LTD decreases surface AMPAR content at the synapse by promoting its internalization via endocytosis and results in a persistent decrease in synaptic efficacy by reducing neuronal excitability. In the CA1 region of the hippocampus, LTD may be induced by a period of low frequency stimulation (LFS). This induction is input specific in the sense that only synapses receiving LFS show LTD, whereas other inputs made onto the same population of postsynaptic neurons are not affected [77]. This is referred to as homosynaptic LTD. LTD is considered to be complementary to LTP as it is important for preventing over-excitation of neurons. Additionally, since continued strengthening of the synapse will eventually reach a maximum efficacy (i.e. become saturated), this would interfere with the acquisition of new information [76]. For this reason, LTD is essential for the selective weakening of synapses so that new information may be encoded.
Although LTD has been less studied in CI, it may have important implications for understanding ischemic resistance expressed in select populations of neurons following ischemia. The link between LTD and ischemic resistance was first made in a very early study by Gao et al. Forty-eight hours after severe ischemia induction in vivo, three types of CA1 neurons, which exhibited different response patterns after contralateral commissural stimulation, were identified [78]. These neurons were classified as late depolarizing post synaptic potential (L-PSP) neurons, non-late depolarizing post synaptic potential (Non L-PSP) neurons, and small excitatory postsynaptic potential neurons (S-EPSP) neurons. Non L-PSP neurons located in the lateral portion of CA1, where neurons are more resistant to ischemia, exhibited a decrease in the EPSP slope 24 hours after reperfusion, indicative of prolonged depression of synaptic transmission, which resembled LTD [78]. The authors postulated that the expression of LTD increases ischemic resistance; however, these neurons may later succumb to late developing apoptotic mechanisms accounting for delayed neuronal death [78].
Interestingly, a more recent study found that administration of a selective inhibitor of endocannabinoid clearance enzymes, JZL195, was able to induce neuroprotection 2 or 12 hours prior to transient global ischemia by inducing LTD at CA3–CA1 synapses [79]. The depression of excitatory synaptic transmission by LTD may serve as a defense mechanism to protect neurons from degeneration and, thus, those that express LTD are more likely to survive. In this manner, LTD induction may serve as a neuroprotective strategy against ischemia and if induced prior to CI may serve as a form of ischemic preconditioning. On the other hand, depression of inhibitory synaptic transmission due to failure of the excitatory input to inhibitory interneurons may also contribute to excitotoxicity with delayed cell death [54]. Taken together, instances of ischemia induce a pathological form of plasticity that may be reversed by LTD (Fig. 1B). There is also a potential therapeutic role for LTD in neuroprotection when induced prior to ischemia (Fig. 1B); however, future studies are required to confirm any beneficial or harmful effects.
Short-term plasticity
Paired-pulse facilitation (PPF) is a form of short-term synaptic plasticity that reflects the probability of transmitter release within the presynaptic cell. PPF is induced by giving a pair of pulses in rapid succession to a synaptic pathway. The response of the postsynaptic cell elicited by the first spike is compared to its response to the second spike measured in terms of EPSPs. If facilitation occurs, the postsynaptic response for the second pulse will be larger than the response for the first. The underlying mechanism is typically attributed to the buildup of residual calcium that remains in the presynaptic terminal following the first spike. However, this theory may be too simplified and studies have proposed that other mechanisms (i.e., saturation of the calcium buffering system) may be involved [80].
Whether PPF is affected following CI is less well understood. Some studies have not detected changes in PPF following cerebral ischemia, although they demonstrate reductions in basal synaptic transmission and LTP deficits. For example, the same study that found LTP impairments in rats following tMCAO did not show any differences in PPF in the CA1 region of the hippocampus between controls [58]. This was also true in a rat model of mild global cerebral ischemia created by four-vessel occlusion [81]. Both studies indicated that presynaptic plasticity was not altered. However, in a model of asphyxial cardiac arrest (ACA), middle-aged rats displayed an increase in the maximum amplitude of the paired-pulse response seven days after ischemia, but LTP was not impaired [82]. PPF dysfunction was attributed to presynaptic calcium dysregulation and was correlated with initial spatial memory deficits observed in rats after ACA [82]. These discrepancies may be accounted for by differences in animal age and strain. Whereas younger Sprague-Dawley or Wistar rats were used in the former studies, middle-aged Fisher rats were used in the latter. Although the extent to which PPF dysfunction is involved in disrupted plasticity mechanisms after CI remains unclear, there is no doubt that this form of synaptic plasticity is important for information processing and any alterations may have a large impact on synaptic and network communication.
NEURAL UNDERPINNINGS OF DISRUPTED PLASTICITY PROCESSES: INVOLVEMENT OF THE PAPEZ CIRCUIT
It is clear that dysfunctional synaptic plasticity processes following CI may contribute to pathological outcomes, such as delayed neuronal death, and cognitive decline as observed in animal models and human patients. As inferred above, the hippocampus is one of the main brain regions affected. On this account, it comes as no surprise that most research focused on ischemia-induced memory impairments to date has been associated with damage restricted to regions of the hippocampal formation [83]; however, damage within these regions may not be solely responsible for memory impairments observed in patients recovering from CI.
It has been demonstrated that damage to other limbic structures, such as the cingulate or parahippocampal cortex, may cause psychosocial behavioral disturbances [84] as well as deficits in spatial learning [85] and memory recognition [3, 86]. Thus, damage within different structures of the limbic system, particularly those belonging to the Papez circuit, may promote cognitive decline associated with CI.
Additionally, studies have detected injury within different structures of the Papez circuit in patients with neurodegenerative disorders, such as traumatic brain injury (TBI) [87–89] and Alzheimer’s disease (AD) [8, 9]. These findings suggest that memory deficits may be related to damage within the Papez circuit; therefore, there is reason to suspect that CI and neurodegenerative diseases associated with cognitive dysfunction may share a common pathogenesis involving damage to the Papez circuit.
Unfortunately, extra-hippocampal brain regions that comprise the Papez circuit are often ignored in terms of understanding cognitive decline in these diseases. This is particularly important considering the fact that anatomical structural lesions within a circuit may result in altered functional connectivity and impaired network communication within the brain. To fully understand where deficits in cognition arise, it is important to elucidate any pathological contributions from all aspects of a given circuit— on the local and global level. For the purposes of this section, we will focus solely on extra-hippocampal components that make up the Papez circuit; however, it should be noted that interactions between distributed neuronal ensembles spread throughout the brain are also essential in understanding the neural basis of cognitive processes [90]. Accordingly, we will first provide experimental evidence highlighting different forms of memory supported by distinct regions within the Papez circuit and scenarios where structural alterations of these regions have been found in CI. We will then discuss the consequences brought upon these alterations, particularly focusing on impaired neural network communication.
The Papez circuit plays an important role in memory function
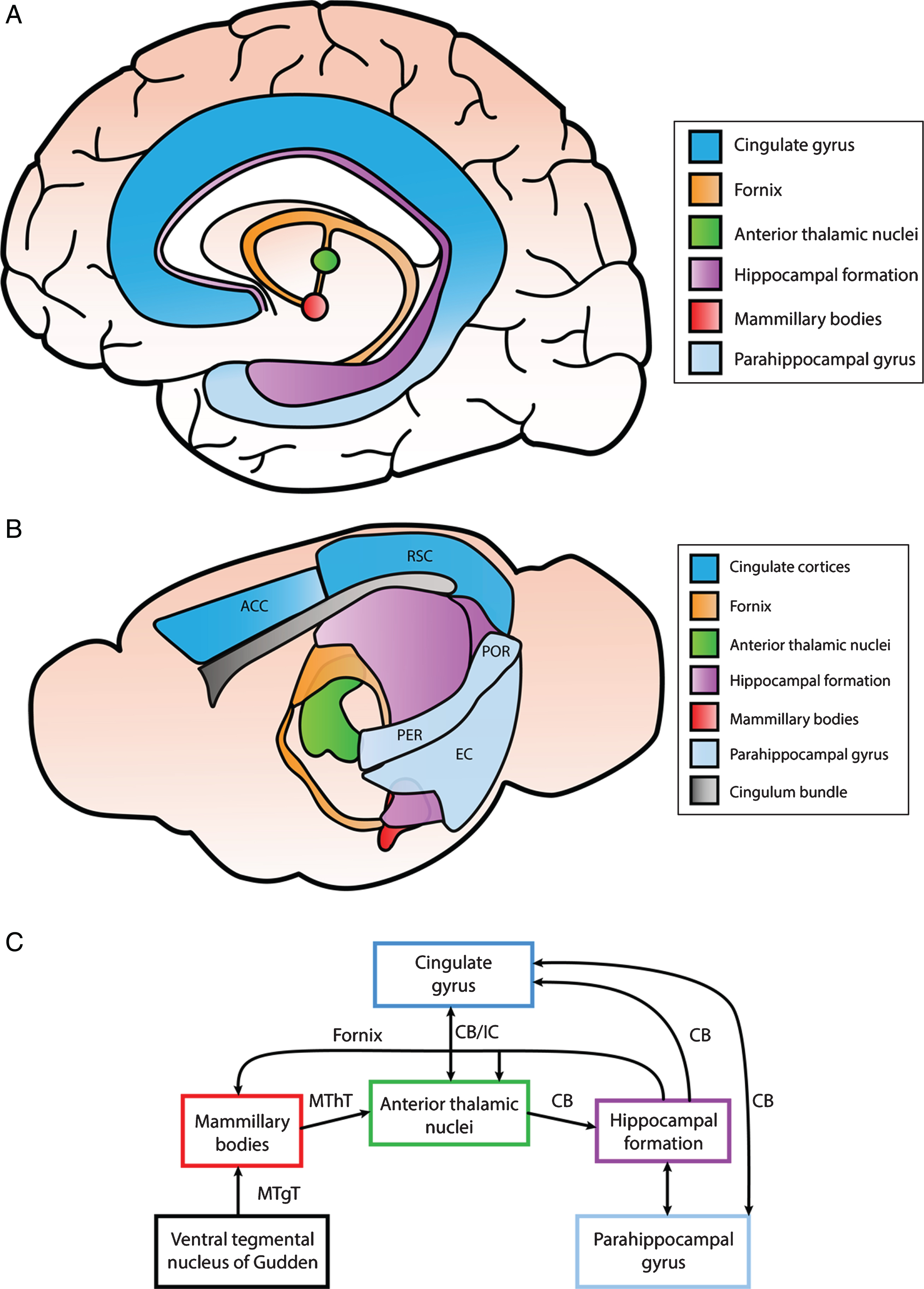
The Papez circuit is a central pathway of the limbic system that is involved in spatial learning and episodic memory, although originally it was thought to be strictly involved in behavioral and emotional expression. This circuit is comprised of the hippocampus, entorhinal cortex, mammillary bodies, anterior thalamic nuclei, cingulate cortices, and parahippocampal gyrus and is interconnected by white matter tracts consisting of the fornix, mammillothalamic tract, and cingulum bundle [51]. The reader is referred to an excellent review by Bubb et al. outlining the connections within this circuit [91]. A simplified schematic of the interconnected brain regions within the Papez circuit are illustrated in Fig. 2. While the hippocampus is one of the most intensely studied brain regions associated with memory impairments in various neurological diseases, other limbic structures within the circuit have been associated with different aspects of learning and memory as well.
Fig.2
Simplified illustration of structures within the Papez circuit and the connections between them. A) Anatomical depiction of structures within the Papez circuit in the human brain. The cingulate gyrus can be divided into the anterior cingulate cortex (ACC), posterior cingulate cortex (PCC), and retrosplenial cortex (RSC). The anterior portion of the parahippocampal gyrus consists of the entorhinal and perirhinal cortex, while the posterior portion includes the postrhinal cortex. B) Anatomical schematic illustrating different regions of the Papez circuit in the mouse brain. The ACC and RSC are indicated as well as the entorhinal cortex (EC), perirhinal cortex (PER), and postrhinal cortex (POR) within the parahippocampal gyrus. C) Interconnections identified among different components of the Papez circuit primarily in rodent brain. These structures are connected via white matter tracts (CB, cingulum bundle; MThT, mammillothalamic tract; MTgT, mammillotegmental tract; IC, internal capsule; and fornix).
For instance, mammillary body (MB) volume has been positively correlated with memory and recall index, both of which are cognitive domains typically affected after CA [92]. Additionally, atrophy of the MBs induced by fornix damage was associated with poor recall of episodic information and patients with bilateral interruption of the fornix displayed deficits in long-term memory [92]. These findings highlight the importance of direct projections from the hippocampus to the MBs on different aspects of memory. Other studies have demonstrated that non-hippocampal inputs to the MBs via the mammillotegmental tract also result in spatial memory impairments and widespread neural hypoactivity [93, 94].
One case study identified an acute lesion within the anterior thalamus using functional magnetic resonance imaging (fMRI) in a patient displaying cognitive dysfunction [95]. The lesion disrupted default mode network (DMN) activity, which is a type of resting state network (RSN) that is more active during passive rest than during task related engagement [95]; however, an exception to this is engagement during recollection [96]. When performing task-directed activities, lower levels of DMN activity are expressed as other task-related brain regions take over. Thus, disrupted DMN activity may impede one’s ability to switch from different modes of brain function (i.e., default mode to a task-related state). As hinted earlier, the DMN is also involved in learning and memory processes, such as mental search important for memory retrieval [97, 98]. In this scenario, the acute lesion within the anterior thalamus disrupted anatomical connections within the circuit, which the authors suggest altered functional network activity and led to acute cognitive dysfunction and persistent short-term memory impairment [95]. It is likely that the anterior thalamus is functionally connected with DMN components to induce this effect. Remarkably, DMN activity has been found within the Papez circuit itself, specifically in the anterior and posterior cingulate cortex (ACC; PCC) [97, 99]. One study found that mnemonic tasks (i.e., episodic word fluency) elicited DMN activity within the PCC, whereas nonmnemonic word fluency had the opposite effect [98]. Furthermore, studies in murine mammals have also demonstrated the importance of the anterior thalamic nuclei in spatial memory and navigation [100–103] as well as non-spatial hippocampal-dependent memory tasks [104]. Additionally, both the parahippocampal cortex and cingulate cortex have been associated with episodic memory [105, 106].
Thus far, we have only briefly touched upon the involvement of extra-hippocampal structures within the Papez circuit in different memory processes. However, it is important to note that no single brain structure operates in isolation and intact circuits are necessary for reliable network communication between different brain regions. This is important for understanding cognitive decline in different neurological diseases from a network perspective.
Structural and functional alterations in connectivity within the Papez circuit after CI
Given the fact that extra-hippocampal components of the Papez circuit play a vital role in memory, there is reason to suspect that following CI, structural alterations within these regions may also occur. Interestingly, injury has been detected in heart failure, with patients displaying greater neural atrophy and structural alterations in virtually every region of the Papez circuit [107]. Although this disease state is separate from CI, it is a well-established risk factor for ischemic insult, such as stroke, that may result in vascular dementia.
There has been less research focused on alterations related to extra-hippocampal structures of the Papez circuit following CI; nonetheless, we will highlight few of the studies that have. A very recent case report detected injury in the Papez circuit of a patient with hypoxic–ischemic brain injury due to cardiac arrest [108]. Utilizing diffusion tensor tractography, the authors found discontinuation of the fornical column in both hemispheres, thinning of the thalamocingulate tract in the right hemisphere, and non-reconstruction in the left hemisphere [108]. Injury of these neural tracts within the Papez circuit was associated with severe memory impairments exhibited by the patient [108]. A previous study found low gray matter volume of several brain regions in patients that survived cardiac arrest [109]. Using MRI, atrophy was observed in the retrosplenial cortex and ventromedial anterior cingulate cortex, which was correlated with clinical memory deficits [109].
It should be noted that cognitive deficits following stroke are not always explained by the size or location of infarction [110]. Lesions in one brain region may result in cortical impairments due to disrupted network activity that results in cognitive symptoms. Thus, alterations within functional networks should also be considered. One network that has been investigated in CI patients and has been found to be important in episodic memory processing [111, 112] is the DMN described earlier. One study found that there was decreased functional connectivity in the PCC within the DMN in acute ischemic stroke patients [113]. Inter-regional functional connectivity was also decreased between the PCC and other nodes of the DMN including the medial prefrontal and left medial temporal lobe [113]. Another study, which utilized multi-modality MRI, examined resting-state functional connectivity in patients with chronic subcortical stroke [114]. They found that regional homogeneity, which measured resting-state regional brain spontaneous activity, was decreased in the PCC within the DMN and was correlated with cognitive decline [114]. Conversely, a recent study reported that functional connectivity was increased between the DMN and PCC in patients with post-stroke memory dysfunction, although in some areas within the DMN connectivity was decreased [115]. The disparity between these studies may be explained by a number of factors including the nature of the brain injury due to stroke and inclusion/exclusion criteria, such as the time frame in which patients were scanned after stroke onset. These findings highlight the importance of the PCC within the Papez circuit and suggest that altered DMN activity may underlie episodic memory impairments after stroke.
The DMN has also been found to be affected after CA. Using resting state fMRI, one study investigated network connectivity in patients who underwent CA 4–7 days prior. DMN functional connectivity within the PCC and precuneus was severely disrupted— and in some instances completely diminished— in comatose CA survivors, particularly those who sustained severe neurological deficits compared to survivors with good outcomes [116]. A more recent study also found decreased functional connectivity in the DMN in CA patients as revealed by MRI taken within 28 days after the event [117]. Interestingly, patients with preserved resting-state functional connectivity (including the DMN) were found to have a favorable outcome one year after CA [117]. While these studies are limited by various factors and have been less explored in the field, it is likely that disruptions within the DMN following CI may play a role in the development of cognitive impairment. However, most of the evidence presented for this claim is correlative. Future studies seeking to elucidate the role of the DMN, particularly within the PCC, and its potential direct effects on different aspects of cognitive function after CI are required to better establish this probable link.
Implications for altered neural circuitry in cognitive dysfunction: The importance of theta oscillations
By altering intrinsic functional connectivity patterns, structural damage confined to local circuits following CI may have widespread effects in other remote non-lesioned regions of the brain, such as the cortex. As a result, cognitive and executive functions become impaired. This idea highlights the importance of interactions between distinct brain regions, which relies on effective communication between widely distributed neuronal groups. However, the question remains: how is effective neuronal communication established?
While anatomical structural connectivity is essential for linking distinct neural ensembles between different brain areas, neuronal communication also depends on neuronal synchronization [118]. Synchronous rhythmic activity of neuronal assemblies gives rise to oscillatory activity within different frequency bands. Whereas local processes are typically associated with higher frequency oscillations at gamma band (25–200 Hz), slower oscillations, such as those in the theta-alpha frequency band (4–10 Hz), are involved in long-range interactions [119, 120]. Neurons undergo an enhanced excitability and reduced excitability phase during an oscillation cycle. In order to enhance coupling between neural ensembles, oscillation frequencies must be adjusted in a way that excitatory inputs onto a post synaptic cell arrive at the peak of the cells enhanced excitability phase where it is most sensitive to excitatory input [90]. This so called “phase synchronization” mediates links between brain regions that are responsible for large-scale integration [121]. Coordinated synchronous activity of multiple neuronal populations is required for information processing and storage by neuronal networks. Disruptions in large-scale network synchronization have been correlated with cognitive dysfunction in different neurological disorders including, but not limited to, AD, Parkinson’s disease, and autism [122].
Theta oscillations in particular have been attributed to the coordination of global network activity in relation to mnemonic processes [123]. Specifically, theta oscillations can group and segregate neural assemblies in order to boost communication between distinct groups of neurons, which is essential for information exchange and processing [124]. In addition, theta rhythm can also modulate synaptic plasticity processes [125–127]. Although these oscillatory patterns are found within different structures throughout the mammalian brain, it is most prominently found in the hippocampus of murine animals where they are generated via complex interactions among multiple rhythm generators and numerous theta current dipoles [123, 124, 128].
Initially, the medial septum and vertical limb of the diagonal band of Broca (MS-vDBB) was considered to be the central rhythm generator— or pacemaker—necessary for phasic modulation of oscillations in the hippocampus. However, recent evidence indicates that intrahippocampal rhythm generators derived from local connectivity also exist, such as the CA3 recurrent collateral system [124, 129, 130]. Additionally, hippocampal neurons exhibit intrinsic resonant membrane properties and subthreshold membrane potential oscillations which contribute to hippocampal theta oscillations [124, 128, 131–133]. Thus, theta currents are likely driven not only by extrinsic synaptic inputs, but also by intrinsic membrane properties of hippocampal neurons. Notably, extrinsic inputs create two distinct dipoles (current sinks and sources), which determine the amplitude and phase versus depth profiles of theta waves [124]. In the CA1 region, for example, the first dipole is formed by periodic excitatory inputs from layer III of the entorhinal cortex and CA3 subfield of the hippocampus to the distal apical dendrites of CA1 pyramidal neurons [124, 132]. The second dipole is generated by inhibitory inputs to the soma derived from the MS-vDBB [124, 132]. Synchronous somatic hyperpolarization and dendritic depolarization results in membrane potential oscillations that are reflected as changes in the local field potential (LFP). Ultimately, the interaction between multiple rhythm generators and intrinsic membrane events will sum together and contribute to the LFP.
Of note, inhibitory hippocampal interneurons play a critical role in this process as they are primary targets of the MS. Cholinergic and glutamatergic neurons from the MS provide tonic excitation to hippocampal inhibitory interneurons and can also modulate excitability in CA1 pyramidal cells [132]. In the latter case, CA1 pyramidal cells may then activate hippocamposeptal projecting interneurons that relay back to the MS and are important for coordinating rhythmic discharges. GABAergic inputs from the MS exclusively target hippocampal interneurons, and thus act as pacemakers of theta generation in CA1 pyramidal cells [132]. The interplay between inhibition and excitation results in the rhythmic discharge of IPSPs from interneurons, which play a critical role in inhibiting and disinhibiting CA1 pyramidal cell bodies at theta frequency [124, 132].
Moreover, cells located in different regions of the Papez circuit, including the mammillary bodies [134, 135], anterior ventral region of the anterior thalamus [103, 136], and cingulate/retrosplenial cortex [137] have also been found to fire rhythmically synchronous with the hippocampal theta rhythm. These findings suggest that theta-rhythmic signals propagate throughout the Papez circuit, which may underlie one aspect of the circuit’s critical role in memory function [136].
This is particularly important in the context of CI, where networks may be damaged following an ischemic insult. In fact, temporal changes in the theta rhythm have been reported after CI. Following 20 minutes of forebrain ischemia, a reduction in theta was observed in the dorsal hippocampal formation, which occurred on the first day post-ischemia [138]. The reduction in amplitude became significantly large on the eighth day and was maintained throughout the duration of the survival period [138]. In line with these findings, an in vitro study using rat neocortical brain slices found that hypoglycemia and/or hypoxia also lead to a decrease in theta oscillation amplitudes [139]. Another study highlighted the importance of prefrontal-hippocampal network synchrony. An early hypoxic-ischemic episode in the developing rat lead to decreased temporal coupling between the prefrontal cortex and hippocampus by disrupting hippocampal theta drive [140]. Although these studies do not explicitly describe which cell types initiate these disruptions in theta, changes in membrane potential caused by energy deprivation will inevitably change the LFP during ischemia. Following ischemia, cell death of interneurons and pyramidal cells in the hippocampus will likely affect theta. Whether extrahippocampal rhythm generators are also affected is not known.
The functional importance of theta oscillations is clear. Network disturbances responsible for cognitive deficits following CI may be due to alterations in oscillatory activity, which effectively disrupt synchronous coupling between neural ensembles within a large-scale network. Thus, research focused on elucidating the involvement of theta oscillations after CI will lead to a greater understanding of the mechanisms underlying dysregulated network activity.
COMBATTING DISRUPTED PLASTICITY MECHANISMS: TREATMENTS/THERAPIES TO RESTORE COGNITION
The current gold standard for acute ischemic stroke treatment is intravenous (IV) administration of recombinant tissue plasminogen activator (rtPA or alteplase). It is the only FDA approved treatment that has been found to be effective when administered within 3 hours of symptom onset, although an expanded time window has been identified of up to 4.5 hours in eligible patients [141, 142]. Unfortunately, because of the relatively narrow time window for treatment, only a small fraction of all acute ischemic stroke patients, about 3.4% to 5.2%, are able to receive IV rtPA [142]. Even of those patients treated within the first 3 hours after stroke, it is unclear whether rtPA improves cognitive outcomes [143]. This is in line with the idea that a reduction in lesion volume does not necessarily correspond to improved cognitive functioning in the long term, as any changes in plasticity or local circuitry may be sufficient to alter network communication. Patients with a proximal large artery occlusion in the anterior circulation also have the option to undergo an endovascular procedure, known as mechanical thrombectomy, within 6 hours after stroke onset [144]. This procedure entails the delivery of a mechanical thrombectomy device, which can remove the occlusive clot through different biomechanical mechanisms, to achieve recanalization [145]. However, the percentage of patients eligible for this treatments is only about 7 to 9% of all acute ischemic stroke patients [146, 147].
In the case of CA, cardiopulmonary resuscitation (CPR) is typically performed to reverse hypoperfusion. Notably, early, high-quality resuscitation interventions are crucial to increase survival and cognitive outcomes after CA. Although therapies, such as mild induced hypothermia, have shown to increase survival and preserve cognitive function after CA [148–150], these findings are somewhat obscure and complications have also been documented [151–153]. Taken together, there is a need for the development of novel treatments that can be applied to a larger cohort of CI patients and can improve cognitive outcomes. In this section, we will discuss recent therapies/targets that hold promise as potential future treatments for CI with respect to improving cognitive outcomes.
Potential therapeutic targets for CI-related deficits in synaptic plasticity
As discussed earlier, both transient and long-term instances of synaptic failure have been reported as a result of CI. Further investigation into the ischemia-related molecular mechanisms that contribute to deficits in synaptic plasticity may provide effective targeted therapies to ameliorate ischemia-related deficits. There exists evidence for preventative therapies to reduce the ischemia-related effects on synaptic plasticity as well as ad hoc treatments to enhance recovery and restore the function of plastic processes, and in some cases cognitive function.
Synaptic plasticity protection from ischemic insult can come from preventative therapies that help confer tolerance to ischemic conditions. Many studies have investigated how different pre-stroke treatments can help to preserve or protect synaptic function from future ischemic insult. Ischemic preconditioning (IPC) is the subjugation to transient non-lethal periods of ischemia that confer adaptive changes within the cell to provide tolerance to future ischemic events. In some cases, adaptive changes to the electrophysiological properties of neurons is resultant from IPC as well as pharmacological mimetics that induce like-wise effects [154]. A summary of the induced electrophysiological effects of some preconditioning molecules and therapies are illustrated in Fig. 3.
Fig.3
The effects of preconditioning molecules/therapies on electrophysiological function and synaptic plasticity. Numerous pharmacological mimetics of IPC (i.e., PKCɛ; brain-derived neurotrophic factor, BDNF; vascular endothelial growth factor-A, VEGF; and ovarian hormone 17β-oestradiol, E2) induce neuroprotection through different mechanisms. This includes the regulation of certain processes (i.e., autophagy), as well as increased expression of certain proteins or receptors on the synaptic membrane. These changes may alter electrophysiological properties of neurons, as seen with PKCɛ, such as decreasing firing frequency. When exposed to conditions of oxygen and glucose deprivation, PKCɛ may also increase the latency to anoxic depolarization. Preconditioning therapies such as physical exercise (PE) training is also protective. PE along with BDNF, VEGF, and E2 have been found to retain both short-term and long-term synaptic plasticity processes after ischemia. Additionally, since iron accumulates in the cell after ischemia and exacerbates oxidative stress, it has also been a target of interest.
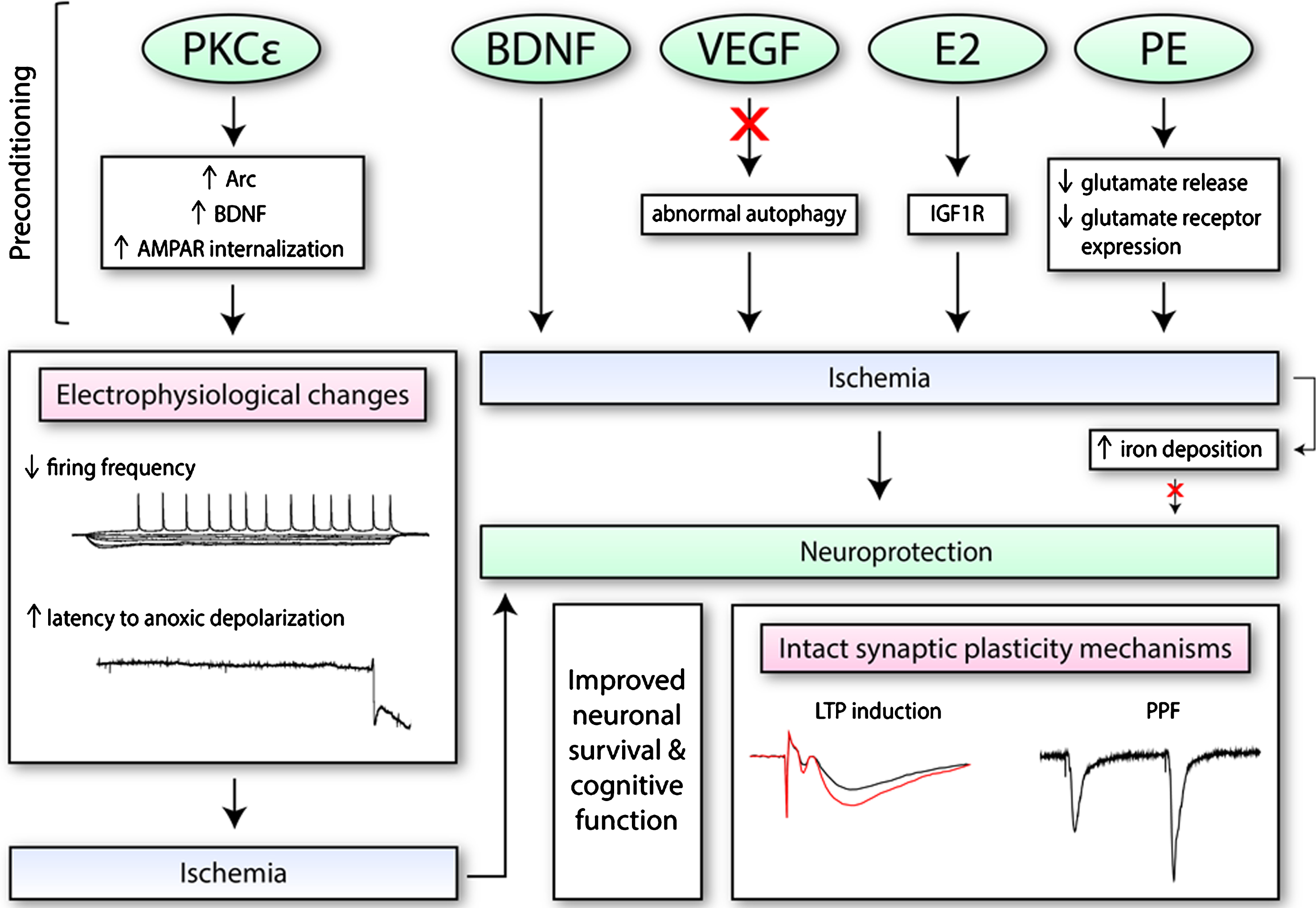
Pharmacological agents that exhibit pleiotropic properties, in particular, have attracted much attention in the field given their potential to target multiple aspects of several molecular and physiological events that take place during ischemia. One potential target is PKCɛ, which upon activation has been shown to confer protection against ischemia in vitro and in vivo [155]. PKCɛ-mediated neuroprotection was found to be dependent on increases in brain-derived neurotrophic factor (BDNF), which enacted electrophysiological changes in rat hippocampal slices during oxygen glucose deprivation [154]. Firing frequency was reduced and threshold potential increased 48 hours after preconditioning in vivo and in vitro, while time to anoxic depolarization, which is an ischemic mechanism of neuronal cell death, was delayed [154]. This evidence suggests that IPC or PKCɛ-preconditioning promotes increased levels of BDNF, which confers changes in synaptic function that provides protection against ischemic injury. Further investigation into the relationship between BDNF and its effect on synaptic function showed changes in post-synaptic receptors [156]. The activation of the BDNF/TrkB pathway resulted in decreased AMPAR current due to the internalization of GluR2/AMPAR [156]. These effects on AMPARs were enacted by activity-regulated cytoskeleton-associated protein (Arc), which was required for PKCɛ induced neuroprotection against oxygen glucose deprivation and delay to anoxic depolarization [156]. Thus, by inducing the internalization of AMPARs through a BDNF/Arc related mechanism, preconditioning with PKCɛ was able to alter synaptic function, which enabled cells to become more tolerant to ischemic conditions.
The importance of BDNF post-stroke was also shown in a study using a juvenile cerebral ischemia model. Mice subjected to CA/CPR displayed hippocampal LTP impairments 7–14 days post ischemia with a recovery by 30 days [157]. BDNF expression was shown to transiently decrease early after ischemic injury and the activation of a BDNF receptor, TrkB, reversed synaptic impairments induced by ischemia [157]. This evidence implicates BDNF as a potential prophylactic target for the protection of synaptic function and plasticity in the face of future ischemic insult. Studies have also shown that BDNF may function as a post-ischemia therapeutic for synaptic recovery [158].
Another potential target is vascular endothelial growth factor-A (VEGF), which is heavily involved in vascular biology and associated with angiogenesis as well as vascular cell survival. Although its major importance in the brain is during development, VEGF is commonly expressed in the adult brain [159]. VEGF has been shown to be protective against ischemia-induced synaptic plasticity deficits. In a chronic cerebral hypoperfusion (CCH) model, exogenous VEGF was applied prior to ischemia to determine its effects on synaptic plasticity of rat hippocampal CA1 neurons [160]. Exogenous application of VEGF prevented CCH-induced learning and memory deficits and prevented CCH-induced changes in basal synaptic transmission, paired-pulse facilitation, and LTP [160]. The proposed mechanism underlying the protective effect of VEGF on synaptic plasticity processes was the inhibition of ischemia induced-abnormal autophagy, which at the synapse could derail normal neurotransmission [160]. Similarly, another study using a total cerebral ischemia model found improved neuronal viability and cell survival when VEGF was applied exogenously prior to ischemia [161]. The improvement of neuronal viability coincided with preservation of hippocampal CA1 membrane potential, excitability, and spontaneous EPSPs after ischemia [161]. These two studies show VEGF as a potential therapeutic target for ischemic preconditioning in terms of synaptic function and plasticity. Further investigation into VEGF and its effects on synaptic plasticity could afford greater understanding of signaling pathways that regulate plasticity and ischemic tolerance.
The ovarian hormone 17β-oestradiol, estradiol or E2, has also been shown to exert protective effects against neurodegenerative diseases and cognitive decline [162]. The receptors for estradiol, the ERs, are expressed in numerous regions of both the male and female brain. Estradiol has been associated with signaling pathways connected to the regulation and maintenance of synaptic plasticity, including BDNF, in regions of the brain that are responsible for cognitive functions like the prefrontal cortex and hippocampus [163]. Estradiol may be an effective therapeutic target to protect mechanisms of synaptic plasticity from ischemic insult. In a model of transient global forebrain ischemia, aged long-term hormone-deprived female rats were treated with E2 immediately after ischemia [164]. Seven days after the ischemic insult, E2 treatment was able to protect and significantly attenuate ischemia-induced CA1 hippocampal neuronal death [164]. Furthermore, bath application of E2 onto hippocampal brain slices derived from aged long-term hormone-deprived female rats also improved LTP and responses were similar to those obtained from younger hormone-normal rats [164]. Another study investigated the effects of long-term (14 days) treatment with E2 prior to global ischemia induction [165]. They showed that basal transmission at the Schaffer collateral to CA1 synapse was impaired from global ischemia three days post-surgery; however, pretreatment with E2 ameliorated LTP deficits at these synapses [165]. Interestingly, estradiol-induced rescue of LTP required insulin-like growth factor 1 receptor (IGF1R) activation and not the typical ER-α or β receptors [165]. These findings identify estradiol as a prime therapeutic target against CI injury, given its role in signaling pathways associated with synaptic plasticity and neuroprotective effects.
Iron has also been investigated as a therapeutic target for CI. Under normal conditions, iron plays a critical role in neuronal function and activity, and is a necessary element for LTP in the hippocampal CA1 region [166–168]. Of note, iron is a pro-oxidant element, which catalyzes the conversion of hydrogen peroxide into the highly reactive hydroxyl radical via the Fenton reaction [169, 170]. Since the production of hydroxyl free radicals is proportional to the concentration of iron, levels must not exceed certain physiological limits. For this reason, iron homeostasis must be regulated to avoid excessive oxidative stress that may lead to neuronal damage. As it turns out, iron has been found to accumulate in the cell after ischemia due to disturbances in iron homeostasis, thereby contributing to ROS production, neuronal death, and cognitive impairments [169, 171–174]. Thus, treatments that maintain iron homeostasis in the face of ischemic injury may prove to be protective. Not surprisingly, iron-chelating agents have been found to elicit neuroprotective effects against CI [169, 171–175, 176]. For example, the iron-chelating agent, deferoxamine, was able to reduce iron deposition, hydroxyl radical generation, and infarct volume in neonatal rats subjected to hypoxia-ischemia [171]. Indirect approaches have also been implemented. For example, targeting free heme— a hemolytic agent that can exert toxic effects by releasing iron— has been shown to be protective. The administration of hemoplexin (HPX), a scavenger of free heme, was able to protect rats against I/R injury as evidenced by sustained BBB integrity and synaptic plasticity mechanisms [177]. Specifically, hippocampal slices derived from rats treated with HPX were capable of LTP induction 3 days after MCAO compared to the non-treated group, which could not induce LTP [177]. Although the former studies did not investigate the effects of chelating agents on synaptic plasticity following CI, this latter study suggests that preventing iron overload may have beneficial effects.
Physical exercise (PE) has also been implicated as a potential prophylactic intervention against ischemia. Many studies have elucidated a neuroprotective role for exercise training in different animal models of CI [178]. PE may exert these neuroprotective effects through several mechanisms including modulating excitatory signaling. Two studies from the same group found that three to four weeks of pre-ischemic treadmill training significantly reduced infarct size and neurological defects in rats subjected to MCAO [179, 180]. These protective effects were attributed to decreased glutamate release and reduced expression of glutamate receptors [179, 180]. Post-ischemia interventions have also been investigated. Long periods of exercise training following ischemia was found to enhance the induction of learning dependent LTP in the hippocampal CA3 region [181]. Additionally, short periods of moderate treadmill exercise in two different models of CI were found to enhance cognitive function in rats as indicated by improved hippocampal-dependent contextual fear memory [182]. Interestingly, exercise has also been shown to enhance short term plasticity, as indicated by increased PPF, as well as upregulate innervation from regions within the brain that are important for spatial memory and theta rhythm generation [183]. Thus, there is potential for PE to be utilized as a therapeutic intervention to aid cognitive recovery by improving synaptic plasticity and promoting the reorganization of new neuron circuits.
Neuromodulation as a novel approach for treating cognitive dysfunction in CI patients
While pharmacological interventions have shown some promise as potential therapeutic treatments, non-pharmacological interventions have also been investigated. More specifically, electrical neuromodulation has become an attractive therapeutic strategy for ameliorating cognitive and motor symptoms associated with several neurological diseases, including Parkinson’s disease [184], epilepsy [185, 186], AD [187], and TBI [132]. Given that oscillatory patterns in the brain become altered after ischemia [188], electrical stimulation may provide a means by which dysfunctional brain circuitry and plasticity processes can be repaired in order to promote the drive of neural networks.
There are currently several forms of non-invasive neuromodulation techniques under study to improve cognitive functions after CI, including transcranial direct-current stimulation (tDCS) and repetitive transcranial magnetic stimulation (rTMS) [189]. Specifically, rTMS, which utilizes a wire coil to generate a magnetic field that can penetrate through the scalp, induces changes in excitability via LTP-LTD-like mechanisms that may induce plasticity [190]. Current studies utilizing rTMS on CI patients have focused primarily on improved motor outcomes [191–193]; however, potential benefits in cognition may also be probable. A meta-analysis demonstrated that low frequency (≤1 Hz) rTMS over the unaffected hemisphere in post stroke patients with aphasia was effective in improving overall language function [194]. Although the underlying mechanisms involved in observed language recovery by rTMS are unclear, it is thought that rTMS can normalize neural activity in the targeted area by eliciting either an excitatory or, in this case, an inhibitory effect [194]. Although these findings are promising, future studies which investigate multiple aspects of cognition and recruit a larger cohort size are required to confirm rTMS as a beneficial therapy to alleviate cognitive symptoms following CI.
Invasive forms of neuromodulation also exist, such as deep brain stimulation (DBS). DBS requires a pair of stimulating electrodes to be implanted in the brain parenchyma, which are connected to a pulse generator implanted in the chest. Similar to rTMS, DBS can target specific brain regions; however, different parameters (i.e., frequency and voltage strength) may also be customized to the clinical status of the patient. More importantly, the delivery of specific patterns of electrical activity via DBS can also influence oscillatory activity [195]. In a rat model of TBI, DBS at 7.7 Hz in the medial septal nucleus was able to increase hippocampal theta power, which was correlated with improved spatial learning and restored normal object exploration [196]. Interestingly, acute DBS of the fornix area was found to modulate protein expression in the hippocampus [197]. Increases in BDNF and VEGF, as well as synaptic markers, including synaptophysin, were found within 2.5 hours after DBS [197]. Given the importance of these factors and markers in plasticity, DBS in the fornix may improve memory function. In this regard, DBS may improve overall network function where dysregulated synaptic plasticity processes and oscillatory activity are present. Despite these positive outcomes, severe consequences have also been reported following DBS [198], evoking caution when considering this form of treatment in patients. Thus, future studies should investigate all of the potential side effects affiliated with DBS.
CONCLUSIONS
CI has debilitating consequences for patients who suffer cognitive deficits following ischemia. While the exact cause of these impairments are unknown, altered network communication has been implicated as a prime suspect. Neural networks within the brain are subject to alterations when changes in synaptic transmission and circuitry arise. In this review we highlight the importance of synaptic plasticity processes, particularly LTP and LTD, as well as the integrity of intact structural and functional connections within the Papez circuit in cognition and how they are affected following CI. We also emphasize the importance of endogenous oscillatory patterns which drive synchronization between neural ensembles— a mechanism that is vital for efficient network communication among distinct brain regions. We pay particular attention to the hippocampus and other extra-hippocampal components of the Papez circuit in these processes, given their role in episodic memory; however, the importance of other brain regions (i.e., cortex) should also be considered since global network disruptions most likely account for impairments in cognitive processes.
Although we primarily focus on CA and ischemic stroke, it is likely that other neurological diseases and subtypes of stroke may cause or increase the risk of dementia through similar mechanisms. For example, deficits in learning and memory following subarachnoid hemorrhage stroke have been attributed to lesions in the Papez circuit as well as impairments in hippocampal LTP [199–201]. There seems to be a common theme among these neurological diseases that indicates dysfunctional plasticity processes and disturbances in circuity contribute to cognitive impairment.
Thus, treatments and/or therapies that improve synaptic plasticity and functional connectivity within circuits may prove to be useful in terms of alleviating cognitive symptoms. Although we suggest a probable link between dysregulated synaptic plasticity and circuitry causing disrupted network activity after CI, future studies are required to delineate a direct relationship. This may help elucidate the exact mechanisms responsible for cognitive decline and may be applicable to other neurological diseases.
ACKNOWLEDGMENTS
This work was financially supported by the NIH/NINDS grants NS45676, NS097658, NS34773, and AHA/ASA Bugher Foundation 14BFSC17690007.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/18-0875r1).
REFERENCES
[1] | Wahul AB , Joshi PC , Kumar A , Chakravarty S ((2018) ) Transient global cerebral ischemia differentially affects cortex, striatum and hippocampus in Bilateral Common Carotid arterial occlusion (BCCAo) mouse model. J Chem Neuroanat 92: , 1–15. |
[2] | Bachevalier J , Meunier M ((1996) ) Cerebral ischemia: Are the memory deficits associated with hippocampal cell loss? Hippocampus 6: , 553–560. |
[3] | Smith ML , Auer RN , Siesjo BK ((1984) ) The density and distribution of ischemic brain injury in the rat following 2-10min of forebrain ischemia. Acta Neuropathol 64: , 319–332. |
[4] | Neumann JT , Cohan CH , Dave KR , Wright CB , Perez-Pinzon MA ((2013) ) Global cerebral ischemia: Synaptic and cognitive dysfunction. Curr Drug Targets 14: , 20–35. |
[5] | Fisher M , Bastan B ((2012) ) Identifying and utilizing the ischemic penumbra. Neurology 79: , S79–85. |
[6] | Cervos-Navarro J , Diemer NH ((1991) ) Selective vulnerability in brain hypoxia. Crit Rev Neurobiol 6: , 149–182. |
[7] | Martone ME , Jones YZ , Young SJ , Ellisman MH , Zivin JA , Hu BR ((1999) ) Modification of postsynaptic densities after transient cerebral ischemia: A quantitative and three-dimensional ultrastructural study. J Neurosci 19: , 1988–1997. |
[8] | Zarei M , Patenaude B , Damoiseaux J , Morgese C , Smith S , Matthews PM , Barkhof F , Rombouts SA , Sanz-Arigita E , Jenkinson M ((2010) ) Combining shape and connectivity analysis: An MRI study of thalamic degeneration in Alzheimer’s disease. Neuroimage 49: , 1–8. |
[9] | Aggleton JP , Pralus A , Nelson AJ , Hornberger M ((2016) ) Thalamic pathology and memory loss in early Alzheimer’s disease: Moving the focus from the medial temporal lobe to Papez circuit. Brain 139: , 1877–1890. |
[10] | Kase CS , Wolf PA , Kelly-Hayes M , Kannel WB , Beiser A , D’Agostino RB ((1998) ) Intellectual decline after stroke: The Framingham Study. Stroke 29: , 805–812. |
[11] | Kokmen E , Whisnant JP , O’Fallon WM , Chu CP , Beard CM ((1996) ) Dementia after ischemic stroke: A population-based study in Rochester, Minnesota (1960-1984). Neurology 46: , 154–159. |
[12] | Desmond DW , Moroney JT , Sano M , Stern Y ((2002) ) Incidence of dementia after ischemic stroke: Results of a longitudinal study. Stroke 33: , 2254–2260. |
[13] | Corraini P , Henderson VW , Ording AG , Pedersen L , Horvath-Puho E , Sorensen HT ((2017) ) Long-term risk of dementia among survivors of ischemic or hemorrhagic stroke. Stroke 48: , 180–186. |
[14] | Mohan KM , Wolfe CD , Rudd AG , Heuschmann PU , Kolominsky-Rabas PL , Grieve AP ((2011) ) Risk and cumulative risk of stroke recurrence: A systematic review and meta-analysis. Stroke 42: , 1489–1494. |
[15] | Pendlebury ST , Rothwell PM ((2009) ) Prevalence, incidence, and factors associated with pre-stroke and post-stroke dementia: A systematic review and meta-analysis. Lancet Neurol 8: , 1006–1018. |
[16] | Moroney JT , Bagiella E , Tatemichi TK , Paik MC , Stern Y , Desmond DW ((1997) ) Dementia after stroke increases the risk of long-term stroke recurrence. Neurology 48: , 1317–1325. |
[17] | Sibolt G , Curtze S , Melkas S , Putaala J , Pohjasvaara T , Kaste M , Karhunen PJ , Oksala NK , Erkinjuntti T ((2013) ) Poststroke dementia is associated with recurrent ischaemic stroke. J Neurol Neurosurg Psychiatry 84: , 722–726. |
[18] | Desmond DW , Moroney JT , Sano M , Stern Y ((2002) ) Mortality in patients with dementia after ischemic stroke. Neurology 59: , 537–543. |
[19] | Gao CY , Lian Y , Zhang M , Zhang LL , Fang CQ , Deng J , Li J , Xu ZQ , Zhou HD , Wang YJ ((2016) ) Association of dementia with death after ischemic stroke: A two-year prospective study. Exp Ther Med 12: , 1765–1769. |
[20] | Pusswald G , Fertl E , Faltl M , Auff E ((2000) ) Neurological rehabilitation of severely disabled cardiac arrest survivors. Part II. Life situation of patients and families after treatment. Resuscitation 47: , 241–248. |
[21] | Jaszke-Psonka M , Piegza M , Scislo P , Pudlo R , Piegza J , Badura-Brzoza K , Leksowska A , Hese RT , Gorczyca PW ((2016) ) Cognitive impairment after sudden cardiac arrest. Kardiochir Torakochirurgia Pol 13: , 393–398. |
[22] | O’Reilly SM , Grubb NR , O’Carroll RE ((2003) ) In-hospital cardiac arrest leads to chronic memory impairment. Resuscitation 58: , 73–79. |
[23] | Drysdale EE , Grubb NR , Fox KA , O’Carroll RE ((2000) ) Chronicity of memory impairment in long-term out-of-hospital cardiac arrest survivors. Resuscitation 47: , 27–32. |
[24] | Buanes EA , Gramstad A , Sovig KK , Hufthammer KO , Flaatten H , Husby T , Langorgen J , Heltne JK ((2015) ) Cognitive function and health-related quality of life four years after cardiac arrest. Resuscitation 89: , 13–18. |
[25] | Perez CA , Samudra N , Aiyagari V ((2016) ) Cognitive and functional consequence of cardiac arrest. Curr Neurol Neurosci Rep 16: , 70. |
[26] | Grubb NR , O’Carroll R , Cobbe SM , Sirel J , Fox KA ((1996) ) Chronic memory impairment after cardiac arrest outside hospital. BMJ 313: , 143–146. |
[27] | Sulzgruber P , Kliegel A , Wandaller C , Uray T , Losert H , Laggner AN , Sterz F , Kliegel M ((2015) ) Survivors of cardiac arrest with good neurological outcome show considerable impairments of memory functioning. Resuscitation 88: , 120–125. |
[28] | Zhou J , Yu JT , Wang HF , Meng XF , Tan CC , Wang J , Wang C , Tan L ((2015) ) Association between stroke and Alzheimer’s disease: Systematic review and meta-analysis. J Alzheimers Dis 43: , 479–489. |
[29] | De Vos A , Bjerke M , Brouns R , De Roeck N , Jacobs D , Van den Abbeele L , Guldolf K , Zetterberg H , Blennow K , Engelborghs S , Vanmechelen E ((2017) ) Neurogranin and tau in cerebrospinal fluid and plasma of patients with acute ischemic stroke. BMC Neurol 17: , 170. |
[30] | Lasek-Bal A , Jedrzejowska-Szypulka H , Rozycka J , Bal W , Kowalczyk A , Holecki M , Dulawa J , Lewin-Kowalik J ((2016) ) The presence of Tau protein in blood as a potential prognostic factor in stroke patients. J Physiol Pharmacol 67: , 691–696. |
[31] | Liu W , Wong A , Au L , Yang J , Wang Z , Leung EY , Chen S , Ho CL , Mok VC ((2015) ) Influence of amyloid-beta on cognitive decline after stroke/transient ischemic attack: Three-year longitudinal study. Stroke 46: , 3074–3080. |
[32] | Randall J , Mortberg E , Provuncher GK , Fournier DR , Duffy DC , Rubertsson S , Blennow K , Zetterberg H , Wilson DH ((2013) ) Tau proteins in serum predict neurological outcome after hypoxic brain injury from cardiac arrest: Results of a pilot study. Resuscitation 84: , 351–356. |
[33] | Rosen C , Rosen H , Andreasson U , Bremell D , Bremler R , Hagberg L , Rosengren L , Blennow K , Zetterberg H ((2014) ) Cerebrospinal fluid biomarkers in cardiac arrest survivors. Resuscitation 85: , 227–232. |
[34] | Mattsson N , Zetterberg H , Nielsen N , Blennow K , Dankiewicz J , Friberg H , Lilja G , Insel PS , Rylander C , Stammet P , Aneman A , Hassager C , Kjaergaard J , Kuiper M , Pellis T , Wetterslev J , Wise M , Cronberg T ((2017) ) Serum tau and neurological outcome in cardiac arrest. Ann Neurol 82: , 665–675. |
[35] | Wisniewski HM , Maslinska D ((1996) ) Beta-protein immunoreactivity in the human brain after cardiac arrest. Folia Neuropathol 34: , 65–71. |
[36] | Zetterberg H , Mortberg E , Song L , Chang L , Provuncher GK , Patel PP , Ferrell E , Fournier DR , Kan CW , Campbell TG , Meyer R , Rivnak AJ , Pink BA , Minnehan KA , Piech T , Rissin DM , Duffy DC , Rubertsson S , Wilson DH , Blennow K ((2011) ) Hypoxia due to cardiac arrest induces a time-dependent increase in serum amyloid beta levels in humans. PLoS One 6: , e28263. |
[37] | Skaper SD , Facci L , Zusso M , Giusti P ((2017) ) Synaptic plasticity, dementia and Alzheimer disease. CNS Neurol Disord Drug Targets 16: , 220–233. |
[38] | Hamos JE , DeGennaro LJ , Drachman DA ((1989) ) Synaptic loss in Alzheimer’s disease and other dementias. Neurology 39: , 355–361. |
[39] | DeKosky ST , Scheff SW ((1990) ) Synapse loss in frontal cortex biopsies in Alzheimer’s disease: Correlation with cognitive severity. Ann Neurol 27: , 457–464. |
[40] | Wang Z , Yang L , Zheng H ((2012) ) Role of APP and Abeta in synaptic physiology. Curr Alzheimer Res 9: , 217–226. |
[41] | Kamat PK , Kalani A , Rai S , Swarnkar S , Tota S , Nath C , Tyagi N ((2016) ) Mechanism of oxidative stress and synapse dysfunction in the pathogenesis of Alzheimer’s disease: Understanding the therapeutics strategies. Mol Neurobiol 53: , 648–661. |
[42] | Jang SS , Chung HJ ((2016) ) Emerging link between Alzheimer’s disease and homeostatic synaptic plasticity. Neural Plast 2016: , 7969272. |
[43] | Tonnies E , Trushina E ((2017) ) Oxidative stress, synaptic dysfunction, and Alzheimer’s disease. J Alzheimers Dis 57: , 1105–1121. |
[44] | ((1894) ) The Croonian Lecture: The minute structure of the nervous centres. Br Med J 1: , 543. |
[45] | Jones EG ((1994) ) Santiago Ramon y Cajal and the Croonian Lecture, March 1894. Trends Neurosci 17: , 190–192. |
[46] | Hoshiba Y , Wada T , Hayashi-Takagi A ((2017) ) Synaptic ensemble underlying the selection and consolidation of neuronal circuits during learning. Front Neural Circuits 11: , 12. |
[47] | Morris RG ((1999) ) D.O. Hebb: The Organization of Behavior, Wiley: New York; 1949. Brain Res Bull 50: , 437. |
[48] | Pulvermuller F ((1996) ) Hebb’s concept of cell assemblies and the psychophysiology of word processing. Psychophysiology 33: , 317–333. |
[49] | Frankland PW , Bontempi B ((2005) ) The organization of recent and remote memories. Nat Rev Neurosci 6: , 119–130. |
[50] | Willshaw DJ , Buckingham JT ((1990) ) An assessment of Marr’s theory of the hippocampus as a temporary memory store. Philos Trans R Soc Lond B Biol Sci 329: , 205–215. |
[51] | Vann SD , Nelson AJ ((2015) ) The mammillary bodies and memory: More than a hippocampal relay. Prog Brain Res 219: , 163–185. |
[52] | Lynch MA ((2004) ) Long-term potentiation and memory. Physiol Rev 84: , 87–136. |
[53] | Zhang S , Boyd J , Delaney K , Murphy TH ((2005) ) Rapid reversible changes in dendritic spine structure in vivo gated by the degree of ischemia. J Neurosci 25: , 5333–5338. |
[54] | Hofmeijer J , van Putten MJ ((2012) ) Ischemic cerebral damage: An appraisal of synaptic failure. Stroke 43: , 607–615. |
[55] | Whitlock JR , Heynen AJ , Shuler MG , Bear MF ((2006) ) Learning induces long-term potentiation in the hippocampus. Science 313: , 1093–1097. |
[56] | Rioult-Pedotti MS , Friedman D , Donoghue JP ((2000) ) Learning-induced LTP in neocortex. Science 290: , 533–536. |
[57] | Cohen Y , Avramoav S , Barkai E , Maroun M ((2011) ) Olfactory learning-induced enhancement of the predisposition for LTP induction. Learn Mem 18: , 594–597. |
[58] | Li W , Huang R , Shetty RA , Thangthaeng N , Liu R , Chen Z , Sumien N , Rutledge M , Dillon GH , Yuan F , Forster MJ , Simpkins JW , Yang SH ((2013) ) Transient focal cerebral ischemia induces long-term cognitive function deficit in an experimental ischemic stroke model. Neurobiol Dis 59: , 18–25. |
[59] | Kiprianova I , Sandkuhler J , Schwab S , Hoyer S , Spranger M ((1999) ) Brain-derived neurotrophic factor improves long-term potentiation and cognitive functions after transient forebrain ischemia in the rat. Exp Neurol 159: , 511–519. |
[60] | Sopala M , Frankiewicz T , Parsons C , Danysz W ((2000) ) Middle cerebral artery occlusion produces secondary, remote impairment in hippocampal plasticity of rats - involvement of N-methyl-D-aspartate receptors? Neurosci Lett 281: , 143–146. |
[61] | Wang S , Kee N , Preston E , Wojtowicz JM ((2005) ) Electrophysiological correlates of neural plasticity compensating for ischemia-induced damage in the hippocampus. Exp Brain Res 165: , 250–260. |
[62] | McEachern JC , Shaw CA ((1999) ) The plasticity-pathology continuum: Defining a role for the LTP phenomenon. J Neurosci Res 58: , 42–61. |
[63] | Calabresi P , Centonze D , Pisani A , Cupini L , Bernardi G ((2003) ) Synaptic plasticity in the ischaemic brain. Lancet Neurol 2: , 622–629. |
[64] | Crepel V , Hammond C , Chinestra P , Diabira D , Ben-Ari Y ((1993) ) A selective LTP of NMDA receptor-mediated currents induced by anoxia in CA1 hippocampal neurons. J Neurophysiol 70: , 2045–2055. |
[65] | Hammond C , Crepel V , Gozlan H , Ben-Ari Y ((1994) ) Anoxic LTP sheds light on the multiple facets of NMDA receptors. Trends Neurosci 17: , 497–503. |
[66] | Di Filippo M , Tozzi A , Costa C , Belcastro V , Tantucci M , Picconi B , Calabresi P ((2008) ) Plasticity and repair in the post-ischemic brain. Neuropharmacology 55: , 353–362. |
[67] | Hsu KS , Huang CC ((1997) ) Characterization of the anoxia-induced long-term synaptic potentiation in area CA1 of the rat hippocampus. Br J Pharmacol 122: , 671–681. |
[68] | Izumi Y , Katsuki H , Benz AM , Zorumski CF ((1998) ) Oxygen deprivation produces delayed inhibition of long-term potentiation by activation of NMDA receptors and nitric oxide synthase. J Cereb Blood Flow Metab 18: , 97–108. |
[69] | Wang S , Zhang J , Sheng T , Lu W , Miao D ((2015) ) Hippocampal ischemia causes deficits in local field potential and synaptic plasticity. J Biomed Res 29: , 370–379. |
[70] | Stein ES , Itsekson-Hayosh Z , Aronovich A , Reisner Y , Bushi D , Pick CG , Tanne D , Chapman J , Vlachos A , Maggio N ((2015) ) Thrombin induces ischemic LTP (iLTP): Implications for synaptic plasticity in the acute phase of ischemic stroke. Sci Rep 5: , 7912. |
[71] | Herreras O ((2016) ) Local field potentials: Myths and misunderstandings. Front Neural Circuits 10: , 101. |
[72] | Orfila JE , McKinnon N , Moreno M , Deng G , Chalmers N , Dietz RM , Herson PS , Quillinan N ((2018) ) Cardiac arrest induces ischemic long-term potentiation of hippocampal CA1 neurons that occludes physiological long-term potentiation. Neural Plast 2018: , 9275239. |
[73] | Jourdain P , Nikonenko I , Alberi S , Muller D ((2002) ) Remodeling of hippocampal synaptic networks by a brief anoxia-hypoglycemia. J Neurosci 22: , 3108–3116. |
[74] | Lenz M , Vlachos A , Maggio N ((2015) ) Ischemic long-term-potentiation (iLTP): Perspectives to set the threshold of neural plasticity toward therapy. Neural Regen Res 10: , 1537–1539. |
[75] | Crepel V , Epsztein J , Ben-Ari Y ((2003) ) Ischemia induces short- and long-term remodeling of synaptic activity in the hippocampus. J Cell Mol Med 7: , 401–407. |
[76] | Meunier CN , Chameau P , Fossier PM ((2017) ) Modulation of synaptic plasticity in the cortex needs to understand all the players. Front Synaptic Neurosci 9: , 2. |
[77] | Bear MF , Abraham WC ((1996) ) Long-term depression in hippocampus. Annu Rev Neurosci 19: , 437–462. |
[78] | Gao TM , Pulsinelli WA , Xu ZC ((1998) ) Prolonged enhancement and depression of synaptic transmission in CA1 pyramidal neurons induced by transient forebrain ischemia in vivo. Neuroscience 87: , 371–383. |
[79] | Wang F , Han J , Higashimori H , Wang J , Liu J , Tong L , Yang Y , Dong H , Zhang X , Xiong L ((2018) ) Long-term depression induced by endogenous cannabinoids produces neuroprotection via astroglial CB1R after stroke in rodents. J Cereb Blood Flow Metab, 271678X18755661. |
[80] | Rozov A , Burnashev N , Sakmann B , Neher E ((2001) ) Transmitter release modulation by intracellular Ca2+buffers in facilitating and depressing nerve terminals of pyramidal cells in layer 2/3 of the rat neocortex indicates a target cell-specific difference in presynaptic calcium dynamics. J Physiol 531: , 807–826. |
[81] | Dai X , Chen L , Sokabe M ((2007) ) Neurosteroid estradiol rescues ischemia-induced deficit in the long-term potentiation of rat hippocampal CA1 neurons. Neuropharmacology 52: , 1124–1138. |
[82] | Cohan CH , Neumann JT , Dave KR , Alekseyenko A , Binkert M , Stransky K , Lin HW , Barnes CA , Wright CB , Perez-Pinzon MA ((2015) ) Effect of cardiac arrest on cognitive impairment and hippocampal plasticity in middle-aged rats. PLoS One 10: , e0124918. |
[83] | Nunn J , Hodges H ((1994) ) Cognitive deficits induced by global cerebral ischaemia: Relationship to brain damage and reversal by transplants. Behav Brain Res 65: , 1–31. |
[84] | Hewlett KA , Kelly MH , Corbett D ((2014) ) ‘Not-so-minor’ stroke: Lasting psychosocial consequences of anterior cingulate cortical ischemia in the rat. Exp Neurol 261: , 543–550. |
[85] | Marston HM , Everitt BJ , Robbins TW ((1993) ) Comparative effects of excitotoxic lesions of the hippocampus and septum/diagonal band on conditional visual discrimination and spatial learning. Neuropsychologia 31: , 1099–1118. |
[86] | Heuer E , Bachevalier J ((2011) ) Effects of selective neonatal hippocampal lesions on tests of object and spatial recognition memory in monkeys. Behav Neurosci 125: , 137–149. |
[87] | Lee HD , Jang SH ((2014) ) Changes of an injured fornix in a patient with mild traumatic brain injury: Diffusion tensor tractography follow-up study. Brain Inj 28: , 1485–1488. |
[88] | Jang SH , Kwon HG ((2018) ) Injury of the Papez circuit in a patient with traumatic spinal cord injury and concomitant mild traumatic brain injury. Neural Regen Res 13: , 161–162. |
[89] | Yang DS , Kwon HG , Jang SH ((2016) ) Injury of the thalamocingulate tract in the Papez circuit in patients with mild traumatic brain injury. Am J Phys Med Rehabil 95: , e34–38. |
[90] | Uhlhaas PJ , Singer W ((2012) ) Neuronal dynamics and neuropsychiatric disorders: Toward a translational paradigm for dysfunctional large-scale networks. Neuron 75: , 963–980. |
[91] | Bubb EJ , Kinnavane L , Aggleton JP ((2017) ) Hippocampal - diencephalic - cingulate networks for memory and emotion: An anatomical guide. Brain Neurosci Adv 1: , 2398212817723443. |
[92] | Tsivilis D , Vann SD , Denby C , Roberts N , Mayes AR , Montaldi D , Aggleton JP ((2008) ) A disproportionate role for the fornix and mammillary bodies in recall versus recognition memory. Nat Neurosci 11: , 834–842. |
[93] | Vann SD ((2013) ) Dismantling the Papez circuit for memory in rats. Elife 2: , e00736. |
[94] | Vann SD ((2010) ) Re-evaluating the role of the mammillary bodies in memory. Neuropsychologia 48: , 2316–2327. |
[95] | Jones DT , Mateen FJ , Lucchinetti CF , Jack CR Jr , Welker KM ((2011) ) Default mode network disruption secondary to a lesion in the anterior thalamus. Arch Neurol 68: , 242–247. |
[96] | Vincent JL , Snyder AZ , Fox MD , Shannon BJ , Andrews JR , Raichle ME , Buckner RL ((2006) ) Coherent spontaneous activity identifies a hippocampal-parietal memory network. J Neurophysiol 96: , 3517–3531. |
[97] | Raichle ME ((2015) ) The brain’s default mode network. Annu Rev Neurosci 38: , 433–447. |
[98] | Shapira-Lichter I , Oren N , Jacob Y , Gruberger M , Hendler T ((2013) ) Portraying the unique contribution of the default mode network to internally driven mnemonic processes. Proc Natl Acad Sci U S A 110: , 4950–4955. |
[99] | Greicius MD , Krasnow B , Reiss AL , Menon V ((2003) ) Functional connectivity in the resting brain: A network analysis of the default mode hyothesis. Proc Natl Acad Sci U S A 100: , 253–258. |
[100] | Warburton EC , Aggleton JP ((1999) ) Differential deficits in the Morris water maze following cytotoxic lesions of the anterior thalamus and fornix transection. Behav Brain Res 98: , 27–38. |
[101] | Warburton EC , Baird AL , Morgan A , Muir JL , Aggleton JP ((2000) ) Disconnecting hippocampal projections to the anterior thalamus produces deficits on tests of spatial memory in rats. Eur J Neurosci 12: , 1714–1726. |
[102] | Warburton EC , Baird A , Morgan A , Muir JL , Aggleton JP ((2001) ) The conjoint importance of the hippocampus and anterior thalamic nuclei for allocentric spatial learning: Evidence from a disconnection study in the rat. J Neurosci 21: , 7323–7330. |
[103] | Jankowski MM , Ronnqvist KC , Tsanov M , Vann SD , Wright NF , Erichsen JT , Aggleton JP , O’Mara SM ((2013) ) The anterior thalamus provides a subcortical circuit supporting memory and spatial navigation. Front Syst Neurosci 7: , 45. |
[104] | Wolff M , Gibb SJ , Dalrymple-Alford JC ((2006) ) Beyond spatial memory: The anterior thalamus and memory for the temporal order of a sequence of odor cues. J Neurosci 26: , 2907–2913. |
[105] | Aminoff EM , Kveraga K , Bar M ((2013) ) The role of the parahippocampal cortex in cognition. Trends Cogn Sci 17: , 379–390. |
[106] | Weible AP ((2013) ) Remembering to attend: The anterior cingulate cortex and remote memory. Behav Brain Res 245: , 63–75. |
[107] | Alosco ML , Hayes SM ((2015) ) Structural brain alterations in heart failure: A review of the literature and implications for risk of Alzheimer’s disease. Heart Fail Rev 20: , 561–571. |
[108] | Jang SH , Kwon HG ((2016) ) Neural injury of the Papez circuit following hypoxic-ischemic brain injury: A case report. Medicine (Baltimore) 95: , e5173. |
[109] | Horstmann A , Frisch S , Jentzsch RT , Muller K , Villringer A , Schroeter ML ((2010) ) Resuscitating the heart but losing the brain: Brain atrohy in the aftermath of cardiac arrest. Neurology 74: , 306–312. |
[110] | Snaphaan L , de Leeuw FE ((2007) ) Poststroke memory function in nondemented patients: A systematic review on frequency and neuroimaging correlates. Stroke 38: , 198–203. |
[111] | Sestieri C , Corbetta M , Romani GL , Shulman GL ((2011) ) Episodic memory retrieval, parietal cortex, and the default mode network: Functional and topographic analyses. J Neurosci 31: , 4407–4420. |
[112] | Huo L , Li R , Wang P , Zheng Z , Li J ((2018) ) The default mode network supports episodic memory in cognitively unimpaired elderly individuals: Different contributions to immediate recall and delayed recall. Front Aging Neurosci 10: , 6. |
[113] | Tuladhar AM , Snaphaan L , Shumskaya E , Rijpkema M , Fernandez G , Norris DG , de Leeuw FE ((2013) ) Default mode network connectivity in stroke patients. PLoS One 8: , e66556. |
[114] | Liu J , Qin W , Wang H , Zhang J , Xue R , Zhang X , Yu C ((2014) ) Altered spontaneous activity in the default-mode network and cognitive decline in chronic subcortical stroke. J Neurol Sci 347: , 193–198. |
[115] | Liu J , Wang Q , Liu F , Song H , Liang X , Lin Z , Hong W , Yang S , Huang J , Zheng G , Tao J , Chen LD ((2017) ) Altered functional connectivity in patients with post-stroke memory impairment: A resting fMRI study. Exp Ther Med 14: , 1919–1928. |
[116] | Koenig MA , Holt JL , Ernst T , Buchthal SD , Nakagawa K , Stenger VA , Chang L ((2014) ) MRI default mode network connectivity is associated with functional outcome after cardiopulmonary arrest. Neurocrit Care 20: , 348–357. |
[117] | Sair HI , Hannawi Y , Li S , Kornbluth J , Demertzi A , Di Perri C , Chabanne R , Jean B , Benali H , Perlbarg V , Pekar J , Luyt CE , Galanaud D , Velly L , Puybasset L , Laureys S , Caffo B , Stevens RD , Neuroimaging for Coma E, Recovery C ((2018) ) Early functional connectome integrity and 1-year recovery in comatose survivors of cardiac arrest. Radiology 287: , 247–255. |
[118] | Fries P ((2015) ) Rhythms for cognition: Communication through coherence. Neuron 88: , 220–235. |
[119] | Buzsaki G , Draguhn A ((2004) ) Neuronal oscillations in cortical networks. Science 304: , 1926–1929. |
[120] | Uhlhaas PJ , Singer W ((2013) ) High-frequency oscillations and the neurobiology of schizophrenia. Dialogues Clin Neurosci 15: , 301–313. |
[121] | Varela F , Lachaux JP , Rodriguez E , Martinerie J ((2001) ) The brainweb: Phase synchronization and large-scale integration. Nat Rev Neurosci 2: , 229–239. |
[122] | Uhlhaas PJ , Singer W ((2006) ) Neural synchrony in brain disorders: Relevance for cognitive dysfunctions and pathophysiology. Neuron 52: , 155–168. |
[123] | Stella F , Treves A ((2011) ) Associative memory storage and retrieval: Involvement of theta oscillations in hippocampal information processing. Neural Plast 2011: , 683961. |
[124] | Buzsaki G ((2002) ) Theta oscillations in the hippocampus. Neuron 33: , 325–340. |
[125] | Huerta PT , Lisman JE ((1993) ) Heightened synaptic plasticity of hippocampal CA1 neurons during a cholinergically induced rhythmic state. Nature 364: , 723–725. |
[126] | Orr G , Rao G , Houston FP , McNaughton BL , Barnes CA ((2001) ) Hippocampal synaptic plasticity is modulated by theta rhythm in the fascia dentata of adult and aged freely behaving rats. Hippocampus 11: , 647–654. |
[127] | Bikbaev A , Manahan-Vaughan D ((2008) ) Relationship of hippocampal theta and gamma oscillations to potentiation of synaptic transmission. Front Neurosci 2: , 56–63. |
[128] | Montgomery SM , Betancur MI , Buzsaki G ((2009) ) Behavior-dependent coordination of multiple theta dipoles in the hippocampus. J Neurosci 29: , 1381–1394. |
[129] | Kowalczyk T , Bocian R , Konopacki J ((2013) ) The generation of theta rhythm in hippocampal formation maintained in vitro. Eur J Neurosci 37: , 679–699. |
[130] | Colgin LL , Moser EI ((2009) ) Hippocampal theta rhythms follow the beat of their own drum. Nat Neurosci 12: , 1483–1484. |
[131] | Pignatelli M , Beyeler A , Leinekugel X ((2012) ) Neural circuits underlying the generation of theta oscillations. J Physiol Paris 106: , 81–92. |
[132] | Pevzner A , Izadi A , Lee DJ , Shahlaie K , Gurkoff GG ((2016) ) Making waves in the brain: What are oscillations, and why modulating them makes sense for brain injury. Front Syst Neurosci 10: , 30. |
[133] | Goutagny R , Jackson J , Williams S ((2009) ) Self-generated theta oscillations in the hippocampus. Nat Neurosci 12: , 1491–1493. |
[134] | Kocsis B , Vertes RP ((1994) ) Characterization of neurons of the supramammillary nucleus and mammillary body that discharge rhythmically with the hippocampal theta rhythm in the rat. J Neurosci 14: , 7040–7052. |
[135] | Kocsis B , Vertes RP ((1997) ) Phase relations of rhythmic neuronal firing in the supramammillary nucleus and mammillary body to the hippocampal theta activity in urethane anesthetized rats. Hippocampus 7: , 204–214. |
[136] | Vertes RP , Albo Z , Viana Di Prisco G ((2001) ) Theta-rhythmically firing neurons in the anterior thalamus: Implications for mnemonic functions of Papez’s circuit. Neuroscience 104: , 619–625. |
[137] | Colom LV , Christie BR , Bland BH ((1988) ) Cingulate cell discharge patterns related to hippocampal EEG and their modulation by muscarinic and nicotinic agents. Brain Res 460: , 329–338. |
[138] | Monmaur P , Thomson MA , M’Harzi M ((1986) ) Temporal changes in hippocampal theta activity following twenty minutes of forebrain ischemia in the chronic rat. Brain Res 378: , 262–273. |
[139] | Rabinovici GD , Lukatch HS , MacIver MB ((2000) ) Hypoglycemic and hypoxic modulation of cortical micro-EEG activity in rat brain slices. Clin Neurophysiol 111: , 112–121. |
[140] | Brockmann MD , Kukovic M , Schonfeld M , Sedlacik J , Hanganu-Opatz IL ((2013) ) Hypoxia-ischemia disrupts directed interactions within neonatal prefrontal-hippocampal networks. PLoS One 8: , e83074. |
[141] | Roth JM ((2011) ) Recombinant tissue plasminogen activator for the treatment of acute ischemic stroke. Proc (Bayl Univ Med Cent) 24: , 257–259. |
[142] | Cheng NT , Kim AS ((2015) ) Intravenous thrombolysis for acute ischemic stroke within 3 hours versus between 3 and 4.5 hours of symptom onset. Neurohospitalist 5: , 101–109. |
[143] | Nys GM , van Zandvoort MJ , Algra A , Kappelle LJ , de Haan EH ((2006) ) Cognitive and functional outcome after intravenous recombinant tissue plasminogen activator treatment in patients with a first symptomatic brain infarct. J Neurol 253: , 237–241. |
[144] | Evans MRB , White P , Cowley P , Werring DJ ((2017) ) Revolution in acute ischaemic stroke care: A practical guide to mechanical thrombectomy. Pract Neurol 17: , 252–265. |
[145] | Raychev R , Saver JL ((2012) ) Mechanical thrombectomy devices for treatment of stroke. Neurol Clin Pract 2: , 231–235. |
[146] | Cohen DL , Kearney R , Griffiths M , Nadesalingam V , Bathula R ((2015) ) Around 9% of patients with ischaemic stroke are suitable for thrombectomy. BMJ 351: , h4607. |
[147] | Chia NH , Leyden JM , Newbury J , Jannes J , Kleinig TJ ((2016) ) Determining the number of ischemic strokes potentially eligible for endovascular thrombectomy: A population-based study. Stroke 47: , 1377–1380. |
[148] | Fugate JE , Moore SA , Knopman DS , Claassen DO , Wijdicks EF , White RD , Rabinstein AA ((2013) ) Cognitive outcomes of patients undergoing therapeutic hypothermia after cardiac arrest. Neurology 81: , 40–45. |
[149] | Che D , Li L , Kopil CM , Liu Z , Guo W , Neumar RW ((2011) ) Impact of therapeutic hypothermia onset and duration on survival, neurologic function, and neurodegeneration after cardiac arrest. Crit Care Med 39: , 1423–1430. |
[150] | Hypothermia after Cardiac Arrest Study Group ((2002) ) Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med 346: , 549–556. |
[151] | Soleimanpour H , Rahmani F , Golzari SE , Safari S ((2014) ) Main complications of mild induced hypothermia after cardiac arrest: A review article. J Cardiovasc Thorac Res 6: , 1–8. |
[152] | Chan PS , Berg RA , Tang Y , Curtis LH , Spertus JA , American Heart Association’s Get With the Guidelines–Resuscitation Investigators ((2016) ) Association between therapeutic hypothermia and survival after in-hospital cardiac arrest. JAMA 316: , 1375–1382. |
[153] | Leao RN , Avila P , Cavaco R , Germano N , Bento L ((2015) ) Therapeutic hypothermia after cardiac arrest: Outcome predictors. Rev Bras Ter Intensiva 27: , 322–332. |
[154] | Neumann JT , Thompson JW , Raval AP , Cohan CH , Koronowski KB , Perez-Pinzon MA ((2015) ) Increased BDNF protein expression after ischemic or PKC epsilon preconditioning promotes electrophysiologic changes that lead to neuroprotection. J Cereb Blood Flow Metab 35: , 121–130. |
[155] | Morris-Blanco KC , Dave KR , Saul I , Koronowski KB , Stradecki HM , Perez-Pinzon MA ((2016) ) Protein kinase C epsilon promotes cerebral ischemic tolerance via modulation of mitochondrial Sirt5. Sci Rep 6: , 29790. |
[156] | Cohan CH , Stradecki-Cohan HM , Morris-Blanco KC , Khoury N , Koronowski KB , Youbi M , Wright CB , Perez-Pinzon MA ((2017) ) Protein kinase C epsilon delays latency until anoxic depolarization through arc expression and GluR2 internalization. J Cereb Blood Flow Metab 37: , 3774–3788. |
[157] | Dietz RM , Orfila JE , Rodgers KM , Patsos OP , Deng G , Chalmers N , Quillinan N , Traystman RJ , Herson PS ((2018) ) Juvenile cerebral ischemia reveals age-dependent BDNF-TrkB signaling changes: Novel mechanism of recovery and therapeutic intervention. J Cereb Blood Flow Metab 38: , 2223–2235. |
[158] | Chen A , Xiong LJ , Tong Y , Mao M ((2013) ) The neuroprotective roles of BDNF in hypoxic ischemic brain injury. Biomed Rep 1: , 167–176. |
[159] | Licht T , Keshet E ((2013) ) Delineating multiple functions of VEGF-A in the adult brain. Cell Mol Life Sci 70: , 1727–1737. |
[160] | Wang L , Wang J , Wang F , Liu C , Yang X , Yang J , Ming D ((2017) ) VEGF-mediated cognitive and synaptic improvement in chronic cerebral hypoperfusion rats involves autophagy process. Neuromolecular Med 19: , 423–435. |
[161] | Yang J , Yao Y , Chen T , Zhang T ((2014) ) VEGF ameliorates cognitive impairment in in vivo and in vitro ischemia via improving neuronal viability and function. Neuromolecular Med 16: , 376–388. |
[162] | Arevalo MA , Azcoitia I , Garcia-Segura LM ((2015) ) The neuroprotective actions of oestradiol and oestrogen receptors. Nat Rev Neurosci 16: , 17–29. |
[163] | Arevalo MA , Azcoitia I , Gonzalez-Burgos I , Garcia-Segura LM ((2015) ) Signaling mechanisms mediating the regulation of synaptic plasticity and memory by estradiol. Horm Behav 74: , 19–27. |
[164] | Inagaki T , Kaneko N , Zukin RS , Castillo PE , Etgen AM ((2012) ) Estradiol attenuates ischemia-induced death of hippocampal neurons and enhances synaptic transmission in aged, long-term hormone-deprived female rats. PLoS One 7: , e38018 . |
[165] | Takeuchi K , Yang Y , Takayasu Y , Gertner M , Hwang JY , Aromolaran K , Bennett MV , Zukin RS ((2015) ) Estradiol pretreatment ameliorates impaired synaptic plasticity at synapses of insulted CA1 neurons after transient global ischemia. Brain Res 1621: , 222–230. |
[166] | Hidalgo C , Carrasco MA , Munoz P , Nunez MT ((2007) ) A role for reactive oxygen/nitrogen species and iron on neuronal synaptic plasticity. Antioxid Redox Signal 9: , 245–255. |
[167] | Salvador GA , Uranga RM , Giusto NM ((2010) ) Iron and mechanisms of neurotoxicity. Int J Alzheimers Dis 2011: , 720658. |
[168] | Munoz P , Humeres A , Elgueta C , Kirkwood A , Hidalgo C , Nunez MT ((2011) ) Iron mediates N-methyl-D-aspartate receptor-dependent stimulation of calcium-induced pathways and hippocampal synaptic plasticity. J Biol Chem 286: , 13382–13392. |
[169] | Selim MH , Ratan RR ((2004) ) The role of iron neurotoxicity in ischemic stroke. Ageing Res Rev 3: , 345–353. |
[170] | Massaad CA , Klann E ((2011) ) Reactive oxygen species in the regulation of synaptic plasticity and memory. Antioxid Redox Signal 14: , 2013–2054. |
[171] | Lu Q , Harris VA , Rafikov R , Sun X , Kumar S , Black SM ((2015) ) Nitric oxide induces hypoxia ischemic injury in the neonatal brain via the disruption of neuronal iron metabolism. Redox Biol 6: , 112–121. |
[172] | Mehta SH , Webb RC , Ergul A , Tawfik A , Dorrance AM ((2004) ) Neuroprotection by tempol in a model of iron-induced oxidative stress in acute ischemic stroke. Am J Physiol Regul Integr Comp Physiol 286: , R283–288. |
[173] | Li Y , He Y , Guan Q , Liu W , Han H , Nie Z ((2012) ) Disrupted iron metabolism and ensuing oxidative stress may mediate cognitive dysfunction induced by chronic cerebral hypoperfusion. Biol Trace Elem Res 150: , 242–248. |
[174] | Millerot-Serrurot E , Bertrand N , Mossiat C , Faure P , Prigent-Tessier A , Garnier P , Bejot Y , Giroud M , Beley A , Marie C ((2008) ) Temporal changes in free iron levels after brain ischemia Relevance to the timing of iron chelation therapy in stroke. Neurochem Int 52: , 1442–1448. |
[175] | Oubidar M , Boquillon M , Marie C , Schreiber L , Bralet J ((1994) ) Ischemia-induced brain iron delocalization: Effect of iron chelators. Free Radic Biol Med 16: , 861–867. |
[176] | Methy D , Bertrand N , Prigent-Tessier A , Mossiat C , Stanimirovic D , Beley A , Marie C ((2008) ) Beneficial effect of dipyridyl, a liposoluble iron chelator against focal cerebral ischemia: in vivo and in vitro evidence of protection of cerebral endothelial cells. Brain Res 1193: , 136–142. |
[177] | Yang Y , Dong B , Lu J , Wang G , Yu Y ((2018) ) Hemopexin reduces blood-brain barrier injury and protects synaptic plasticity in cerebral ischemic rats by promoting EPCs through the HO-1 pathway. Brain Res 1699: , 177–185. |
[178] | Zhang F , Wu Y , Jia J ((2011) ) Exercise preconditioning and brain ischemic tolerance. Neuroscience 177: , 170–176. |
[179] | Zhang F , Wu Y , Jia J , Hu YS ((2010) ) Pre-ischemic treadmill training induces tolerance to brain ischemia: Involvement of glutamate and ERK1/2. Molecules 15: , 5246–5257. |
[180] | Zhang F , Jia J , Wu Y , Hu Y , Wang Y ((2010) ) The effect of treadmill training pre-exercise on glutamate receptor expression in rats after cerebral ischemia. Int J Mol Sci 11: , 2658–2669. |
[181] | Yu Q , Li X , Wang J , Li Y ((2013) ) Effect of exercise training on long-term potentiation and NMDA receptor channels in rats with cerebral infarction. Exp Ther Med 6: , 1431–1436. |
[182] | Stradecki-Cohan HM , Youbi M , Cohan CH , Saul I , Garvin AA , Perez E , Dave KR , Wright CB , Sacco RL , Perez-Pinzon MA ((2017) ) Physical exercise improves cognitive outcomes in 2 models of transient cerebral ischemia. Stroke 48: , 2306–2309. |
[183] | Vivar C , Peterson BD , van Praag H ((2016) ) Running rewires the neuronal network of adult-born dentate granule cells. Neuroimage 131: , 29–41. |
[184] | Okun MS , Gallo BV , Mandybur G , Jagid J , Foote KD , Revilla FJ , Alterman R , Jankovic J , Simpson R , Junn F , Verhagen L , Arle JE , Ford B , Goodman RR , Stewart RM , Horn S , Baltuch GH , Kopell BH , Marshall F , Peichel D , Pahwa R , Lyons KE , Troster AI , Vitek JL , Tagliati M , Group SDS ((2012) ) Subthalamic deep brain stimulation with a constant-current device in Parkinson’s disease: An open-label randomised controlled trial. Lancet Neurol 11: , 140–149. |
[185] | Nagel SJ , Najm IM ((2009) ) Deep brain stimulation for epilepsy. Neuromodulation 12: , 270–280. |
[186] | Laxpati NG , Kasoff WS , Gross RE ((2014) ) Deep brain stimulation for the treatment of epilepsy: Circuits, targets, and trials. Neurotherapeutics 11: , 508–526. |
[187] | Laxton AW , Tang-Wai DF , McAndrews MP , Zumsteg D , Wennberg R , Keren R , Wherrett J , Naglie G , Hamani C , Smith GS , Lozano AM ((2010) ) A phase I trial of deep brain stimulation of memory circuits in Alzheimer’s disease. Ann Neurol 68: , 521–534. |
[188] | Rabiller G , He JW , Nishijima Y , Wong A , Liu J ((2015) ) Perturbation of brain oscillations after ischemic stroke: A potential biomarker for post-stroke function and therapy. Int J Mol Sci 16: , 25605–25640. |
[189] | Azad TD , Veeravagu A , Steinberg GK ((2016) ) Neurorestoration after stroke. Neurosurg Focus 40: , E2. |
[190] | Klomjai W , Katz R , Lackmy-Vallee A ((2015) ) Basic principles of transcranial magnetic stimulation (TMS) and repetitive TMS (rTMS). Ann Phys Rehabil Med 58: , 208–213. |
[191] | Khedr EM , Etraby AE , Hemeda M , Nasef AM , Razek AA ((2010) ) Long-term effect of repetitive transcranial magnetic stimulation on motor function recovery after acute ischemic stroke. Acta Neurol Scand 121: , 30–37. |
[192] | Simonetta-Moreau M ((2014) ) Non-invasive brain stimulation (NIBS) and motor recovery after stroke. Ann Phys Rehabil Med 57: , 530–542. |
[193] | Kubis N ((2016) ) Non-invasive brain stimulation to enhance post-stroke recovery. Front Neural Circuits 10: , 56. |
[194] | Ren CL , Zhang GF , Xia N , Jin CH , Zhang XH , Hao JF , Guan HB , Tang H , Li JA , Cai DL ((2014) ) Effect of low-frequency rTMS on aphasia in stroke patients: A meta-analysis of randomized controlled trials. PLoS One 9: , e102557. |
[195] | Lu M , Wei X , Loparo KA ((2017) ) Investigating synchronous oscillation and deep brain stimulation treatment in a model of cortico-basal ganglia network. IEEE Trans Neural Syst Rehabil Eng 25: , 1950–1958. |
[196] | Lee DJ , Gurkoff GG , Izadi A , Seidl SE , Echeverri A , Melnik M , Berman RF , Ekstrom AD , Muizelaar JP , Lyeth BG , Shahlaie K ((2015) ) Septohippocampal neuromodulation improves cognition after traumatic brain injury. J Neurotrauma 32: , 1822–1832. |
[197] | Gondard E , Chau HN , Mann A , Tierney TS , Hamani C , Kalia SK , Lozano AM ((2015) ) Rapid modulation of protein expression in the rat hippocampus following deep brain stimulation of the fornix. Brain Stimul 8: , 1058–1064. |
[198] | Wang Y , Liu H , Li P , Wang W ((2016) ) Deep brain stimulation could cause delayed and recurrent cerebral ischemia: A case report. Acta Neurochir (Wien) 158: , 2369–2372. |
[199] | Tariq A , Ai J , Chen G , Sabri M , Jeon H , Shang X , Macdonald RL ((2010) ) Loss of long-term potentiation in the hippocampus after experimental subarachnoid hemorrhage in rats. Neuroscience 165: , 418–426. |
[200] | Jang SH , Yeo SS ((2016) ) Injury of the Papez circuit in a patient with provoked confabulation following subarachnoid hemorrhage: A diffusion tensor tractography study. Acta Neurol Belg 116: , 655–658. |
[201] | Han SM , Wan H , Kudo G , Foltz WD , Vines DC , Green DE , Zoerle T , Tariq A , Brathwaite S , D’Abbondanza J , Ai J , Macdonald RL ((2014) ) Molecular alterations in the hippocampus after experimental subarachnoid hemorrhage. J Cereb Blood Flow Metab 34: , 108–117. |


